 Hong Chang
Hong Chang Lei Zhang
Lei Zhang Huanhuan Xie1
Huanhuan Xie1 Jianquan Liu
Jianquan Liu Zhenxiang Xi
Zhenxiang Xi Xiaoting Xu
Xiaoting Xu- 1Key Laboratory of Bio-Resource and Eco-Environment of Ministry of Education, College of Life Sciences, Sichuan University, Chengdu, China
- 2State Key Laboratory of Grassland Agro-Ecosystems, College of Life Sciences, Lanzhou University, Lanzhou, China
Crassulaceae are the largest family in the angiosperm order Saxifragales. Species of this family are characterized by succulent leaves and a unique photosynthetic pathway known as Crassulacean acid metabolism (CAM). Although the inter- and intrageneric relationships have been extensively studied over the last few decades, the infrafamilial relationships of Crassulaceae remain partially obscured. Here, we report nine newly sequenced chloroplast genomes, which comprise several key lineages of Crassulaceae. Our comparative analyses and positive selection analyses of Crassulaceae species indicate that the overall gene organization and function of the chloroplast genome are highly conserved across the family. No positively selected gene was statistically supported in Crassulaceae lineage using likelihood ratio test (LRT) based on branch-site models. Among the three subfamilies of Crassulaceae, our phylogenetic analyses of chloroplast protein-coding genes support Crassuloideae as sister to Kalanchoideae plus Sempervivoideae. Furthermore, within Sempervivoideae, our analyses unambiguously resolved five clades that are successively sister lineages, i.e., Telephium clade, Sempervivum clade, Aeonium clade, Leucosedum clade, and Acre clade. Overall, this study enhances our understanding of the infrafamilial relationships and the conservation of chloroplast genomes within Crassulaceae.
Introduction
The angiosperm family Crassulaceae, also known as the stonecrop family, belongs to the order Saxifragales and include 34 genera and approximately 1,400 species (APG IV, 2016; Messerschmid et al., 2020), which are predominantly perennial herbs, subshrubs, or shrubs (Thiede and Eggli, 2007). The species of Crassulaceae primarily occur in (semi-)arid and mountainous habitats of the temperate and subtropical areas (van Ham and Hart, 1998), and are distributed worldwide with centers of diversity in Mexico, southern Africa, Macaronesia, and the Himalayas (Mort et al., 2001). Physiologically, Crassulaceae are characterized by the Crassulacean acid metabolism (CAM) photosynthetic pathway (Gontcharova and Gontcharov, 2009), which achieves a higher level of water-use efficiency than either the C3 or C4 pathway in water-limited environment (Nobel, 1991). A recent study has identified the crown group of Crassulaceae as one of the 30 core diversification shifts across the angiosperm phylogeny (Magallón et al., 2019). This increased diversification rate may be well associated with the CAM pathway that has been recognized as an evolutionary key innovation (Pilon-Smits et al., 1996; Quezada and Gianoli, 2011; Silvestro et al., 2014).
Until now, numerous phylogenetic studies have been performed to evaluate the infrafamilial relationships of Crassulaceae with a broad taxon sampling (e.g., Berger, 1930; van Ham and Hart, 1998; Mort et al., 2001; Mayuzumi and Ohba, 2004; Gontcharova et al., 2006, 2008; Folk et al., 2019; Messerschmid et al., 2020), which have led to the establishment of three subfamilies by Thiede and Eggli (2007), i.e., Crassuloideae, Kalanchoideae, and Sempervivoideae. Although most of these studies are based on one or a few genetic loci, the monophyly of each of three subfamilies sensu Thiede and Eggli (2007) is well supported. The inter- and intrageneric relationships within each of three subfamilies have been also extensively studied using a variety of chloroplast and nuclear loci. The smallest subfamily Crassuloideae comprise approximately 200 species in a single genus, Crassula L. (Messerschmid et al., 2020), whose intrageneric relationships have been recently addressed by sampling 103 species (Bruyns et al., 2019). The subfamily Kalanchoideae include approximately 240 species in four genera (Smith et al., 2019), i.e., Adromischus Lem., Cotyledon L., Kalanchoe, and Tylecodon Toelken, and the inter- and intrageneric relationships have been exclusively assessed in a few separate studies (Gehrig et al., 2001; Mort et al., 2005; Nowell, 2008). The largest and most complex subfamily Sempervivoideae contain 28 genera and over 1,000 species (Thiede and Eggli, 2007), and the inter- and intrageneric relationships have been investigated by a considerable number of studies (e.g., Mes et al., 1997; Jorgensen and Frydenberg, 1999; Mort et al., 2002; Acevedo-Rosas et al., 2004; Fairfield et al., 2004; Mayuzumi and Ohba, 2004; Gontcharova et al., 2006; Carrillo-Reyes et al., 2008, 2009; Yost et al., 2013; Zhang et al., 2014; Klein and Kadereit, 2015; Nikulin et al., 2016; Vázquez-Cotero et al., 2017; de la Cruz-López et al., 2019). These phylogenetic studies have led to the recognition of five tribes within Sempervivoideae by Thiede and Eggli (2007), i.e., Aeonieae (Aeonium Webb and Berth., Aichryson Webb and Berth., and Monanthes Haw.), Sedeae (Afrovivella A. Berger, Dudleya Britton and Rose, Echeveria DC., Graptopetalum Rose, Lenophyllum Rose, Pachyphytum Link et al., Pistorinia DC., Prometheum H. Ohba, Rosularia Stapf, Sedella Britton and Rose, Sedum L., Thompsonella Britton and Rose, and Villadia Rose), Semperviveae (Petrosedum Grulich and Sempervivum L.), Telephieae (Hylotelephium H. Ohba, Kungia K.T. Fu, Meterostachys Nakai, Orostachys Fisch., and Sinocrassula A. Berger), and Umbiliceae (Phedimus Raf., Pseudosedum A. Berger, Rhodiola, and Umbilicus DC.). Furthermore, based on one nuclear (ITS) and three chloroplast markers (matK, rps16, and trnL-trnF), a recent phylogenetic study of 298 Crassulaceae species has recovered six major clades of Sempervivoideae, i.e., Telephium clade, Petrosedum clade, Sempervivum clade, Aeonium clade, Leucosedum clade, and Acre clade (Messerschmid et al., 2020). Despite the significant progress made in the tribal and generic circumscription of Crassulaceae, phylogenetic relationships among tribes and major clades remain poorly to moderately supported or sometimes contradicted, especially within Sempervivoideae (Supplementary Figure 1).
Chloroplasts are semi-autonomous replication organelles, originating in endosymbiosis between cyanobacteria and non-photosynthetic host, and play crucial roles in photosynthesis and physiology of plants (Gao et al., 2019a; Huo et al., 2019; Li et al., 2021). The chloroplast genome is mostly a typical quadripartite circular DNA genome comprising a small single-copy (SSC), a large single-copy (LSC), and two inverted repeats (IRs) (Hu et al., 2016; Chang et al., 2020). With the development of next−generation sequencing technology, chloroplast genomes have been proven to be powerful tools for resolving vague relationships of many complicated lineages due to many advantages, such as uniparental inheritance, the relatively conserved structure, low recombination and substitution rate (Goremykin et al., 2003, 2004; Twyford and Ness, 2017; Li et al., 2020a). Although the structure and sequence of plastomes are relatively conservative, variations in structure, size, and evolutionary rates of genes have been found in many studies, which in some cases signify phylogenetical information and adaptation to environment (Chumley et al., 2006; Hu et al., 2015; Piot et al., 2018; Gao et al., 2019a; Gruzdev et al., 2019; Shrestha et al., 2019; Zang et al., 2019).
Here, we sequenced fourteen fully annotated chloroplast genomes including nine species of Crassulaceae and five species of Haloragaceae. By taking advantage of the data already available, we analyzed a total of 33 chloroplast genomes and aimed to (i) assess the structural characteristics of the chloroplast genome in a comparative framework, (ii) improve the resolution of the infrafamilial relationships within Crassulaceae, and (iii) investigate the adaptive evolution by selective pressures analysis of protein-coding genes in Crassulaceae.
Materials and Methods
Taxon Sampling, DNA Extraction, and Sequencing
The aim of our taxon sampling was to try to obtain chloroplast genome sequences for at least one representative of each well-supported major clades. The final taxon sampling contained a total of 26 Crassulaceae species (Table 1), representing all three subfamilies sensu Thiede and Eggli (2007), and all five tribes sensu Thiede and Eggli (2007) or five out of the six major clades sensu Messerschmid et al. (2020) within Sempervivoideae. Of these complete chloroplast genomes, nine were newly generated in this study, i.e., Aeonium arboreum (L.) Webb and Berthel., Cotyledon tomentosa Harv., Crassula perforata Thunb., Graptopetalum amethystinum (Rose) E. Walther, Kalanchoe fedtschenkoi Raym.-Hamet and H. Perrier, Orostachys fimbriata (Turcz.) A. Berger, Pachyphytum compactum Rose, Sempervivum tectorum L., and Sinocrassula densirosulata (Praeger) A. Berger. In addition, seven complete chloroplast genomes were included as outgroups, i.e., Myriophyllum spicatum L., Penthorum chinense L., and five new sequenced chloroplast genomes of Haloragis aspera Lindl., Haloragis erecta (Murray) Oken, Glischrocaryon aureum (Lindl.) Orchard, Glischrocaryon glandulosum (Orchard) Christenh. and Byng, Gonocarpus micranthus Thunb. These seven species belong to the family Haloragaceae sensu lato, which are the closest living relatives of Crassulaceae (Jian et al., 2008).
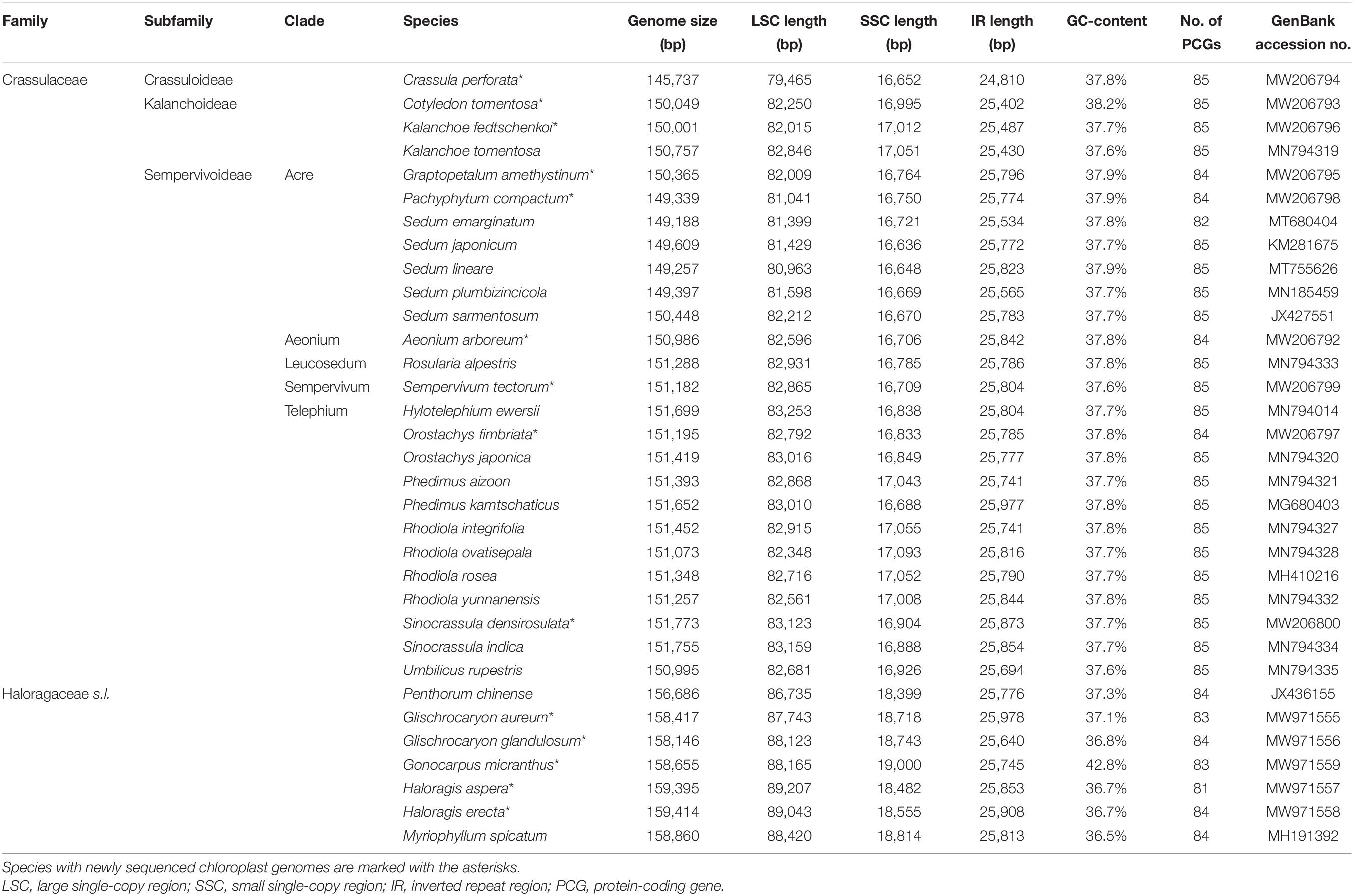
Table 1. Structural information of the chloroplast genomes of Crassulaceae and outgroups.
The DNA materials of four species (Haloragis aspera, Haloragis erecta, Glischrocaryon aureum and Glischrocaryon glandulosum) were provided by DNA Bank of Royal Botanic Gardens, Kew1. Fresh leaves of the other ten species were collected from the field and preserved with silica gel. The total genomic DNA was extracted using a modified CTAB method (Allen et al., 2006). For each species, one paired-end library with an insertion size of ∼350 base pairs (bp) was prepared from the total genomic DNA using the NEBNext Ultra II DNA Library Prep Kit for Illumina (New England Biolabs, MA, United States), which was then sequenced using the HiSeq 2500 System (Illumina, Inc., CA, United States) to obtain paired 150-bp reads. Briefly, (i) the genomic DNA was sonicated using the S220 Focused-ultrasonicator (Covaris, MA, United States), (ii) the fragmented DNA was end-repaired, dA-tailed, adapter ligated, and subjected to 10–12 cycles of PCR amplification, and (iii) the quality of each library was assessed using the 2100 Bioanalyzer system (Agilent, CA, United States).
Chloroplast Genome Assembly and Annotation
The raw Illumina reads were first filtered to remove paired-end reads if either of the reads contained (i) adapter sequences, (ii) more than 10% of N bases, and (iii) more than 50% of bases with a Phred quality score less than five. The filtered reads were then assembled using NOVOPlasty version 2.7.2 (Dierckxsens et al., 2017), and the complete chloroplast genome of Sedum sarmentosum Bunge (Dong et al., 2013) was used as the reference genome. These assemblies were manually inspected using Geneious version 11.0.3 (Kearse et al., 2012).
The assembled chloroplast genomes were annotated using Plann version 1.1 (Huang and Cronk, 2015), and the positions of exons and introns were inspected and adjusted using Sequin version 15.50. In addition, the circular maps of the chloroplast genomes were drawn using OGDRAW version 1.2 (Lohse et al., 2013), and all annotated chloroplast genomes were deposited in GenBank (Sayers et al., 2020).
Comparative Analysis of Chloroplast Genomes
For the 26 chloroplast genomes of Crassulaceae, the complete nucleotide sequences were compared using the glocal alignment algorithm Shuffle-LAGAN (Brudno et al., 2003) as implemented in the program Mvista2 (Frazer et al., 2004). Here, Rhodiola rosea L. was chosen as the reference to evaluate gene content variation following Zhao et al. (2020).
To better determine whether any specific pattern of structural variation exists at the family level, the chloroplast genome of Rhodiola rosea was used as the representative owing to the highly conserved gene content and gene order within Crassulaceae (see section “Results”), and compared with that of Penthorum chinense using progressiveMauve (Darling et al., 2010) as implemented in the software package Mauve version 2.3.1 (Darling et al., 2004). Following Firetti et al. (2017), one of the inverted repeat (IR) regions was manually removed prior to the alignment.
To further identify the hypervariable regions, coding and non-coding regions of the 26 chloroplast genomes were first extracted using PhyloSuite version 1.2.1 (Zhang et al., 2020) and aligned separately using MAFFT version 7.427 (Katoh and Standley, 2013) with default parameters. The nucleotide diversity (π) was then estimated separately for coding and non-coding regions using DnaSP version 6.0 (Rozas et al., 2017).
Moreover, since the size variation of the chloroplast genome may be attributed to the expansion or contraction of the IR region, the boundaries between the IR and single-copy regions were identified using IRscope3 (Amiryousefi et al., 2018) and manually inspected using Geneious.
Phylogenetic Analysis
The nucleotide sequences of the chloroplast protein-coding genes were aligned separately using MAFFT and then concatenated into a supermatrix using PhyloSuite. The optimal partitioning scheme and models of DNA sequence evolution were determined using the relaxed hierarchical clustering algorithm (Lanfear et al., 2014) as implemented in PartitionFinder v2.1.1 (Lanfear et al., 2017).
Phylogenetic relationships were inferred for 26 species of Crassulaceae (i.e., the 33-taxon supermatrix) using both Bayesian inference (BI) and maximum likelihood (ML) methods. For the BI method, four parallel Markov chain Monte Carlo (MCMC) runs were performed using MrBayes version 3.2.7 (Ronquist et al., 2012). The supermatrix was partitioned based on the optimal scheme determined by PartitionFinder, and the best-fitting substitution model was specified as prior for each partition with model parameters unlinked across partitions. A total of 1,000,000 generations were run with sampling every 500 generations, and the first 25% of samples were discarded as burn-in. Convergence of runs was assumed when the average standard deviation of split frequencies dropped below 0.01. The best-scoring ML tree was inferred using RAxML version 8.2.11 (Stamatakis, 2014) with the GTRGAMMAX model for each partition, and branch support was assessed using the rapid bootstrap algorithm (Stamatakis et al., 2008) with 1,000 replicates.
In order to test the potential effect of uneven taxon sampling from each of the genera, we further subsampled the 26 species of Crassulaceae down to a single species as the representative of each genus. Phylogenetic relationships were then estimated from the 20-taxon supermatrix as described above.
Positive Selection Analysis
The likelihood ratio test (LRT) and Bayes empirical Bayes (BEB) based on modified branch-site model (Yang et al., 2005; Zhang et al., 2005; Yang and Dos, 2011) were used to identify positively selected genes. Since the topological structures of phylogenetic trees constructed by ML and BI methods were congruent, ML tree was used to positive selection analysis. The amino acid sequences of the sixty-seven common protein-coding genes were aligned using MAFFT and converted into nucleotide alignments using PAL2NAL version 14 (Suyama et al., 2006). The nucleotide alignments were trimmed to obtain the final alignments for positive selection analysis by trimAL version 1.4 (Capellagutiérrez et al., 2009). The branch-site model was performed by codeml program in PAML version 4.9 (Yang, 2007). The branch-site test of positive selection was run with the ω of foreground lineage fixed to 1 (fix_omega = 1) for the null hypothesis and estimated (fix_omega = 0) for the alternative hypothesis. The LRT values at df = 1 were calculated by Chi Square test in PAML, and genes with p < 0.05 were treated as candidate positives. Finally, BEB was used to identify those positively selected codon sites.
Results
Characteristics of Chloroplast Genomes in Crassulaceae
After quality control and pre-processing, at least four gigabases (Gb) of whole-genome sequencing data were obtained for each of the nine species (Table 1). These clean reads were assembled into high-quality chloroplast genomes using a reference-guided approach, and the resulting coverage ranged from 1,115 × (i.e., Cotyledon tomentosa) to 12,687 × (i.e., Kalanchoe fedtschenkoi). All these newly assembled chloroplast genomes exhibited a typical quadripartite structure, with two IR regions (i.e., IRa and IRb) separating the LSC and SSC regions (Figure 1).
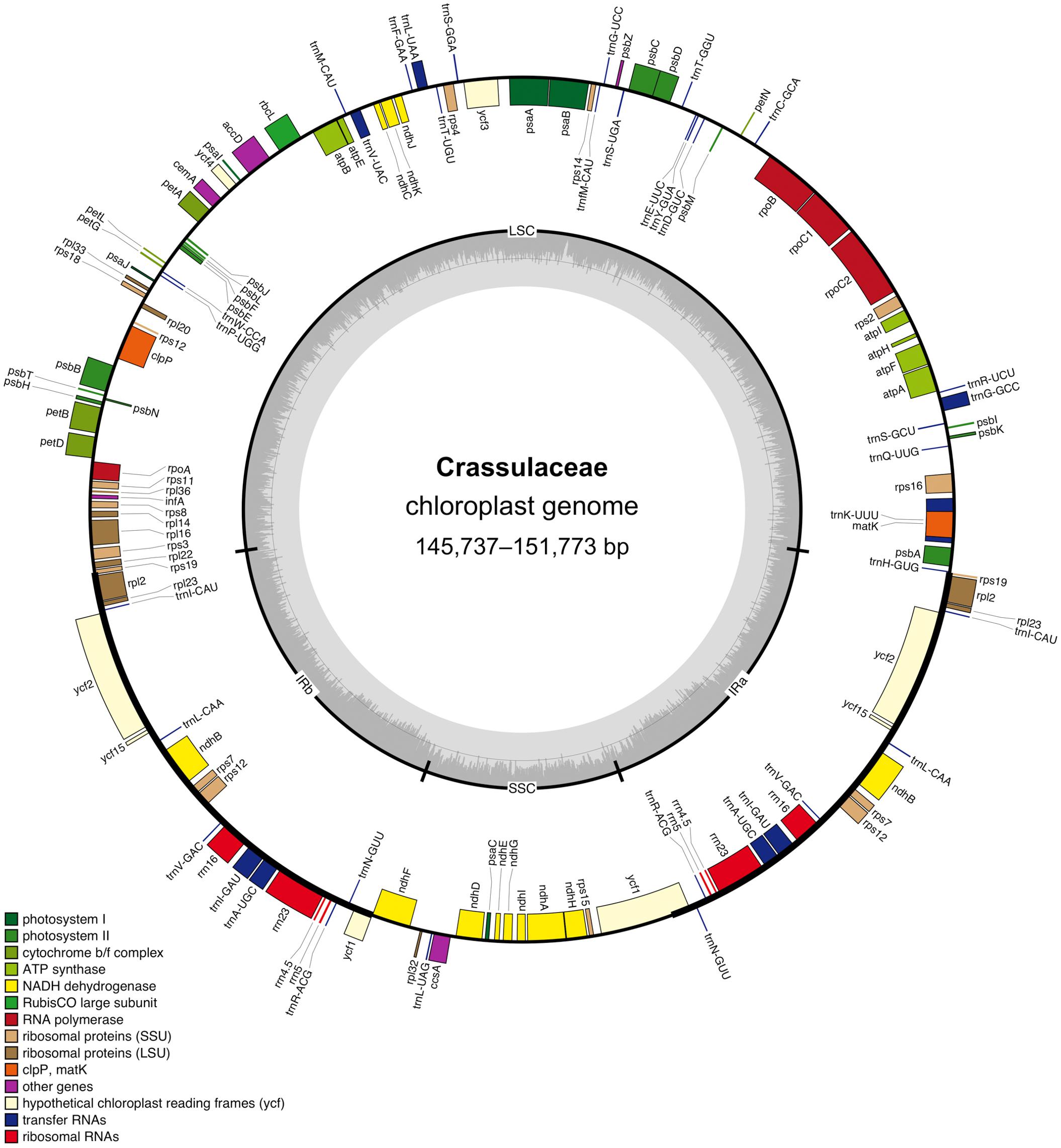
Figure 1. Circular gene map of the Crassulaceae chloroplast genome. The genes labeled inside and outside of the circle are transcribed in clockwise and counterclockwise directions, respectively. The inner circle shows the quadripartite structure, with two IR regions (IRa and IRb) separating the large single-copy (LSC) and small single-copy (SSC) regions. The gray ring marks the GC-content with the inner circle indicating a 50% threshold.
The structure of the chloroplast genome appeared to be largely conserved across the family (Table 1). For each of the 26 Crassulaceae species, the size of the chloroplast genome varied from 145,737 bp (i.e., Crassula perforata) to 151,773 bp (i.e., Sinocrassula densirosulata), and the overall GC-content ranged from 37.6% (i.e., Sempervivum tectorum) to 38.2% (i.e., Cotyledon tomentosa). In addition, the total number of annotated genes in each of these chloroplast genomes ranged from 131 (i.e., Sedum emarginatum Migo) to 134 (i.e., Sedum lineare Thunb.), and all these chloroplast genomes possessed 37 tRNA and four rRNA genes.
Using Rhodiola rosea as the reference, the analysis of mVISTA showed high similarity in gene content and gene order among the 26 chloroplast genomes, and further indicated a fairly high sequence similarity, especially in the coding regions (Supplementary Figure 2). This observation was corroborated by our analysis of nucleotide diversity (Figure 2). The nucleotide diversity in the coding regions ranged from 0 to 0.0794, with an average of 0.0230, which was significantly lower than that in the non-coding regions (0–0.1614, 0.0647; p-value < 2.2 × 10–16, Welch’s t-test). Here, the five coding regions with the highest nucleotide diversity were matK, ycf1, ndhF, rpl22, and rpl32, and the corresponding non-coding regions were trnH-psbA, trnG-trnR, rpl32-trnL, rps16-trnQ, and ccsA-ndhD. Furthermore, no evidence of genomic rearrangement was found in the chloroplast genome of Crassulaceae, when compared with that of Penthorum chinense using progressiveMauve (Supplementary Figure 3).
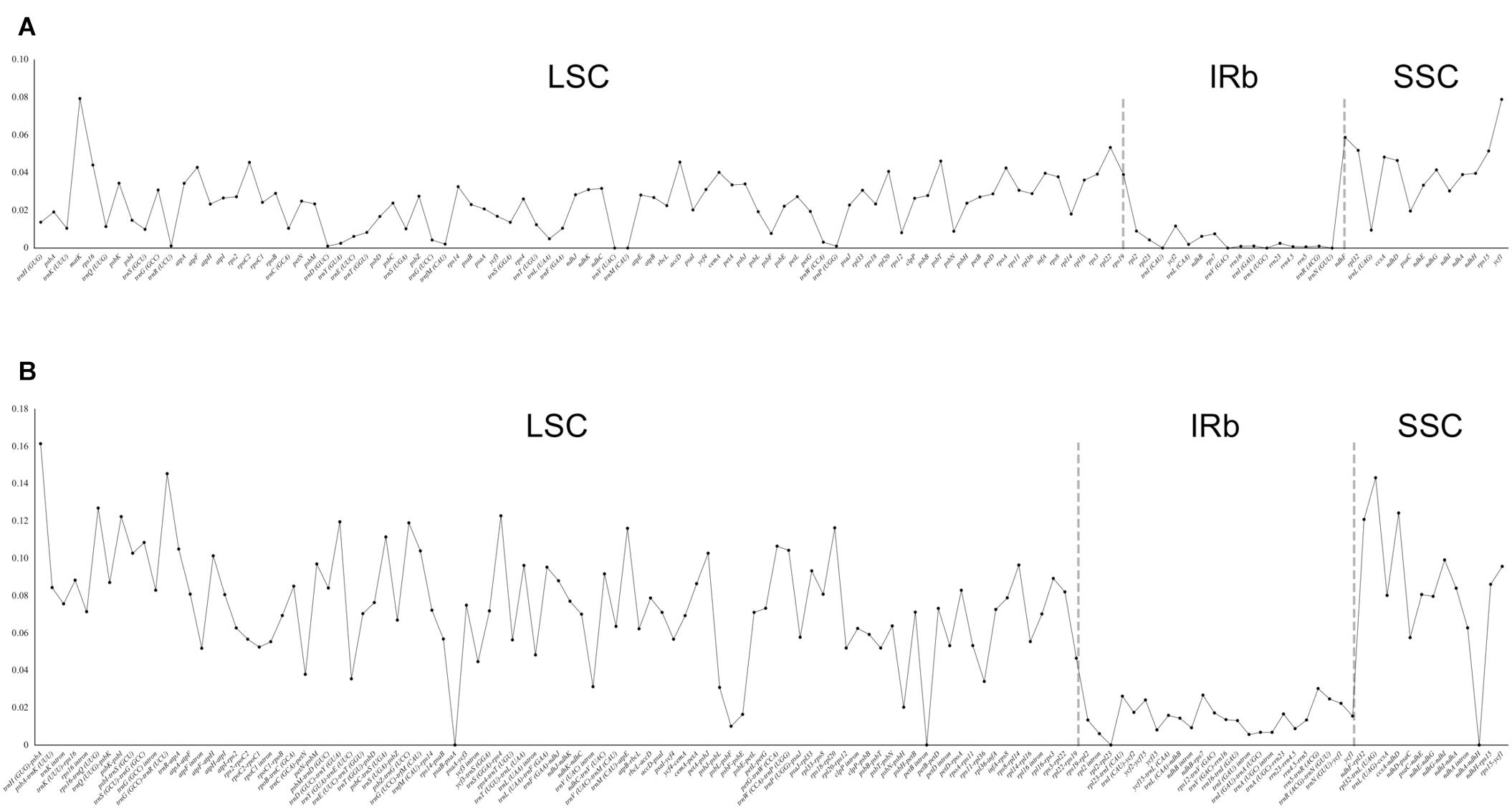
Figure 2. Distribution of nucleotide diversity (π) in coding (A) and non-coding (B) regions of 26 Crassulaceae chloroplast genomes.
The boundaries of the LSC, SSC, and IR regions were highly consistent within the family, and no obvious expansion or contraction of the IR region was detected in the 26 chloroplast genomes (Supplementary Figure 4). Here, trnH was shown to be the first gene in the LSC region at the junction between IRa and LSC (i.e., IRa/LSC). At the other end of the LSC region, the junction LSC/IRb was identified as located within the rps19 gene, which gave rise to a truncated copy of the rps19 gene in the IRa region. For both ends of the SSC region, the junctions IRb/SSC and SSC/IRa were found to be located within ndhF and ycf1 gene, respectively. As a consequence, a truncated copy of the ycf1 gene was retained in the IRb region.
Phylogenetic Relationships of Crassulaceae
The 33-taxon supermatrix contained a total of 79 genes and 70,905 sites, and the amount of missing data (including gaps and undetermined characters) was 4.5%. Phylogenetic analyses of the 33-taxon supermatrix using ML and BI methods yielded an identical topology (Figure 3), and all relationships were strongly supported by both methods, i.e., ≥85 ML bootstrap percentage (BP) and ≥0.99 Bayesian posterior probability (PP).
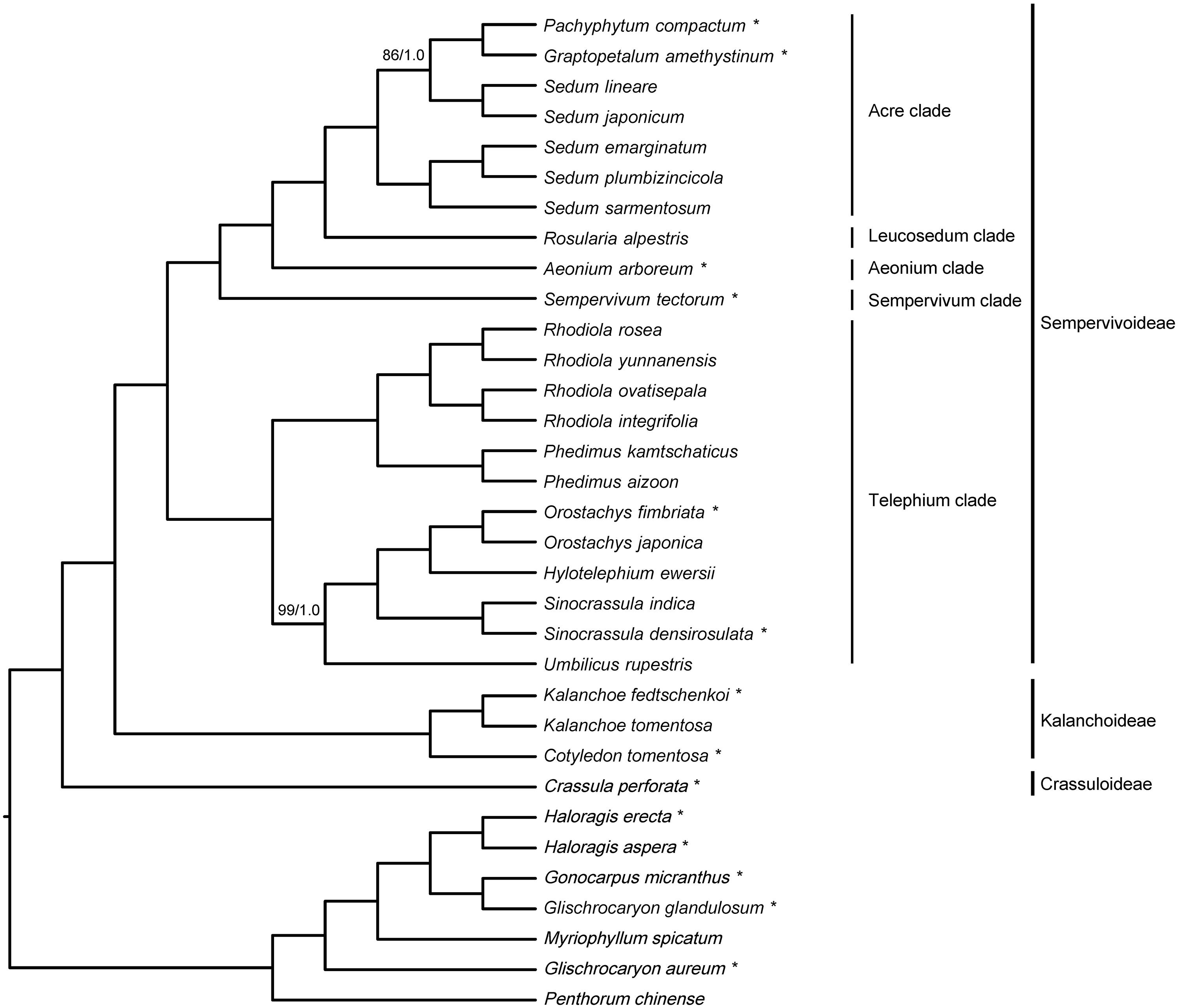
Figure 3. Phylogenetic inference of 33-taxon supermatrix using maximum likelihood (ML) and Bayesian inference (BI) methods. Branch support was assessed using ML bootstrap percentage (BP) and Bayesian posterior probability (PP), and internal branches with less than 100 BP/1.0 PP are indicated with corresponding values. Species with newly sequenced chloroplast genomes are marked with the asterisks, and clade designations are labeled accordingly.
All 26 Crassulaceae species formed a monophyletic group, and were divided into three subclades corresponding to the three subfamilies sensu Thiede and Eggli (2007), i.e., Crassuloideae, Kalanchoideae, and Sempervivoideae (Figure 3). When rooted with Haloragaceae s.l., Crassuloideae (represented by Crassula perforata) were resolved as sister to Kalanchoideae plus Sempervivoideae. Within Kalanchoideae, the two sampled species of Kalanchoe formed a monophyletic group that was sister to Cotyledon tomentosa. Within Sempervivoideae, our sampled species fell into five clades, i.e., Acre clade, Aeonium clade, Leucosedum clade, Sempervivum clade, and Telephium clade (Figure 3). Here, the Telephium clade was established as sister to the rest of Sempervivoideae, and further split into two lineages. One lineage comprised two sampled species of Phedimus and four sampled species of Rhodiola, which formed two reciprocally monophyletic groups, and the other contained the sampled species of Hylotelephium, Orostachys, Sinocrassula, and Umbilicus. Importantly, Umbilicus rupestris (Salisb.) Dandy was recovered as sister to a clade consisting of Hylotelephium, Orostachys, and Sinocrassula. The Sempervivum clade (represented by Sempervivum tectorum) and the Aeonium clade (represented by Aeonium arboreum) were placed as successive sister lineages to the Leucosedum clade [represented by Rosularia alpestris (Kar. and Kir.) Boriss.] plus the Acre clade. Furthermore, the five sampled species of Sedum in the Acre clade were recovered as a paraphyletic group, with Graptopetalum amethystinum and Pachyphytum compactum nested within them. Importantly, except the non-monophyly of Sedum, the same set of intergeneric relationships was recovered from the 20-taxon supermatrix (Supplementary Figure 5), suggesting that our results should be robust to taxon sampling.
Positive Selection Analysis
A total number of sixty-seven common genes were subjected to positive selection analyses (Table 2). The LRTs with p-value > 0.05 suggested that there was no statistical support for positive selection in any genes (Table 2), although the BEB approach identified fourteen genes (atpB, ndhE, ndhJ, petA, psaC, psaJ, psbB, psbD, psbN, rpoC2, rps15, rps3, rps7, and ycf4) with relatively high posterior probabilities of codon sites.
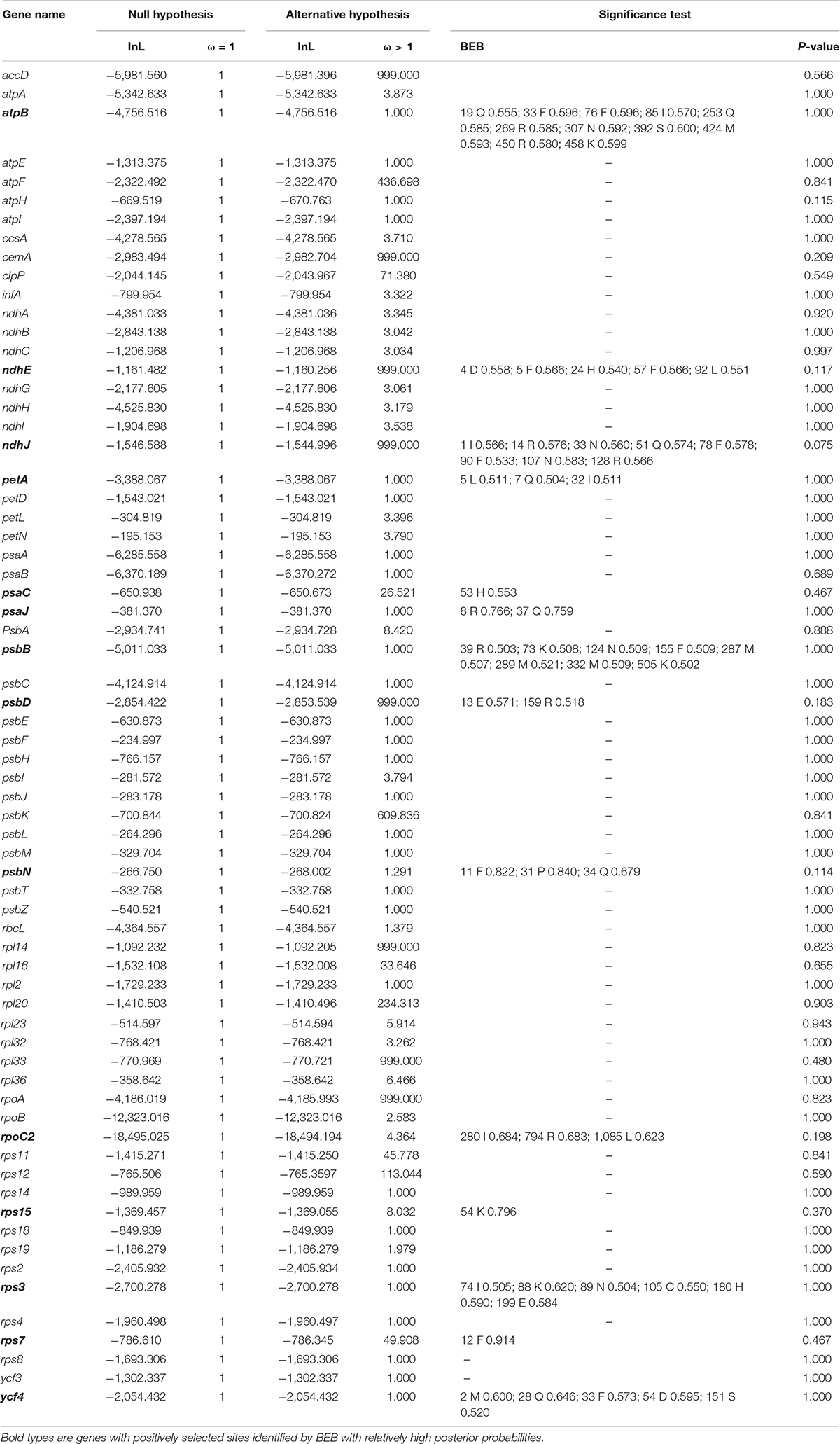
Table 2. The positive selection test based on the branch-site model.
Discussion
In this study, we report nine newly sequenced complete chloroplast genomes of Crassulaceae. Our comparative analyses indicate that the overall gene organization of the chloroplast genome is highly conserved across all 26 Crassulaceae species investigated here. In addition, the results of IRscope analysis reveal no obvious expansion or contraction of the chloroplast IR region. Furthermore, although a recent study has identified a unique 4-kb inversion in the chloroplast genome of the outgroup species Myriophyllum spicatum (Liao et al., 2020), our analyses show a high degree of similarity in chloroplast gene order between Crassulaceae and the outgroup species Penthorum chinense.
Previous studies have demonstrated that the size variation of the angiosperm chloroplast genome is primarily due to variation in the IR region, intergenic region, and gene copy number (Zheng et al., 2017; Amiryousefi et al., 2018; Bedoya et al., 2019). The results of mVISTA analysis suggest that hypervariable intergenic regions in the LSC region, such as petA-psbJ, psbM-trnD, psbZ-trnG, rps16-trnQ, and trnE-trnT (Supplementary Figure 2), contribute most to the chloroplast genome size variation within Crassulaceae. Moreover, phylogenetic studies of Crassulaceae have relied primarily on a limited set of chloroplast markers (e.g., matK, rps16, and trnL-trnF). Our comparative analyses, however, have shown that these chloroplast markers appear to be relatively low in nucleotide diversity (Figure 2), which may be partially responsible for the lack of phylogenetic resolution within Sempervivoideae (Mort et al., 2001; Messerschmid et al., 2020). Thus, to achieve better phylogenetic resolution, future studies of Crassulaceae should focus on molecular markers from more variable regions of the chloroplast genome, such as ccsA-ndhD, rps16-trnQ, rpl32-trnL, and trnH-psbA (Prince, 2015).
With increased taxon sampling of key lineages of Crassulaceae, our phylogenetic analyses of chloroplast genome sequences have substantially improved the phylogenetic resolution, and provided robust inference of the infrafamilial relationships. Among the three subfamilies sensu Thiede and Eggli (2007), our phylogenetic analyses have confirmed the monophyly of Kalanchoideae and of Sempervivoideae, and suggested that Crassuloideae are sister to Kalanchoideae plus Sempervivoideae, corroborating previous studies based on DNA sequences and restriction site variation (e.g., van Ham and Hart, 1998; Mort et al., 2001, 2009; Bruyns et al., 2019; Folk et al., 2019). In addition, the sister relationship between Kalanchoideae and Sempervivoideae is supported by two putative morphological synapomorphies, i.e., leaves with a single apical or subapical hydathode and seeds with costate testa (Thiede and Eggli, 2007).
Numerous studies have attempted to identify major lineages within Sempervivoideae (e.g., van Ham and Hart, 1998; Mort et al., 2001; Mayuzumi and Ohba, 2004; Nikulin et al., 2016; Folk et al., 2019; Messerschmid et al., 2020), but relationships among these major lineages remain uncertain (Supplementary Figure 1). Our analyses have revealed five clades that are successively sister lineages, i.e., Telephium clade, Sempervivum clade, Aeonium clade, Leucosedum clade, and Acre clade. These clades correspond to five out of the six major clades sensu Messerschmid et al. (2020). Of these six major clades, the lone exception here is the Petrosedum clade sensu Messerschmid et al. (2020), for which no chloroplast genome sequence is currently available. The Telephium clade identified here is equivalent to tribes Telephieae plus Umbiliceae sensu Thiede and Eggli (2007). Although it was first proposed by van Ham and Hart (1998), the monophyly of the Telephium clade has only recently been confirmed (i.e., ≥85 BP) by phylogenetic analyses of chloroplast genome sequences (Kim and Kim, 2020) and 301 low-copy nuclear genes (Folk et al., 2019). Our results add further evidence to support the recognition of the Telephium clade. In addition, Umbilicus rupestris is strongly supported by our phylogenetic analyses as sister to the tribe Telephieae (i.e., our sampled species of Hylotelephium, Orostachys, and Sinocrassula), thus corroborating recent findings (Folk et al., 2019; Kim and Kim, 2020; Zhao et al., 2020) and highlighting the paraphyly of the tribe Umbiliceae (i.e., our sampled species of Phedimus, Rhodiola, and Umbilicus). Furthermore, the Sempervivum and Aeonium clades identified here correspond to tribes Semperviveae and Aeonieae sensu Thiede and Eggli (2007), respectively, and the Leucosedum and Acre clades identified here together constitute tribe Sedeae sensu Thiede and Eggli (2007).
It has been indicated that the genes with positive selection played key parts in the adaptation to diverse environments (Moseley et al., 2018; Gao et al., 2019b; Li et al., 2020b). However, no positive selection was statistically supported among sixty-seven chloroplast protein-coding genes in sampled Crassulaceae. This result indicated these protein-coding genes are under strong structural and functional constraints. The absence of positive selection in most protein-coding genes of chloroplast genome was also found in Euonymus (Li et al., 2021) and Quercus (Yin et al., 2018).
In conclusion, by expanding the number of informative molecular characters, we have further improved the resolution of the phylogenetic relationships among major lineages within Crassulaceae, which will facilitate the identification of non-molecular synapomorphies. However, additional sampling of key lineages (e.g., Petrosedum clade) is required to fully resolve the infrafamilial relationships. Furthermore, the boundaries of some of the traditional genera in Crassulaceae remain poorly defined. For example, Sedum, the largest genus with approximately 470 species, has been shown to be highly polyphyletic, and its intrageneric relationships remain largely unresolved (Messerschmid et al., 2020). Thus, chloroplast phylogenomics will continue to enhance our understanding of the evolutionary history of Crassulaceae in the future.
Data Availability Statement
All annotated chloroplast genomes have been deposited in GenBank (https://www.ncbi.nlm.nih.gov/genbank/), and accession numbers are provided in Table 1.
Author Contributions
JL, ZX, and XX designed research. HC, LZ, and HX performed research and analyzed data. HC, JL, ZX, and XX wrote the manuscript. All authors reviewed and revised the manuscript.
Funding
This work was supported by the National Key Research and Development Program of China (2017YFC0505203), the National Natural Science Foundation of China (31770566, 31600172), and Biodiversity Survey, Observation and Assessment Program of Ministry of Ecology and Environment of China.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We would like to thank Quanjun Hu, Minghui Kang, Tao Ma, Deyan Wang, and Xuchen Yang for technical assistance and valuable discussions. We would also like to thank DNA Bank of Royal Botanic Gardens for providing DNA materials of Haloragaceae.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.631884/full#supplementary-material
Footnotes
- ^ https://dnabank.science.kew.org
- ^ http://genome.lbl.gov/vista/mvista/submit.shtml
- ^ https://irscope.shinyapps.io/irapp/
References
Acevedo-Rosas, R., Cameron, K., Sosa, V., and Pell, S. (2004). A molecular phylogenetic study of graptopetalum (Crassulaceae) based on ETS, ITS, rpl16, and trnL-F nucleotide sequences. Am. J. Bot. 91, 1099–1104. doi: 10.3732/ajb.91.7.1099
Allen, G. C., Flores-Vergara, M. A., Krasnyanski, S., Kumar, S., and Thompson, W. F. (2006). A modified protocol for rapid DNA isolation from plant tissues using cetyltrimethylammonium bromide. Nat. Protoc. 1, 2320–2325. doi: 10.1038/nprot.2006.384
Amiryousefi, A., Hyvönen, J., and Poczai, P. (2018). IRscope: an online program to visualize the junction sites of chloroplast genomes. Bioinformatics 34, 3030–3031. doi: 10.1093/bioinformatics/bty220
APG IV. (2016). An update of the angiosperm phylogeny group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 181, 1–20. doi: 10.1111/boj.12385
Bedoya, A. M., Ruhfel, B. R., Philbrick, C. T., Madriñán, S., Bove, C. P., Mesterházy, A., et al. (2019). Plastid genomes of five species of riverweeds (Podostemaceae): structural organization and comparative analysis in Malpighiales. Front. Plant Sci. 10:1035. doi: 10.3389/fpls.2019.01035
Berger, A. (1930). “Crassulaceae,” in Die Natürlichen Pflanzenfamilien, Band 18a, eds A. Engler and K. Prantl (Leipzig: Wilhelm Engelmann), 352–483.
Brudno, M., Malde, S., Poliakov, A., Do, C. B., Couronne, O., Dubchak, I., et al. (2003). Glocal alignment: finding rearrangements during alignment. Bioinformatics 19, i54–i62.
Bruyns, P. V., Hanáček, P., and Klak, C. (2019). Crassula, insights into an old, arid-adapted group of southern African leaf-succulents. Mol. Phylogenet. Evol. 131, 35–47. doi: 10.1016/j.ympev.2018.10.045
Capellagutiérrez, S., Sillamartínez, J. M., and Gabaldón, T. (2009). Trimal: a tool for automated alignment trimming in large-scale phylogenetic analyses. Bioinformatics 25, 1972–1973. doi: 10.1093/bioinformatics/btp348
Carrillo-Reyes, P., Sosa, V., and Mort, M. E. (2008). Thompsonella and the “Echeveria group” (Crassulaceae): phylogenetic relationships based on molecular and morphological characters. Taxon 57, 863–874. doi: 10.1002/tax.573015
Carrillo-Reyes, P., Sosa, V., and Mort, M. E. (2009). Molecular phylogeny of the Acre clade (Crassulaceae): dealing with the lack of definitions for Echeveria and Sedum. Mol. Phylogenet. Evol. 53, 267–276. doi: 10.1016/j.ympev.2009.05.022
Chang, H., Tang, Y.-G., and Xu, X.-T. (2020). The complete chloroplast genome of sedum emarginatum (Crassulaceae). Mitochondrial DNA B Resour. 3, 3082–3083. doi: 10.1080/23802359.2020.1797567
Chumley, T. W., Palmer, J. D., Mower, J. P., Fourcade, H. M., Calie, P. J., Boore, J. L., et al. (2006). The complete chloroplast genome sequence of Pelargonium x hortorum: organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol. Biol. Evol. 23, 2175–2190. doi: 10.1093/molbev/msl089
Darling, A. C. E., Mau, B., Blattner, F. R., and Perna, N. T. (2004). Mauve: multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 14, 1394–1403. doi: 10.1101/gr.2289704
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PLoS One 5:e11147. doi: 10.1371/journal.pone.0011147
de la Cruz-López, L. E., Vergara-Silva, F., Reyes Santiago, J., Espino Ortega, G., Carrillo-Reyes, P., and Kuzmina, M. (2019). Phylogenetic relationships of Echeveria (Crassulaceae) and related genera from Mexico, based on three DNA barcoding loci. Phytotaxa 422, 33–57. doi: 10.11646/phytotaxa.422.1.3
Dierckxsens, N., Mardulyn, P., and Smits, G. (2017). NOVOPlasty: de novo assembly of organelle genomes from whole genome data. Nucleic Acids Res. 45:e18.
Dong, W., Xu, C., Cheng, T., and Zhou, S. (2013). Complete chloroplast genome of Sedum sarmentosum and chloroplast genome evolution in Saxifragales. PLoS One 8:e77965. doi: 10.1371/journal.pone.0077965
Fairfield, K. N., Mort, M. E., and Santos-Guerra, A. (2004). Phylogenetics and evolution of the Macaronesian members of the genus Aichryson (Crassulaceae) inferred from nuclear and chloroplast sequence data. Plant Syst. Evol. 248, 71–83.
Firetti, F., Zuntini, A. R., Gaiarsa, J. W., Oliveira, R. S., Lohmann, L. G., and Van Sluys, M.-A. (2017). Complete chloroplast genome sequences contribute to plant species delimitation: a case study of the Anemopaegma species complex. Am. J. Bot. 104, 1493–1509. doi: 10.3732/ajb.1700302
Folk, R. A., Stubbs, R. L., Mort, M. E., Cellinese, N., Allen, J. M., Soltis, P. S., et al. (2019). Rates of niche and phenotype evolution lag behind diversification in a temperate radiation. Proc. Natl. Acad. Sci. U.S.A. 116, 10874–10882. doi: 10.1073/pnas.1817999116
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., and Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32, W273–W279.
Gao, L.-Z., Liu, Y.-L., Zhang, D., Li, W., Gao, J., Liu, Y., et al. (2019a). Evolution of Oryza chloroplast genomes promoted adaptation to diverse ecological habitats. Commun. Biol. 2:278.
Gao, C.-M., Deng, Y.-F., and Wang, J. (2019b). The complete chloroplast genomes of Echinacanthus species (Acanthaceae): phylogenetic relationships, adaptive evolution, and screening of molecular markers. Front. Plant Sci. 9:1989. doi: 10.3389/fpls.2018.01989
Gehrig, H., Gaußmann, O., Marx, H., Schwarzott, D., and Kluge, M. (2001). Molecular phylogeny of the genus Kalanchoe (Crassulaceae) inferred from nucleotide sequences of the ITS-1 and ITS-2 regions. Plant Sci. 160, 827–835. doi: 10.1016/s0168-9452(00)00447-7
Gontcharova, S. B., Artyukova, E. V., and Gontcharov, A. A. (2006). Phylogenetic relationships among members of the subfamily sedoideae (Crassulaceae) inferred from the ITS region sequences of nuclear rDNA. Russ. J. Genet. 42, 654–661. doi: 10.1134/s102279540606010x
Gontcharova, S. B., and Gontcharov, A. A. (2009). Molecular phylogeny and systematics of flowering plants of the family Crassulaceae DC. Mol. Biol. 43, 794–803. doi: 10.1134/s0026893309050112
Gontcharova, S. B., Gontcharov, A. A., and Stephenson, R. (2008). Analysis of phylogenetic relationships in the family crassulaceae based on comparisons of ITS region of nuclear rDNA. Bot. Zhurn. 93, 97–113.
Goremykin, V. V., Hirsch-Ernst, K. I., Wolfl, S., and Hellwig, F. H. (2003). Analysis of the Amborella trichopoda chloroplast genome sequence suggests that Amborella is not a basal angiosperm. Mol. Biol. Evol. 20, 1499–1505. doi: 10.1093/molbev/msg159
Goremykin, V. V., Hirsch-Ernst, K. I., Wolfl, S., and Hellwig, F. H. (2004). The chloroplast genome of Nymphaea alba: whole-genome analyses and the problem of identifying the most basal angiosperm. Mol. Biol. Evol. 21, 1445–1454. doi: 10.1093/molbev/msh147
Gruzdev, E. V., Kadnikov, V. V., Beletsky, A. V., Mardanov, A. V., and Ravin, N. V. (2019). Extensive plastome reduction and loss of photosynthesis genes in Diphelypaea coccinea, a holoparasitic plant of the family Orobanchaceae. PeerJ. 7:e7830. doi: 10.7717/peerj.7830
Hu, S.-L., Sablok, G., Wang, B., Qu, D., Barbaro, E., Viola, R., et al. (2015). Plastome organization and evolution of chloroplast genes in Cardamine species adapted to contrasting habitats. BMC Genom. 16:306. doi: 10.1186/s12864-015-1498-0
Hu, Y.-H., Woeste, K. E., and Zhao, P. (2016). Completion of the chloroplast genomes of five chinese juglans and their contribution to chloroplast phylogeny. Front. Plant Sci. 7:1955. doi: 10.3389/fpls.2016.01955
Huang, D. I., and Cronk, Q. C. B. (2015). Plann: a command-line application for annotating plastome sequences. Appl. Plant Sci. 3:1500026. doi: 10.3732/apps.1500026
Huo, Y.-L., Gao, L.-M., Liu, B.-J., Yang, Y.-Y., Kong, S. P., Sun, Y.-Q., et al. (2019). Complete chloroplast genome sequences of four Allium species: comparative and phylogenetic analyses. Sci. Rep. 9:12250.
Jian, S., Soltis, P. S., Gitzendanner, M. A., Moore, M. J., Li, R., Hendry, T. A., et al. (2008). Resolving an ancient, rapid radiation in Saxifragales. Syst. Biol. 57, 38–57. doi: 10.1080/10635150801888871
Jorgensen, T. H., and Frydenberg, J. (1999). Diversification in insular plants: inferring the phylogenetic relationship in Aeonium (Crassulaceae) using ITS sequences of nuclear ribosomal DNA. Nord. J. Bot. 19, 613–621. doi: 10.1111/j.1756-1051.1999.tb01150.x
Katoh, K., and Standley, D. M. (2013). MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol. Biol. Evol. 30, 772–780. doi: 10.1093/molbev/mst010
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kim, S.-H., and Kim, S.-C. (2020). Plastid genome of stonecrop hylotelephium verticillatum (Sedoideae; Crassulaceae): insight into structure and phylogenetic position. Mitochondrial DNA B Resour. 5, 2729–2731. doi: 10.1080/23802359.2020.1788464
Klein, J. T., and Kadereit, J. W. (2015). Phylogeny, biogeography, and evolution of edaphic association in the European oreophytes sempervivum and jovibarba (Crassulaceae). Int. J. Plant Sci. 176, 44–71. doi: 10.1086/677948
Lanfear, R., Calcott, B., Kainer, D., Mayer, C., and Stamatakis, A. (2014). Selecting optimal partitioning schemes for phylogenomic datasets. BMC Evol. Biol. 14:82. doi: 10.1186/1471-2148-14-82
Lanfear, R., Frandsen, P. B., Wright, A. M., Senfeld, T., and Calcott, B. (2017). PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34, 772–773.
Li, H.-W., Liu, B., Davis, C. C., and Yang, Y. (2020a). Plastome phylogenomics, systematics, and divergence time estimation of the Beilschmiedia group (Lauraceae). Mol. Phylogenet. Evol. 151:106901. doi: 10.1016/j.ympev.2020.106901
Li, C.-Y., Zhao, Y.-L., Xu, Z.-G., Yang, G.-Y., and Peng, J. (2020b). Initial characterization of the chloroplast genome of Vicia sepium, an important wild resource plant, and related inferences about its evolution. Front. Genet. 11:73. doi: 10.3389/fgene.2020.00073
Li, Y.-T., Dong, Y., Liu, Y.-C., Yu, X.-Y., Yang, M.-S., and Huang, Y.-R. (2021). Comparative analyses of Euonymus chloroplast genomes: genetic structure, screening for loci with suitable polymorphism, positive selection genes, and phylogenetic relationships within Celastrineae. Front. Plant Sci. 11:593984. doi: 10.3389/fpls.2020.593984
Liao, Y.-Y., Liu, Y., Liu, X., Lü, T.-F., Mbichi, R. W., Wan, T., et al. (2020). The complete chloroplast genome of Myriophyllum spicatum reveals a 4-kb inversion and new insights regarding plastome evolution in Haloragaceae. Ecol. Evol. 10, 3090–3102. doi: 10.1002/ece3.6125
Lohse, M., Drechsel, O., Kahlau, S., and Bock, R. (2013). OrganellarGenomeDRAW—a suite of tools for generating physical maps of plastid and mitochondrial genomes and visualizing expression data sets. Nucleic Acids Res. 41, W575–W581.
Magallón, S., Sánchez-Reyes, L. L., and Gómez-Acevedo, S. L. (2019). Thirty clues to the exceptional diversification of flowering plants. Ann. Bot. 123, 491–503. doi: 10.1093/aob/mcy182
Mayuzumi, S., and Ohba, H. (2004). The phylogenetic position of eastern Asian sedoideae (Crassulaceae) inferred from chloroplast and nuclear DNA sequences. Syst. Bot. 29, 587–598. doi: 10.1600/0363644041744329
Mes, T. H. M., Wijers, G.-J., and Hart, H. (1997). Phylogenetic relationships in Monanthes (Crassulaceae) based on morphological, chloroplast and nuclear DNA variation. J. Evol. Biol. 10, 193–216. doi: 10.1046/j.1420-9101.1997.10020193.x
Messerschmid, T. F. E., Klein, J. T., Kadereit, G., and Kadereit, J. W. (2020). Linnaeus’s folly – phylogeny, evolution and classification of sedum (Crassulaceae) and crassulaceae subfamily sempervivoideae. Taxon 69, 892–926. doi: 10.1002/tax.12316
Mort, M. E., Levsen, N., Randle, C. P., Van Jaarsveld, E., and Palmer, A. (2005). Phylogenetics and diversification of Cotyledon (Crassulaceae) inferred from nuclear and chloroplast DNA sequence data. Am. J. Bot. 92, 1170–1176. doi: 10.3732/ajb.92.7.1170
Mort, M. E., Randle, C. P., Burgoyne, P., Smith, G., Jaarsveld, E., and Hopper, S. D. (2009). Analyses of cpDNA matK sequence data place Tillaea (Crassulaceae) within crassula. Plant Syst. Evol. 283, 211–217. doi: 10.1007/s00606-009-0227-z
Mort, M. E., Soltis, D. E., Soltis, P. S., Francisco-Ortega, J., and Santos-Guerra, A. (2001). Phylogenetic relationships and evolution of Crassulaceae inferred from matK sequence data. Am. J. Bot. 88, 76–91. doi: 10.2307/2657129
Mort, M. E., Soltis, D. E., Soltis, P. S., Francisco-Ortega, J., and Santos-Guerra, A. (2002). Phylogenetics and evolution of the macaronesian clade of crassulaceae inferred from nuclear and chloroplast sequence data. Syst. Bot. 27, 271–288.
Moseley, R. C., Mewalal, R., Motta, F., Tuskan, G. A., Haase, S., and Yang, X.-H. (2018). Conservation and diversification of circadian rhythmicity between a model crassulacean acid metabolism plant Kalanchoe fedtschenkoi and a model C3 photo synthesis plant Arabidopsis thaliana. Front. Plant Sci. 9:1757. doi: 10.3389/fpls.2018.01757
Nikulin, V. Y., Gontcharova, S. B., Stephenson, R., and Gontcharov, A. A. (2016). Phylogenetic relationships between Sedum L. and related genera (Crassulaceae) based on ITS rDNA sequence comparisons. Flora 224, 218–229. doi: 10.1016/j.flora.2016.08.003
Nobel, P. S. (1991). Achievable productivities of certain CAM plants: basis for high values compared with C3 and C4 plants. New Phytol. 119, 183–205. doi: 10.1111/j.1469-8137.1991.tb01022.x
Nowell, T. L. (2008). The spatial and temporal dynamics of diversification in Tylecodon, Cotyledon and Adromischus (Crassulaceae) in southern Africa. dissertation/doctor’s thesis. Cape Town, South Africa: University of Cape Town.
Pilon-Smits, E. A. H., Hart, H., and van Brederode, J. (1996). Evolutionary aspects of Crassulacean acid metabolism in the Crassulaceae. Ecol. Stud. 114, 349–359. doi: 10.1007/978-3-642-79060-7_24
Piot, A., Hackel, J., Christin, P.-A., and Besnard, G. (2018). One-third of the plastid genes evolved under positive selection in PACMAD grasses. Planta 247, 255–266. doi: 10.1007/s00425-017-2781-x
Prince, L. M. (2015). Plastid primers for angiosperm phylogenetics and phylogeography. Appl. Plant Sci. 3:1400085. doi: 10.3732/apps.1400085
Quezada, I. M., and Gianoli, E. (2011). Crassulacean acid metabolism photosynthesis in Bromeliaceae: an evolutionary key innovation. Bot. J. Linn. Soc. 104, 480–486. doi: 10.1111/j.1095-8312.2011.01713.x
Ronquist, F., Teslenko, M., van der Mark, P., Ayres, D. L., Darling, A., Hohna, S., et al. (2012). MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Rozas, J., Ferrer-Mata, A., Sánchez-Delbarrio, J. C., Guirao-Rico, S., Librado, P., Ramos-Onsins, S. E., et al. (2017). DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 34, 3299–3302. doi: 10.1093/molbev/msx248
Sayers, E. W., Cavanaugh, M., Clark, K., Ostell, J., Pruitt, K. D., and Karsch-Mizrachi, I. (2020). GenBank. Nucleic Acids Res. 48, D84–D86.
Shrestha, B., Weng, M. L., Theriot, E. C., Gilbert, L. E., Ruhlman, T. A., Krosnick, S. E., et al. (2019). Highly accelerated rates of genomic rearrangements and nucleotide substitutions in plastid genomes of Passiflora subgenus Decaloba. Mol. Phylogenet. Evol. 138, 53–64. doi: 10.1016/j.ympev.2019.05.030
Silvestro, D., Zizka, G., and Schulte, K. (2014). Disentangling the effects of key innovations on the diversification of Bromelioideae (Bromeliaceae). Evolution 68, 163–175. doi: 10.1111/evo.12236
Smith, G. F., Figueiredo, E., and van Wyk, A. E. (2019). Kalanchoe (Crassulaceae) in Southern Africa: Classification, Biology, and Cultivation. Cambridge, MA: Academic Press.
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Stamatakis, A., Hoover, P., and Rougemont, J. (2008). A rapid bootstrap algorithm for the RAxML web servers. Syst. Biol. 57, 758–771. doi: 10.1080/10635150802429642
Suyama, M., Torrents, D., and Bork, P. (2006). PAL2NAL: robust conversion of protein sequence alignments into the corresponding codon alignments. Nucleic Acids Res. 34, W609–W612.
Thiede, J., and Eggli, U. (2007). “Crassulaceae,” in The Families and Genera of Vascular Plants, ed. K. Kubitzki (Berlin: Springer), 83–118.
Twyford, A. D., and Ness, R. W. (2017). Strategies for complete plastid genome sequencing. Mol. Ecol. Resour. 17, 858–868. doi: 10.1111/1755-0998.12626
van Ham, R., and Hart, H. (1998). Phylogenetic relationships in the Crassulaceae inferred from chloroplast DNA restriction-site variation. Am. J. Bot. 85, 123–134. doi: 10.2307/2446561
Vázquez-Cotero, C., Sosa, V., and Carrillo-Reyes, P. (2017). Phylogenetic position of echeveria heterosepala (Crassulaceae): a rare species with diagnostic characters of Pachyphytum. Bot. Sci. 95, 515–526. doi: 10.17129/botsci.1190
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Yang, Z., Wong, W. S. W., and Nielsen, R. (2005). Bayes empirical bayes inference of amino acid sites under positive selection. Mol. Biol. Evol. 22, 1107–1118. doi: 10.1093/molbev/msi097
Yang, Z.-H., and Dos, R.-M. (2011). Statistical properties of the branch-site test of positive selection. Mol. Biol. Evol. 28, 1217–1228. doi: 10.1093/molbev/msq303
Yin, K., Zhang, Y., Li, Y., and Du, F. (2018). Different natural selection pressures on the atpF gene in evergreen Sclerophyllous and Deciduous oak species: evidence from comparative analysis of the complete chloroplast genome of Quercus aquifolioides with other oak species. Int. J. Mol. Sci. 19:1042. doi: 10.3390/ijms19041042
Yost, J. M., Bontrager, M., Mccabe, S. W., Burton, D., Simpson, M. G., Kay, K. M., et al. (2013). Phylogenetic relationships and evolution in Dudleya (Crassulaceae). Syst. Bot. 38, 1096–1104. doi: 10.1600/036364413x674760
Zang, M., Su, Q., Weng, Y., Lu, L., Zheng, X., Ye, D., et al. (2019). Complete chloroplast genome of fokienia hodginsii (Dunn) Henry et Thomas: insights into repeat regions variation and phylogenetic relationships in cupressophyta. Forests 10:528. doi: 10.3390/f10070528
Zhang, D., Gao, F., Jakovlić, I., Zou, H., Zhang, J., Li, W. X., et al. (2020). PhyloSuite: an integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 20, 348–355. doi: 10.1111/1755-0998.13096
Zhang, J.-Q., Meng, S.-Y., Wen, J., and Rao, G.-Y. (2014). Phylogenetic relationships and character evolution of Rhodiola (Crassulaceae) based on nuclear ribosomal ITS and plastid trnL-F and psbA-trnH sequences. Syst. Bot. 39, 441–451. doi: 10.1600/036364414x680753
Zhang, J.-Z., Nielsen, R., and Yang, Z.-H. (2005). Evaluation of an improved branch-site likelihood method for detecting positive selection at the molecular level. Mol. Biol. Evol. 22, 2472–2479. doi: 10.1093/molbev/msi237
Zhao, D.-N., Ren, Y., and Zhang, J.-Q. (2020). Conservation and innovation: plastome evolution during rapid radiation of Rhodiola on the Qinghai-Tibetan Plateau. Mol. Phylogenet. Evol. 144:106713. doi: 10.1016/j.ympev.2019.106713
Keywords: Crassulaceae, chloroplast genome, comparative genomics, phylogenomics, infrafamilial relationships, adaptive evolution
Citation: Chang H, Zhang L, Xie H, Liu J, Xi Z and Xu X (2021) The Conservation of Chloroplast Genome Structure and Improved Resolution of Infrafamilial Relationships of Crassulaceae. Front. Plant Sci. 12:631884. doi: 10.3389/fpls.2021.631884
Received: 21 November 2020; Accepted: 10 June 2021;
Published: 01 July 2021.
Edited by:
Michael R. McKain, University of Alabama, United StatesReviewed by:
Olwen M. Grace, Royal Botanic Gardens, Kew, United KingdomAndrey Gontcharov, Federal Scientific Center of the East Asia Terrestrial Biodiversity, Far Eastern Branch of the Russian Academy of Sciences, Russia
Copyright © 2021 Chang, Zhang, Xie, Liu, Xi and Xu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhenxiang Xi, enhpQHNjdS5lZHUuY24=; Xiaoting Xu, eGlhb3Rpbmd4dUBzY3UuZWR1LmNu
†These authors have contributed equally to this work