 Yue Zhang
Yue Zhang Mei-Fang Song
Mei-Fang Song Yao Li
Yao Li Hui-Fang SunDei-Ying TangAn-Shun XuCui-Yun Yin
Hui-Fang SunDei-Ying TangAn-Shun XuCui-Yun Yin Zhong-Lian Zhang*Li-Xia Zhang*
Zhong-Lian Zhang*Li-Xia Zhang*- Yunnan Key Laboratory of Southern Medicine Utilization, Yunnan Branch of Institute of Medicinal Plant Development, Chinese Academy of Medical Sciences, Peking Union Medical College, Jinghong, China
Most Alpinia species are valued as foods, ornamental plants, or plants with medicinal properties. However, morphological characteristics and commonly used DNA barcode fragments are not sufficient for accurately identifying Alpinia species. Difficulties in species identification have led to confusion in the sale and use of Alpinia for medicinal use. To mine resources and improve the molecular methods for distinguishing among Alpinia species, we report the complete chloroplast (CP) genomes of Alpinia galanga and Alpinia kwangsiensis species, obtained via high-throughput Illumina sequencing. The CP genomes of A. galanga and A. kwangsiensis exhibited a typical circular tetramerous structure, including a large single-copy region (87,565 and 87,732 bp, respectively), a small single-copy region (17,909 and 15,181 bp, respectively), and a pair of inverted repeats (27,313 and 29,705 bp, respectively). The guanine–cytosine content of the CP genomes is 36.26 and 36.15%, respectively. Furthermore, each CP genome contained 133 genes, including 87 protein-coding genes, 38 distinct tRNA genes, and 8 distinct rRNA genes. We identified 110 and 125 simple sequence repeats in the CP genomes of A. galanga and A. kwangsiensis, respectively. We then combined these data with publicly available CP genome data from four other Alpinia species (A. hainanensis, A. oxyphylla, A. pumila, and A. zerumbet) and analyzed their sequence characteristics. Nucleotide diversity was analyzed based on the alignment of the complete CP genome sequences, and five candidate highly variable site markers (trnS-trnG, trnC-petN, rpl32-trnL, psaC-ndhE, and ndhC-trnV) were found. Twenty-eight complete CP genome sequences belonging to Alpinieae species were used to construct phylogenetic trees. The results fully demonstrated the phylogenetic relationship among the genera of the Alpinieae, and further proved that Alpinia is a non-monophyletic group. The complete CP genomes of the two medicinal Alpinia species provides lays the foundation for the use of CP genomes in species identification and phylogenetic analyses of Alpinia species.
Introduction
Alpinia Roxb. is an important genus of Zingiberaceae and includes 250 species mainly distributed in tropical Southeast Asia, but their distribution extends south into Australia and the South Pacific islands, and west into India (Wu and Larsen, 2000; Wu et al., 2016; Li D. M. et al., 2020). There are approximately 50 species in China, mainly in the south (Wu and Larsen, 2000). Most Alpinia species are valued as foods, ornamental plants, or plants with medicinal properties. As such, studies of Alpinia species have focused on their effective chemical components and pharmacological properties. For example, Alpinia oxyphylla is an important food and traditional Chinese herbal medicine; previous studies have demonstrated its antioxidative, anti-inflammatory, anti-apoptotic, and neuropharmacological effects (Zhang et al., 2011; Gao et al., 2019; Li J. et al., 2020). A. galanga and A. zerumbet have been used as traditional medicines and food seasonings for hundreds of years (Chouni and Paul, 2018; Xiao et al., 2020). The rhizomes of A. kwangsiensis are used in Chinese traditional medicine to treat abdominal pain, stomach flu, vomiting, and traumatic injury (Medicinal Materials Company in Yunnan Province, 1993; Wu et al., 2015). Over the years, modern pharmacological research has shown that A. zerumbet has important physiological and pharmacological functions, including antioxidative, antimicrobial, antianxiety effects, and promotes osteoblastic cell differentiation activities (Elzaawely et al., 2007; Sousa et al., 2015; Kumagai et al., 2016; Xuan et al., 2016; Castro et al., 2018). Meanwhile, A. galanga has been used as an antifungal, antimicrobial, anti-inflammatory, antioxidant, and anti-osteoarthritic drug (Chouni and Paul, 2018). However, the medicinal market for Alpinia is chaotic, and source materials are often misidentified due to the similarities in morphological characters between Alpinia species and their medicinal organs; for example, the rhizomes of A. galanga, A. calcarata, and A. officinarum have been adulterated or substituted for one another during the sales process (Upadhye et al., 2018; Li J. Z. et al., 2020). These problems have severely hindered the clinical use of and scientific research related to medicinal Alpinia species.
Alpinia is the largest, most widespread genus in the Zingiberaceae, and it includes many cultivated varieties (Wu and Larsen, 2000; Zhao et al., 2001; Kress et al., 2005). This has led to substantial difficulties in the classification and identification of Alpinia species, and researchers have studied the molecular markers of species in this genus. Kress et al. (2002, 2005) conducted a thorough phylogenetic analysis of Zingiberaceae and Alpinia species using the DNA sequences of the internal transcribed spacer (ITS) and plastid matK regions, and the results indicate that Alpinia is a complex polyphyletic group. A phylogenetic analysis of Zingiberaceae plants, conducted based on the ycf1 barcode, could not accurately classify taxa below the genus level (Zhong et al., 2018). These results indicate that commonly used DNA barcoding sequences are only useful for genus-level identification, and the relationships among species within the genus remain challenging to determine.
Chloroplasts (CPs) are important organelles in plant cells, as they are the location of photosynthesis. CPs convert solar energy into chemical energy and release oxygen while providing essential energy for the growth and reproduction of green plants (Wicke et al., 2011; Bock and Knoop, 2012). The CP genome consists of a closed circular DNA molecule that includes a large single-copy (LSC) region, a small single-copy (SSC) region, and two inverted repeats (IRa and IRb) (Sato et al., 1999; Ferrarini et al., 2013). CP genomes have been widely used for species evolution analysis, species identification, and the development of molecular markers because of their highly conserved gene sequences (Gao et al., 2019; Zhang et al., 2019; Li D. M. et al., 2020). With the rapid development of high-throughput sequencing technologies in recent years, the complete CP genome sequence has been obtained become easier. CP genomes have shown great potential for use in species identification, particularly closely related species (Park et al., 2018; Cui et al., 2019a). The complete CP genome, as a barcode has been widely used to evaluate plant phylogenetic relationships or distinguish species, and selected sequences from high-variation regions of the complete CP genome have been used for species identification (Chen et al., 2018; Cui et al., 2019a; Huo et al., 2019; Zhang et al., 2019). For example, Zhang et al. (2021) identified barcode markers to aid in the accurate identification of raw materials of Dragon’s blood (Dracaena) species in China by comparing the complete CP genome sequences of all species in the genus. In a recent study, a phylogenetic analysis of all genera in the Zingiberaceae was conducted using the complete CP genomes of three medicinal Alpinia species, and the phylogenetic relationship between A. zerumbet and A. oxyphylla was evaluated using the entire CP genome (Gao et al., 2019; Li D. M. et al., 2020). However, these publicly available CP genome data are insufficient to resolve the intraspecific and interspecific differences among Alpinia species, which exhibit only subtle morphological differences. Relative to the rest of the plant kingdom, a very limited amount of CP genome data is available for medicinal plants. It is therefore essential to obtain additional CP genome data to support the effective utilization of medicinal plant resources.
We sequenced the complete CP genomes of A. galanga and A. kwangsiensis sampled from Yunnan, using the Illumina HiSeq4000 sequencing platform. Next, we investigated their essential characteristics [including analyses of molecular structure, simple sequence repeats (SSRs), and long repeats]. Then, we compared the resulting CP genomes with the published CP genomes of A. hainanensis (MK262728), A. oxyphylla (MK262729), A. pumila (MK262731) (Gao et al., 2019; Li D. M. et al., 2020), and A. zerumbet (JX088668), and found potential high-variation region markers for Alpinia. Finally, we collected 28 whole CP genome sequences for Alpinieae species, which we used as a super-barcode to identify species in this group and analyze their phylogenetic positions. Our study provides significant genetic information for species identification and phylogenetic analyses of Alpinia plants. In addition, it can serve as a reference to help alleviate issues with the accurate identification of Alpinia plants in the medicinal market.
Materials and Methods
Plant Materials and DNA Extraction
Fresh A. galanga and A. kwangsiensis leaves were collected from Xishuangbanna City, Yunnan Province. Voucher specimens were deposited in the Yunnan branch of the Institute of Medicinal Plant Development (IMPLAD), Chinese Academy of Medical Sciences herbarium. The fresh leaves were cleaned with 75% ethanol and preserved at −80°C. Total genomic DNA was extracted from frozen clean leaves using the TaKaRa Mini BEST Universal Genomic DNA Extraction Kit with a standard protocol (TaKaRa, Shiga, Japan). The concentration and quality of DNA were checked using electrophoresis in a 1% (w/v) agarose gel and the Nanodrop 2000 instrument (Thermo Scientific, Waltham, MA, United States). The OD260/280 values ranged from 1.8 to 2.2, and ≥2 μg of DNA was equally pooled from individuals of the two species to construct a shotgun library.
Chloroplast Genome Sequencing and Assembly
DNA samples were randomly sheared, incubated with fragmentation buffer, and broken into 300–500-bp fragments in a Covaris M220 focused ultrasonicator (Covaris, Woburn, MA, United States). The DNA library was prepared using the Illumina TruSeq™ Nano DNA Sample Prep Kit (Illumina, San Diego, CA, United States). To complement the fragmented DNA ends, A was added to the 3′ end of double-stranded DNA to form a sticky end, and a sequence adapter index was added. Polymerase chain reaction (PCR) amplification was performed for eight cycles for library enrichment, and the target band was recovered from a 2% agarose gel (Certified Low Range Ultra Agarose). Bridge PCR amplification was performed with the cBot solid phase (Truseq PE Cluster Kit v3-cBot-HS; Illumina) carrier to generate clusters. The library was sequenced at the Biozeron Company (Shanghai, China) using the Illumina HiSeq4000 sequencing platform to obtain 2 × 150 bp paired-end reads. Raw reads were checked, first using FastQC (Brown et al., 2017) and then using Trimmomatic version 0.39 (Bolger et al., 2014). Adapter sequences in the reads were removed, and low-quality reads were filtered from the raw data. Reads containing 10% N were removed, and small fragments of <75 bp were discarded after quality pruning. The CP genomes of A. hainanensis (MK262728), A. oxyphylla (MK262729), and A. pumila (MK262731), which are closely related species, were used as reference genomes. We used NOVOPlasty version 2.7.21 (Dierckxsens et al., 2017) to assemble a contig containing the complete CP genome sequence. Then, using Celera Assembler version 8.0 (Denisov et al., 2008), we assembled and cleaned the data and constructed scaffolds of the CP genome using SSPACE (Boetzer et al., 2011). We optimized the assembly results using GapCloser version 1.12 (Luo et al., 2012), which repairs gaps. Finally, we used the reference genome to correct the starting position of the CP assembly sequence and determine the position and direction of the four CP regions (LSC, IRa, SSC, and IRb), to generate the assembled CP genomic sequence.
Chloroplast Genome Annotation and Structure Analysis
We used the online tool Dual Organellar GenoMe Annotator (DOGMA; University of Texas at Austin, Austin, TX, United States) (Wyman et al., 2004) and the Chloroplast Genome Annotation Software with manual corrections to perform preliminary gene annotation of the CP genomes of both species of interest. We performed a BLASTN search on the National Center for Biotechnology Information (NCBI) website to identify and confirm boundary junctions, introns, exons, and coding regions. The tRNA genes were further verified using tRNAscanSE (Lowe and Chan, 2016) and DOGMA (Wyman et al., 2004) with the default settings. Additionally, the online OrganellarGenomeDRAW (OGDRAW) software version 1.2 (Max Planck Institute of Molecular Plant Physiology, Potsdam, Germany) (Greiner et al., 2019) was used to construct the gene map using the default settings and manual checking. Finally, we generated an.sqn file to submit our findings to NCBI. The high-quality reads were deposited into the NCBI BioProject database. The complete and correct CP genome sequences of A. galanga and A. kwangsiensis were deposited in GenBank under the accession numbers MZ066611 and MZ066612, respectively. CodonW software (University of Texas, Houston, TX, United States) with the relative synonymous codon usage (RSCU) ratio were used to investigate the codon distribution (Sharp and Li, 1987). The guanine–cytosine (GC) content was analyzed using Molecular Evolutionary Genetics Analysis Version X (Kumar et al., 2018).
Repeat Sequence Analysis
Simple sequence repeats were detected using MISA software2, with the parameters set to encompass mononucleotide SSRs with ≥10 repeat units, di- and tri-nucleotide SSRs with ≥5 and 4 repeat units, respectively, and tetra-, penta-, and hexanucleotide SSRs with ≥3 repeat units. We identified the size and location of repeat sequences in the CP genomes of A. galanga and A. kwangsiensis using REPuter (University of Bielefeld, Bielefeld, Germany) (Kurtz et al., 2001) with the following parameters: 90% similarity percentage of scattered repeat copies and a minimum repeat size of 30 bp.
Genome Comparison Analyses and Marker Discovery
The whole CP genomes were initially aligned using the online MAFFT software (Katoh et al., 2019). Conserved sequences between the CP genomes of A. galanga and A. kwangsiensis were identified using BLASTN with an E-value cutoff of 1e-10. The mVISTA (Frazer et al., 2004) program in Shuffle-LAGAN mode was used to compare the two Alpinia CP genomes using the A. galanga CP genome as a reference. Then, we used DnaSP (Rozas et al., 2017) software to determine the nucleotide variability (Pi) with a 200 bp step size and a 600 bp window length. Ten highly variable sites (psbK-psbI, trnS-trnG, trnC-petN, rps4-trnT, rpl32-trnL, psaC-ndhE, ndhC-trnV, ndhF-rpl32, trnT-trnL, and psbE-petL) were selected. We used the primer design tool Primer-BLAST to design labeled primers for the highly variable regions (Ye et al., 2012). Next, a total of seven Alpinia species (A. zerumbet, A. galanga, A. blepharocalyx, A. kwangsiensis, A. hainanensis, A. conchigera, and A. oxyphylla) (Supplementary Table 1) were used to assess the identification efficiency of the highly variable sites. Total genomic DNA was extracted using the TaKaRa MiniBEST Universal Genomic DNA Extraction Kit with a standard protocol (TaKaRa) and 1% agarose gel electrophoresis. We used an ultra-micro ultraviolet spectrophotometer to assess the purity and concentration of the extracted genomic DNA. The PCR reactions were conducted in a total reaction volume of 25 μL, which contained DNA (0.5 μL), 10 × PCR buffer (2.5 μL), dNTPs (2.5 mM, 2 μL), primers (0.5 μL each), Taq DNA polymerase (5 U/μL, 0.5 μL; TaKaRa), and double-distilled water (18.5 μL). For each reaction, we used the following program: an initial 5 min of denaturation at 94°C; 35 cycles of 30 s at 94°C, 30 s of annealing at Tm with different primers, and 15 s of extension at 72°C; and a final extension for 7 min at 72°C. The PCR products were visualized using 2% agarose gels, and the successfully amplified PCR products were sent to Sangon Biotech (Shanghai, China) for bidirectional sequencing. Finally, we used high-quality sequences to construct neighbor-joining (NJ) phylogenetic trees.
Phylogenetic Analyses
To determine the phylogenetic positions of A. galanga and A. kwangsiensis, we downloaded 28 complete CP genomes of Alpinieae species from the NCBI database. The sequences were initially compared using MAFFT (Katoh et al., 2019). Next, we conducted multiple sequence visual analyses and manually delete useless gaps using BioEdit (Hall, 1999). We also used the CP genomes of Curcuma longa (MK262732) and Zingiber officinale (NC_044775) as outgroups. We constructed phylogenetic trees with 32 CP genomes sequences using the NJ, maximum parsimony (MP) and maximum likelihood (ML) methods with MEGA X software and 1000 bootstrap replicates (Kumar et al., 2018). The best-fit substitution models were selected by ModelTest-NG (Darriba et al., 2019) for ML trees.
Results and Discussion
Chloroplast Genome Features of Alpinia Species
The complete CP genome sequences of A. galanga and A. kwangsiensis are 160,100 and 162,323 bp in length, respectively, with both having an obvious quadripartite structure (Figure 1). The whole CP genomes contain a pair of IRs (IRa and IRb) at, respectively, 27,313 and 29,705 bp separated by an SSC region at 17,909 and 15,181 bp, and a LSC region at 87,565 and 87,732 bp. The A. kwangsiensis CP genome (162,323 bp) is 2223 bp longer than that of A. galanga. The GC contents of the CP genomes of A. galanga and A. kwangsiensis are 36.26 and 36.15%, respectively. And the GC content is unevenly distributed across different regions of each CP genome. The GC content in the IR regions is the highest (41.2–42.2%), followed by the LSC region at ∼33.8–33.9%, and the lowest content was found in the SSC region (29.8–30.0%). Moreover, the AT content at the third codon position (71.2–71.4%) is higher than that at the second (62.2–62.6%) and first (55.2–55.4%) positions in the protein-coding genes of the two Alpinia species (Table 1). In the complete CP genomes of both Alpinia species, 133 genes were detected, including 87 distinct protein-coding genes, eight distinct rRNA genes, and 38 distinct tRNA genes (Table 2). The distribution of genes in the two CP genomes is the same: 81 genes are distributed in the LSC region, including 60 protein-coding genes and 21 tRNA genes, whereas the SSC region contains 11 protein-coding genes and one tRNA gene; a total of 20 genes are duplicated in the IR regions, including eight protein-coding genes, eight tRNA genes, and four rRNAs (Figure 1 and Supplementary Table 2). These genomic structure features are similar to those of the other published CP genomes of the family Zingiberaceae (Cui et al., 2019a; Gao et al., 2019; Li D. M. et al., 2020).
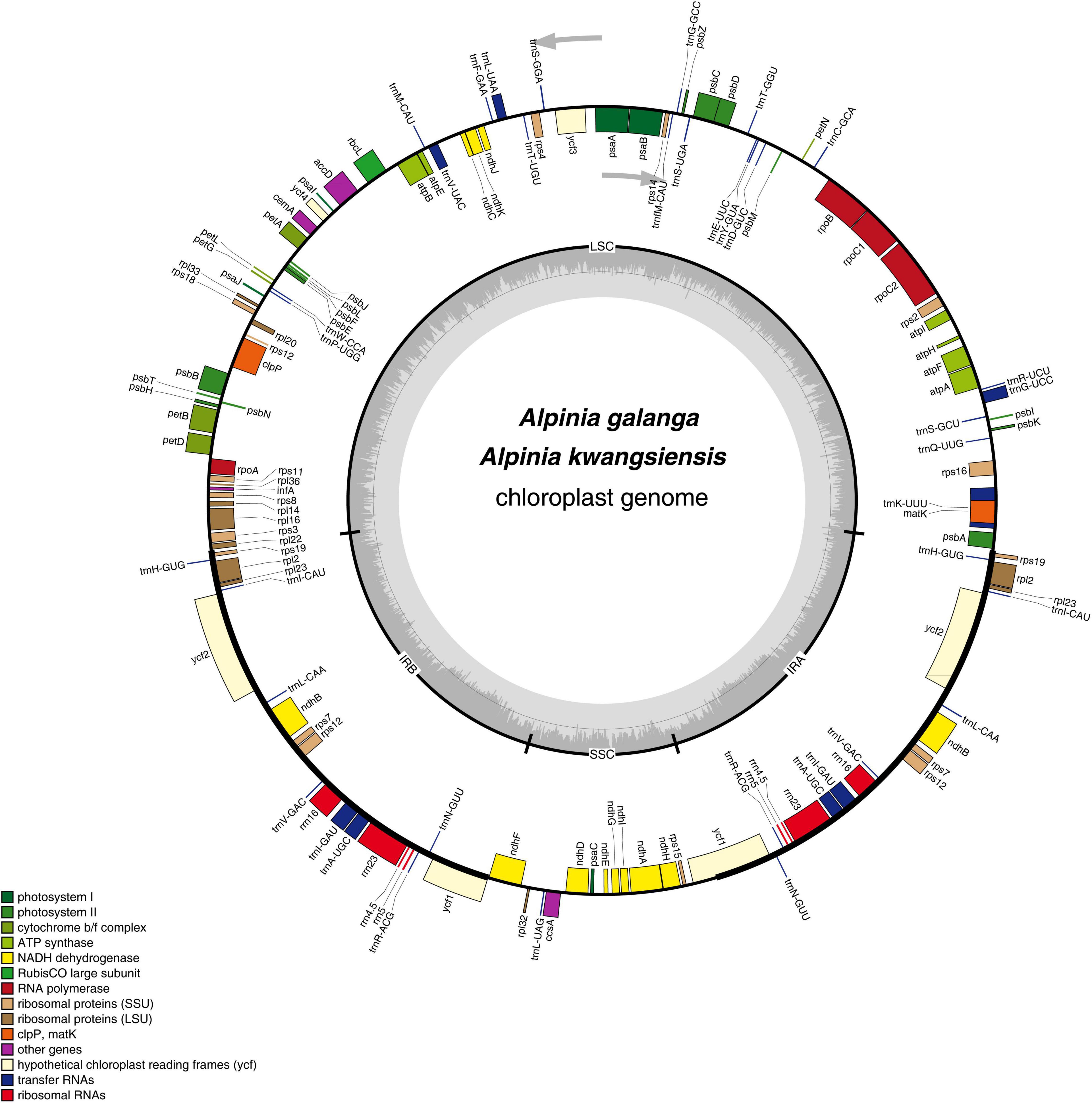
Figure 1. Gene map of the Alpinia complete chloroplast (CP) genome. Genes inside and outside the circle are transcribed clockwise and counterclockwise, respectively. Genes belonging to different functional groups are indicated by different colors. The darker gray area in the inner circle corresponds to the GC content, whereas the lighter gray area corresponds to the AT content.
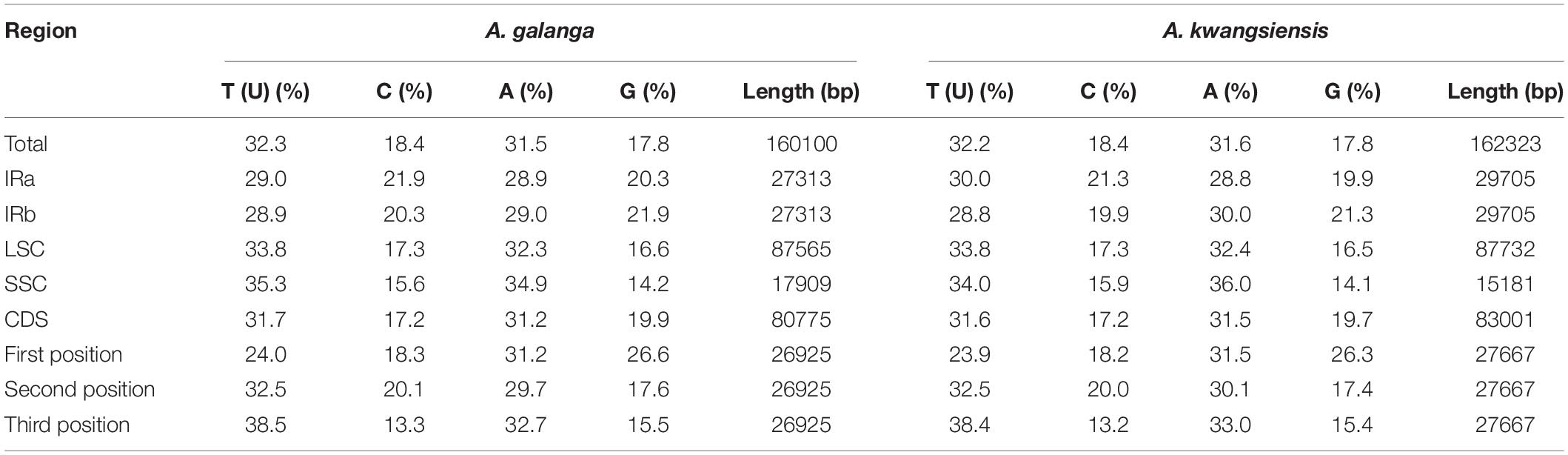
Table 1. The base composition of the A. galanga and A. kwangsiensis CP genomes.

Table 2. Genes present in the CP genomes of two Alpinia species.
We combined the CP genome information of A. galanga and A. kwangsiensis with four CP genomes for Alpinia species published in the NCBI database and performed comparisons and analyses (Table 3). The results showed that the CP genome size in the six species varied from 159,773 bp (A. zerumbet) to 162,387 bp (A. hainanensis). Moreover, the LSC, SSC, and IR regions of the different species exhibited different characteristics. There were some interesting features; for example, A. kwangsiensis contained the longest LSC region (87,732 bp) but the shortest SSC region (15,181 bp), whereas A. zerumbet had the longest SSC region (18,295 bp) but the shortest IR (26,917 bp). In terms of the number of genes, except for A. zerumbet (which contained 132 genes), the remaining five species contained 133 genes; A. zerumbet lacked one protein-coding gene. Among the six species analyzed, the A. zerumbet CP genome had the highest GC content (36.27%), whereas the A. hainanensis and A. kwangsiensis CP genomes had the lowest GC content (36.15%).
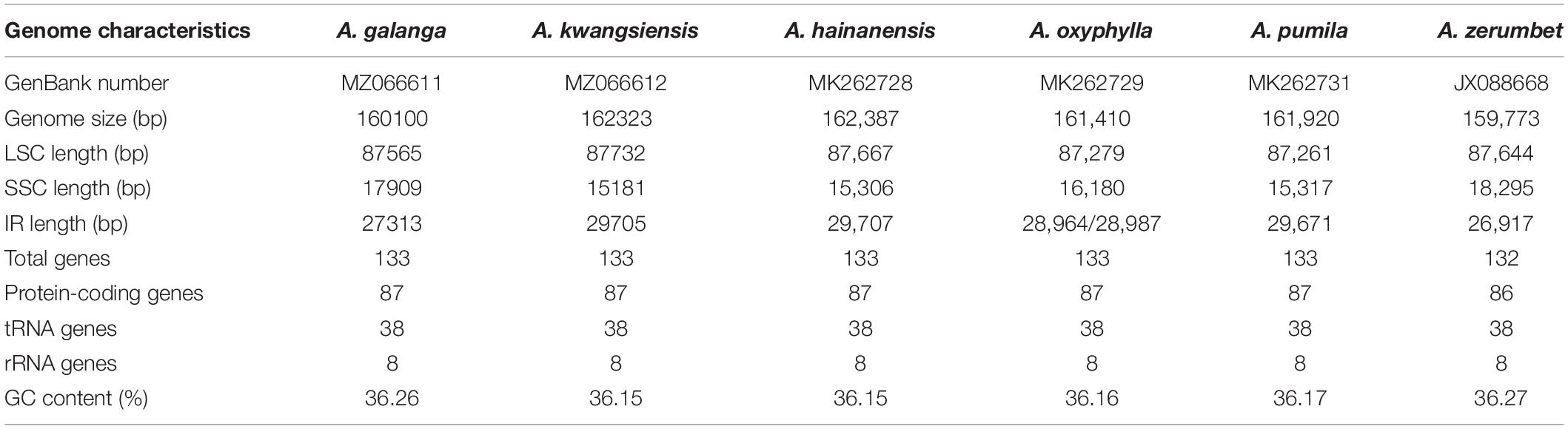
Table 3. Comparison of the general features of the six Alpinia CP genomes.
Codon Usage
The RSCU ratio is a measure of uneven usage of synonymous and non-synonymous codons in a coding sequence. An RSCU ratio <1.00 indicates that the frequency of codon usage is lower than expected, whereas codons used more frequently than expected with a ratio >1.00 (Sharp and Li, 1987; Liu et al., 2018). The codon usage levels of the A. galanga and A. kwangsiensis CP genomes are shown in Figure 2 and Supplementary Table 3. We analyzed the codon usage frequency and RSCU ratios of 87 protein-coding genes in the A. galanga and A. kwangsiensis CP genomes. In total, the genes in the A. galanga and A. kwangsiensis CP genomes contain 26,925 and 27,667 codons, respectively. The codons for leucine, serine, and arginine are the most common in both the A. galanga and A. kwangsiensis CP genomes. In the CP genomes of these Alpinia species, usage of the codons AUG and UGG-encoding methionine and tryptophan, respectively-is not biased (RSCU ratio = 1.00). Usage of the UCC codon encoding serine is also not biased (RSCU ratio = 1.00) in the A. galanga CP genome. Most protein-coding genes in the CP genomes of terrestrial plants use standard AUG initiator codons, and the use of AUG to encode methionine is not biased (RSCU ratio = 1) in the A. galanga and A. kwangsiensis CP genomes. Codons ending in A and/or U account for 71.1 and 71.4% of all the protein-coding genes in the CP genomes of A. galanga and A. kwangsiensis, respectively. Codons ending in A and/or T (U) usually have high RSCU ratios in the two CP genomes, e.g., AGA (1.96) encoding arginine, GCU (1.82) encoding alanine, and UCU (1.74) encoding serine. These results are similar to those observed for Zingiber officinale and Wurfbainia vera (Wu et al., 2017; Cui et al., 2019b). The codon usage pattern can be determined by whether there is a high proportion of A/T component bias. In the CP genomes of other higher terrestrial plants, preferred codon usage tends to be very high, and the preference for A/T is widespread (Kim and Lee, 2004; Qian et al., 2013). Our results also showed that except for Leu-UUG, all types of preferred synonymous codons (RSCU ratio > 1.00) in the two Alpinia species end with A or U. The high RSCU ratio may be attributed to the functions of the amino acids or the structures of peptides needed to avoid transcription errors (Li et al., 2016; Gao et al., 2019). This phenomenon indicates that stable CP genome evolution helps to protect important CP genes from harmful mutations while improving adaptation to selection pressure (Wang et al., 2016; Ivanova et al., 2017; Zuo et al., 2017).
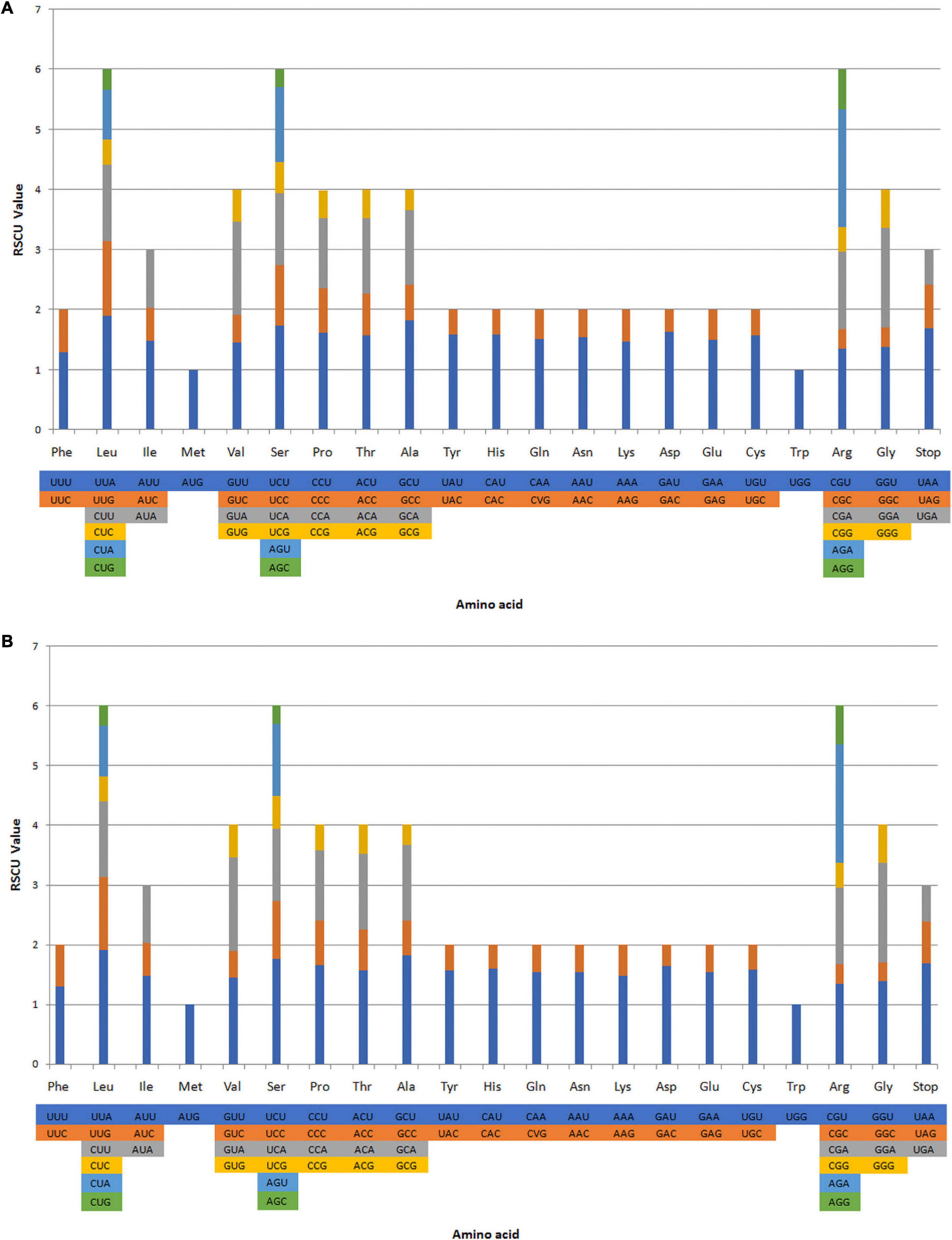
Figure 2. Codon contents in all protein-coding genes in the A. galangal (A) and A. kwangsiensis (B) complete chloroplast (CP) genome. RSCU, relative synonymous codon usage.
Analyses of Simple Sequence Repeats and Long Repeats
Simple sequence repeats, or microsatellites, are tandem repeat sequences consisting of 1–6 nucleotide repeat units and are widely distributed in CP genomes (Wu et al., 2017; Zhou J. et al., 2018). Repeated sequences were divided into tandem repeats and scattered repeats. Scattered repeats can be further divided into four types of repeats: complementary, forward, reverse, and palindromic (Kurtz et al., 2001; Zhang et al., 2019). These repeat structures promote intermolecular recombination and create diversity among CP genomes in the population (Guo et al., 2017; Zhou J. et al., 2018). In this manuscript, we used the Tandem Repeats Finder and REPuter software tools to analyze the repeat sequences and distribution of repeat sequences and SSRs in the CP genomes of six species (A. galanga, A. kwangsiensis, A. hainanensis, A. oxyphylla, A. pumila, and A. zerumbet). Results of the repeat-sequence structural analysis are shown in Figure 3. The results showed that the number of repeat types was very similar among the six Alpinia species; palindromes (24–28) were the most abundant, followed by forward (10–19) and reverse repeats (0–7), and complementary (0–2) repeats were the least abundant. In all six species, most of these repeats are between 30–39 and 40–49 bp in length, with only a few repeats >70 bp in length.
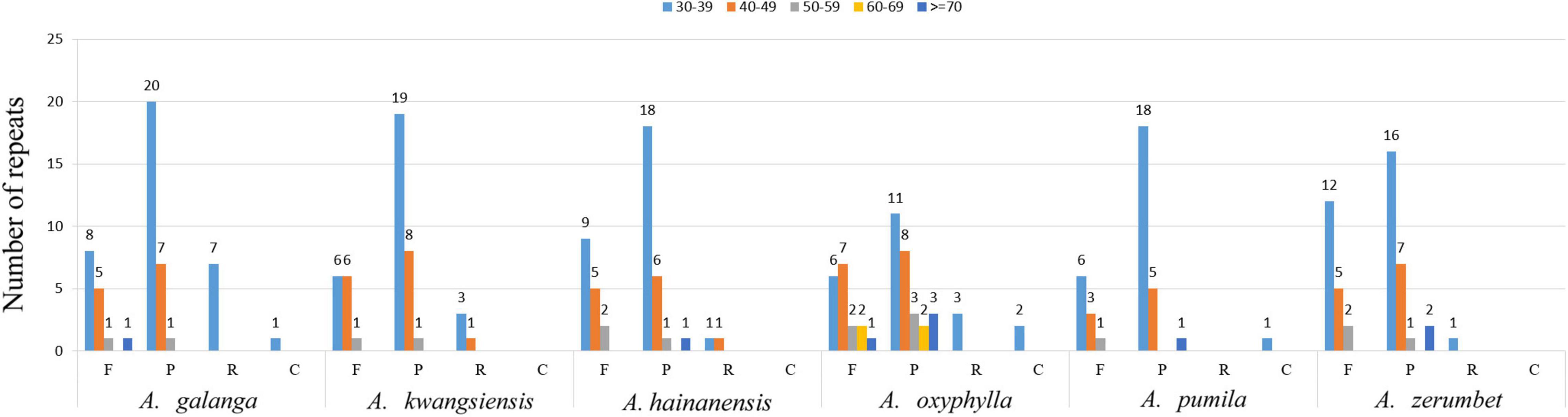
Figure 3. Long repeat sequence analysis of six Alpinia complete chloroplast (CP) genomes. REPuter was used to identify repeat sequences ≥30 bp in length and sequences with ≥90% similarity in the CP genomes. F, P, R, and C indicate the forward, palindromic, reverse, and complementary repeat types, respectively. Repeats with different lengths are indicated by different colors.
Furthermore, we analyzed the distribution and types of SSRs contained in the CP genomes of the six Alpinia species. We identified 110, 125, 113, 122, 121, and 118 SSRs in the CP genomes of A. galanga, A. kwangsiensis, A. hainanensis, A. oxyphylla, A. pumila, and A. zerumbet using MISA software, respectively (Table 4). The SSRs are mainly distributed in the LSC region of the CP genome, followed by the SSC region and the IR region (Supplementary Table 4). The most abundant type is repeated mononucleotides (50–60.17%), which were found 15–16 times in the six Alpinia species. These are followed by dinucleotide (20.03–30.33%), trinucleotide (3.28–5.31%), tetranucleotide (13.22–16.36%), and pentanucleotide repeats (0–3.64%). The A/T (46.36–57.63%) repeat is the most abundant motif in all repeats, followed by AT/TA (20.34–28.68%) dinucleotide repeats and AAAT/ATTT (5.93–8.18%) tetranucleotide repeats. Our results are consistent with previous studies reporting that CP SSRs usually consist of short poly-A or poly-T repeats. A and T are always the most frequently used bases, and tandem G or C repeats are rare in many plants (Kuang et al., 2011; Zhang et al., 2019). Interestingly, except for A. galanga, A. kwangsiensis, A. oxyphylla, and A. pumila, the other two species have no pentanucleotide SSRs, and none of the Alpinia species have any hexanucleotide SSRs. Because CP SSRs have high substitution rates, SSR markers are widely used in genetic diversity and population structure assessments, comparative genomics, the development of genetic maps, and marker-assisted selective breeding (Flannery et al., 2006; Chen et al., 2015; Cui et al., 2019b; Zhang et al., 2019). The repeat sequences identified in this study are a valuable resource for species identification as well as research on the genetic diversity and population structure of Zingiberaceae plants.
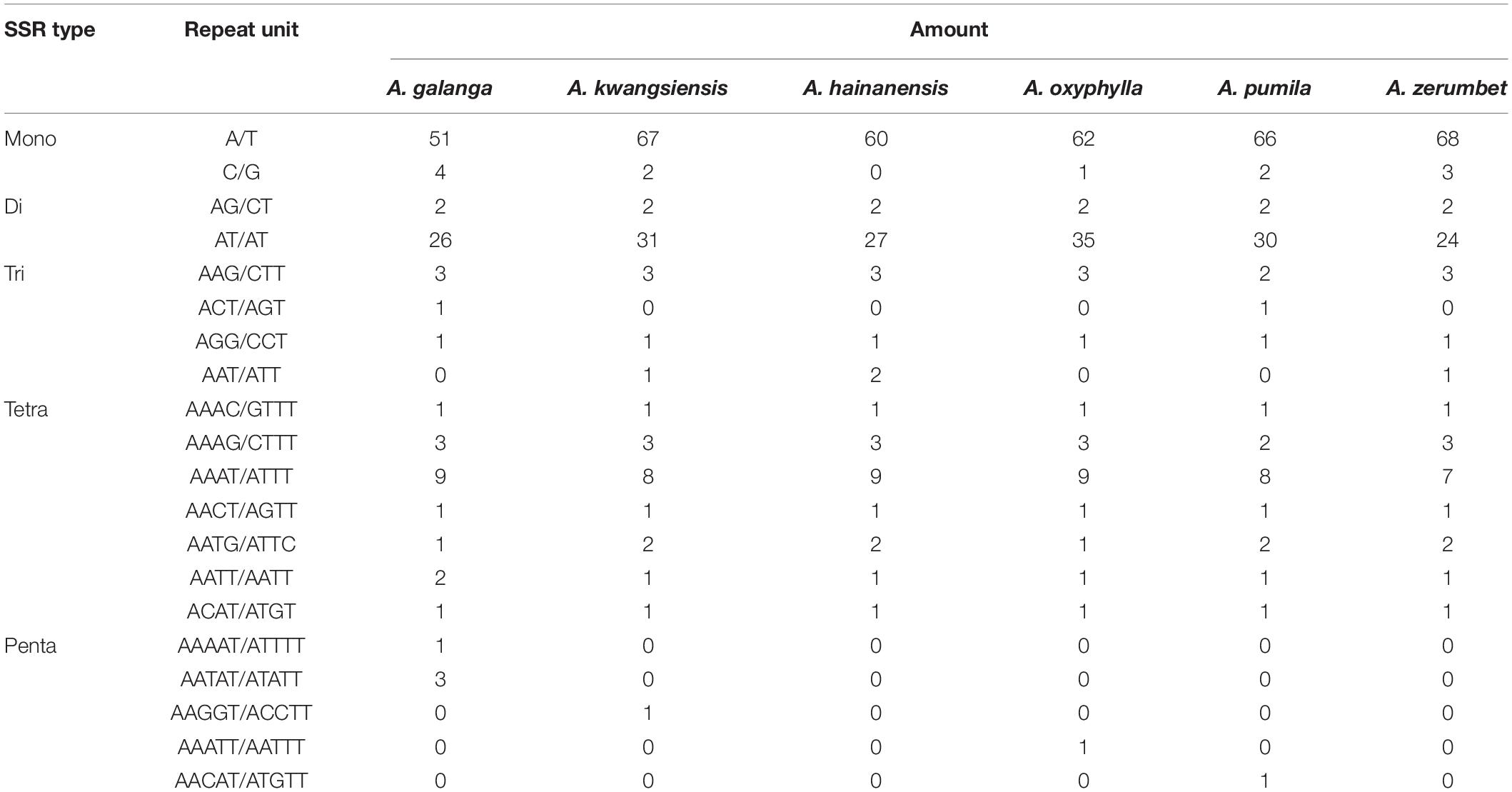
Table 4. The simple sequence repeat (SSR) types of the six CP genomes of Alpinia species.
Genome Comparison and Nucleotide Diversity
A comparison of CP genomes helps with elucidating the genetic structure and evolutionary relationships of plants in different environments (Daniell et al., 2016; Zhang et al., 2019). To determine the level of sequence similarity and genome rearrangements, we used mVISTA software to compare and analyze the sequence homologies of the CP genomes of six Alpinia species with A. galanga as the reference sequence (Figure 4). These analyses revealed few differences among the CP genomes of the six Alpinia species. These differences were mostly found in non-coding regions, such as trnG-UCC, rps4-trnT, psaC-ndhE, rpl32-trnL-UAG, trnC-GCA-petN, trnS-GCU-trnG-UCC, ndhC-trnV, and psbK-psbI. The most divergent coding regions include rpoC2, ycf1, ycf2, atpE, and rpl22. Furthermore, a comparison of the CP genomes of the six Alpinia species showed that most of the sequence variation is in the LSC and SSC regions, and the IR region exhibits the least sequence variation. This result further supports the view that the IR regions are more conserved than the LSC and SSC regions in higher plants (Nazareno et al., 2015; Cui et al., 2019a). Some scholars believe that this may be because gene conversion has corrected the mutations in the IR sequences (Khakhlova and Bock, 2006).
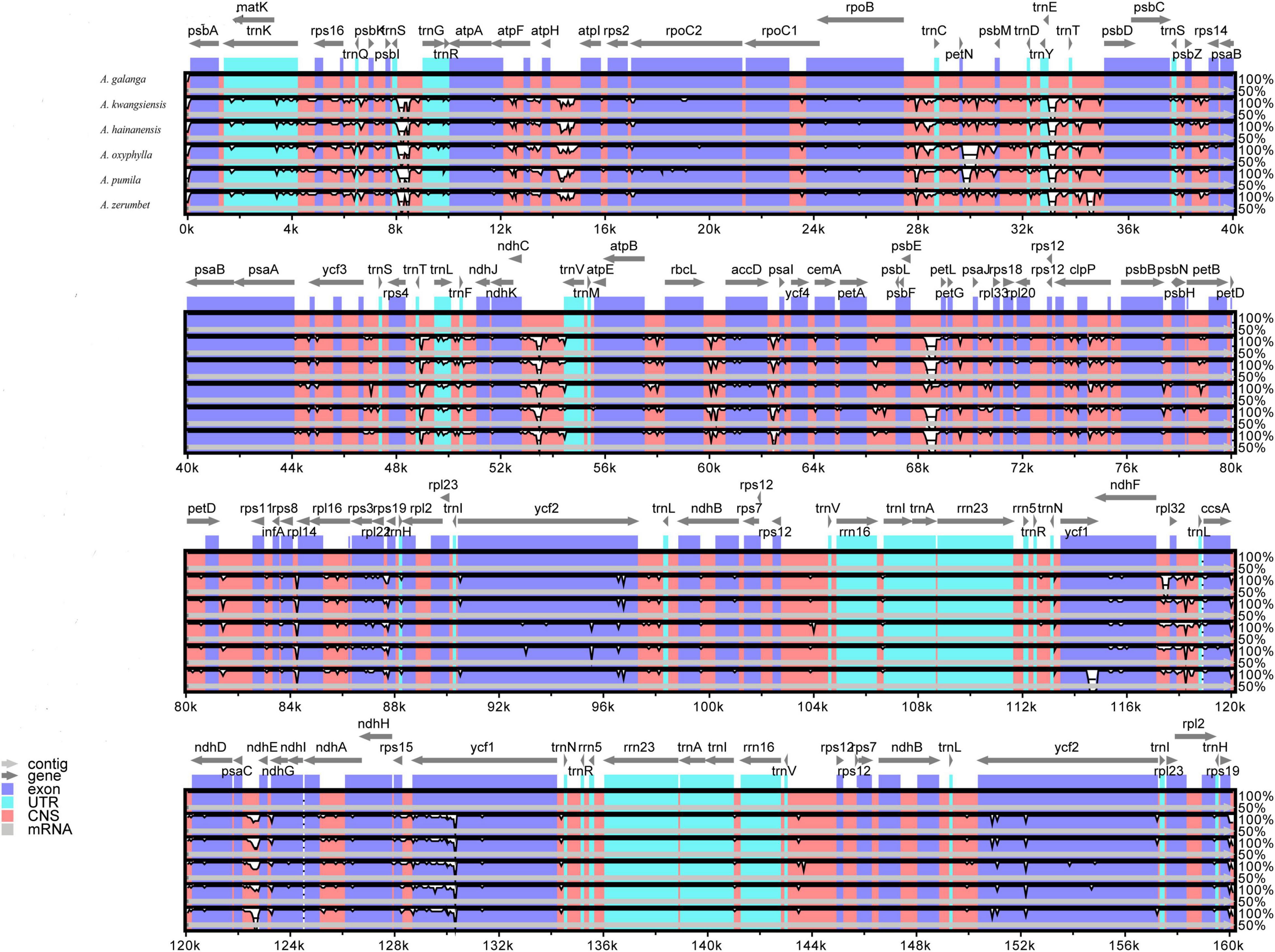
Figure 4. Structural comparison of the complete CP genomes of six Alpinia species using mVISTA. The CP genome of A. galanga was used as a reference. Gray arrows and thick black lines above the alignments indicate gene orientations and the positions of IRs in the genes, respectively. White peaks represent differences among CP genomes. A similarity cut-off value of 70% was used for the plots, and the Y-axis represents the percentage similarity (50–100%).
In addition, we also used DnaSP (Rozas et al., 2017) software to determine nucleotide diversity (PI) to detect sequence level differences in CP genomes of six Alpinia species and detecting highly variable regions (Figure 5). The IR region exhibits lower variability than the LSC and SSC regions, similar to previous studies (Zhou J. et al., 2018; Cui et al., 2019a; Li D. M. et al., 2020). The results showed an average value of Pi in all six Alpinia species of 0.0056 (Supplementary Table 5). In addition, some regions with high Pi values (>0.04) were observed in the LSC and SSC regions; for example, the Pi values of trnG-UCC and ycf1 were 0.0462 and 0.0457, respectively. The results indicate that the LSC and SSC regions may be undergoing rapid nucleotide substitutions in Zingiberaceae species, and this variation plays an important role in species identification and phylogenetic analysis.

Figure 5. Sliding window analysis based on the complete chloroplast (CP) genomes of six Alpinia species. Window length: 600 bp; step size: 200 bp. X-axis: position of the midpoint of a window. Y-axis: nucleotide diversity of each window.
Discovery of Candidate Markers Based on Highly Variable Regions
Previous molecular identification studies of Panax, Zanthoxylum, and Gentiana species showed that CP genetic markers had high identification capabilities (Lee et al., 2017; Nguyen et al., 2017; Zhou T. et al., 2018). Based on the alignment of complete CP genome sequences, 10 highly variable sites were selected to perform PCR amplification in seven species of Alpinia. Ultimately, five markers (trnS-trnG, trnC-petN, rpl32-trnL, psaC-ndhE, and ndhC-trnV) successfully amplified fragments of the expected sizes, and their PCR products were sent to the Sangon Laboratory for sequencing (Supplementary Table 6). An NJ phylogenetic tree was constructed using high-quality sequences to show the ability of the five highly variable regions in species-level identification (Supplementary Figure 1), using 50% as a cut-off value for the condensed tree. Yang et al. (2021) recently published an article on the development of molecular markers for five medicinal Alpinia species based on complete plastome sequences and developed molecular markers based on two highly variable regions (petN-psbM and psaJ-rpl33). We identified different highly variable sites from those of Yang et al. (2021) possibly because we used different Alpinia species in our analyses. And adding more species might be more accurate for finding and developing highly variable markers. In recent years, numerous studies have used plastid genomes to detect regions with high variation that may be used as molecular markers for species authentication (Jiang et al., 2017; Manzanilla et al., 2018; Zhou T. et al., 2018; Zhou Y. et al., 2018); however, this method is still limited to a few taxa. For example, Cui et al. (2019a) conducted a comparison and phylogenetic analysis of the CP genomes of Wurfbainia (= Amomum) species and identified four potential highly divergent region markers, but none provided sufficient discriminatory power to distinguish the eight study species. We infer that the reason for this result may be due to the slow evolution and monophyletic inheritance of cpDNA, so there are certain limitations in using cpDNA among species with frequent gene exchanges. Alpinia is an extremely complex group, and the method of using only a few species to find the identification markers for the genus remains to be verified. In summary, we used CP genome screening to detect highly variable regions that may be used as molecular markers for the authentication of Alpinia species, but their effectiveness of accurate identification remains to be further determined for the complex relationships within Alpinia.
Phylogenetic Relationships of Alpinia Species
The CP sequence is essential for studying phylogenetic relationships and species identification and for determining taxonomic status in angiosperms (Li et al., 2015, 2019; Zhang et al., 2019). Alpinieae is the largest tribes of Zingiberaceae, including 25 genera. We obtained 28 complete CP genome sequences from NCBI for Alpinieae species (Alpinia, Wurfbainia, and Lanxangia). C. longa and Z. officinale (Zingibereae) was used as an outgroup for the construction of the NJ (Figure 6), MP (Supplementary Figure 2), and ML (Supplementary Figure 3) phylogenetic trees. The results of the NJ, MP, and ML phylogenetic trees further validate the phylogenetic relationships in the Zingiberaceae reported in previous studies (Kress et al., 2002, 2005; Meng et al., 2019). The structure of these phylogenetic trees shows the close relationships among species, with support values of >70%. A. galanga and A. nigra are the first to split off into a separate branch, whereas the remaining species are included in another large branch. Then, the two sequences of Lanxangia tsaoko split off into a branch, and the remaining Alpinia and Wurfbainia species split off into a large branch. Next, Alpinia and Wurfbainia species clustered into two branches, respectively. This result further proves the research conclusion of Kress et al. (2002) and de Boer et al. (2018) that Alpinia is a non-monophyletic genus: the genus Alpinia forms six polyphyletic clades (including clades II and IV in our study). A. oxyphylla (NC-035895 and KY985237) from Hainan is closer to A. chinensis and A. oxyphylla (MK262729)/A. oxyphylla (MK940824) from Guangdong/Guangxi is closer to A. officinarum in Alpinia. This result may be because the CP genome sequences of A. oxyphylla individuals representing different regions and varieties exhibit relatively obvious variations, resulting in greater intraspecific than interspecific variation in the CP genome of A. oxyphylla. Furthermore, we believe that the changes in the relationships between some species are due to addition of more samples therefore species relationships in this genus will be clearer when more species are included in the analyses. Due to the wide variety of Alpinia plants, the taxonomic status and phylogenetic relationships of many species have been difficult to determine, and future phylogenetic analyses should include more CP genome samples. Our results provide a valuable reference and a foundation for using CP genomes in species identification, and aid in improving the understanding of the phylogeny of Alpinia plants.

Figure 6. Phylogenetic tree constructed using the neighbor-joining method based on the 32 complete chloroplast (CP) genomes. Numbers at branch nodes are the bootstrap support values.
Conclusion
We used high-throughput sequencing to sequence the complete CP genomes of A. galanga (160,100 bp) and A. kwangsiensis (162,323 bp), both of which exhibited an obvious quadripartite structure. We then combined these data with publicly available CP genome data for four species (A. hainanensis, A. oxyphylla, A. pumila, and A. zerumbet) and found five candidate highly variable marker sites (trnS-trnG, trnC-petN, rpl32-trnL, psaC-ndhE, and ndhC-trnV) based on the alignment of complete CP genome sequences. Next, we obtained existing CP genome sequences for Alpinieae from NCBI to construct phylogenetic trees and revealed limited phylogenetic relationships within the groups. The complete CP genomes of the two medicinal Alpinia species provides lays the foundation for the use of CP genomes in species identification and phylogenetic analyses of Alpinia species.
Data Availability Statement
The original contributions presented in the study are publicly available. This data can be found here: The complete and correct CP genome sequences of A. galanga and A. kwangsiensis were deposited in GenBank under the accession numbers MZ066611 and MZ066612, respectively.
Author Contributions
Z-LZ and YZ conceived and designed the manuscript. YZ, M-FS, C-YY, and H-FS analyzed the experiments data. YZ executed the manuscript. Z-LZ revised the manuscript. Z-LZ, YZ, D-YT, and A-SX collected the samples. L-XZ and YL provided technical support. All authors approved the final manuscript.
Funding
This study was supported by the Yunnan Science and Technology Talents Platform Program (202105AG070011), National Key R&D Program of China (2019YFC1712301 and 2019YFC1712304), CAMS Initiative for Innovative Medicine (2021-1-I2M-032), and Yunnan Provincial Science and Technology Major Projects: Digitalization, Development, and Application of Biotic Resource (202002AA100007).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.705892/full#supplementary-material
Footnotes
References
Bock, R., and Knoop, V. (2012). Genomics of chloroplasts and mitochondria (advances in photosynthesis and respiration). Adv. Photosynth. Respir. 35, 377–377. doi: 10.1007/978-94-007-2920-9
Boer, H. D., Newman, M., Poulsen, A. D., Droop, A. J., Fér, T., Hiên, L. T. T., et al. (2018). Convergent morphology in Alpinieae (Zingiberaceae): Recircumscribing Amomum as a monophyletic genus. Taxon 67, 6–36. doi: 10.12705/671.2
Boetzer, M., Henkel, C. V., Jansen, H. J., Butler, D., and Pirovano, W. (2011). Scaffolding pre-assembled contigs using SSPACE. Bioinformatics 27, 578–579. doi: 10.1093/bioinformatics/btq683
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Brown, J., Pirrung, M., and Lee, A. M. (2017). FQC Dashboard: integrates FastQC results into a web-based, interactive, and extensible FASTQ quality control tool. Bioinformatics 33, 3137–3139. doi: 10.1093/bioinformatics/btx373
Castro, K. N. C., Canuto, K. M., Brito, E. S., Costa, J. L. M., Andrade, I. M., Magalhães, J. A., et al. (2018). In vitro efficacy of essential oils with different concentrations of 1,8-cineole against Rhipicephalus (Boophilus) microplus. Rev. Bras. Parasitol. Vet. 27, 203–210. doi: 10.1590/s1984-296120180015
Chen, L. Y., Cao, Y. N., Yuan, N., Nakamura, K., Wang, G. M., and Qiu, Y. X. (2015). Characterization of transcriptome and development of novel EST-SSR makers based on next-generation sequencing technology in Neolitsea sericea (Lauraceae) endemic to East Asian land-bridge islands. Mol. Breed. 35:187. doi: 10.1007/s11032-015-0379-1
Chen, X. L., Zhou, J. G., Cui, Y. X., Wang, Y., Duan, B. Z., and Yao, H. (2018). Identification of Ligularia Herbs Using the Complete Chloroplast Genome as a Super-Barcode. Front. Pharmacol. 9:695. doi: 10.3389/fphar.2018.00695
Chouni, A., and Paul, S. (2018). A Review on Phytochemical and Pharmacological Potential of Alpinia galanga. Pharmacognosy J. 10, 9–15. doi: 10.5530/pj.2018.1.2
Cui, Y. X., Chen, X. L., Nie, L. P., Sun, W., Hu, H. Y., Lin, Y. L., et al. (2019a). Comparison and Phylogenetic Analysis of Chloroplast Genomes of Three Medicinal and Edible Amomum Species. Int. J. Mol. Sci. 20:4040. doi: 10.3390/ijms20164040
Cui, Y. X., Nie, L. P., Sun, W., Xu, Z. C., Wang, Y., Yu, J., et al. (2019b). Comparative and Phylogenetic Analyses of Ginger (Zingiber officinale) in the Family Zingiberaceae Based on the Complete Chloroplast Genome. Plants 8:283. doi: 10.3390/plants8080283
Daniell, H., Lin, C. S., Yu, M., and Chang, W. J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17:134. doi: 10.1186/s13059-016-1004-2
Darriba, D., Posada, D., Kozlov, A. M., Stamatakis, A., Morel, B., and Flouri, T. (2019). ModelTest-NG: A New and Scalable Tool for the Selection of DNA and Protein Evolutionary Models. Mol. Biol. Evol. 37, 291–294. doi: 10.1093/molbev/msz189
Denisov, G., Walenz, B., Halpern, A. L., Miller, J., Axelrod, N., Levy, S., et al. (2008). Consensus generation and variant detection by Celera Assembler. Bioinformatics 24, 1035–1040. doi: 10.1093/bioinformatics/btn074
Dierckxsens, N., Mardulyn, P., and Smits, G. (2017). NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45:e18. doi: 10.1093/nar/gkw955
Elzaawely, A. A., Xuan, T. D., Koyama, H., and Tawata, S. (2007). Antioxidant activity and contents of essential oil and phenolic compounds in flowers and seeds of Alpinia zerumbet (Pers.) B.L. Burtt. & R.M. Sm. Food Chem. 104, 1648–1653. doi: 10.1016/j.foodchem.2007.03.016
Ferrarini, M., Moretto, M., Ward, J. A., Surbanovski, N., Stevanovic, V., Giongo, L., et al. (2013). An evaluation of the PacBio RS platform for sequencing and de novo assembly of a chloroplast genome. BMC Genom. 14:670. doi: 10.1186/1471-2164-14-670
Flannery, M. L., Mitchell, F. J., Coyne, S., Kavanagh, T. A., Burke, J. I., and Salamin, N. (2006). Plastid genome characterisation in Brassica and Brassicaceae using a new set of nine SSRs. Theor. Appl. Genet. 113, 1221–1231. doi: 10.1007/s00122-006-0377-0
Frazer, K. A., Lior, P., Alexander, P., Rubin, E. M., and Inna, D. (2004). VISTA: Computational tools for comparative genomics. Nucleic Acids Res. 32:W273. doi: 10.1093/nar/gkh458
Gao, B. M., Yuan, L., Tang, T. L., Hou, J., Pan, K., and Wei, N. (2019). The complete chloroplast genome sequence of Alpinia oxyphylla Miq. and comparison analysis within the Zingiberaceae family. PLoS One. 14:e0218817. doi: 10.1371/journal.pone.0218817
Greiner, S., Lehwark, P., and Bock, R. (2019). Organellar Genome DRAW (OGDRAW) version 1.3.1: Expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Res. 47, W59–W64. doi: 10.1093/nar/gkz238
Guo, H. J., Liu, J. S., Luo, L., Wei, X. P., Zhang, J., Qi, Y. D., et al. (2017). Complete chloroplast genome sequences of Schisandra chinensis: Genome structure, comparative analysis, and phylogenetic relationship of basal angiosperms. Sci. China Life Sci. 60, 1286–1290. doi: 10.1007/s11427-017-9098-5
Hall, T. A. (1999). BioEdit: A user-friendly biological sequence alignment editor and analysis program for windows 95/98/NT. Nucleic Acids Symp. Ser. 41, 95–98. doi: 10.1021/bk-1999-0734.ch008
Huo, Y. M., Gao, L. M., Liu, B. J., Yang, Y. Y., Kong, S. P., Sun, Y. Q., et al. (2019). Complete chloroplast genome sequences of four Allium species: comparative and phylogenetic analyses. Sci. Rep. 9:12250. doi: 10.1038/s41598-019-48708-x
Ivanova, Z., Sablok, G., Daskalova, E., Zahmanova, G., Apostolova, E., Yahubyan, G., et al. (2017). Chloroplast Genome Analysis of Resurrection Tertiary Relict Haberlea rhodopensis Highlights Genes Important for Desiccation Stress Response. Front. Plant Sci. 8:204. doi: 10.3389/fpls.2017.00204
Jiang, D., Zhao, Z., Zhang, T., Zhong, W., Liu, C., Yuan, Q., et al. (2017). The Chloroplast Genome Sequence of Scutellaria baicalensis Provides Insight into Intraspecific and Interspecific Chloroplast Genome Diversity in Scutellaria. Genes 8:227. doi: 10.3390/genes8090227
Katoh, K., Rozewicki, J., and Yamada, K. D. (2019). MAFFT online service: multiple sequence alignment, interactive sequence choice and visualization. Brief Bioinform. 20, 1160–1166. doi: 10.1093/bib/bbx108
Khakhlova, O., and Bock, R. (2006). Elimination of deleterious mutations in plastid genomes by gene conversion. Plant J. 46, 85–94. doi: 10.1111/j1365-313x.2006.02673.x
Kim, K. J., and Lee, H. L. (2004). Complete Chloroplast Genome Sequences from Korean Ginseng (Panax schinseng Nees) and Comparative Analysis of Sequence Evolution among 17 Vascular Plants. DNA Res. 11, 247–261. doi: 10.1093/dnares/11.4.247
Kress, W. J., Liu, A. Z., Newman, M. F., and Li, Q. J. (2005). The molecular phylogeny of Alpinia (Zingiberaceae): a complex and polyphyletic genus of gingers. Am. J. Bot. 92, 167–178. doi: 10.3732/ajb.92.1.167
Kress, W. J., Prince, L. M., and Williams, K. J. (2002). The phylogeny and a new classification of the gingers (Zingiberaceae) evidence from molecular data. Am. J. Bot. 89, 1682–1696. doi: 10.3732/ajb.89.10.1682
Kuang, D. Y., Wu, H., Wang, Y. L., Gao, L. M., Zhang, S. Z., and Lu, L. (2011). Complete chloroplast genome sequence of Magnolia kwangsiensis (Magnoliaceae): implication for DNA barcoding and population genetics. Genome 54, 663–673. doi: 10.1139/G11-026
Kumagai, M., Mishima, T., Watanabe, A., Harada, T., Yoshida, I., Fujita, K., et al. (2016). 5,6-Dehydrokawain from Alpinia zerumbet promotes osteoblastic MC3T3-E1 cell differentiation. Biosci. Biotechnol. Biochem. 80, 1425–1432. doi: 10.1080/09168451.2016.1153959
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2001). Reputer: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Lee, H. J., Koo, H. J., Lee, J., Lee, S. C., Lee, D. Y., Giang, V. N. L., et al. (2017). Authentication of Zanthoxylum species based on integrated analysis of complete chloroplast genome sequences and metabolite profiles. J. Agric. Food Chem. 65, 10350–10359. doi: 10.1021/acs.jafc.7b04167
Li, D. M., Zhu, G. F., Xu, Y. C., Ye, Y. J., and Liu, J. M. (2020). Complete Chloroplast Genomes of Three Medicinal Alpinia Species: Genome Organization, Comparative Analyses and Phylogenetic Relationships in Family Zingiberaceae. Plants 9:286. doi: 10.3390/plants9020286
Li, H. T., Yi, T. S., Gao, L. M., Ma, P. F., Zhang, T., Yang, J. B., et al. (2019). Origin of angiosperms and the puzzle of the Jurassic gap. Nat. Plants 5, 461–470. doi: 10.1038/s41477-019-0421-0
Li, J. Z., Yang, H. B., Liu, Y., Chen, C., and Luo, Y. S. (2020). Identification of Alpinia officinarum and Alpinia Galanga by HS-SPME-GC-MS. Hubei. Agr. Sci. 59, 135–138. doi: 10.14088/j.cnki.issn0439-8114.2020.01.030
Li, J., Du, Q. Z., Li, N., Du, S. Z., and Sun, Z. (2020). Alpiniae oxyphyllae Fructus and Alzheimer’s disease: An update and current perspective on this traditional Chinese medicine. Biomed. Pharmacother. 135:111167. doi: 10.1016/j.biopha.2020.111167
Li, X. W., Yang, Y., Henry, R. J., Rossetto, M., Wang, Y. T., and Chen, S. L. (2015). Plant DNA barcoding: from gene to genome. Biol. Rev. Camb. Philos. Soc. 90, 157–166. doi: 10.1111/brv.12104
Li, Y., Kuang, X. J., Zhu, X. X., Zhu, Y. J., and Sun, C. (2016). Codon usage bias of Catharanthus roseus. China J. Chin. Mater. Med. 41, 4165–4168. doi: 10.4268/cjcmm20162213
Liu, X., Li, Y., Yang, H., and Zhou, B. (2018). Chloroplast genome of the folk medicine and vegetable plant Talinum paniculatum (Jacq.) Gaertn.: Gene organization, comparative and phylogenetic analysis. Molecules 23:857. doi: 10.3390/molecules23040857
Lowe, T. M., and Chan, P. P. (2016). tRNAscan-SE On-line: Search and Contextual Analysis of Transfer RNA Genes. Nucleic Acids Res. 44, W54–W57. doi: 10.1093/nar/gkw413
Luo, R. B., Liu, B. H., Xie, Y. L., Li, Z. Y., Huang, W. H., Yuan, J. Y., et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. Gigascience 1:18. doi: 10.1186/2047-217X-1-18
Manzanilla, V., Kool, A., Nguyen, N. L., Nong, V. H., Le, T. T. H., and Boer, H. J. (2018). Phylogenomics and barcoding of Panax: toward the identification of ginseng species. BMC Evol. Biol. 18:44. doi: 10.1186/s12862-018-1160-y
Medicinal Materials Company in Yunnan Province (1993). List of Traditional Chinese Medicine Resources in Yunnan Province. China: Science Press.
Meng, J., Jiang, Hui, He, J., He, Y. H., Zhang, Y. W., and Zhao, Y. (2019). The first complete chloroplast genome sequence of Lanxangia tsaoko and phylogenetic analysis. Mitochondrial. DNA B Resour. 4, 2320–2321. doi: 10.1080/23802359.2019.1629354
Nazareno, A. G., Carlsen, M., and Lohmann, L. G. (2015). Complete chloroplast genome of Tanaecium tetragonolobum: the first Bignoniaceae plastome. PLoS One. 10:e0129930. doi: 10.1371/journal.pone.0129930
Nguyen, V. B., Park, H. S., Lee, S. C., Lee, J., Park, J. Y., and Yang, T. J. (2017). Authentication markers for five major Panax species developed via comparative analysis of complete chloroplast genome sequences. J. Agric. Food Chem. 65, 6298–6306. doi: 10.1021/acs.jafc.7b00925
Park, I., Yang, S., Kim, W. J., Noh, P., Lee, H. O., and Moon, B. C. (2018). Authentication of Herbal Medicines Dipsacus asper and Phlomoides umbrosa Using DNA Barcodes, Chloroplast Genome, and Sequence Characterized Amplified Region (SCAR) Marker. Molecules 23:1748. doi: 10.3390/molecules23071748
Qian, J., Song, J. Y., Gao, H. H., Zhu, Y. J., Xu, J., Pang, X., et al. (2013). The Complete Chloroplast Genome Sequence of the Medicinal Plant Salvia miltiorrhiza. PLoS One. 8:e57607. doi: 10.1371/journal.pone.0057607
Rozas, J., Ferrermata, A., Sánchezdelbarrio, J. C., Guiraorico, S., Librado, P., Ramosonsins, S. E., et al. (2017). DnaSP 6: DNA Sequence Polymorphism Analysis of Large Datasets. Mol. Biol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Sato, S., Nakamura, Y., Kaneko, T., Asamizu, E., and Tabata, S. (1999). Complete structure of the chloroplast genome of Arabidopsis thaliana. DNA Res. 6, 283–290. doi: 10.1093/dnares/6.5.283
Sharp, P. M., and Li, W. H. (1987). The codon Adaptation Index-a measure of directional synonymous codon usage bias, and its potential applications. Nucleic Acids Res. 15, 1281–1295. doi: 10.1093/nar/15.31281
Sousa, D. P., Hocayen, P. A. S., Andrade, L. N., and Andreatini, R. (2015). A Systematic Review of the Anxiolytic-Like Effects of Essential Oils in Animal Models. Molecules 20, 18620–18660. doi: 10.3390/molecules201018620
Upadhye, A. S., Rajopadhye, A., and Dias, L. (2018). Development and validation of HPTLC fingerprints of three species of Alpinia with biomarker Galangin. BMC Compl. Altern. Med. 18:16. doi: 10.1186/s12906-017-2033-4
Wang, Y., Zhan, D. F., Jia, X., Mei, W. L., Dai, H. F., Chen, X. T., et al. (2016). Complete chloroplast genome sequence of Aquilaria sinensis (Lour.) Gilg and evolution analysis within the malvales order. Front. Plant Sci. 7:280. doi: 10.3389/fpls.2016.00280
Wicke, S., Schneeweiss, G. M., Pamphilis, C. W., Muller, K. F., and Quandt, D. (2011). The evolution of the plastid chromosome in land plants: Gene content, gene order, gene function. Plant Mol. Biol. 76, 273–297. doi: 10.1007/s11103-011-9762-4
Wu, D., Liu, N., and Ye, Y. (2016). The Zingiberaceous Resources in China*. China: Huazhong University of Science and Technology University Press.
Wu, M. L., Li, Q., Hu, Z. G., Li, X. W., and Chen, S. L. (2017). The Complete Amomum kravanh Chloroplast Genome Sequence and Phylogenetic Analysis of the Commelinids. Molecules 22:1875. doi: 10.3390/molecules22111875
Wu, Y., Zhang, W. J., Huang, D. Y., Wang, Y., Wei, J. Y., Li, Z. H., et al. (2015). Chemical Compositions and Insecticidal Activities of Alpinia kwangsiensis Essential Oil against Lasioderma serricorne. Molecules 20, 21939–21945. doi: 10.3390/molecules201219818
Wyman, S. K., Jansen, R. K., and Boore, J. L. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Xiao, T., Huang, J. Y., Wang, X. W., Wu, L. J., Zhou, X., Jiang, F., et al. (2020). Alpinia zerumbet and Its Potential Use as an Herbal Medication for Atherosclerosis: Mechanistic Insights from Cell and Rodent Studies. Lifestyle Genom. 13, 138–145. doi: 10.1159/000508818
Xuan, T. D., Khanh, T. D., Khang, D. T., Quan, N. T., and Elzaawely, A. A. (2016). Changes in Chemical Composition, Total Phenolics and Antioxidant Activity of Alpinia (Alpinia zerumbet) Leaves Exposed to UV. Int. Lett. Nat. Sci. 55, 25–34. doi: 10.18052/www.scipress.com/ILNS.55.25
Yang, H. Y., Wang, L. Q., Chen, H. M., Jiang, M., Wu, W. W., Liu, S. Y., et al. (2021). Phylogenetic analysis and development of molecular markers for five medicinal Alpinia species based on complete plastome sequences. BMC Plant Biol. 21:431. doi: 10.1186/s12870-021-03204-1
Ye, J., Coulouris, G., Zaretskaya, I., Cutcutache, I., Rozen, S., and Madden, T. L. (2012). Primer-BLAST: a tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 13:134. doi: 10.1186/1471-2105-13-134
Zhang, X. L., Shi, G. F., Liu, X. Z., An, L. J., and Guan, S. (2011). Anti-ageing effects of protocatechuic acid from Alpinia on spleen and liver antioxidative system of senescent mice. Cell Biochem. Funct. 29, 342–347. doi: 10.1002/cbf.1757
Zhang, Y., Song, M. F., Li, H. T., Sun, H. F., and Zhang, Z. L. (2021). DNA barcoding identification of original plants of a rare medicinal material Resina Draconis and related Dracaena species. China J. Chin. Mater. Med. 46, 2173–2181. doi: 10.19540/j.cnki.cjcmm.20210124.104
Zhang, Z. L., Zhang, Y., Song, M. F., Guan, Y. H., and Ma, X. J. (2019). Species Identification of Dracaena Using the Complete Chloroplast Genome as a Super-Barcode. Front. Pharmacol. 11:1441. doi: 10.3389/fphar.2020.00051
Zhao, Z. L., Wang, Z. T., Dong, H., Xu, L. S., and Lin, X. F. (2001). Advances in studies on medicinal plants and pharmacognosy of Aplinia Roxb [J]. Chin. Tradit. Herbal Drugs 32, 171–173. doi: 10.7501/j.issn.0253-2670.2001.2.087
Zhong, Z. M., Lai, X. P., Huang, S., and Zhang, G. F. (2018). Identification and clustering analysis of Zingiberaceae plant based on ycf1 barcode[J]. Chin. Tradit. Herbal Drugs. 33, 4089–4092.
Zhou, J., Cui, Y., Chen, X., Li, Y., Xu, Z., Duan, B., et al. (2018). Complete chloroplast genomes of Papaver rhoeas and Papaver orientale: Molecular structures, comparative analysis and phylogenetic analysis. Molecules 23:437. doi: 10.3390/molecules23020437
Zhou, T., Wang, J., Jia, Y., Li, W., Xu, F., Wang, X., et al. (2018). Comparative Chloroplast Genome Analyses of Species in Gentiana section Cruciata (Gentianaceae) and the Development of Authentication Markers. Int. J. Mol. Sci. 19:1962. doi: 10.3390/ijms19071962
Zhou, Y., Nie, J., Xiao, L., Hu, Z., and Wang, B. (2018). Comparative Chloroplast Genome Analysis of Rhubarb Botanical Origins and the Development of Specific Identification Markers. Molecules 23:2811. doi: 10.3390/molecules23112811
Keywords: Alpinia galanga, Alpinia kwangsiensis, chloroplast genome, Zingiberaceae, phylogenetic relationship
Citation: Zhang Y, Song M-F, Li Y, Sun H-F, Tang D-Y, Xu A-S, Yin C-Y, Zhang Z-L and Zhang L-X (2021) Complete Chloroplast Genome Analysis of Two Important Medicinal Alpinia Species: Alpinia galanga and Alpinia kwangsiensis. Front. Plant Sci. 12:705892. doi: 10.3389/fpls.2021.705892
Received: 06 May 2021; Accepted: 18 November 2021;
Published: 15 December 2021.
Edited by:
Isabel Larridon, Royal Botanic Gardens, Kew, United KingdomReviewed by:
Surabhi Ranavat, Royal Botanic Garden Edinburgh, United KingdomYannick Woudstra, University of Oxford, United Kingdom
Copyright © 2021 Zhang, Song, Li, Sun, Tang, Xu, Yin, Zhang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhong-Lian Zhang, enpsMDYwNUAxNjMuY29t; Li-Xia Zhang, ODcwNTAyMzNAcXEuY29t