
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 04 June 2021
Sec. Plant Development and EvoDevo
Volume 12 - 2021 | https://doi.org/10.3389/fpls.2021.660803
This article is part of the Research Topic Petal Development: From Cell Biology to EvoDevo View all 10 articles
 Yves Deveaux1*†
Yves Deveaux1*† Natalia Conde e Silva1†
Natalia Conde e Silva1† Domenica Manicacci1Martine Le Guilloux1
Domenica Manicacci1Martine Le Guilloux1 Véronique Brunaud2Harry Belcram1Johann Joets1Ludivine Soubigou-Taconnat2,3Etienne Delannoy2,3Hélène Corti1Sandrine Balzergue2,4Jose Caius2,3Sophie Nadot5
Véronique Brunaud2Harry Belcram1Johann Joets1Ludivine Soubigou-Taconnat2,3Etienne Delannoy2,3Hélène Corti1Sandrine Balzergue2,4Jose Caius2,3Sophie Nadot5 Catherine Damerval1
Catherine Damerval1Even though petals are homoplastic structures, their identity consistently involves genes of the APETALA3 (AP3) lineage. However, the extent to which the networks downstream of AP3 are conserved in species with petals of different evolutionary origins is unknown. In Ranunculaceae, the specificity of the AP3-III lineage offers a great opportunity to identify the petal gene regulatory network in a comparative framework. Using a transcriptomic approach, we investigated putative target genes of the AP3-III ortholog NdAP3-3 in Nigella damascena at early developmental stages when petal identity is determined, and we compared our data with that from selected eudicot species. We generated a de novo reference transcriptome to carry out a differential gene expression analysis between the wild-type and mutant NdAP3-3 genotypes differing by the presence vs. absence of petals at early stages of floral development. Among the 1,620 genes that were significantly differentially expressed between the two genotypes, functional annotation suggested a large involvement of nuclear activities, including regulation of transcription, and enrichment in processes linked to cell proliferation. Comparing with Arabidopsis data, we found that highly conserved genes between the two species are enriched in homologs of direct targets of the AtAP3 protein. Integrating AP3-3 binding site data from another Ranunculaceae species, Aquilegia coerulea, allowed us to identify a set of 18 putative target genes that were conserved between the three species. Our results suggest that, despite the independent evolutionary origin of petals in core eudicots and Ranunculaceae, a small conserved set of genes determines petal identity and early development in these taxa.
Petals are floral organs that play a major role in pollinator attraction. In flowers with a differentiated perianth, petals are defined as the second whorl of sterile organs surrounding the androecium; the first whorl of sterile organs is composed of sepals, which protect the fertile organs during development. A recent reconstruction of the ancestral angiosperm flower suggests that the ancestral flower has an undifferentiated perianth and floral organs inserted in whorls or along a spiral (Sauquet et al., 2017). Perianth differentiation has evolved several times independently, and consequently, the term “petal” is not indicative of organ homology across angiosperms (Irish, 2009; Ronse de Craene and Brockington, 2013).
The analysis of floral mutants in two core eudicot species, Antirrhinum majus and Arabidopsis thaliana, has been a turning point for understanding the genetic control of floral organ identity by proposing a model based on four main functions: A, B, C, and E. The A-function specifies sepals, A‐ and B-functions determine petals, B‐ and C-functions specify stamens, and C-function specifies carpels (Coen and Meyerowitz, 1991). The E-function combines with the three other functions to specify floral identity (Pelaz et al., 2001). Most genes involved in A-, B-, C-, and E-functions, encode transcription factors belonging to the MIKC-type MADS-box gene family (reviewed in Theißen and Gramzow, 2016). They are known to act in tetrameric protein complexes (“floral quartets”; Theißen et al., 2016; Yan et al., 2016). In particular, B-function relies on two proteins APETALA3/DEFICIENS (AP3) and PISTILLATA/GLOBOSA (PI) in Arabidopsis/Antirrhinum, respectively, which function as an obligate heterodimer within tetrameric complexes (Goto and Meyerowitz, 1994).
AP3 and PI belong to two paralogous gene lineages that result from an ancient duplication preceding the angiosperm radiation. Additional duplications took place in the AP3 and PI lineages during angiosperm evolution, at different points in time (Lee and Irish, 2011). In the AP3 lineage, an ancient duplication event occurred at the base of the core eudicots, resulting in the TM6 and euAP3 gene lineages. The euAP3 lineage is characterized by a C-terminal motif that differs from the paleoAP3 motif present in TM6 and other AP3 lineages (Kramer et al., 1998, 2006). EuAP3 genes are thought to play an important role in the formation and diversification of petals in core eudicots (Irish, 2009). Independently, two successive duplications of AP3 have occurred at the base of the order Ranunculales, the sister group to all other eudicots, giving rise to three paralogous lineages: AP3-I, AP3-II, and AP3-III (Rasmussen et al., 2009). The PI lineage evolution is also complex and dynamic with cases of multiple recent duplications (Zahn et al., 2005). In Ranunculales, the B-function involves generally the three AP3 paralogous and at least one PI paralogous genes. Heterodimerization of PI proteins with AP3 proteins of the different lineages has been observed in several species (Kramer et al., 2007; Wang et al., 2016; Galimba et al., 2018). While PI gene expression is essential for both stamen and petal identity (Kramer et al., 2007; Wang et al., 2016), the function of the different paralogous AP3 genes with regard to stamen and petal identity has diverged (Kramer et al., 2007; Sharma and Kramer, 2013; Wang et al., 2016). In particular, it has been shown that the presence of petals is strongly correlated with the expression of AP3-III orthologs (Kramer et al., 2003; Di Stilio et al., 2005; Rasmussen et al., 2009), and that the absence of petals in some species is linked to a disruption of the AP3-3 locus (Zhang et al., 2013). In two Ranunculaceae species where functional studies can be conducted using Virus-Induced Gene Silencing (VIGS), namely, Aquilegia coerulea and Nigella damascena, it has been shown that expression of AP3-3 genes is required for petal identity (Sharma et al., 2011; Gonçalves et al., 2013; Wang et al., 2016). These lines of evidence indicate that AP3-3 orthologs play a crucial role in determining petal identity in Ranunculaceae.
Petals, with their multiple independent origins, have been consistently associated with the expression of B-function genes (Ambrose et al., 2000; Drea et al., 2007; Mondragón-Palomino and Theissen, 2008). Therefore, they are a good case study of the conservation of gene networks downstream of B-function proteins. While Arabidopsis AP3 gene is involved in both petal and stamen development, the petal specificity of the Ranunculaceae AP3-3 genes offers a good opportunity to unravel a petal-specific gene regulatory network. The target genes of MADS-box proteins are beginning to be identified in model species, mainly in Arabidopsis (Bey et al., 2004; Kaufmann et al., 2009, 2010; Wuest et al., 2012; Ó’Maoiléidigh et al., 2013; Pajoro et al., 2014). High throughput sequencing methods now open the way for comparing possible target genes in a wider range of non-model species representing diverse organ morphologies and evolutionary origins (Thomson et al., 2017).
Expression of B-function genes is required at every stage of development to obtain full organ identity. In N. damascena, we observed that the presence and morphology of petals differed according to the timing of inactivation of NdAP3-3 by VIGS (Gonçalves et al., 2013). Early inactivation results in the absence of petal formation, resulting in a phenocopy of the spontaneous mutant lacking petals known since the 16th century (Toxopéus, 1927). When inactivation takes place at later stages, a range of intermediate morphologies between petals and sepals are observed. In this paper we intended to characterize the early target genes of NdAP3-3 that determine the full petal identity. Because of the large size of the N. damascena genome (~10 Gb), no genomic data are available for this species yet. To circumvent this limiting factor, we assembled and annotated a floral reference transcriptome and characterized differential gene expression between wild-type and mutant genotypes at the NdAP3-3 locus differing by the presence vs. absence of petals. The wild type vs. mutant comparison gives the unique opportunity to investigate the early stages of floral development when petals are initiated or not. Therefore, differentially expressed genes are candidates for direct or indirect targets of the NdAP3-3 protein involved in petal identity and early development. Genes were annotated and compared to known or suspected targets of B-function genes in other eudicot species (Bey et al., 2004; Wuest et al., 2012; Jiang et al., 2020), and the conservation of the petal gene network is discussed.
Segregating populations were obtained by selfing plants issued from two commercial seed lots obtained from horticultural companies (Royal Fleur and Truffaut) as described in Gonçalves et al. (2013). The heterozygosity level of these mother plants is unknown. For analysis of the early stages, the progeny of F2 plants (F3 offspring), which were homozygous either for the wild-type P allele (P morph, with petals) or the mutant p allele (T morph, without petals), were sown in three replicates. Each of the three replicates consisted of a mix of eight seeds from the same six PP or pp F2 mother plants. Plants were grown in pots in randomized blocks in a growth chamber under controlled conditions (18 h day at 25°C, 6 h night at 16°C). Another set of PP plants was grown independently to collect late developmental stages. The genotype at the NdAP3-3 locus was checked for all plants by PCR using primers flanking the MITE element insertion site in intron 2, which is a marker for the p allele (Gonçalves et al., 2013).
Floral buds were collected at different stages, based on the developmental schedule previously described by Jabbour et al. (2015). Stage 1 corresponds to 1 day after floral transition marked by the beginning of stem extension; at this stage, only sepals are initiated in both genotypes. Stage 2 occurs 3 days later, and corresponded to petal initiation (PP plants) or internal sepal initiation (pp plants). Approximately 20 terminal flower meristems were dissected under a stereomicroscope allowing stage inspection for each condition (morph × stage × replicate). Flower buds were collected from 32 PP plants at three late stages (bud diameter 5–6 mm, 6–8 mm, and just before anthesis), and the organs were dissected and kept separately (sepals, petals, stamens, and carpels). All dissected floral tissues were immediately frozen in liquid nitrogen and stored at −80°C until RNA extraction.
Total RNA from floral meristems and dissected organs was extracted using the RNeasy Plant Mini Kit (Qiagen) with the additional DNAse I step according to the manufacturer’s instructions. Total RNA from each tissue was checked for integrity on an RNA_Nano chip, using an Agilent 2,100 bioanalyzer (Agilent Technologies, Waldbroon, Germany). Twelve libraries were constructed for the two morphs at the two earliest developmental stages in three replicates. Two additional libraries were constructed from the dissected floral organs of the P morph, the first one by pooling equal amounts of RNA from the three non-petal organs at the three late developmental stages, and the second one by pooling RNA from petals only at these three stages. The libraries were constructed with the TruSeq-stranded mRNA library Prep kit (Illumina, California, USA) with 300-bp size for libraries from early-stage material and 260-bp size for dissected material. Libraries were paired-end (PE) sequenced with a read length of 150 bp for early-stage material and 100 bp for dissected material using an Illumina HiSeq2000 at the Genoscope Laboratory (Evry, France). Lane distribution and barcoding gave approximately 20–30 million PE reads per sample. For each sample, raw data (fastq) were trimmed with Trimmomatic (Bolger et al., 2014) with a Phred Quality Score Qscore > 20, and read lengths > 30 bases. Ribosome sequences were removed with the sortMeRNA tool (Kopylova et al., 2012).
All steps of the study, from growth conditions to bioinformatic analyses, were managed in a CATdb database (Gagnot et al., 2007, http://tools.ips2.u-psud.fr/CATdb/) with ProjectID NGS2013_09_AAP-IDEEV and NGS2015_16_Ranunculaceae. This project was submitted from CATdb to the international public repository GEO (Gene Expression Omnibus, Edgar et al., 2002, http://www.ncbi.nlm.nih.gov/geo, GSE159429).
Assembly of the reference transcriptome was made using Trinity (version 2.4, Grabherr et al., 2011). To balance between the different genotypes, developmental stages, and organs, we combined the following six samples for a total of ~130 million oriented PE reads: four libraries were from floral buds of the P and T morphs at stages 1 and 2, and two libraries were from petal and non-petal tissues from the P morph at later stages. Trinity was run with default parameters and a kmer size of 32. Contigs that were smaller than 200 bases were removed, and 81,194 contigs were finally obtained. iAssembler (version 1.3, Zheng et al., 2011) was then used for scaffolding contigs and reducing redundancy, with 97% identity for sequence clustering. For simplicity, hereafter we use the term transcript or gene to refer to the contigs, which may include paralogs, alleles, and alternative splicing of the same gene.
Before annotating the transcriptome, we predicted all coding sequences. First, the TransDecoder software (v5.0.2; Perina et al., 2016; Roy et al., 2018) was used to predict all putative open reading frames (ORFs) among assembled transcript sequences (default parameters). These predicted ORFs were then searched for Pfam domain hits (v28) using the HMMER software (3.1b2, default settings; Eddy and Pearson, 2011). Final coding region predictions were achieved by running TransDecoder a second time taking into account the Pfam domain search output. Functional annotations were performed by searching for sequence similarity and protein domains. Similarity of the predicted proteins with sequences from the UniRef90 (Suzek et al., 2015) and TAIR databases (Huala et al., 2001; Rhee et al., 2003) was determined using Blast (ncbi-blast-2.6.0+, E-value < 1.10–3; Altschul et al., 1990). InterProScan software (5.29–68.0; Zdobnov and Apweiler, 2001; Jones et al., 2014) was used to search for InterPro domain hits (default settings). We extracted Gene Ontology (GO) and GO slim terms (functions, locations, and biological roles) from Blast and InterProScan outputs with a lab script (Ashburner et al., 2000; The Gene Ontology Consortium, 2001).
BlastP tool was used to compare N. damascena predicted proteins to the 43,550 proteins of A. coerulea from Phytozome (322-v3.1). We kept only the best hit for each Nigella protein (E-value ≤ 0.01). Blastn was also used to search for the homology of some unannotated transcripts in the A. coerulea genome.
Nine transcripts with differential expression between morphs and without functional annotation were chosen randomly to check whether they could correspond to novel potentially Nigella-specific sequences or were misassembled transcripts. Primers were designed based on the full sequences of these nine transcripts and used to amplify from cDNA and/or gDNA (Supplementary Table S1).
Reads corresponding to the three replicates of stages 1 and stage 2 floral buds of the P and T morphs were mapped on the reference transcriptome. Every PE read in each sample was mapped against the complete list of contigs with Bowtie2 (Langmead and Salzberg, 2012, local option). Ninety six percent of the reads could be mapped, demonstrating the quality of transcriptome assembly. All PE reads with multi-hits, meaning mapped on sequence regions shared by isoforms, were removed. We kept only PE reads associated with a unique contig, ensuring specific quantification of gene expression. The method generated quantitative data for 70,491 contigs.
Low-expression genes were filtered to include only contigs (genes) that have at least 1 read count per million (CPM, counts per million) after normalization, in at least two samples. 34,614 transcripts (that include NdAP3-3) were thus retained for quantitative analysis. The coefficients of determination calculated on normalized counts between pairs of replicates for each stage–organ combination were high (R2 > 0.95). To assess how the overall variability is structured among the 12 datasets (2 morphs × 2 stages × 3 replicates), a principal component analysis was carried out on the normalized counts for these 34,614 informative transcripts using the FactoMineR package. Using the EdgeR package v3.8.6 (Robinson et al., 2010), we then fitted the normalized count data to a model that considered the effect of plant morph (2 levels), developmental stage (2 levels), replicate (3 levels), as well as the morph by stage and morph by replicate interactions. We performed contrasts to (i) compare the P and T morphs at stage 1, at stage 2, and at both stages on average; (ii) compare the developmental stages in the P morph, the T morph, and in both morphs on average; and (iii) test the morph by stage interaction. In each case, p-values for statistical significance were adjusted following the Benjamini and Hochberg’s (1995) FDR method.
To quantify the level of differential expression between morphs, we calculated the log2 ratio of the mean of the normalized counts of T over P for the three replicates (hereafter M_LFC) at stage 1, stage 2, or both together. A heatmap of the log2-normalized counts of the two morphs at the two developmental stages in the three replicates was drawn with the heatmap function in R.
As a complementary approach to identify possible targets of the NdAP3-3 transcription factor, we correlated the expression of the remaining 34,613 transcripts for quantitative analysis with the expression of the NdAP3-3 gene in the P morph. Because the effect of the developmental stage was high for NdAP3-3 and for most transcripts, we normalized the data per stage. The Pearson’s correlation coefficient was then calculated for each of the 34,613 transcripts and the corresponding FDR was calculated. Although this analysis may be of very low power (normalization and correlation based on 6 data points – three replicates for each developmental stage) and some statistical hypotheses may not be fulfilled (normality of expression data), we used it to strengthen the evidence of a functional interaction between differentially expressed genes and NdAP3-3. We chose to focus on genes with a Pearson’s correlation coefficient ≥ 0.8, which corresponded to an FDR value ≤ 0.26.
Functional category enrichment/depletion among differentially expressed vs. non-differentially expressed genes were tested using Chi-squared contingency tests (p < 0.001).
A set of 15 genes were randomly selected for comparison of quantitative expression measured by RNAseq-normalized counts and quantitative RT-PCR. The RNA pools from the three replicates used for RNA-seq were used to synthesize cDNAs for qRT-PCR. Primer pairs were designed from the transcript sequences (Supplementary Table S1) and validated by qRT-PCR on a dilution series. Four genes were initially chosen as possible references, based on their stable expression within the range of expression of the 15 genes of interest. After qRT-PCR analysis, we finally selected RN20658, a pentatricopeptide repeat (PPR)-containing protein as the best reference. Relative expression was calculated using the 2−ΔΔCt method.
First, we searched the N. damascena annotated transcriptome for homologs of the A. thaliana genes (AGI-ID) whose expression is correlated with that of MADS-box genes of the ABCE model (Supplementary Table S4). This was done using the Expression Angler program and the AtGenExpress Plus ‐ Extended tissue Compendium data set (Toufighi et al., 2005), as previously described (Damerval et al., 2019).
Second, we focused on genes involved in petal formation in Arabidopsis and searched for their homologs in the Nigella transcriptome. These genes are often members of gene families and functional diversification may be species specific. Therefore, for each gene of the Nigella transcriptome, we sorted the 10 best AGI-ID hits to identify homology with any member of Arabidopsis petal gene families. The obtained dataset was then filtered using two different AGI-ID lists to identify Nigella homologs of Arabidopsis genes that were (i) co-expressed with AtAP3, and/or (ii) differentially expressed between the wild type and the ap3.3 mutant in flowers. The first AGI-ID list was composed of the 100 best positively and 100 best negatively correlated genes to AtAP3 in Arabidopsis (Toufighi et al., 2005), in addition to AtPI and AtAP3 (AtAP3-BCG). The second AGI-ID list contained differentially expressed genes between wild type and mutant regardless of whether they are considered in the literature to be putative direct targets of AtAP3 or not (AtDEG, Mara and Irish, 2008; Wuest et al., 2012). Whereas the first dataset aimed to identify genes associated with petal and stamen development in a broad sense, the second dataset was more specific to the AtAP3 downstream network. Both lists were then used to identify the Nigella genes that were homologous, respectively, to the AtAP3-BCG (NdHBCG) and the AtDEG (NdH). Finally, these genes were characterized as (i) being differentially expressed between the two Nigella morphs (NdHBCG-DEG and NdH-DEG, respectively) and (ii) having as best homolog, an AtAP3 direct target gene or a paralog of an AtAP3 direct target gene.
Third, to identify additional putative target genes in the set of genes differentially expressed in Nigella, we took advantage of the ChIP-sequence analysis of the whole genome of A. coerulea for the detection of AP3-3 protein (AcAP3-3) target sequences that was conducted at a late floral developmental stage (Jiang et al., 2020).
The floral reference transcriptome of N. damascena was assembled de novo from a pool of transcripts obtained from early developmental stages when organs are initiated to later stages when all organs are formed and are growing (see Material and Methods). 71,319 contigs with a mean length of 1,160 bp and an N50 length of 1843 bp were generated. These parameters were in the same range as those of another N. damascena transcriptome elaborated from floral buds, bracts, and leaves (95,758 unigenes, mean length 933 bp, N50 of 1711 bp, Zhang et al., 2020). As expected, compared to the data from the annotated whole genome of another Ranunculaceae species, A. coerulea (43,550 transcripts, mean gene length; 1755 bp, N50 length: 2133 bp, https://phytozome.jgi.doe.go), the contigs of our Nigella transcriptome were shorter and more numerous. Two factors may account for this difference: our material was heterozygous, and classical assembly tools tend to fall short of complete gene coverage and generate too many smaller than expected contigs.
In the absence of a fully sequenced and annotated genome of N. damascena, a total of 53,133 peptides were predicted from 35,409 different transcripts. InterProScan identified 19,347 transcripts encoding peptides with at least one protein domain and a GO annotation. A BLAST search of UniRef90 allowed 5,603 genes to be annotated against 5,751 proteins. Selecting the GO:0003700 (DNA-binding transcription factor activity), we found 355 genes with at least one domain corresponding to 20 types of transcription factors (Supplementary Figure S1). Among these, the K-box, AP2/ERF, and basic leucine zipper were the most represented (20, 19, and 15%, respectively). Homologs of all ABCE MADS-box floral genes were found, except for AP1.
A high percentage of coding regions could be annotated against Arabidopsis (29,877–84%) and A. coerulea (30,700–87%) proteins. The GO slim annotation of the N. damascena transcriptome was based on homology with Arabidopsis genes annotated with Arabidopsis GO slim terms (see Material and Methods). The two best-represented categories within biological process were “other cellular processes” (29%) and “other metabolic processes” (20%); within cellular components, the four best-represented categories were “nucleus” (17%), “other cytoplasmic components” (17%), “other intracellular components” (15%), and “chloroplast” (11%); among molecular functions, the best-represented categories were “protein binding” (12%), “hydrolase activity” (11%), and “transferase activity” (13%; Figure 1).
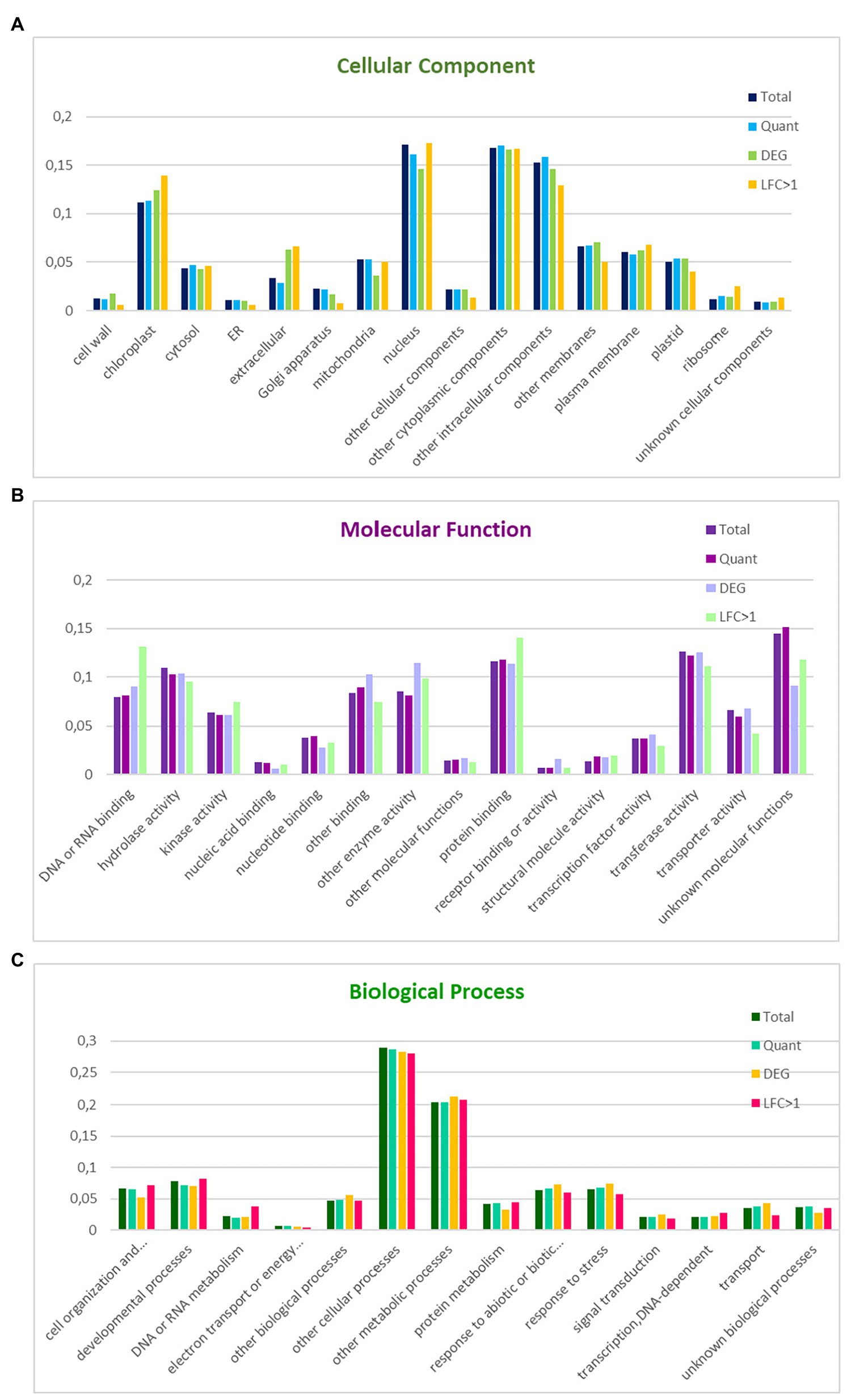
Figure 1. Distribution of GO slim terms in the floral reference transcriptome, the set of informative transcripts used in the quantitative analysis, and the genes differentially expressed between floral morphs in Nigella damascena. Distributions are compared within GO cellular component (A), molecular function (B), and biological process (C). Total: reference transcriptome; Quant: transcripts in quantitative analysis; DEG: genes differentially expressed between the two morphs; LFC > 1: most differentially expressed genes between the two morphs.
Quantitative RNA-seq was conducted at two early flower developmental stages framing petal initiation, as defined in Jabbour et al. (2015). We compared gene expression in two pools of full-sib F3 genotypes homozygous for the NdAP3-3 alleles determining the presence (PP genotype, P morph) or absence (pp genotype, T morph) of petals. Genes that are expressed differentially between the two morphs should therefore include genes from the petal initiation network downstream of NdAP3-3, but also a limited number of genes in close linkage disequilibrium with NdAP3-3 that are expressed at these floral developmental stages.
Based on informative read counts, 34,614 transcripts, which include NdAP3-3, were included in the quantitative analysis (Supplementary Table S2). We tested the consistency of gene expression patterns observed with RNA-seq counts using qRT-PCR and by randomly choosing a few genes and replicates. Among a set of 14 randomly chosen transcripts and the NdAP3-3 gene, the expression patterns were similar between the two methods for NdAP3-3 and 11 of the 14 genes (Supplementary Figure S2).
To assess how variability is structured among the 12 (2 morphs x 2 stages x 3 replicates) datasets, a principal component analysis (PCA) was carried out on the expression of the 34,614 genes. The variation in expression between replicates accounted for 31% of the overall variability, while the second axis of the PCA separated developmental stages 1 and 2 (15% of the variability) and the third axis separated the two morphs (9.7%; Figure 2; Supplementary Figure S3). The relatively high part of variability between replicates may be accounted for by the residual heterozygosity in the F3 generation, even though we tried to homogenize the background by using pools of plants. The developmental stage was the major factor for differential expression. 4,959 genes were differentially expressed (DEGs) between stages 1 and 2 (FDR < 0.05) in at least one of the two genotypes; about half of these (2,133, with twice as many genes showing an increased expression in stage 2 compared with stage 1 than vice versa) were differentially expressed in both genotypes, suggesting that they participate in a general “flower” or “sepal” developmental program but not in petal specification.
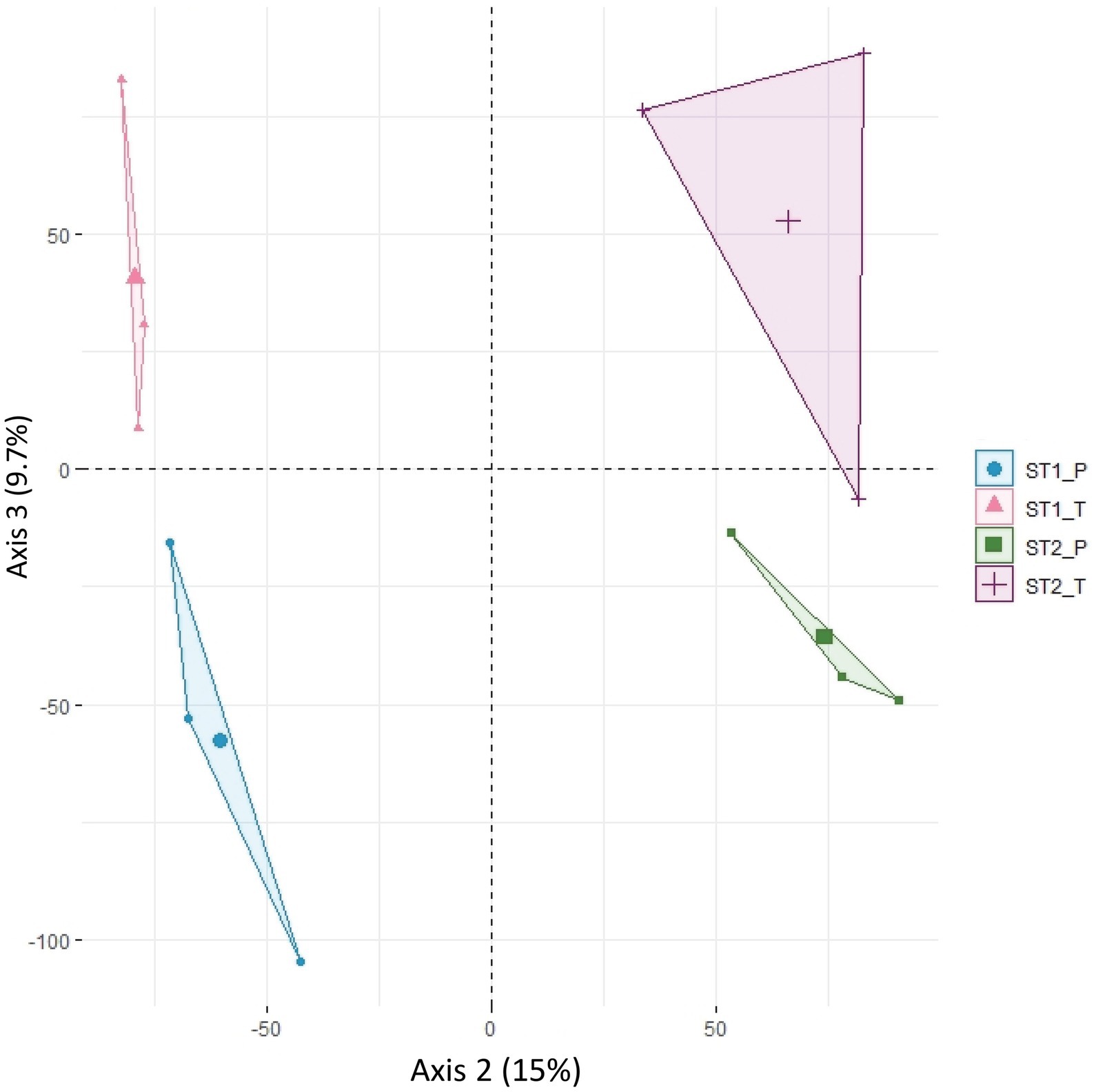
Figure 2. Principal component analysis (PCA) of normalized counts for 34,614 informative transcripts. PCA was done on the four-stage x morph combinations and the three biological replicates. Axis 2 (15% of the variability) separates individuals according to developmental stage (stage 1 vs. stage 2) and axis 3 (9.7% of the variability) separates them according to morph (P morph vs. T morph). ST1_P, ST1_T: individuals at stage 1 of P and T morph, respectively; ST2_P, ST2_T: individuals at stage 2 of P and T morph, respectively.
To investigate the possible direct or indirect targets of NdAP3-3, we focused on the 1,620 genes that were found to be differentially expressed between morphs at either stage or over both stages on average (FDR < 0.05, Supplementary Table S3; Supplementary Figure S4). This DEG set between morphs was highly enriched in genes also exhibiting a stage effect (652 genes, 40% vs. 14% of all genes analyzed). The number of DEGs between the two morphs was higher at stage 2 (884 genes) than at stage 1 (580 genes), which is consistent with petal initiation at stage 2 in the P morph, while at stage 2 the T morph produces additional sepal-like primordia. Interestingly, a significant morph by stage interaction was found for 74 transcripts (FDR < 0.05), most of which also exhibited a significant morph effect at one or both stages (63 out of 74). Among the B-function genes, only the three NdAP3 paralogs were found differentially expressed between the two morphs, NdAP3-1 (RN021161) at stage 2 but not stage 1, NdAP3-2 (RN002991) at stage 1 but not stage 2, and NdAP3-3 (RN035793) at both stages, as expected.
Among these 1,620 DEGs, 739 exhibited at least two-fold difference in expression level between morphs (|M_LFC| > 1) at one or both developmental stages. The most represented GO term within the 243 annotated genes in this set was “nucleus” (GO:0005634, 74 genes), including DNA binding (GO:0003677), protein binding (GO:0005515), and regulation of transcription activity (GO:0006355). Other than NdAP3-3 (RN035793), homologs of transcription factors, such as CRABS CLAW (NdCRC, RN058291); REDUCED VERNALIZATION RESPONSE 1 (NdVRN1, RN059576); OBP3-RESPONSIVE GENE 2 (NdORG2 with two isoforms, RN057454 and RN012090), and AGAMOUS-LIKE 15 (NdAGL15, RN008012), as well as histones and proteins involved in the maintenance of cell division (RN047478 and RN003149) and several F-Box proteins were found (Supplementary Table S3). Differential expression between morphs generally showed the same trend at both stages: 400 genes were more expressed in the T morph than in the P morph (e.g., NdCRC), while 275 genes were more expressed in the P morph than in the T morph (e.g., NdAP3-3, RN047478 and RN003149) at both developmental stages (Supplementary Table S3; Figure 3). For 64 transcripts, including homologs of ORG2 genes, the difference in expression between the two morphs was reversed between the two developmental stages (e.g., P > T at stage 1 and T > P at stage 2, Figure 3).
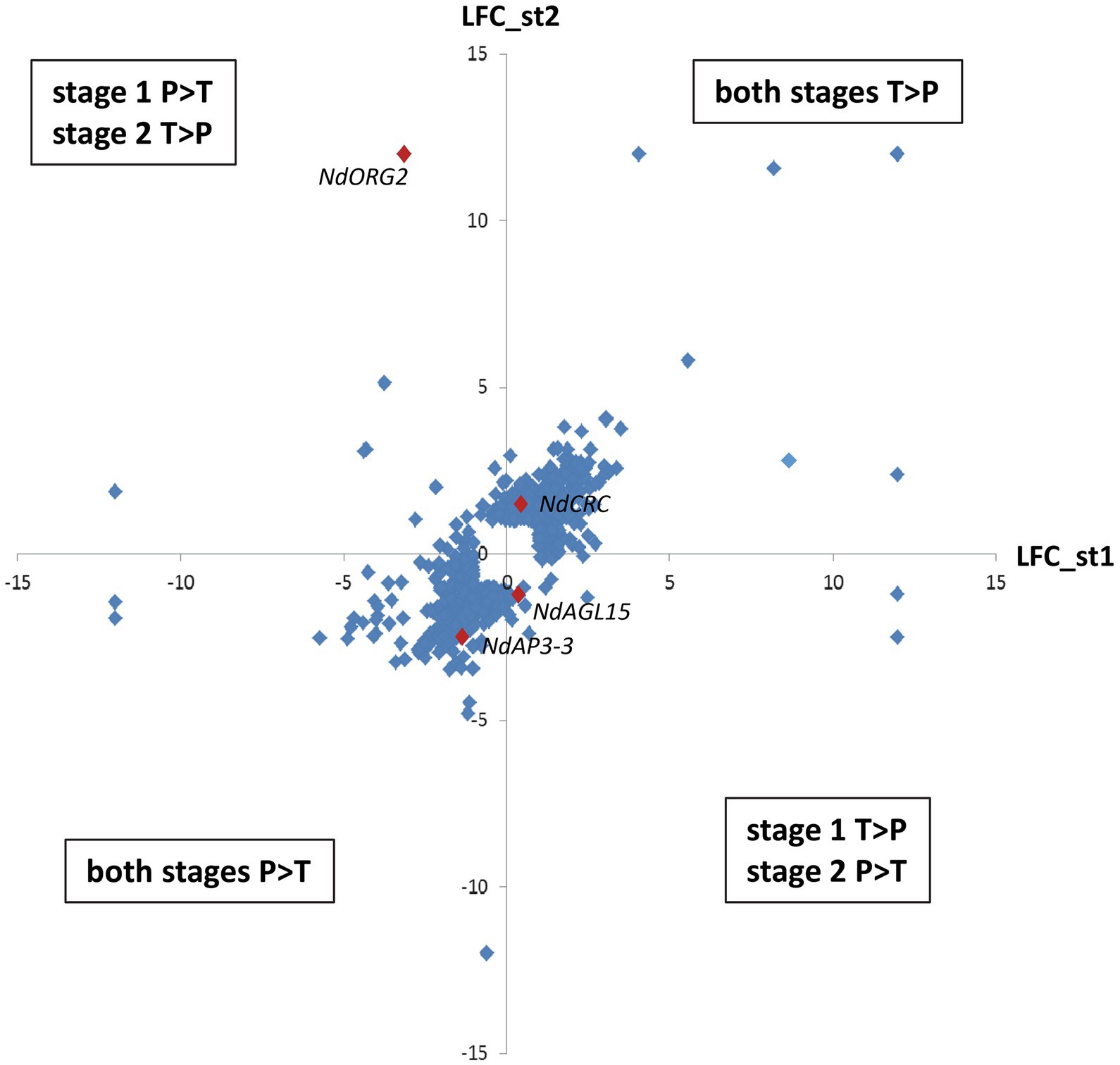
Figure 3. Differential expression between morphs at the two developmental stages for the most differentially expressed genes. Differential expression is expressed by Log2fold change, and only genes with |M_LFC| > 1 at one or both developmental stages are represented. The y-axis represents differential expression between the two morphs at stage 2 and the x-axis represents differential expression at stage 1. The upper right quadrant includes genes more expressed in the T morph at both stages, while the lower left quadrant includes genes more expressed in the P morph at both stages. The two other quadrant group genes that have reversed differential expression between the two morphs at the two stages. Examples of genes illustrating these situations are represented by red diamonds. Genes not expressed at one stage have been arbitrarily attributed M_LFC = +12: (T morph) or −12: (P morph).
To identify the petal gene network in Nigella, we searched for the DEGs that were most correlated with wild-type NdAP3-3 gene expression. We calculated the correlation between the expression of the 34,613 transcripts and the expression of NdAP3-3 in the P morph, then focused on the correlation values among the set of 739 transcripts with a |M_LFC| > 1. We found 94 transcripts with a Pearson’s correlation coefficient ≥ 0.8, which corresponded to an FDR of 26%. Among these, about one-third could be annotated against the Arabidopsis proteome. The four most correlated transcripts (|r| > 0.99, FDR < 0.01) were unannotated and likely non-coding (Supplementary Table S3).
Forty-nine percent of the differentially expressed genes could not be annotated (792 out of 1,620, Supplementary Table S3), mostly because they did not include a coding phase longer than 100 amino acids. This proportion appears significantly less than the proportion in the whole set of quantitatively analyzed transcripts (57%, p < 0.00001). The alignment of the unannotated transcripts against the genomic sequence of A. coerulea revealed a significant percentage of similarity for 90 transcripts. Additionally, we verified a set of nine transcripts by PCR amplification to check whether they could correspond to N. damascena sequences with unknown homology or misassembled transcripts. Three sequences could not be amplified and one gave a mix of two different sequences. For the remaining five sequences, the amplified fragments covered 80.8–89.6% of the total length and corresponded to the expected sequence, with a few single-nucleotide polymorphisms. Among these validated unannotated transcripts, one included a putative coding phase of at least 100 amino acids and four were likely to be non-coding.
18,883 of the 34,614 transcripts included in the quantitative analysis, including 771 DEGs between the two morphs and 243 genes with a |M_LFC| > 1 could be annotated with GO terms. The proportion of genes represented in the different GO terms was compared between the different gene sets and with the annotation of the whole transcriptome within each of the three major GO slim categories (Figure 1). Compared with genes that are not differentially expressed between morphs, the morph DEG set was enriched in the extracellular, cell wall and chloroplast components, other enzyme activities, and receptor binding or receptor activity, and was depleted in mitochondrial components, cell organization, and biogenesis and protein metabolism processes. Interestingly, among the morph DEGs, those with a |M_LFC| > 1 were enriched in ribosome components, DNA or RNA binding, cell organization and biogenesis, and/or DNA or RNA or protein metabolism, and were depleted in transport and response to stress processes (Figure 1).
The shared expression patterns that result from functional constraints imposed by the developmental process and the cell microenvironment are good indicators of the conservation of floral gene regulatory networks (Davila-Velderrain et al., 2014). To determine the extent to which the gene network involved in floral organ development is conserved in eudicots, we searched for homologs of genes in Nigella with expression patterns that are best correlated (BCGs) with that of ABCE MADS-box genes in A. thaliana, as done previously in Grevillea juniperina (Damerval et al., 2019). Analysis of the annotated N. damascena transcriptome revealed a rate of conservation of BCGs that was identical to that observed in G. juniperina using the same approach (Supplementary Figure S5A). In addition, N. damascena and G. juniperina have 75% of their BCGs in common with an r-value ≥ 0.75 (Supplementary Table S5; Damerval et al., 2019). Homologs of all ABCE genes except AP1 were found in N. damascena, and most of the genes that were highly correlated with them were also conserved between Nigella and Arabidopsis (Supplementary Table S5). Individual analyses of the 25 BCGs showed that on average, 49% of homologs of the ABCE MADS-box correlated genes were conserved. Homologs of AP1 and AP3 correlated genes were slightly over-represented, whereas homologs of AG and SEP1/3 correlated genes were under-represented (Supplementary Figure S5B). Overall, good conservation of the floral genes is observed between the species.
To be considered as a direct target of a transcription factor, a gene must have a binding signal in its regulatory regions and its expression must be regulated by this protein. In Arabidopsis, such a study combining genome-wide site detection and wild type vs. mutant comparison was conducted to identify the target genes of the floral transcription factor AtAP3 (Wuest et al., 2012). Because no genomic data are available in Nigella, we opted for a comparative approach, using the Arabidopsis data, to identify the putative target genes of NdAP3-3. We searched for homologous genes in Arabidopsis and Nigella whose expression is altered by the inactivation of their respective AP3 petal identity genes and identify those that are homologs to AtAP3 target genes. Only 20% of the 1,075 DEGs between the Arabidopsis ap3.3 mutant and wild type (AtDEG, Supplementary Table S6A) were found to be direct targets of AtAP3 (Wuest et al., 2012). Among the 4,910 Nigella transcripts that are homologous to AtDEG (NdH), 172 were differentially expressed between the two Nigella floral morphs (NdH-DEG). Twenty-eight percent of the NdH-DEG were homologous to AtAP3 direct target genes (48 out 172), either as best hits to Arabidopsis target genes (23 out 172, 13%) or as a paralog of the target genes (25 out 172, 15%, Figure 4A).
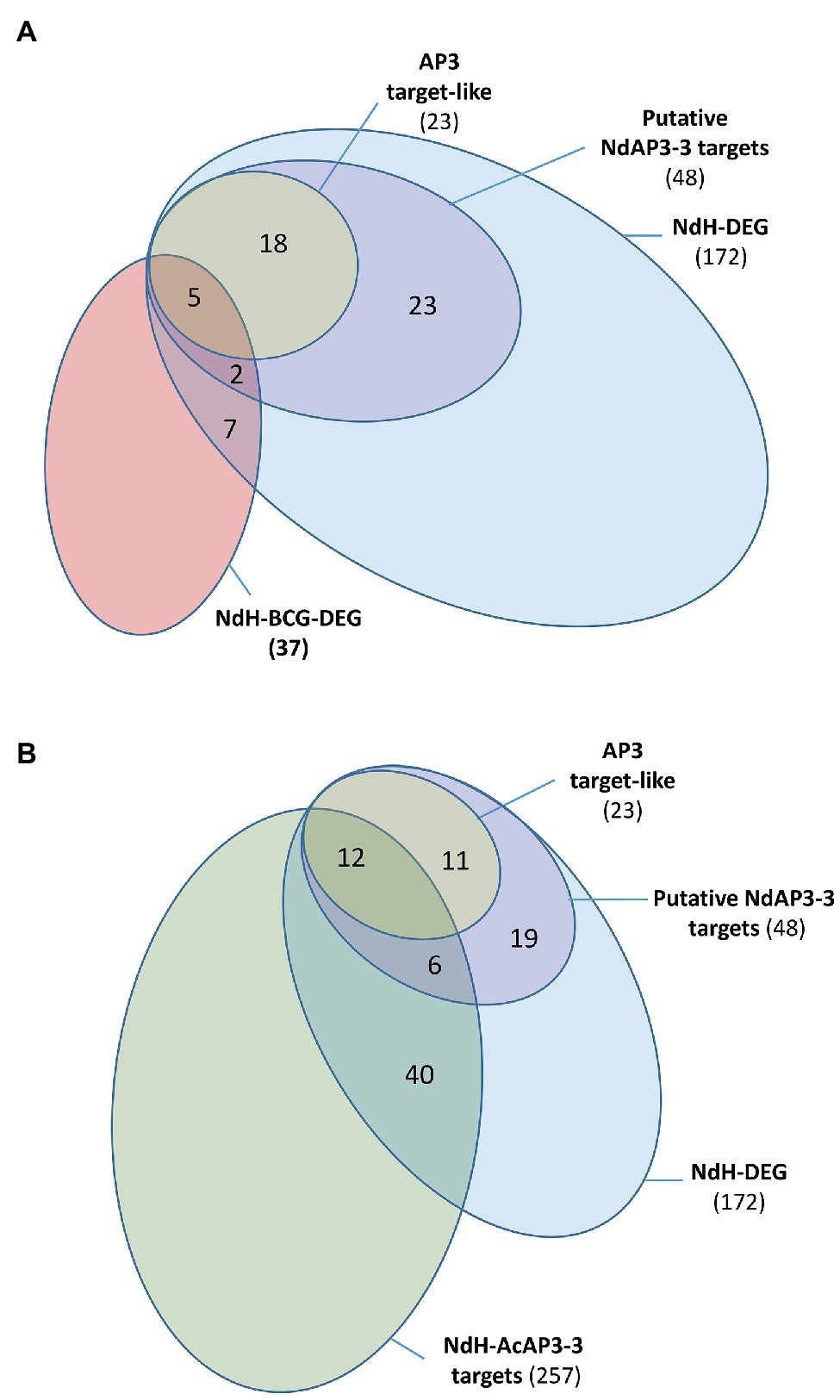
Figure 4. Venn diagram representing the distribution of the Nigella transcripts into subcategories. (A) NdH-DEG and NdH-BCG-DEG are Nigella genes which are differentially expressed between the two morphs and homologous to Arabidopsis genes which are differentially expressed between the wild type and the ap3.3 mutant and the best correlated genes to AtAP3, respectively. Putative AP3-3 target genes include the best homologs of the AtAP3 target genes (AP3 target-like) and paralogs of AtAP3 target genes in Nigella (see details in Supplementary Table S6). (B) NdH-AcAP3-3 are the Nigella best homologs of the Aquilegia AcAP3-3 target genes (see details in Supplementary Table S8). The other categories are the same as panel (A).
The overlapping of the AtAP3-BCG set with that of the conserved DEG set between Arabidopsis and Nigella (NdH-DEG) was also analyzed to identify the core gene set of the petal development network. When considering the 202 AtAP3 BCGs (AtAP3-BCG, Supplementary Table S6B), only 9% were differentially expressed between mutant and wild type (19 AtBCG-DEG), but almost half (9 out of 19, 47%) were classified as putative direct targets of the AtAP3 protein (Supplementary Table S6B). Among the 1,310 Nigella transcripts that were homologous to the AtAP3-BCG (NdH-BCG), ~3% (37) were differentially expressed between the two Nigella morphs (NdH-BCG-DEG). Within this group, the proportion of homologs of AtAP3 direct target genes was 13% (5 out 37) when considering only the best hits, and 18% (7 out 37) if paralogs of target genes are included (Figure 4A).
When restricting the analysis to the set of Nigella DEG genes that were the best hits of Arabidopsis AtBCG-DEGs and AtDEGs, the percentage of homologs of AtAP3-target genes increased to 45% (5 out of 11, Supplementary Table S6B) and 49% (23 out of 47, Supplementary Table S6A), respectively. Thus, the highly conserved gene set between the two species seems to be enriched in direct targets of AtAP3 protein. In addition, the expression of the majority of highly conserved homologs is affected in the same way in both the Arabidopsis ap3.3 and Nigella NdAP3-3 null mutants, as shown by the LFC in expression level during the early stages of flower development (Supplementary Table S7). This further suggests that these homologs of AtAP3 target genes could be putative direct targets of the NdAP3-3 protein in N. damascena. Among these putative conserved target genes, we found the AP3 paralogs themselves, other transcription factor genes, such as CRC, DRNL, AINTEGUMENTA-like 5, and genes involved in cell wall biogenesis, gibberellin biosynthesis, and signal transduction (Supplementary Table S7).
Finally, to identify additional target genes in the set of genes differentially expressed between the two floral morphs in Nigella, we carried out a comparative analysis with the AP3-3 (AcAP3-3) target genes detected in the full genome of A. coerulea, another Ranunculaceae species (Jiang et al., 2020). Among the 1,620 DEGs found in Nigella, 257 were homologous to AcAP3-3 target genes (NdH-AcAP3-3; Figure 4B). When considering the set of 172 homologs that are differentially expressed in both Arabidopsis and Nigella (NdH-DEG), 18 (12+6) putative targets were found conserved between the three species, 30 (19+11) were specific to Arabidopsis and Nigella, and 239 were specific to Nigella and Aquilegia (Supplementary Table S8). Interestingly, among the target genes that were conserved between the three species, one-third of the Nigella genes homologous to a paralog of AtAP3-target genes were found to be the best homologs of an AcAP3-3 target gene (6 out of 18, Supplementary Table S8). Among the 239 putative conserved target genes between Nigella and Aquilegia, 40 genes were homologous to genes that were differentially expressed between the Arabidopsis wild type and ap3.3 mutant although not direct targets of the AtAP3 protein.
Petals in Ranunculaceae are believed to have evolved independently of petals in core eudicots (Irish, 2009; Carrive et al., 2020). In both Ranunculaceae and core eudicots, however, B-function genes are instrumental for their identity and development. In Ranunculaceae, of the three AP3 paralogous lineages, the AP3-III orthologs have shown to be key genes for petal identity (Sharma et al., 2011; Gonçalves et al., 2013; Wang et al., 2016). In this study, we took advantage of the spontaneous NdAP3-3 mutation in N. damascena that results in a floral morph without petals to characterize early target genes of NdAP3-3, which determine petal identity and first steps of development. Then we adopted a comparative perspective to address the question of the conservation of this early gene network, using the wealth of knowledge available in Arabidopsis as a reference point for core eudicots, and the recently available results obtained from developing petals in A. coerulea (Jiang et al., 2020).
We found in our transcriptome all the MADS-box genes involved in the N. damascena ABCE model of floral organ identity (Wang et al., 2016). In Arabidopsis, AP1 is one of the two genes involved in the A-function. In N. damascena, no AP1 homolog was found, but several isoforms of the NdAGL6 gene, which from functional analysis is suspected to be an A-function gene (Wang et al., 2016) were identified. The high proportion of homologs of the Arabidopsis AP1 best correlated genes in Nigella suggests that the A-function network is as represented as the networks of the other functions, even though it may have a different master control gene, possibly NdAGL6. The slight overrepresentation of homologs of AP1-BCGs, as well as of AP3-BCGs, could be due to a bias toward early developmental–stage genes and a preponderance of perianth developmental genes at the expense of reproductive organ genes in the Nigella transcriptome (Supplementary Figure S5B). This hypothesis could also account for the comparatively slight underrepresentation of homologs of AG and possibly SEP1/3-correlated genes.
In Arabidopsis, the B-function AtAP3 and AtPI proteins interact as a heterodimer to maintain their expression at a high level at late stages. However, their initial expressions are independent (Goto and Meyerowitz, 1994). In N. damascena, all the B-function genes are more or less expressed in petal depending on the developmental stage (Gonçalves et al., 2013; Wang et al., 2016). NdPI1, which encodes a protein shown to interact with NdAP3-3 in yeast two-hybrid (Wang et al., 2016), was not differentially expressed between morphs during early petal development; no differential expression was observed either for its paralog NdPI2 (Supplementary Table S2). These data further support that their initial expression does not depend on AP3/PI protein–protein interaction, as shown in Arabidopsis. By contrast, NdAP3-2 appeared significantly more expressed in the T morph than the P morph at stage 1, but the increase in expression between stages was higher in the P morph than in the T morph, resulting in no significant difference between morphs at stage 2. The reverse situation was observed for the NdAP3-1 isoform with no differential expression between morphs at stage 1 but a greater increase in expression in the P morph than in the T morph resulting in a significant difference at stage 2. Thus, the differential expression profiles of NdAP3-1 and NdAP3-2 suggest either a regulatory control of their expression by NdAP3-3 (Wang et al., 2016; Jiang et al., 2020), or some kind of interaction between all three NdAP3 proteins that may affect their expression.
Because petals are completely suppressed in the homozygous NdAP3-3 mutant, we assume that differentially expressed genes between the two homozygous PP and pp genotypes would include good candidates for petal identity and early development gene network in N. damascena. In Arabidopsis, AP3/PI heterodimer is bifunctional, acting either as activator or repressor of downstream target genes (Wuest et al., 2012). Interestingly, among the genes differentially expressed between the two floral morphs in N. damascena, we found more genes with a higher expression in the T morph than in the P morph, suggesting that NdAP3-3 and the complex it belongs to act more often as a repressor than as an activator at the early stages that we investigated.
Almost 50% of the morph DEGs could not be functionally annotated (Supplementary Table S3). Part of these transcripts could be non-coding sequences, or possibly misassembled sequences, a limitation of transcriptome assembly without a reference genome. Based on the results of homology searches in the genome of the closely related species A. coerulea and of experimental validation of a small set of unannotated sequences, we make the assumption that about 50% of these transcripts, possibly including non-coding transcripts, would be specific to N. damascena and could possibly be involved in species-specific processes. Annotation and GO-term assignment in the most differentially expressed gene set suggested a large involvement of nuclear activities, including regulation of transcription, and an enrichment in processes linked to cell proliferation (ribosome components, DNA or RNA binding, cell organization and biogenesis, and/or DNA or RNA or protein metabolism), which is consistent with the initiation of a novel organ. Most Arabidopsis homologous genes specific to or preferentially expressed in the Nigella petal at late developmental stages (Zhang et al., 2020) were found in our transcriptome. However, they were not differentially expressed between morphs at the early stages we investigated, supporting the hypothesis that different gene sets and networks operate during development to build the elaborate petal. Within the annotated set of DEGs between morphs, we found a homolog of CRC, a carpel developmental gene known to be repressed by B-function genes in early stages of floral development in Arabidopsis (Wuest et al., 2012). Consistent with this, we found that NdCRC was more expressed in the T morph than in the P morph at both stages. We found a few homologs of genes known to play a role in Arabidopsis petal organogenesis sensu stricto (Huang and Irish, 2016). A homolog of a DRNL transcription factor, one of the earliest markers of petal initiation, was found to be downregulated in the T morph, and to be a putative target of NdAP3-3 (Supplementary Table S7). Conversely, a homolog of KIP-RELATED PROTEIN 7, an inhibitor of petal initiation and cell proliferation, was found upregulated in the mutant. Although a homolog of BIGPETAL was found in the Nigella transcriptome, it was not differentially expressed between the two morphs. This low number of homologs in N. damascena may suggest a lack of functional conservation, but it may also be that the developmental stages investigated are not appropriate to observe an expression and/or an effect of NdAP3-3 on these genes. Indeed, it can be noted that a few of these genes are differentially expressed between the wild type and the ap3.3 mutant in Arabidopsis at developmental stages that are similar to ours (Wuest et al., 2012).
To further investigate the conservation of the petal gene network, we took advantage of the knowledge of the AtAP3 gene network and the large amount of expression data available in Arabidopsis. Because the inactivation of NdAP3-3 alone is sufficient to lose petal identity without affecting stamen identity in N. damascena, unlike the AP3 mutation in Arabidopsis, we expected only partial conservation of the gene network between the two species. Among the most conserved set of genes between Nigella and Arabidopsis (11 NdH-BCG-DEG and 47 NdH-DEG, Supplementary Table S6) that are regulated by the AP3 genes, almost half were homologs of direct targets of the AtAP3 protein, indicating an enrichment of AtAP3 target genes. Potential NdAP3-3 target genes included the three NdAP3 paralogs, the NdCRC gene, as well as genes involved in hormone signaling and morphogenesis (Supplementary Table S7). Early repression of carpel developmental genes could be a conserved function of AP3 genes (Wuest et al., 2012).
The percentage of target genes of AP3-3 homologous to AtAP3 target genes are in the same range in A. coerulea (Jiang et al., 2020) and N. damascena (Figure 4A). Combining these results, we found a set of 18 potential target genes that were conserved between the three species. Because the developmental stage investigated in A. coerulea was quite late, part of early target genes that could be conserved between species could have been missed. Nevertheless, our analyses point to a core set of conserved genes between three species belonging to two divergent clades of eudicots with independently derived petals (Carrive et al., 2020). These genes might encode conserved protein functions and be targeted by the common ancestor protein of the AP3-III and euAP3 lineages. Among these conserved genes, we found again the homologs of AP3 and other transcription factors, as well as enzymes involved in cell wall biosynthesis (Supplementary Table S8). Interestingly, when comparing Arabidopsis and the two Ranunculaceae species, we noticed that different paralogs of a gene family could be recruited as the direct target of the AP3 proteins to fulfill a similar function (6 out 18, Supplementary Table S8). To confirm these conserved genes as part of the AP3 gene network at an early stage, both additional ChIP‐ and RNA-seq analyses should be carried out in A. coerulea, as well as ChIP-sequence experiments in N. damascena when the genome sequence will become available.
The comparative analysis of the Nigella differentially expressed genes and the putative target genes of AcAP3-3 revealed a higher conservation of the petal gene network between the two Ranunculaceae species than between them and Arabidopsis (Figure 4B). This is expected, because petals in Ranunculaceae have been evolved once independently from those of core eudicots (Carrive et al., 2020), and Ranunculaceae and core eudicots diverged around 130 million years ago (Magallón et al., 2015). Interestingly, among these conserved Ranunculaceae putative target genes, we found 40 genes that have Arabidopsis homologs with a differential expression between the wild type and the ap3.3 mutant, but no AP3-binding sites in their regulatory regions, suggesting that they are not direct target genes of the AtAP3 protein. One hypothesis would be that these genes would have a flexible position in the petal gene network and could shift from an AP3 direct-to-indirect target position, depending on the species. New plant models for flower development chosen across the angiosperm phylogeny, and advanced DNA sequencing and analyses methods, should help addressing these hypotheses in the near future.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found at: https://www.ncbi.nlm.nih.gov/geo/, GSE159429.
YD, NC, CD, DM, and SN designed the study. MG and HC performed the molecular work. LS-T, SB, and JC produced the RNA-seq data. VB, HB, JJ, and ED performed the bioinformatic work. YD, NC, DM, and CD performed the analyses. All authors contributed to the article and approved the submitted version.
This project received financial support from the Institut Diversité, Ecologie et Evolution du Vivant (IDEEV; AAP2013 and AAP2015).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The GQE-Le Moulon and Institute of Plant Sciences Paris-Saclay benefit from the support of the LabExSaclay Plant Sciences-SPS (ANR-10-LABX-0040-SPS). The authors acknowledge H Citerne for helpful comments, and M-L Martin-Magniette and G Rigaill for their advice on the statistical analyses of transcriptomic quantitative data.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.660803/full#supplementary-material
AtAP3-BCG, 200 Arabidopsis genes whose expression is best correlated (100 positively and 100 negatively) with AtAP3 expression; AtDEG, Arabidopsis genes whose expression differs between wild type and ap3.3 mutant; AtBCG-DEG, Arabidopsis genes that are both AtDEG and AtAP3-BCG; DEG, differentially expressed genes; M_LFC, Log-fold change between the two morphs; NdH, Nigella genes homologous to AtDEG; NdH-BCG, Nigella genes homologous to AtAP3-BCG; NdH-BCG-DEG, NdH-BCG that are differentially expressed between the Nigella floral morphs; NdH-DEG, NdH that are differentially expressed between the Nigella floral morphs.
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Ambrose, B. A., Lerner, D. R., Ciceri, P., Padilla, C. M., Yanofsky, M. F., and Schmidt, R. J. (2000). Molecular and genetic analyses of the Silky1 gene reveal conservation in floral organ specification between eudicots and monocots. Mol. Cell 5, 569–579. doi: 10.1016/S1097-2765(00)80450-5
Ashburner, M., Ball, C. A., Blake, J. A., Botstein, D., Butler, H., Cherry, J. M., et al. (2000). Gene ontology: tool for the unification of biology. The gene ontology consortium. Nat. Genet. 25, 25–29. doi: 10.1038/75556
Benjamini, Y., and Hochberg, Y. (1995). Controlling the false discovery rate: a practical and powerful approach to multiple testing. J. Roy. Soc. 57, 289–300.
Bey, M., Stüber, K., Fellenberg, K., Schwarz-Sommer, Z., Sommer, H., Saedler, H., et al. (2004). Characterization of Antirrhinum petal development and identification of target genes of the class B MADS box gene DEFICIENS. Plant Cell 16, 3197–3215. doi: 10.1105/tpc.104.026724
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Carrive, L., Domenech, B., Sauquet, H., Jabbour, F., Damerval, C., and Nadot, S. (2020). Insights into the ancestral flowers of Ranunculales. Bot. J. Linn. Soc. 194, 23–46. doi: 10.1093/botlinnean/boaa031
Coen, E., and Meyerowitz, E. M. (1991). The war of the whorls: genetic interactions controlling flower development. Nature 353, 31–37. doi: 10.1038/353031a0
Damerval, C., Citerne, H. C., Conde e Silva, N., Deveaux, Y., Delannoy, E., Joets, J., et al. (2019). Unravelling the developmental and genetic mechanisms underpinning floral architecture in proteaceae. Front. Plant Sci. 10:18. doi: 10.3389/fpls.2019.00018
Davila-Velderrain, J., Servin-Marquez, A., and Alvarez-Buylla, E. R. (2014). Molecular evolution constraints in the floral organ specification gene regulatory network module across 18 angiosperm genomes. Mol. Biol. Evol. 31, 560–573. doi: 10.1093/molbev/mst223
Di Stilio, V. S., Kramer, E. M., and Baum, D. A. (2005). Floral MADS box genes and homeotic gender dimorphism in Thalictrum dioicum (Ranunculaceae) – a new model for the study of dioecy. Plant J. 41, 755–766. doi: 10.1111/j.1365-313X.2005.02336.x
Drea, S., Hileman, L. C., de Martino, G., and Irish, V. F. (2007). Functional analyses of genetic pathways controlling petal specification in poppy. Development 134, 4157–4166. doi: 10.1242/dev.013136
Eddy, S. R., and Pearson, W. R. (2011). Accelerated profile HMM searches. PLoS Comput. Biol. 7:e1002195. doi: 10.1371/journal.pcbi.1002195
Edgar, R., Domrachev, M., and Lash, A. E. (2002). Gene expression omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 30, 207–210. doi: 10.1093/nar/30.1.207
Gagnot, S., Tamby, J.-P., Martin-Magniette, M.-L., Bitton, F., Taconnat, L., Balzergue, S., et al. (2007). CATdb: a public access to Arabidopsis transcriptome data from the URGV-CATMA platform. Nucleic Acids Res. 36, D986–D990. doi: 10.1093/nar/gkm757
Galimba, K. D., Martínez-Gómez, J., and Di Stilio, V. S. (2018). Gene duplication and transference of function in the paleoAP3 lineage of floral organ identity genes. Front. Plant Sci. 9:334. doi: 10.3389/fpls.2018.00334
Gonçalves, B., Nougué, O., Jabbour, F., Ridel, C., Morin, H., Laufs, P., et al. (2013). An Apetala3 homolog controls both petal identity and floral meristem patterning in Nigella damascena L. (Ranunculaceae). Plant J. 76, 223–235. doi: 10.1111/tpj.12284
Goto, K., and Meyerowitz, E. M. (1994). Function and regulation of the Arabidopsis floral homeotic gene PISTILLATA. Genes Dev. 8, 1548–1560. doi: 10.1101/gad.8.13.1548
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Huala, E., Dickerman, A., Garcia-Hernandez, M., Weems, D., Reiser, L., LaFond, F., et al. (2001). The Arabidopsis information resource (TAIR): a comprehensive database and web-based information retrieval, analysis, and visualization system for a model plant. Nucleic Acids Res. 29, 102–105. doi: 10.1093/nar/29.1.102
Huang, T., and Irish, V. F. (2016). Gene networks controlling petal organogenesis. J. Exp. Bot. 67, 61–68. doi: 10.1093/jxb/erv444
Irish, V. F. (2009). Evolution of petal identity. J. Exp. Bot. 60, 2517–2527. doi: 10.1093/jxb/erp159
Jabbour, F., Udron, M., Le Guilloux, M., Gonçalves, B., Manicacci, D., Nadot, S., et al. (2015). Flower development schedule and AGAMOUS-like gene expression patterns in two morphs of Nigella damascena (Ranunculaceae) differing in floral architecture. Bot. J. Linn. Soc. 178, 608–619. doi: 10.1111/boj.12297
Jiang, Y., Wang, M., Zhang, R., Xie, J., Duan, X., Shan, H., et al. (2020). Identification of the target genes of AqAPETALA3-3 (AqAP3-3) in Aquilegia coerulea (Ranunculaceae) helps understand the molecular bases of the conserved and nonconserved features of petals. New Phytol. 227, 1235–1248. doi: 10.1111/nph.16601
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Kaufmann, K., Muiño, J. M., Jauregui, R., Airoldi, C. A., Smaczniak, C., Krajewski, P., et al. (2009). Target genes of the MADS transcription factor SEPALLATA3: integration of developmental and hormonal pathways in the Arabidopsis flower. PLoS Biol. 7:e1000090. doi: 10.1371/journal.pbio.1000090
Kaufmann, K., Wellmer, F., Muiño, J. M., Ferrier, T., Wuest, S. E., Kumar, V., et al. (2010). Orchestration of floral initiation by APETALA1. Science 328, 85–89. doi: 10.1126/science.1185244
Kopylova, E., Noé, L., and Touzet, H. (2012). SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscritomic data. Bioinformatics 28, 3211–3217. doi: 10.1093/bioinformatics/bts611
Kramer, E. M., Di Stilio, V. S., and Schlüter, P. M. (2003). Complex patterns of gene duplication in APETALA3 and PISTILLATA lineages of the Ranunculaceae. Int. J. Plant Sci. 164, 1–11. doi: 10.1086/344694
Kramer, E. M., Dorit, R. L., and Irish, V. F. (1998). Molecular evolution of genes controlling petal and stamen development: duplication and divergence within the APETALA3 and PISTILLATA MADS-box gene lineages. Genetics 149, 765–783. doi: 10.1093/genetics/149.2.765
Kramer, E. M., Holappa, L., Gould, B., Jaramillo, M. A., Setnikov, D., and Santiago, P. M. (2007). Elaboration of B gene function to include the identity of novel floral organs in the lower eudicot Aquilegia. Plant Cell 19, 750–766. doi: 10.1105/tpc.107.050385
Kramer, E. M., Su, H. J., Wu, C. C., and Hu, J. M. (2006). A simplified explanation for the frameshift mutation that created a novel C-terminal motif in the APETALA3 gene lineage. BMC Evol. Biol. 6:30. doi: 10.1186/1471-2148-6-30
Langmead, B., and Salzberg, S. (2012). Fast gapped-read alignment with Bowtie2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Lee, H. L., and Irish, V. F. (2011). Gene duplication and loss in a MADS box gene transcription factor circuit. Mol. Biol. Evol. 28, 3367–3380. doi: 10.1093/molbev/msr169
Magallón, S., Gómez-Acevedo, S., Sánchez-Reyes, L. L., and Hernández-Hernández, T. (2015). A metacalibrated time-tree documents the early rise of flowering plant phylogenetic diversity. New Phytol. 207, 437–453. doi: 10.1111/nph.13264
Mara, C. D., and Irish, V. F. (2008). Two GATA transcription factors are downstream effectors of floral homeotic gene action in Arabidopsis. Plant Physiol. 147, 707–718. doi: 10.1104/pp.107.115634
Mondragón-Palomino, M., and Theissen, G. (2008). MADS about the evolution of orchid flowers. Trends Plant Sci. 13, 51–59. doi: 10.1016/j.tplants.2007.11.007
Ó’Maoiléidigh, D. S., Wuest, S. E., Rae, L., Raganelli, A., Ryan, P. T., Kwasniewska, K., et al. (2013). Control of reproductive floral organ identity specification in Arabidopsis by the C function regulator AGAMOUS. Plant Cell 25, 2482–2503. doi: 10.1105/tpc.113.113209
Pajoro, A., Biewers, S., Dougali, E., Valentim, F. L., Mendes, M. A., Porri, A., et al. (2014). The (r)evolution of gene regulatory networks controlling Arabidopsis plant reproduction: a two decades history. J. Exp. Bot. 65, 4731–4745. doi: 10.1093/jxb/eru233
Pelaz, S., Tapia-Lopez, R., Alvarez-Buylla, E. R., and Yanofsky, M. F. (2001). Conversion of leaves into petals in Arabidopsis. Curr. Biol. 11, 182–184. doi: 10.1016/S0960-9822(01)00024-0
Perina, A., González-Tizón, A. M., Meilán, I. F., and Martínez-Lage, A. (2016). De novo transcriptome assembly of shrimp Palaemon serratus. Genom. Data 11, 89–91. doi: 10.1016/j.gdata.2016.12.009
Rasmussen, D. A., Kramer, E. M., and Zimmer, E. A. (2009). One size fits all? Molecular evidence for a commonly inherited petal identity program in Ranunculales. Am. J. Bot. 96, 96–109. doi: 10.3732/ajb.0800038
Rhee, S. Y., Beavis, W., Berardini, T. Z., Chen, G., Dixon, D., Doyle, A., et al. (2003). The Arabidopsis information resource (TAIR): a model organism database providing a centralized, curated gateway to Arabidopsis biology, research materials and community. Nucleic Acids Res. 31:224. doi: 10.1093/nar/gkg076
Robinson, M. D., McCarthy, D. J., and Smyth, G. K. (2010). EdgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. doi: 10.1093/bioinformatics/btp616
Ronse de Craene, L. P., and Brockington, S. F. (2013). Origin and evolution of petals in angiosperms. Plant Ecol. Evol. 146, 5–25. doi: 10.5091/plecevo.2013.738
Roy, N. S., Kim, J. A., Choi, A. Y., Ban, Y. W., Park, N. I., Park, K. C., et al. (2018). RNA-seq de novo assembly and differential transcriptome analysis of Korean medicinal herb Cirsium japonicum var. spinossimum. Genomics Inform. 16:e34. doi: 10.5808/GI.2018.16.4.e34
Sauquet, H., von Balthazar, M., Magallón, S., Doyle, J. A., Endress, P. K., Bailes, E. J., et al. (2017). The ancestral flower of angiosperms and its early diversification. Nat. Commun. 8:16047. doi: 10.1038/ncomms16047
Sharma, B., Guo, C., Kong, H., and Kramer, E. M. (2011). Petal-specific subfunctionalization of an APETALA3 paralog in the Ranunculales and its implications for petal evolution. New Phytol. 191, 870–883. doi: 10.1111/j.1469-8137.2011.03744.x
Sharma, B., and Kramer, E. M. (2013). Sub‐ and neo-functionalization of APETALA3 paralogs have contributed to the evolution of novel floral organ identity in Aquilegia (columbine, Ranunculaceae). New Phytol. 197, 949–957. doi: 10.1111/nph.12078
Suzek, B. E., Wang, Y., Huang, H., McGarvey, P. B.,Wu, C. H., and the UniProt Consortium (2015). UniRef clusters: a comprehensive and scalable alternative for improving sequence similarity searches. Bioinformatics 31, 926–932. doi: 10.1093/bioinformatics/btu739
The Gene Ontology Consortium (2001). Creating the gene ontology resource: design and implementation. Genome Res. 11, 1425–1433. doi: 10.1101/gr.180801
Theißen, G., and Gramzow, L. (2016). “Structure and evolution of plant MADS domain transcription factors,” in Plant Transcription Factors: Evolutionary, Structural and Functional Aspects. Vol. 2016. ed. D. H. Gonzalez (Philadelphia: Elsevier), 127–138.
Theißen, G., Melzer, R., and Rümpler, F. (2016). MADS-domain transcription factors and the floral quartet model of flower development: linking plant development and evolution. Development 143, 3259–3271. doi: 10.1242/dev.134080
Thomson, B., Zheng, B., and Wellmer, F. (2017). Floral organogenesis: when knowing your ABCs is not enough. Plant Physiol. 173, 56–64. doi: 10.1104/pp.16.01288
Toufighi, K., Brady, S. M., Austin, R., Ly, E., and Provart, N. J. (2005). The botany Array resource: e-northern, expression angling, and promoter analyses. Plant J. 43, 153–163. doi: 10.1111/j.1365-313X.2005.02437.x
Toxopéus, H. J. (1927). Erblichkeitsuntersuchungen an Nigella damascena L. Genetica 9, 341–440. doi: 10.1007/BF01508296
Wang, P., Liao, H., Zhang, W., Yu, X., Zhang, R., Shan, H., et al. (2016). Flexibility in the structure of spiral flowers and its underlying mechanisms. Nat. Plants 2:15188. doi: 10.1038/NPLANTS.2015.188
Wuest, S. E., O’Maoileidigh, D. S., Rae, L., Kwasniewska, K., Raganellia, A., Hanczaryk, K., et al. (2012). Molecular basis for the specification of floral organs by APETALA3 and PISTILLATA. Proc. Natl. Acad. Sci. U. S. A. 109, 13452–13457. doi: 10.1073/pnas.1207075109
Yan, W., Chen, D., and Kaufmann, K. (2016). Molecular mechanisms of floral organ specification by MADS domain proteins. Curr. Opin. Plant Biol. 29, 154–162. doi: 10.1016/j.pbi.2015.12.004
Zahn, L. M., Leebens-Mack, J., dePamphilis, C. W., Ma, H., and Theissen, G. (2005). To B or not to B a flower: the role of DEFICIENS and GLOBOSA Orthologs in the evolution of the angiosperms. J. Hered. 96, 225–240. doi: 10.1093/jhered/esi033
Zdobnov, E. M., and Apweiler, R. (2001). InterProScan ‐ an integration platform for the signature-recognition methods in InterPro. Bioinformatics 17, 847–848. doi: 10.1093/bioinformatics/17.9.847
Zhang, R., Fu, X., Zhao, C., Cheng, J., Liao, H., Wang, P., et al. (2020). Identification of the key regulatory genes involved in elaborate petal development and specialized character formation in Nigella damascena (Ranunculaceae). Plant Cell 32, 3095–3311. doi: 10.1105/tpc.20.00330
Zhang, R., Guo, C., Zhang, W., Wang, P., Li, L., Duan, X., et al. (2013). Disruption of the petal identity gene APETALA3-3 is highly correlated with loss of petals within the buttercup family (Ranunculaceae). Proc. Natl. Acad. Sci. U. S. A. 13, 5077–5079. doi: 10.1073/pnas.1219690110
Keywords: APETALA3, RNA-seq, AP3 target genes, petal, Nigella damascena, floral mutant, Ranunculaceae, Arabidopsis
Citation: Deveaux Y, Conde e Silva N, Manicacci D, Le Guilloux M, Brunaud V, Belcram H, Joets J, Soubigou-Taconnat L, Delannoy E, Corti H, Balzergue S, Caius J, Nadot S and Damerval C (2021) Transcriptome Analysis Reveals Putative Target Genes of APETALA3-3 During Early Floral Development in Nigella damascena L. Front. Plant Sci. 12:660803. doi: 10.3389/fpls.2021.660803
Edited by:
Elena M. Kramer, Harvard University, United StatesReviewed by:
Verónica S. Di Stilio, University of Washington, United StatesCopyright © 2021 Deveaux, Conde e Silva, Manicacci, Le Guilloux, Brunaud, Belcram, Joets, Soubigou-Taconnat, Delannoy, Corti, Balzergue, Caius, Nadot and Damerval. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yves Deveaux, eXZlcy5kZXZlYXV4QHVuaXZlcnNpdGUtcGFyaXMtc2FjbGF5LmZy
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.