 Alejandro Calle
Alejandro Calle Jérôme Grimplet
Jérôme Grimplet Loïck Le Dantec
Loïck Le Dantec Ana Wünsch
Ana Wünsch- 1Unidad de Hortofruticultura, Centro de Investigación y Tecnología Agroalimentaria de Aragón, Zaragoza, Spain
- 2Instituto Agroalimentario de Aragón-IA2, CITA-Universidad de Zaragoza, Zaragoza, Spain
- 3Univ. Bordeaux, INRAE, Biologie du Fruit et Pathologie, UMR 1332, Villenave d'Ornon, France
Dormancy release and bloom time of sweet cherry cultivars depend on the environment and the genotype. The knowledge of these traits is essential for cultivar adaptation to different growing areas, and to ensure fruit set in the current climate change scenario. In this work, the major sweet cherry bloom time QTL qP-BT1.1m (327 Kbs; Chromosome 1) was scanned for candidate genes in the Regina cv genome. Six MADS-box genes (PavDAMs), orthologs to peach and Japanese apricot DAMs, were identified as candidate genes for bloom time regulation. The complete curated genomic structure annotation of these genes is reported. To characterize PavDAMs intra-specific variation, genome sequences of cultivars with contrasting chilling requirements and bloom times (N = 13), were then mapped to the ‘Regina’ genome. A high protein sequence conservation (98.8–100%) was observed. A higher amino acid variability and several structural mutations were identified in the low-chilling and extra-early blooming cv Cristobalina. Specifically, a large deletion (694 bp) upstream of PavDAM1, and various INDELs and SNPs in contiguous PavDAM4 and -5 UTRs were identified. PavDAM1 upstream deletion in ‘Cristobalina’ revealed the absence of several cis-acting motifs, potentially involved in PavDAMs expression. Also, due to this deletion, a non-coding gene expressed in late-blooming ‘Regina’ seems truncated in ‘Cristobalina’. Additionally, PavDAM4 and -5 UTRs mutations revealed different splicing variants between ‘Regina’ and ‘Cristobalina’ PavDAM5. The results indicate that the regulation of PavDAMs expression and post-transcriptional regulation in ‘Cristobalina’ may be altered due to structural mutations in regulatory regions. Previous transcriptomic studies show differential expression of PavDAM genes during dormancy in this cultivar. The results indicate that ‘Cristobalina’ show significant amino acid differences, and structural mutations in PavDAMs, that correlate with low-chilling and early blooming, but the direct implication of these mutations remains to be determined. To complete the work, PCR markers designed for the detection of ‘Cristobalina’ structural mutations in PavDAMs, were validated in an F2 population and a set of cultivars. These PCR markers are useful for marker-assisted selection of early blooming seedlings, and probably low-chilling, from ‘Cristobalina’, which is a unique breeding source for these traits.
Introduction
Adequate blooming and pollination are essential for fruit set in sweet cherry (Prunus avium L.) and other fruit tree species. Temperate climate fruit trees such as sweet cherry go through a dormancy period in which meristem growth is inactive (Lang et al., 1987; Rohde and Bhalerao, 2007). This occurs before the blooming season to prevent winter damage due to frost and low temperatures. Dormancy is divided into three stages: paradormancy and endodormancy, in which bud growth is inhibited during autumn and winter seasons, and ecodormancy, in which bud growth is resumed under more favorable climatic conditions in late winter and early spring (Lang et al., 1987). The length of the dormant period depends on the environmental temperatures since determined amounts of chill and heat (Chilling and Heat requirements) are needed to complete endodormancy and ecodormancy before bud burst (Cooke et al., 2012). These requirements are specific to each genotype and vary according to the environmental conditions (Alburquerque et al., 2008). Both chilling and heat requirements influence blooming, however, several studies in Prunus species have reported that chilling requirement is the major determinant of bloom time (Alburquerque et al., 2008; Fan et al., 2010; Campoy et al., 2011; Castède et al., 2014).
Dormancy release, chilling requirement, and bloom time are relevant traits for cultivar adaptation to the growing area and to ensure an adequate fruit set. As many cherry cultivars are self-incompatible, blooming has to be synchronized between cultivars planted in the same vicinity. Late season blooming allows for the avoidance of spring frosts in cold regions. Cultivars with low chilling requirements are useful to adapt to temperature rise in the actual context of climate change. Additionally, cultivars with low chilling requirements can be used to extend cultivation to warmer areas, thus extending cultivation further away from traditional cultivation regions. Several works have investigated the physiology and the genetics of these traits in sweet cherry and other fruit tree species (reviewed in Abbott et al., 2015; and Fadón and Rodrigo, 2018). In sweet cherry, genetic analyses have revealed that bloom time is a quantitative trait with very high heritability (Dirlewanger et al., 2012; Castède et al., 2014; Calle et al., 2020). In this species, major quantitative trait loci (QTLs) associated with bloom time have been identified on linkage groups (LGs) 1, 2, and 4 (Dirlewanger et al., 2012; Castède et al., 2014; Calle et al., 2020). In other Prunus species, like almond (Prunus amygdalus L.), peach [Prunus persica (L). Batsch] and Japanese apricot (Prunus mume L.), main bloom time QTLs have also been mapped on the orthologous regions of LG1 (Fan et al., 2010; Zhebentyayeva et al., 2014; Bielenberg et al., 2015) and LG4 (Dirlewanger et al., 2012; Sánchez-Pérez et al., 2012; Kitamura et al., 2018). In the same region of LG1, stable and significant QTLs associated with chilling requirements in almond, peach, and sweet cherry have also been detected (Fan et al., 2010; Sánchez-Pérez et al., 2012; Castède et al., 2014; Bielenberg et al., 2015). This LG1 QTL region overlaps with a deletion in the evergrowing (EVG) peach mutant, which does not enter dormancy (Rodriguez et al., 1994). A tandem repeat of six MADS-box genes, named dormancy-associated MADS-box (DAM), was identified in this LG1 region, four of them being deleted in the EVG mutant (Bielenberg et al., 2008), reveling the potential involvement of these genes in dormancy control of Prunus species. In sweet cherry, DAM5 and -6 have also been mapped on LG1, overlapping with the main bloom time and chilling requirement QTL of this LG (Castède et al., 2015). In other Rosaceous species, like apple and pear, a variable number of DAM gene have also been reported (Saito et al., 2013; Mimida et al., 2015), some of them overlapping with regions in which bloom time QTLs for these species were found (Allard et al., 2016).
In different plant species, MADS-box transcription factors have been reported as strong candidate genes for the genetic control of blooming and temperature responses (Gramzow and Theissen, 2010). MADS-box genes play fundamental roles in pathways involved in the transition from vegetative to reproductive phases, growth, floral organ determination, and other processes related to root, leaf, fruit, and gametophyte development (Becker and Theißen, 2003; Messenguy and Dubois, 2003; Smaczniak et al., 2012). The DAM genes reported in sweet cherry, peach, Japanese apricot, and European plum (Prunus domestica L.) belong to MIKCc Type II of MADS-box genes and are phylogenetically related to Arabidopsis SHORT VEGETATIVE PHASE (SVP) and AGAMOUS-LIKE 24 (AGL24) genes, which have been reported as main floral regulators (Jiménez et al., 2009; Sasaki et al., 2011; Quesada-Traver et al., 2020; Wang et al., 2020). Analyses of DAM gene expression levels in Prunus species have shown a similar pattern in different years and correlations with photoperiod and temperature changes (Falavigna et al., 2019), suggesting that these genes are the main regulators of the dormancy cycle in Prunus species (Yamane, 2014). Maximum expression levels of DAM1 to -4 were observed during bud set, suggesting a role in the regulation of growth cessation and bud formation in peach and Japanese apricot (Li et al., 2009; Sasaki et al., 2011; Zhang et al., 2018). On the other side, DAM5 and -6 showed the highest expression level in the winter season during induction and maintenance of dormancy and minimal or absent expression during the budbreak and bloom time (Jiménez et al., 2010; Yamane et al., 2011; Leida et al., 2012; Prudencio et al., 2018). Therefore, down-regulation of DAM5 and -6 during the winter season, with minimum expression level when chilling requirements are fulfilled, is compatible with the role of dormancy release repressor of DAM genes in Prunus species (Sasaki et al., 2011). In sweet cherry, expression patterns of these genes have been reported. DAM1, -3, and -6 were highly expressed during paradormancy and at the beginning of endodormancy, whereas DAM4 and -5 showed an expression peak at the end of endodormancy and chilling requirement fulfillment (Vimont et al., 2019, 2020; Villar et al., 2020; Wang et al., 2020). In the low chilling requirement, sweet cherry cultivar ‘Cristobalina’, a low expression level of PavDAM1, PavDAM4, and PavDAM5 during the dormancy period was observed compared with the expression pattern of high chill-requirements cultivars for these genes (Vimont et al., 2019). Similarly, in other low chilling sweet cherry cultivar, ‘Royal Lee’, a low expression level of PavDAM1, especially during the chilling accumulation, compared with the expression pattern of high chill-requirement cultivar was reported (Wang et al., 2020). Epigenetic modification and the evolution of transcript levels during dormancy were evaluated for DAM3 and -5 in the sweet cherry cultivar ‘Bing’ (Rothkegel et al., 2017), revealing the involvement of siRNAs and DNA methylations in the silencing of DAM3 during chilling accumulation and dormancy release.
In Calle et al. (2020), bloom time in sweet cherry was evaluated during 4 years using a multi-family QTL approach. In this work plant materials included populations that descend from cultivars with very low to high chilling requirements. These populations derive from self- and cross-pollination of ‘Cristobalina’, a cultivar with very low chilling requirement (<500 h) and extra-early flowering and maturity dates (Tabuenca, 1983; Alburquerque et al., 2008; Calle and Wünsch, 2020; Calle et al., 2020). This cultivar is of breeding interest due to these traits and other relevant characters like self-compatibility (Wünsch and Hormaza, 2004; Ono et al., 2018). Bloom time QTL analysis for these plant materials revealed that the highest percentage of phenotypic variation was explained by QTLs on LGs 1 (qP-BT1.1m) and 2 (qP-BT2.1m). The QTL detected on LG1 overlaps with a chilling requirement QTL previously reported on Prunus LG1 (Fan et al., 2010; Sánchez-Pérez et al., 2012; Castède et al., 2014; Bielenberg et al., 2015), and with DAM genes mapped in this region in sweet cherry, Japanese apricot and peach (Bielenberg et al., 2008; Sasaki et al., 2011; Castède et al., 2015). Moreover, haplotype analyses of this QTL showed that ‘Cristobalina’ was the only cultivar with alleles contributing to early blooming (Calle et al., 2020). Since early blooming in this plant material is believed to be due to low chilling requirements in ‘Cristobalina’, candidate genes from these QTLs may be involved in chilling requirement control.
The objective of this work is to confirm and characterize DAM genes as candidate genes in sweet cherry major bloom time QTL on LG1 using the sweet cherry genome sequence recently available, to investigate the genomic structure of these genes in the species, and to uncover variation of these candidate genes in cultivars with contrasting chilling requirements and bloom times (including ‘Cristobalina’). Intraspecific variation of PavDAM genes has not been previously investigated in sweet cherry and this study may allow us to identify polymorphisms associated with the phenotypic variation in the plant material studied. This knowledge may be further used to develop markers for assisted selection of these traits from this plant material. Furthermore, these results may help to broaden the understanding of dormancy regulation in sweet cherry and other Prunus species by improving our knowledge of these candidate genes.
Materials and Methods
Plant Materials and Sequence Resources
Sequence Resources
Sweet cherry cultivar ‘Regina’ genome (Le Dantec et al., 2020) was used for QTL candidate gene mining and annotation. This genome was also employed in the rest of the experiments as a sweet cherry reference genome. For phylogenetic analysis, nucleotide sequences of peach DAM genes [PpeDAM1 (ABJ96361), PpeDAM2 (ABJ96363), PpeDAM3 (ABJ96364), PpeDAM4 (ABJ96358), PpeDAM5 (ABJ96359), and PpeDAM6 (ABJ96360)] and Japanese apricot DAM genes [PmuDAM1 (BAK78921), PmuDAM2 (BAK78922), PmuDAM3 (BAK78923), PmuDAM4 (BAK78924), PmuDAM5 (BAK78920), and PmuDAM6 (BAH22477)] were compared. For the study of intraspecific variation of PavDAMs, the genome sequences of 13 cultivars (Illumina HiSeq 2500 and 4000 systems; DDBJ; SRA bioproject ID PRJDB6734), previously generated by Ono et al. (2018), were downloaded and aligned to the reference genome.
Plant Materials
For PavDAM mutation characterization and marker validation, two sets of plant materials were used. One is a collection of sweet cherry cultivars (N = 72; Table 1), from “CITA de Aragón” cultivar and germplasm collection (Zaragoza, Spain). This sample includes landraces and bred cultivars from different genetic backgrounds and variable chilling requirements and bloom dates. Some of these cultivars were only utilized for marker validation, while others were also analyzed for PavDAM mutation characterization (see Table 1). The other set of plant materials is an F2 population (B×C2; N = 61) from the self-pollination of selection ‘BC8’ (‘Brooks’ × ‘Cristobalina’; Calle et al., 2020). This population was only used for marker validation. Genomic DNA from all plant materials evaluated for genetic analyses was extracted from young leaves using DNeasy Plant Mini kit (Qiagen, MD, USA). DNA quantity and quality were assayed using NanoDrop ND-1000 spectrophotometer (Thermo Scientific, DE, USA).
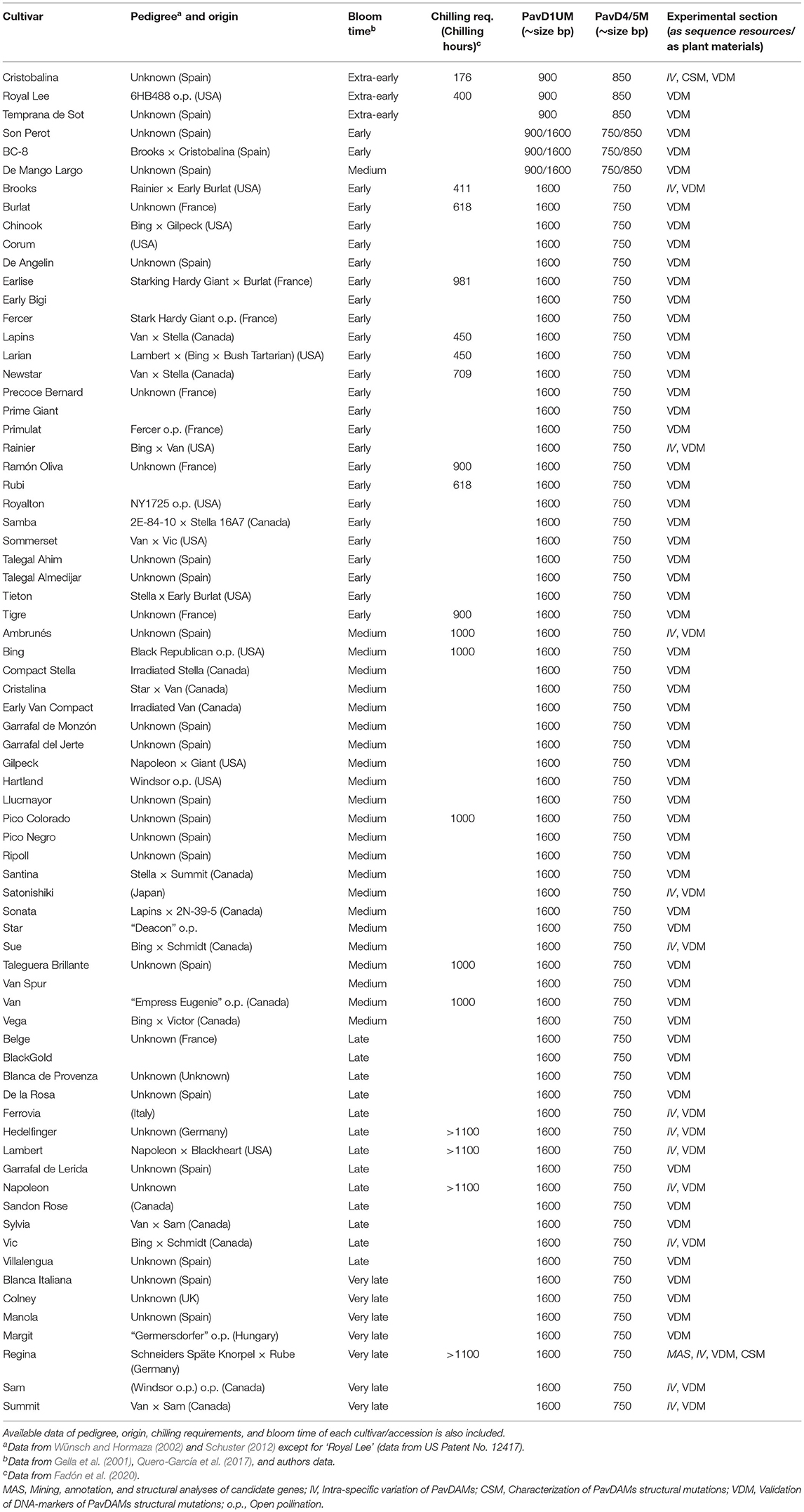
Table 1. PavD1UM and PavD4/5M (PavDAM1 and PavDAM4,-5 structural mutations, respectively) PCR marker genotypes of 72 sweet cherry cultivars and accessions.
Mining, Annotation, and Structural Analyses of Candidate Genes in Major Bloom Time QTL (qP-BT1.1m)
Coding DNA sequences of predicted genes in region Chr01_49296241:49622837 (326,596 bp) were extracted from ‘Regina’ sweet cherry genome. This region spans a previous main bloom time QTL in sweet cherry, qP-BT1.1m (Calle et al., 2020). The protein sequences of the predicted genes annotated in this region were blasted against the NCBI non-redundant protein sequences (nr) database using the BLASTP algorithm to obtain the corresponding gene ontologies. For each gene, we searched for bibliographic evidence (annotation and predicted function) that led to information associated with their potential involvement in bloom time and chilling requirement. Curation of the structural annotation was performed from the ‘Regina’ genome annotation using BLAST analysis, motif detection, and public ‘Regina’ RNAseq data from Vimont et al. (2019). The original gene nomenclature was conserved.
Sweet cherry DAM genes sequences and the GFF (General Feature Format) annotation file containing the exon-intron structure of these genes were retrieved from the ‘Regina’ genome database. These files were uploaded into the Integrative Genomics Viewers (IGV) software (Thorvaldsdóttir et al., 2013) to double-check structure with their ortholog genes in peach genome v2.0.a1 (Verde et al., 2017). Manual sequence editing was done to correct the automatic annotation if needed, conserving an adequate intron splicing prediction.
Phylogenetic Analysis of PavDAMs
Phylogenetic analysis of dormancy-associated MADS-box genes (DAM1 to 6) from peach (Bielenberg et al., 2008), Japanese apricot (Sasaki et al., 2011), and sweet cherry (this work) was conducted using MEGA X (Kumar et al., 2018). Multiple sequence alignment was carried out before tree construction using the MUSCLE algorithm (Edgar, 2004). The evolutionary history was inferred by using the Maximum Likelihood method and Tamura-Nei model (Tamura and Nei, 1993). Phylogenetic analysis was estimated using a bootstrap value of 1000, and the tree with the highest log likelihood was selected. Heuristic search for the initial tree was automatically obtained by using Neighbor Joining (NJ) and BioNJ algorithms to a matrix of pairwise distances estimated by the Maximum Composite Likelihood (MCL) approach, then the topology with superior log likelihood value was selected.
Intraspecific Variation of PavDAMs Sequences in Cultivars With Large Phenotypic Variation
Genome sequences of 13 sweet cherry cultivars (‘Ambrunés’, ‘Brooks’, ‘Cristobalina’, ‘Ferrovia’, ‘Hedelfingen’, ‘Lambert’, ‘Napoleon’, ‘Rainier’, ‘Sam’, ‘Satonishiki’, ‘Sue’, ‘Summit’, and ‘Vic’) were used for genome sequence alignment. Genomic DNA-seq libraries (Ono et al., 2018), were downloaded and aligned using the Galaxy software framework (Afgan et al., 2018). Raw sequence data were processed using the SLIDINGWINDOW operation from Trimmomatic v0.36.6 (Bolger et al., 2014) to remove adapter sequences and to obtain clean sequence data. A FASTQ file for each cultivar containing clean reads was then aligned to the ‘Regina’ genome. The whole-genome sequence was targeted for alignment using the Bowtie 2 tool (Langmead and Salzberg, 2012) with default parameters. The consensus sequence of each cultivar was extracted from Binary Alignment Map (BAM) file using Geneious 11.1.5 software (Biomatters Ltd, Auckland, NZ). A target region of 69,179 bp in the ‘Regina’ genome, spanning the PavDAM genes (1,500 bp upstream of PavDAM1 start codon to 1,500 bp downstream of PavDAM6 stop codon), was analyzed in all the cultivars. Visual inspection was carried out to search for putative structural mutations. The full-length amino acid sequence of the six DAM genes from the 13 aligned sweet cherry cultivars was deduced and compared. The comparison was carried out by multiple amino acid sequence alignment using the ClustalW algorithm implemented in Geneious 11.1.5 software (Biomatters Ltd, Auckland, NZ). The percentage of identity between DAM genes of each cultivar was calculated as the percentage of identical amino acids between each pair of cultivars.
Characterization of PavDAMs Structural Mutations in Low-Chilling and Early Blooming Cultivars
To confirm the presence of the putative structural mutations detected in ‘Cristobalina’ PavDAMs by in silico sequence comparison, primers flanking these regions were designed (PavD1UM: PavDAM1 Upstream Mutation; PavD4/5M: PavDAM4 and -5 mutation). These PCR primers were designed in conserved regions observed in multiple cultivar alignments of these genes. PCR analyses using primer combinations PavD1UMr-PavD1UMf and PavD4/5Mr-PavD4/5Mf (Supplementary Table 1) were initially carried out in 14 cultivars (Table 1). The cultivars analyzed were those for which genome sequences were available (‘Ambrunés’, ‘Brooks’, ‘Cristobalina’, ‘Ferrovia’, ‘Hedelfingen’, ‘Lambert’, ‘Napoleon’, ‘Rainier’, ‘Regina’, ‘Sam’, ‘Satonishiki’, ‘Sue’, ‘Summit’, and ‘Vic’). PCR analysis was carried out as described in Cachi and Wünsch (2014) using the following program: 4 min at 94°C; 35 cycles of 45 s at 94°C, 45 s 59°C, and 2 min at 72°C; and a final step of 7 min at 72°C. PCR products were analyzed by agarose gel electrophoresis in 1.7% TBE and stained with GelRed® Nucleic Acid Stain (Biotium, CA, USA).
To characterize the genomic mutations identified in ‘Cristobalina’ PavDAMs, Sanger sequencing of PCR products (D1Sf-D1Sr and PavD4/5Mr-PavD4/5Mf; Supplementary Table 1) was carried out using ‘Cristobalina’ and ‘Regina’ DNA. PCR reactions were performed as described above. PCR products were purified and sequenced by STAB VIDA (Lisbon, Portugal). Sequencing of PCR products of each cultivar was repeated at least twice with each forward and reverse primer. All sequences were trimmed to eliminate low-quality nucleotides, and sequences from each cultivar were aligned to construct the consensus sequence of each cultivar (‘Cristobalina’ and ‘Regina’). These consensus sequences were then aligned for comparison. All sequences visualizing, editing, and alignments, as well as primers design, were carried out using Geneious 11.1.5 (Biomatters Ltd, Auckland, NZ).
Plant cis-acting regulatory DNA elements were searched upstream of ‘Regina’ PavDAM1, in the region covering the large deletion in ‘Cristobalina’. This search was performed using the PLACE database (https://www.dna.affrc.go.jp/PLACE/?action=newplace; Higo et al., 1999). ‘Regina’ published genome sequence was used as the template. Additionally, to compare PavDAMs expression in ‘Cristobalina’ and ‘Regina’, RNAseq data from both cultivars (Vimont et al., 2019) were aligned to the ‘Regina’ genome sequence (upstream of PavDAM1, PavDAM4, and -5) and on the Sanger sequencing for both cultivars. This analysis was carried out using HISAT2 (Kim et al., 2012).
Validation of DNA-Markers of PavDAMs Structural Mutations (PavD1UM and PavD4/5M)
PavD1UM and PavD4/5M genotyping was carried out by PCR using primers PavD1UMf-PavD1UMr and PavD4/5Mf-PavD4/5Mr as described above (Supplementary Table 1). PavD1UM and PavD4/5M markers were validated in F2 population B×C2, which is expected to segregate for these markers because the parental genotype (‘BC8’) is heterozygous for both markers (Table 1). Description and mean bloom dates over 4 years (2015–2018) of B×C2 have been previously published (Calle et al., 2020). QTL haplotypes for major bloom time QTL on LG1 (qP-BT1.1m) of this population were also published in the same work. QTL haplotypes obtained then were compared with marker genotypes observed in this work. Deviation of marker segregation from expected Mendelian segregation in this population was evaluated by Chi-square goodness-of-fit (χ2). Statistical analysis was done using SPSS statistics v21.0.0 software (IMB, IL, USA) and R v3.4.1 (R Core Team, 2017). The two markers, PavD1UM and PavD4/5M, were also assayed in a diverse set of sweet cherry cultivars and accessions (Table 1).
Results
Mining, Annotation, and Structural Analyses of Candidate Genes in Major Bloom Time QTL (qP-BT1.1m)
Functional analysis in sweet cherry Chromosome 1 region (Chr1:49,296,241-49,622,837) of ‘Regina’ sweet cherry genome was carried out to identify candidate genes for bloom time and chilling requirement in sweet cherry. This genomic region spans major bloom time QTL qP-BT1.1m. In total, 47 predicted genes (Supplementary Table 2) were retrieved. Predicted amino acid sequences of seven of these genes (14.9%) resulted in BLAST hits in the NCBI gene database with uncharacterized proteins, while another six (12.8%) had no significant similarity with any other sequence (Supplementary Table 2). The rest of the predicted genes (34 genes; 72.3%), revealed hits with proteins involved in different pathways. Most relevant finding was eight contiguous genes, localized close to the QTL cofactor marker, which are sequentially annotated as PAV01_g0075081, PAV01_g0075091, PAV01_g0075101, PAV01_g0075111, PAV01_g0075121, PAV01_g0075131, PAV01_g0075141, and PAV01_g0075151 (Supplementary Table 2). Blastx, revealed these genes match MADS-box proteins, with percentages of similarity ranging from 86 to 100% (Supplementary Table 2). Due to their genetic similarity with type II SVP subclass of MADS-box proteins sequences, these eight sequences may correspond to DAM genes in sweet cherry (PavDAM), and they are therefore strong candidate genes for chilling requirement and bloom time regulation in this QTL region.
Sequence inspection of these eight candidate genes revealed flaws in the automatic annotation of the initial gene models when compared to peach gene models. Besides, the expected structure of MADS-box domains was not complete. Only two predicted proteins (PAV1_g0075081 and PAV1_g0075151) contained domains MADS (M), Intervening (I), Keratin-like (K), and C-terminal (C), which are characteristics of type II MADS-box genes (Figure 1A). In another predicted gene (PAV01_g0075091), exon 3 was not annotated, and in PAV01_g0075121, two additional exons before the M domain were present. Similarly, PAV01_g0075101 and PAV01_g0075111 were automatically annotated as two different MADS-box, although domain structure revealed that both sequences were two separated fragments of the same MADS-box protein. The same was observed for PAV01_g0075131 and PAV01_g0075141 sequences, which correspond to the same MADS-box gene but had been automatically annotated as two different gene sequences.
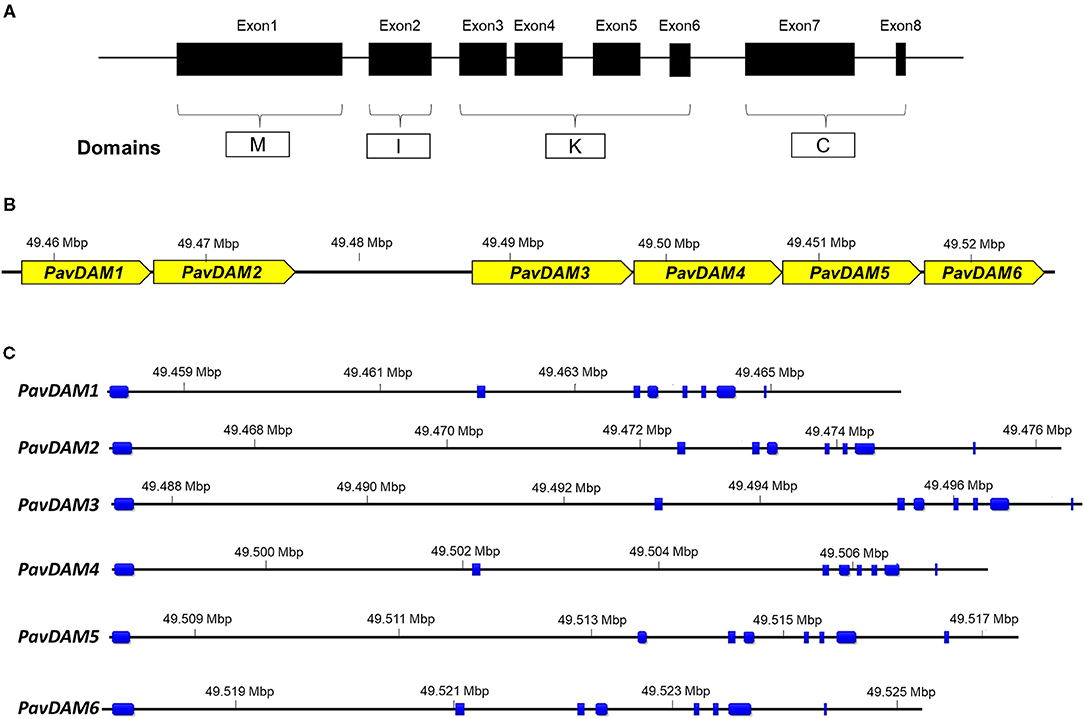
Figure 1. Characterization of sweet cherry PavDAM genes. (A) Schematic overview of the intron-exon structure of MADS-box genes and M, I, K, and C domains. (B) Diagram of size and position of PavDAM genes in chromosome 1 of the sweet cherry genome (Le Dantec et al., 2020). (C) Distribution of exons (blue boxes) and introns in the six PavDAM genes in ‘Regina’ sweet cherry genome.
The corrected annotation of the retrieved sequences revealed six MADS-box genes instead of the eight automatically predicted in the ‘Regina’ genome. The alignment of transcriptome data confirmed these coding structures and allowed an extensive curation of the UTR regions. Curated data can be accessed in Supplementary Table 3. Six DAM genes have also been previously reported in peach, Japanese apricot, and European plum in the syntenic genomic region. Thus, the six MADS-box sequences were identified as PAV1_g0075081, PAV1_g0075091, PAV1_g0075101, PAV1_g0075121, PAV1_g0075131, and PAV1_g0075151 in the ‘Regina’ genome, were named PavDAM1 to -6, respectively (Figure 1). These genes are tandemly located in the ‘Regina’ genome (Chr01_49457863:49524699 bp) with a larger gap (11,433 bp) between PavDAM2 and -3 (Figure 1B). Gene structure analysis of the six genes revealed an identical structure of eight exons and seven introns in each gene, as well as, the conserved M, I, K, and C domains (Figure 1C). Genomic gene length ranged from 7,672 (PavDAM6) to 10,438 bp (PavDAM3), whereas the predicted genes coding regions ranged from 667 (PavDAM4) to 730 (PavDAM5) bp. Variable sizes were observed in the six introns of each gene, while exon sizes were highly conserved (Figure 1C).
Phylogenetic Analysis of PavDAMs
A phylogenetic analysis of peach, Japanese apricot (Bielenberg et al., 2008; Sasaki et al., 2011), and sweet cherry (this work) DAM genes was carried out using the maximum likelihood of the gene coding sequences (Figure 2). DAM genes orthologs (DAM1 to DAM6) of the three species clustered together with a high bootstrap value (99; Figure 2). Within these sub-clades, in all cases, peach and Japanese apricot DAM genes were phylogenetically closer to each other than to sweet cherry DAM genes (Figure 2). Additionally, two major clades of DAM orthologs were observed, one includes DAM1, -2, and -3; and the other includes DAM4, -5, and -6, suggesting a common ancestor for each of them (Figure 2). Within these clades, DAM1 and -2 were closer to each other than to DAM3, and -4 and -6 were closer to each other than to DAM5.
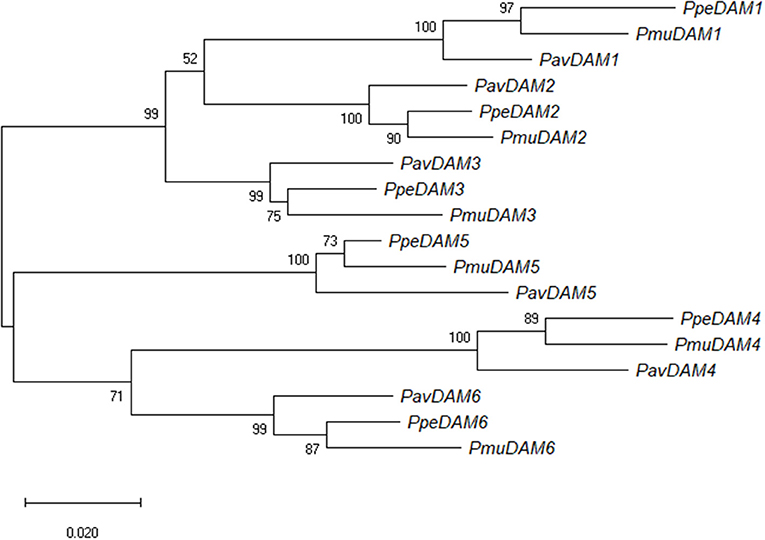
Figure 2. Maximum likelihood phylogenetic tree of nucleotide DAM sequences of sweet cherry (PavDAM1, PavDAM2, PavDAM3, PavDAM4, PavDAM5, and PavDAM6; Bielenberg et al., 2008) and its orthologs in Japanese apricot (PmuDAM1, PmuDAM2, PmuDAM3, PmuDAM4, PmuDAM5, and PmuDAM6; Sasaki et al., 2011) and peach (PpeDAM1, PpeDAM2, PpeDAM3, PpeDAM4, PpeDAM5, and PpeDAM6). The numbers at branch nodes indicate the percentage of bootstrap support at 1,000 replicates.
Intraspecific Variation of PavDAMs Sequences in Cultivars With Large Phenotypic Variation
The ‘Regina’ genome was used as a reference to map the genome sequence reads of 13 sweet cherry cultivars with variable chilling requirements and bloom times (Table 1). Of these cultivars, ‘Cristobalina’ shows extra early blooming while the rest show midseason to late flowering (Table 1). From this sequence mapping, the PavDAM genes consensus sequences of each cultivar were obtained. From these sequences, the complete amino acid sequence of each of the six PavDAM genes of each cultivar was predicted (Supplementary Figure 1). Comparison of PavDAM amino acid sequences amongst the different cultivars revealed a high degree of conservation (Supplementary Figure 1; Supplementary Tables 4, 5). The exon-intron structure was conserved in the six genes in all the cultivars. Also, the similarity between cultivars for the six PavDAM amino acid sequences was very high (98.8 to 100% identity; Supplementary Table 4). ‘Cristobalina’ was the cultivar with lower similarity to the rest (98.8–99.0%; Supplementary Table 4), while the remaining cultivars had higher similarities (99.7–100%). Complete amino acid identity (100% similarity) was observed for PavDAM sequences of ‘Ambrunés’ and ‘Summit’; ‘Vic’ and ‘Brooks’; and ‘Regina’, ‘Sam’, and ‘Sue’ (Supplementary Table 4).
Alignment of the PavDAM amino acid sequences of all the cultivars (Supplementary Figure 1) revealed 24 amino acid substitutions (Supplementary Figure 1; Supplementary Table 5). Of these, 20 were unique to specific cultivars, and the remaining four were common to various cultivars. ‘Cristobalina’ was the cultivar with the largest number of unique amino acid substitutions (14; Supplementary Figure 1; Supplementary Table 5). ‘Ferrovia’, ‘Lambert’, ‘Hedelfingen’, ‘Satonishiki’, and ‘Rainier’ showed 1–2 unique amino acid substitutions (Supplementary Table 5). PavDAM1 and PavDAM4 presented the largest number of polymorphisms (Supplementary Figure 1; Supplementary Table 5). Unique amino acid substitutions were found on all domains, with a large number found on domain C. Only ‘Cristobalina’ presented a substitution in the M domain (PavDAM2 and -5).
Visual inspection of cultivars sequence reads mapping to the ‘Regina’ genome revealed two genomic regions where no sequence reads from ‘Cristobalina’ were mapped. These regions are located upstream of PavDAM1, and between PavDAM4 and -5 coding regions, spanning ~700 and 400 bp respectively (Supplementary Figures 2, 3). These regions seemed to contain putative structural mutations in the ‘Cristobalina’ genome.
Characterization of PavDAMs Structural Mutations in Low-Chilling and Early Blooming Cultivars
To investigate putative mutations in ‘Cristobalina’ PavDAMs (Supplementary Figures 2, 3), PCR primers flanking these regions (PavD1UM and PavD4/5M) were designed. These markers were used to analyze 13 cultivars with sequences available and ‘Regina’ (Table 1). For the PavD1UM marker, a fragment of the same size as in ‘Regina’ (~1,600 bp) was amplified in all the sweet cherry cultivars, except in ‘Cristobalina’, in which a shorter fragment (~900 bp) was obtained (Figure 3). The amplification of a smaller fragment in ‘Cristobalina’ supports the presence of a putative deletion of ~700 bp upstream of ‘Cristobalina’ PavDAM1. For the PavD4/5M marker, a fragment of 850 bp was amplified only in ‘Cristobalina’, whereas the remaining cultivars, including ‘Regina’, presented a 750 bp fragment (Figure 3). This result supports the presence of a putative insertion in the ‘Cristobalina’ genome, found between PavDAM4 and -5 coding regions.
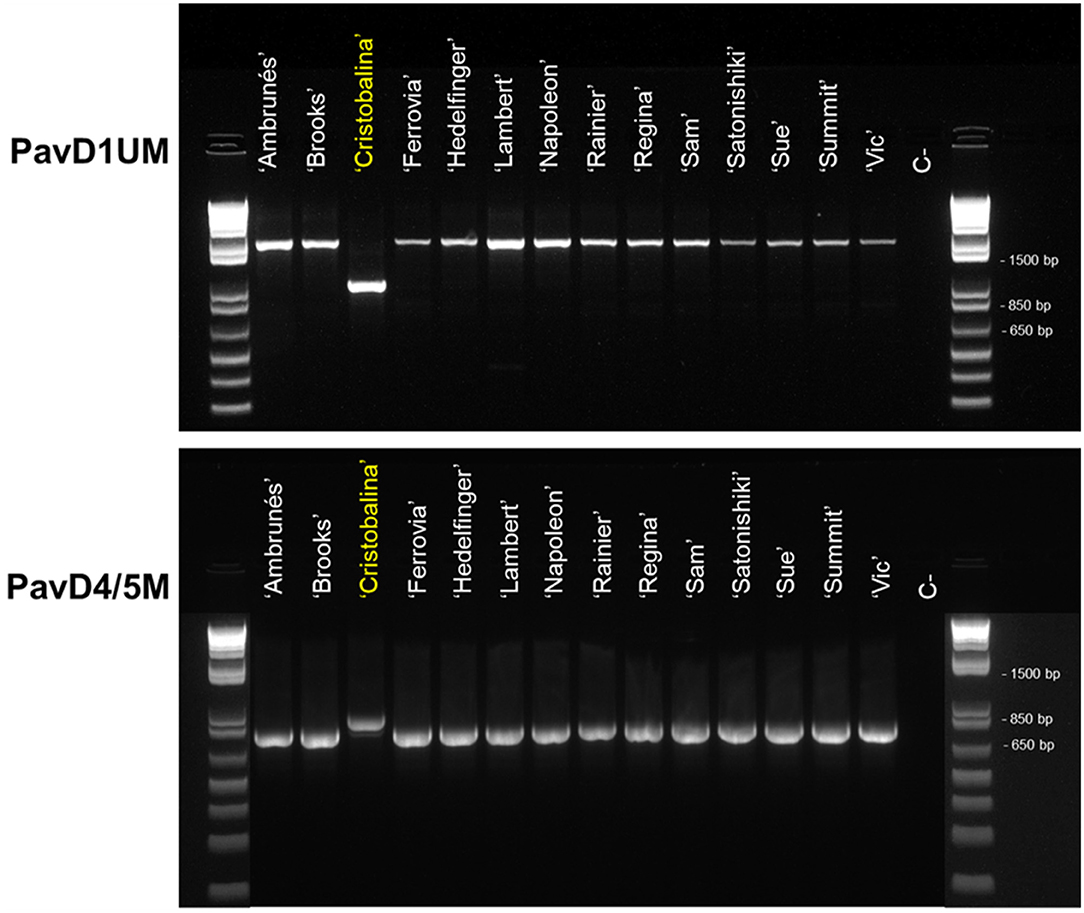
Figure 3. PCR analysis of PavDAM1 and PavDAM4 and -5 structural mutations (PavD1UM and PavD4/5M, respectively) with primers PavD1UMf-PavD1UMr and PavD4/5Mf-PavD4/5Mr in 14 sweet cherry cultivars. C-: Negative control.
To confirm these mutations, these genomic regions were Sanger sequenced from PCR fragments using ‘Cristobalina’ and ‘Regina’ genomic DNA. The obtained sequences were compared (Supplementary Figures 4, 5), revealing a deletion of 694 bp in the ‘Cristobalina’ genome, 736 bp upstream of PavDAM1 start codon of ‘Regina’ genome (Figure 4; Supplementary Figure 4). The rest of the sequence compared was highly similar except for a few SNPs (Supplementary Figure 4). For the PavDAM4 and -5 region, sequence comparison between ‘Cristobalina’ and ‘Regina’ revealed various polymorphisms (Figure 4; Supplementary Figure 5). These included four short insertions (21, 22, 30, and 46 bp) and one short deletion (18 bp) in ‘Cristobalina’; and 41 SNPs between both cultivars (Figure 4; Supplementary Figure 5).
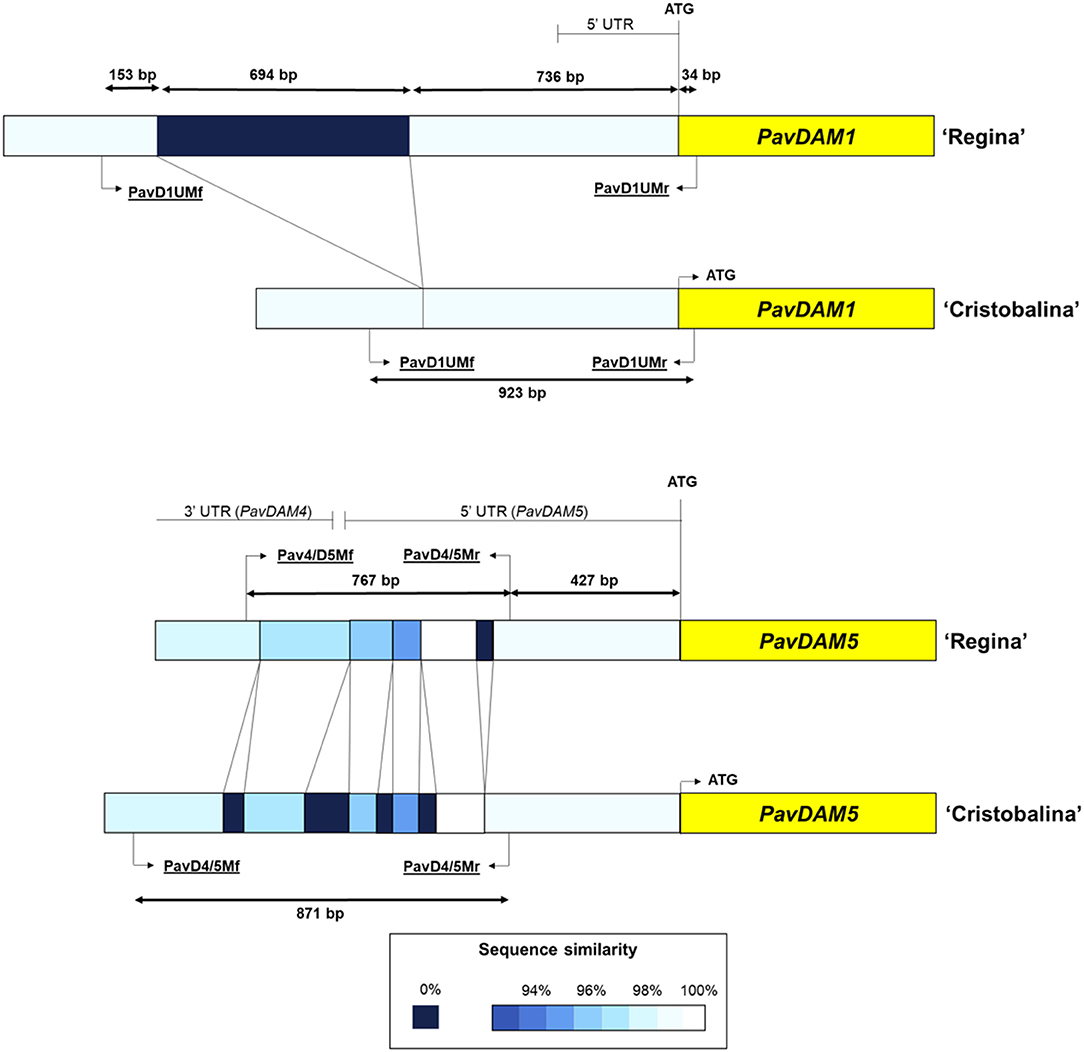
Figure 4. Schematic overview of PavDAM1 and PavDAM4 and -5 structural mutations (PavD1UM and PavD4/5M, respectively) in ‘Cristobalina’ and ‘Regina’. PCR primers positions are shown (underlined text). The percentage of similarity between sequences is shown in different colors.
Analysis of cis-acting regulatory sites in the ‘Regina’ sequence upstream of PavDAM1, which is absent in ‘Cristobalina’, revealed the presence of 60 unique sites (Supplementary Table 6). These include motifs like ARFAT, MYC, CArG, site II, TATA box, and WUSATAg that are associated with dormancy, bloom, flower development, and hormone regulation, among others (Supplementary Table 6; Supplementary Figure 4). Additionally, RNAseq data (Vimont et al., 2019) analysis in this genome region in both cultivars, ‘Regina’ and ‘Cristobalina’, revealed the alignment of short reads in ‘Regina’, but not in ‘Cristobalina’. The level of expression and the number of these reads were high enough to identify a putative non-coding gene that is expressed in ‘Regina’, but not in ‘Cristobalina’. On the other side, analysis of the highly variable region between PavDAM4 and -5 in ‘Cristobalina’ and ‘Regina’, revealed this region spans part of contiguous PavDAM4 3′UTR and PavDAM5 5′UTR (Figure 4; Supplementary Figure 5), with INDELs located in both UTRs. Splice junction coverage of RNAseq data in this region, in the two cultivars, revealed differences in splicing variants between ‘Regina’ and ‘Cristobalina’ for PavDAM5.
Validation of DNA-Markers of PavDAMs Structural Mutations (PavD1UM and PavD4/5M)
PavD1UM and PavD4/5M analysis in F2 population B×C2 revealed three segregating classes for both markers. The same individuals were in the same segregating classes for both markers, confirming both markers are linked. For both markers the segregating classes were: homozygous like ‘Regina’ and ancestor ‘Brooks’ (genotype nn), heterozygous like the parental cultivar ‘BC8’ (pn), and homozygous like ‘Cristobalina’ (pp; Figure 5). For marker PavD1UM, these genotypes correspond to PCR fragments of ~1600, 950/1600, and 900 bp, respectively. In the case of PavD4/5M, the corresponding PCR genotypes are 750, 750/850, and 850 bps for nn, pn, and pp, respectively. The estimated exact expected sizes of these PCR fragments are 1638 and 944 bp for PavD1UM, and 766 and 867 bp for PavD4/5M. Segregation of the three classes occurred in the proportion 24:33:4 (pp:pn:nn), which significantly differs from the expected 1:2:1 ratio (χ2 = 1.87; Supplementary Table 7).
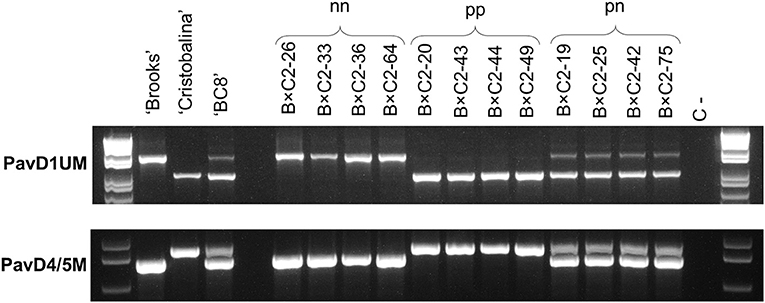
Figure 5. PCR analysis of PavDAM1 and PavDAM4 and -5 structural mutations (PavD1UM and PavD4/5M, respectively) in B×C2 population with primers PavD1UMf-PavD1UMr and PavD4/5Mf-PavD4/5Mr, respectively. Four individuals from each segregating class are shown. Population B×C2 parental genotype (‘BC8’), and ‘BC8’ parental genotypes (‘Brooks’ and ‘Cristobalina’) are also shown. C-: Negative control.
PavD1UM and PavD4/5M genotypes identified herein, and QTL qP-BT1.1m genotypes previously reported for the same population B×C2 were compared (Supplementary Table 7). The comparison revealed that individuals with QTL haplotypes cc, ac, and aa (Calle et al., 2020) were the same as those belonging to PavD1UM and PavD4/5M segregating classes pp, pn, and nn, respectively (Supplementary Table 7). This result confirms that the mutations in PavDAMs in ‘Cristobalina’ show complete correlation with the QTL haplotypes associated with bloom date (Calle et al., 2020). Specifically, QTL haplotype cc is associated with earlier blooming than ac, and both correspond to extra-early and intermediate blooming phenotypes, respectively (Supplementary Table 7; Calle et al., 2020). Therefore, results herein confirm that PavD1UM and PavD4/5M markers are valid for identifying different bloom time QTL haplotypes, and therefore for identifying earlier and later blooming phenotypes from these plant materials. Additionally, PavD1UM and PavD4/5M markers genotyping allowed identifying the genotype of 14 recombinant individuals for this QTL in B×C2. From the 14 recombinants, three individuals corresponded to genotype pp and 11 to pn (Supplementary Table 7). Estimation of mean bloom date of each segregating class for both markers confirmed a significant difference of 7 days in mean bloom time between individuals of classes pp and pn (p < 0.001; Student's T-test; Supplementary Table 7). This is the same difference observed for QTL qP-BT1.1m haplotypes in the same family. No phenotype data for nn individuals are available to estimate the phenotypic value of this segregating class (Supplementary Table 7).
Markers validation in a sweet cherry cultivar collection (Table 1) showed that only the low chilling and extra-early bloom time cultivars, ‘Temprana de Sot’ and ‘Royal Lee’ showed the same genotype as ‘Cristobalina’. These cultivars were homozygous for the early bloom allele (pp) for both markers (900 bp for PavD1UM, and 850 bp for PavD4/5M; Table 1). Additionally, the two local Spanish cultivars ‘De Mango Largo’ (midseason bloom) and ‘Son Perot’ (early bloom) were heterozygous (pn) for both markers PavD1UM (950/1600) and PavD4/5M (750/850) (Table 1). As described above, the selection ‘BC8’ (‘Brooks’ × ‘Cristobalina’), which shows early bloom time is also heterozygous for the markers. The rest of the cultivars, which show early to late bloom time, were homozygous (nn), and hence they had the same genotype as Regina (Table 1).
Discussion
Annotation, Structural, and Phylogenetic Analysis of Candidate Genes (PavDAMs) in Major Bloom Time QTL
In this study, six MADS-box genes, PavDAM, were identified in sweet cherry major bloom time QTL, qP-BT1.1m. This bloom time QTL was previously detected in populations derived from the low chilling and extra-early blooming cultivar ‘Cristobalina’ (Calle et al., 2020). This chromosome 1 genome region is determinant in the genetic control of chilling requirements and bloom time in sweet cherry, as other QTLs for these traits were also previously reported on the same location in sweet cherry populations from different genetic backgrounds (Dirlewanger et al., 2012; Castède et al., 2014). Six tandemly arranged MICKc-type MADS-box, denoted DAM genes, have been previously identified in the syntenic region of chromosome 1 in the almond, peach, Japanese apricot, and European plum genomes (Xu et al., 2014; Wells et al., 2015; Quesada-Traver et al., 2020). In sweet cherry, PavDAM genes have been recently cloned and sequenced from flower bud RNA, and their cDNA and predicted amino acid sequences have been reported (Wang et al., 2020). In the present work, the genomic sequence and structure of these genes were characterized and annotated from the sweet cherry genome sequence of Regina cv (Le Dantec et al., 2020).
The amino acid sequence of PavDAMs recently predicted in ‘Royal Lee’ and ‘Hongdeng’ cultivars (Wang et al., 2020) is highly similar to that reported in this work for the ‘Regina’ genome (99.0 and 99.9%, respectively). The sequence of each PavDAM reported in this work includes the four characteristic domains of MIKC type II MADS-box, as reported earlier in peach, Japanese apricot, or plum (Jiménez et al., 2009; Xu et al., 2014; Quesada-Traver et al., 2020). Furthermore, we observed in this study, that each PavDAM comprises eight exons making the genomic structure of the six genes very similar to that of the DAM genes previously reported in other Prunus species, namely peach, Japanese apricot, European plum (Jiménez et al., 2009; Sasaki et al., 2011; Quesada-Traver et al., 2020). Thus, the six MADS-box (PavDAM) genes identified within the major bloom time QTL in this work, as expected, are solid candidate genes for chilling requirement and bloom time regulation in sweet cherry.
Like in earlier works (Rothkegel et al., 2017; Wang et al., 2020), phylogenetic analysis in this work revealed that PavDAMs are orthologs to the peach and Japanese apricot corresponding DAM genes. Within each DAM gene clade, peach and Japanese apricot genes appeared phylogenetically closer to each other than to sweet cherry genes, reflecting the species phylogeny. Peach and Japanese apricot, belong to Amygdalus and Prunus subgenus, respectively, phylogenetically closer to each other than to the sweet cherry subgenus (Cerasus) (Potter et al., 2007). The detection of six clades of DAM ortholog groups indicates that DAM diversification occurred before Prunus speciation. Additionally, the six DAM genes may be paralogs (outparalogs), as earlier duplication events may have led to the six tandemly arranged genes (Koonin, 2005). As suggested before (Jiménez et al., 2009; Li et al., 2009), posterior subfunctionalization and/or neofunctionalization may have resulted in their actual function. The clustering of the DAM orthologs in two major clades, namely DAM1, -2, and -3; and DAM4, -5, and -6, as previously observed (Prudencio et al., 2018; Balogh et al., 2019; Quesada-Traver et al., 2020; Wang et al., 2020), agrees with previous transcriptomic studies of DAM genes in peach and Japanese apricot, in which two different expression patterns have been observed for the two groups of genes. DAM1, -2, and -3 have a maximum expression during bud set, while DAM4, -5, and -6 show maximum expression when chilling requirement are satisfied (Falavigna et al., 2019).
Intraspecific Variation of PavDAMs Sequences in Cultivars With Large Phenotypic Variation
PavDAMs predicted amino acid sequences of 13 sweet cherry cultivars revealed a high degree of similarity despite their different genetic backgrounds and contrasting phenotypes (Table 1). Also, these sequences are highly similar to those previously reported (‘Royal Lee’ and ‘Hongdeng’; Wang et al., 2020). This high degree of conservation may indicate that PavDAMs proteins’ phenotypic effect may be more dependent on expression regulation than on protein structure. Most amino acid differences among the cultivars studied were found in the same positions, confirming also the presence of highly variable amino acids. However, no correlation of these amino acid polymorphisms could be associated with the chilling requirement and/or bloom time of these cultivars. Nevertheless, it cannot be discarded that these amino acid substitutions may be associated with phenotypic differences. Single amino acid substitutions in MADS-box genes in Arabidopsis have been associated with the loss of function leading to early flowering phenotypes (Hartmann et al., 2000; Méndez-Vigo et al., 2013).
‘Cristobalina’ PavDAM genes showed the lowest similarity with the rest of the cultivars and accumulated the largest number of unique amino acid substitutions. ‘Cristobalina’ was the only cultivar that has a unique amino substitution in the M domain of PavDAM2 and -5, whereas more substitutions were observed in the C domain in all DAMs. It has been reported that the M domain is the most conserved of all MADS-box domains; and that the C domain, which is related to protein complex formation and transcriptomic activation, is the most variable (Honma and Goto, 2001; Kaufmann et al., 2005). The differences observed between the PavDAM genes coding sequences of ‘Cristobalina’ and the other cultivars analyzed may be associated with the phenotypic differences in chilling requirements and/or bloom time. ‘Cristobalina’ has lower chilling requirements and earlier blooming than the other cultivars analyzed (Table 1; Tabuenca, 1983; Alburquerque et al., 2008; Calle et al., 2020). It has also been observed that ‘Cristobalina’ enters endodormancy later and fulfills its chilling requirements before medium to late bloom time cultivars (Fadón et al., 2018). The genetic differences may be due to a different genetic origin and adaptation to different eco-geographic regions. ‘Cristobalina’ is a local Spanish cultivar from the Mediterranean region and is genetically well-differentiated from the rest of the cultivars analyzed (Wünsch and Hormaza, 2002; Martínez-Royo and Wünsch, 2014).
It was also observed that, despite the large variability exhibited by ‘Cristobalina’ PavDAMs predicted amino acid sequences, these are identical to those reported for ‘Royal Lee’ (Wang et al., 2020). These two cultivars seemed unrelated, as there is no proof of a relationship between them. ‘Royal Lee’ is also a low-chill cultivar, which derives from a breeding program in California (Zaiger's Inc Genetics; US patent N° 12417), while ‘Cristobalina’ is a local Spanish landrace. The chilling requirements of ‘Royal Lee’ (Wang et al., 2020) are also similar to those of ‘Cristobalina’ (approx. 400 chilling hours; Tabuenca, 1983). A possible explanation for this unexpected genetic and phenotypic similarity is that ‘Cristobalina’ is an ancestor of ‘Royal Lee’. In fact, the contribution of a low-chilling cultivar of unknown origin is described in the ‘Royal Lee’ pedigree (US patent N° 12417). In any case, the similarities observed reinforce the hypothesis that the genetic differences identified in ‘Cristobalina’ PavDAMs may be the cause of low chilling and extra early blooming.
PavDAMs Structural Mutations in Low-Chilling and Early Blooming Cultivars
Greater variation upstream of ‘Cristobalina’ PavDAM1 and between PavDAM4 and -5 was also identified in this work. Specifically, a 694 bp deletion, 736 bp upstream of the PavDAM1 coding sequence, and a highly polymorphic region, which includes various INDELs, in the UTRs of PavDAM4 and -5, were detected. These mutations were detected in ‘Cristobalina’ by sequence reads mapping to the ‘Regina’ genome sequence (Le Dantec et al., 2020), and confirmed by Sanger sequencing of PCR fragments spanning the mutations. PCR markers (PavD1UM, PavD4/5M) were designed to detect these mutations and to validate their association with low chilling and early blooming in an F2 segregating population and in a cultivar collection.
Analysis of PCR fragments from PavD1UM and PavD4/5M markers in the only available segregating population for these mutations (F2 population B×C2) revealed a complete correlation with the linkage group 1 bloom time QTL qP-BT1.1m segregating classes (Calle et al., 2020). These results indicate a correlation between the presence of the mutation and earlier blooming (7 days) in homozygous genotypes (pp). Furthermore, analyses of the markers in a sweet cherry cultivar collection with genotypes with large phenotypic differences for chilling requirements and bloom time also revealed an association of the mutations in homozygosis (pp), with low chilling and extra-early blooming. The other cultivars for which PavD1UM and PavD4/5M mutations were identified were other local Spanish cultivars (‘Temprana de Sot’, ‘Son Perot’, and ‘De Mango Largo’) and the bred cultivar ‘Royal Lee’. The presence in other local Spanish cultivars confirms that the putative origin of this PavDAM haplotype is the southern European Mediterranean region. The presence of these mutations also in ‘Royal Lee’ confirms that the PavDAM genotype is the same in ‘Cristobalina’ and ‘Royal Lee’, as discussed above for the PavDAMs protein sequences. It also confirms the correlation of this genotype with low chilling and extra-early blooming. The result also reinforces the hypothesis that ‘Cristobalina’ may be part of the pedigree of ‘Royal Lee’.
In two other local Spanish cultivars (‘Son Perot’ and ‘De Mango Largo’), the ‘Cristobalina’ PavDAMs mutations were found in heterozygosity (pn genotypes). These cultivars have early and medium bloom date phenotypes and their chilling requirements are not known. Similarly, in other individuals from ‘Cristobalina’-derived populations, which are heterozygous for these mutations (data not shown), different bloom time phenotypes have been observed (Calle et al., 2020), but none of them show such early-blooming as those in which the mutations are in homozygosity (pp, like in ‘Cristobalina’). In fact, in the B×C2 population, the heterozygous individuals (np) are not as early blooming as the homozygous ones (pp) (see Calle et al., 2020). The phenotypic effect associated with these mutations is more evident in those individuals homozygous for the mutations probably due to the additive effect of each PavDAM haplotype. The rest of the cultivars analyzed with the PavDAM markers, PavD1UM and PavD4/5M, are homozygous for the absence of the mutation (nn). Among these, there are cultivars of medium to high chilling requirements from early to late blooming. This result also indicates that not all early blooming cultivars in sweet cherry have the same mutation as ‘Cristobalina’, and therefore, that there are additional sources of early blooming in sweet cherry. But only, the extra-early cultivars analyzed do have the described PavDAM mutations and protein sequences. Therefore, the markers developed in this work correlate with earlier bloom time (and probably low chilling) and are useful for the selection of this trait from ‘Cristobalina’ and likely from ‘Royal Lee’ too.
‘Cristobalina’ PavDAM genotype has revealed several unique polymorphisms in its predicted protein sequences and large structural mutations upstream of PavDAM1 and in contiguous PavDAM4 and -5 UTR sequences. These structural mutations were shown to correlate with extra-early blooming. Although further research is needed, it cannot be discarded that these mutations may be the cause of low-chilling and extra-early blooming in this cultivar. Protein variability in relevant conserved regions of PavDAMs may be altering protein function in this genotype due to variation in oligomerization in conserved regions (Lai et al., 2019). Alternatively, the structural mutations observed may result in differential gene expression of PavDAMs in this cultivar. Differential expression of PavDAMs in ‘Cristobalina’ (and ‘Royal Lee’) has been observed in transcriptomic analyses during dormancy when compared with high-chilling cultivars (Vimont et al., 2019; Supplementary Figure 6; Wang et al., 2020). Most evident differences have been observed for PavDAM1,−4, and -5 (Vimont et al., 2019; Supplementary Figure 6; Wang et al., 2020). Specifically, the expression of PavDAM1 of ‘Royal Lee’ has been shown to decrease much earlier than in the high chilling cultivar ‘Hongdeng’ (Wang et al., 2020). A similar result has been observed for ‘Cristobalina’ compared with ‘Regina’ (Vimont et al., 2019). For PavDAM4 and -5, large differences have been observed between ‘Cristobalina’ and ‘Regina’, especially for PavDAM4 in which much lower expression was observed in ‘Cristobalina’ (Vimont et al., 2019).
The deletion upstream of PavDAM1 in ‘Cristobalina’ results in the absence of potentially relevant cis-acting binding sites. DAM genes have been observed to be regulated by proteins related to the response of environmental signals and the cold response pathway that can bind to DAM promoters (Zhao et al., 2015). This is the case of C-Repeat Binding Factors (CBF), which have been reported in some Rosaceae species binding DAM promoter and to regulate these genes expression in apple, Japanese apricot, and pear (Mimida et al., 2015; Saito et al., 2015; Wisniewski et al., 2015; Zhao et al., 2018). Besides, CArG box motif is the target region of MADS-box transcription factor, but also their own regulation (Zhu and Perry, 2005; Gregis et al., 2013). More recently, it was shown that the site II motif was recognized by the PpeTCP20 transcription factor, down-regulating the expression of DAM5 and -6 in peach (Wang et al., 2020). Motifs CArG and site II, among others, are missing in the deleted region upstream of PavDAM1 in ‘Cristobalina’. The putative involvement of any missing cis-acting elements in PavDAMs expression would be compatible with the differential expression of ‘Cristobalina’ (and ‘Royal Lee’) PavDAMs (Vimont et al., 2019; Supplementary Figure 6; Wang et al., 2020). Additionally, in the same genomic region, the expression of a non-coding gene in ‘Regina’ seems truncated in ‘Cristobalina’. Blast analysis indicates the existence of this ncRNA in peach but it has not been detected in other organisms, and therefore, could be Prunus-specific. As no other relevant information could be obtained from this non-coding gene, further analyses are required to confirm the potential involvement of this gene in the ‘Cristobalina’ phenotype.
The variation observed in PavDAM4 and -5 UTRs between ‘Regina’ and ‘Cristobalina’ may also have implications in PavDAMs expression, and/or in PavDAMs transcripts variability. UTRs can influence gene expression in plants (Srivastava et al., 2018). Noticeably, we observed a predominance of specific splicing variants in each cultivar, ‘Cristobalina’ seems to have a shorter 5′UTR than ‘Regina’. UTR length could influence expression levels as well as play a role in various post-transcriptional processes (Mignone et al., 2002), which can result in PavDAM5 differential transcription, translation, and/or function. Additionally, these mutations in PavDAM4 and -5 could also affect the expression of the other PavDAMs, as previously observed in the EVG peach mutant (with four deleted DAM genes), where the two intact genes (DAM1 and -2) were not expressed (Bielenberg et al., 2008). It is, therefore, necessary to further investigate these mutations, to identify their potential effect in PavDAMs differential transcription, and their correlation with the contrasting phenotypes.
PavD1UM and PavD4/5M, Markers for Breeding for Early Blooming and Low Chilling Requirements
‘Cristobalina’ is a relevant cultivar for breeding, due to self-compatibility, low chilling requirements, and extra-early bloom time. The PavD1UM and PavD4/5M markers, developed here, are a useful tool for sweet cherry breeding of low chilling requirement and early bloom time from ‘Cristobalina’ using marker-assisted selection. These markers revealed a complete correlation with the haplotypes of bloom time QTL (qP-BT1.1m), which accounts for up to 50.1% of the phenotypic variation in ‘Cristobalina’ derived populations (Calle et al., 2020). The large correlation between QTL and marker genotypes, as well as the large amount of phenotypic variation explained by this QTL, makes these markers useful tools for discriminating individuals with lower chilling requirement and earlier blooming, which will be associated with the presence of the mutations in homozygosity or heterozygosity. Earlier blooming is expected to be associated with the presence of the deletion in homozygosity and later blooming and higher chilling requirement will be associated with the absence of the mutations. Besides, the identification of these mutations also in the low chill cultivar ‘Royal Lee’, indicate these markers may also useful for selection from cultivars from different genetic backgrounds other than ‘Cristobalina’.
In the present study, the analysis of candidate genes in a previously reported main bloom time QTL in sweet cherry has allowed for the characterization and annotation of PavDAM genes in the species. This work thus confirms PavDAMs as candidate genes for bloom time regulation in sweet cherry. Protein sequence polymorphisms and structural mutations identified in PavDAMs of low-chilling and extra-early blooming cv. Cristobalina were shown to correlate with earlier blooming in a segregating population and with extra-early blooming in a diverse set of cultivars. These results indicate that the ‘Cristobalina’ PavDAM genotype may be the genetic causal variation of the phenotypic differences exhibited by ‘Cristobalina’, low chilling requirement, and extra-early bloom time, although further research is needed to confirm this hypothesis. PCR DNA-markers based on these structural mutations (useful for the selection of early blooming from this plan material) were designed and validated.
Data Availability Statement
The data presented in this study are deposited in the CITA online repository (https://citarea.cita-aragon.es/citarea/) accesion number https://citarea.cita-aragon.es/citarea/handle/10532/5397. This is an updated model based on the raw sequences deposited at genbank DDBJ (PRJDB6734).
Author Contributions
AC carried out experimental work, data analysis and interpretation, and manuscript writing and revision. JG participated in the bioinformatics analyses, experimental design, data interpretation, and manuscript writing and editing. LL provided reference genome and participated in manuscript revision. AW participated in experimental design, data analysis supervision, manuscript writing and editing, and work coordination. All authors contributed to the article and approved the submitted version.
Funding
This work had been funded by Spanish Government Ministerio de Ciencia e Innovacion Agencia Estatal de Investigación (AEI) (project PID2019-103985RR-I00); Instituto Nacional de Investigación y Tecnología Agraria y Alimentaria (INIA) (projects RTA2015-00027-00-00 and RFP2015-00015-00-00); and FEDER funds. Addiccional funding was obtained from Grupo de Investigación de la Comunidad de Aragón A12-17R (Fruticultura. Caracterización, Adaptación y Mejora Genética) of Departamento de Innovación, Investigación y Universidad (Gobierno de Aragón). AC was funded by Departamento de Innovación, Investigación y Universidad, Gobierno de Aragón, and by the Ph.D. program Subvenciones destinadas a la contratación de personal investigador en formación 2015–2019.
Conflict of Interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2021.621491/full#supplementary-material
Abbreviations
AGL24, AGAMOUS-LIKE 24; BAM, Binary Alignment Map; CBF, C-Repeat Binding Factors; cv, cultivar; DAM, Dormancy associated MADS-box; DDBJ, DNA Data Bank of Japan; EVG, ever-growing peach mutant; GFF, General File Format; IGV, Integrative Genomics Viewers; LG, linkage group; MCL, Maximum Composite Likelihood; NCBI, National Center for Biotechnology Information; PavD1UM, PavDAM1 Upstream Mutation; PavD4/5, PavDAM4 and -5 mutation; PCR, polymerase chain reaction; QTL, quantitative trait locus; SVP, SHORT VEGETATIVE PHASE; TBE, Tris-Borate-EDTA buffer.
References
Abbott, A. G., Zhebentyayeva, T., Barakat, A., and Liu, Z. A. (2015). The genetic control of bud-break in trees. Adv. Bot. Res. 7, 201–228. doi: 10.1016/bs.abr.2015.04.002
Afgan, E., Baker, D., Batut, B., Van Den Beek, M., Bouvier, D., Cech, M., et al. (2018). The Galaxy platform for accessible, reproducible, and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 46, 537–544. doi: 10.1093/nar/gky379
Alburquerque, N., García-Montiel, F., Carrillo, A., and Burgos, L. (2008). Chilling and heat requirements of sweet cherry cultivars and the relationship between altitude and the probability of satisfying the chill requirements. Environ. Exp. Bot. 64, 162–170. doi: 10.1016/j.envexpbot.2008.01.003
Allard, A., Bink, M. C. A. M., Martinez, S., Kelner, J. J., Legave, J. M., di Guardo, M., et al. (2016). Detecting QTLs and putative candidate genes involved in budbreak and flowering time in apple multiparental population. J. Exp. Bot. 67, 2875–2888. doi: 10.1093/jxb/erw130
Balogh, E., Halász, J., Soltész, A., Erös-Honti, Z., Gutermuth, A., Szalay, L., et al. (2019). Identification, structural and functional characterization of dormancy regulator genes in apricot (Prunus armeniaca L.). Front. Plant Sci. 10:402. doi: 10.3389/fpls.2019.00402
Becker, A., and Theißen, G. (2003). The major clades of MADS-box genes and their role in the development and evolution of flowering plants. Mol. Phylogenet. Evol. 29, 464–489. doi: 10.1016/S1055-7903(03)00207-0
Bielenberg, D. G., Rauh, B., Fan, S., Gasic, K., Abbott, A. G., Reighard, G. L., et al. (2015). Genotyping by sequencing for SNP-based linkage map construction and QTL analysis of chilling requirement and bloom date in peach [Prunus persica (L.) Batsch]. PLoS ONE 10:e0139406. doi: 10.1371/journal.pone.0139406
Bielenberg, D. G., Wang, Y., Li, Z. G., Zhetentyayeva, T., Fan, S. H., Reighard, G. L., et al. (2008). Sequencing and annotation of the evergrowing locus in peach Prunus persica (L.) Batsch reveals a cluster of six MADS-box transcription factors as candidate genes for regulation of terminal bud formation. Tree Genet. Genomes 4, 495–507. doi: 10.1007/s11295-007-0126-9
Bolger, A. M., Lohse, M., and Usadel, B. (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. doi: 10.1093/bioinformatics/btu170
Cachi, A. M., and Wünsch, A. (2014). Characterization of self-compatibility in sweet cherry varieties by crossing experiments and molecular genetic analysis. Tree Genet. Genomes 10, 1205–1222. doi: 10.1007/s11295-014-0754-9
Calle, A., Cai, L., Iezzoni, A., and Wünsch, A. (2020). Genetic dissection of bloom time in low chilling sweet cherry (Prunus avium L.) using a multi-family QTL approach. Front. Plant Sci. 10:1647. doi: 10.3389/fpls.2019.01647
Calle, A., and Wünsch, A. (2020). Multiple-population QTL mapping of maturity and fruit-quality traits reveals LG4 region as a breeding target in sweet cherry (Prunus avium L.). Horticult. Res. 7:120. doi: 10.1038/s41438-020-00349-2
Campoy, J. A., Ruiz, D., Allderman, L., Cook, N., and Egea, J. (2011). The fulfilment of chilling requirements and the adaptation of apricot (Prunus armeniaca L) in warm winter climates: an approach in Murcia (Spain) and the Western Cape (South Africa). Eur. J. Agron. 37, 43–55. doi: 10.1016/j.eja.2011.10.004
Castède, S., Campoy, J. A., Le Dantec, L., Quero-García, J., Barreneche, T., Weden, B., et al. (2015). Mapping of candidate genes involved in bud dormancy and flowering time in sweet cherry (Prunus avium). PLoS ONE 10:e0143250. doi: 10.1371/journal.pone.0143250
Castède, S., Campoy, J. A., Quero-García, J., Le Dantec, L., Lafargue, M., Barreneche, T., et al. (2014). Genetic determinism of phenological traits highly affected by climate change in Prunus avium: flowering date dissection into chilling and heat requirements. New Phytol. 202, 703–715. doi: 10.1111/nph.12658
Cooke, J. E. K., Eriksson, M. E., and Junttila, O. (2012). The dynamic nature of bud dormancy in trees: environmental control and molecular mechanisms. Plant Cell Environ. 35, 1707–1728. doi: 10.1111/j.1365-3040.2012.02552.x
Dirlewanger, E., Quero-García, J., Le Dantec, L., Lambert, P., Ruiz, D., Dondini, L., et al. (2012). Comparison of the genetic determinism of two key phenological traits, flowering and maturity dates, in three Prunus species: peach, apricot and sweet cherry. Heredity 109, 280–292. doi: 10.1038/hdy.2012.38
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Fadón, E., Herrera, S., Guerrero, B. I., Guerra, M. E., and Rodrigo, J. (2020). Chilling and heat requirements of temperate stone fruit trees (Prunus sp.). Agronomy 10:409. doi: 10.3390/agronomy10030409
Fadón, E., and Rodrigo, J. (2018). Unveiling winter dormancy through empirical experiments. Environ. Exp. Bot. 152, 28–36. doi: 10.1016/j.envexpbot.2017.11.006
Fadón, E., Rodrigo, J., and Herrero, M. (2018). Is there a specific stage to rest? morphological changes in flower primordia in relation to endodormancy in sweet cherry (Prunus avium L.). Trees 32, 1583–1594. doi: 10.1007/s00468-018-1735-7
Falavigna, V., Guitton, B., Costes, E., and Andrés, F. (2019). I want to (bud) break free: the potential role of DAM and SVP-like genes in regulating dormancy cycle in temperate fruit trees. Front. Plant Sci. 9, 1–17. doi: 10.3389/fpls.2018.01990
Fan, S., Bielenberg, D. G., Zhebentyayeva, T. N., Reighard, G. L., Okie, W. R., Holland, D., et al. (2010). Mapping quantitative trait loci associated with chilling requirement, heat requirement and bloom date in peach (Prunus persica). New Phytol. 185, 917–930. doi: 10.1111/j.1469-8137.2009.03119.x
Gella, R., Fustero, R., and Rodrigo, J. (2001). Variedades de Cerezo. Servicio de Investigación Agroalimentaria. Zaragoza: Diputación General de Aragón.
Gramzow, L., and Theissen, G. (2010). A hitchhiker's guide to the MADS world of plants. Genome Biol. 11:214. doi: 10.1186/gb-2010-11-6-214
Gregis, V., Andres, F., Sessa, A., Guerra, R. F., Simonini, S., Mateos, J. L., et al. (2013). Identification of pathways directly regulated by SHORT VEGETATIVE PHASE during vegetative and reproductive development in Arabidopsis. Genom. Biol. 14:R56. doi: 10.1186/gb-2013-14-6-r56
Hartmann, U., Hohmann, S., Nettesheim, K., Wisman, E., Saedler, H., and Huijser, P. (2000). Molecular cloning of SVP a negative regulator of the floral transition in Arabidopsis. Plant J. 21, 351–360. doi: 10.1046/j.1365-313x.2000.00682.x
Higo, K., Ugawa, Y., Iwamoto, M., and Korenaga, T. (1999). Plant cis-acting regulatory DNA elements (PLACE) database. Nucleic Acids Res. 27, 297–300. doi: 10.1093/nar/27.1.297
Honma, T., and Goto, K. (2001). Complexes of MADS-box proteins are sufficient to convert leaves into floral organs. Nature 409, 525–529. doi: 10.1038/35054083
Jiménez, S., Lawton-Rauh, A. L., Reighard, G. L., Abbott, A. G., and Bielenberd, D. G. (2009). Phylogenetic analysis and molecular evolution of the dormancy associated MADS-box genes from peach. BMC Plant Biol. 9:81. doi: 10.1186/1471-2229-9-81
Jiménez, S., Reighard, G. L., and Bielenberg, D. G. (2010). Gene expression of DAM5 and DAM6 is suppressed by chilling temperatures and inversely correlated with bud break rate. Plant Mol. Biol. 73, 157–167. doi: 10.1007/s11103-010-9608-5
Kaufmann, K., Melzer, R., and Theißen, G. (2005). MIKC-type MADS-domain proteins: structural modularity, protein interactions, and network evolution in land plants. Gene 347, 183–198. doi: 10.1016/j.gene.2004.12.014
Kim, D., Langmead, B., and Salzberg, S. L. (2012). HISAT: a fast spliced aligner with low memory requirements. Nat. Methods 2, 357–360. doi: 10.1038/nmeth.3317
Kitamura, Y., Habu, T., Yamane, H., Nishiyama, S., Kajita, K., Soube, T., et al. (2018). Identification of QTLs controlling chilling and heat requirements for dormancy release and bud break in Japanese apricot (Prunus mume). Tree Genet. Genomes 14:33. doi: 10.1007/s11295-018-1243-3
Koonin, E. V. (2005). Orthologs, paralogs, and evolutionary genomics. Annu. Rev. Genet. 39, 309–338. doi: 10.1146/annurev.genet.39.073003.114725
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lai, X., Daher, H., Galien, A., Hugouvieux, V., and Zubieta, C. (2019). Structural basis for plant MADS transcription factor oligomerization. Comput. Struct. Biotechnol. J. 17, 946–953. doi: 10.1016/j.csbj.2019.06.014
Lang, G. A., Early, J. D., Martin, G. C., and Darrel, R. L. (1987). Endo-, para-, and endodormancy: physiological terminology and classification for dormancy research. Hortscience 22, 37–377.
Langmead, B., and Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Le Dantec, L., Girollet, N., Gouzy, J., Sallet, E., Carrère, M., Fouché, M., et al. (2020). Assembly and annotation of 'Regina' sweet cherry genome. Portail Data INRAE, V1. Paris: INRAE.
Leida, C., Conejero, A., Arbona, V., Gómez-Cadenas, A., Llácer, G., Badenes, M. L., et al. (2012). Chilling-dependent release of seed and bud dormancy in peach associates to common changes in gene expression. PLoS ONE 7:e35777. doi: 10.1371/journal.pone.0035777
Li, Z., Reighard, G. L., Abbott, A. G., and Bielenberg, D. G. (2009). Dormancy-associated MADS genes from the EVG locus of peach [Prunus persica (L.) Batsch] have distinct seasonal and photoperiodic expression patterns. J. Exp. Bot. 60, 3521–3530. doi: 10.1093/jxb/erp195
Martínez-Royo, A., and Wünsch, A. (2014). “Genetic structure of sweet cherry with the 6K SNP Array v1,” in 7th International Rosaceae Genomics Conference (Seattle, WA).
Méndez-Vigo, B., Martínez-Zapater, J. M., and Alonso-Blanco, C. (2013). The flowering repressor SVP underlies a novel Arabidopsis thaliana QTL interacting with the genetic background. PLoS Genet. 9:e1003289. doi: 10.1371/journal.pgen.1003289
Messenguy, F., and Dubois, E. (2003). Role of MADS-box proteins and their cofactors in combinatorial control of gene expression and cell development. Gene 316, 1–21. doi: 10.1016/S0378-1119(03)00747-9
Mignone, F., Gissi, C., Liuni, S., and Pesole, G. (2002). Untranslated regions of mRNAs. Genome Biol. 3:reviews0004.1. doi: 10.1186/gb-2002-3-3-reviews0004
Mimida, N., Saito, T., Moriguchi, T., Suzuki, A., Komori, S., and Wada, M. (2015). Expression of DORMANCY-ASSOCIATED MADS-BOX (DAM)-like genes in apple. Biol. Plant. 59, 237–244. doi: 10.1007/s10535-015-0503-4
Ono, K., Akagi, T., Morimoto, T., Wünsch, A., and Tao, R. (2018). Genome resequencing of diverse sweet cherry (Prunus avium) individuals reveals a modifier gene mutation conferring pollen-part self-compatibility. Plant Cell Physiol. 59, 1265–1275. doi: 10.1093/pcp/pcy068
Potter, D., Eriksson, T., Evans, R. C., Oh, S., Smedmark, J. E. E., Morgan, D. R., et al. (2007). Phylogeny and classification of Rosaceae. Plant Syst. Evol. 266, 5–43. doi: 10.1007/s00606-007-0539-9
Prudencio, A. S., Dicenta, F., and Martínez-Gómez, P. (2018). Monitoring dormancy transition in almond [Prunus dulcis (Miller) Webb] during cold and warm Mediterranean seasons through the analysis of a DAM (Dormancy-Associated MADS-Box) gene. Horticulturae 4:41. doi: 10.3390/horticulturae4040041
Quero-García, J., Schuster, M., López-Ortega, G., and Charlot, G. (2017). “Sweet cherry varieties and improvement,” in Cherries: Botany, Production, and Uses, eds J. Quero-García, A. Iezzoni, and G. Lang (Boston, MA: CAB International), 60–94.
Quesada-Traver, C., Guerrero, B. I., Badenes, M. L., Rodrigo, J., Ríos, G., and Lloret, A. (2020). Structure and expression of bud dormancy-associated MADS-box genes (DAM) in European plum. Front. Plant Sci. 11:1288. doi: 10.3389/fpls.2020.01288
R Core Team (2017). R: A Language and Environment for Statistical Computing. Vienna: R Foundation for Statistical Computing. Available online at: http://www.R-project.org/ (accessed November 12, 2018).
Rodriguez, A. J., Sherman, W. B., Scorza, R., Wisniewski, M., and Okie, W. R. (1994). ‘Evergreen' peach, its inheritance and dormant behavior. J. Amer. Soc. Horti. Sci. 119, 789–792. doi: 10.21273/JASHS.119.4.789
Rohde, A., and Bhalerao, R. P. (2007). Plant dormancy in the perennial context. Trends Plant Sci. 12, 217–223. doi: 10.1016/j.tplants.2007.03.012
Rothkegel, K., Sánchez, E., Montes, C., Greve, M., Tapia, S., Bravo, S., et al. (2017). DNA methylation and small interference RNAs participate in the regulation of MADS-box genes involved in dormancy in sweet cherry (Prunus avium L.). Tree Physiol. 37, 1739–1751. doi: 10.1093/treephys/tpx055
Saito, T., Bai, S., Imai, T., Ito, A., Nakajima, I., and Moriguchi, T. (2015). Histone modification and signaling cascade of the dormancy-associated MADS-box gene, PpMADS 13-1, in Japanese pear (Pyrus pyrifolia) during endodormancy. Plant Cell Environ. 38, 1157–1166. doi: 10.1111/pce.12469
Saito, T., Bai, S., Ito, A., Sakamoto, D., Saito, T., Ubi, B. E., et al. (2013). Expression and genomic structure of the dormancy-associated MADS-box genes MADS13 in Japanese pears (Pyrus pyrifolia Nakai) that differ in their chilling requirement for endodormancy release. Tree Physiol. 33, 654–667. doi: 10.1093/treephys/tpt037
Sánchez-Pérez, R., Dicenta, F., and Martinez-Gomez, P. (2012). Inheritance of chilling and heat requirements for flowering in almond and QTL analysis. Tree Genet. Genomes 8, 379–389. doi: 10.1007/s11295-011-0448-5
Sasaki, R., Yamane, H., Ooka, T., Jotatsu, H., Kitamura, Y., Akagi, T., et al. (2011). Functional and expressional analyses of PmDAM genes associated with endodormancy in Japanese apricot. Plant Physiol. 157, 485–497. doi: 10.1104/pp.111.181982
Schuster, M. (2012). Incompatilbe (S-) genotypes of sweet cherry cultivars (Prunus avium L.). Sci. Hortic. 148, 59–73. doi: 10.1016/j.scienta.2012.09.012
Smaczniak, C., Immink, R. G. H., Angenent, G. C., and Kaufmann, K. (2012). Developmental and evolutionary diversity of plant MADS-domain factors: insights from recent studies. Development 139, 3081–3098. doi: 10.1242/dev.074674
Srivastava, A. K., Lu, Y., Zinta, G., Lang, Z., and Zhu, J. K. (2018). UTR-dependent control of gene expression in plants. Trends Plant Sci. 23, 248–259. doi: 10.1016/j.tplants.2017.11.003
Tabuenca, M. C. (1983). “Winter chilling requirements of cherry varieties,” in I Congreso Nacional de la Sociedad Española de Ciencias Hortícolas (Valencia), 661–667.
Tamura, K., and Nei, M. (1993). Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 10, 512–526.
Thorvaldsdóttir, H., Robinson, J. T., and Mesirov, J. P. (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192. doi: 10.1093/bib/bbs017
Verde, I., Jenkins, J., Dondini, L., Micali, S., Pagliarani, G., Vendramin, E., et al. (2017). The Peach v2.0 release: high-resolution linkage mapping and deep resequencing improve chromosome-scale assembly and contiguity. BMC Genom. 18, 1–18. doi: 10.1186/s12864-017-3606-9
Villar, L., Lienqueo, I., Llanes, A., Rojas, P., Perez, J., Correa, F., et al. (2020). Comparative transcriptomic analysis reveals novel roles of transcription factors and hormones during the flowering induction and floral bud differentiation in sweet cherry trees (Prunus avium L. cv. Bing). PLoS ONE 15:e0230110. doi: 10.1371/journal.pone.0230110
Vimont, N., Fouché, M., Campoy, J. A., Tong, M., Arkoun, M., Yvin, J. C., et al. (2019). From bud formation to flowering: transcriptomic state defines the cherry developmental phases of sweet cherry bud dormancy. BMC Genom. 20:974. doi: 10.1186/s12864-019-6348-z
Vimont, N., Quah, F. X., Schöepfer, D. G., Roudier, F., Dirlewanger, E., Wigge, P. A., et al. (2020). ChIP-seq and RNA-seq for complex and low-abundance tree buds reveal chromatin and expression co-dynamics during sweet cherry bud dormancy. Tree Genet. Genomes 16:9. doi: 10.1007/s11295-019-1395-9
Wang, J., Gao, Z., Li, H., Jiu, S., Qu, Y., Wang, L., et al. (2020). Dormancy-Associated MADS-Box (DAM) genes influence chilling requirement of sweet cherries and co-regulate flower development with SOC1 gene. Int. J. Mol. Sci. 21:921. doi: 10.3390/ijms21030921
Wells, C. E., Vendramin, E., Jimenez Tarodo, S., Verde, I., and Bielenberg, D. G. (2015). A genome-wide analysis of MADS-box genes in peach [Prunus persica (L.) Batsch]. BMC Plant Biol. 15:41. doi: 10.1186/s12870-015-0436-2
Wisniewski, M., Norelli, J., and Artlip, T. (2015). Overexpression of a peach CBF gene in apple: a model for understanding the integration of growth, dormancy, and cold hardiness in woody plants. Front. Plant Sci. 6:85. doi: 10.3389/fpls.2015.00085
Wünsch, A., and Hormaza, J. I. (2002). Molecular characterization of sweet cherry (Prunus avium L.) cultivars using peach (Prunus persica L. Batsch.) SSR sequences. Heredity 89, 56–63. doi: 10.1038/sj.hdy.6800101
Wünsch, A., and Hormaza, J. I. (2004). Genetic and molecular analysis in Cristobalina sweet cherry, a spontaneous self-compatible mutant. Sex. Plant Reprod. 17, 203–210. doi: 10.1007/s00497-004-0234-8
Xu, Z., Zhang, Q., Sun, L., Du, D., Cheng, T., Pan, H., et al. (2014). Genome-wide identification, characterization and expression analysis of the MADS-box gene family in Prunus mume. Mol. Genet. Genomics 289, 903–920. doi: 10.1007/s00438-014-0863-z
Yamane, H. (2014). Regulation of bud dormancy and bud break in Japanese apricot (Prunus mume Siebold and Zucc.) and peach [Prunus persica (L.) Batsch]: a summary of recent studies. J. Jap. Soc. Hortic. Sci. 83, 187–202. doi: 10.2503/jjshs1.CH-Rev4
Yamane, H., Ooka, T., Jotatsu, H., Hosaka, Y., Sasaki, R., and Tao, R. (2011). Expressional regulation of PpDAM5 and PpDAM6 peach (Prunus persica) dormancy-associated MADS-box genes, by low temperature and dormancy-breaking reagent treatment. J. Exp. Bot. 62, 3481–3488. doi: 10.1093/jxb/err028
Zhang, Z., Zhuo, X., Zhao, K., Zheng, T., Han, Y., Yuan, C., et al. (2018). Transcriptome profiles reveal the crucial roles of hormone and sugar in the bud dormancy of Prunus mume. Sci. Rep. 8, 1–15. doi: 10.1038/s41598-018-23108-9
Zhao, C., Lang, Z., and Zhu, J. K. (2015). Cold responsive gene transcription become more complex. Trends Plant Sci. 20, 466–468. doi: 10.1016/j.tplants.2015.06.001
Zhao, K., Zhou, Y., Ahmad, S., Yong, X., Xie, X., Han, Y., et al. (2018). PmCBFs synthetically affect PmDAM6 by alternative promoter binding and protein complexes towards the dormancy of bud for Prunus mume. Sci. Rep. 8:4527. doi: 10.1038/s41598-018-22537-w
Zhebentyayeva, T. N., Fan, S., Chandra, A., Bielenberg, D. G., Reighard, G. L., Okie, W. R., et al. (2014). Dissection of chilling requirement and bloom date QTLs in peach using a whole genome sequencing of sibling trees from an F2 mapping population. Tree Genet. Genomes 10, 35–51. doi: 10.1007/s11295-013-0660-6
Keywords: Prunus avium L, chill requirement, blooming, DAMs, gene expression regulation, non-coding gene, UTRs, breeding
Citation: Calle A, Grimplet J, Le Dantec L and Wünsch A (2021) Identification and Characterization of DAMs Mutations Associated With Early Blooming in Sweet Cherry, and Validation of DNA-Based Markers for Selection. Front. Plant Sci. 12:621491. doi: 10.3389/fpls.2021.621491
Received: 26 October 2020; Accepted: 06 May 2021;
Published: 08 July 2021.
Edited by:
Fernando Andres, INRA UMR Amélioration Génétique et Adaptation des Plantes Méditerranéennes et Tropicales, FranceReviewed by:
Pedro Martinez-Gomez, Center for Edaphology and Applied Biology of Segura, Spanish National Research Council, SpainTetyana Zhebentyayeva, Pennsylvania State University (PSU), United States
Copyright © 2021 Calle, Grimplet, Le Dantec and Wünsch. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Ana Wünsch, YXd1bnNjaCYjeDAwMDQwO2FyYWdvbi5lcw==
†Present address: Alejandro Calle, Department of Plant and Environmental Sciences, Clemson University, Clemson, SC, United States