 Shiyong Chen
Shiyong Chen Hao Yan2
Hao Yan2- 1College of Animal and Veterinary Sciences, Southwest Minzu University, Chengdu, China
- 2Triticeae Research Institute, Sichuan Agricultural University, Chengdu, China
- 3Key Laboratory of Crop Genetic Resources and Improvement, Ministry of Education, Sichuan Agricultural University, Ya’an, China
Kengyilia is a group of allohexaploid species that arose from two hybridization events followed by genome doubling of three ancestral diploid species with different genomes St, Y, and P in the Triticeae. Estimating the phylogenetic relationship in resolution of the maternal lineages has been difficult, owing to the extremely low rate of sequence divergence. Here, phylogenetic reconstructions based on the plastome sequences were used to explore the role of maternal progenitors in the establishment of Kengyilia polyploid species. The plastome sequences of 11 Kengyilia species were analyzed together with 12 tetraploid species (PP, StP, and StY) and 33 diploid taxa representing 20 basic genomes in the Triticeae. Phylogenomic analysis and genetic divergence patterns suggested that (1) Kengyilia is closely related to Roegneria, Pseudoroegneria, Agropyron, Lophopyrum, Thinopyrum, and Dasypyrum; (2) both the StY genome Roegneria tetraploids and the PP genome Agropyron tetraploids served as the maternal donors during the speciation of Kengyilia species; (3) the different Kengyilia species derived their StY genome from different Roegneria species. Multiple origins of species via independent polyploidization events have occurred in the genus Kengyilia, resulting in a maternal haplotype polymorphism. This helps explain the rich diversity and wide adaptation of polyploid species in the genus Kengyilia.
Introduction
Polyploidy, defined as the possession of two or more sets of homologous chromosomes following whole-genome duplication, is a major mechanism in plant evolution and speciation (Otto, 2007; Spoelhof et al., 2017). Recent studies even suggested that multiple origins (including independent origin) of polyploid species are the rule rather than the exception (Soltis and Soltis, 2000; Symonds et al., 2010; Fan et al., 2013; Sha et al., 2017a). Polyploidy promotes variability through the change in the chromosomal number per se, increased genetic diversity, and genomic reorganization, leading to benefits in new phenotypes and evolutionary innovation in physiological and ecological flexibility (Ramsey and Ramsey, 2014; Sha et al., 2017b). However, a clear and appropriate identification of phylogenetic relationships among taxa and genomes is needed (Fan et al., 2013).
Kengyilia Yen et J. L. Yang, a polyploid perennial genus in the wheat tribe (Poaceae: Triticeae), includes about 22 perennial species that were distributed in a wide range of natural habitats over the upper and middle mountain ranges of Central Asia and the Qinghai-Tibetan Plateau (Yang et al., 1992). Kengyilia was also often classified in Elymus L. sensu lato, which was the largest taxon in Triticeae, including Roegneria, Kengyilia, and Elymus sensu stricto (Löve, 1984). A comparison of the morphological features among the genera Kengyilia, Roegneria, Elymus, and Agropyron suggested that species in Kengyilia are intermediate between the species of Roegneria C. Koch and Agropyron Gaertn., but with some distinct morphological divergence in the spikelet characters between Kengyilia and Roegneria and between Kengyilia and Agropyron (Baum et al., 1995). Kengyilia species exhibit variation with high (K. grandiglumis) to low (K. thoroldiana) plants, lax (K. rigidula) to dense (K. hirsuta) spikes, adnate (K. longilumis) to incohesive (K. stenachyra) spikelets attached to the rachis, and yellow (K. gobicola) to black (K. melanthera) anthers.
All the species of Kengyilia are allohexaploids (2n = 6x = 42) with StYP genomes (Yang et al., 1992; Yen et al., 2006). The St and P genomes have originated from Pseudoroegneria (Nevski) Á. Löve and Agropyron Gaertn., respectively (Löve, 1984). It is unknown where the Y genome originates, although it is a fundamental Kengyilia genome (Yen et al., 2006; Fan et al., 2013). Cytogenetic evidence suggested that speciation of the Kengyilia polyploid was derived from hybridization between tetraploid Roegneria species (2n = 4x = 28, StY) and diploid Agropyron species (2n = 2x = 14, P) (Yen et al., 2006; Fan et al., 2013). Analysis of nuclear single-copy Pgk1 gene sequences suggested that Kengyilia species from Central Asia and the Qinghai-Tibetan Plateau have independent origins with geographically differentiated P genome donors (Fan et al., 2012). Data from chloroplast trnL-F, matK, rbcL, trnH-psbA, and mitochondrial CoxII suggested that different species of Kengyilia have derived their maternal lineages either from the species of Pseudoroegneria or the species of Agropyron or an unknown donor (Zhang et al., 2009; Zeng et al., 2010; Luo et al., 2012). Analysis of trnL-F, matK, and rbcL sequences showed that four species of Kengyilia (K. kokonorica, K. melanthera, K. mutica, and K. thoroldiana) were related to species of Agropyron, and the remaining sampled species were close to species of Pseudoroegneria (Zhang et al., 2009; Luo et al., 2012). In the trnH-psbA tree, four species of Kengyilia (K. grandiglumis, K. hirsuta, K. laxiflora, and K. rigidula) were grouped with Agropyron, which is inconsistent with the maternal relationship presented by the trnL-F, matK, and rbcL sequence data (Zhang et al., 2009; Luo et al., 2012). Moreover, analysis of CoxII suggested that some species of Kengyilia (e.g., K. batalinii), Agropyron, and Pseudoroegneria formed a paraphyletic grade with zero-length branches (Zeng et al., 2010). While these studies added to our understanding of the phylogenetic relationships of Kengyilia, the molecular phylogenies based on published chloroplast DNA (trnL-F, matK, rbcL, and trnH-psbA) and mitochondrial sequences in resolution of the maternal lineages of Kengyilia species are still in dispute either due to the unresolved gene tree with polytomies or incongruence among the cytoplasmic gene data (Zhang et al., 2009; Zeng et al., 2010; Luo et al., 2012). Moreover, the processes that have driven polyploid diversification and speciation, especially with regard to which tetraploid and diploid species, as maternal progenitors, were involved in the hexaploid evolution in Kengyilia, remain unclear. Thus, to better understand the maternal contribution to the evolution of the species of Kengyilia, it is essential to conduct a good comparative study of chloroplast genome-wide in Kengyilia and its relatives, covering nearly all of the genomic combinations in Triticeae.
By integrating 38 newly and 18 previously sequenced plastomes representing the StYP genomes and its related tetraploid and diploid genomic types in Triticeae, this study applies phylogenetic reconstruction methods in combination with an estimate of the genetic distance among the coding region to clarify maternal lineage relationships. Our objectives are to demonstrate a phylogenomic framework for illustrating the maternal donor of Kengyilia polyploids and to explore the role of maternal progenitors in the establishment of Kengyilia polyploids.
Materials and Methods
Plant Materials
A total of 23 polyploids, comprising 11 Kengyilia (StYP genomes) species, eight Agropyron (PP) tetraploids, one Douglasdeweya (StP genomes) species, and three Roegneria (StY genomes) species, were analyzed together with 33 diploid taxa representing 20 basic genomes in the Triticeae (Supplementary Table 1). Brachypodium distachyon was used as an outgroup. The chloroplast genome sequences of Triticum–Aegilops complex, Secale, Pseudoroegneria cognata, Pseudoroegneria stipifolia, Pseudoroegneria strigosa, and Pseudoroegneria tauri were from the published data (Gornicki et al., 2014; Bernhardt et al., 2017). The remaining sequences in Supplementary Table 1 in Triticeae were newly sequenced. Sample information including accession numbers, origins, genome type, ploidy, and GenBank accession data were also listed in Supplementary Table 1. The seed materials with PI and W6 numbers were graciously provided by the American National Plant Germplasm System (Pullman, WA, United States). The seed materials with ZY and Y numbers were collected by the authors of this article. The plants used for sequencing and voucher specimens are deposited at the Herbarium of Triticeae Research Institute, Sichuan Agricultural University, China (SAUTI).
Plastome Sequencing and Data Assembly
DNA extractions were performed on young leaves dried in silica using the DNeasy Plant Mini kit (QIAGEN, Valencia, CA, United States) according to the instructions of the manufacturer after homogenization with liquid nitrogen. The plastomes were amplified in overlapping fragments using the long-range PCR method of Yang et al. (2014). The PCR products were fragmented into short inserts (400–600 bp) to construct the sequencing paired-end library according to the NEBNext® protocol. DNA from each individual was indexed using tags and pooled together in one lane of an Illumina Hiseq 4000 PE150 for sequencing. The raw reads were trimmed using Fastp v0.20.0 (Chen et al., 2018) and the clean reads were subjected to de novo assembly using NOVOPlasty 4.0 (Dierckxsens et al., 2016) with default parameters. Approximately, 100 million reads were randomly selected and aligned to the close-related sequences of Agropyron cristatum using BWA v0.7.15 (Li and Durbin, 2009) (MEM algorithm), and a perfect matched read to the psbA gene was selected as the seed input. NOVOPlasty produced two optional sequences which were simply different in small single-copy region (SSC) direction. The sequence with the same SSC direction to Agropyron cristatum (GenBank No. KY126307) and Pseudoroegneria libanotica (GenBank No. KX822019) was selected and subjected to annotation.
Sequence Alignment and Analysis
The complete chloroplast sequences were aligned with MAFFT v. 7 (Katoh and Standley, 2013) using the default settings. All alignments were visually inspected in MEGA 6.0 (Tamura et al., 2013) and manually adjusted where needed. We also conducted a co-linear analysis using the software LASTZ, and the results were visualized using AliTV (Ankenbrand et al., 2017). Multiple alignment of the protein-coding sequence was conducted using ClustalW in MEGA 6 (Tamura et al., 2013), with manual adjustment. Amino acid translations were used to guide the nucleotide alignments. The sequence statistics, including nucleotide substitutions, Kimura 2-parameter (K2-p) distances, transition/transversion ratio, and variability of the sequences, were calculated by MEGA 6 (Tamura et al., 2013).
To estimate the genetic differentiation of the protein-coding sequences between Kengyilia and its close relatives, the K2-p model was used to calculate the genetic distances of the protein-coding sequences of 52 genes (76 unique genes, excluding 24 which have no variable nucleotide sites and/or are <200 bp). A total of 1,664 (32 samples and 52 protein-coding genes) genetic distances were used to estimate the genome relationship employing hierarchical agglomerative clustering in R (Version 3.4.2; Vienna, Austria). The Hopkins statistic was used for the evaluation of the clustering tendency. The optimal number of clusters was determined by the “fviz_dend” algorithm in the R package “factoextra” Version 1.0.5. Bivariate cluster (k-means clustering) analysis based on genetic distances and agglomerative hierarchical clustering was performed by the “clustplot” function in the R package “cluster” package (Version 2.0.6).
Phylogenetic Analysis
Because complete chloroplast genome sequences offer the greatest phylogenetic resolution (Ma et al., 2014), phylogenomic trees were generated from the complete genomes of all sampled chloroplasts. Phylogenomic analyses were conducted using maximum likelihood (ML) and Bayesian inference (BI). The ML analysis was performed using the IQ-Tree software.1 The TVM + F + R3 model was selected for the ML analysis according to ModelFinder implemented in IQ-Tree. To assess branch support, the IQ-Tree analyses used the ultrafast bootstrap approximation (UFboot) with 10,000 replicates (Minh et al., 2013) and the SH-like approximate likelihood ratio test (SH-aLRT) with 1000 bootstrap replicates (Guindon et al., 2010).
The evolutionary model used for BI analysis was determined using ModelTest v3.7 (Ronquist et al., 2012) with Akaike information criterion (AIC). The BI analysis was performed using MrBayes v3.2 (Posada and Crandall, 1998) under the GTR + G + I model that was identified as the best fit by ModelTest. Four Markov Chain Monte Carlo (MCMC) chains (one cold and three heated), applying MrBayes default heating values (t = 0.2), were run 1,000,000 generations for plastome data, with each sampled in each data set every 100 generations. Majority-rule (>50%) consensus trees were generated with a relative burn-in of 25%. The statistical confidence in nodes was estimated by posterior probabilities (PP). A PP-value less than 90% was not included in the figures.
Results
Characteristics of Chloroplast Genomes and Genes
All sequenced genomes are very similar to the published chloroplast genomes of Triticeae (Gornicki et al., 2014; Bernhardt et al., 2017) and are rather conservative in genome structure and gene content. Their genome size ranges from 134,985 in Roegneria ciliaris to 135,489 bp in A. cristatum (ZY09064). All plastomes exhibited a typical quadripartite structure that included a pair of IRs separated by a large single-copy region (LSC) and a SSC and contained a total of 109 genes (including 76 protein coding genes, 29 tRNA genes and 4 rRNA genes). Assemblies in the genus Kengyilia averaged 135,113 bp, with an estimated 0.064% insertion data (compared to P. libanotica reference); genus Roegneria assemblies averaged just less than 135,079 bp (0.039% estimated insertion data, compared to P. libanotica reference).
Analysis of co-linearity is inferred for two diploid taxa representing St and P genomes, one tetraploid species with the StP genome, one tetraploid species with the StY genome, and three hexaploid species with the StYP genome (Figure 1). Despite a high degree of co-linearity among these genomes due to the conservation in chloroplast genome structure and gene content, five big indels (at positions 17,819–18,278, 56,172–56,963, 62,664–63,130, 83,590–84,338, and 130,804–131,592 bp, respectively) were detected between the St- and P-containing lineages, which is indicative of high genetic divergence between them.
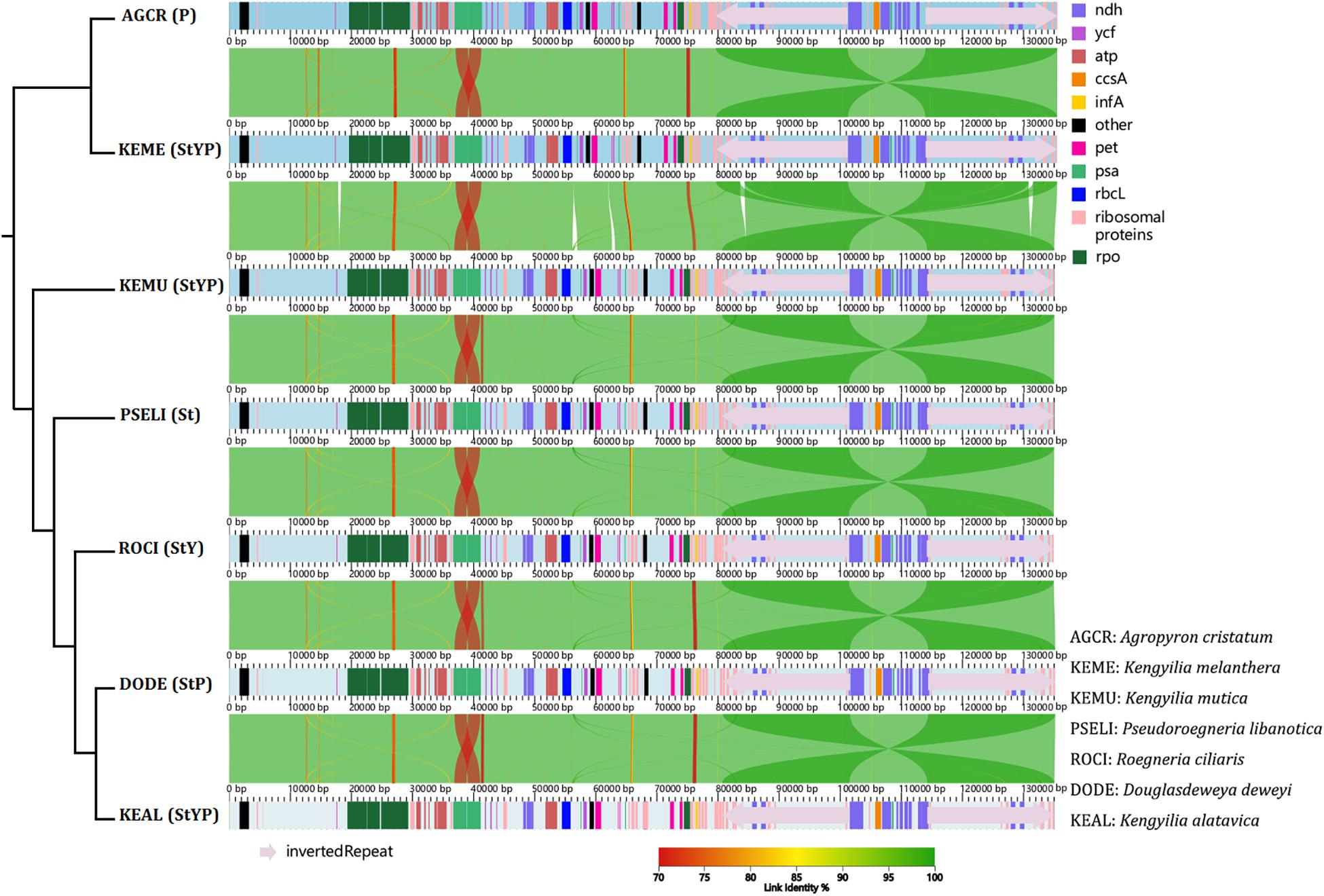
Figure 1. The co-linear analysis of the chloroplast genomes of Kengyilia species and its close relatives. The phylogenetic tree was constructed based on the complete chloroplast genomes using ML.
The features of each of the 76 protein-coding genes in the diploid–polyploid plastome data are summarized in Supplementary Table 2. The lengths of each gene ranged from 90 (petN) to 4,440 (rpoC2) bp. The proportion of the variable sites (variable sites/total sites, V/T) varied from 0 (e.g., petG) to 3.36% (rpl32). The ratio of parsimony-informative characters per total aligned characters was greatest for petL (2.08%) and lowest for petG, psbF, and rpl23 (0).
Phylogenetic Analyses
Bayesian phylogenetic reconstruction of the plastome data under the GTR + G + I model resulted in a tree with high posterior probability support across most clades. The ML analyses in IQ-Tree under the TVM + F + R3 model recovered the same topology as the Bayesian analyses (Supplementary Figure 1).
The tree illustrated in Figure 2 was the BI tree with statistical support (UFboot, SH-aLRT, and PP) above branches. The phylogenetic tree showed that plastome sequences of Kengyilia were split into two major clades (Clade I and II) with consistent statistical support (100% UFboot and SH-aLRT; 1.0 PP). The Clade I included Thinopyrum (Eb), Lophopyrum (Ee), Dasypyrum (V), and Pseudoroegneria (St), as well as all the sampled St-containing (Douglasdeweya, StP; Roegneria, StY; and Kengyilia, StYP) polyploid species (except for K. melanthera), which also had the consistent statistical support (100% UFboot and SH-aLRT; 1.0 PP). In this clade, Thinopyrum, Lophopyrum, Dasypyrum, Douglasdeweya, four species of Pseudoroegneria (P. stipifolia, P. cognata, P. Libanotica, and P. tauri), two species of Roegneria (R. grandis and R. ciliaris), and four species of Kengyilia (K. alatavica, K. hirsuta, K. laxiflora, and K. batalinii) were in one subclade (99.8% UFboot, 100% SH-aLRT, and 1.0 PP). K. alatavica from Central Asia formed a paraphyletic grade with Thinopyrum, Lophopyrum, Dasypyrum, Douglasdeweya, and two species of Pseudoroegneria (P. stipifolia and P. cognata). Two Kengyilia species from Central Asia (K. hirsuta and K. batalinii) and one Kengyilia species from the Qinghai-Tibetan Plateau (K. laxiflora) were clustered with two species of Roegneria (R. grandis and R. ciliaris) (95% UFboot, 95% SH-aLRT, and 1.0 PP). K. kokonorica from the Qinghai-Tibetan Plateau and two species of Pseudoroegneria (P. libanotica and P. tauri) formed a paraphyletic grade in the subclade. Five species of Kengyilia from the Qinghai-Tibetan Plateau (K. thoroldiana, K. grandiglumis, K. mutica, K. stenachyra, and K. rigidula) were grouped with one species of Roegneria (R. longearistata) (100% UFboot, 100% SH-aLRT, and 1.0 PP), and this group was a sister group to two accessions of Pseudoroegneria spicata (99.6% UFboot, 100% SH-aLRT, and 1.0 PP). The clade II contained all sampled Agropyron species (A. cristatum and A. mongolicum) and K. melanthera from the Qinghai-Tibetan Plateau (100% UFboot, 100% SH-aLRT, and 1.0 PP).
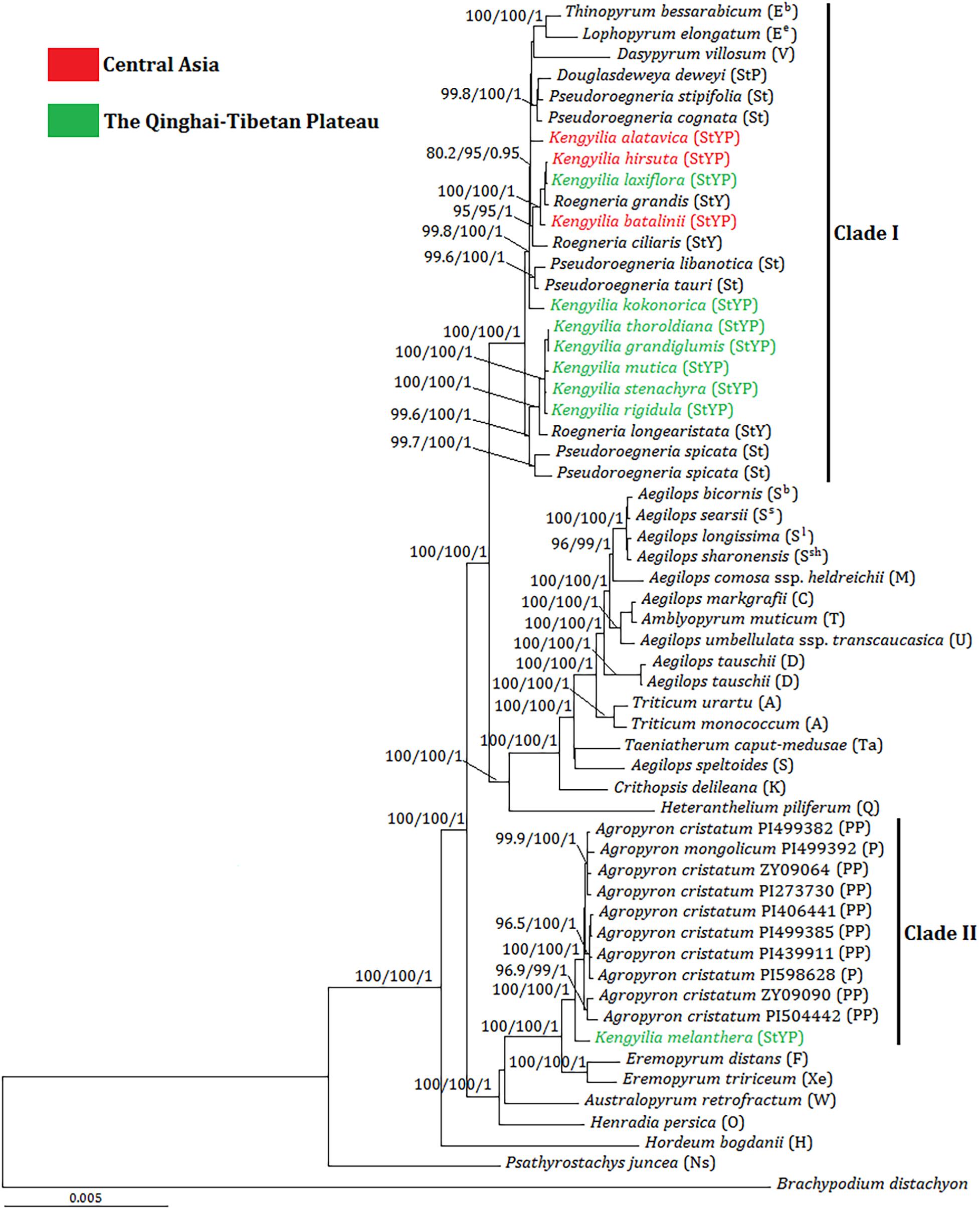
Figure 2. Bayesian tree inferred from the complete chloroplast genome sequences for Kengyilia species and its diploid relatives in the Triticeae. Numbers above branches are values of the statistical support values, and values are indicated only if deemed robust as follows: UFboot ≥ 95%/SH-aLRT ≥ 80%/PP ≥ 0.9. The capital letters in brackets indicate the genome type of the species. Different colors indicated the geographic information of Kengyilia species.
Statistic of K2-p Distance Matrix
A distance matrix including 1,664 genetic values was generated to investigate the relationship between the plastomes of Kengyilia and those of its close relatives (Supplementary Table 3). The Hopkins statistic was found to be 0.2057, indicating that data provides information to cluster the samples. Analysis of hierarchical agglomerative clustering shows four major clusters (Figure 3), which correspond to the four genomic types (P/StYP, Ee/Eb, StY/StYP, and St/StP/StY/StYP). This is also well congruent with the groupings in the phylogenomic tree inferred from the plastome data including all sampled Triticeae plants.
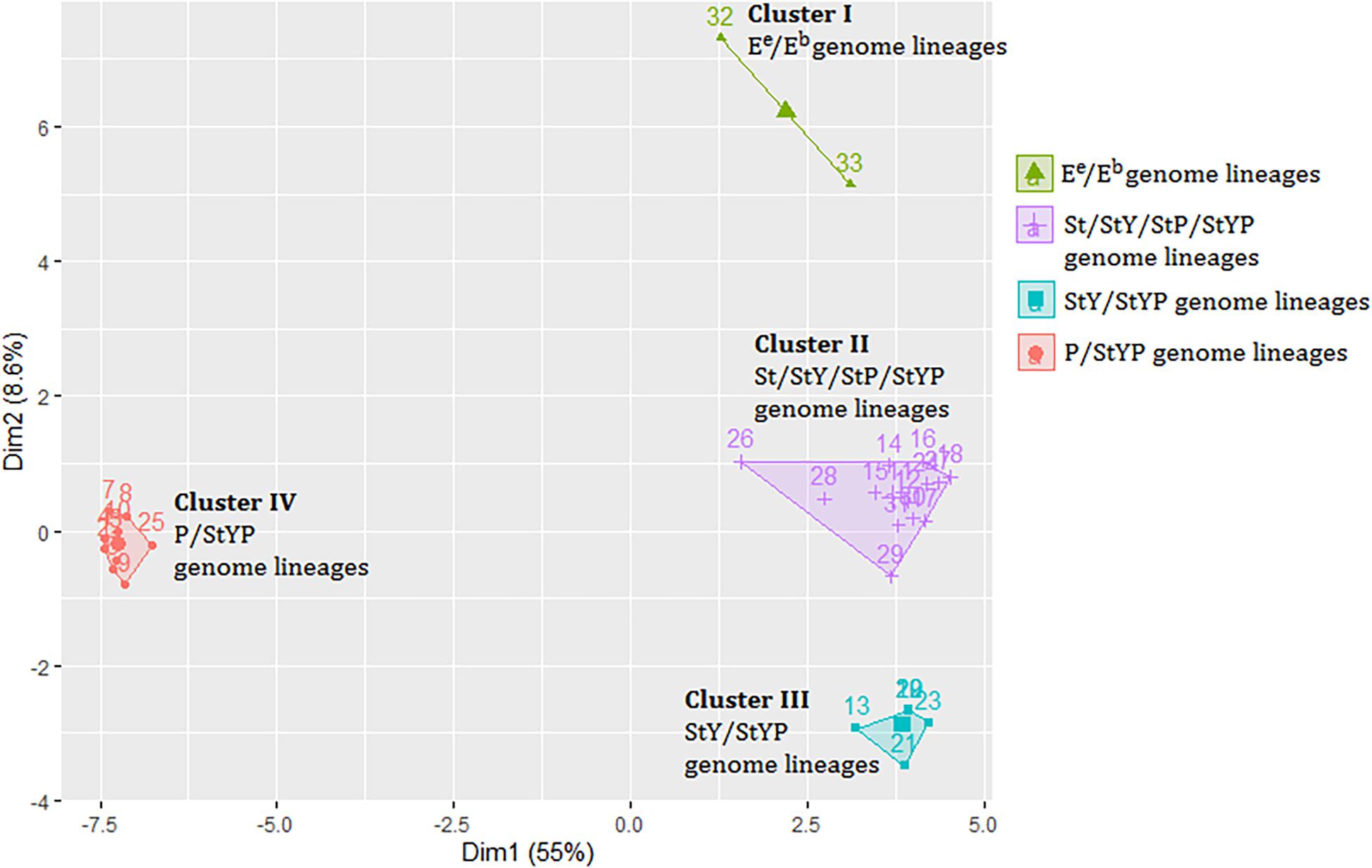
Figure 3. The cluster plot based on a distance matrix of Kengyilia species and its close relatives. Numbers represent lineages listed in Supplementary Table 3.
Discussion
The cpDNA-based (trnL-F, matK, rbcL, and trnH-psbA) phylogenies of the genus Kengyilia, especially with regard to the origin of the maternal donor during hexaploid polyploidization events, were largely unresolved due to the occurrence of many polytomies and incongruence among the published gene trees (Zhang et al., 2009; Luo et al., 2012). Ma et al. (2014) pointed out that despite missing samples, phylogenetic analysis of plastome sequences can offer the greatest phylogenetic resolution. In this study, a resolved tree with high statistical support was inferred from the plastome sequences of Kengyilia and those of its relatives in Triticeae, allowing the relationship regarding the maternal lineages of Kengyilia to be clarified.
In the phylogenomic tree, 10 species of Kengyilia (K. alatavica, K. hirsuta, K. laxiflora, K. batalinii, K. kokonorica, K. thoroldiana, K. grandiglumis, K. mutica, K. stenachyra, and K. rigidula), Roegneria, and Pseudoroegneria were in one group with consistent support, indicating that Pseudoroegneria is likely to be the maternal donor of these 10 StYP genome Kengyilia species and the sampled StY genome Roegneria species. Since Kengyilia species arose from two hybridization events followed by three genome doublings (the St, Y, and P genomes), with one first generating the StY genome Roegneria and the other forming the StYP genome (Yen et al., 2006; Fan et al., 2012); Roegneria served as the maternal donor during the speciation of the 10 Kengyilia species.
Analysis of trnL-F suggested that four species of Kengyilia (K. kokonorica, K. melanthera, K. mutica, and K. thoroldiana) were closely related to species of Agropyron (Zhang et al., 2009). A similar deep-level relationship regarding the maternal lineages is also presented by Luo et al. (2012), although molecular characters (including matK, rbcL, and trnH-psbA) and more taxa were sampled from Kengyilia. In this study, only K. melanthera was grouped with the species of Agropyron, and the remaining three species (K. kokonorica, K. mutica, and K. thoroldiana) were placed into the clade including St-containing species. Moreover, the plastome sequence of K. melanthera and Agropyron are obviously distinct from those of the St-containing species. Thus, the molecular phylogenies based on the published cpDNA fragments and the present plastome sequence data in resolution of the placement of K. kokonorica, K. mutica, and K. thoroldiana led to contradictory results, apparently. Discordances among the phylogenetic trees result from methodological artifacts (e.g., sampling error and/or a failure of molecular characters) and the complex dynamics of the evolutionary processes in organisms (e.g., hybridization and/or ancestral polymorphisms) (Betancur-R et al., 2014; Sha et al., 2017a). Sampling error is likely to be the candidate for the current incongruences because our samples for the comparative phylogenies with Kengyilia species included nearly all of the monogenomic genera accepted in genome-based classifications of the Triticeae, and most monogenomic genera were not covered in the previous study (Zhang et al., 2009; Luo et al., 2012). It is well known that molecular characters can affect the accuracy of phylogenetic estimates (Ma et al., 2014). Incongruences would also be the result of a lack of molecular characters. Fewer molecular characters in cpDNA regions, as indicated by our estimate for the variable features of each chloroplast protein-coding gene (Supplementary Table 3), together with their slowly evolving rates in the chloroplast genome, would not only provide lower variable information for the accuracy of the phylogenetic reconstruction but also result in the occurrence of polytomies in the phylogenetic tree. On the contrary, the plastome data offer enough molecular characters for the accuracy of phylogenetic estimates with a well-supported topology. Both hybridization and ancestral polymorphisms acting alone or in concert can generate discordance and therefore are the principal processes to explain the phylogenetic incongruence in Triticeae species (Mason-Gamer, 2013; Sha et al., 2017a).
The analysis of the genetic distance matrix based on the 52 protein-coding genes suggested that Lophopyrum and Thinopyrum are closely related to the St-containing species. In the phylogenomic tree inferred from the complete chloroplast genome, Lophopyrum, Thinopyrum, Dasypyrum, and two species of Pseudoroegneria (P. stipifolia and P. congnata) form a monophyletic group. These results indicated Lophopyrum, Thinopyrum, Dasypyrum, and Pseudoroegneria (most likely P. stipifolia and P. congnata) shared ancestral polymorphisms due to the incomplete diversification of the common maternal ancestry. Such ancestral polymorphisms could be genetically transmitted to certain polyploid species (e.g., StP, StY, and StYP) via the hybridization between Pseudoroegneria as the female parent and the donors with Y and/or P genomes. The hypothesis of hybridization is also a likely candidate to explain the conflict because different polyploid species with the same genotypes could be derived from different parental donors via independent hybridization events, generating a diverse array of polyploid genotypes in Triticeae (Sun et al., 2008; Fan et al., 2013). The present plastome data also provides support for the independent origin of certain polyploid species, which can be shown by different Kengyilia species that were grouped with different Roegneria species in a phylogenetic tree. For example, in the clade I of the phylogenomic tree, three Kengyilia species (K. hirsuta, K. laxiflora, and K. batalinii) were clustered with R. grandis with strong statistical support (100% UFboot, 100% SH-aLRT, and 1.0 PP), and five Kengyilia species (K. thoroldiana, K. grandiglumis, K. mutica, K. stenachyra, and K. rigidula) were grouped with R. longearistata (100% UFboot, 100% SH-aLRT, and 1.0 PP). The analysis of genetic distances based on the 52 protein-coding sequences also presented similar results. Sympatric distribution among R. grandis, R. longearistata, and Agropyron species have provided an opportunity in physical proximity for hybridization events. It is thus suggested that different Kengyilia species derived their StY genome from different Roegneria species. Our data also indicated that Agropyron species served as the maternal donor during the speciation of K. melanthera, providing additional support for the independent origins of different Kengyilia species. However, it seems unlikely that maternal Agropyron lineage in K. melanthera resulted from hybridization between high ploidy Roegneria species with StY genomes (served as paternal donor) and diploid P genome Agropyron species. One possible explanation is that the P genome of K. melanthera originated from the tetraploid Agropyron lineage as the female parent (Figure 4). Given the present data, multiple origins of polyploid species result in a maternal haplotype polymorphism and could explain the rich diversity and wide adaptation of polyploid species in the genus Kengyilia (Yen et al., 2006).
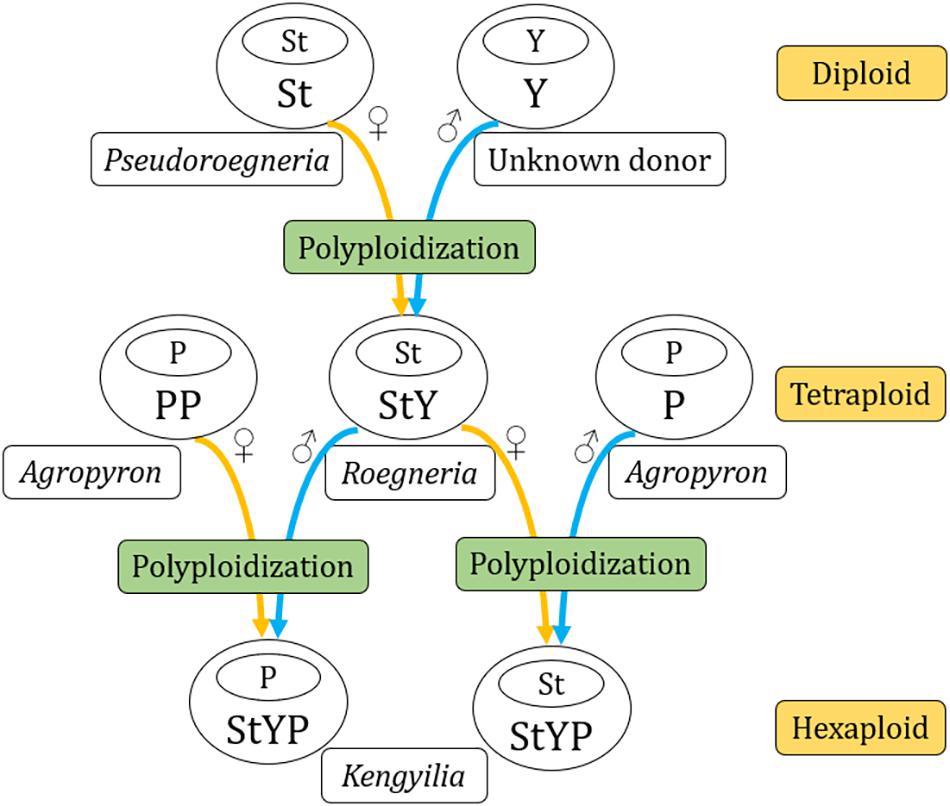
Figure 4. A diagram of lineage relationships among genera Kengyilia, Roegneria, and Agropyron.
Conclusion
The present analysis of phylogenetic relationships in Kengyilia based on the plastome sequences revealed that both Roegneria and Agropyron tetraploid species served as the maternal donor during the speciation of Kengyilia species, and different Roegneria species contributed their StY genome to different Kengyilia species. This is indicative of the independent origin of different Kengyilia species, which shed new light on our understanding of the maternal lineages, polyploidization events, and speciation process of Kengyilia.
Data Availability Statement
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found in the article/Supplementary Material.
Author Contributions
XF, SC, and YZ designed the research. SC, HY, LS, and NC performed the research. LS, HZ, and YZ collected and identified the plant materials. XF, SC, and HY analyzed the data and wrote the manuscript. XF edited the manuscript. All authors contributed to the article and approved the final manuscript.
Funding
This study was supported by the National Natural Science Foundation of China (Nos. 31900280 and 31870360) and the Second Tibetan Plateau Scientific Expedition and Research Program (No. 2019QZKK0303).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s Note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Acknowledgments
We are grateful to the American National Plant Germplasm System (Pullman, WA, United States) for providing the part seed materials for this study.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.682040/full#supplementary-material
Footnotes
References
Ankenbrand, M. J., Hohlfeld, S., Hackl, T., and Förster, F. (2017). AliTV—interactive visualization of whole genome comparisons. Peer J. Comput. Sci. 3:e116. doi: 10.7717/peerj-cs.116
Baum, B. R., Yang, J. L., and Yen, C. (1995). Taxonomic separation of Kengyilia (Poaceae: Triticeae) in relation to nearest related Roegneria, Elymus, and Agropyron, based on some morphological characters. Plant Syst. Evol. 194, 123–132. doi: 10.1007/BF00982851
Bernhardt, N., Brassac, J., Kilian, B., and Blattner, F. R. (2017). Dated tribe-wide whole chloroplast genome phylogeny indicates recurrent hybridizations within Triticeae. BMC Evol. Biol. 17:141. doi: 10.1186/s12862-017-0989-9
Betancur-R, R., Naylor, G. J. P., and Orti, G. (2014). Conserved genes, sampling error, and phylogenomic inference. Syst. Biol. 63, 257–262. doi: 10.1093/sysbio/syt073
Chen, S. F., Zhou, Y. Q., Chen, Y. R., and Gu, J. (2018). fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34, i884–i890. doi: 10.1093/bioinformatics/bty560
Dierckxsens, N., Mardulyn, P., and Smits, G. (2016). NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45:e18. doi: 10.1093/nar/gkw955
Fan, X., Sha, L. N., Dong, Z. Z., Zhang, H. Q., Kang, H. Y., Wang, Y., et al. (2013). Phylogenetic relationships and Y genome origin in Elymus L. sensu lato (Triticeae; Poaceae) based on single-copy nuclear Acc1 and Pgk1 gene sequences. Mol. Phylogenet. Evol. 69, 919–928. doi: 10.1016/j.ympev.2013.06.012
Fan, X., Sha, L. N., Zeng, J., Kang, H. Y., Zhang, H. Q., Wang, X. L., et al. (2012). Evolutionary dynamics of the Pgk1 gene in the polyploid genus Kengyilia (Triticeae: Poaceae) and its diploid relatives. PLoS One 7:e31122. doi: 10.1371/journal.pone.0031122
Gornicki, P., Zhu, H., Wang, J., Challa, G. S., Zhang, Z., Gill, B. S., et al. (2014). The chloroplast view of the evolution of polyploid wheat. New Phytol. 204, 704–714. doi: 10.1111/nph.12931
Guindon, S., Dufayard, J. F., Lefort, V., Anisimova, M., Hordijk, W., and Gascuel, O. (2010). New algorithms and methods to estimate maximum- likelihood phylogenies: assessing the performance of PhyML 3.0. Syst. Biol. 59, 307–321. doi: 10.1093/sysbio/syq010
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Luo, X., Tinker, N. A., Fan, X., Zhang, H. Q., Sha, L. N., Kang, H. Y., et al. (2012). Phylogeny and maternal donor of Kengyilia species (Poaceae: Triticeae) based on three cpDNA (matK, rbcL and trnH-psbA) sequences. Biochem. Syst. Ecol. 44, 61–69. doi: 10.1016/j.bse.2012.04.004
Ma, P. F., Zhang, Y. X., Zeng, C. X., Guo, Z. H., and Li, D. Z. (2014). Chloroplast phylogenomic analyses resolve deep-level relationships of an intractable bamboo tribe Arundinarieae (Poaceae). Syst. Biol. 63, 933–950. doi: 10.1093/sysbio/syu054
Mason-Gamer, R. J. (2013). Phylogeny of a genomically diverse group of Elymus (Poaceae) allopolyploids reveals multiple levels of reticulation. PLoS One 8:e78449. doi: 10.1371/journal.pone.0078449
Minh, B. Q., Nguyen, M. A. T., and von Haeseler, A. (2013). Ultrafast approximation for phylogenetic bootstrap. Mol. Biol. Evol. 30, 1188–1195. doi: 10.1093/molbev/mst024
Otto, S. P. (2007). The evolutionary consequences of polyploidy. Cell 131, 452–462. doi: 10.1016/j.cell.2007.10.022
Posada, D., and Crandall, K. A. (1998). MODELTEST: testing the model of DNA substitution. Bioinformatics 14, 817–818. doi: 10.1093/bioinformatics/14.9.817
Ramsey, J., and Ramsey, T. S. (2014). Ecological studies of polyploidy in the 100 years following its discovery. Philos. Trans. R. Soc. Lond. B Biol. Sci. 369:20130352. doi: 10.1098/rstb.2013.0352
Ronquist, F., Teslenko, M., van Der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Boil. 61, 539–542. doi: 10.1093/sysbio/sys029
Sha, L. N., Fan, X., Li, J., Liao, J. Q., Zeng, J., Wang, Y., et al. (2017a). Contrasting evolutionary patterns of multiple loci uncover new aspects in the genome origin and evolutionary history of Leymus (Triticeae. Poaceae). Mol. Phylogenet. Evol. 114, 175–188. doi: 10.1016/j.ympev.2017.05.015
Sha, L. N., Fan, X., Wang, X. L., Dong, Z. Z., Zeng, J., Zhang, H. Q., et al. (2017b). Genome origin, historical hybridization and genetic differentiation in Anthosachne australasica (Triticeae; Poaceae), inferred from chloroplast rbcL, trnH-psbA and nuclear Acc1 gene sequences. Ann. Bot. 119, 95–107. doi: 10.1093/aob/mcw222
Soltis, P. S., and Soltis, D. E. (2000). The role of genetic and genomic attributes in the success of polyploids. Proc. Natl. Acad. Sci. U.S.A. 97, 7051–7057. doi: 10.1073/pnas.97.13.7051
Spoelhof, J. P., Soltis, P. S., and Soltis, D. E. (2017). Pure polyploidy: closing the gaps in autopolyploid research. J. Syst. Evol. 55, 340–352. doi: 10.1111/jse.12253
Sun, G. L., Ni, Y., and Daley, T. (2008). Molecular phylogeny of RPB2 gene reveals multiple origin, geographic differentiation of H genome, and the relationship of the Y genome to other genomes in Elymus species. Mol. Phylogenet. Evol. 46, 897–907. doi: 10.1016/j.ympev.2007.12.024
Symonds, V. V., Soltis, P. S., and Soltis, D. E. (2010). Dynamics of polyploid formation in Tragopogon (Asteraceae): recurrent formation, gene flow, and population structure. Evolution 64, 1984–2003. doi: 10.1111/j.1558-5646.2010.00978.x
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Yang, J. B., Li, D. Z., and Li, H. T. (2014). Highly effective sequencing whole chloroplast genomes of angiosperms by nine novel universal primer pairs. Mol. Ecol. Resour. 14, 1024–1031. doi: 10.1111/1755-0998.12251
Yang, J. L., Yen, C., and Baum, B. R. (1992). Kengyilia: synopsis and key to species. Hereditas 116, 25–28. doi: 10.1111/j.1601-5223.1992.tb00795.x
Yen, C., Yang, J. L., and Baum, B. R. (2006). Biosystematics of Triticeae, Vol. 3. (Beijing: Chinese Agriculture Press), 1–40.
Zeng, J., Fan, X., Zhang, L., Wang, X. L., Zhang, H. Q., and Zhou, Y. H. (2010). Molecular phylogeny and maternal progenitor implication in the genus Kengyilia (Triticeae: Poaceae): evidence from coxII intron sequences. Biochem. Syst. Ecol. 38, 202–209. doi: 10.1016/j.bse.2009.12.033
Keywords: polyploid, Triticeae, Kengyilia, multiple origins, maternal donor
Citation: Chen S, Yan H, Sha L, Chen N, Zhang H, Zhou Y and Fan X (2021) Chloroplast Phylogenomic Analyses Resolve Multiple Origins of the Kengyilia Species (Poaceae: Triticeae) via Independent Polyploidization Events. Front. Plant Sci. 12:682040. doi: 10.3389/fpls.2021.682040
Received: 17 March 2021; Accepted: 01 July 2021;
Published: 06 August 2021.
Edited by:
Jorge Duitama, University of Los Andes, ColombiaReviewed by:
Matheus Bianconi, The University of Sheffield, United KingdomSean Vincent Burke, University of Chicago, United States
Copyright © 2021 Chen, Yan, Sha, Chen, Zhang, Zhou and Fan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shiyong Chen, Y2hlbmdzaGk4ODI3QDE2My5jb20=; Yonghong Zhou, emhvdXloQHNpY2F1LmVkdS5jbg==; Xing Fan, ZmFueGluZzk5ODhAMTYzLmNvbQ==