 Qian Yang
Qian Yang Gao-Fei Fu3
Gao-Fei Fu3 Zhi-Qiang Wu
Zhi-Qiang Wu Jian-Li Zhao
Jian-Li Zhao Qing-Jun Li
Qing-Jun Li- 1Yunnan Key Laboratory of Plant Reproductive Adaptation and Evolutionary Ecology, Yunnan University, Kunming, China
- 2Laboratory of Ecology and Evolutionary Biology, School of Ecology and Environmental Science, Yunnan University, Kunming, China
- 3Guangdong Laboratory of Lingnan Modern Agriculture, Genome Analysis Laboratory of the Ministry of Agriculture and Rural Affairs, Agricultural Genomics Institute at Shenzhen, Chinese Academy of Agricultural Sciences, Shenzhen, China
Chloroplasts are critical to plant survival and adaptive evolution. The comparison of chloroplast genomes could provide insight into the adaptive evolution of closely related species. To identify potential adaptive evolution in the chloroplast genomes of four montane Zingiberaceae taxa (Cautleya, Roscoea, Rhynchanthus, and Pommereschea) that inhabit distinct habitats in the mountains of Yunnan, China, the nucleotide sequences of 13 complete chloroplast genomes, including five newly sequenced species, were characterized and compared. The five newly sequenced chloroplast genomes (162,878–163,831 bp) possessed typical quadripartite structures, which included a large single copy (LSC) region, a small single copy (SSC) region, and a pair of inverted repeat regions (IRa and IRb), and even though the structure was highly conserved among the 13 taxa, one of the rps19 genes was absent in Cautleya, possibly due to expansion of the LSC region. Positive selection of rpoA and ycf2 suggests that these montane species have experienced adaptive evolution to habitats with different sunlight intensities and that adaptation related to the chloroplast genome has played an important role in the evolution of Zingiberaceae taxa.
Introduction
Even though the chloroplast genome is typically far smaller than most plant nuclear genomes, chloroplasts play a crucial role in plant survival, adaptation, and evolution (Wicke et al., 2011; Gao et al., 2019; Zhao C. et al., 2019; Dopp et al., 2021). In angiosperms, chloroplast genomes typically exhibit a conserved quadripartite structure, which includes two inverted repeat regions (IRs), a small single copy (SSC) region, and a large single copy (LSC) region (Shinozaki et al., 1986), as well as a relatively conserved set of genes, which can be categorized according to their involvement in photosynthesis, transcription, translation, and biosynthesis (Sassenrath-Cole, 1998). Chloroplast genes, usually 110–130, include two sets of four ribosomal RNA genes and 30 tRNA genes, which are capable of interacting with all mRNA codons by wiggle (Rogalski et al., 2008; Sibah et al., 2012). The stable genetic structure of chloroplast genomes facilitates a low mutation rate during evolution, which is uniparental inheritance (most angiosperms are maternally inherited), guaranteeing the stability of the chloroplast genome during evolution. Therefore, the chloroplast genome provides an ideal system for investigating species and genomic evolution (Dong et al., 2013).
The gene content of chloroplast genomes can change to facilitate the adaptation of species to specific habitats or life strategies. For example, the absence of the ndh gene and one of the IR regions in the chloroplast genome of Cassytha (Lauraceae) taxa and the absence of almost all photosynthesis-related genes in Aeginetia indica (Orobanchaceae) are associated with parasitic lifestyles (Song et al., 2017; Chen et al., 2020), and many chloroplasts are absent from the chloroplast genome of Gastrodia elata (Orchidaceae), which is mycoheterotrophic and does not rely on photosynthesis, thereby resulting in a relatively small chloroplast genome (35,326 bp; Yuan et al., 2018). These extreme examples suggest that changes in chloroplast gene content are closely associated with plant adaptation. The gene content, number, and structure of most autotrophic land plant chloroplast genomes are much more conserved. The main manifestation is that some special genes have been subjected to different selection pressures during adaptive evolution. For example, positive selection has been reported to play an important role in driving the functional diversification of CHS genes during the speciation of Quercus (Fagaceae; Yang et al., 2016). However, the adaptive evolution of most angiosperm groups, especially the Zingiberaceae, remains largely unknown.
Variation in chloroplast genomes provides plentiful and specific markers that can be used to resolve phylogenetic relationships at various levels (Wu and Ge, 2012; Li et al., 2019; Zhang R. et al., 2020). Moreover, as chloroplasts are maternally inherited in most angiosperms (Corriveau and Coleman, 1988), its conflict with nuclear phylogenetic relationships can provide insight into speciation processes, such as hybridization and incomplete lineage sorting (Degnan and Rosenberg, 2009; Joly et al., 2009; Petit and Excoffier, 2009). Thus, the comparative analysis of chloroplast genomes can be used to explore the evolution of plants.
Members of the Zingiberaceae are pantropically distributed (Wu and Larsen, 2001; Kress et al., 2002), and the family includes the genera Cautleya, Roscoea, Rhynchanthus, and Pommereschea, which are distributed in the mountains of southern Asia. The origin and evolution of these four genera have been linked to the orogeny caused by the collision of the Indian and Eurasian plates (Zhao et al., 2016), and phylogenetic reconstruction, using both chloroplast and nuclear markers, suggests that Cautleya and Roscoea are sister genera, as are Rhynchanthus and Pommereschea (Kress et al., 2002). Furthermore, field studies have revealed that Cautleya and Rhynchanthus taxa are epiphytic on rocks or tree trunks and inhabit shaded forest understories, whereas Roscoea and Pommereschea taxa are terrestrial and inhabit higher-altitude open habitats on the ground. In terms of morphology, the epiphytic genera (Cautleya and Rhynchanthus) are taller than the terrestrial genera (Roscoea and Pommereschea; Wu and Larsen, 2001; Kress et al., 2002). However, no studies have investigated the adaptive evolution of these genera. Previous studies have suggested that several chloroplast genes in Zingiber and Curcuma of Zingiberaceae, such as clpP, ycf1, ycf2, psbA, psbD, petA, and rbcL, are related to adaptative evolution (Gui et al., 2020; Li et al., 2020).
This study aimed to investigate the hypothesis that two pairs of sister genera (Cautleya vs. Roscoea and Rhynchanthus vs. Pommereschea) have common chloroplast genes associated with adaptive divergence to contrast habitats. Therefore, 13 newly sequenced and previously reported chloroplast genomes from Cautleya, Pommereschea, Rhynchanthus, Hedychium, and Roscoea taxa were collected to (1) analyze the characteristics and genes associated with adaptive evolution of these four montane genera, (2) reconstruct a chloroplast genome-based phylogeny of the Zingiberaceae and compare it with a nuclear marker-based phylogenetic reconstruction, and (3) explore possible adaptive evolution of these four montane genera based on associated chloroplast genes and phylogenies.
Materials and Methods
Sample Collection and Chloroplast Genome Assembly
Fresh leaves were collected from Cautleya gracilis (99.70°E, 24.18°N), Rhynchanthus beesianus (99.50°E, 22.48°N), Pommereschea lackneri (101.23°E, 21.99°N), Hedychium coronarium (planted variety, 102.72°E, 25.05°N), and H. villosum (101.23°E, 21.99°N) in Yunnan, China, and 45G sequence data were generated for each species using the Illumina Hiseq 2500 platform (San Diego, CA, United States). A total of 277,483,161, 691,955,913, 631,731,352, 309,816,484, and 309,816,484 reads were generated for C. gracilis, R. beesianus, P. lackneri, H. coronarium, and H. villosum, respectively. GetOrganelle was used to execute the de novo assembly of the five chloroplast genomes (− R 15 − k 105,121; Jin et al., 2020), and several previously reported chloroplast genomes from the members of the Zingiberaceae were used as references for automatic annotation and manual adjustment, which were performed using GeSeq and DOGMA, respectively (Wyman et al., 2004; Michael et al., 2017). To ensure accuracy, the coding sequences were further confirmed by online BLAST searches in NCBI. Finally, a circular map of each annotated complete chloroplast genome was drawn using Organellar Genome DRAW (Lohse et al., 2007).
Genome Structure and Sequence Variation Analysis
A total of 13 representative chloroplast genomes, including the five newly sequenced ones, were aligned using the Mauve plugin (Darling et al., 2004) in Geneious R8 (Biomatters Ltd., Auckland, New Zealand), with the default parameters to detect inversions and rearrangements. As the chloroplast genome borders of different species typically exhibit varying degrees of contraction and expansion, SC/IR boundary maps and sequence differences were plotted according to the length differences of the four regions and the distribution of related genes.
Even though chloroplast genomes are relatively conserved, structural differences and internal mutations exist between species. To determine the sequence variation of protein-coding genes, we aimed to identify potential DNA barcode genes that may be available in the future. Protein-coding sequences were aligned using MAFFT version 7.308 (Standley, 2013), and genome divergence and variation hotspots were identified using mVISTA (Frazer et al., 2004). Finally, nucleotide diversity (π) was calculated through sliding window analysis using DnaSP version 5 (Librado and Rozas, 2009), with a window length of 600 bp and step size of 50 bp.
Molecular Evolution Analysis
Mean amino acid usage frequency was mapped using Circos version 0.69 (Krzywinski et al., 2009), and amino acids were calculated using Geneious R8 (Biomatters Ltd.). To calculate rates of synonymous (Ks) and non-synonymous (Ka) substitution and their ratio (Ka/Ks), the nucleotide sequences of protein-coding genes shared among the four species (C. gracilis, R. tibetica, P. lackneri, and R. beesianus) were extracted and aligned separately using MAFFT version 7.308. Before calculation, gaps and stop codons between the compared sequences were removed. As the YN model considers sequence evolution characteristics (e.g., transition/transversion ratio and codon usage frequency), it has been used increasingly in molecular evolution research (Yang and Nielsen, 2000; Zeng et al., 2017; Zhang R. T. et al., 2020). Thus, the YN algorithm was chosen in KaKs_calculator (Zhang et al., 2006) to illustrate the Ka/Ks value and perform selective pressure analysis. Genes with evidence of positive selection (Ka/Ks > 1) along each branch were identified using the improved branch-site model in PAML (Yang, 2007). The targeted branch(es) was assigned as the foreground branch and the remains served as background branches (Zhang et al., 2005). Finally, a likelihood ratio test (LRT) was used to compare a model (model = 2, NSsites = 2, omega = 1, fix_omega = 0) of positive selection on the foreground branch with a null model (model = 2, NSsites = 2, omega = 1, fix_omega = 1), where no positive selection occurred on the foreground branch. The LTR and corresponding P values were calculated using the chi-squared module in PAML.
Previous studies have suggested that chloroplast RNA editing can improve transcript stability, contribute to the regulation of chloroplast gene expression, and enable genes to produce multiple protein products, thereby expanding the original genetic information (Hanson et al., 1996). To investigate the role of RNA editing mechanisms in the evolution of the Zingiberaceae, PREP-cp (Mower, 2009) was used to predict RNA editing sites, with a parameter threshold (cutoff value) of 0.8 to ensure prediction accuracy.
Phylogenetic Analysis
The Zingiberaceae phylogeny was reconstructed using the chloroplast genome (whole genome or protein-coding only) and internal transcribed spacer (ITS) sequences. In addition to the five newly sequenced chloroplast genomes, other chloroplast genomes and all ITS sequences were downloaded from the NCBI database (Supplementary Table 1). In total, 47 chloroplast genomes and 54 ITS sequences, which each represented 20 genera were selected and aligned using MAFFT. Sequences from species in the Costaceae and Musaceae were also obtained for use as ingroups and outgroups, respectively. Modeltest version 3.7 (Posada and Crandall, 1998) was used to determine the best fitting model, based on Akaike Information Criterion (AIC) score (David and Buckley, 2004). Maximum-likelihood (ML) phylogenetic analysis was conducted using RAxML version 8 (Alexandros, 2014), with 1,000 bootstrap replicates, and Bayesian inference (BI) analysis was performed using the Markov Chain Monte Carlo (MCMC) algorithm in MrBayes version 3.2 (Ronquist and Huelsenbeck, 2003), with 1,000,000 generations and sampling once every 1,000 generations. The first 25% of trees from all runs were discarded as burn-in, and the remaining trees were used to construct a majority-rule consensus tree.
Results
Chloroplast Genome Characterization and Structure
The five newly sequenced chloroplast genomes (162,878–163,831 bp, 36.0–36.1% GC content) possessed the typical quadripartite structure, including an LSC region (87,918–89,237 bp, 33.8–33.9% GC content), SSC region (15,707–16,720 bp, 29.3–29.6% GC content), and a pair of IR regions (IRa and IRb; 28,994–29838 bp, 41.0–41.4% GC content). Except for C. gracilis, which was missing the rps19 gene, the chloroplast genomes contained 133 genes, including 87 protein-coding genes, eight ribosomal RNA genes, and 38 tRNA genes (Supplementary Table 2). Of the 133 genes, 15 (atpF, petB, petD, ndhA, ndhB, rpoC1, rps16, rpl2, rpl16, trnA-UGC, trnI-GAU, trnV-UAC, trnL-UAA, trnG-UCC, and trnK-UUU) contained a single intron and 3 (rps12, clpP, and ycf3) contained two introns. The annotated complete chloroplast genome sequences were deposited in NCBI (GenBank accession numbers: MW769779–MW769783). Meanwhile, the lengths and GC contents of chloroplast genomes from all Zingiberaceae taxa (13 species and 12 genera) ranged from 161,920 bp (Alpinia pumila) to 164,068 bp (Wurfbainia longiligularis; Figure 1 and Supplementary Figure 1) and from 36.0 to 36.2%, respectively. More specifically, the lengths and GC contents of the LSC regions ranged from 86,982 bp (Curcuma amarissima) to 89,237 bp (C. gracilis) and from 33.7 to 34.0%, whereas those of the SSC regions ranged from 15,317 bp (A. pumila) to 16,720 bp (R. beesianus) and from 29.2 to 30.0%, and those of the IR regions ranged from 28,994 bp (R. beesianus) to 30,117 bp (Stahlianthus involucratus) and from 40.9 to 41.4% (Supplementary Table 3).

Figure 1. Comparison of chloroplast genome structure in Zingiberaceae. IR (inverted repeat), LSC (large single copy) and SSC (small single copy) regions and border genes are indicated.
Moreover, variation at the SC-IR boundary and contraction and expansion were observed (Figure 1). The rpl22 and rps19 were located at the LSC-IRb junction, and ycf1 and ndhF were located at the SSC-IRb junction. In R. beesianus, P. lackneri, H. villosum, and H. coronarium, the rpl22 gene crossed the LSC-IRb boundary, with 52, 41, 53, and 53 bp located in the IRb region, respectively. Interestingly, in C. gracilis, the rps19 gene, which was represented by a copy in both the IRa and IRb regions of the other genomes, was only represented by a single copy in the LSC region. In P. lackneri and S. involucratus, the ndhF gene in crossed the SSC-IRb boundary, with 39 and 14 bp in the IRb region, respectively, the ycf1 gene crossed the SSC-IRa boundary in all 13 chloroplast genomes, with variable sequence lengths in the SSC region. The IRa-LSC boundary was relatively stable, except that the C. gracilis genome lacked an rps19 gene (Figure 1). No gene rearrangements or inversions were observed (Supplementary Figure 2).
Sequence mutates indicated that the chloroplast genomes of Zingiberaceae taxa were highly conserved (Figure 2). The coding regions were more conserved than the non-coding regions, and the IR regions were less variable than the single-copy regions. Four protein-coding regions (psbM, rps12, rpl22, and ycf1), which possessed > 25% variability (Supplementary Figure 3) could be used for DNA barcode research in the future.
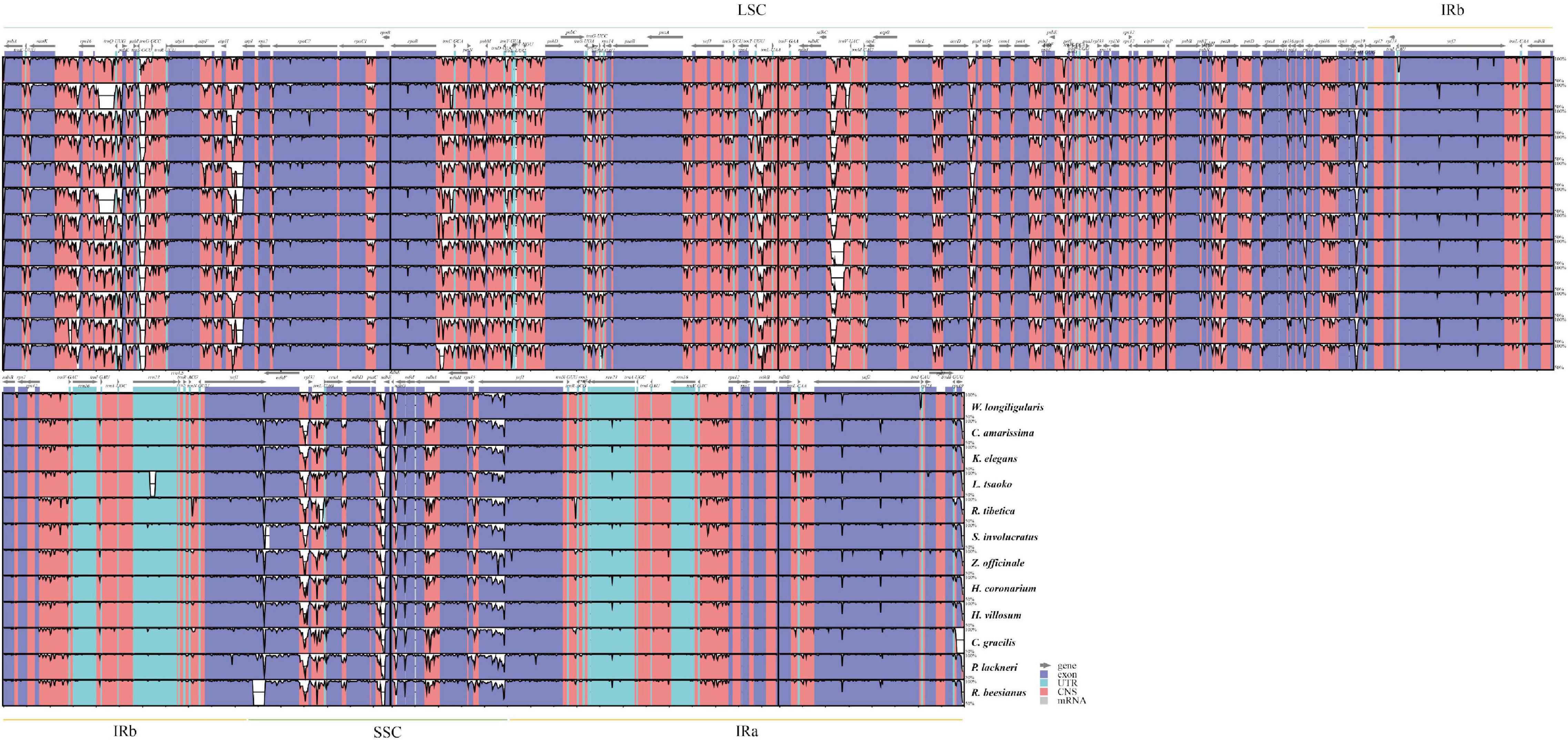
Figure 2. Variation level of the Zingiberaceae chloroplast genome sequences, the y-axis indicates the level of variation (between 50 and 100%) and the x-axis represents the coordinate in the chloroplast genome.
Selection and Evolution of the Protein-Coding Genes
Leucine (10.3%), isoleucine (8.8%), and serine (7.9%) were the most frequently used amino acids, whereas cysteine (1.1%) and tryptophan (1.7%) were the least frequently used amino acids (Supplementary Figure 4 and Supplementary Table 4). The nucleotide diversity of the four montane taxa was ∼0.01 (Supplementary Figure 5).
As some genes yielded Ks values of 0, which resulted in invalid Ka/Ks ratios, only 49 genes were included in the Ka/Ks analysis. KaKs_calculator suggested that four genes (atpF, rpoA, rps15, and ycf2) possessed Ka/Ks ratios of > 1 in at least one pairwise comparison among the four montane taxa (Figure 3). The genes atpF and rpoA were detected in P. lackneri and R. beesianus, respectively, whereas rps15 was detected in R. tibetica and P. lackneri. The gene ycf2 was detected in C. gracilis and R. beesianus. Further verification of the branch-site model revealed that the P-values of the targeted branches (rpoA and ycf2) were significant and retrieved sites under positive selection using the Bayes Empirical Bayes (BEB) method (Supplementary Table 5).
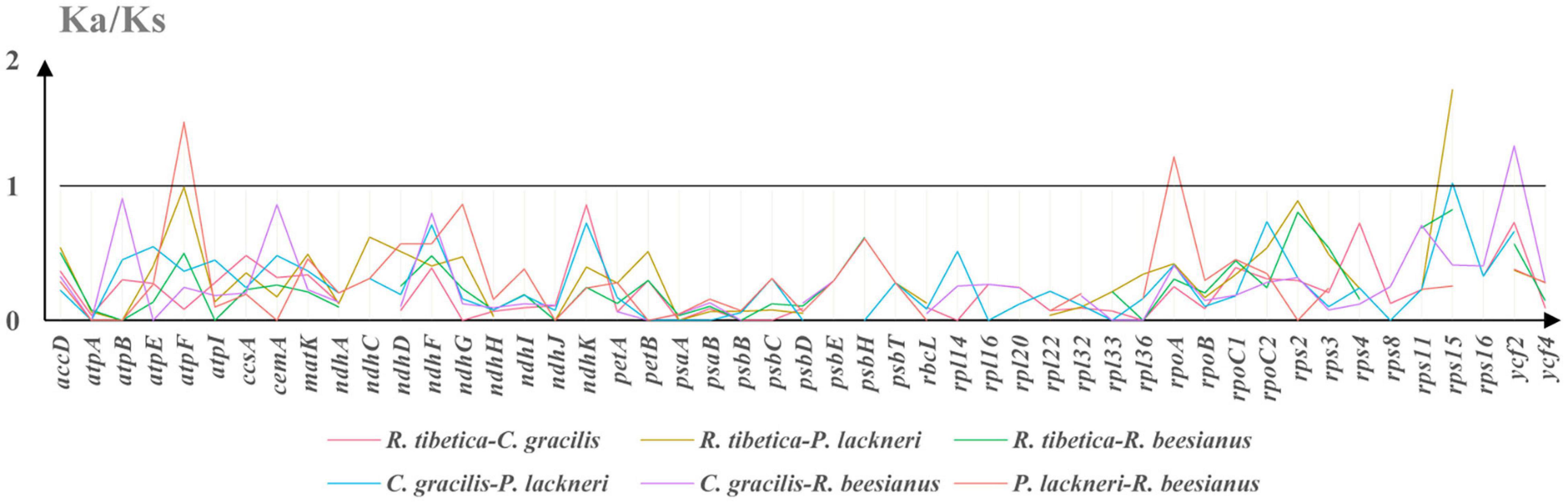
Figure 3. The Ka/Ks ratio of protein-coding genes of four species chloroplast genomes, and Ka/Ks > 1 suggests positive selection.
A total of 76–81 RNA editing sites were predicted in 25–27 genes (Supplementary Table 6). The ndhB gene contained the most predicted editing sites (9–11), which is consistent with findings in other plants, such as rice, maize, and tomato (Freyer et al., 1995). Meanwhile, ndhD contained 7–9 predicted editing sites, whereas ndhF contained 5–7 predicted editing sites, and the other genes contained between 0 and 7 predicted editing sites (ndhA, 4–7; rpoB, accD, 4–5; ycf3, 4; rpoC2, matK, 3–5; rpl20, rpoA, rps14, 3; ndhG, 2–3; petB, rpoC1, 2; atpB, atpI, psbB, rps16, 1–2; atpA, atpF, ccsA, psbF, rps8, 1; clpP, rpl2, rps2, 0–1). All predicted editing sites were C-to-U transitions, and most of the editing sites were predicted to greatly increase protein hydrophobicity but maintain the original function. While maintaining stability, it also provided a basis for adapting to different environments. More work is needed in this area in the future.
Phylogenetic Relationships Analysis
Pommereschea, Rhynchanthus, Cautleya, Roscoea, and Hedychium formed a monophyletic clade in the chloroplast genome tree, with BI support of 0.8, and the taxa were also closely related in the ITS tree (Figure 4). In both trees, the sister relationship of Pommereschea and Rhynchanthus was strongly supported (100% ML support and 1.0 BI support), and Roscoea was closely related to the Pommereschea-Rhynchanthus clade in the chloroplast genome tree, and the sister relationship of Cautleya and Roscoea was strongly supported in the ITS tree (100% ML support and 1.0 BI support).
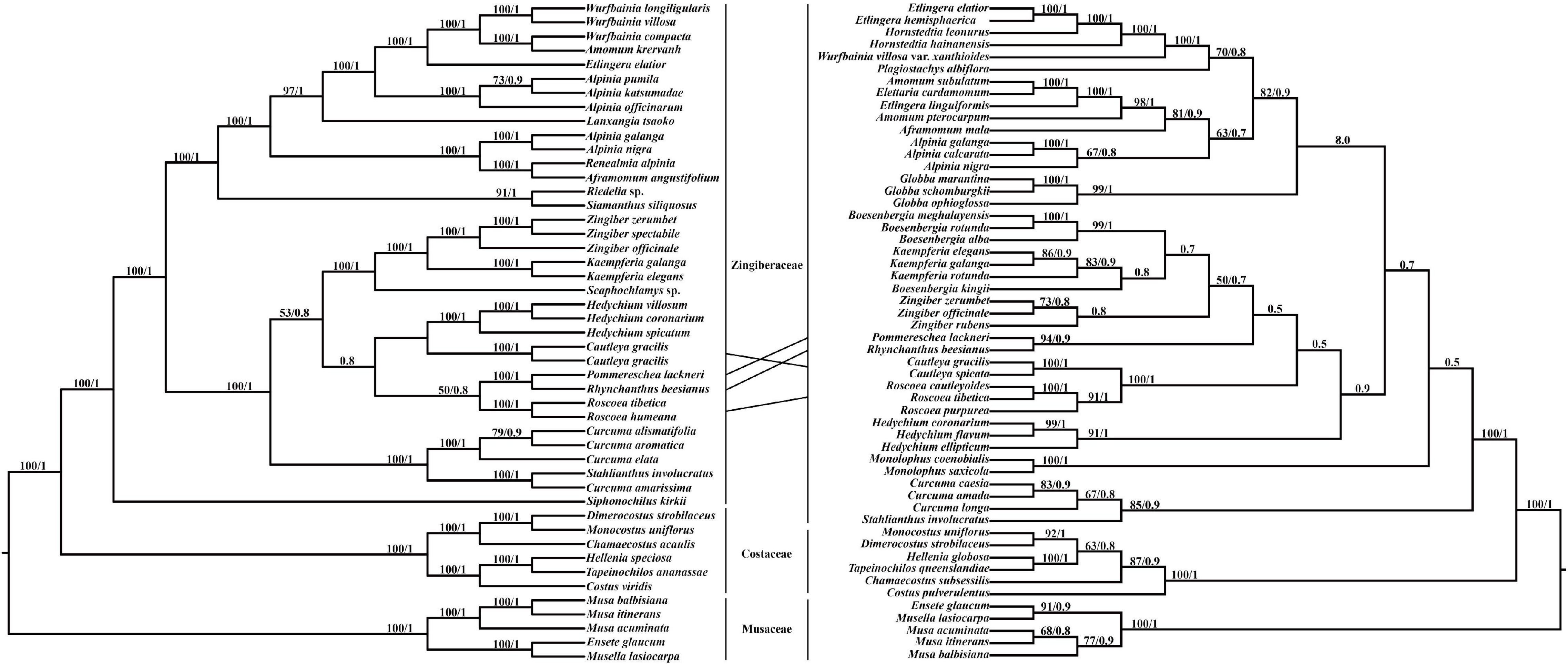
Figure 4. The phylogenetic tree ML (maximum likelihood) and BI (Bayesian Inference) based on 47 complete chloroplast genomes (left) and 54 ITS (internal transcribed spacer) sequences (right). Supporting values of > 50% and > 0.5 for ML and BI, respectively, were shown on the branch.
Discussion
In this study, chloroplast genomes from 13 species (12 genera) in the Zingiberaceae were compared to investigate the sequence structural variation and the evolution of protein-coding genes, and 47 chloroplast genomes and 54 ITS sequences were used to reconstruct phylogenetic relationships among the family. This analysis provided insight into the evolution of montane Zingiberaceae taxa.
Loss of rps19 Copy in Cautleya
Previous studies have reported that the chloroplast genomes of herbaceous plants have undergone rapid evolution, with certain structural changes, such as inversions (Doyle et al., 1992) and gene losses (Takayuki et al., 2004; Saski et al., 2005). No inversions or gene rearrangements were detected in the chloroplast genomes of the Zingiberaceae taxa included in this study. However, although most angiosperms, including most members of the Zingiberaceae, possess two copies of the rps19 gene at the boundaries of the LSC and IR regions (Xu et al., 2015), the Cautleya chloroplast genome only contained a single copy of the rps19 gene in the LSC region. Changes in rps19 genes have been reported in several other genera, including Dianthus (Caryophyllaceae; Raman and Park, 2015), Cardiocrinum (Liliaceae; Lu et al., 2016), Prunus (Rosaceae; Zhao et al., 2019), and Colobanthus (Caryophyllaceae; Androsiuk et al., 2020). However, the changes observed in the rps19 copies of Cautleya were different from those reported in other genera in two respects. First, the rps19 copy in the IRa region of Cautleya was completely lost, whereas those in the IRa regions of other genera were reportedly shortened and pseudogenized. Second, the rps19 gene in the IRb region of Cautleya was located in the LSC region, whereas in other taxa, the rps19 gene remained in the IRb region.
The rps19 protein is a component of the 40S small ribosomal subunit and is essential to both the maturation of the 3′-end of 18S rRNA and the assembly and maturation of pre-40S particles, which are related to chloroplast transcription and translation (Soulet et al., 2001; Matsson et al., 2004). The loss of rps19 has also been observed in a few other dicot taxa (e.g., Morus, Nicotiana, Vitis, and Tetrastigma) but is relatively rare in monocots (Ravi et al., 2006; Li et al., 2015), which suggests that rps19 is more likely to be lost or pseudogenized in dicots. The changes in rps19 could be due to (1) partial gene duplication (Lu et al., 2016; Zhao X. et al., 2019) or (2) the contraction and expansion of IR regions (Zhao X. et al., 2019). It was suggested that there are two evolutionary mechanisms of the IR region boundary: the small amplitude amplification of the boundary gene and the recombination repair of the boundary of the LSC region. The former is an important factor for maintaining the stability of IR regions (Goulding et al., 1996). The expansion and contraction of chloroplast IR regions are relatively common (Hansen et al., 2007). Except for Cautleya, other Zingiberaceae taxa included in this study possessed two complete rps19 copies, which suggests that the presence of two copies is the ancestral state within the Zingiberaceae. Cautleya also possesses the longest LSC region among the included taxa, which suggests that large changes in the rps19 of Cautleya should be the result of LSC region expansion and repair. Previous studies have suggested that rps19 cannot be completely removed from the IRa region through the expansion of LSC or IR regions (Raman and Park, 2015; Lu et al., 2016; Zhao X. et al., 2019; Androsiuk et al., 2020). Therefore, the complete loss of rps19 in Cautleya is more likely than the suppression of rps19 duplication by the LSC region expansion.
Positive Selection of rpoA and ycf2
The rpoA and ycf2 genes are commonly associated with positive selection, which suggests that the chloroplast genomes of Cautleya, Roscoea, Rhynchanthus, and Pommereschea have undergone adaptive evolution. Notable adaptive divergence was noted for rpoA in the chloroplast genomes of the sister genera Rhynchanthus and Pommereschea. The rpoA gene encodes the α subunit of plastid-encoded RNA polymerase, which is responsible for the expression of most genes involved in photosynthesis and is essential for chloroplast gene expression and chloroplast development (Purton and Gray, 1989; Hajdukiewicz et al., 2014; Zhang et al., 2018). The evolution of rpoA is complicated in angiosperms. In the Annonaceae, Passifloraceae, and Geraniaceae, rpoA divergence was caused by structural rearrangement and purifying selection (Blazier et al., 2016). In Passiflora (Passifloraceae), rpoA is subject to either positive or purifying selection, depending on the specific clade (Shrestha et al., 2020). In Rehmannia (Orobanchaceae), rpoA is under positive selection (Zeng et al., 2017). In this study, Rhynchanthus, members of which are typically epiphytic on limestone or tree trunks in forest understories at lower elevations, when compared with Pommereschea. Habitat differentiation, in regard to sunlight exposure, suggests that these sister genera have experienced selection based on the utilization of different light intensities.
In angiosperms, ycf2 is the largest chloroplast gene (Huang et al., 2010) and is subjected to positive or purifying selection (Yan et al., 2019; Zhong et al., 2019). Even though previous studies have suggested that ycf2 has been lost from the chloroplast genomes of monocots (Drescher et al., 2000; Wang et al., 2018; Mishra et al., 2019), two ycf2 copies were present in the chloroplast genomes of the Zingiberaceae taxa included in this study. Furthermore, even though the specific function and role of ycf2 remain unclear, studies have suggested that the gene is not essential to either photosynthesis (Drescher et al., 2000; Zhang Y. et al., 2020) or leaf patterning and is, instead, related to cell survival and possibly ATPase metabolism (Kikuchi et al., 2018; Wang et al., 2018; Zhang et al., 2018; Zhang Y. et al., 2020). The ycf2 gene was also reported to contribute to encoding the 2-MD AAA-ATPase complex, which is a motor protein for generating ATP required for inner membrane translocation (Kikuchi et al., 2018), and to plant cell survival (Drescher et al., 2000). The positive selection of ycf2 suggests that the gene is involved in the adaptive evolution of the montane investigated here.
Phylogenetic Analysis
Even though the chloroplast-based Zingiberaceae phylogeny reconstruction was strongly supported and consistent with previous systematic studies (Kress et al., 2002), the phylogenetic positions of Cautleya and Roscoea in the chloroplast genome and ITS trees were inconsistent. Hybridization and incomplete lineage sorting are the most likely factors to underly phylogenetic conflict between nuclear and chloroplast genome signals (Degnan and Rosenberg, 2009; Joly et al., 2009; Petit and Excoffier, 2009). For example, Roscoea could be a hybrid descendant of Cautleya and the ancestor of Rhynchanthus and Pommereschea (Figure 4). However, incomplete lineage sorting is also possible because incomplete lineage sorting could be present at deeper-divergence lineages in angiosperms (Yang et al., 2020). Either way, this study confirmed the close phylogenetic relationships of the genera Pommereschea, Rhynchanthus, Cautleya, and Roscoea.
Conclusion
This study reports five newly sequenced chloroplast genomes (H. coronarium, H. villosum, C. gracilis, P. lackneri, and R. beesianus). Even though the loss of a rps19 gene in Cautleya may be associated with expansion of the LSC region and positive selection was observed for several genes in the four montane species, the functions of these genes in the adaptive evolution of this group remain unclear. Nevertheless, this study provides an important foundation for further investigation of the adaptive evolution of Pommereschea, Rhynchanthus, Cautleya, and Roscoea.
Data Availability Statement
The original contributions presented in the study are publicly available. This data can be found here: NCBI (GenBank accessions: MW769779-MW769783).
Author Contributions
QY, J-LZ, and Q-JL conceived and designed the study. QY, J-LZ, and LL collected and analyzed the data. QY, G-FF, Z-QW, J-LZ, and Q-JL wrote the manuscript. All authors have directly contributed to this manuscript.
Funding
This work was financially supported by the National Natural Science Foundation of China (Grant Numbers 41871047 and U1602263).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.774482/full#supplementary-material
Supplementary Figure 1 | Gene map of the Zingiberaceae chloroplast genomes. Dashed area in the inner circle indicates the GC content of the chloroplast genome.
Supplementary Figure 2 | MAUVE alignment of Zingiberaceae chloroplast genomes.
Supplementary Figure 3 | Percentage of variable characters in aligned protein-coding regions of the chloroplast genomes.
Supplementary Figure 4 | Average amino acid use frequency of chloroplast genomes in Zingiberaceae.
Supplementary Figure 5 | Comparative analysis of nucleotide diversity in protein-coding regions of four species: (A) R. tibetica-C. gracilis; (B) P. lackneri-R. beesianus; (C) R. tibetica-P. lackneri; (D) R. tibetica-P. lackneri; (E) C. gracilis-P. lackneri; (F) C. gracilis-R. beesianus.
References
Alexandros, S. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 9, 1312–1313. doi: 10.1093/bioinformatics/btu033
Androsiuk, P., Jastrzbski, J. P., Paukszto, U., Makowczenko, K., and Giewanowska, I. (2020). Evolutionary dynamics of the chloroplast genome sequences of six Colobanthus species. Sci. Rep. 10:11522. doi: 10.1038/s41598-020-68563-5
Blazier, J. C., Ruhlman, T. A., Weng, M. L., Rehman, S. K., Sabir, J., and Jansen, R. K. (2016). Divergence of RNA polymerase α subunits in angiosperm plastid genomes is mediated by genomic rearrangement. Sci. Rep. 6:24595. doi: 10.1038/srep24595
Chen, J., Yu, R., Dai, J., Liu, Y., and Zhou, R. (2020). The loss of photosynthesis pathway and genomic locations of the lost plastid genes in a holoparasitic plant Aeginetia indica. BMC Plant Biol. 1:199. doi: 10.1186/s12870-020-02415-2
Corriveau, J. L., and Coleman, A. W. (1988). Rapid screening method to detect potential biparental inheritance of plastid DNA and results for over 200 angiosperm species. Am. J. Bot. 75, 1443– 1458.
Darling, A., Mau, B., Blattner, F. R., and Perna, A. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
David, P., and Buckley, T. R. (2004). Model selection and model averaging in phylogenetics: advantages of akaike information criterion and Bayesian approaches over likelihood ratio tests. Syst. Biol. 53, 793–808. doi: 10.1080/10635150490522304
Degnan, J. H., and Rosenberg, N. A. (2009). Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 24, 332–340. doi: 10.1016/j.tree.2009.01.009
Dong, W., Xu, C., Cheng, T., Lin, K., and Zhou, S. (2013). Sequencing angiosperm plastid genomes made easy: a complete set of universal primers and a case study on the phylogeny of Saxifragales. Genome Biol. Evol. 5, 989–997. doi: 10.1093/gbe/evt063
Dopp, I. J., Yang, X., and Mackenzie, S. A. (2021). A new take on organelle-mediated stress sensing in plants. New Phytol. 230, 2148–2153. doi: 10.1111/nph.17333
Doyle, J. J., Davis, J. I., Soreng, R. J., and Anderson, M. J. (1992). Chloroplast DNA inversions and the origin of the grass family (Poaceae). Proc. Natl. Acad. Sci. USA 89, 7722–7726. doi: 10.1073/pnas.89.16.7722
Drescher, A., Ruf, S., Calsa, T., Carrer, H., and Bock, R. (2000). The two largest chloroplast genome-encoded open reading frames of higher plants are essential genes. Plant J. 22, 97–104. doi: 10.1046/j.1365-313x.2000.00722.x
Frazer, K. A., Lior, P., Alexander, P., Rubin, E. M., and Inna, D. (2004). Vista: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279. doi: 10.1093/nar/gkh458
Freyer, R., López, C., Maier, R. M., Martín, M., and Kssel, H. (1995). Editing of the chloroplast ndhB encoded transcript shows divergence between closely related members of the grass family (Poaceae). Plant Mol. Biol. 29, 679–684. doi: 10.1007/BF00041158
Gao, L. Z., Liu, Y. L., Zhang, D., Li, W., and Eichler, E. E. (2019). Evolution of Oryza chloroplast genomes promoted adaptation to diverse ecological habitats. Commun. Biol. 1:278. doi: 10.1038/s42003-019-0531-2
Goulding, S. E., Wolfe, K. H., Olmstead, R. G., and Morden, C. W. (1996). Ebb and flow of the chloroplast inverted repeat. Mol. General Genet. 252, 195–206. doi: 10.1007/BF02173220
Gui, L., Jiang, S., Xie, D., Yu, L., and Liu, Y. (2020). Analysis of complete chloroplast genomes of Curcuma and the contribution to phylogeny and adaptive evolution. Gene 732:144355. doi: 10.1016/j.gene.2020.144355
Hajdukiewicz, P., Allison, L. A., and Maliga, P. (2014). The two RNA polymerases encoded by the nuclear and the plastid compartments transcribe distinct groups of genes in tobacco plastids. Embo J. 16, 4041–4048. doi: 10.1093/emboj/16.13.4041
Hansen, D. R., Dastidar, S. G., Cai, Z., Penaflor, C., Kuehl, J. V., Boore, J. L., et al. (2007). Phylogenetic and evolutionary implications of complete chloroplast genome sequences of four early-diverging angiosperms: Buxus (Buxaceae), Chloranthus (Chloranthaceae), Dioscorea (Dioscoreaceae), and Illicium (Schisandraceae). Mol. Phylogenet. Evol. 2, 547–563. doi: 10.1016/j.ympev.2007.06.004
Hanson, M. R., Sutton, C., and Lu, B. (1996). Plant organelle gene expression: altered by RNA editing. Trends Plant Sci. 1, 57–64.
Huang, J. L., Sun, G. L., and Zhang, D. M. (2010). Molecular evolution and phylogeny of the angiosperm ycf2 gene. J. Syst. Evol. 48, 240–248. doi: 10.1111/j.1759-6831.2010.00080.x
Jin, J. J., Yu, W. B., Yang, J. B., Song, Y., and Li, D. Z. (2020). Getorganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biol. 21:241. doi: 10.1186/s13059-020-02154-5
Joly, S., Mclenachan, P. A., and Lockhart, P. J. (2009). A statistical approach for distinguishing hybridization and incomplete lineage sorting. Am. Naturalist 174, E54–E70. doi: 10.1086/600082
Kikuchi, S., Asakura, Y., Imai, M., Nakahira, Y., Kotani, Y., Hashiguchi, Y., et al. (2018). A ycf2-FtsHi heteromeric AAA-ATPase complex is required for chloroplast protein import. Plant Cell 30, 2677–2703. doi: 10.1105/tpc.18.00357
Kress, W. J., Prince, L. M., and Williams, K. J. (2002). The phylogeny and a new classification of the gingers (Zingiberaceae): evidence from molecular data. Am. J. Bot. 89, 1682–1696. doi: 10.3732/ajb.89.10.1682
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Li, D. M., Ye, Y. J., Xu, Y. C., Liu, J. M., and Zhu, G. F. (2020). Complete chloroplast genomes of Zingiber montanum and Zingiber zerumbet: genome structure, comparative and phylogenetic analyses. PLoS One 15:e236590. doi: 10.1371/journal.pone.0236590
Li, H. T., Yi, T. S., Gao, L. M., Ma, P. F., Zhang, T., Yang, J. B., et al. (2019). Origin of angiosperms and the puzzle of the Jurassic gap. Nat. Plants 5, 461–470. doi: 10.1038/s41477-019-0421-0
Li, M., Chen, Q., Yang, B. M. J., and Li, B. (2015). The complete chloroplast genome sequence of tetrastigma hemsleyanum Diels at Gilg. Mitochondrial DNA Part A 27, 3729–3730. doi: 10.3109/19401736.2015.1079878
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Lohse, M., Drechsel, O., and Bock, R. (2007). OrganellarGenomeDRAW (OGDRAW): a tool for the easy generation of high-quality custom graphical maps of plastid and mitochondrial genomes. Curr. Genet. 52, 267–274. doi: 10.1007/s00294-007-0161-y
Lu, R. S., Pan, L., and Qiu, Y. X. (2016). The complete chloroplast genomes of three Cardiocrinum (Liliaceae) species: comparative genomic and phylogenetic analyses. Front. Plant Sci. 7:2054. doi: 10.3389/fpls.2016.02054
Matsson, H., Davey, E. J., Draptchinskaia, N., Hamaguchi, I., Ooka, A., Levéen, P., et al. (2004). Targeted disruption of the ribosomal protein s19 gene is lethal prior to implantation. Mol. Cell. Biol. 24, 4032–4037. doi: 10.1128/MCB.24.9.4032-4037.2004
Michael, T., Pascal, L., Tommaso, P., Ulbricht-Jones, E. S., Axel, F., Ralph, B., et al. (2017). GeSeq–versatile and accurate annotation of organelle genomes. Nucleic Acids Res. 45, W6–W11. doi: 10.1093/nar/gkx391
Mishra, L. S., Kati, M., Raik, W., and Christiane, F. (2019). Reduced expression of the proteolytically inactive FtsH members has impacts on the Darwinian fitness of Arabidopsis thaliana. J. Exp. Bot. 70, 2173–2184. doi: 10.1093/jxb/erz004
Mower, J. P. (2009). The PREP suite: predictive RNA editors for plant mitochondrial genes, chloroplast genes and user-defined alignments. Nucleic Acids Res. 37, W253–W259. doi: 10.1093/nar/gkp337
Petit, R. J., and Excoffier, L. (2009). Gene flow and species delimitation. Trends Ecol. Evol. 24, 386–393. doi: 10.1016/j.tree.2009.02.011
Posada, D., and Crandall, K. A. (1998). MODELTEST: testing the model of DNA substitution. Bioinformatics 14, 817–818. doi: 10.1093/bioinformatics/14.9.817
Purton, S., and Gray, J. C. (1989). The plastid rpoA gene encoding a protein homologous to the bacterial RNA polymerase alpha subunit is expressed in pea chloroplasts. Mol. General Genet. 217, 77–84. doi: 10.1007/BF00330945
Raman, G., and Park, S. J. (2015). Analysis of the complete chloroplast genome of a medicinal plant, Dianthus superbus var. longicalyncinus, from a comparative genomics perspective. PLoS One 10:e141329. doi: 10.1371/journal.pone.0141329
Ravi, V., Khurana, J. P., Tyagi, A. K., and Khurana, P. (2006). The chloroplast genome of mulberry: complete nucleotide sequence, gene organization and comparative analysis. Tree Genet. Genomes 3, 49–593.
Rogalski, M., Karcher, D., and Bock, R. (2008). Superwobbling facilitates translation with reduced tRNA sets. Nat. Struct. Mol. Biol. 15, 192–198. doi: 10.1038/nsmb.1370
Ronquist, F., and Huelsenbeck, J. P. (2003). MrBayes 3: Bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Saski, C., Lee, S. B., Daniell, H., Wood, T. C., Tomkins, J., Kim, H. G., et al. (2005). Complete chloroplast genome sequence of Glycine max and comparative analyses with other legume genomes. Plant Mol. Biol. 59, 309–322. doi: 10.1007/s11103-005-8882-0
Shinozaki, K., Ohme, M., Tanaka, M., Wakasugi, T., Hayashida, N., Matsubayashi, T., et al. (1986). The complete nucleotide sequence of the tobacco chloroplast genome: its gene organization and expression. EMBO J. 5, 2043–2049. doi: 10.1002/j.1460-2075.1986.tb04464.x
Shrestha, B., Gilbert, L. E., Ruhlman, T. A., and Jansen, R. K. (2020). Rampant nuclear transfer and substitutions of plastid genes in Passiflora. Genome Biol. Evol. 12, 1313–1329. doi: 10.1093/gbe/evaa123
Sibah, A., Fleischmann, T. T., Scharff, L. B., and Ralph, B. (2012). Evolutionary constraints on the plastid tRNA set decoding methionine and isoleucine. Nucleic Acids Res. 40, 6713–6724. doi: 10.1093/nar/gks350
Song, Y., Yu, W. B., Tan, Y. H., Liu, B., Yao, X., Jin, J. J., et al. (2017). Evolutionary comparisons of the chloroplast genome in Lauraceae and insights into loss events in the magnoliids. Genome Biol. Evol. 9, 2354–2364. doi: 10.1093/gbe/evx180
Soulet, F., Saati, T., Roga, S., Amalric, F., and Bouche, G. (2001). Fibroblast growth factor-2 interacts with free ribosomal protein s19. Biochem. Biophys. Res. Commun. 289, 591–596. doi: 10.1006/bbrc.2001.5960
Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Takayuki, A., Takahiko, T., Sakiko, T., Hiroaki, S., and Koh-Ichi, K. (2004). Complete nucleotide sequence of the sugarcane (Saccharum officinarum) chloroplast genome: a comparative analysis of four monocot chloroplast genomes. DNA Res. 11, 93–99. doi: 10.1093/dnares/11.2.93
Wang, Q., Cui, J., Dai, H., Zhou, Y., Li, N., and Zhang, Z. (2018). Comparative transcriptome profiling of genes and pathways involved in leaf-patterning of Clivia miniata var. variegata. Gene 677, 280–288. doi: 10.1016/j.gene.2018.07.075
Wicke, S., Schneeweiss, G. M., De Pamphilis, C. W., Kai, F. M., and Quandt, D. (2011). The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Wu, T. L., and Larsen, K. (2001). “Zingiberaceae” in Flora of China, eds Z. Y. Wu, P. H. Raven, and D. Y. Hong (Beijing: Science Press and Missouri Botanical Garden Press), 322–377.
Wu, Z. Q., and Ge, S. (2012). The phylogeny of the BEP clade in grasses revisited: evidence from the whole-genome sequences of chloroplasts. Mol. Phylogenet. Evol. 62, 573–578. doi: 10.1016/j.ympev.2011.10.019
Wyman, S., Jansen, R., and Boore, J. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Xu, J., Liu, Q., Hu, W., Wang, T., Xue, Q., and Messing, J. (2015). Dynamics of chloroplast genomes in green plants. Genomics 106, 221–231. doi: 10.1016/j.ygeno.2015.07.004
Yan, C., Du, J., Gao, L., Li, Y., and Hou, X. (2019). The complete chloroplast genome sequence of watercress (Nasturtium officinale R. Br.): genome organization, adaptive evolution and phylogenetic relationships in Cardamineae. Gene 699, 24–36. doi: 10.1016/j.gene.2019.02.075
Yang, L., Su, D., Chang, X., Foster, C., Sun, L., Huang, C., et al. (2020). Phylogenomic insights into deep phylogeny of angiosperms based on broad nuclear gene sampling. Plant Commun. 1:100027. doi: 10.1016/j.xplc.2020.100027
Yang, Y., Zhou, T., Duan, D., Yang, J., Feng, L., and Zhao, G. (2016). Comparative analysis of the complete chloroplast genomes of five Quercus species. Front. Plant Sci. 7:959. doi: 10.3389/fpls.2016.00959
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yang, Z., and Nielsen, R. (2000). Estimating synonymous and nonsynonymous substitution rates under realistic evolutionary models. Mol. Biol. Evol. 17, 32–43. doi: 10.1093/oxfordjournals.molbev.a026236
Yuan, Y., Jin, X., Liu, J., Zhao, X., Zhou, J., Wang, X., et al. (2018). The Gastrodia elata genome provides insights into plant adaptation to heterotrophy. Nat. Commun. 9:1615. doi: 10.1038/s41467-018-03423-5
Zeng, S., Tao, Z., Han, K., Yang, Y., Zhao, J., and Liu, Z. L. (2017). The complete chloroplast genome sequences of six Rehmannia species. Genes 8:103. doi: 10.3390/genes8030103
Zhang, J., Nielsen, R., and Yang, Z. (2005). Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 22, 2472–2479. doi: 10.1093/molbev/msi237
Zhang, R., Wang, Y. H., Jin, J. J., Stull, G. W., Anne, B., Domingos, C., et al. (2020). Exploration of plastid phylogenomic conflict yields new insights into the deep relationships of Leguminosae. Syst. Biol. 69, 613–622. doi: 10.1093/sysbio/syaa013
Zhang, R. T., Xu, B., Li, J., Zhao, Z., Han, J., Lei, Y., et al. (2020). Transit from autotrophism to heterotrophism: sequence variation and evolution of chloroplast genomes in Orobanchaceae species. Front. Genet. 11:542017. doi: 10.3389/fgene.2020.542017
Zhang, Y., An, D., Li, C., Zhao, Z., and Wang, W. (2020). The complete chloroplast genome of greater duckweed (Spirodela polyrhiza 7498) using PacBio long reads: insights into the chloroplast evolution and transcription regulation. BMC Genom. 21:76. doi: 10.1186/s12864-020-6499-y
Zhang, Y., Cui, Y., Zhang, X., Yu, Q., Wang, X., Yuan, X., et al. (2018). A nuclear-encoded protein, mTERF6, mediates transcription termination of rpoA polycistron for plastid-encoded RNA polymerase-dependent chloroplast gene expression and chloroplast development. Sci. Rep. 8:11929. doi: 10.1038/s41598-018-30166-6
Zhang, Z., Li, J., Zhao, X. Q., Wang, J., Wong, K. S., and Yu, J. (2006). KaKs_calculator: calculating Ka and Ks through model selection and model averaging. Genom. Proteomics Bioinform. 4, 259–263. doi: 10.1016/s1672-0229(07)60007-2
Zhao, C., Wang, Y., Chan, K. X., Marchant, D. B., Franks, P. J., Randall, D., et al. (2019). Evolution of chloroplast retrograde signaling facilitates green plant adaptation to land. Proc. Natl. Acad. Sci. U.S.A. 116, 5015–5020. doi: 10.1073/pnas.1812092116
Zhao, J. L., Xia, Y. M., Cannon, C. H., Kress, W. J., and Li, Q. J. (2016). Evolutionary diversification of alpine ginger reflects the early uplift of the Himalayan–Tibetan Plateau and rapid extrusion of Indochina. Gondwana Res. 32, 232–241. doi: 10.1016/j.gr.2015.02.004
Zhao, X., Yan, M., Yu, D., Huo, Y., and Yuan, Z. (2019). Characterization and comparative analysis of the complete chloroplast genome sequence from Prunus avium ‘summit’. Peerj 7:e821010. doi: 10.7717/peerj.8210
Keywords: adaptive evolution, chloroplast genome, gene loss, genomic variation, Zingiberaceae
Citation: Yang Q, Fu G-F, Wu Z-Q, Li L, Zhao J-L and Li Q-J (2022) Chloroplast Genome Evolution in Four Montane Zingiberaceae Taxa in China. Front. Plant Sci. 12:774482. doi: 10.3389/fpls.2021.774482
Received: 12 September 2021; Accepted: 08 November 2021;
Published: 10 January 2022.
Edited by:
Sonia Garcia, Consejo Superior de Investigaciones Científicas, Spanish National Research Council (CSIC), SpainReviewed by:
Jinming Chen, Wuhan Botanical Garden, Chinese Academy of Sciences (CAS), ChinaSunil Kumar Sahu, Beijing Genomics Institute (BGI), China
Copyright © 2022 Yang, Fu, Wu, Li, Zhao and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jian-Li Zhao, amlhbmxpLnpoYW9AeW51LmVkdS5jbg==