 Tao Liu1,2,3
Tao Liu1,2,3 Bo Zhang
Bo Zhang- 1Key Laboratory of Adaptation and Evolution of Plateau Biota (AEPB), Northwest Institute of Plateau Biology, Chinese Academy of Sciences, Xining, China
- 2Qinghai Province Key Laboratory of Crop Molecular Breeding, Xining, China
- 3University of Chinese Academy of Sciences, Beijing, China
- 4Xining No. 8 Junior High School, Xining, China
Selenium is an essential microelement for humans and animals. The specific processing technique of oats can maximize the preservation of its nutrients. In this study, to understand the genetic response of oats in a high-selenium environment, oats were treated with sodium selenate for 24 h, and transcriptome analysis was performed. A total of 211,485,930 clean reads composing 31.30 Gb of clean data were retained for four samples. After assembly, 186,035 unigenes with an average length of 727 bp were generated, and the N50 length was 1,149 bp. Compared with that in the control group, the expression of 7,226 unigenes in the treatment group was upregulated, and 2,618 unigenes were downregulated. Based on the sulfur assimilation pathway and selenocompound metabolic pathway, a total of 27 unigenes related to selenate metabolism were identified. Among them, the expression of both key genes APS (ATP sulfurylase) and APR (adenosine 5′-phosphosulfate reductase) was upregulated more than 1,000-fold under selenate treatment, while that of CBL (cystathionine-β-synthase) was upregulated 3.12-fold. Based on the transcriptome analysis, we suspect that the high-affinity sulfur transporter Sultr1;2 plays a key role in selenate uptake in oats. A preliminary regulatory mechanism explains the oat response to selenate treatment was ultimately proposed based on the transcriptome analysis and previous research.
Introduction
Avena sativa L. (oats) ranks sixth in global cereal production statistics, and oats are grown as multipurpose crops for grain, pasture, and forage or as rotation crops in many parts of the world (Stevens et al., 2004). Cultivated oats are known for their economic value for human nutrition and health care (Singh and Kumar, 2016). Compared to food products such as wheat flour, rice or corn meal, oatmeal retains nearly all the components of the seed, with the micronutrients highly retained (Winkel et al., 2012). Furthermore, oats are rich in protein, fat, vitamins, antioxidants and minerals, which can effectively regulate blood lipids, lower cholesterol and delay the body’s aging (Sadiq Butt et al., 2008). Cultivated oat is an allohexaploid (2n = 6x = 42), consisting of three different genomes with a total size of 12.5 Gbps (Bekele et al., 2018). With the development of next generation sequencing technologies, RNA sequencing (RNA-Seq) could provide an economic and convenient approach for discovering new genes and studying the molecular mechanism on plant abiotic stress, plant development, and diseases resistance (Margulies et al., 2005; Kushwaha et al., 2019). Cheng et al. (2020) found the mechanism of the changes of energy production and AsA-GSH cycle in oat embryos during seed aging by using RNA-seq. Zhang et al. (2021) compared the transcriptome data of an oat male sterile material and a fertile material at different developmental stages, and the results revealed the dynamic expression profile of pollen abortion male sterile and male fertile in oats firstly. It is worth mentioning that PepsiCo and Corteva Agriscience announced the first-ever sequencing of the full oat genome and proposed a new chromosome nomenclature in 20201. This work will greatly promote the vigorous development of the oat industry and accelerate the analysis of various mechanisms in oats while conducive to cultivating oat varieties with improved sustainability, taste and nutrition.
Selenium (Se) is an essential micronutrient for humans and animals (Hatfield and Gladyshev, 2002). It is a key component of more than 25 mammalian selenoenzymes or selenoproteins with important biological functions (Kryukov et al., 2003; Rayman, 2012). Approximately 15% of the global population is affected by Se deficiency due to the consumption of food crops with low Se concentrations (White and Broadley, 2009). For humans, a lack of selenium will lead to a weakened immune system, hypothyroidism, Keshan disease, Kaschin-Beck disease, hair loss, male infertility and so on (Rayman, 2012; White, 2016). In China, one of the 40 countries whose population is deficient in Se, more than 105 million people face adverse health impacts due to Se deficiency (Xu et al., 2012; Dinh et al., 2018). Selenium resources are unevenly distributed in the global environment, and selenium intake for many people is significantly lower than the national recommended standard (Winkel et al., 2012). The eastern region (Ping’an-Ledu area) of Qinghai Province is a selenium-rich area (Zhang et al., 2012; Liu et al., 2014). The humid, cool, and long-day characteristics of this region are very suitable for oat growth. Se biofortification of food crops by means of Se fertilization or breeding Se-enriched cultivars serve as powerful approaches to reduce Se deficiency (Broadley et al., 2007; Hussain et al., 2010; Kumar et al., 2011; Joy et al., 2015). Elucidating the genetic basis of selenium accumulation and metabolism is important for crop biofortification improvement.
In the present study, a comparison of the gene expression profiles of oat seedlings cultured in Hoagland media supplemented with selenium with those cultured in normal Hoagland solution was expected to reflect the metabolic changes resulting in Se accumulation in oats. The final goal of this study was to identify selenium-related genes and to elucidate the relevant metabolic pathways in oats. The assembled and annotated transcriptome will help to understand the genetic basis of selenium uptake, assimilation, transportation and accumulation in oats.
Materials and Methods
Plant Material and Se Treatments
Avena sativa L. cv. Jiayan 2 (Jiayan 2 for short) was selected in this research. This cultivar is a hulled oat widely grown in Qinghai Province, and it produces high yields, displays good quality and is strongly reproducible. The seeds were first soaked in double distilled water (ddH2O) for 24 h and then germinated in sterilized petri dishes with moist filter paper at a constant temperature (25°C) in darkness (Wu et al., 2019). Five days later, the seedlings were transplanted to a new container containing 1/2-strength Hoagland’s nutrient solution, under daily conditions of 25°C and a 16/8 h photoperiod (light/dark). Half of the 2-week-old seedlings were treated with 20 μM Na2SeO4 for 24 h, and the other (as a control group) was not treated. Both the control and treatment groups included two biological replications. The roots of all samples were taken, placed into corresponding numbered centrifuge tubes, immediately frozen in liquid nitrogen, and then stored at −80°C for RNA extraction.
RNA Extraction, Library Construction, and Sequencing
Total RNA from the roots of oats in the treatment (T) and control groups (CK) was extracted by using a Trizol Plant RNA Extraction Kit according to the user manual (Takara Bio, Japan). The quality of the total RNA was then detected using a 1% agarose gel, and its concentration was measured with a NanoDrop 2000C micro-UV detector (Thermo Fisher Scientific, United States). mRNA was enriched by oligo(dT) beads and fragmented into short fragments according to the manufacturer’s protocol. In conjunction with random primers, mRNA was reverse transcribed into cDNA (Illumina, Inc., San Diego, United States). The cDNA library products were subsequently sequenced in paired-end sequencing technology with read lengths of 150 bp by Gene Denovo Biotechnology Co. (Guangzhou, China) by using an Illumina HiSeqTM 4000 instrument.
Filtering of Sequencing Data and de novo Assembly
The raw data contained low-quality sequences, and adapters were obtained from the sequencing machine. Before assembly and analysis, the low-quality reads were removed and trimmed by software Trimmomatic v0.39 (Zheng et al., 2015). The clean reads were then mapped to ribosomal RNA (rRNA) to remove residual rRNA reads and used for further analysis. After purity filtering, the high-quality reads were de novo assembled by Trinity, with the default parameters (Grabherr et al., 2011). Sequencing reads were mapped back to the assembled transcripts for assessing the quality of the transcriptome assembly using the Bowtie2 v2.3.4.3 (Li et al., 2009). To evaluate completeness of the de novo assembled, BUSCO analysis was performed at default parameters (Kushwaha et al., 2019), and blast was used to validate the assembled contigs with OT3098 genome sequence v1. The longest transcripts of the same genes were screened as unigenes.
Gene Functional Annotations
With the purpose of acquiring the functional annotations of the unigenes, all the assembled unigenes were searched against the Nr (non-redundant) protein database (Pruitt et al., 2007), SwissProt database (Bairoch and Boeckmann, 1992), KOG (Eukaryotic Orthologous Group) database (Tatusov et al., 2003), and KEGG (Kyoto Encyclopedia of Genes and Genomes) protein pathway database (Kanehisa et al., 2008) by using BLASTX, with an E-value < 0.00001 (Altschul et al., 1997).
Analysis of DEGs
FPKM (fragments per kilobase per million reads) values were calculated and normalized to estimate the unigene expression abundances by software ERANGE (Mortazavi et al., 2008). The differentially expressed genes (DEGs) between the control group and treatment groups of Jiayan No. 2 were analyzed by the edgeR v3.32.1 (Law et al., 2018). The genes with an FDR (false discovery rate) < 0.001 and an absolute value of the log2(ratio) > 1 were considered DEGs (Liu et al., 2017; Song et al., 2017).
To further determine the biological function of the differentially expressed genes, all of them were mapped to the GO (Gene Ontology) database using Blast2GO v4.1.9 (Conesa et al., 2005). The significantly enriched GO terms in the DEGs were analyzed on the online platform “omicshare” with a p-value < 0.052. Statistical enrichment of differential expression genes in KEGG pathways with a p-value and Q-value < 0.05 was performed by KOBAS v3.0 software (Mao et al., 2005).
qRT-PCR Verification of DEGs
Two microgram purified total RNA from each tissue was reverse transcribed into cDNA using a PrimeScriptTM RT reagent kit (Takara Bio Inc.) according to the manufacturer. Then, the cDNA was diluted to 100 ng/μl for relative quantitative real-time RT-PCR analysis. For qRT-PCR validation, a TB Green® Premix Ex TaqTM II kit (Takara Bio Inc.) was used on an ABI ViiA 7 real-time PCR system (Applied Biosystems, United States) with three replicates per sample. The relative expression level of each unigene was calculated by the 2–ΔΔCT method (Livak and Schmittgen, 2001).
Results
Illumina Sequencing, Unigene Assembly, and Functional Annotation
To determine the root transcriptome of oat under selenate, a cDNA library was constructed and sequenced. From the four samples, named CK-1 (control check repeat 1), CK-2 (control check repeat 2), T-1 (treated with Na2SeO4 repeat 1), and T-2 (treated with Na2SeO4 repeat 2), 216,038,006 raw reads were obtained. After filtering, a total of 211,485,930 clean reads composing 31.30 Gb of clean data were retained, and both the Q20 and Q30 percentages were greater than 95% (Table 1). These data indicated that the sequencing results are of high quality and can be used for further analysis.
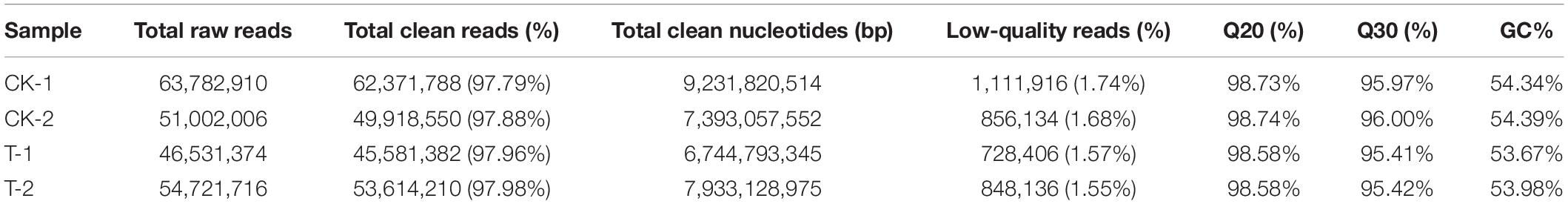
Table 1. Output of sequencing data.
High-quality clean reads were assembled as contigs and further processed as unigenes with Trinity. A total of 186,035 unigenes with an average length of 727 bp were generated, and the length of N50 was 1,149 bp. Of these, 36,734 (19.75%) unigenes surpassed 1 kb, and 13,169 (7.08%) unigenes were > 2,000 bp (Figure 1). In the transcriptome assembly, BUSCO yielded 2,679 (81.7%) complete BUSCO genes with liliopsida_odb10 database (Supplementary Table 1), and 64.72% contigs could mapped to OT3098 genome v1. The obtained oat transcriptome clean data of 6.7∼9.2 Gb in the present study is less than that predicted oat genome of 12.5Gb. In addition, OT3098 genome v1 could not sufficiently represent its real situation. Above two reasons could explain the lower mapping rate of 64.72%.
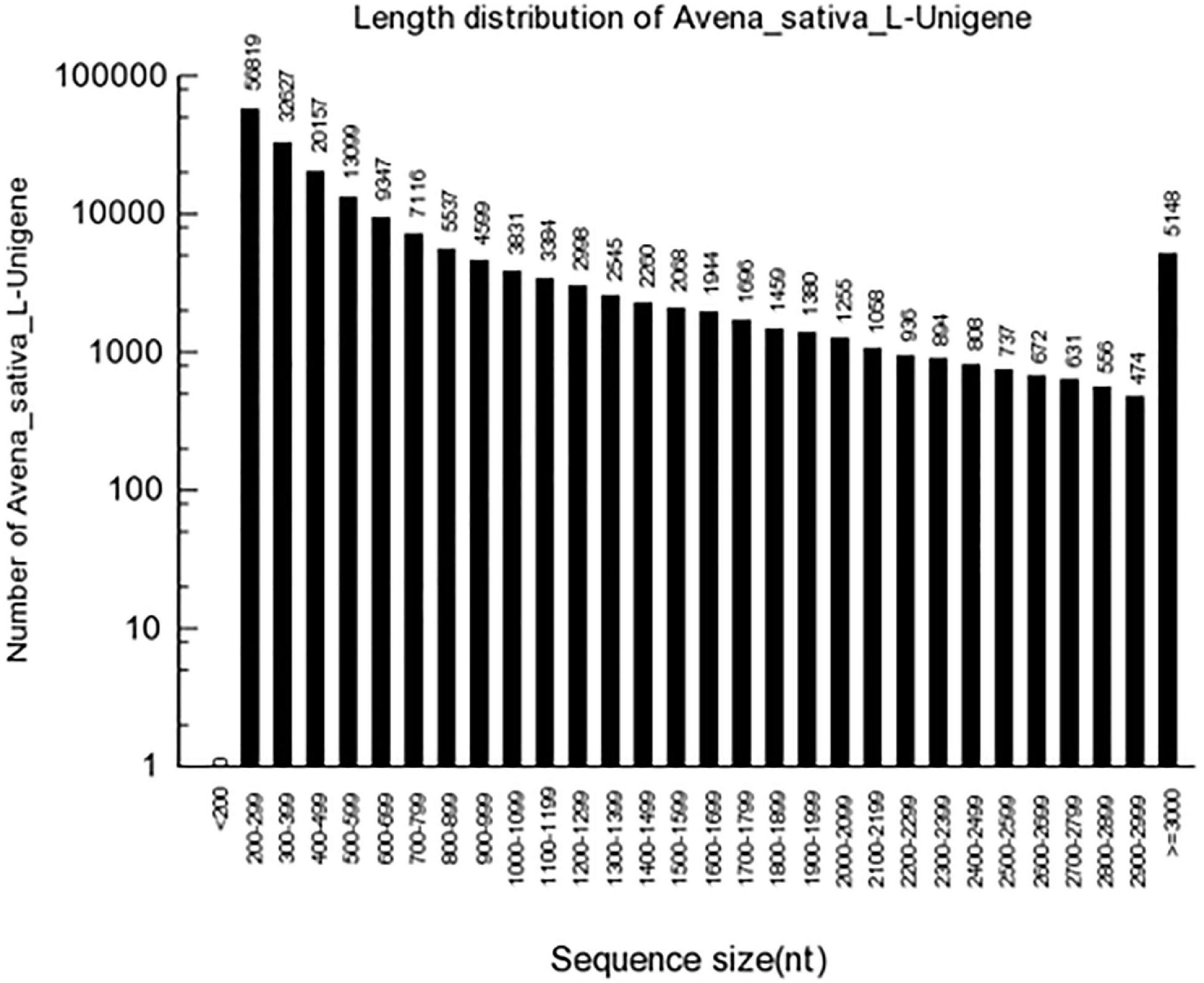
Figure 1. Length distribution of the assembled unigenes.
All 186,035 unigenes were compared with the sequences in several protein databases, including the Nr, SwissProt, KOG, and KEGG databases, by BLAST, wherein 90,542 unigenes showed high homology with sequences in at least one of the above databases (Figure 2). There were 88,441 (97.68%), 65,606 (72.46%), 43,660 (48.22%), and 58,161 (64.24%) unigenes annotated by the Nr, SwissProt, KEGG, and KOG databases, respectively.
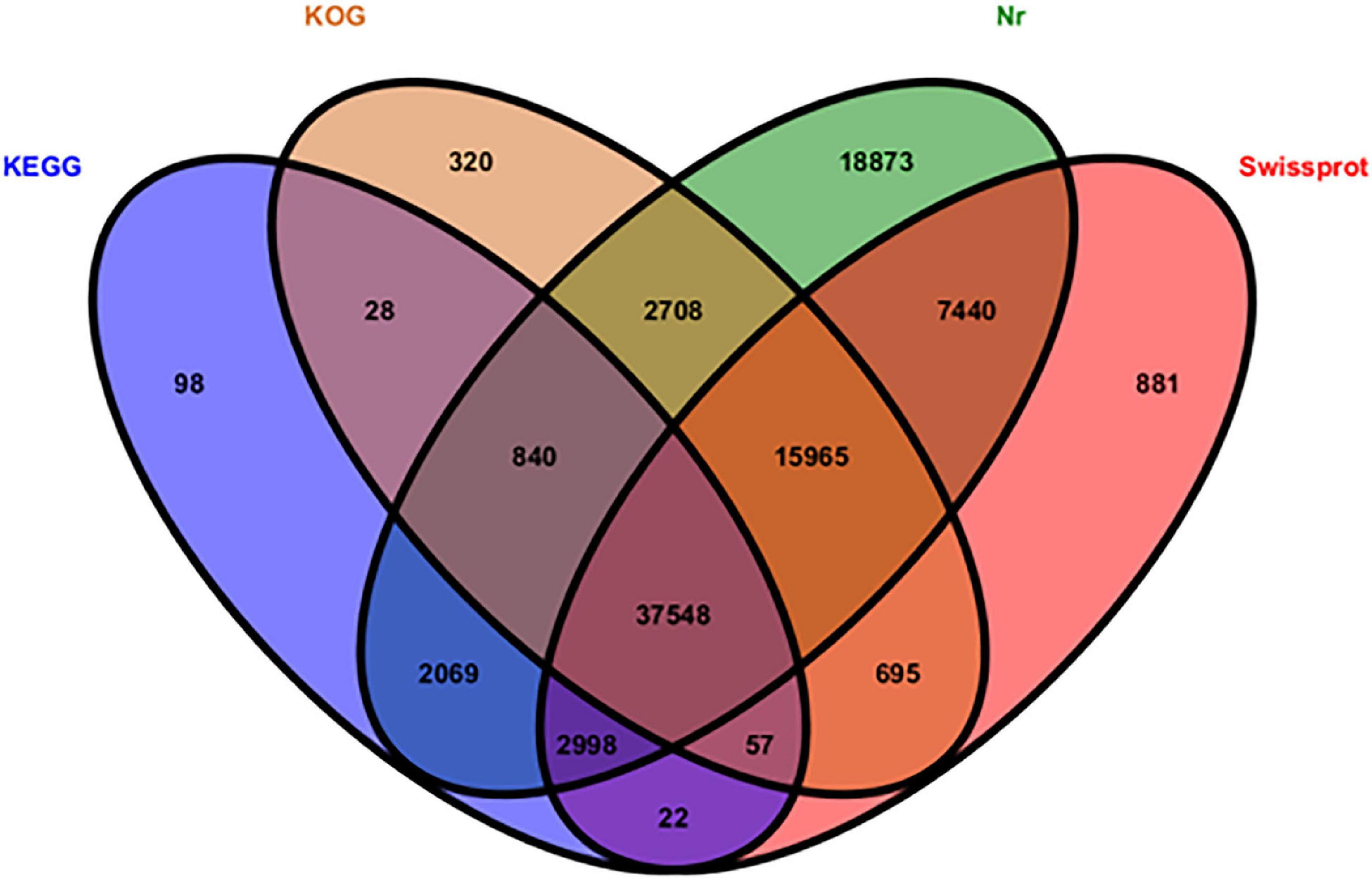
Figure 2. Distribution of functionally annotated unigenes in the KEGG, KOG, Nr, and SwissProt databases.
A statistical analysis was performed, and the results showed that 21,111 (23.87%) unigenes matched genes of Aegilops tauschii, 8,789 (9.94%) unigenes matched genes of Brachypodium distachyon, and 6,183 (6.99%) unigenes matched genes of Oryza sativa (Figure 3). We had supposed that more Avena sativa L. unigenes would have matched, but that is not the case. The results reflected the truth that little oat genomic information was registered in the NCBI database.
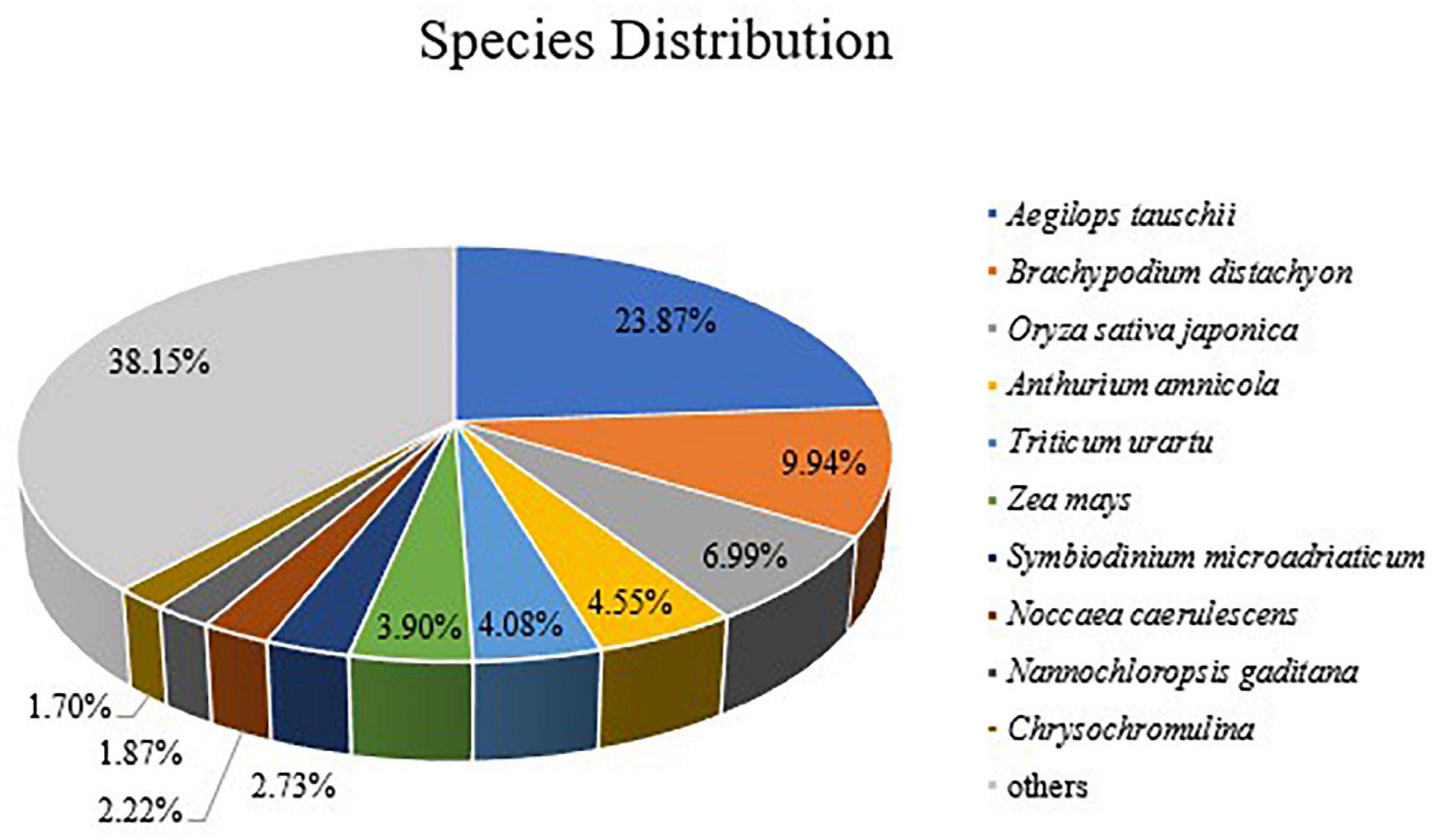
Figure 3. Species distribution of homologous proteins of unigenes. The different color blocks represent different species.
Analysis of DEGs and GO Classification
On the basis of the FPKM value of each unigene, the edgeR was used to determine the differentially expressed genes (DEGs) with an FDR (false discovery rate) of < 0.05 and a | log2 fold change| of > 1. A total of 9,844 genes were significantly differentially expressed between the CK and Jiayan 2 treatment groups. In total, 7,226 DEGs were upregulated and another 2618 downregulated in the treatment group compared with the control group (Figure 4). The log2 fold changes ranged from 1 to 14.70.
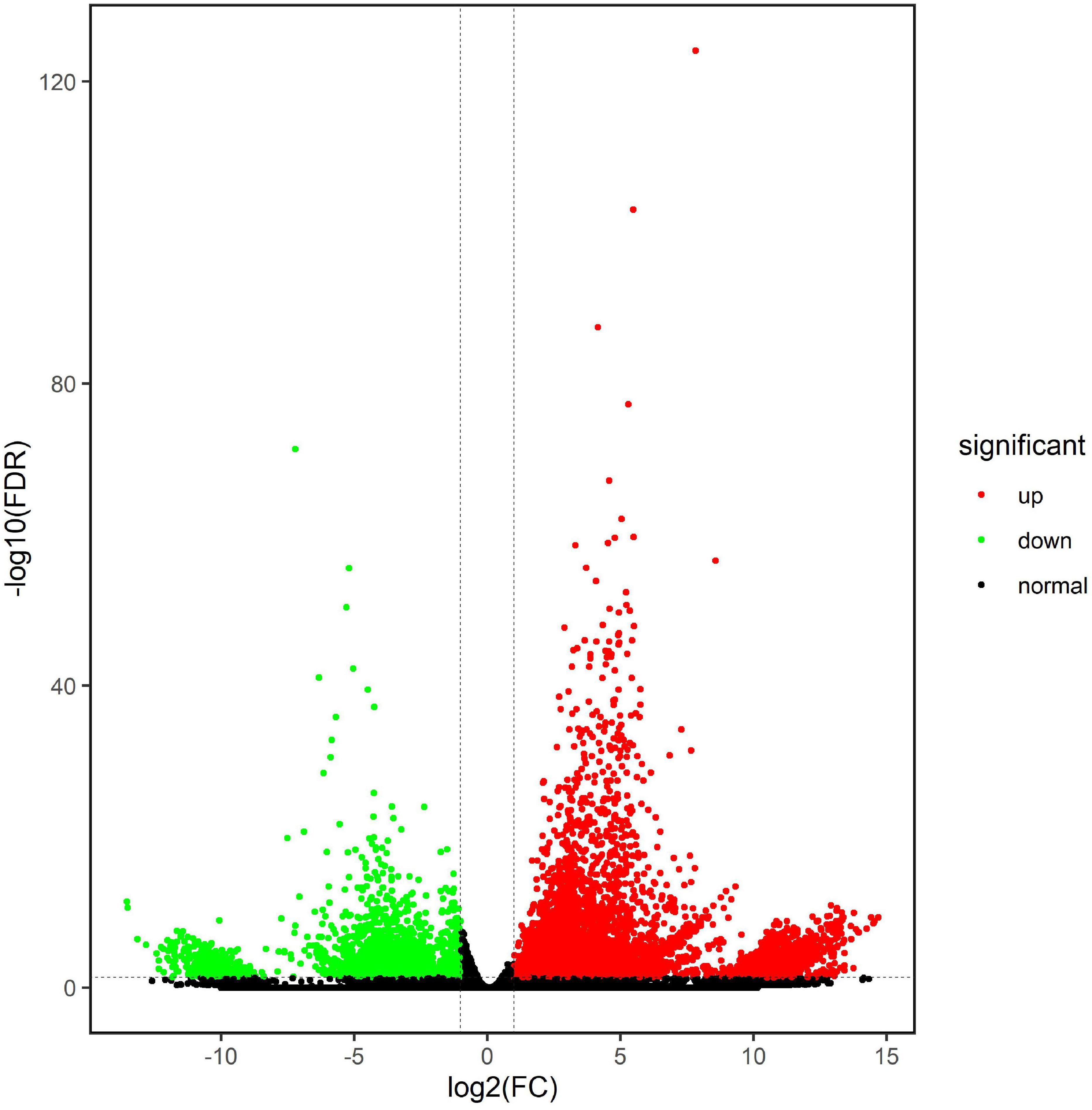
Figure 4. Differentially expressed genes between the CK and treatment groups. The green points represent the downregulated unigenes in the T group, and the red points represent the upregulated unigenes in the T group. The black points indicate that the unigenes have no expression difference between the two groups.
Based on the Nr database, all DEGs were classified into 46 functional groups (Figure 5). Among these unigenes, 5,671 were related to biological processes, 2,132 were involved in molecular functions, and 7,811 unigenes were associated with cellular components. Metabolic processes (GO:0008152), cellular component processes (GO:0071840), and single-organism processes (GO:0044699) were the dominant groups in the biological process category. In the cellular component category, a high percentage of the DEGs were related to cell (24.04% of the total DEGs) (GO:0005623), cell part (24.03% of the total DEGs) (GO:0044464), and organelle (14.95% of the total DEGs) (GO:0043226). Catalytic activity (GO:0003824), binding (GO:0005488), and structural molecule activity (GO:0005198) were the three top terms in the molecular function ontology. Subsequently, the top20 GO terms in each category were analyzed and found that a large number of genes were enriched in gene expression (GO:0010467), macromolecule metabolic process (GO:0043170), organic substance metabolic process (GO:0071704), cell part (GO:0044464), macromolecular complex (GO:0032991), structural molecular activity (GO:0005198), heterocyclic compound binding (GO:1901363), and organic cyclic compound binding (GO:0097159). Compared with the control group, a large number of genes were enriched in GO terms related to macromolecular compounds in the selenium-treated oats. In other words, selenate is rapidly converted into selenium-containing macromolecular compounds in oats.
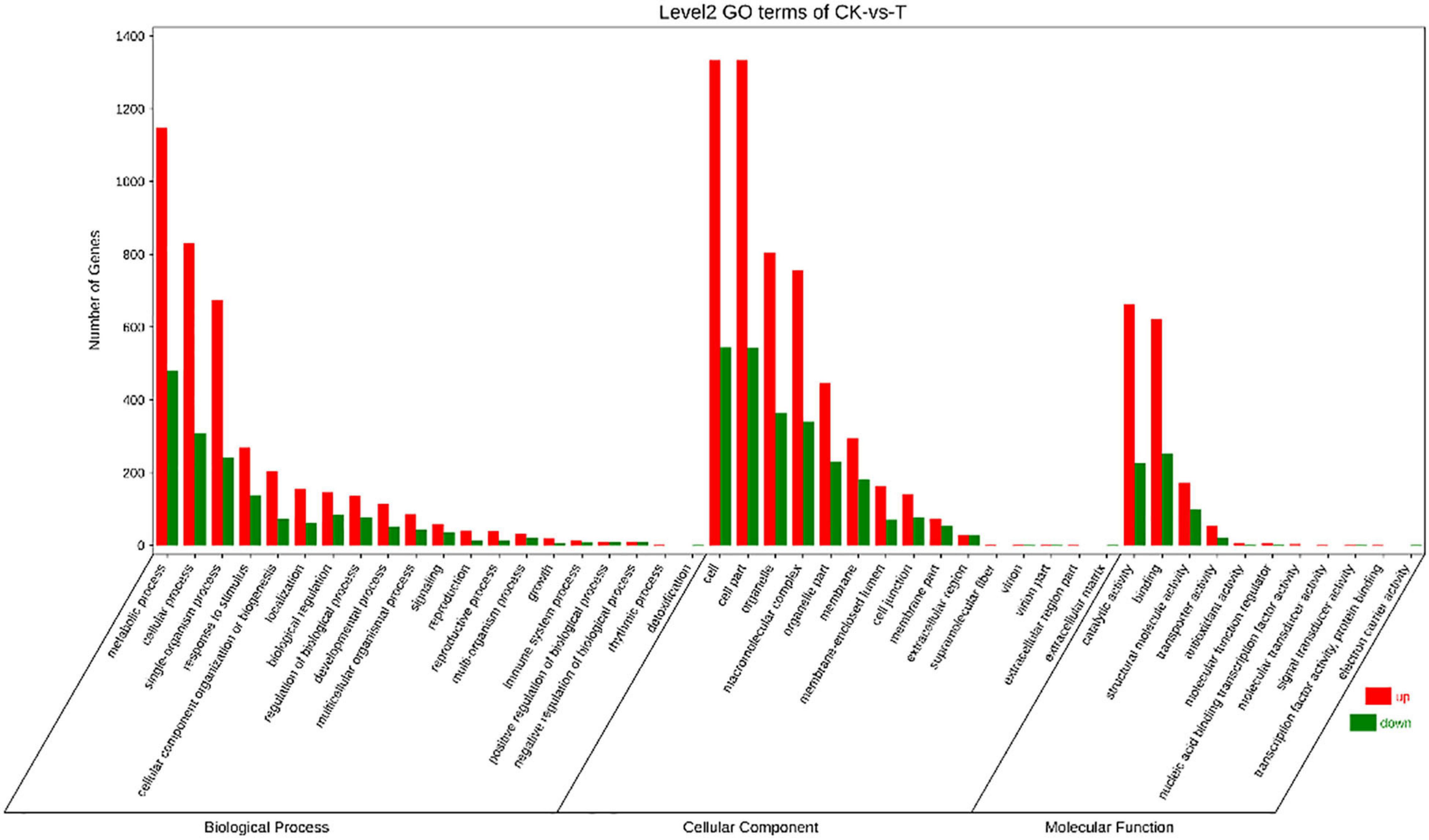
Figure 5. Gene Ontology (GO) classifications of the DEGs between CK and T. Unigenes were assigned to three categories: biological processes, cellular components, and molecular functions.
All the DEGs were annotated in five categories with KEGG pathway enrichment analysis (Figure 6). “Global and overview maps,” “transition,” and “signal transport” were enriched in the most unigenes in their respective classifications. Due to the similarity of chemical properties between Se and sulfur, when Se accumulates inside plants, it is incorporated into selenocompounds through the sulfur assimilation pathway (Mostofa et al., 2020). Cysteine and methionine metabolism (ko00270) pertain to amino acid metabolism, and 216 unigenes were associated with this metabolic pathway. Selenocompound metabolism (ko00450) belongs to the category involving the metabolism of other amino acids in the metabolic classification. A total of 120 differentially expressed unigenes were found to participate in the metabolism of other amino acids (Figure 6).
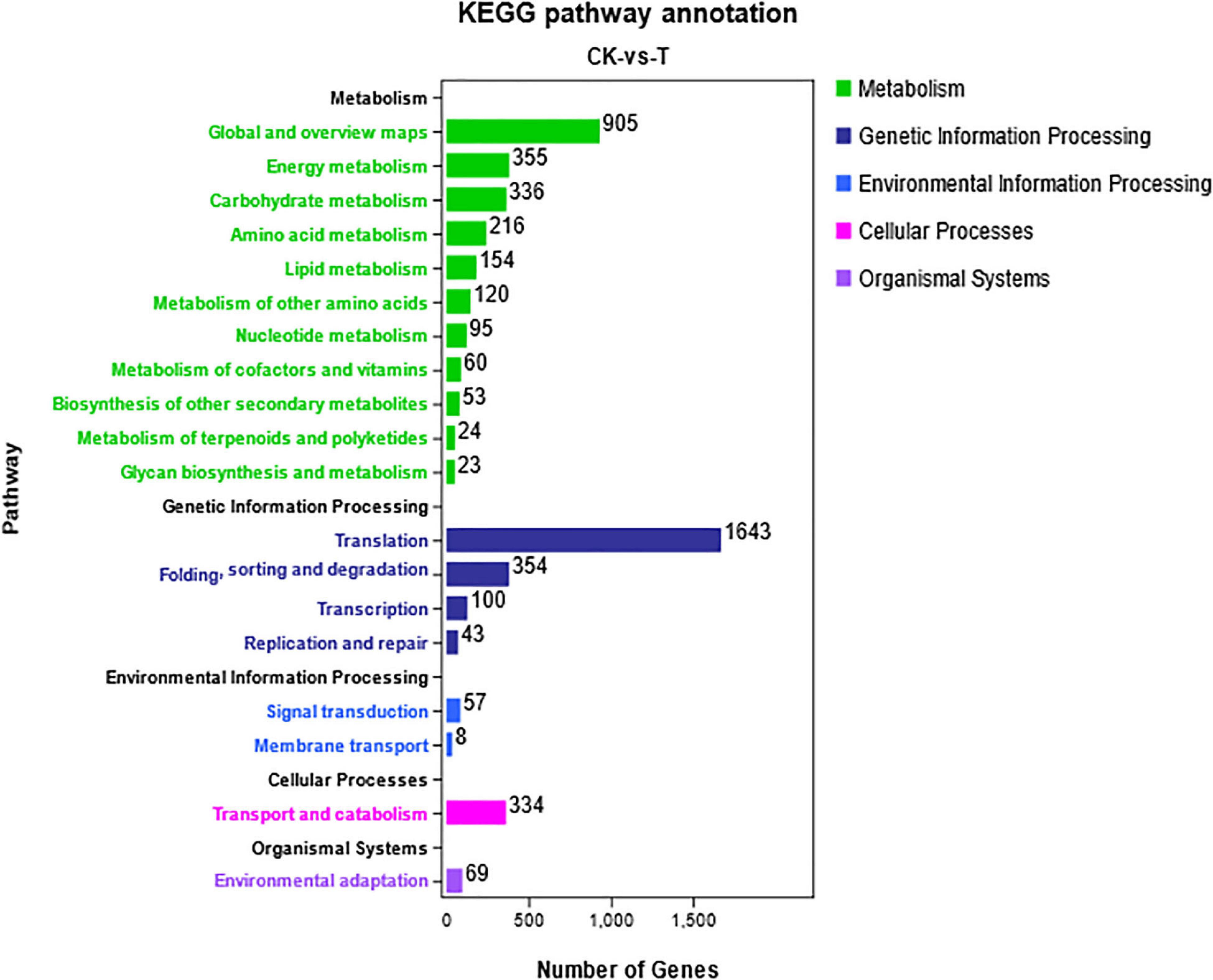
Figure 6. KEGG pathway analysis of differentially expressed unigenes. The DEGs were assigned to five categories: metabolism, genetic information processing, environmental information processing, cellular processes, and organismal systems.
Expression of Genes Involved in Selenate Metabolism in Oats
Selenocysteine (SeCys), considered the 21st amino acid, was proven to be the active site of all known selenoproteins (Rayman, 2000). Based on the sulfur assimilation pathway and the selenocompound metabolic pathway in higher plants (Ellis and Salt, 2003; Zhu et al., 2009), a total of 27 unigenes related to selenate metabolism were identified by comparison of the gene expression levels in the two groups (Table 2). These unigenes were found to be sequence similarity to the 11 selenate metabolism structural genes: Sultr1;2 (sulfate transporter), APS (ATP sulfurylase), APR (adenosine 5′-phosphosulfate reductase), SAT/OAS-TL (cysteine synthase complex), CGS (cystathionine-γ-synthase), CBL (cystathionine-β-synthase), HMT (homocysteine methyltransferase), SAMS (S-adenosylmethionine synthase), SAM-Mtase (S-adenosyl-methionine-dependent methyltransferase), AHCY (adenosyl-homocysteinase), and MARS (methionyl-tRNA synthetase). El Kassis et al. (2007) conducted selenate resistance studies on 13 Arabidopsis mutants and found that sultr1;2 is the only carrier involved in selenate transport in plants. The expression of Sultr1;2 was 5.06 times higher in the T group than in the CK group, which indicated that selenate was taken by oats and could participate in subsequent metabolic pathways (Figure 7). APS, APR, SiR, and SAT/OAS-TL are key enzymes that catalyze the conversion of inorganic selenium into an organic form (Akbudak and Filiz, 2019; Chen et al., 2019; Harun-Ur-Rashid et al., 2019). Among them, the expression of both APS and APR increased more than 1,000-fold under selenate treatment, while the expression of SiR did not change significantly.
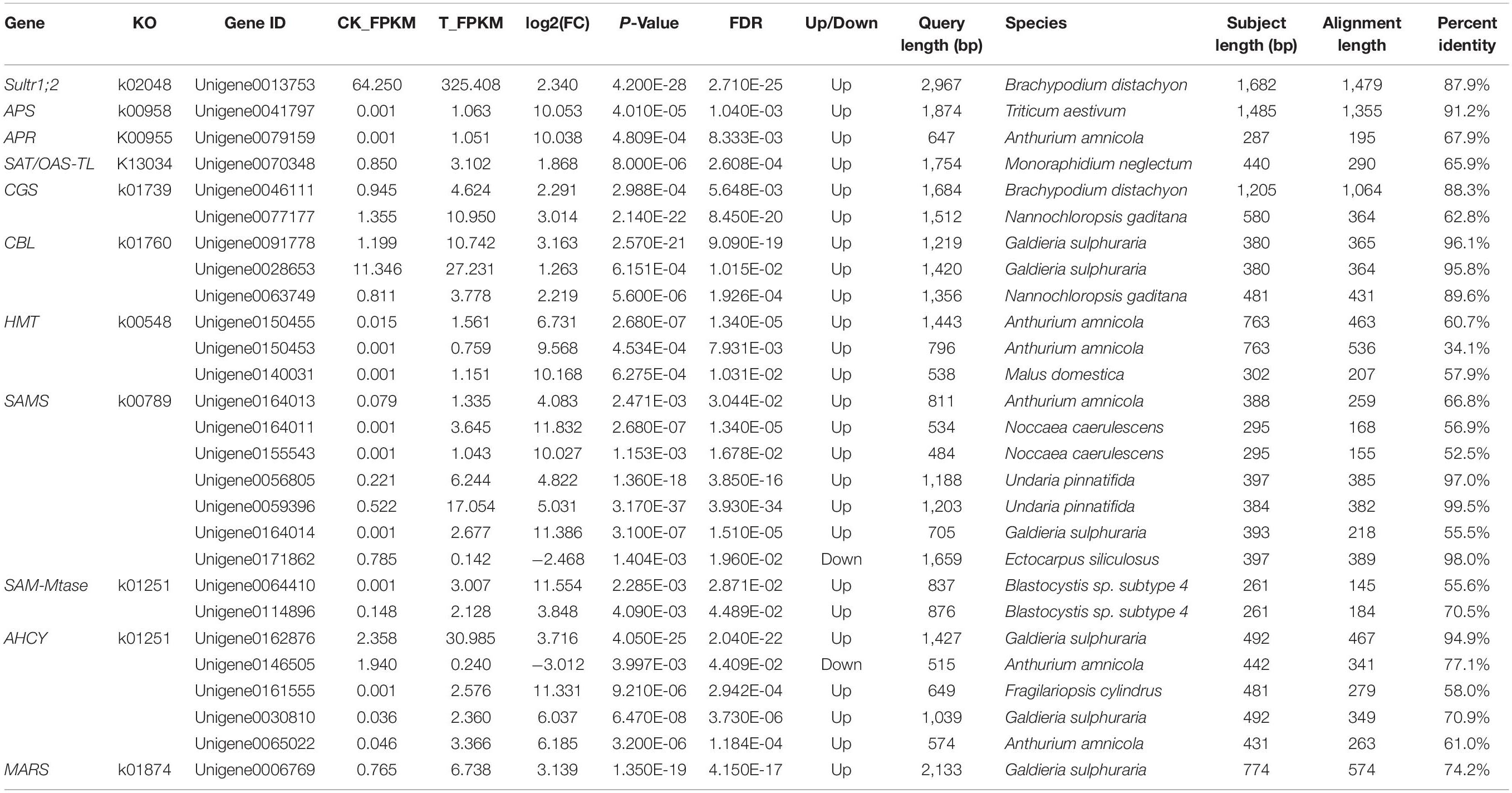
Table 2. Differential expression analysis of genes related to selenate metabolism in oats.
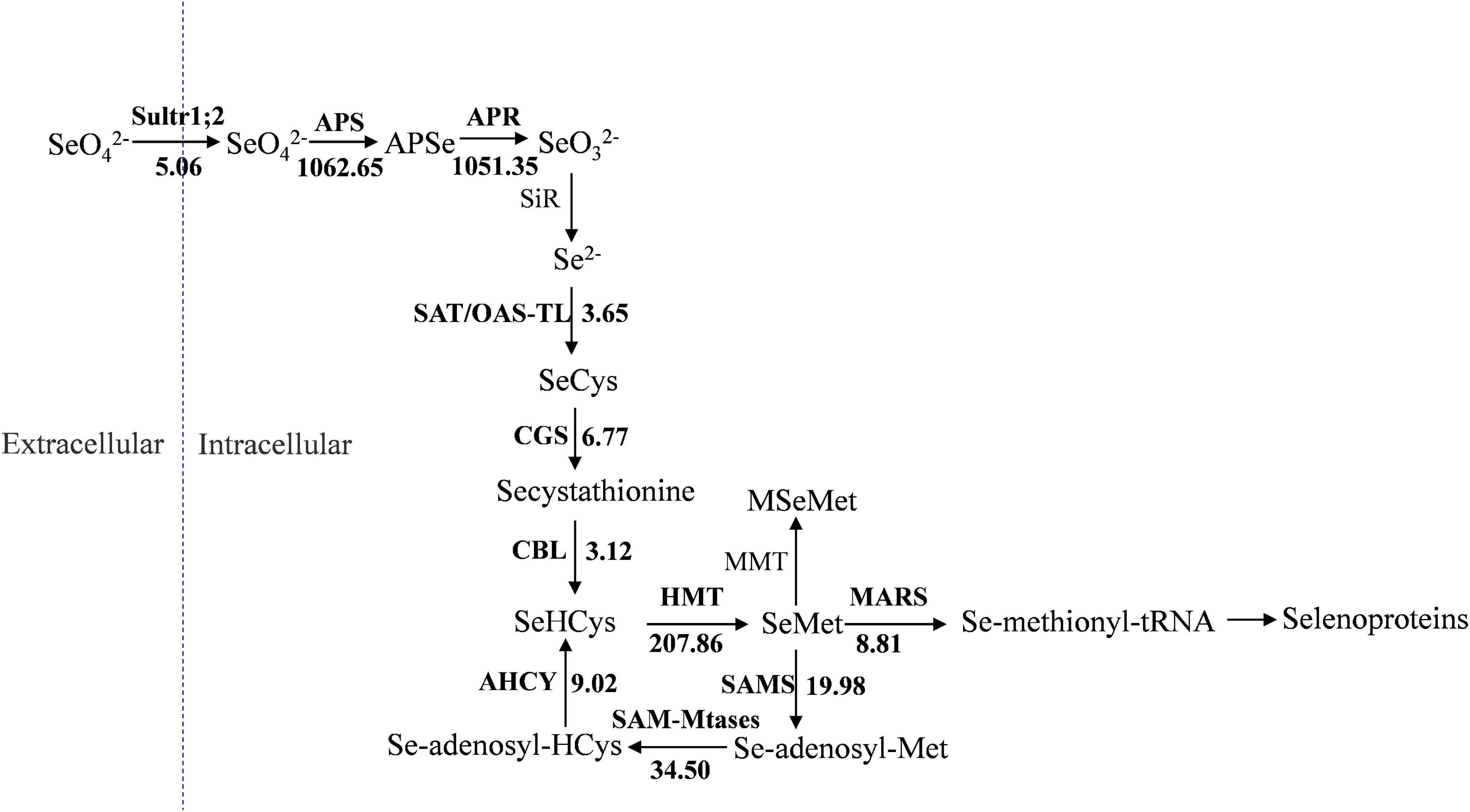
Figure 7. Presumptive expression model of structural genes involved in selenium metabolism in oats. The arrows show the metabolic stream, the short names on the left or above of arrows represent the genes catalyzing the reaction, and the number represents the fold increases of the gene expression after selenium treatment.
SeCys is the first molecule for synthesizing selenium-harboring metabolites. Selenomethionine (SeMet) biosynthesis is performed in plants by the sequential action of three enzymes: CGS, CBL, and HMT (Van Huysen et al., 2003; Yang et al., 2004). CGS and CBL, as the rate-limiting enzymes, play crucial roles in the reaction, and their genes are expressed 6.77-fold and 3.12-fold more in the T group than in the CK group, respectively. Three unigenes were found to be homologous to HMT, and they had a significant change in expression in oats after Se treatment. Unlike Se-enriched plant species such as onion, garlic, and broccoli, where Se-methylselenocysteine (SeMeSeCys) is a major Se compound, SeMet is the predominant Se form in cereal crop species (Lyons et al., 2005; Zhu et al., 2009). Based on the selenocompound metabolic pathway, it can be concluded that there are multiple catalytic pathways using SeMet as a substrate. MMT (methionine S-methyltransferase) presented no significantly different expression between the CK group and the T group (Figure 7), indicating that oats might not synthesize selenium-methyl selenomethionine (MSeMet) under selenium treatment. By the activity of MARS, SeMet is catalyzed to produce selenoproteins and subsequently participates in plant metabolic regulation.
Validation of DEGs by qRT-PCR
To validate the RNA-seq results, qRT-PCR was used to quantified analysis the expression of 11 DEGs, Sultrl1;2, APS, APR, SAT/OAS-TL, CGS, CBL, HMT, SAMS, SAM-Mtases, AHCY, MARS, which were related to selenium metabolism. As shown in Figure 8, the qRT-PCR results (log2fc) of these DEGs were basically consistent with those of RNA-seq data. The correlation coefficient (R2 = 0.8445) indicated that the DEGs expression pattern obtained by RNA-seq and qRT-qPCR results were in good concordance, which supports the reliability of the RNA-seq data.
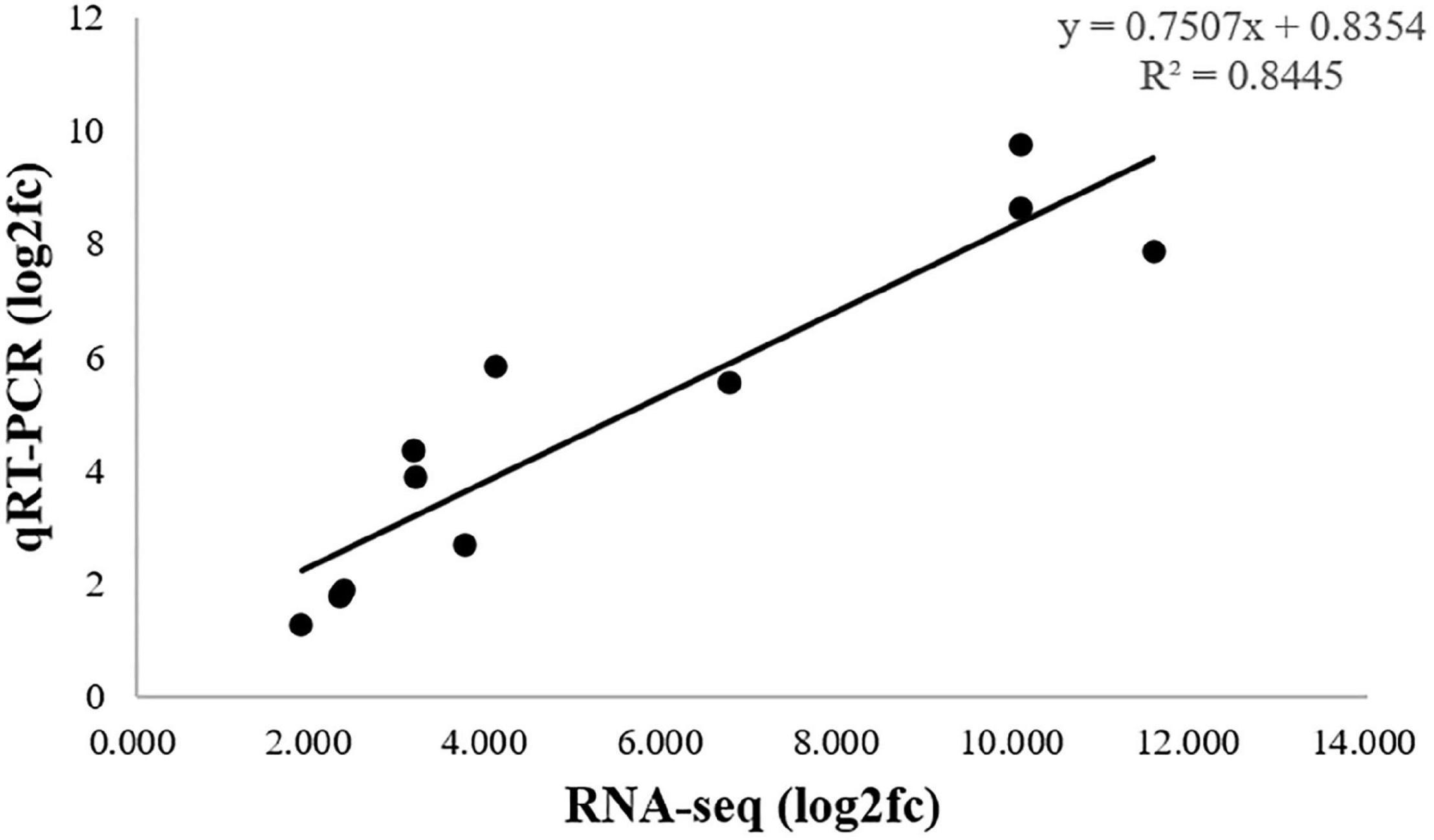
Figure 8. Comparison of expression patterns by RNA-seq and qRT-PCR. The R2-value is 0.8445.
Discussion
Oats are important grain and forage crops worldwide, and oats are grown on 14.1 million hectares, with a grain production that reached 28.3 million metric tons in 2005 (USDA, Foreign Agricultural Service, Commodity production, supply and disposition database). Oatmeal’s unique processing technique allows nutrient elements to be stored in numerous finished products. With more in-depth research on oats, scientists have found that the amino acid composition of oats is more suitable for human nutrition than that of other cereal species and have begun to highly recommend it for human consumption (Zimmer et al., 2019; Zhao et al., 2020). Se from oats is highly bioavailable and regarded as a good dietary source of Se (Yan and Johnson, 2011). Therefore, oats are likely to become one of the dominant food crops in the future. Elucidating the molecular basis and finding key enzymes involved in Se metabolism will be more significant for creating selenium-enriched oat cultivars. Next-generation sequencing technologies have become a powerful technology to illuminate new genes and their involved biochemical pathways in non-model plant species.
In this study, after stimulation with Na2SeO4 for 24 h, a total of 9,844 DEGs were detected in the CK and T groups after RNA-seq analysis. To elucidate the molecular mechanisms underlying salt tolerance in oat, Wu et al. (2017) utilized 100 mM NaCl to treat hull-less oats and performed a transcriptome analysis; many more (65,000) differentially expressed unigenes were identified. Their results showed that 100 mM NaCl could not only induce Na+ and Cl– metabolic pathway genes but also stimulate Na+ and Cl– stress-related genes. In our study, 20 μM Na2SeO4 was used to stimulate related gene expression, which is not a-inducing stress concentration for oats. The differentially expressed unigenes in the present work are mainly focused on selenium uptake, assimilation, transportation, and accumulation. Among them, 7226 DEGs were upregulated in the treatment group, and another 2618 DEGs were downregulated. The absolute log2(fold change) values ranged from 1 to 14.70. With the help of functional annotation and KEGG pathway analysis, 27 unigenes related to selenium metabolism were identified, and expression models of related structural genes in oats in response to selenate metabolism were constructed. From the expression model, it could be inferred that MSeMet would not be synthesized and that dimethylselenide (DMSe) would not be synthesized, either (Figure 7); DMSe is one of the major volatile forms of Se produced by plants (Tagmount et al., 2002), and these results are not consistent with those of previous research (Wu et al., 2019). Selenate will be completely converted into selenoproteins after being assimilated by oats and will not form gaseous selenium for release into the environment. Numerous long-term studies have shown that supplemental Se is associated with significant reductions in lung, colorectal and prostate cancers (Ellis and Salt, 2003). Kryukov et al. (2003) found that the human selenoproteome is composed of 25 selenoproteins. They suggested that selenium plays a crucial role in fighting cancer, improving immunity, and maintaining male reproduction because selenoproteins are responsible for biomedical effects in the body.
Because of the similarity of the chemical properties of selenium and sulfur, the absorption of inorganic selenium by plant roots via high-affinity sulfur transporters has been demonstrated by a large number of documents (Sors et al., 2005; Luo et al., 2019). Shinmachi et al. (2010) found that Sultr1;1 and Sultr4;1 transporters in wheat roots played crucial roles in selenate acquisition by S starvation treatment (Shinmachi et al., 2010). Luo et al. (2019) found that TaSultr1;1, TaSultr1;3, and TaSultr2;1 gene expression is significantly upregulated after supplying selenide to wheat roots (Luo et al., 2019). In the present study, the expression of sultrl1;2 was upregulated 5.06-fold under selenate treatment compared with that in the CK group, and the other high-affinity sulfur transporters in oats exhibited no obvious differences. We concluded that sultrl1;2 played a key role in selenate uptake in oats.
In this research, we utilized the transcriptome to analyze the genetic changes in oats under selenium treatment. The expression pattern of selenium-related structural genes in oats was constructed through KEGG pathway analysis. We speculate that volatile selenium would not be generated in selenium metabolism in oats. To the best of our knowledge, this is the first study to identify the differentially expressed genes related to oat selenium metabolism through combined sequencing in oats. These findings will enrich the understanding of selenium metabolism and lay a foundation for the subsequent identification of key candidate genes for breeding selenium-enriched oat cultivars.
Data Availability Statement
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author/s.
Author Contributions
HZ and BZ designed the study. TL conducted the experiments and analyzed the data and drafted the manuscript. XL, RZ, HC, and BZ critically reviewed and improved the manuscript. All authors have read and approved the final version of the manuscript.
Funding
This research was funded by the Project from Qinghai Province (No. 2020-HZ-807), the Strategic Priority Research Program of the Chinese Academy of Sciences (No. XDA26030202), and joint Research Project of Three-River- Resource National Park funded by Chinese Academy of Sciences and Qinghai Provincial People’s Government (No. LHZX-2020-08).
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer ZL declared a shared affiliation, with no collaboration, with the authors to the handling editor at the time of the review.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.625520/full#supplementary-material
Footnotes
References
Akbudak, M. A., and Filiz, E. (2019). Genome-wide analyses of ATP sulfurylase (ATPS) genes in higher plants and expression profiles in sorghum (Sorghum bicolor) under cadmium and salinity stresses. Genomics 111, 579–589. doi: 10.1016/j.ygeno.2018.03.013
Altschul, S. F., Madden, T. L., Schäffer, A. A., Zhang, J. H., Zhang, Z., Miller, W., et al. (1997). Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. doi: 10.1093/nar/25.17.3389
Bairoch, A., and Boeckmann, B. (1992). The SWISS-PROT protein sequence data bank. Nucleic Acids Res. 19, 2247–2249. doi: 10.1093/nar/19.suppl.2247
Bekele, W. A., Wight, C. P., Chao, S., Howarth, C. J., and Tinker, N. A. (2018). Haplotype-based genotyping-by-sequencing in oat genome research. Plant Biotechnol. J. 16, 1452–1463. doi: 10.1111/pbi.12888
Broadley, M. R., White, P. J., Bryson, R. J., Meacham, M. C., Bowen, H. C., Johnson, S. E., et al. (2007). Biofortification of UK food crops with selenium. Proc. Nutr. Soc. 65, 169–181. doi: 10.1079/pns2006490
Chen, F. F., Chien, C. Y., Cho, C. C., Chang, Y. Y., and Hsu, C. H. (2019). C-terminal redox domain of Arabidopsis APR1 is a non-canonical thioredoxin domain with glutaredoxin function. Antioxidants (Basel) 8:461. doi: 10.3390/antiox8100461
Cheng, H., Ma, X. Q., Jia, S. G., Li, M. L., and Mao, P. S. (2020). Transcriptomic analysis reveals the changes of energy production and asa-gsh cycle in oat embryos during seed ageing. Plant Physiol. Bioch. 153, 40–52. doi: 10.1016/j.plaphy.2020.03.054
Conesa, A., Götz, S., García-Gómez, J. M., Terol, J., and Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Dinh, Q. T., Cui, Z., Huang, J., Tran, T. A. T., Wang, D., Yang, W., et al. (2018). Selenium distribution in the Chinese environment and its relationship with human health: a review. Environ. Int. 112, 294–309. doi: 10.1016/j.envint.2017.12.035
El Kassis, E., Cathala, N., Rouached, H., Fourcroy, P., Berthomieu, P., Terry, N., et al. (2007). Characterization of a selenate-resistant Arabidopsis mutant. Root growth as a potential target for selenate toxicity. Plant Physiol. 143, 1231–1241. doi: 10.1104/pp.106.091462
Ellis, D. R., and Salt, D. E. (2003). Plants, selenium and human health. Curr. Opin. Plant Biol. 6, 273–279. doi: 10.1016/s1369-5266(03)00030-x
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., Amit, I., et al. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29, 644–652. doi: 10.1038/nbt.1883
Harun-Ur-Rashid, M., Oogai, S., Parveen, S., Inafuku, M., Iwasaki, H., Fukuta, M., et al. (2019). Molecular cloning of putative chloroplastic cysteine synthase in Leucaena leucocephala. J. Plant Res. 133, 95–108. doi: 10.1007/s10265-019-01158-y
Hatfield, D. L., and Gladyshev, V. N. (2002). How selenium has altered our understanding of the genetic code. Mol. Cell. Biol. 22, 3565–3576. doi: 10.1128/mcb.22.11.3565-3576.2002
Hussain, A., Larsson, H., Kuktaite, R., and Johansson, E. (2010). Mineral composition of organically grown wheat genotypes: contribution to daily minerals intake. Int. J. Environ. Res. Public Health 7, 3442–3456. doi: 10.3390/ijerph7093442
Joy, E. J., Broadley, M. R., Young, S. D., Black, C. R., Chilimba, A. D., Ander, E. L., et al. (2015). Soil type influences crop mineral composition in Malawi. Sci. Total. Environ. 505, 587–595. doi: 10.1016/j.scitotenv.2014.10.038
Kanehisa, M., Araki, M., Goto, S., Hattori, M., Hirakawa, M., Itoh, M., et al. (2008). KEGG for linking genomes to life and the environment. Nucleic Acids Res. 36, D480–D484. doi: 10.1093/nar/gkm882
Kryukov, G. V., Castellano, S., Novoselov, S. V., Lobanov, A. V., Zehtab, O., Guigo, R., et al. (2003). Characterization of mammalian selenoproteomes. Science 300, 1439–1443. doi: 10.1126/science.1083516
Kumar, P., Yadava, R., Gollen, B., Verma, S. K. R., and Yadav, S. (2011). Nutritional contents and medicinal properties of wheat: a review. Life Sci. Med. Res. 22, 1–10.
Kushwaha, S. K., Grimberg, Å, Carlsson, A. S., and Hofvander, P. (2019). Charting oat (Avena sativa) embryo and endosperm transcription factor expression reveals differential expression of potential importance for seed development. Mol. Genet. Genomics 294, 1183–1197. doi: 10.1007/s00438-019-01571-x
Law, C. W., Alhamdoosh, M., Su, S., Dong, X. Y., and Ritchie, M. E. (2018). RNA-seq analysis is easy as 1-2-3 with limma, Glimma and edgeR. F1000Research 5:1408. doi: 10.12688/f1000research.9005.3
Li, R. Q., Yu, C., Li, Y. R., Lam, T., and Wang, J. (2009). SOAP2: an improved ultrafast tool for short read alignment. Bioinformatics 25, 1966–1967. doi: 10.1093/bioinformatics/btp336
Liu, C., Wang, J. M., Wei, T. Z., Wei, M. Q., Yang, C. J., and Xiong, H. Y. (2014). Selenum enrichment capacities of six major vegetables in selenium-rich region of Ledu, Qinghai Province. J. Northwest A F Univ. 42, 157–161.
Liu, Y., Zhang, Z., Fu, J., Wang, G., Wang, J., and Liu, Y. (2017). Transcriptome analysis of maize immature embryos reveals the roles of cysteine in improving agrobacterium infection efficiency. Front. Plant Sci. 8:1778. doi: 10.3389/fpls.2017.01778
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Luo, W., Li, J., Ma, X., Niu, H., Hou, S., and Wu, F. (2019). Effect of arbuscular mycorrhizal fungi on uptake of selenate, selenite, and selenomethionine by roots of winter wheat. Plant Soil 438, 71–83. doi: 10.1007/s11104-019-04001-4
Lyons, G. H., Genc, Y., Stangoulis, J. C., Palmer, L. T., and Graham, R. D. (2005). Selenium distribution in wheat grain, and the effect of postharvest processing on wheat selenium content. Biol. Trace Elem. Res. 103, 155–168. doi: 10.1385/bter:103:2:155
Mao, X., Cai, T., Olyarchuk, J. G., and Wei, L. (2005). Automated genome annotation and pathway identification using the KEGG Orthology (KO) as a controlled vocabulary. Bioinformatics 21, 3787–3793. doi: 10.1093/bioinformatics/bti430
Margulies, M., Egholm, M., Altman, W. E., Attiya, S., Bader, J. S., and Rothberg, J. M. (2005). Genome sequencing in microfabricated high-density picolitre reactors. Nature 43, 376–380. doi: 10.1038/nature03959
Mortazavi, A., Williams, B. A., McCue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628. doi: 10.1038/nmeth.1226
Mostofa, M. G., Rahman, M. M., Siddiqui, M. N., Fujita, M., and Tran, L. P. (2020). Salicylic acid antagonizes selenium phytotoxicity in rice: selenium homeostasis, oxidative stress metabolism and methylglyoxal detoxification. J. Hazard Mater. 394:122572. doi: 10.1016/j.jhazmat.2020.122572
Pruitt, K. D., Tatusova, T., and Maglott, D. R. (2007). NCBI reference sequences (RefSeq): a curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 35, D61–D65. doi: 10.1093/nar/gkl842
Rayman, M. P. (2000). The importance of selenium to human health. Lancet 356, 233–241. doi: 10.1016/s0140-6736(00)02490-9
Rayman, M. P. (2012). Selenium and human health. Lancet 379, 1256–1268. doi: 10.1016/s0140-6736(11)61452-9
Sadiq Butt, M., Tahir-Nadeem, M., Khan, M. K., Shabir, R., and Butt, M. S. (2008). Oat: unique among the cereals. Eur. J. Nutr. 47, 68–79. doi: 10.1007/s00394-008-0698-7
Shinmachi, F., Buchner, P., Stroud, J. L., Parmar, S., Zhao, F. J., McGrath, S. P., et al. (2010). Influence of sulfur deficiency on the expression of specific sulfate transporters and the distribution of sulfur, selenium, and molybdenum in wheat. Plant Physiol. 153, 327–336. doi: 10.1104/pp.110.153759
Singh, M., and Kumar, S. (2016). Broadening the Genetic Base of Grain Cereals. New Delhi: Springer Nature.
Song, K., Kim, H. C., Shin, S., Kim, K. H., Moon, J. C., Kim, J. Y., et al. (2017). Transcriptome analysis of flowering time genes under drought stress in maize leaves. Front. Plant Sci. 8:267. doi: 10.3389/fpls.2017.00267
Sors, T. G., Ellis, D. R., and Salt, D. E. (2005). Selenium uptake, translocation, assimilation and metabolic fate in plants. Photosynth Res. 86, 373–389. doi: 10.1007/s11120-005-5222-9
Stevens, E. J., Armstrong, K. W., Bezar, H. J., Griffin, W. B., and Hampton, J. G. (2004). Fodder Oats: an Overview. Rome: Food and Agriculture Organization of the United Nations.
Tagmount, A., Berken, A., and Terry, N. (2002). An essential role of s-adenosyl-L-methionine:L-methionine s-methyltransferase in selenium volatilization by plants. Methylation of selenomethionine to selenium-methyl-L-selenium- methionine, the precursor of volatile selenium. Plant Physiol. 130, 847–856. doi: 10.1104/pp.001693
Tatusov, R. L., Fedorova, N. D., Jackson, J. D., Jacobs, A. R., Kiryutin, B., Koonin, E. V., et al. (2003). The COG database: an updated version includes eukaryotes. BMC Bioinformatics 4:41. doi: 10.1186/1471-2105-4-41
Van Huysen, T., Abdel-Ghany, S., Hale, K. L., LeDuc, D., Terry, N., and Pilon-Smits, E. A. (2003). Overexpression of cystathionine-gamma-synthase enhances selenium volatilization in Brassica juncea. Planta 218, 71–78. doi: 10.1007/s00425-003-1070-z
White, P. J. (2016). Selenium accumulation by plants. Ann. Bot. 117, 217–235. doi: 10.1093/aob/mcv180
White, P. J., and Broadley, M. R. (2009). Biofortification of crops with seven mineral elements often lacking in human diets–iron, zinc, copper, calcium, magnesium, selenium and iodine. New Phytol. 182, 49–84. doi: 10.1111/j.1469-8137.2008.02738.x
Winkel, L. H., Johnson, C. A., Lenz, M., Grundl, T., Leupin, O. X., Amini, M., et al. (2012). Environmental selenium research: from microscopic processes to global understanding. Environ. Sci. Technol. 46, 571–579. doi: 10.1021/es203434d
Wu, B., Hu, Y., Huo, P., Zhang, Q., Chen, X., and Zhang, Z. (2017). Transcriptome analysis of hexaploid hulless oat in response to salinity stress. PLoS One 12:e0171451. doi: 10.1371/journal.pone.0171451
Wu, L., Liu, T., Xu, Y., Chen, W., Liu, B., Zhang, L., et al. (2019). Comparative transcriptome analysis of two selenium-accumulating genotypes of Aegilops tauschii Coss. in response to selenium. BMC Genet. 20:9. doi: 10.1186/s12863-018-0700-1
Xu, Z. C., Shao, H. F., Li, S., and Zheng, C. (2012). Relationships between the selenium content in flue-cured tobacco leaves and the selenium content in soil in Enshi, China tobacco-growing area. Pak. J. Bot. 44, 1563–1568.
Yan, L., and Johnson, L. K. (2011). Selenium bioavailability from naturally produced high-selenium peas and oats in selenium-deficient rats. J. Agric. Food Chem. 59, 6305–6311. doi: 10.1021/jf201053s
Yang, G., Cao, K., Wu, L., and Wang, R. (2004). Cystathionine gamma-lyase overexpression inhibits cell proliferation via a H2S-dependent modulation of ERK1/2 phosphorylation and p21Cip/WAK-1. J. Biol. Chem. 279, 49199–49205. doi: 10.1074/jbc.M408997200
Zhang, L., Ma, M., Cui, L., and Liu, L. L. (2021). Deciphering the dynamic gene expression patterns of pollen abortion in a male sterile line of Avena sativa through transcriptome analysis at different developmental stages. BMC Plant Biol. 21:101. doi: 10.1186/s12870-021-02881-2
Zhang, Y., Zhang, R., Sun, X. F., Gao, X. S., and Wu, L. J. (2012). Reflection on exploitation and utilization of selenium resources in Qinghai Province. J. Anhui Agri. Sci. 40, 942–943. doi: 10.13989/j.cnki.0517-6611.2012.02.027
Zhao, J., Kebede, A. Z., Bekele, W. A., Menzies, J. G., Chong, J., Mitchell Fetch, J. W., et al. (2020). Mapping of the oat crown rust resistance gene Pc39 relative to single nucleotide polymorphism markers. Plant Dis. 104, 1507–1513. doi: 10.1094/PDIS-09-19-2002-RE
Zheng, G., Qin, Y., Clark, W., Dai, Q., and Pan, T. (2015). Efficient and quantitative high-throughput tRNA sequencing. Nat. Methods 12, 835–837. doi: 10.1038/nmeth.3478
Zhu, Y. G., Pilon-Smits, E. A., Zhao, F. J., Williams, P. N., and Meharg, A. A. (2009). Selenium in higher plants: understanding mechanisms for biofortification and phytoremediation. Trends Plant Sci. 14, 436–442. doi: 10.1016/j.tplants.2009.06.006
Keywords: selenium, oats, transcriptome analysis, selenoproteins, selenium metabolism
Citation: Liu T, Liu X, Zhou R, Chen H, Zhang H and Zhang B (2021) De novo Transcriptome Assembly and Comparative Analysis Highlight the Primary Mechanism Regulating the Response to Selenium Stimuli in Oats (Avena sativa L.). Front. Plant Sci. 12:625520. doi: 10.3389/fpls.2021.625520
Received: 19 November 2020; Accepted: 27 May 2021;
Published: 18 June 2021.
Edited by:
Dianella G. Howarth, St. John’s University, United StatesReviewed by:
Zhiyong Liu, Institute of Genetics and Developmental Biology, Chinese Academy of Sciences, ChinaHaixiao Hu, University of California, Davis, United States
Copyright © 2021 Liu, Liu, Zhou, Chen, Zhang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Huaigang Zhang, aGd6aGFuZ0Bud2lwYi5jYXMuY24=; Bo Zhang, emhhbmdib0Bud2lwYi5jYXMuY24=