 Anastasia McKinlay
Anastasia McKinlay Dalen Fultz
Dalen Fultz Feng Wang
Feng Wang Craig S. Pikaard
Craig S. Pikaard
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
METHODS article
Front. Plant Sci., 28 April 2021
Sec. Plant Cell Biology
Volume 12 - 2021 | https://doi.org/10.3389/fpls.2021.656049
This article is part of the Research TopicMolecular Organization, Evolution, and Function of Ribosomal DNAView all 15 articles
Large regions of nearly identical repeats, such as the 45S ribosomal RNA (rRNA) genes of Nucleolus Organizer Regions (NORs), can account for major gaps in sequenced genomes. To assemble these regions, ultra-long sequencing reads that span multiple repeats have the potential to reveal sets of repeats that collectively have sufficient sequence variation to unambiguously define that interval and recognize overlapping reads. Because individual repetitive loci typically represent a small proportion of the genome, methods to enrich for the regions of interest are desirable. Here we describe a simple method that achieves greater than tenfold enrichment of Arabidopsis thaliana 45S rRNA gene sequences among ultra-long Oxford Nanopore Technology sequencing reads. This method employs agarose-embedded genomic DNA that is subjected to restriction endonucleases digestion using a cocktail of enzymes predicted to be non-cutters of rRNA genes. Most of the genome is digested into small fragments that diffuse out of the agar plugs, whereas rRNA gene arrays are retained. In principle, the approach can also be adapted for sequencing other repetitive loci for which gaps exist in a reference genome.
Many eukaryotic genomes have chromosomal loci that consist of hundreds, if not thousands, of nearly identical repeats, sometimes spanning millions of basepairs. Examples include the AT-rich satellites of pericentromeric regions (Aldrup-Macdonald and Sullivan, 2014), ribosomal RNA (rRNA) gene repeats (Gerbi, 1985; Flavell, 1986) and tandemly repeated transposable element (TE)-derived sequences (Ahmed and Liang, 2012). Distinguishing one repeat from the next can be difficult, precluding the easy determination of how individual repeats are arranged at the locus. As a result, repetitive loci are often miss-assembled or absent from assemblies of eukaryotic genomes (Biscotti et al., 2015).
Two long-read sequencing technologies have greatly improved the ability to close gaps in sequenced genomes, namely Pacific Biosciences (PacBio) SMRT sequencing and Oxford Nanopore MinION sequencing (Besser et al., 2018; Michael et al., 2018). PacBio sequencing can yield reads that are 10–100 kb in length, with the potential to obtain multiple reads of the same DNA fragment. This allows one to obtain consensus sequence reads whose accuracy rivals that of short-read Illumina or Sanger sequencing. By obtaining highly accurate long reads, single nucleotide polymorphisms (SNPs) can be identified among repeats that are nearly identical in sequence. For instance, PacBio sequencing has been used to identify subtle sequence differences among Arabidopsis thaliana rRNA genes (Havlova et al., 2016) that are each ∼10 kb in length. However, PacBio sequencing reads are not long enough for assembly of rRNA genes into long contigs due to the paucity of variation that is unique and thus not shared by numerous genes.
Sequencing using Oxford Nanopore Technology (ONT) yields ultra-long reads that can be hundreds of kilobases in length, but with an accuracy of only 75–95% (Rang et al., 2018). The technology is especially well-suited to identifying chromosomal deletions, insertions, or rearrangements. However, the high error rate of ONT sequencing is problematic for assembly of repetitive regions in which there are few sequence differences to discriminate each repeat (Michael et al., 2018). For successful assembly of these repetitive regions, having multiple overlapping ONT reads is necessary, thus allowing consensus sequences to be deduced to improve the accuracy and confidently identify SNPs and other subtle variation (Ebler et al., 2019).
Targeted enrichment aims to increase sequencing coverage for a region of interest (reviewed in Good, 2011; Kozarewa et al., 2015). Current methods are mostly designed for short-read sequencing, but some are amenable to ultra-long sequencing of large repetitive genomic regions. For instance, the clustered regularly interspaced short palindromic repeats (CRISPR) Cas9 system has been used for targeted sequencing (Bennett-Baker and Mueller, 2017; Gabrieli et al., 2018; Nachmanson et al., 2018). In this approach, megabase-sized genomic regions of interest are cleaved and purified from the rest of the genome by pulsed-field gel electrophoresis. Although compatible with PacBio and ONT sequencing, the strategy poses technical challenges and requires large amounts of starting DNA.
Nucleolus Organizer Regions (NORs) are missing from current genome assemblies of multicellular eukaryotes. The number of NORs in a genome varies between species, and within a species the number of rRNA genes within a NOR can vary between individuals and even among cells of an individual (Stults et al., 2008; Tucker et al., 2010; McStay, 2016). Due to the lack of substantial sequence variation among rRNA genes repeats, NORs of reference genomes are sometimes represented by a single rRNA gene repeat, with actual copy numbers and NOR sizes remaining unknown (Treangen and Salzberg, 2011). ONT sequencing holds promise for the assembly of NORs but has not yet been used to assemble complete NORs (Michael et al., 2018). This poses an obstacle to studies of NOR recombination, replication and repeat homogenization as well as studies of large-scale rRNA gene regulation. For instance, our laboratory is interested in understanding why the two NORs of the Arabidopsis thaliana strain Col-0 differ in expression, with one being constitutively active and the other falling silent during development (Chandrasekhara et al., 2016; Mohannath et al., 2016), an epigenetic phenomenon known as nucleolar dominance (McStay, 2006; Tucker et al., 2010). Each Arabidopsis thaliana NOR is composed of hundreds of tandemly repeated rRNA genes that are each ∼10 kb in length, such that both span several million basepairs of DNA (Copenhaver and Pikaard, 1996b). Evidence suggests that chromosomal context or position, rather than rRNA gene sequence variation, is responsible for the differential expression of the two NORs (Chandrasekhara et al., 2016; Mohannath et al., 2016), but the chromosomal basis for nucleolar dominance remains unknown. The possibility exists that one of more locus control elements might be embedded within the NORs, thus their complete sequence is desirable.
Here, we describe a simple method for enrichment of ultra-high molecular weight rRNA gene tandem arrays using a cocktail of restriction endonucleases predicted not cut an rRNA gene reference sequence. When used to treat genomic DNA embedded in an agarose plug, the enzymes digest most of the genome into small fragments that passively diffuse out of the agarose plug (Fritz and Musich, 1990). This depletes the plug of unwanted DNA fragments while retaining large DNA fragments that include rRNA gene arrays. Using A. thaliana rRNA genes as our example, the strategy yields a tenfold increase in ONT sequencing reads corresponding to rRNA genes. In principle, the method should also be adaptable for the enrichment of other target sequences, simply by altering the choice of restriction endonucleases.
Arabidopsis thaliana Col-0 plants (Arabidopsis Biological Resource Center stock #CS 70000) were surface-sterilized and grown on agar plates containing 0.5X Murashige and Skoog medium (MS). Plants were harvested after 2 weeks of growth under short-day conditions (8 h light, 16 h dark).
Ultra-high molecular weight DNA was purified from Arabidopsis thaliana Col-0 plants by following the Bionano Prep Plant Tissue DNA Isolation, Liquid Nitrogen Grinding Protocol (Bionano document number 30177)1 (summarized in Supplementary Figure 1). Briefly, 2 g of fresh tissue was placed in a pre-chilled (overnight at −80°C) mortar and ground in liquid nitrogen using a pre-chilled pestle then resuspended in 40 mL of ice-cold Bionano Prep Plant Tissue Homogenization Buffer (Part #20283) supplemented with 2-mercaptoethanol (0.2% final concentration) and 1 mM spermine-spermidine (known as Plant Tissue Homogenization Buffer plus). The suspension was passed sequentially through 100 μm (VWR Cat# 21008-950) and 40 μm cell strainers (VWR Cat# 21008-949) into a pre-chilled 50 mL conical tube on ice. Nuclei were then pelleted by centrifugation at 3,500 × g for 20 min at 4°C using a swinging bucket rotor. After discarding the supernatant, the pellet was resuspended in 1 mL of Plant Tissue Homogenization Buffer plus buffer with the assistance of a small paint brush that had been presoaked in the buffer. The resuspended nuclei were further diluted with 40 mL of ice-cold Plant Tissue Homogenization Buffer plus buffer and then subjected to centrifugation at 60 × g for 2 min at 4°C using a swinging bucket rotor to remove cell debris, with no braking during rotor deceleration. The supernatant was then subjected to another 40 μm filtration step (VWR Cat# 21008-949). Nuclei were collected by centrifugation at 3,500 × g for 20 min at 4°C using a swinging bucket rotor and washed three times by resuspension in in 30 mL of ice-cold Plant Tissue Homogenization Buffer plus and re-pelleting at 3,500 × g for 20 min at 4°C. The final nuclei pellet was resuspended in 3 mL of ice-cold Plant Tissue Homogenization Buffer plus and applied on top of the Density Gradient (Bionano Prep Density Gradient, catalog numbers 20281 and 20280). After centrifugation at 4,500 × g for 40 min at 4°C in a swinging bucket rotor, with no braking during deceleration, nuclei were recovered from the gradient and collected into a pre-chilled 15 mL conical tube on ice. Nuclei were then diluted with 14 mL of ice-cold Plant Tissue Homogenization Buffer plus and collected by centrifugation at 2,500 × g for 10 min at 4°C in a swinging bucket rotor, with no braking during deceleration. After carefully decanting the supernatant, nuclei were resuspended in 50 μL of ice-cold Density Gradient Buffer (Bionano Prep Density Gradient Buffer Cat #20280). The nuclei were then equilibrated to 43°C for 3 min and mixed with 30 μL of molten 2% agarose equilibrated at 43°C (CleanCut Low Melting Point, Bio-Rad, Cat# 1703594) using a wide-bore tip, and pipetted into a Bio-Rad CHEF Disposable Plug Mold (Bio-Rad, Cat# 170-3713). The final agarose concentration of the plugs was 0.82%. Plug molds were incubated at 4°C for 15 min to solidify the agarose.
Plugs containing embedded nuclei were then subjected to Proteinase K (20 mg/mL; 0.8 mg/plug; QIAGEN, Cat# 19131) and RNase A (100 μg/mL; 1 μg/plug; QIAGEN, Cat# 19101) digestion and washed according to the Bionano Prep Plant Tissue DNA Isolation, Liquid Nitrogen Grinding Protocol (document #30177). For rRNA gene enrichment, embedded nuclei were treated with a restriction endonuclease cocktail composed of six enzymes predicted to be rRNA gene non-cutters. Briefly, agarose plugs were placed in 50 mL conical tubes and were first incubated in 10 mL of T10E10 buffer (10 mM Tris-HCl, 10 mM EDTA, pH 8.0) supplemented with 2 mM PMSF for 1 h at 4°C. Plugs were then washed four times, 30 min each, at room temperature in 10 mL of T10E10 buffer without PMSF. Next, individual agarose plugs were washed twice, 1 h, at room temperature, with 1 ml of 1× restriction enzyme buffer [1× CutSmart buffer (NEB)]. After a second wash, the plug was incubated with 200 μL of 1× CutSmart buffer (NEB) containing 50 U each of the restriction endonucleases BstZ17I-HF (NEB #R3594), SpeI-HF (NEB #R3133), BclI-HF (NEB #R3160), SnaBI (NEB #R0130), MscI (NEB #R0534), and PvuII-HF (NEB #R3151) at 37°C overnight. The buffer was then removed and replaced with 500 μL of 20 mM Tris-HCl, 50 mM EDTA, pH 8.0 and incubated at 10 min at room to stop further digestion. The agarose plug was then subjected to 5 wash steps, each 15 min at room temperature, with 10 mL of TE buffer in order to deplete the plugs of short DNA digestion products fragments that can diffuse from the plugs, unlike large DNA fragments that are retained.
Plugs were then melted at 70°C for 2 min, equilibrated at 43°C for 5 min, and then incubated with 2 μL of 0.5 U/μL agarase (ThermoFisher Scientific, Cat# EO0461) per plug at 43°C for 45 min to digest the agar and liberate the encapsulated DNA. The resulting solution was then subjected to drop dialysis by applying genomic DNA on a 0.1μm dialysis membrane (Millipore, Cat# VCWP04700) floating on the surface of 15 mL of TE buffer inside a 6 cm petri dish. Dialysis was at room temperature for 45 min. DNA was assessed for quantity and quality using a Qubit dsDNA BR Assay kit and by agarose gel electrophoresis.
Genomic DNA was subjected to electrophoresis using a 0.7% agarose gel in 0.5× TBE buffer for 1 h and 15 min at 100 V. The gel was then stained with GelRed diluted 1:10,000 in water (GoldBio # G-725-100). The intensively stained DNA band >23 kb, consisting of all DNA fragments greater than the resolving limit of the gel, was then excised and the DNA extracted using a QIAEX II Gel Extraction Kit (QIAGEN #20021). The resulting DNA was assessed by qPCR for the presence of 25S rRNA gene sequences and actin genes using the following forward (F) and reverse (R) primers:
25S_F: GAGTGCTTGAAATTGTCGGGAGGGAAG;
25S_R: CGAATCTTAGCGACAAAGGGCTGAATC;
actin_F: GAGAGATTCAGATGCCCAGAAGTC;
actin_R: TGGATTCCAGCAGCTTCCA.
DNA sequencing library preparation was performed using the Oxford Nanopore Rapid Library Kit (RAD-004). Sequencing was performed using a MinION sequencer with an R9.4.1 flow cell. Base-calling of raw ONT sequencing data was performed using Albacore v2.3.1. General FASTQ read statistics were calculated by NanoPlot (v 1.13). Length count distribution was analyzed using an R ggplot2 package. Statistical analysis was performed with GraphPad Prizm8 software. Percent sequence identity was calculated using minimap2 and the following Perl script (Li, 2018): <minimap2 -c reference.fasta query.fasta | perl -ane “if(/tp:A:P/&&/NM:i:(\d+)/){$n+ = $1; $m+ = $1 while/(\d+)M/g;$g+ = $1,++$o while/(\d+)[ID]/g} END{print(($n-$g+$o)/($m+$o),“\n”)}” >.
Arabidopsis thaliana plants have two NORs that are located on the short arm of chromosomes 2 and 4 (Copenhaver et al., 1995; Copenhaver and Pikaard, 1996a). Each NOR is estimated to span ∼4 Mbp and consist of ∼350 to 400 rRNA genes that are each ∼10 kb in length (Tucker et al., 2010). We used the New England Biolabs (NEB) cutter tool (Vincze et al., 2003) to examine a full-length Arabidopsis thaliana (ecotype Col-0) 45S rRNA gene sequence (Chandrasekhara et al., 2016) and identify a list of 24 restriction endonucleases (RE) whose recognition sites are missing within the reference rRNA gene sequence. We then performed virtual in silico digestions of the Arabidopsis thaliana Col-0 reference genome (TAIR10) to identify the subset of these enzymes that cut most frequently in the genome (Supplementary Figures 2, 3), selecting six that are each predicted to digest genomic DNA to a median fragment length of 5 kb or smaller (Figure 1A) and that display 100% activity in NEB CutSmart buffer. In silico digestion using a cocktail of all six enzymes predicted that they would cut genomic A. thaliana (ecotype Col-0) into DNA fragments with a mean length of 522 bp (Figure 1B).
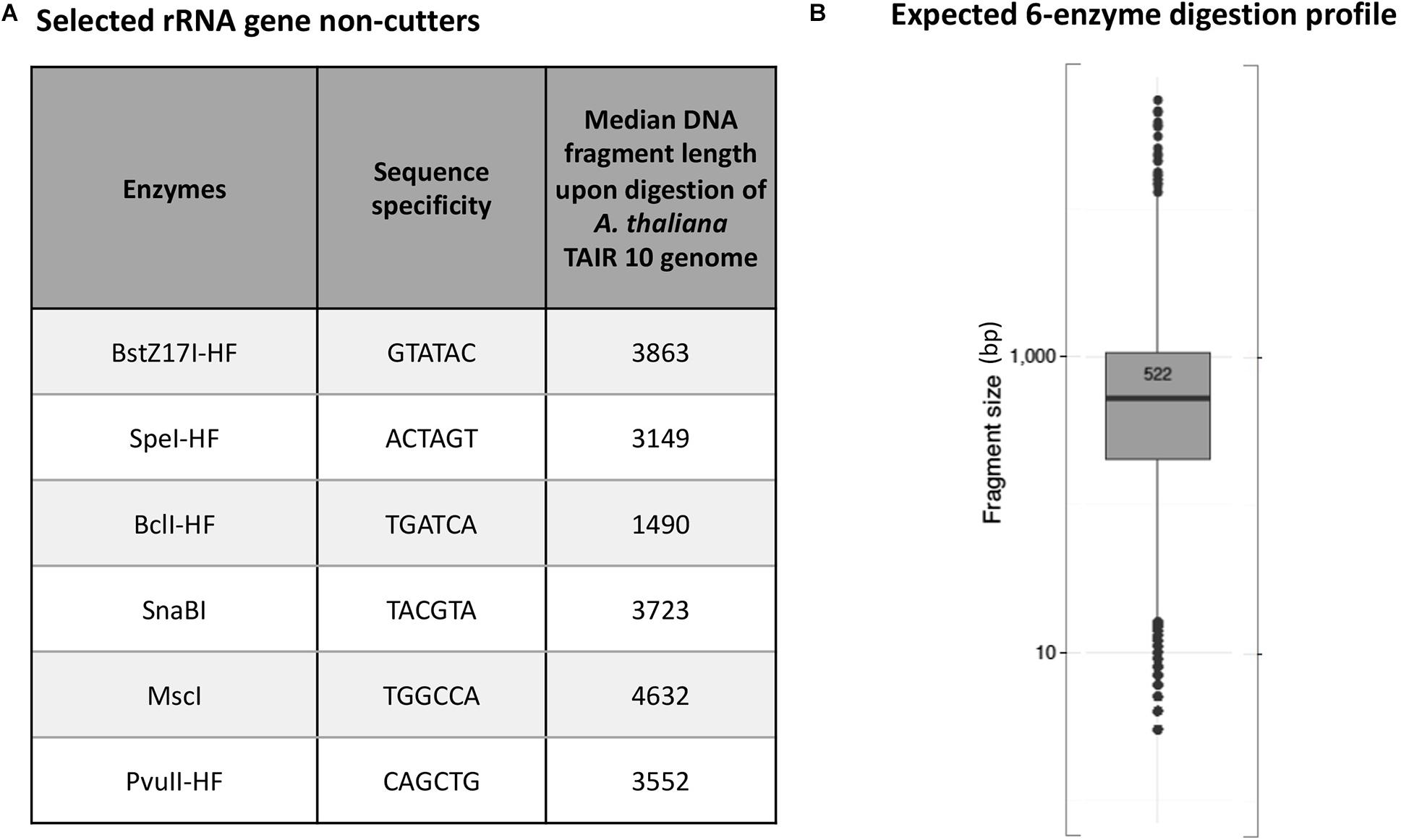
Figure 1. Predicted 45S rRNA gene non-cutting restriction endonucleases used as a cocktail for digestion of genomic DNA. (A) Names and sequence specificities of the enzymes and median fragment sizes obtained upon in silico digestion of A. thaliana Col-0 genomic DNA. (B) Expected in silico digestion mean size and distribution for Col-0 genomic DNA subjected to digestion by the six-enzyme cocktail.
To test the restriction endonuclease cocktail, Col-0 genomic DNA immobilized in agarose plugs was subjected to in-plug digestion as described in Mohannath et al. (2016). Sizes of genomic DNA fragments were visualized following electrophoresis through a 0.7% agarose gel in TBE buffer. As shown in Figure 2A, digestion of genomic DNA with the enzyme cocktail resulted in a significant reduction of high molecular weight DNA (top band) and the appearance of DNA fragments mostly smaller than 5 kb (Figure 2A, lane 3). In contrast, treatment of genomic DNA with the rRNA gene-specific endonuclease I-PpoI, which cleaves once per rRNA gene (Muscarella et al., 1990; Copenhaver and Pikaard, 1996a), resulted in a band of ∼10 kb, the expected rRNA gene unit length (Figure 2A, lane 2). Quantitative PCR analysis of genomic DNA extracted from the top gel band revealed similar quantities of rRNA gene sequences in undigested genomic DNA and DNA cut by the six-enzyme cocktail. By contrast, digestion with I-PpoI depletes rRNA gene sequences, as expected (Figure 2B, top panel). The six-enzyme cocktail reduced the level of control actin gene DNA by 1,000-fold relative to undigested DNA (Figure 2B, bottom panel).
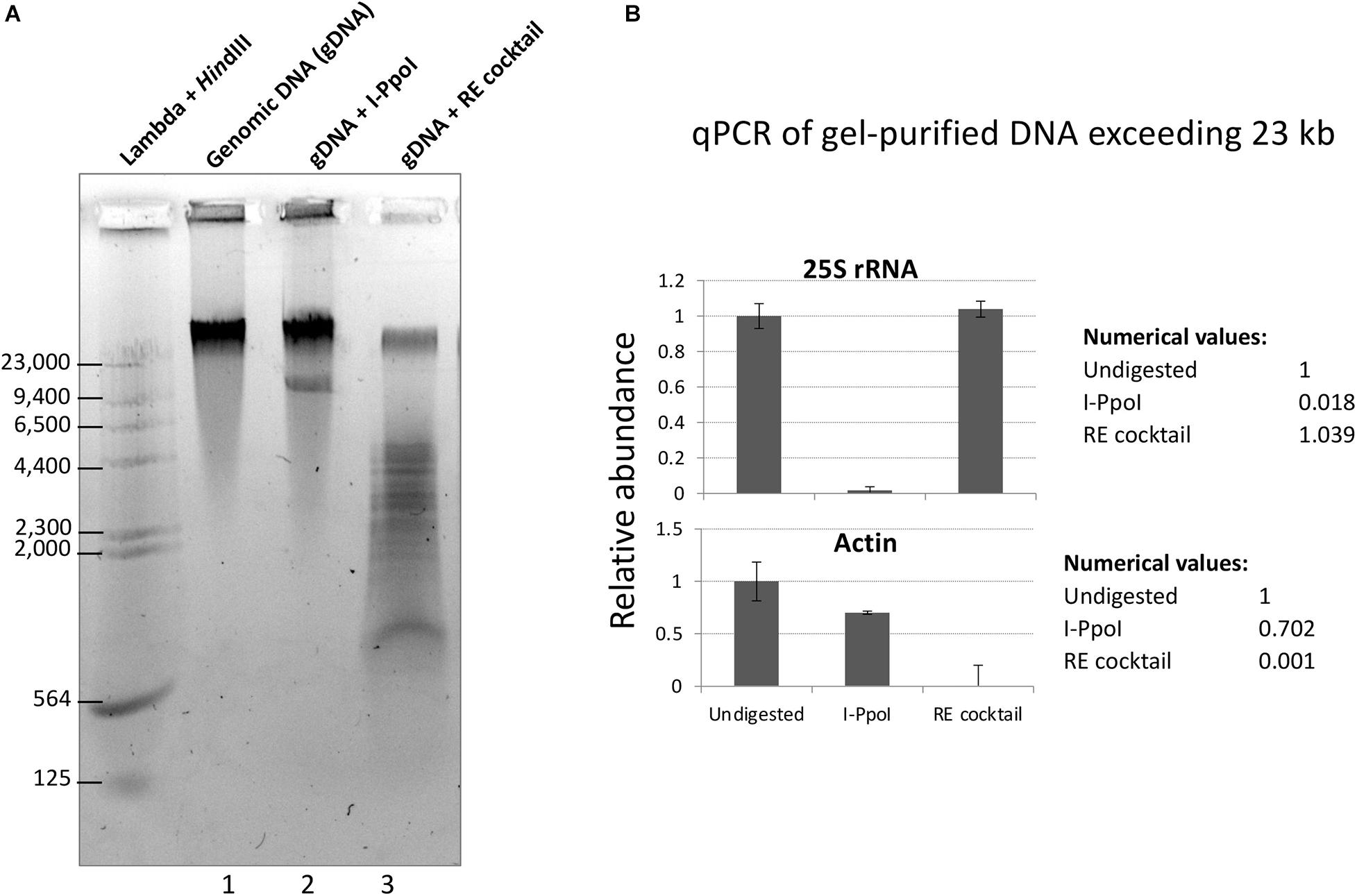
Figure 2. Digestion of genomic DNA with a cocktail of six restriction endonucleases (REs) predicted not to cut rRNA genes. (A) Col-0 gDNA that was either undigested (lane 1) or digested with I-PpoI (Lane 2) or the RE cocktail (lane 3) was separated on a 0.7% agarose gel and stained with GelRed dye. Bacteriophage Lambda DNA digested with HindIII provided size markers. (B) Summary of quantitative PCR (qPCR) results for rRNA gene (top panel) and actin (bottom panel) detection in undigested, I-PpoI, or RE cocktail-digested samples. qPCR was performed on gDNA extracted from the top gel band, containing DNA > 23 kb.
Next, we performed ONT sequencing to test the degree to which digestion with the six-enzyme cocktail enriches for reads containing rRNA gene repeats. For this experiment, agarose-embedded nuclei from Arabidopsis thaliana Col-0 plants (denoted as whole genome, or WG nuclei in Figure 3A) were first subjected to digestion with Proteinase K and RNase A. Half of the sample was then incubated with the cocktail of six restriction endonucleases (RE nuclei) and the other half received only buffer. The resulting DNA was prepared via ONT’s rapid library kit (RAD-004) and sequenced on a MinION. Reads were mapped to the Col-0 reference genome (version TAIR10) with the alignment program ngmlr, using the default cutoffs (minimum identity of 65% and at least 25% of the read length aligned to the reference sequence) (Sedlazeck et al., 2018). The resulting FASTQ files (total sequenced DNA) were aligned to a rRNA gene consensus sequence (Chandrasekhara et al., 2016) in order to identify ribosomal gene DNA reads and separate them from remaining TAIR10-mappable sequences (non-ribosomal DNA reads). The sequence identity of the basecalled reads when aligned to the A. thaliana nuclear genome (TAIR10) was 85.78%. The sequence identity of the ribosomal gene reads was 86.12% for the 45S rRNA gene region (excluding the variable 3’ETS region). Sequencing statistics are shown in Figure 3A.
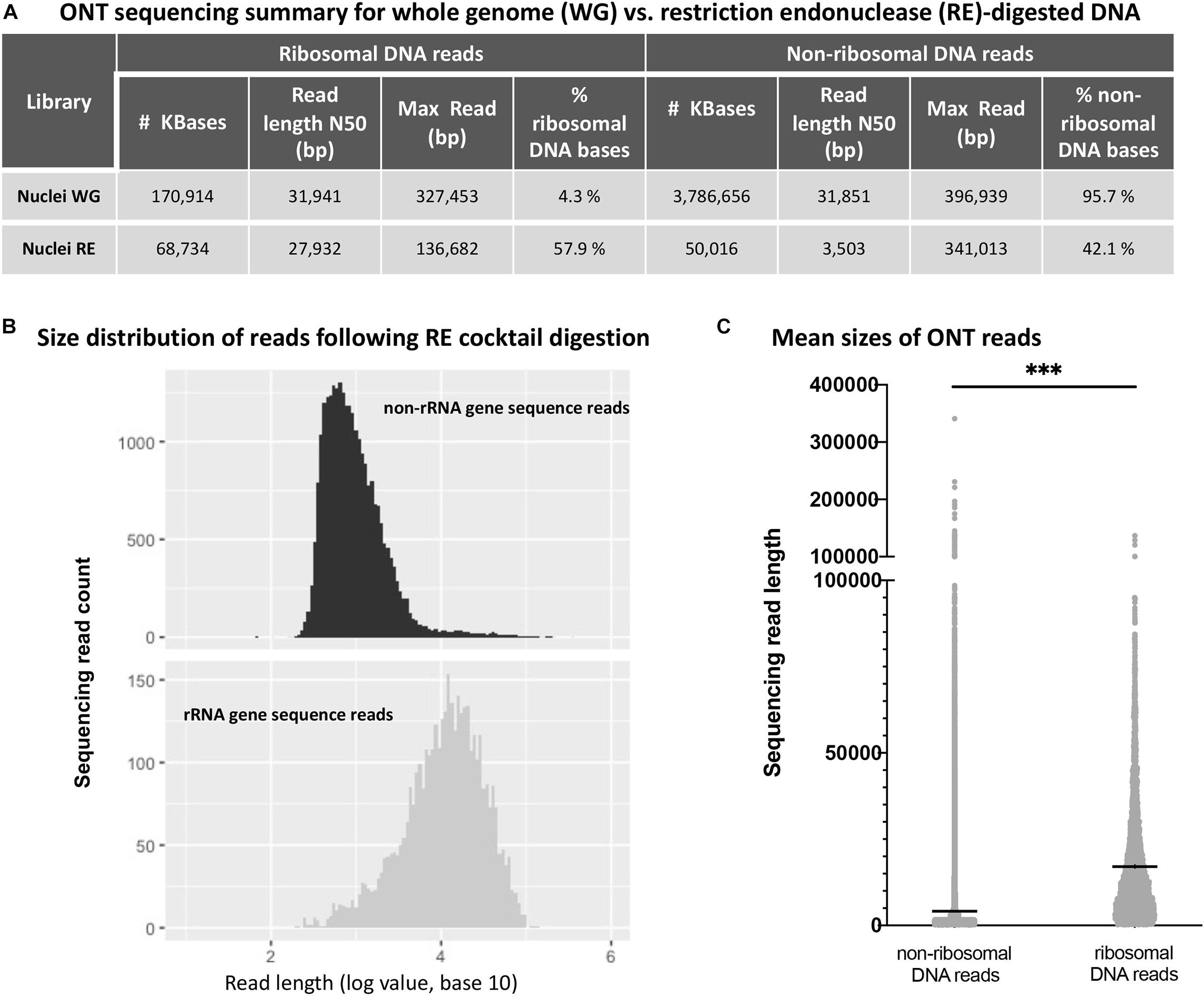
Figure 3. Oxford Nanopore sequencing results for whole genome (WG) vs. restriction endonuclease (RE)-digested DNA. (A) Effect of restriction endonuclease cocktail digestion on read distribution for non-ribosomal and ribosomal RNA gene sequences. (B) Non-ribosomal DNA (top) and ribosomal RNA gene DNA (bottom) read numbers are plotted as a function of read length. (C) Sequencing read mean length (horizontal black lines within the distributions) for non-ribosomal and ribosomal RNA gene sequences are significantly different (*** denotes unpaired t-test values of p < 0.0001).
By definition, the N50 value of a sequencing run indicates a read length at which half of the total yield is in read lengths equal to or greater than this value. Consistent with targeted digestion of genomic DNA other than rRNA genes, the six-enzyme cocktail treatment greatly reduced the N50 read length for non-ribosomal DNA reads, whereas the N50 value for ribosomal DNA reads was less affected. Importantly, the percentage of sequenced rRNA gene bases (% ribosomal DNA bases) increased 13.5-fold, from 4.3% in the undigested control sample (Nuclei WG) to 57.9% in the sample digested with the six-enzyme cocktail (Nuclei RE). Additionally, the read length distribution of the restriction enzyme digested sample shows a statistically significant difference (unpaired t-test, p-value < 0.0001) between the non-ribosomal DNA reads and ribosomal RNA gene reads (Figure 3).
Gaps in published reference genomes can be millions of basepairs in size and can consist of repeats with nearly identical sequences, as is the case for NORs and pericentromeric repeats. Assembly of these regions can benefit from ultra-long ONT sequencing that yields reads that span multiple repeat units. However, a high depth of coverage is needed to assure accuracy and continuity of the assembly. Obtaining the needed coverage can be costly when the repeat region represents only a fraction of the genome to be sequenced.
In our proof-of-concept approach described in this brief report, we explored whether targeted enrichment of highly repetitive ribosomal RNA gene arrays can be combined with Oxford Nanopore Technology (ONT) sequencing in order to increase read depth coverage for A. thaliana NOR regions. Without enrichment, ribosomal RNA gene sequences are expected to account for ∼4.3% of the sequencing data, based on an estimated size of ∼8 Mbp for the two NORs (Copenhaver and Pikaard, 1996b). Restriction endonuclease-mediated sequence enrichment increased the proportion of rRNA gene reads by ∼13.5-fold. In our test, we used an RE cocktail chosen based on the sequence of a reference consensus gene sequence. A caveat to this approach is that rRNA gene sequence variants that can be cut by one or more of these enzymes may occur at low frequency. Thus, rRNA gene reads obtained by direct sequencing of genomic DNA, without restriction endonuclease digestion, should also be conducted. The latter can provide unbiased “scaffold reads” to which the “enriched” reads can be matched to increase the depth of sequence coverage. Alternatively, two or more different restriction endonuclease cocktails could be employed, designed to account for rare variants that might be cut using one cocktail but not another. These and other strategies for improving ultra-long sequencing coverage will likely be needed to achieve complete de novo assembly of NORs (Rang et al., 2018).
ONT sequencing of bacterial artificial chromosomes (BACs) is another way to obtain sequences for cloned arrays of tandem repeats, as recently demonstrated for BAC-cloned Arabidopsis thaliana rRNA gene arrays (Sims et al., 2021). An advantage of BACs is the ability to achieve high sequence coverage for the cloned insert, allowing high per-base accuracy. However, unlike direct genomic DNA sequencing, BACs tend to be ∼100 kb in size, which may not be long enough to identify sufficient variation for overlapping sequences to be identified and longer contigs assembled. BACs are also known to recombine, especially BACs containing cloned repetitive regions (Mozo et al., 1998). Thus, secondary confirmation of gene arrangements determined by BAC sequencing, obtained by direct genomic DNA sequencing to obtain even longer reads, is desirable and can have synergistic benefits, with ultra-long genomic DNA sequences serving as scaffolds for contig assembly and BAC sequences providing high accuracy at each nucleotide position within the contig.
Despite their overall conservation, 45S rRNA gene sequences are diverse in eukaryotes such that restriction endonuclease enrichment strategies must be adapted on a species-by-species, and even strain-by-strain, basis (Rabanal et al., 2017). However, the large selection of commercially available restriction enzymes makes it likely that the strategy be adapted for most species simply by altering the mix of restriction endonucleases. Exceptionally long read lengths will also likely be required to assemble NORs in species such as humans, in which individual rRNA gene repeats are four times longer (∼42 kb) than the ∼10 kb rRNA gene repeats of Arabidopsis. Thus, it is noteworthy that some of the longest reported ONT read lengths been obtained using human genomic DNA (Jain et al., 2018), far surpassing the ONT read lengths we have obtained thus far for Arabidopsis. Keeping these considerations in mind, if one has preliminary knowledge of repeat size and sequence variation, and the length of ONT reads possible for the species and cells being studied, enrichment strategies can likely be designed to obtain long reads to help assemble repetitive loci composed of highly similar genes or DNA elements.
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
CP conceived the experiments. AM designed and performed the experiments. FW performed in silico digestion of Col-0 gDNA by restriction enzymes. AM and DF analyzed results of ONT sequencing runs. AM and CP wrote the manuscript. All authors contributed to the article and approved the submitted version.
This work was supported by funds to CP as an Investigator of the Howard Hughes Medical Institute and by endowment funds of the Carlos O. Miller Professorship at Indiana University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank our fellow Pikaard lab colleagues for valuable discussions.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.656049/full#supplementary-material
Ahmed, M., and Liang, P. (2012). Transposable elements are a significant contributor to tandem repeats in the human genome. Comp. Funct. Genomics 2012:947089. doi: 10.1155/2012/947089
Aldrup-Macdonald, M. E., and Sullivan, B. A. (2014). The past, present, and future of human centromere genomics. Genes (Basel) 5, 33–50. doi: 10.3390/genes5010033
Bennett-Baker, P. E., and Mueller, J. L. (2017). CRISPR-mediated isolation of specific megabase segments of genomic DNA. Nucleic Acids Res. 45:e165. doi: 10.1093/nar/gkx749
Besser, J., Carleton, H. A., Gerner-Smidt, P., Lindsey, R. L., and Trees, E. (2018). Next-generation sequencing technologies and their application to the study and control of bacterial infections. Clin. Microbiol. Infect. 24, 335–341. doi: 10.1016/j.cmi.2017.10.013
Biscotti, M. A., Olmo, E., and Heslop-Harrison, J. S. (2015). Repetitive DNA in eukaryotic genomes. Chromosome Res. 23, 415–420. doi: 10.1007/s10577-015-9499-z
Chandrasekhara, C., Mohannath, G., Blevins, T., Pontvianne, F., and Pikaard, C. S. (2016). Chromosome-specific NOR inactivation explains selective rRNA gene silencing and dosage control in Arabidopsis. Genes Dev. 30, 177–190. doi: 10.1101/gad.273755.115
Copenhaver, G. P., Doelling, J. H., Gens, S., and Pikaard, C. S. (1995). Use of RFLPs larger than 100 kbp to map the position and internal organization of the nucleolus organizer region on chromosome 2 in Arabidopsis thaliana. Plant J. 7, 273–286. doi: 10.1046/j.1365-313x.1995.7020273.x
Copenhaver, G. P., and Pikaard, C. S. (1996a). RFLP and physical mapping with an rDNA-specific endonuclease reveals that nucleolus organizer regions of Arabidopsis thaliana adjoin the telomeres on chromosomes 2 and 4. Plant J. 9, 259–272. doi: 10.1046/j.1365-313x.1996.09020259.x
Copenhaver, G. P., and Pikaard, C. S. (1996b). Two-dimensional RFLP analyses reveal megabase-sized clusters of rRNA gene variants in Arabidopsis thaliana, suggesting local spreading of variants as the mode for gene homogenization during concerted evolution. Plant J. 9, 273–282. doi: 10.1046/j.1365-313x.1996.09020273.x
Ebler, J., Haukness, M., Pesout, T., Marschall, T., and Paten, B. (2019). Haplotype-aware diplotyping from noisy long reads. Genome Biol. 20:116. doi: 10.1186/s13059-019-1709-0
Flavell, R. B. (1986). Repetitive DNA and chromosome evolution in plants. Philos. Trans. R. Soc. Lond. 312, 227–242. doi: 10.1098/rstb.1986.0004
Fritz, R. B., and Musich, P. R. (1990). Unexpected loss of genomic DNA from agarose gel plugs. Biotechniques 9, 542, 544, 546–550.
Gabrieli, T., Sharim, H., Fridman, D., Arbib, N., Michaeli, Y., and Ebenstein, Y. (2018). Selective nanopore sequencing of human BRCA1 by Cas9-assisted targeting of chromosome segments (CATCH). Nucleic Acids Res. 46:e87. doi: 10.1093/nar/gky411
Gerbi, S. A. (1985). “Evolution of ribosomal DNA,” in Molecular Evolutionary Genetics, ed. R. J. McIntyre (New York, NY: Plenum Press), 419–517. doi: 10.1007/978-1-4684-4988-4_7
Good, J. M. (2011). Reduced representation methods for subgenomic enrichment and next-generation sequencing. Methods Mol. Biol. 772, 85–103. doi: 10.1007/978-1-61779-228-1_5
Havlova, K., Dvorackova, M., Peiro, R., Abia, D., Mozgova, I., Vansacova, L., et al. (2016). Variation of 45S rDNA intergenic spacers in Arabidopsis thaliana. Plant Mol. Biol. 92, 457–471. doi: 10.1007/s11103-016-0524-1
Jain, M., Koren, S., Miga, K. H., Quick, J., Rand, A. C., Sasani, T. A., et al. (2018). Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 36, 338–345. doi: 10.1038/nbt.4060
Kozarewa, I., Armisen, J., Gardner, A. F., Slatko, B. E., and Hendrickson, C. L. (2015). Overview of target enrichment strategies. Curr. Protoc. Mol. Biol. 112, 7.21.1–7.21.23. doi: 10.1002/0471142727.mb0721s112
Li, H. (2018). On the Definition of Sequence Identity. Available online at: https://lh3.github.io/2018/11/25/onthe-definition-of-sequence-identity
McStay, B. (2006). Nucleolar dominance: a model for rRNA gene silencing. Genes Dev. 20, 1207–1214. doi: 10.1101/gad.1436906
McStay, B. (2016). Nucleolar organizer regions: genomic ‘dark matter’ requiring illumination. Genes Dev. 30, 1598–1610. doi: 10.1101/gad.283838.116
Michael, T. P., Jupe, F., Bemm, F., Motley, S. T., Sandoval, J. P., Lanz, C., et al. (2018). High contiguity Arabidopsis thaliana genome assembly with a single nanopore flow cell. Nat. Commun. 9:541. doi: 10.1038/s41467-018-03016-2
Mohannath, G., and Pikaard, C. S. (2016). Analysis of rRNA gene methylation in Arabidopsis thaliana by CHEF-conventional 2D gel electrophoresis. Methods Mol. Biol. 1455, 183–202. doi: 10.1007/978-1-4939-3792-9_14
Mohannath, G., Pontvianne, F., and Pikaard, C. S. (2016). Selective nucleolus organizer inactivation in Arabidopsis is a chromosome position-effect phenomenon. Proc. Natl. Acad. Sci. U.S.A. 113, 13426–13431. doi: 10.1073/pnas.1608140113
Mozo, T., Fischer, S., Shizuya, H., and Altmann, T. (1998). Construction and characterization of the IGF Arabidopsis BAC library. Mol. Gen. Genet. 258, 562–570. doi: 10.1007/s004380050769
Muscarella, D. E., Ellison, E. L., Ruoff, B. M., and Vogt, V. M. (1990). Characterization of I-Ppo I, an intron-encoded endonuclease that mediates homing of a group I intron in the ribosomal DNA of Physarum polycephalum. Mol. Cell. Biol. 10, 3386–3396. doi: 10.1128/mcb.10.7.3386
Nachmanson, D., Lian, S., Schmidt, E. K., Hipp, M. J., Baker, K. T., Zhang, Y., et al. (2018). Targeted genome fragmentation with CRISPR/Cas9 enables fast and efficient enrichment of small genomic regions and ultra-accurate sequencing with low DNA input (CRISPR-DS). Genome Res. 28, 1589–1599. doi: 10.1101/gr.235291.118
Rabanal, F. A., Mandakova, T., Soto-Jimenez, L. M., Greenhalgh, R., Parrott, D. L., Lutzmayer, S., et al. (2017). Epistatic and allelic interactions control expression of ribosomal RNA gene clusters in Arabidopsis thaliana. Genome Biol. 18:75. doi: 10.1186/s13059-017-1209-z
Rang, F. J., Kloosterman, W. P., and de Ridder, J. (2018). From squiggle to basepair: computational approaches for improving nanopore sequencing read accuracy. Genome Biol. 19:90. doi: 10.1186/s13059-018-1462-9
Sedlazeck, F. J., Rescheneder, P., Smolka, M., Fang, H., Nattestad, M., von Haeseler, A., et al. (2018). Accurate detection of complex structural variations using single-molecule sequencing. Nat. Methods 15, 461–468. doi: 10.1038/s41592-018-0001-7
Sims, J., Sestini, G., Elgert, C., von Haeseler, A., and Schlogelhofer, P. (2021). Sequencing of the Arabidopsis NOR2 reveals its distinct organization and tissue-specific rRNA ribosomal variants. Nat. Commun. 12:387. doi: 10.1038/s41467-020-20728-6
Stults, D. M., Killen, M. W., Pierce, H. H., and Pierce, A. J. (2008). Genomic architecture and inheritance of human ribosomal RNA gene clusters. Genome Res. 18, 13–18. doi: 10.1101/gr.6858507
Treangen, T. J., and Salzberg, S. L. (2011). Repetitive DNA and next-generation sequencing: computational challenges and solutions. Nat. Rev. Genet. 13, 36–46. doi: 10.1038/nrg3117
Tucker, S., Vitins, A., and Pikaard, C. S. (2010). Nucleolar dominance and ribosomal RNA gene silencing. Curr. Opin. Cell Biol. 22, 351–356. doi: 10.1016/j.ceb.2010.03.009
Keywords: Oxford Nanopore sequencing, Arabidopsis thaliana, ribosomal RNA gene enrichment, Nucleolus Organizer Region, NOR
Citation: McKinlay A, Fultz D, Wang F and Pikaard CS (2021) Targeted Enrichment of rRNA Gene Tandem Arrays for Ultra-Long Sequencing by Selective Restriction Endonuclease Digestion. Front. Plant Sci. 12:656049. doi: 10.3389/fpls.2021.656049
Received: 20 January 2021; Accepted: 06 April 2021;
Published: 28 April 2021.
Edited by:
Sònia Garcia, Consejo Superior de Investigaciones Científicas, Spanish National Research Council (CSIC), SpainReviewed by:
Martina Dvorackova, Central European Institute of Technology (CEITEC), CzechiaCopyright © 2021 McKinlay, Fultz, Wang and Pikaard. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Craig S. Pikaard, Y3Bpa2FhcmRAaW5kaWFuYS5lZHU=
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.