
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 24 November 2021
Sec. Plant Systematics and Evolution
Volume 12 - 2021 | https://doi.org/10.3389/fpls.2021.767478
This article is part of the Research TopicPhylogenomic Discordance in Plant SystematicsView all 15 articles
 Jeffrey P. Rose1,2*
Jeffrey P. Rose1,2* Ricardo Kriebel2
Ricardo Kriebel2 Larissa Kahan2Alexa DiNicola2Jesús G. González-Gallegos3
Larissa Kahan2Alexa DiNicola2Jesús G. González-Gallegos3 Ferhat Celep4
Ferhat Celep4 Emily M. Lemmon5
Emily M. Lemmon5 Alan R. Lemmon6
Alan R. Lemmon6 Kenneth J. Sytsma2
Kenneth J. Sytsma2 Bryan T. Drew1
Bryan T. Drew1Next-generation sequencing technologies have facilitated new phylogenomic approaches to help clarify previously intractable relationships while simultaneously highlighting the pervasive nature of incongruence within and among genomes that can complicate definitive taxonomic conclusions. Salvia L., with ∼1,000 species, makes up nearly 15% of the species diversity in the mint family and has attracted great interest from biologists across subdisciplines. Despite the great progress that has been achieved in discerning the placement of Salvia within Lamiaceae and in clarifying its infrageneric relationships through plastid, nuclear ribosomal, and nuclear single-copy genes, the incomplete resolution has left open major questions regarding the phylogenetic relationships among and within the subgenera, as well as to what extent the infrageneric relationships differ across genomes. We expanded a previously published anchored hybrid enrichment dataset of 35 exemplars of Salvia to 179 terminals. We also reconstructed nearly complete plastomes for these samples from off-target reads. We used these data to examine the concordance and discordance among the nuclear loci and between the nuclear and plastid genomes in detail, elucidating both broad-scale and species-level relationships within Salvia. We found that despite the widespread gene tree discordance, nuclear phylogenies reconstructed using concatenated, coalescent, and network-based approaches recover a common backbone topology. Moreover, all subgenera, except for Audibertia, are strongly supported as monophyletic in all analyses. The plastome genealogy is largely resolved and is congruent with the nuclear backbone. However, multiple analyses suggest that incomplete lineage sorting does not fully explain the gene tree discordance. Instead, horizontal gene flow has been important in both the deep and more recent history of Salvia. Our results provide a robust species tree of Salvia across phylogenetic scales and genomes. Future comparative analyses in the genus will need to account for the impacts of hybridization/introgression and incomplete lineage sorting in topology and divergence time estimation.
It has long been recognized that when generating multilocus nucleotide sequence data, different datasets may generate alternative gene tree topologies and, by extension, differing hypotheses of relationships among species (Pamilo and Nei, 1988; Rieseberg and Soltis, 1991; Maddison, 1997). The underlying causes for why such differing gene tree topologies may exist (apart from analytical artifacts) have been well-discussed in the literature, and include gene duplication, incomplete lineage sorting (ILS), lateral gene transfer, and introgression/hybridization (Degnan, 2018). These processes are not mutually exclusive, and the history of one locus may be shaped by multiple processes. For many years, the solution to deal with such discordance was to analyze incongruent datasets separately, attempt to reconcile these topologies into a consensus tree, or concatenate all loci together to generate a “total-evidence” hypothesis of the species relationships (Ané et al., 2007). The concept of genomic concordance, coupled with new methods for estimating species trees while taking into account ILS and/or horizontal gene flow have been important advances in the field of systematic biology (Ané et al., 2007; Baum, 2007; Heled and Drummond, 2009; Mirarab et al., 2014; Yu and Nakhleh, 2015; Edwards et al., 2016; Solís-Lemus and Ané, 2016).
Contemporaneous with these computational advances, new sequencing technologies have facilitated relatively easy and cost-effective sequencing of complete organellar genomes and hundreds to thousands of nuclear loci. The confluence of these two areas of biology has made for an exciting time for studies in systematic biology but has also presented challenges as to how to best analyze these datasets. For example, individual loci may have relatively little phylogenetic information and thus, confound analyses that rely on individual gene trees. In addition, the computational ability of many current algorithms are challenged by the number of terminals present in the species tree and especially the phylogenetic network that a researcher wishes to estimate (Hejase and Liu, 2016; Solís-Lemus and Ané, 2016; Rose et al., 2021). Despite these challenges, an ever-increasing proportion of phylogenomic studies employ methods that account for sources of intra-genomic discordance, especially due to ILS. While methods that employ the multispecies coalescent only are relatively fast and tractable on datasets with dozens to hundreds of terminals, it is increasingly clear that hybridization and introgression are important processes at both shallow and deep phylogenetic scales, and this affects all branches of the Tree of Life (Folk et al., 2018). If horizontal gene flow has been operative, the species tree estimated by methods that only account for ILS may differ substantially from the “true” species tree not only topologically, but also in branch lengths (Leaché et al., 2014). The misestimation of both properties may impact myriad downstream analyses.
Apart from discordance among nuclear loci, gene trees may differ among genomes. This phenomenon is well-known and often referred to as “cytonuclear discordance” (Rieseberg and Soltis, 1991). In plants, this is best demonstrated in cases of putative “chloroplast capture” which have been documented for decades (e.g., Smith and Sytsma, 1990). Such discordance has generally been taken as evidence of horizontal gene flow, even though organellar genomes are also susceptible to ILS, albeit with a much faster expected time to coalescence, relative to nuclear loci. Simulation studies have generally confirmed that most cases of chloroplast (technically plastid) capture are indeed best explained by horizontal gene flow, rather than ILS (Folk et al., 2017; Morales-Briones et al., 2018; Rose et al., 2021).
Therefore, a better understanding of the evolutionary history of clades requires an assessment of the contribution of each of the multiple processes responsible for the discordance among loci. This assessment is important not only for producing a robust phylogenetic hypothesis, but also for selecting methods, taxa, and loci appropriate for the downstream analyses of trait evolution, historical biogeography, and diversification rates. Robust phylogenetic hypotheses are also crucial for making informed decisions to ensure an accurate and stable taxonomic circumscription, from the species level to higher-level classifications.
Sage and its relatives (Salvia L.) comprise ∼1,000 species, with a subcosmopolitan distribution across a diversity of habitat types (Kriebel et al., 2019). It is the largest genus within the mint family (Lamiaceae) and one of the largest genera of plants. There are three broadly defined centers of diversity of Salvia (Walker et al., 2004): East Asia (∼100 spp.; subgenus (subg.) Glutinaria; Hu et al., 2018), the Mediterranean (∼250 spp.; subg. Salvia, Sclarea), and especially, Mexico, Central, and South America (∼580 spp., subg. Calosphace; González-Gallegos et al., 2020). Salvia is not only of interest from an economic perspective, given its culinary use (e.g., chia: S. hispanica L.; rosemary: S. rosmarinus (L.) Spenn.; sage: S. officinalis L.), but also in its horticultural importance (e.g., blue sage: S. nemorosa L.; pineapple sage: S. elegans Vahl; Russian sage: S. yangii B.T.Drew).
Salvia is florally diverse (Kriebel et al., 2019, 2020) and easily characterized by the presence of two stamens with an elongate (or swollen) anther connective, in addition to several micromorphological synapomorphies (Drew et al., 2017). In many species of Salvia, the connective has been variously modified – possibly multiple times – into a staminal lever mechanism to facilitate effective pollination (Claßen-Bockhoff et al., 2003, 2004; Walker and Sytsma, 2007; Wester and Claßen-Bockhoff, 2007; Celep et al., 2020).
As a result of its practical importance to humans, distribution, size and taxonomic complexity, and unique pollination biology, Salvia has received considerable attention from systematists and pollination biologists. Early in the study of the phylogenetic placement of Salvia, it was realized that the genus was polyphyletic or broadly paraphyletic, with several smaller genera embedded within it (Walker and Sytsma, 2007). Subsequent phylogenetic analyses have confirmed that five previously recognized small genera (Dorystaechas Boiss. & Heldr., Meriandra Benth., Perovskia Kar., Rosmarinus L., and Zhumeria Rech.f. & Wendelbo) are nested within several clades of Salvia (Walker and Sytsma, 2007; Drew and Sytsma, 2012; Will and Claßen-Bockhoff, 2014, 2017; Drew et al., 2017). To accommodate these small genera, Drew et al. (2017) and Kriebel et al. (2019) presented an expanded concept of Salvia, recognizing a total of 11 subgenera, although their informal circumscription of subg. “Heterosphace” represents a geographically diverse assemblage of lineages.
To date, most phylogenetic studies of Salvia have relied on plastid or nuclear ribosomal external transcribed spacer (ETS) and especially internal transcribed spacer (ITS) sequences (Walker and Sytsma, 2007; Jenks et al., 2013; Will and Claßen-Bockhoff, 2014, 2017; Dizkirici et al., 2015; Walker et al., 2015; Fragoso-Martínez et al., 2018; Hu et al., 2018). The resolution at multiple phylogenetic scales with these markers is variable by clade and, while there has been evidence for several deeper-level clades along the backbone of Salvia, relationships among them have usually either not been resolved or well supported. Discordance among plastid and nuclear ribosomal loci is generally found but not well-discussed or quantified (but see Walker et al., 2015). In cases where relationships differ across studies and marker sets, it is not clear if the differences are due to a true discordance in genealogical history or are from errors in the phylogenetic estimation (cf. Will and Claßen-Bockhoff, 2017: Figure 1). Drew et al. (2017) further investigated the backbone relationships in Salvia using two low-copy nuclear loci. While several key nodes remained unresolved and there was a clear conflict between the loci, they found an increased resolution for the backbone relationships. More recently, Zhao et al. (2020) used complete plastomes from seven of 11 subgenera and recovered a nearly fully resolved backbone across Salvia except for uncertainty in the placements of subg. Perovskia and Rosmarinus.
Multilocus nuclear datasets from anchored hybrid enrichment (AHE) have been successfully used to resolve deep and shallow level relationships across multiple angiosperm lineages, including Salvia (Fragoso-Martínez et al., 2017; Kriebel et al., 2019). Previously, we presented a species tree of 35 Salvia exemplars from 10 of 11 subgenera based on 316 nuclear loci using concatenation and one coalescent method (Kriebel et al., 2019). While this topology is congruent with the plastome phylogeny of Zhao et al. (2020) in areas where the two studies overlap in subgeneric sampling, several factors bear further consideration in Kriebel et al. (2019). First, the branching order of subg. “Heterosphace”, Salvia, and Sclarea is not fully supported. Second, the monophyly of subg. Audibertia is not fully supported. Third, within subg. Calosphace, section (sect.) Axillares was recovered as a sister to the “Hastatae clade”. instead of sister to all other Calosphace, in conflict with several previous studies (Jenks et al., 2013; Drew et al., 2017; Fragoso-Martínez et al., 2018). The placement of sect. Axillares has important implications for understanding character evolution in subg. Calosphace (e.g., Fragoso-Martínez et al., 2018; Kriebel et al., 2019, 2020, 2021).
Given the relatively sparse sampling of Salvia diversity from previous phylogenomic analyses, as well as the limited exploration of any discordance surrounding the backbone relationships in Salvia, we aimed to sample the species diversity in Salvia better using AHE to fulfill several goals. (1) Fully resolve the backbone of Salvia and assess the monophyly of the subgenera, quantifying discordance and accounting for both ILS and horizontal gene flow. (2) Generate a species tree for a much broader species-level sampling of Salvia, testing the efficacy of the AHE data for resolving shallow-level relationships. (3) Examine cytonuclear discordance at multiple phylogenetic scales by mining off-target organellar reads.
In total, the analyses consisted of 190 samples, including 179 Salvia and all subgenera recognized by Kriebel et al. (2019) except for the small subg. Meriandra (Benth.) J.B.Walker, B.T.Drew & J.G.Gonz lez. For ease of discussion, we will refer to the “Heterosphace” clade as a subgenus, although we acknowledge that this is not a formally named taxon. Within the subtribe Salviinae, we sampled the clade sister to Salvia (six species of Lepechinia and one species of Melissa: Drew and Sytsma, 2011, 2012, 2013). The ultimate outgroup consisted of samples from all the remaining subtribes of Mentheae, which represents a monophyletic group (Drew and Sytsma, 2012): Lycopinae (Lycopus uniflorus Michx.), Menthinae (Clinopodium mexicanum (Benth.) Govaerts), Nepetinae (Agastache pallida (Lindl.) Cory), and Prunellinae (Prunella vulgaris L.).
Total DNA was extracted from the silica gel-dried or fresh leaf tissue using a DNeasy Plant Mini Kit (Qiagen, Valencia, CA, United States). The DNA concentrations were verified using a Qubit® 2.0 Fluorometer (Life Technologies, Eugene, OR, United States). We used the AHE method (Lemmon and Lemmon, 2012). As the samples were sequenced across several years of study, we enriched them using slightly different probe sets (Supplementary Data Sheet S1). The samples sequenced early on in our studies utilized a generic angiosperm kit that targets 517 loci (Buddenhagen et al., 2016; Mitchell et al., 2017) or the Salvia-specific probes utilized in Kriebel et al. (2019), designed using the genome skimming of several Salvia species. The Salvia-specific probes targeted the same regions as the generic angiosperm kit, only neighboring exons could be combined and target regions extended, resulting in a target of 291 moderately conserved, low copy nuclear loci and their variable flanks. The library preparation, enrichment, assembly, and alignment of nuclear loci were performed at the Florida State University Center for Anchored Phylogenomics1 and are described in detail in Kriebel et al. (2019). Because of the large number of samples, a substantial number of loci were lost during the orthology assessment, resulting in 123 recovered loci. Nine additional loci were lost during trimming and masking due to excessive missing data, resulting in a final dataset of 114 loci.
To examine the monophyly of the subgenera (when represented by multiple samples) and assess shallow-level relationships, we assembled species trees using all accessions. First, we concatenated all loci and generated a maximum likelihood species tree in RAxML v.8.2.11 (Stamatakis, 2014) under GTR + Γ. We assessed the branch support with 500 rapid bootstrap (BS) replicates.
Second, we generated a species tree under the multispecies coalescent using ASTRAL-III (Zhang et al., 2018). Using a batch Perl script, we generated individual gene trees using RAxML under GTR + Γ, assessing the branch support for each locus with 100 rapid BS replicates. We analyzed all the maximum likelihood gene trees in ASTRAL, measuring the branch support in two ways: by using the RAxML BS trees as input to ASTRAL with 100 replicates, and also by calculating the ASTRAL local posterior probability (LPP) (Sayyari and Mirarab, 2016) for each quadripartition.
To detail the gene tree conflict/support for each clade in the species tree, we used Phyparts (Smith et al., 2015). Phyparts takes an estimate of a species tree and set of rooted gene trees and provides four numbers for each clade: the number of loci supporting a clade, the number of loci supporting the main conflicting clade, the number of loci supporting all other conflicting clades, and the number of loci without information for a relationship. Trees were optimally rooted with our outgroups outside of Salviinae, but in cases where these were missing, we rooted trees with Melissa and/or Lepechinia. Gene trees that contained only Salvia were excluded from the Phyparts analysis. Note that rooting with Salviinae may inflate the gene tree support for the monophyly of Salvia and possibly also show misleading support or conflict for the relationships among Lepechinia, Melissa, and Salvia. However, we allowed this potentially incorrect rooting because our chief interest was in the relationships within Salvia. To mitigate the effects of uncertainty in the gene tree estimation providing artificial conflict (or support) for clades, we collapsed the branches in each gene tree with <33% BS.
We further examined the patterns of gene tree discordance to test if the observed gene tree discordance across Salvia and its constituent subgenera are consistent with the expectation under ILS alone. To do this, we first generated 1,000 gene trees under the multispecies coalescent using the treesim.contained_coalescent function in DendroPy v.4.5.2 (Sukumaran and Holder, 2010) using the ASTRAL species tree as the “true” tree. To compare the observed discordance with what would be expected under ILS, we measured the pairwise tree distance of each gene tree (expected and observed) from the ASTRAL species tree using three metrics, considering branching order alone and ignoring branch lengths: the Robinson–Foulds distance (RF) (Robinson and Foulds, 1981), the method proposed by Nye et al. (2006), and the clustering information (CI) metric proposed by Smith (2020a). Calculations were made on complete gene trees or gene trees pruned to the subgenus of interest, as appropriate. Since some of the observed gene trees were missing terminals, we only used observed gene trees which contained >75% of all terminals in the clade of interest. Gene tree distances were calculated using the “TreeDist” package in R (Smith, 2020b), collapsing all the branches in the observed gene trees with <33% BS. The distances were normalized so that they ranged from 0 to 1, with 0 indicating complete agreement between the gene tree and species tree. We tested for mean differences observed in the gene tree discordance among clades using a one-way ANOVA with post hoc testing using the Tukey Test with the aov and glht functions in the “stats” and “multcomp” (Hothorn et al., 2008) R packages, respectively. We tested for differences in the mean gene tree discordance between observed and expected gene trees using a two-tailed Welch’s t-test.
Our second nuclear dataset investigated deeper phylogenetic relationships in Salvia, accounting for both ILS and horizontal gene flow. Because the existing methods for inferring phylogenetic networks are computationally demanding for datasets with more than several dozen terminals, we constructed a dataset of one representative for each subgenus. For each subgenus placeholder, we selected the sample with the greatest number of captured loci, and in the case of ties, the total number of aligned bp. We did not allow any missing data, yielding a matrix of 57 loci for 10 species of Salvia plus Lepechinia chamaedryoides (Balb.) Epling as the outgroup. To reconstruct the phylogenetic networks, we first generated concordance factors for each possible quartet. Using a batch script, we ran MrBayes v.3.2.6 (Ronquist et al., 2012) to find the best gene tree for each locus. The gene trees were inferred under GTR + I + Γ using three runs of three chains each for five million generations each with sampling every 5,000 generations with a chain temperature of 0.4, swap frequency of 500 generations, and a 30% burnin. Following the MrBayes analysis, a Bayesian concordance analysis on the posterior sample of gene trees was conducted in BUCKy v.1.4.4 (Ané et al., 2007; Larget et al., 2010) with 100,000 post-burnin generations and the amount of a piori discordance among loci set to the default of 1. This analysis calculates all possible quartets and prunes on the MrBayes gene trees to all but the four terminals of interest. Then, BUCKy is run on each pruned gene tree to generate a table of all quartet concordance factors (CFs) and their SEs. Using these CFs, we generated a preliminary population tree using Quartet MaxCut (Snir and Rao, 2012).
Using the BUCKy CFs and the Quartet MaxCut tree, we calculated a phylogenetic network with the SNaQ function in the Julia package PhyloNetworks (Solís-Lemus and Ané, 2016; Solís-Lemus et al., 2017). This package uses maximum pseudo-likelihood to fit a network while also accounting for ILS. PhyloNetworks considers quartet topologies only and does not take into account information from branch lengths in individual gene trees. Furthermore, PhyloNetworks assumes a level-1 network: a network where each hybrid node only has one lineage transferring genetic material horizontally. We first tested the fit of models allowing from 0–5 reticulation events (h) and compared the models using their pseudo-likelihood score. The best network model was selected by examining at which value of h the pseudo-likelihood score plateaus, following the recommendation of Solís-Lemus et al. (2017). For each value of h, we selected the best network over 30 search replicates. We examined the branch support on the best phylogenetic network using the bootsnaq function with 50 runs of 10 replicates each.
We assembled the nearly complete plastomes of the Salviinae samples by mapping the off-target reads to previously published plastomes of Salvia for the ingroup or Melissa for the outgroup Salviinae. The assembly of the plastomes was conducted in Geneious v.10.2.3 (Kearse et al., 2012), following the procedure of Rose et al. (2021). For outgroup Salviinae, we used the whole plastome sequence of Melissa yunnanensis C.Y.Wu & Y.C.Huang (GenBank accession MT634148.1) as a reference. For Salvia, we constructed a “super” reference sequence based on the strict consensus of 18 GenBank plastomes (Supplementary Data Sheet S2) aligned with MAFFT v.7.023b (Katoh and Standley, 2013) under default parameters.
We used Geneious to map all the forward and reverse reads from our sequences by first trimming all raw reads, and then assembling them to the appropriate reference using an iterative refinement of up to five times with the default Geneious mapper and medium sensitivity. Consensus sequences were generated using the strict consensus approach. If the coverage for a particular site was <7, the consensus nucleotide was scored as a gap. Unmapped regions were treated as missing data and reads mapped to multiple positions were excluded from consensus calculations. Newly generated plastomes were aligned with the aforementioned GenBank sequences using MAFFT with default parameters. Ambiguously aligned regions were removed manually and were generally distinguished by putative inversions, repeat regions, an abundance of gaps, and/or uncertain base calls.
A plastome tree was inferred in RAxML under GTR + Γ with 500 rapid BS replicates. As described above in Section “Nuclear Dataset 1: Gene Tree Distances”, we measured the tree-to-tree distances between the entire plastome tree and its subclades to the ASTRAL species tree.
The aligned locus length for the 114 loci ranged from 105–3,671 bp, with a mean length of 1,133 bp. The samples contained sequence data for an average of 96.25 loci, with most locus dropout in the non-Salviinae outgroups. We were able to extract the majority of the plastome, with aligned plastomes totaling 157,683 bp.
We were able to root 101 of the 114 gene trees. Species trees resulting from concatenation and accounting for ILS with ASTRAL are completely congruent in the major backbone relationships in Salvia, although support for these relationships sometimes varies across the approach and support metrics (Figure 1 and Supplementary Figures S1–S4). The ASTRAL normalized quartet score, or proportion of the gene tree quartet trees satisfied by the species tree, is 0.91, suggesting a clear underlying topology despite some discordance. The monophyly of Salvia is strongly supported by all measures of support (ASTRAL LPP = 1.0/ASTRAL BS = 100/concatenated BS = 100). In addition, the monophyly of each subgenus for which we had multiple samples is strongly supported by BS/LPP and by the vast majority of loci, with two exceptions. First, subg. Heterosphace, while unambiguously supported by measures of statistical support (ASTRAL LPP = 1.0/ASTRAL BS = 100/concatenated BS = 100), has 23/84 (27%) informative loci conflicting its monophyly. Second and more strikingly, the monophyly of subg. Audibertia is poorly supported by ASTRAL LPP (0.69) with more loci conflicting its monophyly than supporting it (55/73, 75%). However, its monophyly is more strongly supported by the other metrics (ASTRAL BS = 91/concatenated BS > 99), although both sections of subg. Audibertia: sects. Audibertia (S. columbariae Benth., S. mellifera Greene, S. munzii Epling) and Echinosphace (S. californica Brandegee, S. funerea M.E. Jones) are more strongly supported as monophyletic.
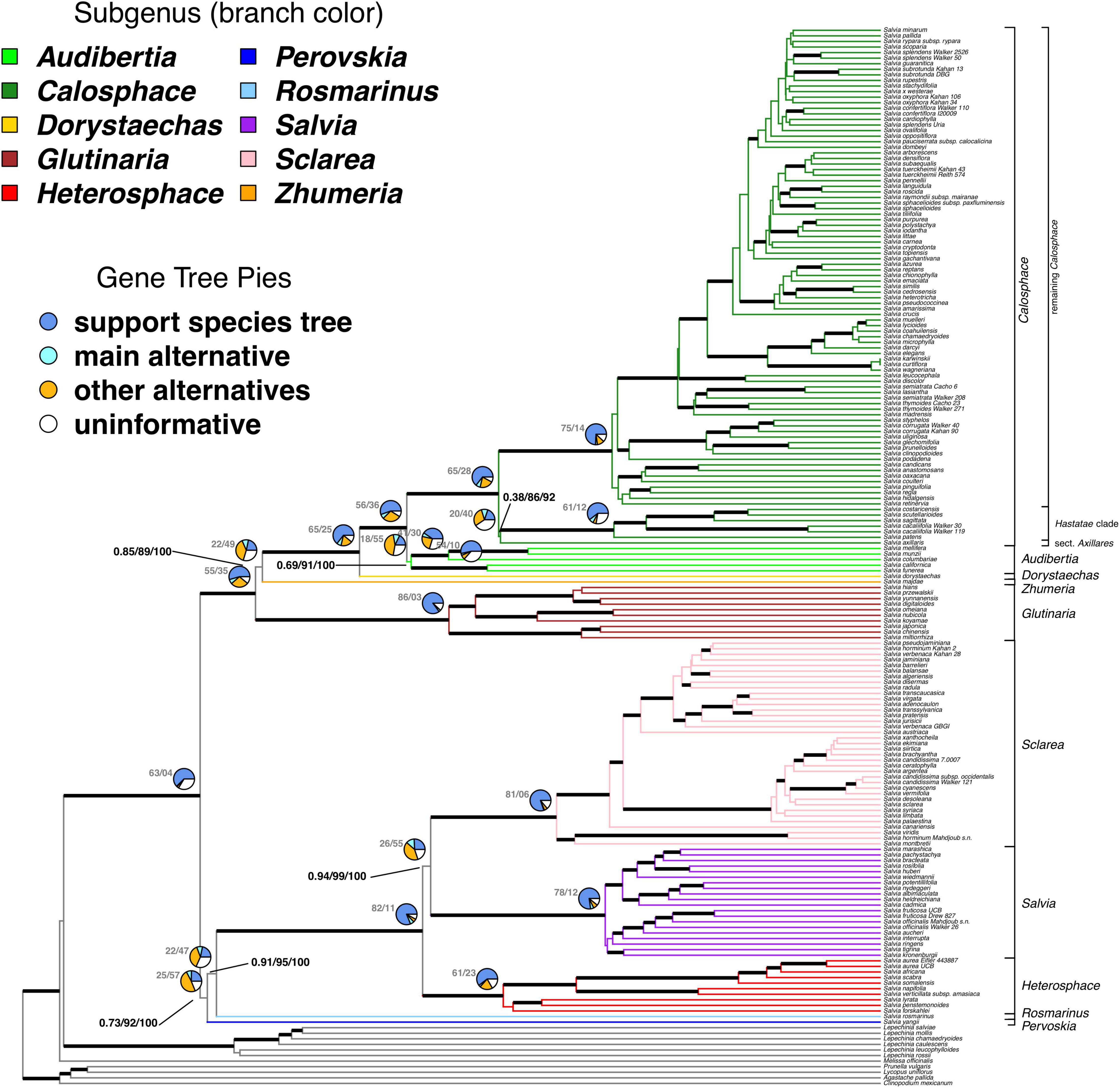
Figure 1. The ASTRAL species tree of Salvia and outgroups. The ingroup branches are colored by subgenus and the subgenera are also labeled to the right. The major clades as discussed in the text are also indicated for subg. Calosphace. Thickened branches denote those with >0.95 ASTRAL local posterior probability. Pies at major nodes summarize the percentage of various phylogenetic signals across 101 gene trees which can be rooted. The numbers at the left of the pies show the total number of gene trees in which the clade is found, followed by the total number of gene trees that conflict with that clade. The remainder of the gene trees, if any, do not provide information on that particular relationship. The numbers at selected, incompletely supported nodes show the ASTRAL local posterior probability followed by the ASTRAL bootstrap support and the bootstrap support from the concatenated maximum likelihood analysis, summarized on the ASTRAL species tree. For support across all branches, see Supplementary Figures S1, S2, S4. For support on the best-scoring maximum likelihood tree, see Supplementary Figure S3.
The earliest divergence in Salvia involves two major clades. First is a clade formed by the most recent common ancestor (MRCA) of subg. Glutinaria and Calosphace (ASTRAL LPP = 1.0/ASTRAL BS = 100/concatenated BS = 100). Subgenus Glutinaria is sister to all remaining subgenera, with a grade formed by the successive sisters of subg. Zhumeria and Dorystaechas, and subg. Audibertia, sister to subg. Calosphace. All of these relationships are strongly supported with the exception of the placement of subg. Glutinaria (ASTRAL LPP = 0.85/ASTRAL BS = 89/concatenated BS > 99; 22/71 informative gene trees).
The second major clade is formed by the MRCA of subg. Perovskia and Salvia. The monophyly of this clade is not fully supported (ASTRAL LPP = 0.73/ASTRAL BS = 92/concatenated BS = 100; 25/82 informative gene trees), nor are many of the intersubgeneric relationships within it (Figure 1). Subgenus Perovskia is sister to subg. Rosmarinus + Heterosphace + Salvia + Sclarea (ASTRAL LPP = 0.91/ASTRAL BS = 95/concatenated BS = 100; 22/69 informative gene trees). While the monophyly of subg. Heterosphace + Salvia + Sclarea is fully supported, relationships among the subgenera are slightly less certain, with subg. Salvia sister to Sclarea being the best resolution of relationships (ASTRAL LPP = 0.94/ASTRAL BS = 99/concatenated BS = 100; 26/81 informative gene trees).
The backbone of the plastome tree is nearly identical to that of the nuclear species trees (Supplementary Figure S5), with all major nodes and monophyly of the subgenera receiving maximal support except for the placement of subg. Glutinaria (BS = 89). The only major topological difference is that subg. Perovskia is weakly supported as sister to Rosmarinus (BS = 47).
Within subg. Calosphace, our nuclear data suggest that sect. Axillares is sister to the Hastatae clade, but with strongly conflicting support (ASTRAL LPP = 0.38/ASTRAL BS = 86/concatenated BS = 92; 20/60 informative gene trees), while our plastid data place sect. Axillares as sister to all other Calosphace (BS = 100). There is also uncertainty about the deepest divergences in subg. Salvia, with weak support based on the ASTRAL and concatenated analyses.
Overall, there is fairly strong support (>90% support across all metrics) for many shallow-scale relationships, but support is notably very low or non-existent for some ASTRAL clades which do not appear in the best tree in the concatenated analysis or are in the low frequency in the BS replicates (Supplementary Figure S6), especially within the radiation of core Calosphace (e.g., relationships among S. chamaedryoides Cav., S. coahuilensis Fern., S. microphylla Kunth, and S. muelleri Epling), subg. Salvia (e.g., if S. officinalis s.s. is monophyletic or not), and subg. Sclarea (e.g., the placement of S. sclarea L.). There is a much more obvious infrageneric gene tree conflict between the nuclear loci and the plastome, with many shallower relationships conflicting between the two datasets, especially in subg. Calosphace, Salvia, and Sclarea (Figure 2).
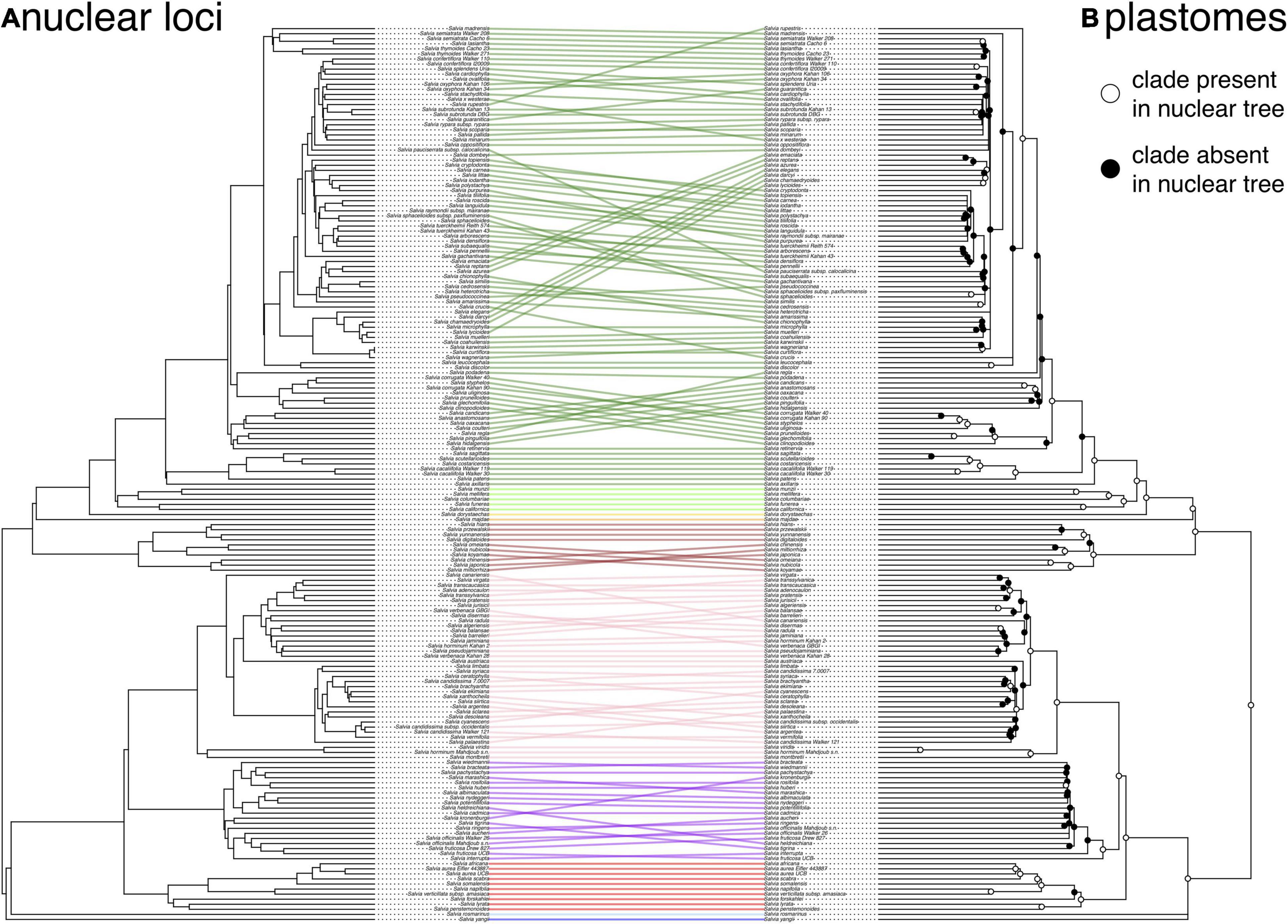
Figure 2. Tanglegram illustrating the cytonuclear discordance in Salvia based on the nuclear ASTRAL species tree (A) and the plastome tree (B). Links connect identical tips, with nodes rotated to minimize the link overlap. Ingroup links are colored by subgenus. Clades that differ between the two trees are indicated by filled circles on the plastome tree. To minimize the discordance caused by the error in the gene tree estimation, clades in the plastome tree with <75% bootstrap support have been collapsed into polytomies.
Gene tree distances demonstrate significant gene tree conflicts within subgenera, both within and across genomic compartments (Table 1), and there are also significant differences in the gene tree distances among the subgenera for all three metrics (RF: F5,454 = 145.7, p < 0.0001; Nye: F5,454 = 98.6, p < 0.0001; CI: F5,454 = 92.4, p < 0.0001). Post hoc testing suggests that compared with the ASTRAL species tree, subg. Glutinaria and Heterosphace have significantly less discordant nuclear gene trees on average than all other subgenera, while subg. Salvia is more discordant than Calosphace for the Nye and CI metrics, and subg. Sclarea is more discordant than Calosphace for CI alone (Table 1). Likewise, although they are only point estimates, the discordance between the plastome and the ASTRAL tree is elevated for subg. Sclarea and especially, Salvia relative to all other subgenera (Table 1).
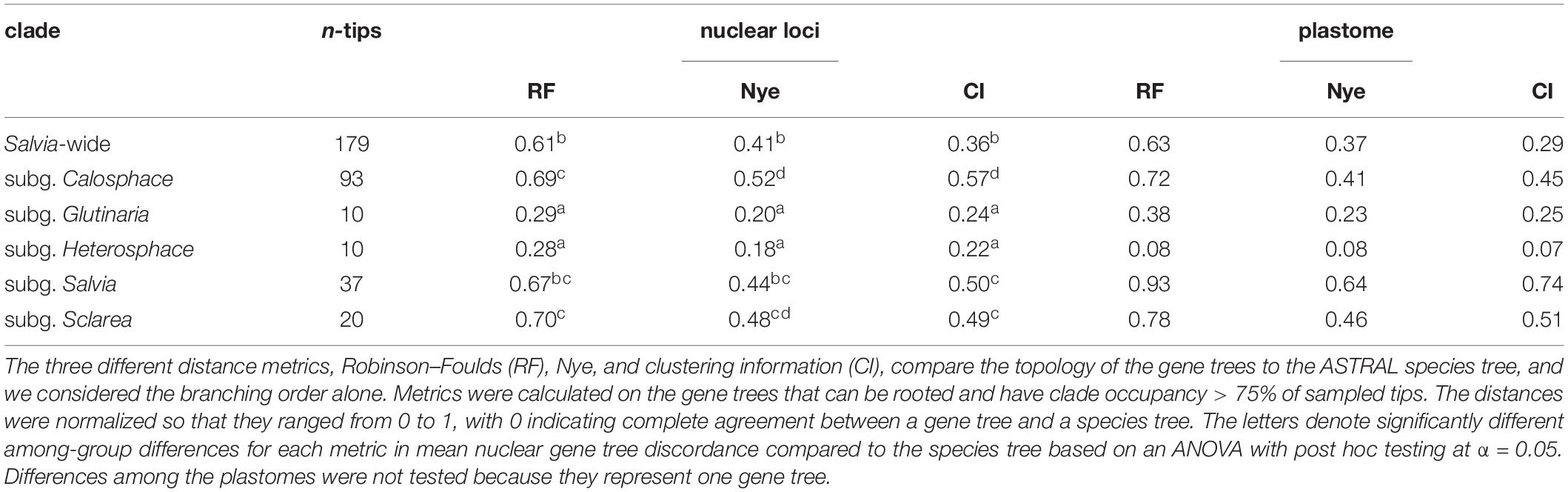
Table 1. Summary of the mean gene tree distances in Salvia and the selected subgenera within and across genomes.
Compared with the expectation under ILS alone, the nuclear gene tree discordance in the observed gene trees is generally on par with or is less than what would be expected for under ILS based on RF distance but is greater than what would be expected in subg. Calosphace, Salvia, and Sclarea based on both the Nye and CI metrics (Table 2).
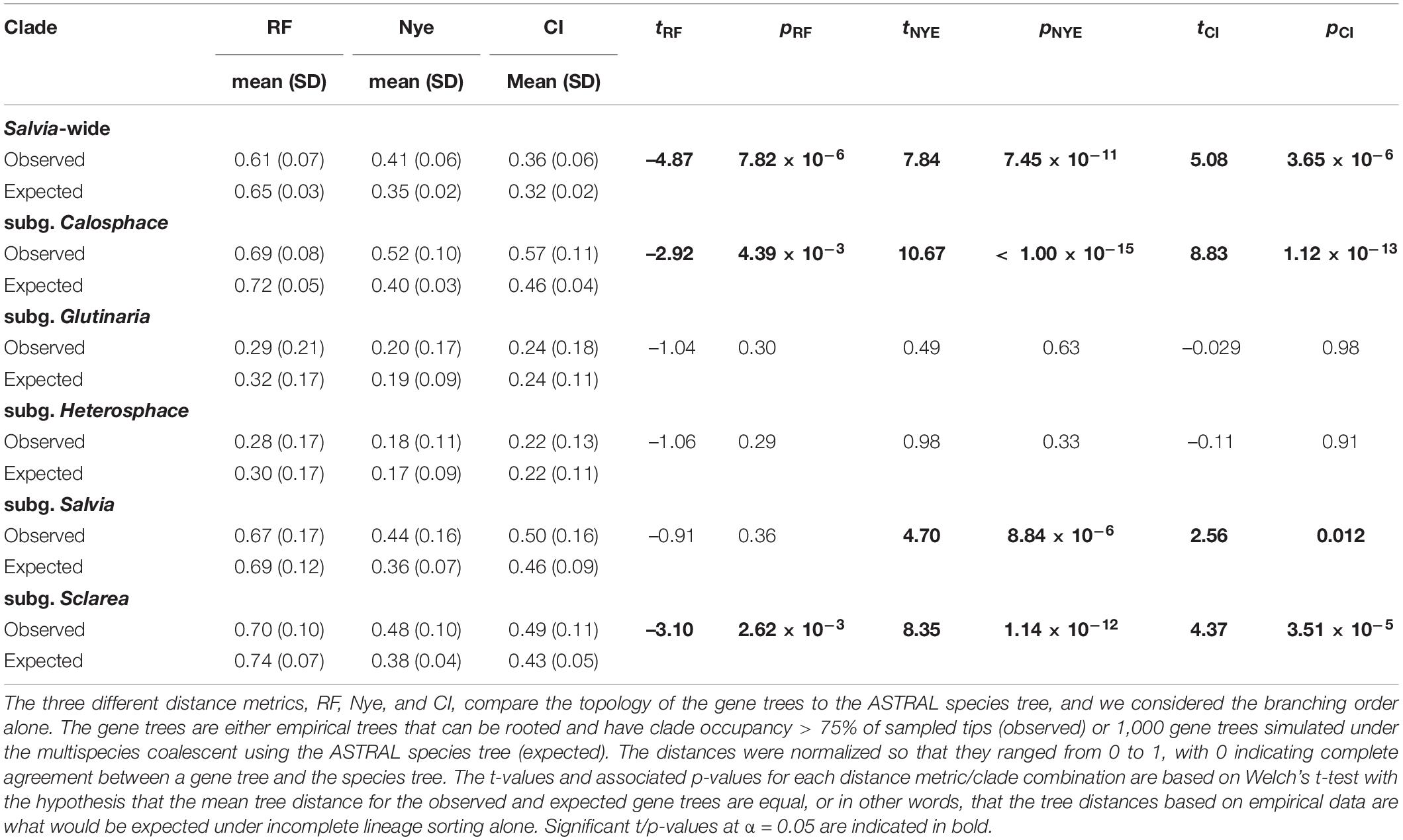
Table 2. Nuclear gene tree distances in Salvia and selected subgenera.
The best phylogenetic network contained four reticulation events along the backbone of Salvia (hmax = 4). The major topology (i.e., bifurcating backbone) was identical to that recovered by the ASTRAL and concatenated analysis of all nuclear loci (Figure 3). Quartet CF along the backbone were generally > 0.50, and these edges received full BS support with the exception of the placement of subg. Zhumeria (CF = 0.42, BS = 96) and the sister relationship of subg. Salvia and Sclarea (CF = 0.42, BS = 86). While the BS analysis found evidence for horizontal gene flow, there was considerable uncertainty regarding the number and placement of reticulation events, with BS replicates recovering either three (52%) or four reticulation events. Inheritance probabilities (γ, the fraction of the nuclear genome involved in a reticulation event) for the four reticulation events on the best-fitting network ranged from 7 to 36% (Figure 3). The best network recovered gene flow from the stem of subg. Salvia to Heterosphace (γ = 0.36, BS = 66), stem subg. Rosmarinus to stem MRCA of Calosphace + Glutinaria (γ = 0.26, BS = 66), stem MRCA of subg. Calosphace + Glutinaria to Zhumeria (γ = 0.10, BS = 10), and stem subg. Dorystaechas to Audibertia (γ = 0.07, BS = 34). Alternative reticulation events found in frequency > 10% in the BS replicates involved the stem of subg. Salvia and Sclarea (BS = 22, with alternative relationships among Heterosphace, Salvia, and Sclarea), stem MRCA subg. Rosmarinus + Salvia and MRCA Calosphace + Glutinaria (BS = 28), stem subg. Glutinaria and stem Audibertia + Calosphace (BS = 40), and stem subg. Glutinaria and stem MRCA Calosphace + Dorystaechas (BS = 40).
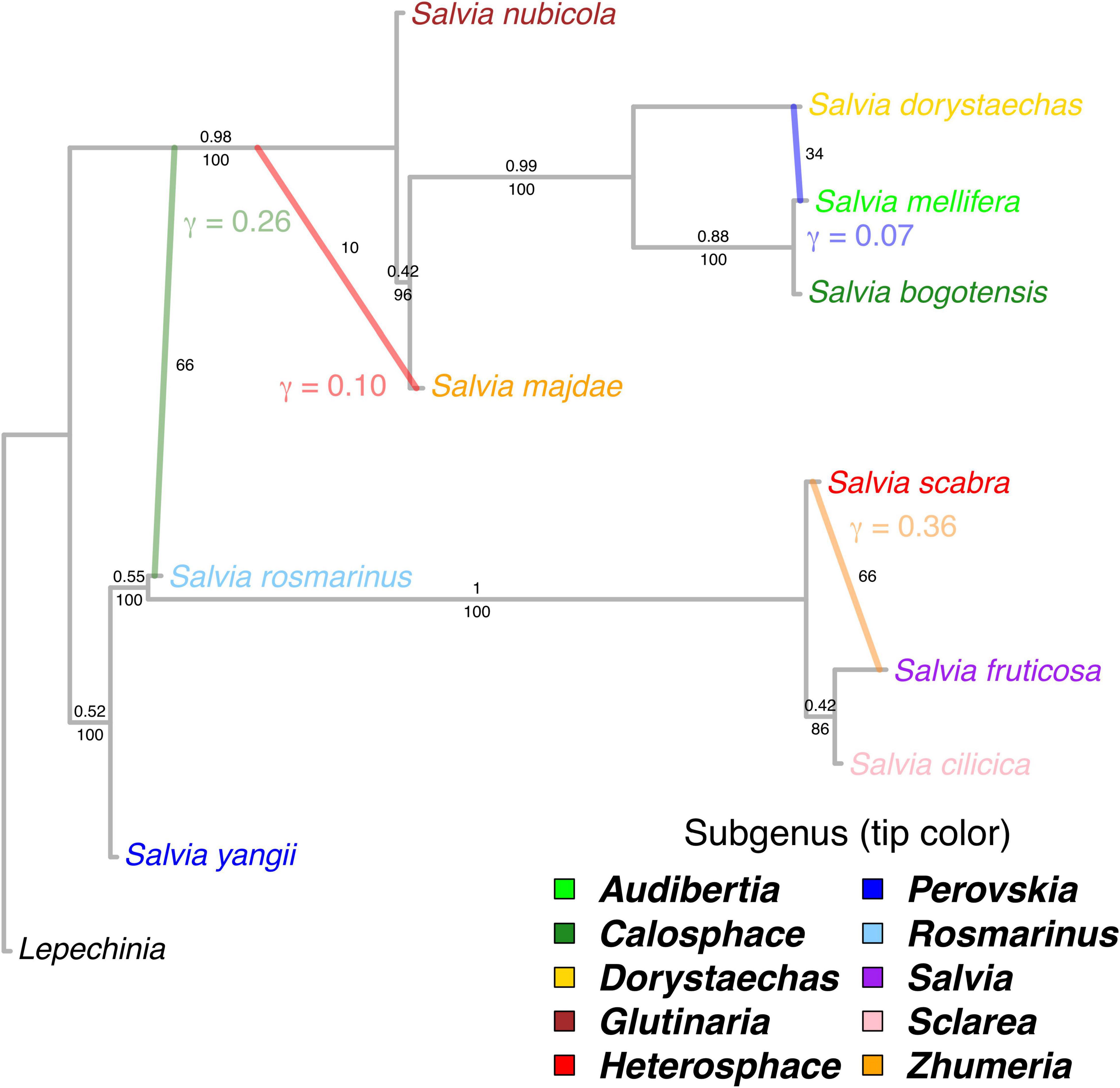
Figure 3. Phylogenetic network based on 57 nuclear genes depicting backbone relationships in Salvia with one exemplar per subgenus, with internal branch lengths in coalescent units. The best-fitting network has four reticulation events. Inheritance probabilities (γ) are indicated next to the lineage which is inferred to have received genetic material. The numbers above the branches on the bifurcating major topology are quartet concordance factors or the proportion of the genome supporting each quartet. The numbers below the major topology branches and to the right of hybrid branches are bootstrap support values. Ingroup tips are colored by subgenus.
Our results demonstrate that despite the gene tree discordance, the backbone relationships of Salvia are identical using nuclear and plastid data. These genomes support the monophyly of all currently recognized subgenera and are largely concordant regarding the intersubgeneric relationships, where supported. Discordance among nuclear loci can largely be reconciled by invoking ILS and horizontal gene flow.
Our results largely corroborate our previous analysis involving a larger number of loci with fewer terminals (Kriebel et al., 2019). Despite the fewer loci examined in this study, the average locus length is nearly twice as long (623 vs. 1,133 bp), presumably increasing the accuracy in the gene tree estimation. Additionally, compared with Kriebel et al. (2019) we found increased ASTRAL BS support for the monophyly of subg. Audibertia (>0.99 vs. 0.79) and for the sister relationship of subg. Salvia and Sclarea (0.99 vs. 0.79). However, we did not find convincing support for subg. Audibertia based on the ASTRAL LPP, and the monophyly for this subgenus based on the nuclear data has been somewhat unclear (Walker et al., 2004, 2015; Walker and Sytsma, 2007; Drew et al., 2017; Will and Claßen-Bockhoff, 2017), although it is clearly monophyletic based on the datasets with a good sampling of plastid loci (Walker et al., 2015; Supplementary Figure S5).
Unexpectedly, we found some uncertainty regarding the placements of subg. Perovskia, Rosmarinus, and Zhumeria, which were previously placed with BS = 100 in Kriebel et al. (2019). Nevertheless, the placement of all these subgenera, especially Perovskia and Rosmarinus, has varied widely across previous molecular studies incorporating low-copy nuclear loci (Drew et al., 2017), transcriptomes (Mint Evolutionary Genomics Consortium, 2018), nuclear ribosomal ITS/ETS (Drew and Sytsma, 2012; Will and Claßen-Bockhoff, 2017; Kriebel et al., 2019), and plastid data (Walker and Sytsma, 2007; Drew and Sytsma, 2012; Drew et al., 2017; Will and Claßen-Bockhoff, 2017; Zhao et al., 2020, 2021). The varying placement of subg. Rosmarinus across studies–and indeed across the loci examined in this study–can be explained by a combination of ILS and ancient horizontal gene flow (see below). This may be true for subg. Zhumeria as well, although it is less likely that horizontal gene flow has been involved in that case given the low BS support for any such gene flow.
While the backbone topology of the plastome of Salvia does not contradict that of the nuclear tree, it is still incompletely supported. However, it is unclear how much more information about the major relationships in Salvia, especially the relationships of subg. Perovskia and Rosmarinus, can be garnered from it. Analyses incorporating complete or nearly complete plastomes have failed to recover a fully supported backbone (Zhao et al., 2020; Supplementary Figure S5). Therefore, adding the portions of the plastome that we excluded does not seem to provide a viable solution. Mitogenomes, possibly combined with plastomes, are a possible avenue of research for a fully supported organellar phylogeny.
A final open question regarding the deeper-level phylogeny of Salvia is the relationship of the unsampled subg. Meriandra. We expect this subgenus to be closely related to subg. Dorystaechas and possibly even sister to it based on previous molecular results (Walker and Sytsma, 2007; Drew and Sytsma, 2012; Will and Claßen-Bockhoff, 2017; Kriebel et al., 2019). The placement of subg. Meriandra has important implications, not only for the historical biogeography of the genus but also implications for the timing and geographic location of any gene flow between the ancestors of subg. Audibertia and Dorystaechas, if present (see below).
While the major topology of our phylogenetic network in Salvia is clear and strongly recovers the same bifurcating backbone found across other analyses, each with different assumptions, it also suggests that such a relatively simple tree may not be the best model of the phylogenetic history of Salvia (Figure 3). While all of our BS trees recovered at least three gene flow events, there is considerable uncertainty regarding which clades were involved in some of the horizontal gene flow events. We are, however, fairly certain that one gene flow event involved the stem MRCA of subg. Glutinaria and Calosphace, with the gene flow involving either the ancestor of subg. Rosmarinus alone (BS = 66) or the MRCA of subg. Rosmarinus and Salvia (BS = 28), and this likely explains the uncertainty regarding the placement of subg. Perovskia and Rosmarinus.
Likewise, it seems probable that the uncertainty regarding the branching order of subg. Heterosphace, Salvia, and Sclarea is the result of the gene flow. Although our phylogenetic network favors horizontal gene flow between subg. Heterosphace and Salvia as a better explanation for the discordance (BS = 66), an alternative resolution of the relationships with the gene flow between subg. Salvia and Sclarea is also possible (BS = 22). Given this result, it is also possible that the constraint of the level-1 network is not an appropriate model, and subg. Salvia may be a hybrid between subg. Heterosphace and Sclarea. This hypothesis requires further testing.
On the other hand, we found poor support for the remaining two inferred horizontal gene flow events on our best network, especially for the gene flow involving subg. Zhumeria. While slightly better supported, the horizontal gene flow between subg. Audibertia and Dorystaechas seem implausible since it necessitates the gene flow between their ancestors in North America and Southwest Asia (Kriebel et al., 2019). Despite its absence from the best network, the BS replicates suggest a strong possibility of gene flow involving subg. Glutinaria, especially with the MRCA of Audibertia + Calosphace (possibly extended to Dorystaechas), which would be more consistent with our current understanding of the historical biogeography of Salvia.
Apart from a level-1 network possibly being an unreasonable restriction to our dataset, the uncertainty in the placement of horizontal gene flow may be due to the relatively few loci employed here. For example, SNAq may recover false positive hybridization events in the datasets with < 100 loci (Solís-Lemus and Ané, 2016). Overall, our results highlight that it is essential to complement searches for best-fitting networks with BS analyses.
Our AHE data provides good support for many shallow-scale relationships in Salvia (Figure 1 and Supplementary Figures S1–S3). We clarify the placement of sect. Axillares within subg. Calosphace, with the nuclear evidence slightly favoring a hypothesis of the sister relationship of sect. Axillares and the Hastatae clade (sects. Blakea, Hastatae, and Standleyana), although the support based on ASTRAL LPP is noticeably weak. This relationship is identical to that suggested by the nuclear ribosomal DNA (Fragoso-Martínez et al., 2018; Kriebel et al., 2019, Appendix S7), but not the plastid data (Drew et al., 2017; Will and Claßen-Bockhoff, 2017; Fragoso-Martínez et al., 2018) or one low copy nuclear marker (PPR: Drew et al., 2017), which instead placed sect. Axillares as sister to the remainder of subg. Calosphace. It is still unclear if the uncertainty in the placement is due to ILS or gene flow, which should be investigated in future studies focused on subg. Calosphace, especially since the placement of sect. Axillares has important macroevolutionary implications.
Where our sampling of the Old World lineages permits, we corroborated relationships among deep subgeneric splits which were strongly supported in previous studies (subg. Glutinaria: Hu et al., 2018, subg. Heterosphace: Will and Claßen-Bockhoff, 2014, 2017). Within the other Old World subgenera, the support for relationships in previous studies has not been robust enough to warrant a discussion of the major relationships (Will and Claßen-Bockhoff, 2014, 2017), and thus our results, where it is well supported, are novel.
From a taxonomic perspective, it is encouraging that for the few species for which we have multiple accessions, morphologically defined species are monophyletic, paraphyletic due to the inclusion of only one other species, or essentially form a polytomy with other morphologically similar species, rather than polyphyletic and/or found in large polytomies (Figure 1 and Supplementary Figures S1–S3). This suggests that our AHE dataset has the power to not only resolve deeper relationships in Salvia but also to provide information pertinent to species delimitation.
While phylogenomic datasets show great promise to resolve relationships in previously intractable angiosperm lineages, the irony is that many of these groups have undergone rapid radiations (Larson et al., 2020; Shee et al., 2020; Rose et al., 2021; Thomas et al., 2021), which increases the chance of gene tree heterogeneity due to ILS (Pamilo and Nei, 1988; Maddison, 1997; Oliver, 2013). Rampant gene tree discordance need not mean that species trees are poorly supported, provided that the discordance is consistent with the underlying model used to generate the species tree. Indeed, our analysis suggests that much of the gene tree discordance is at least consistent with ILS, given the high support for many infrageneric relationships based on our ASTRAL analysis (Figure 1 and Supplementary Figures S1, S2). Conversely, the low support for relationships in approaches that only take ILS into account may be due to either the lack of information about a given relationship in the underlying sequence data, or a more complex model of relationships (i.e., one involving horizontal gene flow). Our results demonstrate that while the average discordance of nuclear gene trees is consistent with what would be expected under ILS alone in the relatively under-sampled subg. Glutinaria and Heterosphace, it exceeds what would be expected under ILS in subg. Calosphace, Salvia, and Sclarea (Table 2). More strikingly, since that under ILS gene tree discordance should increase simply as a function of taxon sampling, it is notable that subg. Salvia and Sclarea have observed mean nuclear gene tree discordance on par with or slightly lower than that for subg. Calosphace, despite being represented by many fewer tips (Table 1).
One possible explanation for this is that the increased ILS results from the very rapid radiations of these clades, in combination with much younger crown ages for the MRCAs of what this study samples in subg. Sclarea (13.4 My) and Salvia (<7.8 My) relative to Calosphace (20.1 My) (Kriebel et al., 2019). However, based on the excess of the nuclear gene tree discordance in the aforementioned clades relative to the expectation under the multispecies coalescent, we suggest that in these clades, especially in subg. Salvia and Sclarea, the multispecies coalescent does not provide an ideal model of phylogenetic relationships. Instead, a model with horizontal gene flow in these lineages is likely a better explanation for the excess of gene tree discordance observed in our data. While another possibility for this pattern is that error in the gene tree estimation adds artificial discordance, we reject this as a major complicating factor given that we collapsed very poorly supported edges in observed gene trees.
The stark discordance present between the ASTRAL tree and the plastome tree at many shallow nodes in subg. Calosphace, Salvia, and Sclarea, with especially large distances between the nuclear and plastid trees in subg. Salvia, is also highly suggestive of an important contribution from horizontal gene flow, either hybridization or introgression (Figure 2). However, depending on the amount of past backcrossing, a signal for past gene flow may be absent from the nuclear genome in some cases. While we did not test it here, we do not think ILS is a likely explanation for the intergenomic gene tree conflict given the results from other angiosperm systems (Folk et al., 2017; Morales-Briones et al., 2018; Lee-Yaw et al., 2019; Rose et al., 2021), although error in the species tree estimation is a possible explanation. In future studies, we expect that relatively under-sampled subgenera should show increasing levels of cytonuclear discordance as we increase species sampling, especially within the subg. Glutinaria (Hu et al., 2018). The potentially confounding effects of polyploidy and whole-genome duplications (WGD) were not evaluated here but are being investigated.
Finally, it is worth a brief note concerning why we found that the mean RF tree distance often conflicts with the other distance metrics by demonstrating that the mean discordance is either on par with expectations under the multispecies coalescent or observed gene trees are, in some cases, less discordant. Despite being a widely used metric, RF distance is probably too conservative in penalizing against relatively minor topological differences (Smith, 2020a), as the movement of a single tip may result in maximum tree-to-tree distances even though all other tips show the same branching pattern. Thus, collapsing poorly supported edges of observed gene trees into polytomies downplays discordance, while any minor topological differences in the fully-resolved expected gene trees are penalized.
Our updated AHE dataset provides evidence for a well-supported backbone of Salvia and indicates that there is an emerging consensus of relationships in the genus that extends across genomic compartments. Past difficulty in inferring relationships has likely been caused by a combination of uninformative markers, ILS, and horizontal gene flow. To the latter point, while our dataset clearly shows evidence of horizontal gene flow at deep and shallow scales in Salvia, we are presently unable to confidently demonstrate how many ancient gene flow events occurred and where they are placed. This highlights the importance of assessing the support for the best-fitting phylogenetic network, rather than only presenting the best network.
Several issues still need clarification, especially in the placement of subg. Meriandra and in the monophyly of subg. Audibertia. We are confident that future analyses using this same or expanded set of loci, in concert with an evaluation of polyploidy and WGD processes, will resolve these issues. Additionally, targeted analyses of clades or further methodological advances will allow us to tease apart horizontal gene flow at shallower scales. Our phylogenetic hypothesis, as well as future, time-calibrated phylogenetic hypotheses of the entire genus Salvia, its constituent subgenera, and targeted clades, will provide an invaluable framework for which to conduct multiple comparative analyses in this fascinating genus.
The data presented in the study are deposited in the NCBI Sequence Read Archive (SRA) as BioProject PRJNA773953.
JR, RK, BD, and KS conceived and undertook the project. JR, RK, LK, AD, JG-G, FC, EL, AL, and BD assisted with the data collection. JR analyzed the data and led the writing with contributions from all authors. All the authors contributed to the article and approved the submitted version.
This manuscript was funded in part by the University of Wisconsin Botany Department Hofmeister Endowment, NSF-DEB collaborative grant to KS and BD (DEB-1655606 and DEB-1655611), and TUBITAK project number 2219 to FC for postdoctoral studies in the United States. JG-G appreciates the financial support provided by CONACYT by means of the project CB-2015-01-255165.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
We gratefully acknowledge Holly Forbes from the UC-Berkeley Botanical Garden, and Cindy Newlander and Mike Kintgen from the Denver Botanical Garden for granting permission to collect garden specimens and assisting us. We are grateful to Jay Walker, N. Ivalú Cacho, Eleftherios Dariotis, and Rolando Uría for their help in collecting the specimen.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.767478/full#supplementary-material
Supplementary Figure S1 | The ASTRAL species tree of Salvia and outgroups, with local posterior probabilities on branches. Ingroup branches are colored by subgenus.
Supplementary Figure S2 | The ASTRAL species tree of Salvia and outgroups, with bootstrap support on branches. Ingroup branches are colored by subgenus.
Supplementary Figure S3 | The RAxML maximum likelihood tree of Salvia and outgroups based on the concatenated nuclear matrix, with bootstrap support on branches. Ingroup branches are colored by subgenus.
Supplementary Figure S4 | Phyparts summary of gene trees. Pies at major nodes summarize the percentage of various phylogenetic signals across 101 gene trees which can be rooted. The numbers at the left of the pies show the total number of gene trees in which the clade is found, followed by the total number of gene trees that conflict with that clade. The remainder of the gene trees, if any, do not provide information on that particular relationship. Ingroup branches are colored by subgenus.
Supplementary Figure S5 | The RAxML maximum likelihood tree of Salvia and outgroups based on entire plastomes, with bootstrap support on branches. GenBank accessions are removed so that the plastome tree matches the nuclear tree in the tip composition. Ingroup branches are colored by subgenus.
Supplementary Figure S6 | Tanglegram illustrating the disagreement between the ASTRAL (A) and concatenated maximum likelihood (B) species trees based on nuclear data. Links connect identical tips, with nodes rotated to minimize link overlap. Ingroup branches are colored by subgenus. Clades that differ between the two trees are indicated by filled circles on the concatenated maximum likelihood tree.
Ané, C., Larget, B., Baum, D. A., Smith, S. D., and Rokas, A. (2007). Bayesian estimation of concordance among gene trees. Mol. Biol. Evol. 24, 412–426. doi: 10.1093/molbev/msl170
Baum, D. A. (2007). Concordance trees, concordance factors, and the exploration of reticulate genealogy. Taxon 56, 417–426. doi: 10.1002/tax.562013
Buddenhagen, C. E., Lemmon, A. R., Lemmon, E. M., Bruhl, J., Cappa, J., Clement, W. L., et al. (2016). Anchored phylogenomics of angiosperms I: assessing the robustness of phylogenetic estimates. BioRxiv 086298. doi: 10.1101/086298
Celep, F., Atalay, Z., Dikmen, F., Doǧan, M., Sytsma, K. J., and Claßen-Bockhoff, R. (2020). Pollination ecology, specialization, and genetic isolation in sympatric bee-pollinated Salvia (Lamiaceae). Intl. J. Plant Sci. 181, 800–811. doi: 10.1086/710238
Claßen-Bockhoff, R., Speck, T., Tweraser, E., Wester, P., Thimm, S., and Reith, M. (2004). The staminal lever mechanism in Salvia L. (Lamiaceae): a key innovation for adaptive radiation? Org. Diver. Evol. 4, 189–205. doi: 10.1016/j.ode.2004.01.004
Claßen-Bockhoff, R., Wester, P., and Tweraser, E. (2003). The staminal lever mechanism in Salvia L. (Lamiaceae) - a review. Plant Biol. 5, 33–41. doi: 10.1093/aob/mcr011
Degnan, J. H. (2018). Modeling hybridization under the network multispecies coalescent. Syst. Biol. 67, 786–799. doi: 10.1093/sysbio/syy040
Dizkirici, A., Celep, F., Kansu, C., Kahraman, A., Dogan, M., and Kaya, Z. (2015). A molecular phylogeny of Salvia euphratica sensu lato (Salvia L., Lamiaceae) and its closely related species with a focus on the section Hymenosphace. Plant Syst. Evol. 301, 2313–2323. doi: 10.1007/s00606-015-1230-1
Drew, B. T., González-Gallegos, J. G., Xiang, C. L., Kriebel, R., Drummond, C. P., Walker, J. B., et al. (2017). Salvia united: the greatest good for the greatest number. Taxon 66, 133–145. doi: 10.12705/661.7
Drew, B. T., and Sytsma, K. J. (2011). Testing the monophyly and placement of Lepechinia in the tribe Mentheae (Lamiaceae). Syst. Bot. 36, 1038–1049.
Drew, B. T., and Sytsma, K. J. (2012). Phylogenetics, biogeography, and staminal evolution in the tribe Mentheae (Lamiaceae). Am. J. Bot. 99, 933–953. doi: 10.3732/ajb.1100549
Drew, B. T., and Sytsma, K. J. (2013). The South American radiation of Lepechinia (Lamiaceae): phylogenetics, divergence times and evolution of dioecy. Bot. J. Linn. Soc. 171, 171–190. doi: 10.1111/j.1095-8339.2012.01325.x
Edwards, S. V., Xi, Z., Janke, A., Faircloth, B. C., McCormack, J. E., Glenn, T. C., et al. (2016). Implementing and testing the multispecies coalescent model: a valuable paradigm for phylogenomics. Mol. Phylogen. Evol. 94, 447–462. doi: 10.1016/j.ympev.2015.10.027
Folk, R. A., Mandel, J. R., and Freudenstein, J. V. (2017). Ancestral gene flow and parallel organellar genome capture result in extreme phylogenomic discord in a lineage of angiosperms. Syst. Biol. 66, 320–337. doi: 10.1093/sysbio/syw083
Folk, R. A., Soltis, P. S., Soltis, D. E., and Guralnick, R. (2018). New prospects in the detection and comparative analysis of hybridization in the tree of life. Am. J. Bot. 105, 364–375. doi: 10.1002/ajb2.1018
Fragoso-Martínez, I., Martínez-Gordillo, M., Salazar, G. A., Sazatornil, F., Jenks, A. A., García-Peña, M. D. R., et al. (2018). Phylogeny of the Neotropical sages (Salvia subg. Calosphace; Lamiaceae) and insights into pollinator and area shifts. Plant Syst. Evol. 304, 43–55.
Fragoso-Martínez, I., Salazar, G. A., Martínez-Gordillo, M., Magallón, S., Sánchez-Reyes, L., Lemmon, E. M., et al. (2017). A pilot study applying the plant Anchored Hybrid Enrichment method to New World sages (Salvia subgenus Calosphace. Lamiaceae). Mol. Phylogen. Evol. 117, 124–134. doi: 10.1016/j.ympev.2017.02.006
González-Gallegos, J. G., Bedolla-García, B. Y., Cornejo-Tenorio, G., Fernández-Alonso, J. L., Fragoso-Martínez, I., García-Peña, M. D. R., et al. (2020). Richness and distribution of Salvia subg. Calosphace (Lamiaceae). Intl. J. Plant Sci. 181, 831–856. doi: 10.1086/709133
Hejase, H. A., and Liu, K. J. (2016). A scalability study of phylogenetic network inference methods using empirical datasets and simulations involving a single reticulation. BMC Bioinform. 17:422. doi: 10.1186/s12859-016-1277-1
Heled, J., and Drummond, A. J. (2009). Bayesian inference of species trees from multilocus data. Mol. Biol. Evol. 27, 570–580. doi: 10.1093/molbev/msp274
Hothorn, T., Bretz, F., and Westfall, P. (2008). Simultaneous inference in general parametric models. Biom. J. 50, 346–363. doi: 10.1002/bimj.200810425
Hu, G. X., Takano, A., Drew, B. T., Liu, E. D., Soltis, D. E., Soltis, P. S., et al. (2018). Phylogeny and staminal evolution of Salvia (Lamiaceae, Nepetoideae) in East Asia. Annals Bot. 122, 649–668. doi: 10.1093/aob/mcy104
Jenks, A. A., Walker, J. B., and Kim, S. C. (2013). Phylogeny of new world Salvia subgenus Calosphace (Lamiaceae) based on cpDNA (psb A-trn H) and nrDNA (ITS) sequence data. J. Plant Res. 126, 483–496. doi: 10.1007/s10265-012-0543-1
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kriebel, R., Drew, B. T., Gonzá lez-Gallegos, J. G., Celep, F., Antar, G. M., Pastore, J. F. B., et al. (2021). Stigma shape shifting in sages (Salvia: Lamiaceae) – hummingbirds guided the evolution of New World floral features. Bot. J. Linn. Soc. (in press).
Kriebel, R., Drew, B., González-Gallegos, J. G., Celep, F., Heeg, L., Mahdjoub, M. M., et al. (2020). Pollinator shifts, contingent evolution, and evolutionary constraint drive floral disparity in Salvia (Lamiaceae): evidence from morphometrics and phylogenetic comparative methods. Evolution 74, 1335–1355. doi: 10.1111/evo.14030
Kriebel, R., Drew, B. T., Drummond, C. P., González-Gallegos, J. G., Celep, F., Mahdjoub, M. M., et al. (2019). Tracking temporal shifts in area, biomes, and pollinators in the radiation of Salvia (sages) across continents: leveraging anchored hybrid enrichment and targeted sequence data. Am. J. Bot. 106, 573–597. doi: 10.1002/ajb2.1268
Larget, B. R., Kotha, S. K., Dewey, C. N., and Ané, C. (2010). BUCKy: gene tree/species tree reconciliation with Bayesian concordance analysis. Bioinformatics 26, 2910–2911. doi: 10.1093/bioinformatics/btq539
Larson, D. A., Walker, J. F., Vargas, O. M., and Smith, S. A. (2020). A consensus phylogenomic approach highlights paleopolyploid and rapid radiation in the history of Ericales. Am. J. Bot. 107, 773–789. doi: 10.1002/ajb2.1469
Leaché, A. D., Harris, R. B., Rannala, B., and Yang, Z. (2014). The influence of gene flow on species tree estimation: a simulation study. Syst. Biol. 63, 17–30. doi: 10.1093/sysbio/syt049
Lee-Yaw, J. A., Grassa, C. J., Joly, S., Andrew, R. L., and Rieseberg, L. H. (2019). An evaluation of alternative explanations for widespread cytonuclear discordance in annual sunflowers (Helianthus). N. Phytol. 221, 515–526. doi: 10.1111/nph.15386
Lemmon, A. R., and Lemmon, E. M. (2012). High-throughput identification of informative nuclear loci for shallow-scale phylogenetics and phylogeography. Syst. Biol. 61, 745–761. doi: 10.1093/sysbio/sys051
Mint Evolutionary Genomics Consortium (2018). Phylogenomic mining of the mints reveals multiple mechanisms contributing to the evolution of chemical diversity in Lamiaceae. Mol. Plant 11, 1084–1096. doi: 10.1016/j.molp.2018.06.002
Mirarab, S., Reaz, R., Bayzid, M. S., Zimmermann, T., Swenson, M. S., and Warnow, T. (2014). ASTRAL: genome-scale coalescent-based species tree estimation. Bioinformatics 30, i541–i548.
Mitchell, N., Lewis, P. O., Moriarty Lemmon, E., Lemmon, A. R., and Holsinger, K. E. (2017). Anchored phylogenomics resolves the evolutionary relationships in the rapid radiation of Protea L. (Proteaceae). Am. J. Bot. 104, 102–115. doi: 10.3732/ajb.1600227
Morales-Briones, D. F., Liston, A., and Tank, D. C. (2018). Phylogenomic analyses reveal a deep history of hybridization and polyploidy in the Neotropical genus Lachemilla (Rosaceae). N. Phytol. 218, 1668–1684. doi: 10.1111/nph.15099
Nye, T. M., Lio, P., and Gilks, W. R. (2006). A novel algorithm and web-based tool for comparing two alternative phylogenetic trees. Bioinformatics 22, 117–119. doi: 10.1093/bioinformatics/bti720
Oliver, J. C. (2013). Microevolutionary processes generate phylogenomic discordance at ancient divergences. Evolution 67, 1823–1830. doi: 10.1111/evo.12047
Pamilo, P., and Nei, M. (1988). Relationships between gene trees and species trees. Mol. Biol. Evol. 5, 568–583.
Rieseberg, L. H., and Soltis, D. E. (1991). Phylogenetic consequences of cytoplasmic gene flow in plants. Evol. Trends Plants 5, 65–84.
Robinson, D. F., and Foulds, L. R. (1981). Comparison of phylogenetic trees. Math. Biosci. 53, 131–147.
Ronquist, F., Teslenko, M., Van Der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Rose, J. P., Toledo, C. A., Lemmon, E. M., Lemmon, A. R., and Sytsma, K. J. (2021). Out of sight, out of mind: widespread nuclear and plastid-nuclear discordance in the flowering plant genus Polemonium (Polemoniaceae) suggests widespread historical gene flow despite limited nuclear signal. Syst. Biol. 70, 162–180. doi: 10.1093/sysbio/syaa049
Sayyari, E., and Mirarab, S. (2016). Fast coalescent-based computation of local branch support from quartet frequencies. Mol. Biol. Evol. 33, 1654–1668. doi: 10.1093/molbev/msw079
Shee, Z. Q., Frodin, D. G., Cámara-Leret, R., and Pokorny, L. (2020). Reconstructing the complex evolutionary history of the Papuasian Schefflera radiation through herbariomics. Front. Plant Sci. 11:258. doi: 10.3389/fpls.2020.00258
Smith, M. R. (2020a). Information theoretic generalized Robinson–Foulds metrics for comparing phylogenetic trees. Bioinformatics 36, 5007–5013. doi: 10.1093/bioinformatics/btaa614
Smith, M. R. (2020b). TreeDist: distances Between Phylogenetic Trees. R package version 2.0.3. Comprehensive R Archive Network. doi: 10.5281/zenodo.3528124
Smith, R. L., and Sytsma, K. J. (1990). Evolution of Populus nigra (sect. Aigeiros): introgressive hybridization and the chloroplast contribution of Populus alba (sect. Populus). Am. J. Bot. 77, 1176–1187.
Smith, S. A., Moore, M. J., Brown, J. W., and Yang, Y. (2015). Analysis of phylogenomic datasets reveals conflict, concordance, and gene duplications with examples from animals and plants. BMC Evol. Biol. 15:150. doi: 10.1186/s12862-015-0423-0
Snir, S., and Rao, S. (2012). Quartet MaxCut: a fast algorithm for amalgamating quartet trees. Mol. Phylogenet. Evol. 62, 1–8.
Solís-Lemus, C., and Ané, C. (2016). Inferring phylogenetic networks with maximum pseudolikelihood under incomplete lineage sorting. PLoS Genet. 12:e1005896. doi: 10.1371/journal.pgen.1005896
Solís-Lemus, C., Bastide, P., and Ané, C. (2017). PhyloNetworks: a package for phylogenetic networks. Mol. Biol. Evol. 34, 3292–3298.
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313.
Sukumaran, J., and Holder, M. T. (2010). DendroPy: a Python library for phylogenetic computing. Bioinformatics 26, 1569–1571.
Thomas, A. E., Igea, J., Meudt, H. M., Albach, D. C., Lee, W. G., and Tanentzap, A. J. (2021). Using target sequence capture to improve the phylogenetic resolution of a rapid radiation in New Zealand Veronica. Am. J. Bot. 108, 1289–1306.
Walker, J. B., Drew, B. T., and Sytsma, K. J. (2015). Unravelling species relationships and diversification within the iconic California Floristic Province sages (Salvia subgenus Audibertia, Lamiaceae). Syst. Bot. 40, 826–844.
Walker, J. B., and Sytsma, K. J. (2007). Staminal evolution in the genus Salvia (Lamiaceae): molecular phylogenetic evidence for multiple origins of the staminal lever. Annals Bot. 100, 375–391.
Walker, J. B., Sytsma, K. J., Treutlein, J., and Wink, M. (2004). Salvia (Lamiaceae) is not monophyletic: implications for the systematics, radiation, and ecological specializations of Salvia and tribe Mentheae. Am. J. Bot. 91, 1115–1125.
Wester, P., and Claßen-Bockhoff, R. (2007). Floral diversity and pollen transfer mechanisms in bird-pollinated Salvia species. Ann. Bot. 100, 401–421.
Will, M., and Claßen-Bockhoff, R. (2014). Why Africa matters: evolution of old world Salvia (Lamiaceae) in Africa. Ann. Bot. 114, 61–83.
Will, M., and Claßen-Bockhoff, R. (2017). Time to split Salvia s.l. (Lamiaceae) – new insights from Old World Salvia phylogeny. Mol. Phylogenet. Evol. 109, 33–58.
Yu, Y., and Nakhleh, L. (2015). A maximum pseudo-likelihood approach for phylogenetic networks. BMC Genomics 16:S10. doi: 10.1186/1471-2164-16-S10-S10
Zhang, C., Rabiee, M., Sayyari, E., and Mirarab, S. (2018). ASTRAL-III: polynomial time species tree reconstruction from partially resolved gene trees. BMC Bioinform. 19:153. doi: 10.1186/s12859-018-2129-y
Zhao, F., Chen, Y. P., Salmaki, Y., Drew, B. T., Wilson, T. C., Scheen, A. C., et al. (2021). An updated tribal classification of Lamiaceae based on plastome phylogenomics. BMC Biol. 19:2. doi: 10.1186/s12915-020-00931-z
Keywords: anchored hybrid enrichment, cyto-nuclear discordance, distance metrics, incongruence, Lamiaceae, Robinson–Foulds distance, Salvia
Citation: Rose JP, Kriebel R, Kahan L, DiNicola A, González-Gallegos JG, Celep F, Lemmon EM, Lemmon AR, Sytsma KJ and Drew BT (2021) Sage Insights Into the Phylogeny of Salvia: Dealing With Sources of Discordance Within and Across Genomes. Front. Plant Sci. 12:767478. doi: 10.3389/fpls.2021.767478
Received: 30 August 2021; Accepted: 22 October 2021;
Published: 24 November 2021.
Edited by:
Stefan Wanke, Dresden University of Technology, GermanyReviewed by:
Itzi Fragoso-Martínez, Instituto de Ecología (INECOL), MexicoCopyright © 2021 Rose, Kriebel, Kahan, DiNicola, González-Gallegos, Celep, Lemmon, Lemmon, Sytsma and Drew. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeffrey P. Rose, cm9zZWpAdW5rLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.