
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 04 September 2020
Sec. Plant Systematics and Evolution
Volume 11 - 2020 | https://doi.org/10.3389/fpls.2020.564414
This article is part of the Research Topic Understanding Plant Diversity and Evolution in the Mediterranean Basin View all 17 articles
 Unai López de Heredia1
Unai López de Heredia1 Fernando Mora-Márquez1
Fernando Mora-Márquez1 Pablo G. Goicoechea2
Pablo G. Goicoechea2 Laura Guillardín-Calvo1†
Laura Guillardín-Calvo1† Marco C. Simeone3
Marco C. Simeone3 Álvaro Soto1*
Álvaro Soto1*Hybridization and its relevance is a hot topic in ecology and evolutionary biology. Interspecific gene flow may play a key role in species adaptation to environmental change, as well as in the survival of endangered populations. Despite the fact that hybridization is quite common in plants, many hybridizing species, such as Quercus spp., maintain their integrity, while precise determination of genomic boundaries between species remains elusive. Novel high throughput sequencing techniques have opened up new perspectives in the comparative analysis of genomes and in the study of historical and current interspecific gene flow. In this work, we applied ddRADseq technique and developed an ad hoc bioinformatics pipeline for the study of ongoing hybridization between two relevant Mediterranean oaks, Q. ilex and Q. suber. We adopted a local scale approach, analyzing adult hybrids (sensu lato) identified in a mixed stand and their open-pollinated progenies. We have identified up to 9,251 markers across the genome and have estimated individual introgression levels in adults and seedlings. Estimated contribution of Q. suber to the genome is higher, on average, in hybrid progenies than in hybrid adults, suggesting preferential backcrossing with this parental species, maybe followed by selection during juvenile stages against individuals with higher Q. suber genomic contribution. Most discriminating markers seem to be scattered throughout the genome, suggesting that a large number of small genomic regions underlie boundaries between these species. In adult hybrids 273 markers (3%) showed allelic frequencies very similar to one of the parental species, and very different from the other; these loci could be relevant for understanding the hybridization process and the occurrence of adaptive introgression. Candidate marker databases developed in this study constitute a valuable resource to design large scale re-sequencing experiments in Mediterranean sclerophyllous oak species and could provide insight into species boundaries and adaptive introgression between Q. suber and Q. ilex.
The relevance of hybridization as an adaptive and evolutionary driving force is a controversial topic. Even the different concepts of species are tightly linked to the relevance authors concede to gene exchange among species (reviewed in Harrison and Larson, 2014). The Biological Species Concept (Mayr, 1982) relies mainly on reproductive isolation among species, while other perspectives admit the “porous” nature of genomes and stress ecological differentiation instead (f. i., Mallet, 2007). Actually, hybridization and introgression are common in plants, where interspecific gene flow is supposed to be an ongoing process in approximately 25% of species (Mallet et al., 2016). Hybridization is especially frequent in certain taxa, as is the case of Quercus species, which have been proposed as models for the Ecological Species Concept (Burger, 1975; van Valen, 1976), and are commonly regarded as “syngameons” (Cannon and Petit, 2020; Kremer and Hipp, 2020). Temperate oaks from Europe and North America have become references for hybridization studies and they have shown that hybridization might be crucial to understand their successful evolutionary trajectories (f.i., Petit et al., 2003; Lepais et al., 2009; Eaton et al., 2015; Goicoechea et al., 2015; Ortego et al., 2018; Crowl et al., 2019).
Hybridization can allow one species to incorporate genes or alleles from another one. Some of these transferred genes can confer a selective advantage to the first species, improving its performance or increasing its ecological range (Sexton et al., 2009). This process is known as adaptive introgression, recently reviewed for plants by Suárez-González et al. (2018). Human-mediated hybridization has been largely used in agriculture, and analysis of spontaneous introgression to search for useful adaptations for crop breeding has been recently claimed, for instance, by Burgarella et al. (2019). Additionally, hybridization has probably played a key role in plant species demography. Threats to small populations can be intensified by lower individual fitness (Allee effect), including reproductive success, maybe related to inbreeding depression. Recurrent hybridization can increase effective population size, overrunning this effect. Moreover, hybridization can also act as a colonization mechanism over other species range (Potts and Reid, 1988; Petit et al., 2003).
Notwithstanding this ample capacity of interspecific gene flow, Quercus species still maintain their integrity (f.i., Valbuena-Carabaña et al., 2005; Moran et al., 2012). Cannon and Scher (2017) presented a well-constructed hypothesis accounting for this apparent paradox. The authors focused on the behavior of male hybrid gametes in the diploid stigma of a pure individual of one of the parental species. In such a situation, it is argued that hybrid gametes most similar to those of that parental species have advantage over the other hybrid gametes. Thus, resulting backcrosses would be quite similar to parental species, bearing, at maximum, a low proportion of introgressed alleles. This would be the reason why species boundaries do not get blurred so often even when there is hybridization. However, such hypothesis does not consider the opposite situation, with hybrids acting as mother trees. In this case, in order to parallel Cannon and Scher hypothesis, it would not be male gametes (assumed to come from parental species), but the female gametes and/or the almost pure zygotes which should show better fitness on a hybrid ovary.
Quercus ilex (holm oak) and Q. suber (cork oak) constitute a very suitable experimental system for hybridization analysis. These two Mediterranean species are easily distinguishable due to their phenotype, mainly according to the characteristic corky bark of Q. suber. Hybridization between both species is well known since ancient times, but it is considered to be rather infrequent. Therefore, their introgression should be more easily traceable than, for instance, the more frequent hybridization and introgression between white oaks, which often hampers correct identification of individuals (Muir et al., 2000; Valbuena-Carabaña et al., 2005). Hybridization is supposed to have played a key role in the survival of Q. suber in Southern France and Eastern Spain, in a general landscape of calcareous soils, not suitable for Q. suber (Lumaret et al., 2002; Jiménez et al., 2004; López de Heredia et al., 2007). In 2009, current hybridization was estimated in less than 2%, in a study using eight nSSR and including natural pure and mixed stands, as well as provenance trials covering the whole distribution range of Q. suber (Burgarella et al., 2009). Nevertheless, recent studies have shown that classification/identification power of the set of nSSR used is too low, and thus, hybridization and introgression could have been underestimated (López de Heredia et al., 2018a; Soto et al., 2018). Therefore, more powerful marker sets are needed in order to obtain accurate estimations.
In recent years, rapid advancement and affordability of next generation sequencing (NGS) methodologies have largely increased the scope and accuracy of genome-wide analysis and its application in phylogenetics, adaptation, hybridization, and breeding studies. For oak species, significant efforts have been made in the white oak complex, producing valuable genomic resources: ESTs, QTLs, SNP panels, or linkage maps, among others (Saintagne et al., 2004; Ueno et al., 2010; Lepoittevin et al., 2015; Lesur et al., 2015; Bodénès et al., 2016; Lesur et al., 2018; Lang et al., 2020). The ongoing progress in NGS technologies has resulted in a first draft genome assembly of Q. suber (Ramos et al., 2018), and in a more complete assembly for Q. robur (Plomion et al., 2018). The latter is the first oak genome assembly that contains information at the chromosome level, thus easing comparative genomic analysis in less studied oak species (see for instance Goicoechea et al., 2015).
The unavailability of reference genome assemblies constrains the scope of genomic studies in non-model species, as is the case for the vast majority of woody plant species (Falk et al., 2018). In this context, the use of reduced representation genotyping methodologies, such as double-digested RAD sequencing (ddRADseq; Peterson et al., 2012), largely facilitates genomic analysis (Hirsch et al., 2014), and has been already used in the phylogenetic analysis (Hipp et al., 2020) and genetic mapping of oak species (Konar et al., 2017). The use of these methodologies, together with the aforementioned NGS-based genomic resources and specifically designed bioinformatic pipelines, opens up new perspectives in oak species genomics.
Here we report the application of ddRADseq to the study of introgression and ongoing hybridization between Q. ilex and Q. suber, using adult hybrids identified in a natural population and their progenies, since natural hybrids can be quite useful for mapping differences and reproductive barriers in non-model organisms (Baack and Rieseberg, 2007). The specific aims of the study were: (1) to identify candidate markers for the study of ongoing hybridization and adaptive introgression and (2) to provide insight into the reproductive behavior of hybrids as mother trees.
The sampling area is located in Fregenal de la Sierra (Badajoz province, southwestern Spain). Twenty-two adult hybrid individuals were identified according to morphological characters, especially those regarding bark, described by Natividade (1936). Leaves from these hybrids, along with leaves from 98 Q. suber and 99 Q. ilex adult individuals were collected and stored at −80°C for further DNA extraction.
In addition, in November 2016, we collected acorns from nine of these hybrid trees, eight of them located in the same estate, in a mixed Q. ilex-Q. suber stand. A total of 1,270 acorns (492 from hybrids, 397 from four cork oaks, and 381 from four holm oaks, presumably unrelated) were collected directly from the canopies, so as to be sure of the identity of mother trees. Acorns were first sown on perlite and seedlings were transplanted to 3 l. containers, in peat:perlite (3:1), and grown in nursery conditions. Hybrid acorns showed lower germination rate, so that the final sampling size for DNA extraction and genotyping consisted of 302 hybrids (22 adults and 280 seedlings), 467 cork oaks (98 adults and 369 seedlings), and 439 holm oaks (99 adults and 340 seedlings) (Table 1).
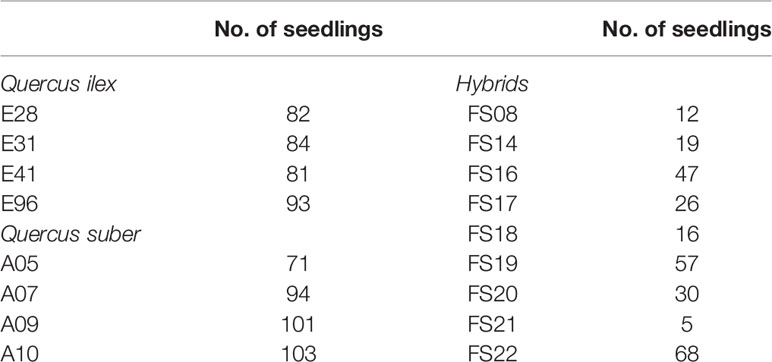
Table 1 Number of seedlings in open-pollinated families, identified by the name of each mother tree.
Leaf discs (6 mm diameter) from adults and seedlings (N = 1,208 samples) were used for DNA extraction on a Genespin™ platform, and for further library preparation and sequencing (LGC Genomics GmbH, Germany).
The trnH-psbA intergenic spacer of chloroplast DNA was amplified and sequenced following Simeone et al. (2016) in the four Q. suber and the four Q. ilex from which acorns were sampled, the 22 adult hybrids and 18 of the hybrid progeny individuals. Four additional adult hybrid individuals from Central Spain (López de Heredia et al., 2018a) were also analyzed. Electropherograms were edited and eye-checked with Chromas 2.6.2 (http://technelysium.com.au). The obtained dataset was enriched with all GenBank accessions showing 100% identity in a nucleotide BLAST search on NCBI (https://www.ncbi.nlm.nih.gov/), and with the trnH-psbA haplotypes representing all the known evolutionary lineages of the West Eurasian Quercus sections Ilex and Cerris (Simeone et al., 2016; Vitelli et al., 2017; Simeone et al., 2018). A multiple alignment was built with MEGA7 (Kumar et al., 2016) and a haplotype list was computed with DNASP 5.1 (Librado and Rozas, 2009). A median-joining (MJ) haplotype network of the entire dataset was inferred with NETWORK 4.6.1.1 (http://www.fluxus-engineering.com/). The MJ algorithm was invoked with default parameters (equal weight of transversion/transition), treating gaps as 5th state.
For ddRADseq analysis of all samples, a pilot study was conducted using the protocol implemented on RADdesigner (Guillardín-Calvo et al., 2019) to assess the optimal enzyme combination, type of library, and number of reads per sample for the aims of the study. Based on this pilot study, PstI/MspI were selected as restriction enzymes. The use of methylation-sensitive restriction enzyme combinations such as PstI/MspI increases the fraction of coding DNA among the sequenced fragments, which is of particular interest in complex plant genomes (Pootakham et al., 2015). ddRADseq libraries were constructed and sequenced by LGC Genomics GmbH (Germany), including a barcode to identify the fragments corresponding to each sample. Libraries were prepared in 14 96-well PCR plates, with 96 inline-barcoded adaptors, and 10 ul of each library were amplified using MyTaq (Bioline GmbH, Germany) and TrueSeq primers (Illumina, USA) in 20 ul PCR reactions. Five ul of each amplified library from the same PCR plate were pooled and cleaned up using Agencourt AMPure XP bead purifications (Beckman Coulter, USA) and MinElute columns (QIAGEN, Netherlands). Normalization was done using Evrogen Trimmer kit (Evrogen, Russia) and reamplified in 100 ul using MyTaq (Bioline GmbH, Germany). Two hundred to 400 bp fragments were selected on Blue Pippin and LMP-agarose gel, in order to produce fewer, yet sufficient, fragments at higher depth. Sequencing was performed on an Illumina NextSeq 500 V2 platform (Illumina Inc., USA) and several runs were performed in order to obtain ~1.5 M 150 bp single-ended reads per sample.
All the scripts to perform the bioinformatic analysis detailed below (Figure 1) are stored in the ddRAD-CORK-HYB package (https://github.com/GGFHF/ddRAD-CORK-HYB). A README file provides instructions to install software and dependencies, and includes information about each script parameter and how to run scripts properly.
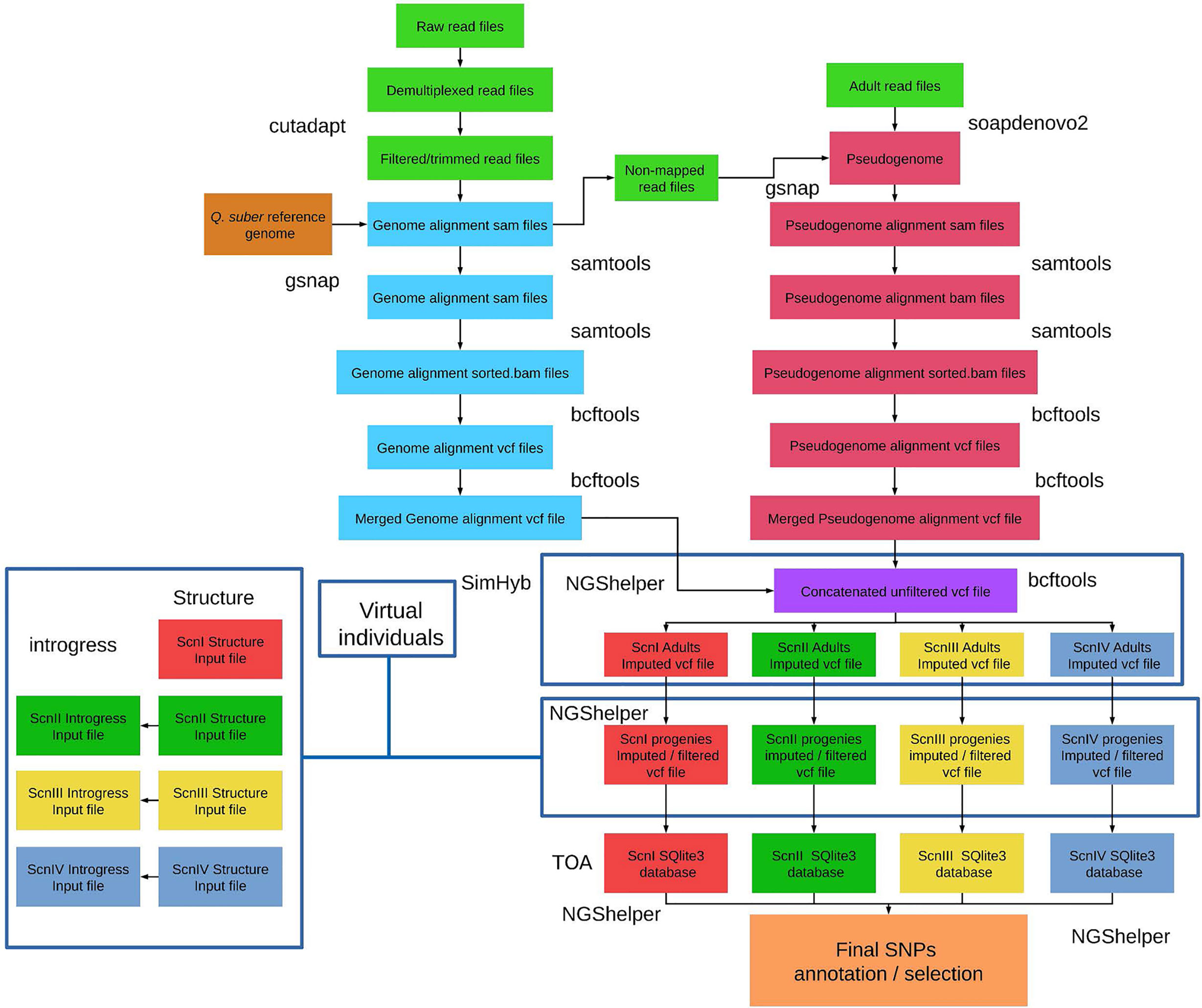
Figure 1 Bioinformatics strategy for ddRADseq data analysis. BASH/R scripts and instructions to install software and dependencies are available by downloading the ddRAD-CORK-HYB package at https://github.com/GGFHF/ddRAD-CORK-HYB.
Read preprocessing consisted in a demultiplexing step of all library groups using the Illumina BCLFASTQC 2.17.1.14 software (http://support.illumina.com/downloads/bcl2fastqconversion-software-v217.html). A maximum of two mismatches or Ns in the barcode read for demultiplexing between libraries was set when the barcode distances between all libraries on the lane allowed for it. Demultiplexing of library groups into samples was performed according to their inline barcodes. No mismatches or Ns were allowed.
Filtering was performed with CUTADAPT (Martin, 2011), and sequencing adapter remnants were clipped from all reads. Reads with final length <20 bp and reads with 5′ ends not matching the restriction enzyme site were discarded. Finally, reads were filtered by quality, removing reads containing Ns and trimming those reads at the 3′-end to get a minimum average Phred quality score of 20 over a ten-base window. Again, reads with a final length <20 bp were discarded. Quality of pre-processed read files was checked with FASTQC (Andrews, 2010).
GSNAP (Wu et al., 2016) was used to align pre-processed and filtered reads to the genome assembly of Q. suber (Ramos et al., 2018). Reads that did not map to the Q. suber genome assembly were aligned back with GSNAP to a pseudogenome built with SOAPDENOVO2 genomic assembler (Luo et al., 2012) using the reads from the adult individuals, and filtered by size to keep only contigs >100 bp.
Genome and pseudogenome alignments in sam format were converted into compressed bam files and sorted with SAMTOOLS (Li et al., 2009). Variant calling was performed individually with SAMTOOLS mpileup and BCFTOOLS (Li, 2011) call options. Variant calling files of each sample from genome and pseudogenome alignments were concatenated with BCFTOOLS to obtain individual vcf files. Finally, all samples were merged with BCFTOOLS merge to obtain a single concatenated-merged unfiltered variant calling file for all the samples (adults, progenies, and replicates).
Multi-fasta sequences of the flanking regions 200 bp to each side of the selected loci positions were extracted from the Q. suber reference genome assembly and from the pseudogenome using the get-flanking-regions.py utility from NGSHELPER (https://github.com/GGFHF/NGShelper). The resulting multifasta file was subjected to a nucleotide BLAST search against Q. robur v. PM1N genome assembly with the local version of BLAST+ (Camacho et al., 2009) using an e-value threshold of 1E-6, to obtain the genomic positions in the Q. robur genome of the identified markers.
Further, we built a linear function to correlate the genomic coordinates in bp of the SNPs used to build linkage groups in Q. robur (Bodénès et al., 2016) with the positions of those SNPs in 12 Q. robur linkage groups in cM. All this information was freely available at the oak genome sequencing web (http://www.oakgenome.fr) and at the Quercus portal (https://arachne.pierroton.inra.fr/cgi-bin/cmap/). Thus, we were able to identify the putative location of our marker loci in the linkage groups of Q. robur, and to visualize them with the R package LINKAGEMAPVIEW (Ouellette et al., 2017).
SQLITE3 databases for each imputation scenario (see below) were constructed with NGSHELPER including information parsed from vcf files (type of marker, genomic coordinates, etc.) and functional annotation of selected markers. Functional annotations of the markers identified in the genome were collected from Q. suber genome gff files and descriptions of gene IDs was obtained from NCBI ‘All_Plants.gene.info’ file. The markers corresponding to the pseudogenome were annotated with TOA (Mora-Márquez et al., 2020), through nucleotide BLAST searches against NCBI NT databases.
Missing data must be considered for vcf files processing steps. There is no standard missing data cutoff for excluding loci, and filtering criteria need to be established depending on the aims of the study (Huang and Knowles, 2016). Excessive data filtration can have unforeseen consequences, such as the truncation of loci with higher mutation rates or missing relevant loci. Besides, filtering protocols must ensure sufficient depth coverage of the remaining loci (Eaton et al., 2017). A common practice is to test for different scenarios encompassing more restrictive and more relaxed filtering criteria.
The procedures to perform filtering and imputation to account for missing data and/or null alleles are implemented in NGSHELPER. Input data consist of the single concatenated-merged unfiltered variant calling file generated in the previous step. The basic procedure included two steps. Firstly, the proportion of missing data in adult individuals of each parental species was considered. For loci with a low proportion of missing data in one of the parental species (md < 0.05) but with a high proportion in the other (md > 0.90), a null allele was imputed to missing data of the adult individuals of the latter species. Successive filtrations of imputed loci were performed, resulting in different imputation scenarios (see Results section). In a second step, genotypes of the seedlings were corrected according to their mother tree imputation state.
Estimation of the introgression level of adult individuals and progenies was performed using the Bayesian approach implemented in STRUCTURE v. 2.3.4 (based on Pritchard et al., 2000), as well as by means of the hybrid index provided by INTROGRESS (Gompert and Buerkle, 2010). Firstly, we checked the performance and accuracy of both classification methods using virtual individuals with known introgression levels. These individuals were simulated with SIMHYB (Soto et al., 2018; https://github.com/GGFHF/SimHyb), based on the allele frequencies of the adult Q. ilex and Q. suber populations. Later on, we estimated contribution of parental species to the genome of each individual in two scenarios of prevalence of hybrids: 1 and 10%. In both cases, additional virtual Q. ilex and Q. suber individuals were included as reference. Number of real and virtual individuals used, according to their introgression level, are provided in Supplementary Information (Supplementary Table 1). In all the cases, we allowed STRUCTURE to estimate admixture proportions in the unknown samples. For all jobs, we used 10,000 burn-in steps followed by 100,000 repetitions of the MCMC chains, and the number of populations was set to K = 2, following the methodology described by Evanno et al. (2005).
Twenty-two adult individuals, identified as Q. ilex–Q. suber hybrids according to their phenotypic characters, were localized in the field, in different estates in the south of Badajoz (SW Spain). Acorns were collected from the canopy of these hybrid trees, as well as from unrelated Q. ilex and Q. suber adults (four of each species). While parent species showed high germination rates (89.24% for Q. ilex and 92.95% for Q. suber), only 56.92% of hybrid acorns germinated successfully.
Chloroplast DNA analysis of these trees revealed that the four cork oak adult individuals gathered in a single haplotype (H12), corresponding to a widespread, likely ancestral haplotype, shared by multiple Cerris section species extending from Anatolia to the central Mediterranean. The four holm oak adult samples grouped with all hybrid adults and progenies in the ‘Euro-Med’ haplotype group (sub-lineage V) (Simeone et al., 2018), i.e. an exclusive lineage of the central and western Mediterranean members of section Ilex (Q. ilex and Q. coccifera) (Figure 2 and Supplementary List 1). This result confirms the directionality in Q. ilex–Q. suber hybridization, in which holm oak acts as mother tree, and therefore hybrids carry holm oak chloroplasts. The trnH-psbA haplotype sequences generated in this study are available on GenBank under accession numbers LR797857-LR797862.
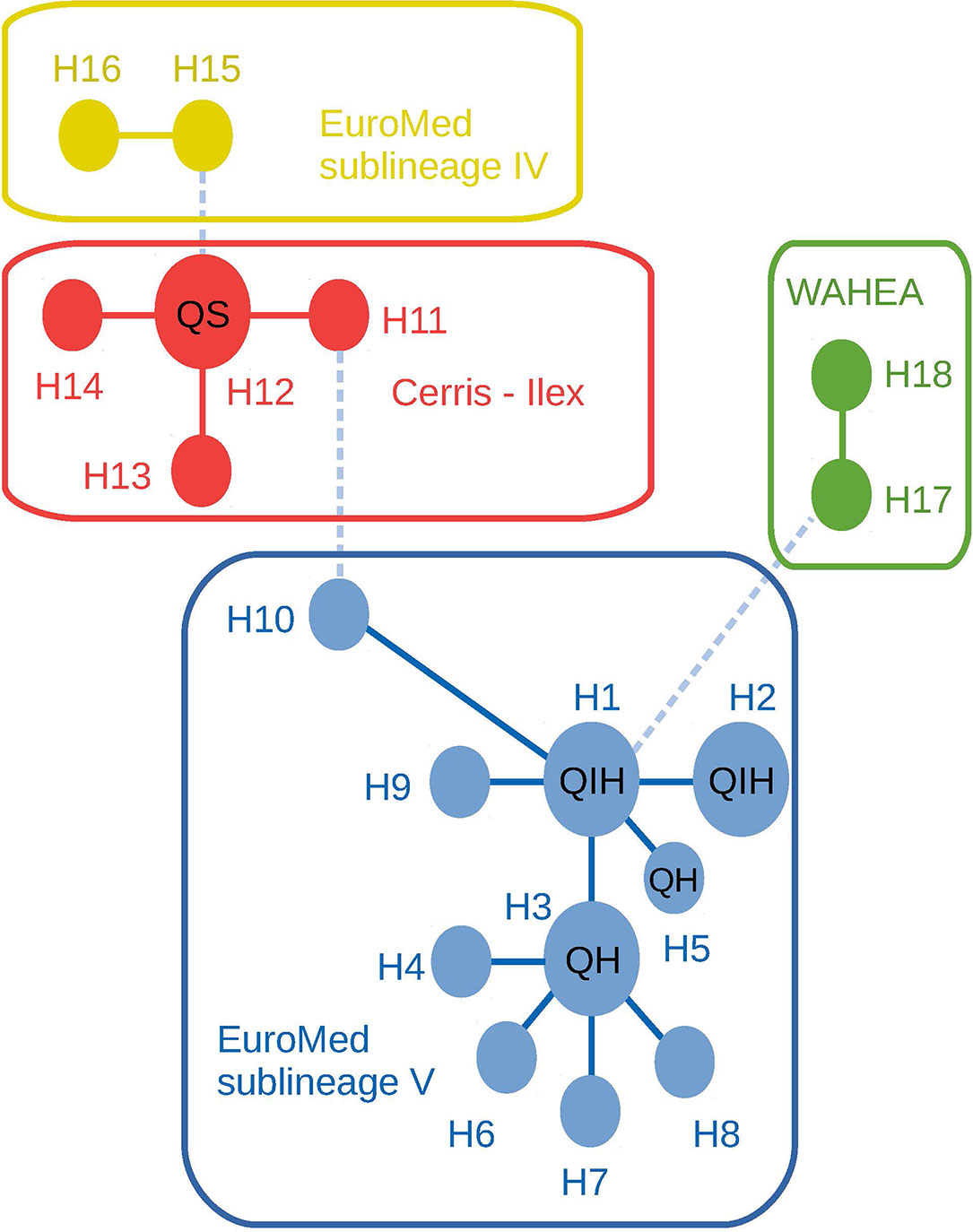
Figure 2 Median-joining haplotype network of trnH-psbA marker combining the investigated samples and published haplotypes of each major Ilex and Cerris lineages. Lineage and sub-lineage identity according to Vitelli et al., 2017 and Simeone et al., 2018. WAHEA: haplotypes from the West Asian, Himalayan, and East Asian lineages. Line types correspond to the number of mutations separating each haplotype: solid lines = 1–3 mutations; dashed lines 6–10 mutations. Coding within haplotype circles indicate the presence of samples of the present work: QS, Adult Q. suber individuals; QIH, Adult Q. ilex and hybrid individuals; QH, only hybrid individuals.
A mean number of 1.74 million single-ended reads per sample was obtained after removal of adapters and read filtering by quality. Sequences are available at NCBI SRA database (BioProject: PRJNA628590). From these, 64.9% of all filtered reads mapped uniquely to the Q. suber genome assembly (Figure 3). However, we found noticeable differences in the total number of aligned reads among progenies and adult individuals of each species. As expected, Q. suber libraries aligned better than Q. ilex, while the hybrids showed intermediate behavior. Reads showing ambiguous mapping were discarded, and reads that did not map to the Q. suber genome assembly were aligned to the pseudogenome, improving total alignment percentages to 67.8% of all filtered reads. The best alignments to the pseudogenome were obtained for those Q. ilex libraries with the poorest mapping to the Q. suber genome.
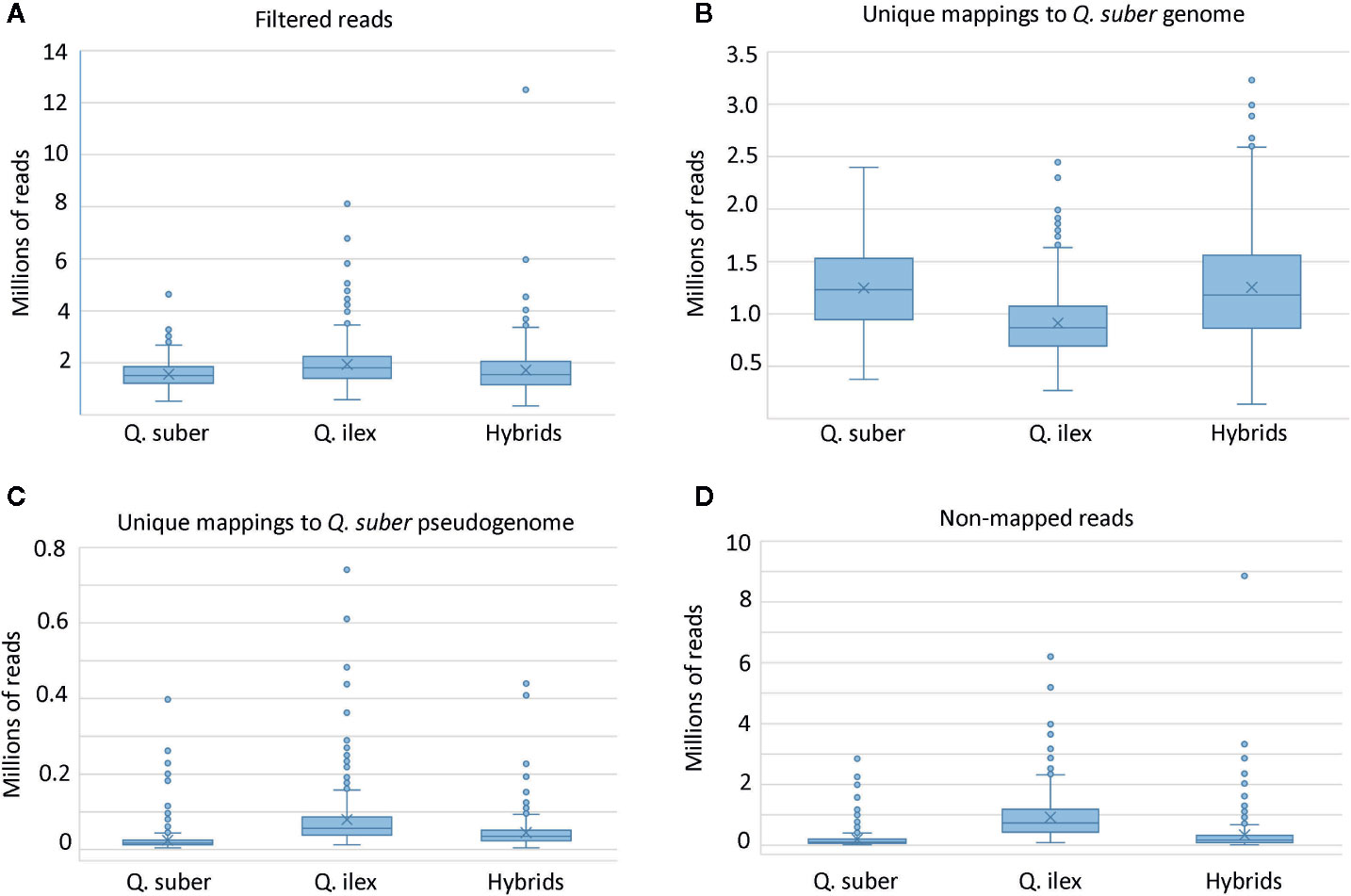
Figure 3 Box and whisker plots of mapping sequences. (A) Total number of reads employed in the ddRADseq pipeline after adapter removal and quality filtering. (B) Number of reads uniquely mapped to Q. suber genome assembly (Ramos et al., 2018). (C) Number of reads uniquely mapped to the pseudogenome generated with SoapDeNovo2. (D) Final number of non-mapped reads.
After individual variant calling, the number of variants ranged between 14,593 and 524,458 for the genome and between 212 and 66,680 for the pseudogenome alignments. The final concatenated-merged variant calling file had 16,234,798 variants, of which 97.4 % were SNPs and <5% multi-allelic sites. This massive vcf file was subjected to filtering and imputation according to four scenarios.
As a basic filtering, we removed loci with low coverage (DP, total number of reads across all samples, <6000). On average, ~70 reads per sample were obtained for each remaining locus. Additionally, for those loci with a low proportion of missing data in one of the parental species (md < 0.05) but with a very high proportion in the other parental species (md > 0.90), a null allele was assigned to such missing data and to one of the alleles in homozygous individuals in the latter species; this was considered the scenario I (ScnI). These loci (hereinafter referred to as imputed loci) can correspond to alterations in one or both restriction sites (including methylation), yielding no scorable fragments in the sequencing phase, or to larger indels in the genome of one of the species; in any case, they can be highly informative. Nevertheless, in order to avoid an excessive impact of imputed loci, we conceived a second scenario (ScnII), in which we kept only one imputed locus per gene or intergenic fragment of the Q. suber genome (Ramos et al., 2018). The location of a locus within an intergenic fragment was assessed considering a proximity criterion using a 10 kb sliding window. This way, ScnII kept approximately 80% of the loci imputed under ScnI. In ScnIII we removed all the imputed alleles, and considered them as missing data. Finally, in the most restrictive scenario, we discarded all the loci with a proportion of missing data >90% in either of the adult Q. suber or Q. ilex populations (ScnIV). Later on, we also corrected the genotypes of the progenies according to each imputation scenario, and considered as missing data those genotypes incompatible with that of the corresponding mother tree.
The number of final recovered loci varied depending on the scenario (Figure 4A). For ScnI and ScnIII we obtained up to 9,251 loci, with 2,026 (21.9%) imputed ones in ScnI. Under ScnII we considered 8,901 loci, with 18.8% of them imputed. The more restrictive ScnIV kept 7,225 unimputed loci. The annotation to the Q. suber genome allowed precise location of many of these loci in specific genes of known function (Ramos et al., 2018) and intergenic regions (Figure 4B). Loci fromScnI, ScnII and ScnIII were located in 3,396 fragments, of which 2,566 were genic and 811 intergenic. For Scn IV the number of fragments dropped to 1,829, of which 1,540 (84.2%) corresponded to genic regions and 279 (15.3%) to intergenic ones. In all the scenarios, loci corresponding to genic regions were mostly exonic c. 72%), although a significant percentage of loci (c. 28%) occurred in introns (Figure 4B). The remaining loci (0.3%) were located in 29 fragments that could not be mapped to the Q. suber genome assembly. Databases of markers are available at Zenodo Repository (López de Heredia et al., 2020).
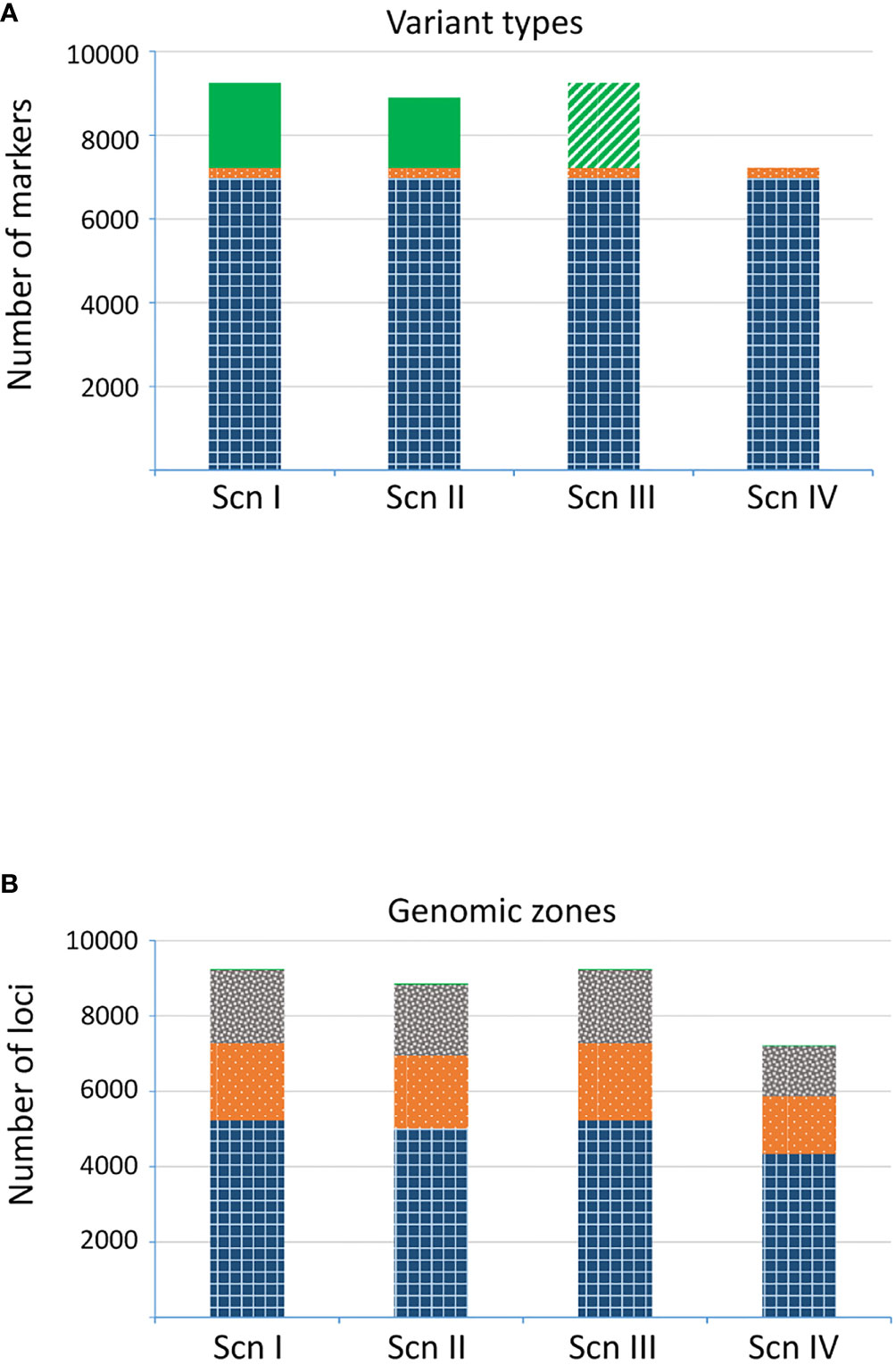
Figure 4 Number of loci detected for each scenario. (A) Variant types: SNP (gridded blue), indel (dotted orange), and imputed loci (plain green; treated as missing data in ScnIII). (B) Genomic region: known exonic region (gridded blue), known intronic region (dotted orange), intergenic region (grey confetti), not assigned region (plain green).
In order to evaluate their distribution across the genome, and provided there is no information regarding chromosomal distribution of Q. suber nor Q. ilex genomes, we checked the homology of these loci on the Q. robur genome, for which 12 linkage groups (corresponding to the 12 chromosomes) have been identified (Bodénès et al., 2012; Bodénès et al., 2016). A total of 8,774 loci were successfully mapped against Q. robur genome; of these, 8,004 showed homology with loci included in the 12 linkage groups. These loci belong to 2,932 genomic fragments: 2,264 genic and 668 intergenic. We found a rather even distribution of these loci among the 12 linkage groups, with an average distribution of almost 670 loci per linkage group, approximately 10.55 loci/Mb (Figure 5). Assuming a high synteny among species of the genus (Bodénès et al., 2012; Konar et al., 2017), these results suggest an unbiased distribution of our loci across Q. suber and Q. ilex genomes.
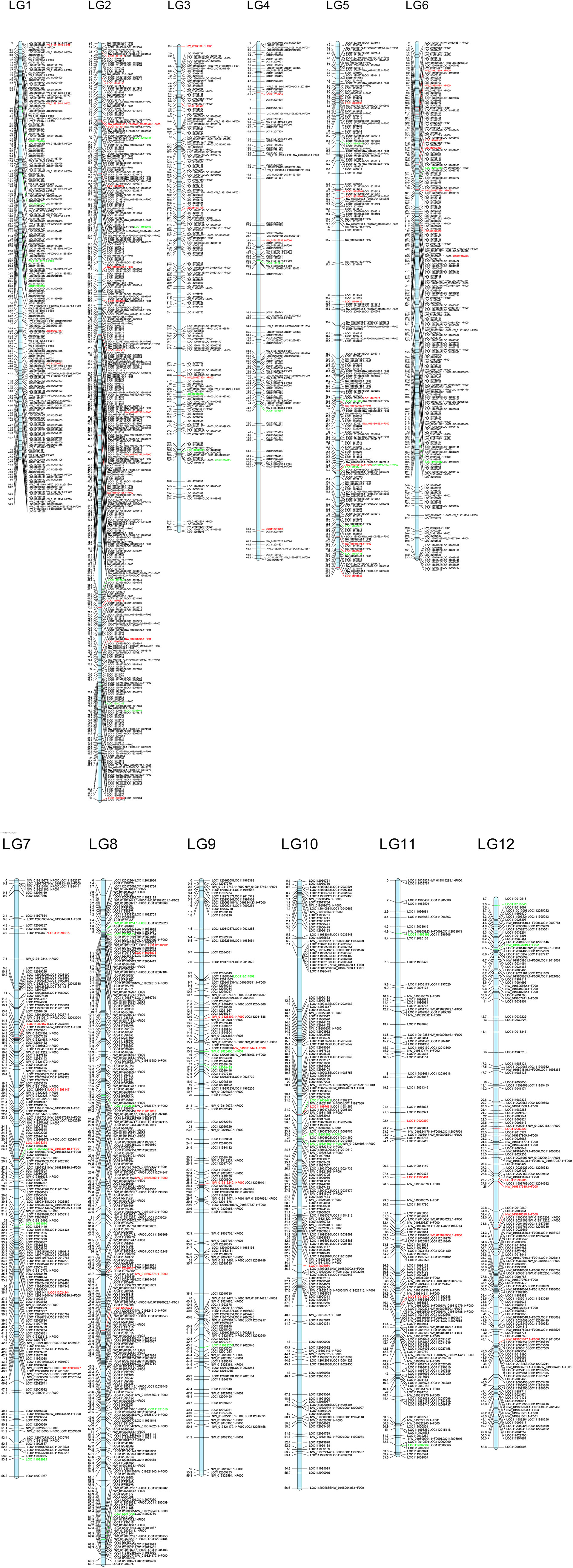
Figure 5 Putative location in the Q. robur linkage groups of the genomic fragments including markers from ScnI. Location of markers with allelic frequencies in the adult hybrids very similar to one of the parental species is highlighted in red (Q. ilex) or in green (Q. suber).
The Bayesian approach implemented in STRUCTURE v. 2.3.4 (Pritchard et al., 2000), as well as the hybrid index provided by the INTROGRESS R-package (Gompert and Buerkle, 2010), were then used to classify individuals according to their introgression levels. Firstly, and following the procedure established by Burgarella et al. (2009), accuracy of the classification was evaluated using virtual individuals of known pedigree created with SIMHYB (Soto et al., 2018). ScnIII yielded the same results as ScnIV, due to the distribution of missing data among species and the way both programs consider them. Therefore, ScnIII was discarded for further analysis of real individuals (Figure 6).
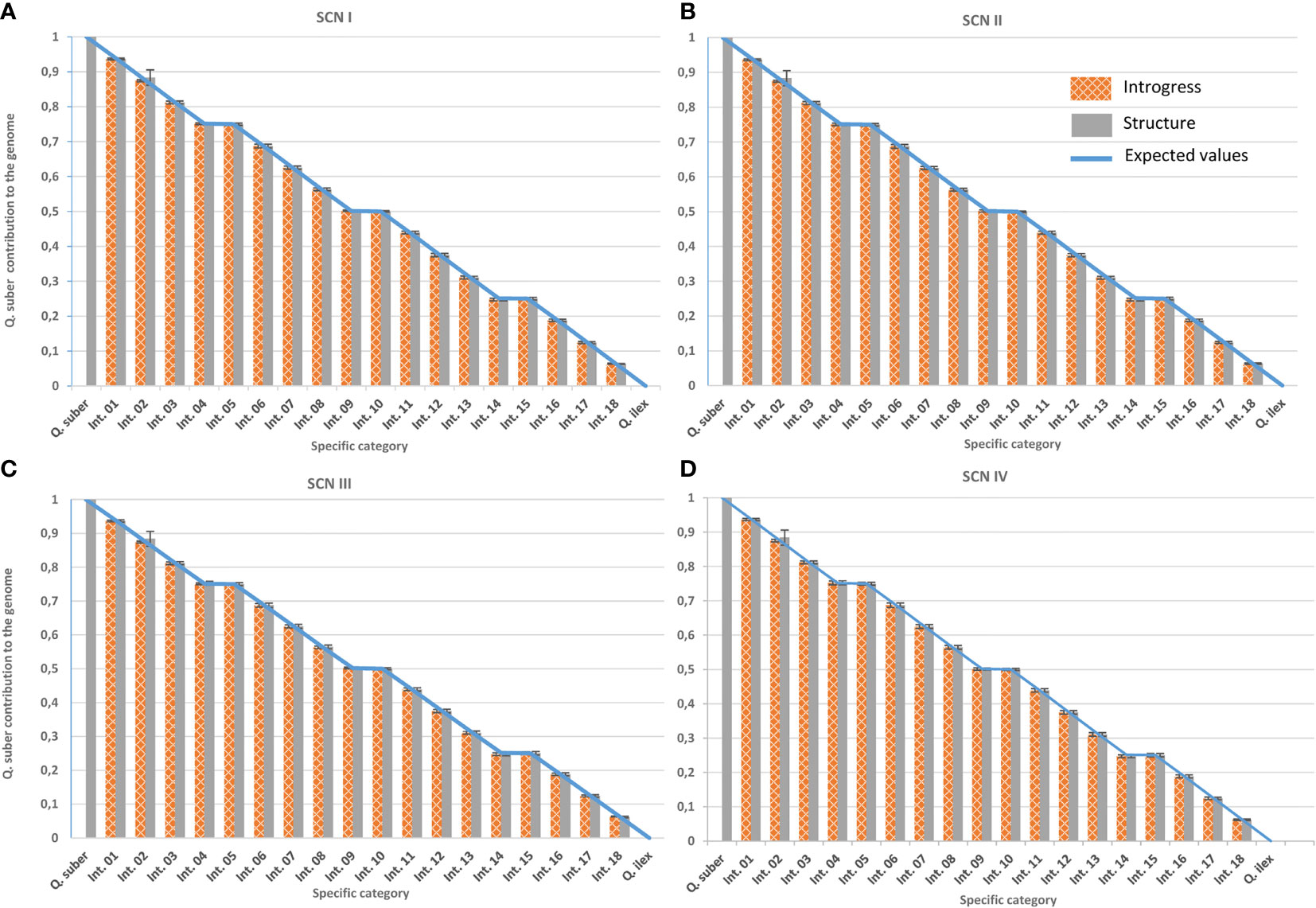
Figure 6 Performance of classification tools under the different imputation scenarios. Genomic contribution of Q. suber is estimated using Structure’s qs (plain grey) and Introgress hybrid index (gridded orange) on virtual individuals generated with SimHyb (pure species and 18 intermediate categories). Expected values are represented by a blue line. Standard deviations are also indicated. (A) Imputation scenario I; (B) imputation scenario II; (C) imputation scenario III; (D) imputation scenario IV.
Once accuracy and reliability of classification tools had been checked, introgression levels of real hybrid adults were assessed. Estimation was performed considering 1% and 10% of hybrids in the analyzed population. INTROGRESS and STRUCTURE yielded similar results in each situation, and very small differences were detected between both hybrid prevalence situations. Under the four imputation scenarios, estimations for adult individuals were roughly compatible with F1 hybrids (except for FS-01, which could be classified as a backcross with Q. suber) (Figure 7). As for the hybrid progenies, a large variability of introgression levels was detected, although we scored, on average, a higher contribution of Q. suber to the genome of the seedlings for all the hybrid families, compared to their mother trees (Table 2).
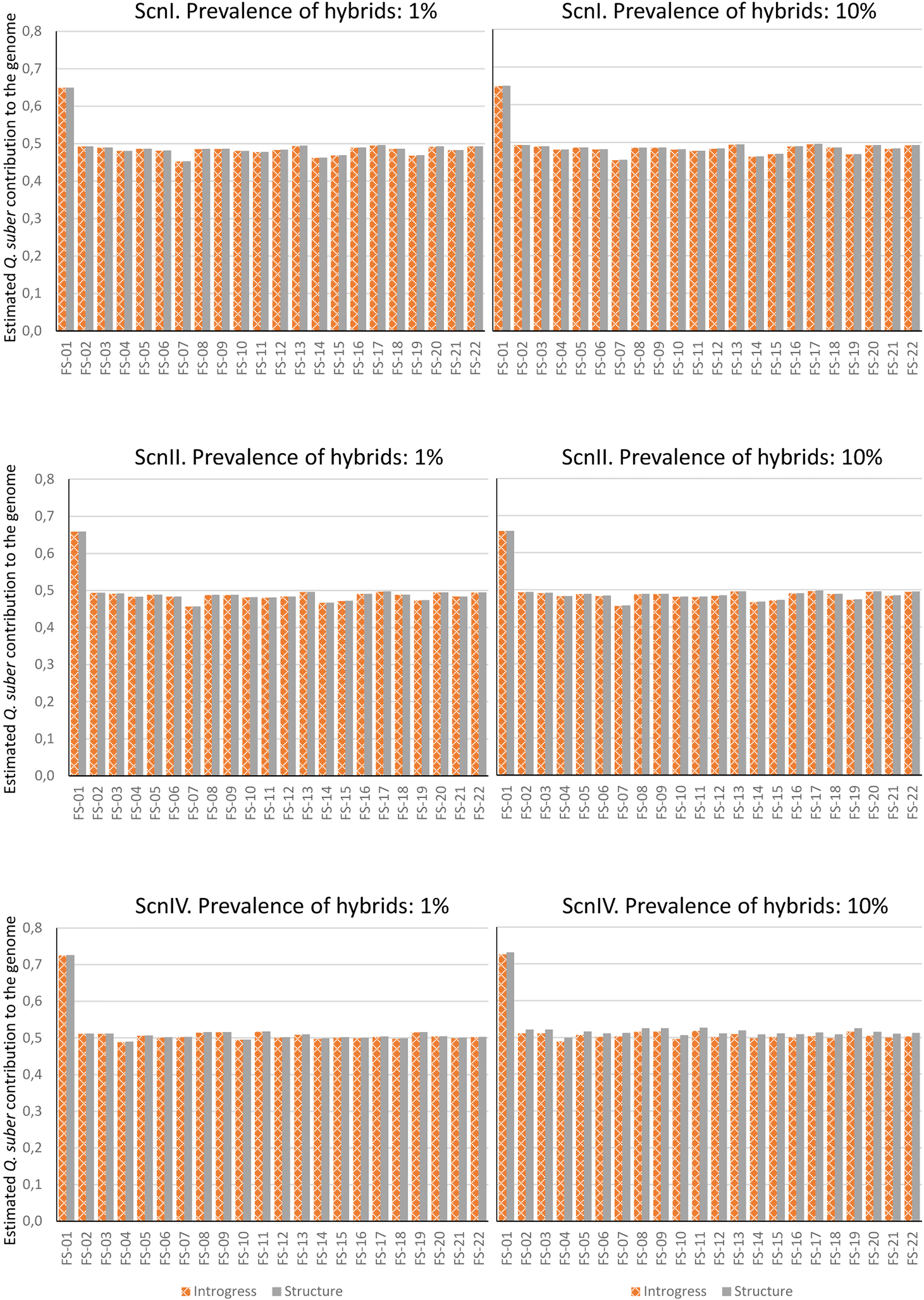
Figure 7 Contribution of Q. suber to the genome of adult hybrids under each scenario, estimated by means of STRUCTURE's qs and Hybrid Index of INTROGRESS, with 1% and 10% of hybrids.
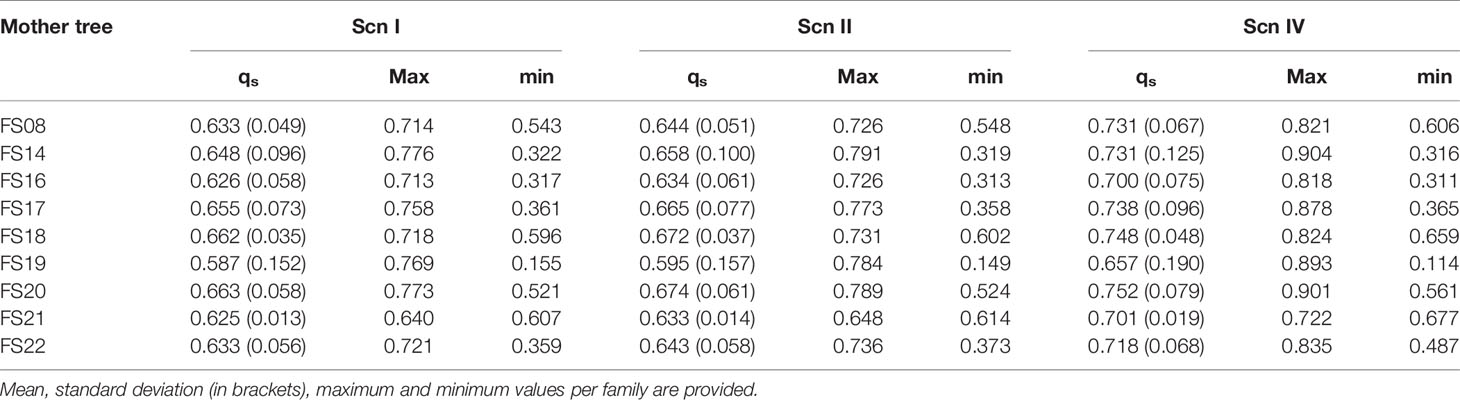
Table 2 STRUCTURE’s qs and INTROGRESS hybrid index estimates for the progenies of the open-pollinated hybrid families under each scenario.
Following López de Heredia et al. (2018a), we compared STRUCTURE estimates of the Q. suber contribution to the genome of each hybrid seedling to that of its mother tree (qsoffspring/qsmother). Values higher than 1 point to likely fertilizations by Q. suber, while values below 1 suggest fertilization by Q. ilex. This comparison suggests that hybrid mother trees can be fertilized by both Q. ilex and Q. suber pollen, although more frequently by the latter species, at least in the studied season (Table 3).
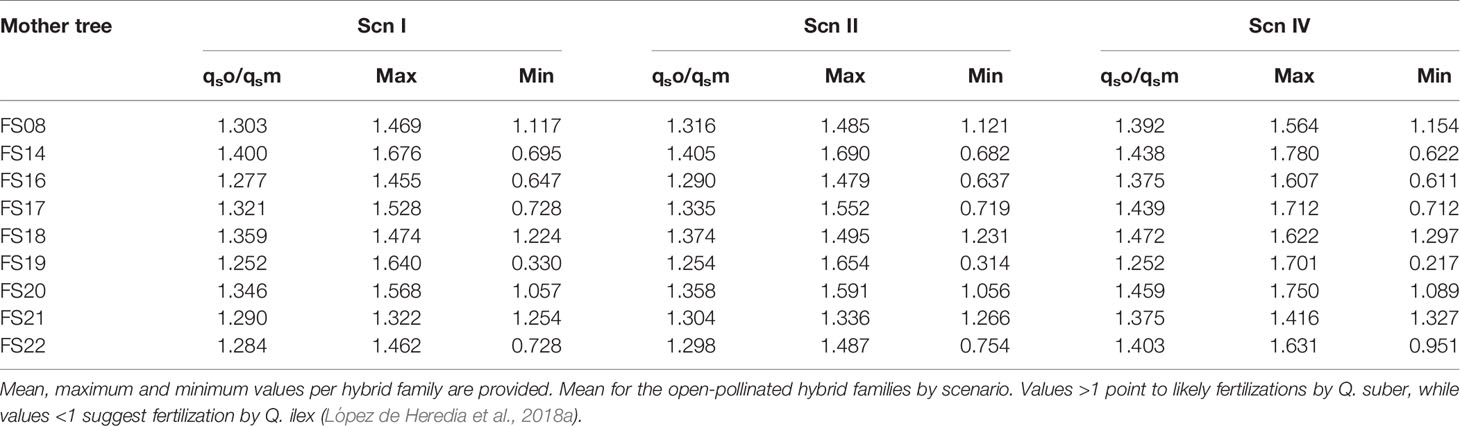
Table 3 Ratio between STRUCTURE’s qs of the offspring and qs of their mothers under each scenario.
Recent advances in NGS methodologies along with the release of reference genome assemblies have opened up new perspectives in the study of open pollinated plant hybridizing systems, such as the Quercus syngameon (Kremer and Hipp, 2020). In this work, we have used ddRADseq methodology and have developed ad hoc bioinformatic pipelines to identify genomic variants that may shed light in the study of ongoing hybridization and genomic boundaries between Q. suber and Q. ilex. This insight may complement the current view derived from other presumably more introgressed Quercus complexes, such as the intensively studied European white oaks.
Most hybridization studies in woody plant species have been conducted to assess ancient introgression from a phylogenetic perspective (McVay et al., 2017) or ongoing introgression by sampling individuals exploring ample hybrid zones (Lepais et al., 2009; Zeng et al., 2011). In this work, we have adopted a local scale approach, aiming to block the impact of environmental or IBD related variation in candidate loci inference. The sampling scheme of adults and progenies of both parental species and hybrids used here has revealed powerful to identify candidate gene markers for the study of ongoing hybridization in Mediterranean sclerophyllous oaks.
Obtaining candidate loci in reduced genome representation studies (ddRADseq), particularly if they are aimed to perform individual classification of hybrids, requires careful experimental design to ensure sufficient coverage of the genome of the focal species and rigorous filtering of variants in order to minimize the potential sources of error in marker identification (O’Leary et al., 2018). In the present work, an experimental design derived from a pilot study (Guillardín-Calvo et al., 2019), together with an ad hoc bioinformatic pipeline has enabled the identification of a significant number of genomic markers to analyze ongoing hybridization between Q. ilex and Q. suber.
We employed an enzyme combination (PstI/MspI) that prioritizes the representation of loci within or in the proximity of genic regions that could be relevant for the study of introgression patterns, enriching for hypomethylated gene space and reducing the number of fragments from highly repetitive genomic regions (Emberton et al., 2005; Nelson et al., 2008; Poland and Rife, 2012). Moreover, a combination of methylation-sensitive rare- and common-cutting enzymes, such as PstI/MspI, has proven to show greater uniformity of read depth across loci providing high quality genotype information in plants (Pootakham et al., 2016). We have also optimized ddRADseq protocols by performing a size selection step to ensure the collection of robust polymorphic loci at sufficient depth. Actually, genome mapping and variant calling using Q. suber genome assembly as a reference have confirmed that most candidate polymorphic markers (c. 80%) correspond to genic regions, more than 55% of loci are located in exons and c. 22% in introns. Approximately 20% of loci were located in intergenic regions, and, comparatively few candidate loci (0.3%) were obtained from the pseudogenome mapping. However, we cannot discard that some of these regions could contain genes or regulatory regions in our focal species and further re-sequencing experimental work will be necessary to confirm this point.
To gain robustness in candidate marker inference, we have applied a filtering-imputation procedure to yield four different scenarios. Using restrictive filteringcriteria (ScnIV), we have obtained 7,225markers that correspond to 1,540 genic fragments of known function, 279 intergenic fragments and 10 fragments that could not be assigned to Q. suber genome assembly. Nevertheless, strict application of these criteria could have discarded informative loci, for instance, those present in the genome of one of the parental species but absent in the other. To avoid missing this information, we designed an imputation procedure, giving rise to what we have called scenarios I and II. This way we identified up to 2,026 additional loci, with imputed null alleles, under ScnI. These loci, which could be highly informative for introgression studies, belonged to 1,264 genic fragments of known function, 584 intergenic fragments and 9 fragments that could not be assigned to Q. suber genome assembly. It is noteworthy that many of these null alleles were imputed to Q. ilex.
The markers identified in this work provide a well-distributed coverage of the whole genome, assuming synteny among genomes of the genus (Bodénès et al., 2012; Konar et al., 2017), since their orthologs appear distributed throughout the 12 linkage groups described for Q. robur (Plomion et al., 2018) (Figure 5). Furthermore, we have investigated the distribution of the most discriminating genomic regions between Q. suber and Q. ilex. Thus, in addition to the 2,026 imputed loci, up to 2,830 non-imputed ones show very different patterns in both species, with frequencies of the most common allele 0.9 in one of the species and 0.2 in the other one. Regarding the hybrids, under ScnI 190 loci (167 imputed) show allelic frequencies in the hybrids quite similar to those of Q. ilex and very different from Q. suber, while 83 loci (17 imputed) show frequencies in the hybrids very similar to Q. suber and different from Q. ilex. These loci, presumably linked to introgression directionality and genomic barriers between Q. suber and Q. ilex, are scattered throughout the 12 linkage groups (Figure 5). This feature seems to be consistent with previous results for Q. robur and Q. petraea based on QTL (Saintagne et al., 2004), suggesting that species boundaries are underpinned by a large number of small genomic regions, rather than on few large blocks.
Most adult hybrids could be classified as F1 hybrids under all the imputation scenarios considered. Inclusion of imputed loci does not entail a significant difference in the estimation of the contribution of parental species to the genome of hybrid individuals. For the adult hybrids, only slightly lower values of Q. suber contribution are obtained under ScnI and ScnII. Different results are observed for the hybrid progenies. Individuals with higher estimated Q. suber contributions under ScnIV show lower values when imputed loci are considered, while the opposite is observed for individuals with lower estimations. Since most of the null alleles are imputed to Q. ilex, this latter result must be due to a higher proportion of non-imputed, “suber” alleles in heterozygosity in these loci in these individuals. Taking into account PstI/MspI sensitivity to methylation, hybridization-mediated alteration of epigenetic characters could also contribute to these results. This way, methylated epialleles in the restriction sites, which would yield no scorable reads and, therefore, would have been imputed with a null allele, could have turned out to be unmethylated and therefore scorable in hybrids, or vice-versa. This could be the case at least of the 184 markers for which very high frequencies of the imputed allele are recorded in adult hybrids ( 0.75), no matter their global classification as F1 hybrids. Such an alteration of epigenetic characters has been reported, for instance, in Spartina (Salmon et al., 2005) or in Solanum (Marfil et al., 2006). On the contrary, Radosavljevic et al. (2019) obtained similar estimations of introgression levels using markers sensitive or insensitive to methylation in Salvia officinalis and S. fruticosa hybrids. Further research will be needed to disentangle the effects of Q. ilex and Q. suber hybridization in epigenetic patterns.
On the other hand, estimated contribution of Q. suber increases in the genome of hybrid offspring compared with their mother trees, for all the families and in all the scenarios considered. These results suggest a preferential fertilization of hybrid trees by Q. suber pollen. Different factors, including phenology can account for this feature. In fact, earlier flowering of Q. ilex (Gómez-Casero et al., 2007) favors that initial hybridization takes place with Q. ilex acting as mother tree; additionally, pre-zygotic incompatibilities for Q. ilex pollen in Q. suber pistil have also been reported (Boavida et al., 2001). Consistently, all the 22 adult hybrid individuals included in this study carry ilex chloroplasts. Actually, ilex chloroplasts have been found in all hybrid trees analyzed to date (Lumaret et al., 2002; Burgarella et al., 2009; López de Heredia et al., 2018a). Thus, hybrid mother trees could be preferentially fertilized by Q. suber. The only study performed to date, to our knowledge, reports a similar reproductive phenology for hybrid trees and Q. suber (Perea Garcia-Calvo, 2006), so that backcrossing with this species would be more likely. Additionally, pre- and post-zygotic incompatibilities or a better performance of Q. suber pollen tubes in the hybrid pistil could also account for such a preferential fertilization. In spite of this, López de Heredia et al. (2018a) reported that hybrids can be effectively pollinated both by Q. suber and by Q. ilex and that backcrossing directionality is largely driven by pollen availability. Consistently, and notwithstanding the preferential fertilization by Q. suber, in the present work we also report some hybrid seedlings presumably derived from Q. ilex pollen in most families. This result is also in accordance with the detection of putatively introgressed holm oaks reported by Burgarella et al. (2009). Moreover, our analyses yield intermediate contribution of parental species to most hybrid seedlings (according to both STRUCTURE and INTROGRESS). This feature in hybrid mother trees contrasts with Cannon and Scher (2017) hypothesis for hybrid pollen and should not be disregarded. Actually, it may be more likely that hybrid trees scattered in stands of parental species reproduce acting as mother trees, pollinated by parental species, than as pollen donors. In that case, there is no reason to suppose any advantage for pure parental gametes in a diploid, hybrid stigma or for “pure” or “almost pure” zygotes and seed development in the hybrid sporophyte (mother tree).
The larger contribution from Q. suber to hybrid progenies’ genomes compared to the adults could be due to greater availability of Q. suber pollen in the only campaign analyzed in the present study. However, a selective advantage for individuals more similar to Q. ilex, from the seedling to the adult stage cannot be discarded either. Such a selection would not be unexpected. Fertilization of hybrids can give rise to a huge amount of genomic combinations. Large part of them can be incompatible since the first developmental stages, hampering the formation of embryos. Later on, other combinations can also be deleterious, as confirmed by the low germination rate of hybrid acorns in this study (56.92% in hybrids, compared to 89.24% in Q. suber and up to 92.95% in Q. ilex), consistent with previous studies in other areas (López de Heredia et al., 2018a). Although many morphological anomalies already described in Q. suber x ilex hybrid seedlings (López de Heredia et al., 2017; López de Heredia et al., 2018b) were present in part of the seedlings, we have scored low mortality in nursery conditions over three years since seedlings emergence. Nevertheless, selection against certain genomic combinations—late-acting genetic incompatibilities—during the juvenile phases of hybrid individuals with major Q. suber ancestry could also account for the higher contribution of Q. ilex to the genome of surviving hybrids. In addition, extrinsic selection during pre-adult stages, mitigated under nursery conditions, cannot be discarded.
Our work reports a case study of hybridization and introgression in two non-model forest tree species, Q. suber and Q. ilex, using genome-wide NGS techniques, and provides a pipeline and scripts for this kind of studies. Out of the 9,251 marker loci identified in this study, 4,856 are highly discriminant between both species, and 2,026 of these are apparently absent in one of the species (Q. ilex in most cases). This can be due to alterations in restriction enzyme target sites or to real indels. Interestingly, for 9.1%of them adult hybrids show patterns quite similar to one of the parental species (8.3% to Q. ilex, while only 0.8% to Q. suber), suggesting selection of those alleles in backcrosses or hybridization-mediated alterations of the methylation patterns. In any case, these loci deserve further attention, since they could be linked to viability of hybrid individuals and/or to selective advantages.
Most discriminant loci and their adjacent regions, as well as those putatively involved in selection of hybrids, should be analyzed both in hybrid individuals and in parental species. These genomic regions are good candidates to be involved in genome permeability, interspecific barriers and, eventually, in adaptive introgression. Further characterization of these loci, as well as of those with different allele frequencies in hybrid adults and seedlings could provide insight in species boundaries and on adaptive introgression between Q. suber and Q. ilex.
The datasets presented in this study can be found in online repositories. The names of the repository/repositories and accession number(s) can be found below: https://www.ncbi.nlm.nih.gov/genbank/, LR797857-LR797862 https://www.ncbi.nlm.nih.gov/, PRJNA628590.
AS conceived the idea and drafted the manuscript. AS and ULH designed the experiments and collected the plant material. LG-C handled the plant material. MS performed the cpDNA analysis. FM-M, PGG, ULH, and AS performed the simulations and bioinformatics analysis. All the authors contributed to the article and approved the submitted version.
This work was funded by the projects AGL2015-67495-C2-2-R (Spanish Ministry of Economy and Competitiveness) and PID2019-110330GB-C22 (Spanish Ministry of Science and Innovation).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer JM declared a past co-authorship with one of the authors MS to the handling Editor.
The authors thank Francisco Martínez Moreno for his help in hybrids localization and sampling in the field.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2020.564414/full#supplementary-material
Andrews, S. (2010). FastQC: a quality control tool for high throughput sequence data. Available at: http://www.bioinformatics.babraham.ac.uk/projects/fastqc (Accessed March 15, 2019).
Baack, E. J., Rieseberg, L. H. (2007). A genomic view of introgression and hybrid speciation. Curr. Opin. Genet. Dev. 17, 513–518. doi: 10.1016/j.gde.2007.09.001
Boavida, L. C., Silva, J. P., Feijó, J. A. (2001). Sexual reproduction in the cork oak (Quercus suber L). II. Crossing intra- and interspecific barriers. Sex Plant Reprod. 14 (3), 143–152. doi: 10.1007/s004970100100
Bodénès, C., Chancerel, E., Gailing, O., Vendramin, G. G., Bagnoli, F., Durand, J., et al. (2012). Comparative mapping in the Fagaceae and beyond with EST-SSRs. BMC Plant Biol. 12:153. doi: 10.1186/1471-2229-12-153
Bodénès, C., Chancerel, E., Ehrenmann, F., Kremer, A., Plomion, C. (2016). High-density linkage mapping and distribution of segregation distortion regions in the oak genome. DNA Res. 23-2, 115–124. doi: 10.1093/dnares/dsw001
Burgarella, C., Lorenzo, Z., Jabbour-Zahab, R., Lumaret, R., Guichoux, E., Petit, R. J., et al. (2009). Detection of hybrids in nature: application to oaks (Quercus suber and Q.ilex). Heredity 102, 442–452. doi: 10.1038/hdy.2009.8
Burgarella, C., Barnaud, A., Kane, N. A., Jankowski, F., Scarcelli, N., Billot, C., et al. (2019). Adaptive introgression: an untapped evolutionary mechanism for crop adaptation. Front. Plant Sci. 10:4. doi: 10.3389/fpls.2019.00004
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: Architecture and applications. BMC Bioinform. 10, 1–9. doi: 10.1186/1471-2105-10-421
Cannon, C. H., Petit, R. J. (2020). The oak syngameon: more than the sum of its parts. New Phytol. 226 (4), 978–983. doi: 10.1111/nph.16091
Cannon, C. H., Scher, C. L. (2017). Exploring the potential of gametic reconstruction of parental genotypes by F1 hybrids as a bridge for rapid introgression. Genome 60 (9), 713–719. doi: 10.1139/gen-2016-0181
Crowl, A. A., Manos, P. A., McVay, J. D., Lemmon, A. R., Moriarty Lemmon, E., Hipp, A. L. (2019). Uncovering the genomic signature of ancient introgression between white oak lineages (Quercus). New Phytol. 226 (4), 1158–1170. doi: 10.1111/nph.15842
Eaton, D. A. R., Hipp, A. L., González-Rodríguez, A., Cavender-Bares, J. (2015). Historical introgression among the American live oaks and the comparative nature of tests for introgression. Evolution 69 (10), 2587–2601. doi: 10.1111/evo.12758
Eaton, D. A. R., Spriggs, E. L., Park, B., Donoghue, M. J. (2017). Misconceptions on Missing Data in RAD-seq Phylogenetics with a Deep-scale Example from Flowering Plants. Syst. Biol. 66 (3), 399–412. doi: 10.1093/sysbio/syw092
Emberton, J, Ma, J., Yuan, Y., SanMiguel, P., Bennetzen, J. L. (2005). Gene enrichment in maize with hypomethylated partial restriction (HMPR) libraries. Genome Res. 15, 1441–1446. doi: 10.1101/gr.3362105
Evanno, G., Regnaut, S., Goudet, J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620. doi: 10.1111/j.1365-294X.2005.02335.x
Falk, T., Herndon, N., Grau, E., Buehler, S., Richter, P., Zaman, S., et al. (2018). Growing and cultivating the forest genomics database, TreeGenes. Database (Oxford) 2018, 1–11. doi: 10.1093/database/bay084
Goicoechea, P., Herrán, A., Durand, J., Bodènés, C., Plomion, C., Kremer, A. (2015). A linkage disequilibrium perspective on the genetic mosaic of speciation in two hybridizing Mediterranean white oaks. Heredity 114, 373–386. doi: 10.1038/hdy.2014.113
Gómez-Casero, M. T., Galán, C., Domínguez-Vilches, E. (2007). Flowering phenology of Mediterranean Quercus species in different locations (Córdoba, SW Iberian Peninsula). Acta Bot. Malac. 32, 127–146. doi: 10.24310/abm.v32i0.7033
Gompert, Z., Buerkle, C. A. (2010). Introgress: a software package for mapping components of isolation in hybrids. Mol. Ecol. Resour. 10, 374–384. doi: 10.1111/j.1755-0998.2009.02733.x
Guillardín-Calvo, L., Mora-Márquez, F., Soto, Á., López de Heredia, U. (2019). RADdesigner: a workflow to select the optimal sequencing methodology in genotyping experiments on woody plant species. Tree Genet. Genomes 15, 64. doi: 10.1007/s11295-019-1372-3
Harrison, R. G., Larson, E. L. (2014). Hybridization, introgression, and the nature of species boundaries. J. Hered. 105, 795–809. doi: 10.1093/jhered/esu033
Hipp, A. L., Manos, P. S., Hahn, M., Avishai, M., Bodénès, C., Cavender-Bares, J., et al. (2020). Genomic landscape of the global oak phylogeny. New Phytol. 226 (4), 1198–1212. doi: 10.1111/nph.16162
Hirsch, C. D., Evans, J., Buell, C. R., Hirsch, C. N. (2014). Reduced representation approaches to interrogate genome diversity in large repetitive plant genomes. Brief. Funct. Genomics 13 (4), 257–267. doi: 10.1093/bfgp/elt051
Huang, H., Knowles, L. L. (2016). Unforeseen Consequences of Excluding Missing Data from Next-Generation Sequences: Simulation study of RAD Sequences. Syst. Biol. 65, 357–365. doi: 10.1093/sysbio/syu046
Jiménez, M. P., López de Heredia, U., Collada, C., Lorenzo, Z., Gil, L. (2004). High variability of chloroplast DNA in three Mediterranean evergreen oaks indicates complex evolutionary history. Heredity 93, 510–515. doi: 10.1038/sj.hdy.6800551
Konar, A., Choudhury, O., Bullis, R., Fiedler, L., Kruser, J. M., Stephens, M. T., et al. (2017). High-quality genetic mapping with ddRADseq in the non-model tree Quercus rubra. BMC Genomics 18, 417. doi: 10.1186/s12864-017-3765-8
Kremer, A., Hipp, A. L. (2020). Oaks: an evolutionary success story. New Phytol. 226 (4), 987–1011. doi: 10.1111/nph.16274
Kumar, S., Stecher, G., Tamura, K. (2016). MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 33 (7), 1870–1874. doi: 10.1093/molbev/msw054
Lang, T., Abadie, P., Leger, V., Decourcelle, T., Frigerio, J. M., Burban, C., et al. (2020). High-quality SNPs from genic regions highlight instrogression patterns among European white oaks (Quercus petraea and Q.robur). bioRxiv. doi: 10.1101/388447
Lepais, O., Petit, R., Guichoux, E., Lavabre, J., Alberto, F., Kremer, A., et al. (2009) Species relative abundance and direction of introgression in oaks. Mol. Ecol. 18, 2228–2242. doi: 10.1111/j.1365-294X.2009.04137.x
Lepoittevin, C., Bodénés, E., Chancerel, E., Villate, L., Lang, T., Lesur, I., et al. (2015). Single-nucleotide polymorphism discovery and validation in high-density SNP array for genetic analysis in European white oaks. Mol. Ecol. Resour. 15 (6), 1446–1459. doi: 10.1111/1755-0998.12407
Lesur, I., Le Provost, G., Bento, P., Da Silva, C., Leplé, J. C., Murat, F., et al. (2015). The oak gene expression atlas: insights into Fagaceae genome evolution and the discovery of genes regulated during bud dormancy release. BMC Genomics 16, 112. doi: 10.1186/s12864-015-1331-9
Lesur, I., Alexandre, H., Boury, C., Chancerel, E., Plomion, C., Kremer, A. (2018). Development of target sequence capture and estimation of genomic relatedness in a mixed oak stand. Front. Plant Sci. 9, 996. doi: 10.3389/fpls.2018.00996
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The Sequence alignment/map (SAM) format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, H. (2011). A statistical framework for SNP calling, mutation discovery, association mapping and population genetical parameter estimation from sequencing data. Bioinformatics 27 (21), 2987–2993. doi: 10.1093/bioinformatics/btr509
Librado, P., Rozas, J. (2009). DnaSP v5: A Software for Comprehensive Analysis of DNA Polymorphism Data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
López de Heredia, U., Jiménez, P., Collada, C., Simeone, M. C., Bellarosa, R., Schirone, B., et al. (2007). Multi-Marker Phylogeny of Three Evergreen Oaks Reveals Vicariant Patterns in the Western Mediterranean. Taxon. 56 (4), 1209–1220. doi: 10.2307/25065912
López de Heredia, U., Vázquez, F. M., Soto, Á. (2017). “The role of hybridization on the adaptive potential of Mediterranean sclerophyllous oaks: the case of the Quercus ilex x Q . suber complex,” in Oaks Physiological Ecology. Exploring the Functional Diversity of Genus. Eds. Quercus, L., Gil-Pelegrín, E., Peguero-Pina, J. J., Sancho-Knapik, D. (Cham, Switzerland: Springer International Publishing), 239–260.
López de Heredia, U., Sánchez, H., Soto, Á. (2018a). Molecular evidence of bidirectional introgression between Quercus suber and Quercus ilex. iForest 11, 338–343. doi: 10.3832/ifor2570-011
López de Heredia, U., Duro-García, M. J., Soto, Á. (2018b). Leaf morphology of progenies in Q.suber, Q. ilex, and their hybrids using multivariate and geometric morphometric analysis. iForest 11, 90–98. doi: 10.3832/ifor2577-010
López de Heredia, U., Mora-Márquez, F., Goicoechea, P. G., Guillardín-Calvo, L., Simeone, M. C., Soto, Á. (2020). SQLite3 databases of candidate marker loci from ddRADseq in Quercus suber, Quercus ilex and their hybrids. Zenodo Repos. doi: 10.5281/zenodo.3786309
Lumaret, R., Mir, C., Michaud, H., Raynal, V. (2002). Phylogeographical variation of chloroplast DNA in holm oak (Quercus ilex L.). Mol. Ecol. 11, 2327–2336. doi: 10.1046/j.1365-294X.2002.01611.x
Luo, R., Liu, B., Xie, Y., Li, Z., Huang, W., Yuan, J., et al. (2012). SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 1 (1):18. doi: 10.1186/2047-217X-1-18
Mallet, J., Besansky, N., Hahn, M. W. (2016). How reticulated are species? Bioessays 38, 140–149. doi: 10.1002/bies.201500149
Marfil, C. F., Masuelli, R. W., Davison, J., Comai, L. (2006). Genomic instability in Solanum tuberosum x Solanum kurtzianum interspecific hybrids. Genome 49, 104–113. doi: 10.1139/g05-088
Martin, M. (2011). Cutadapt removes adapter sequences from high-throughput sequencing reads. EMBnet.journal 17 (1), 10–12. doi: 10.14806/ej.17.1.200
Mayr, E. (1982). The growth of biological thought: diversity, evolution, and inheritance (Cambridge (MA), USA: Belknap Press).
McVay, J. D., Hipp, A. L., Manos, P. S. (2017). A genetic legacy of introgression confounds phylogeny and biogeography in oaks. Proc. Biol. Sci. 284 (1854), 20170300. doi: 10.1098/rspb.2017.0300
Mora-Márquez, F., Chano, V., Vázquez-Poletti, J., López de Heredia, U. (2020). TOA: a software package for automated functional annotation in non-model plant species. Authorea. doi: 10.22541/au.159611047.70067764
Moran, E. V., Willis, J., Clark, J. S. (2012). Genetic evidence for hybridization in red oaks (Quercus sect. Lobatae, Fagaceae). MAm. J. Bot. 99 (1), 92–100. doi: 10.3732/ajb.1100023
Muir, G., Flemming, C. C., Schlötterer, C. (2000). Species status of hybridizing oaks. Nature 405 (6790), 1016. doi: 10.1038/35016640
Natividade, J. V. (1936). Estudo histológico das peridermes do híbrido Quercus ilex x suber, P. Cout. Publ. Dir. G. Serv. Flor. III 1, 32 p. Lisbon, Portugal.
Nelson, W., Luo, M., Ma, J., Estep, M., Estill, J., He, R., et al. (2008). Methylation-sensitive linking libraries enhance gene-enriched sequencing of complex genomes and map DNA methylation domains. BMC Genomics 9, 621. doi: 10.1186/1471-2164-9-621
O’Leary, S. J., Puritz, J. B., Willis, S. C., Hollenbeck, C. M., Portnoy, D. S. (2018). These aren’t the loci you’e looking for: Principles of effective SNP filtering for molecular ecologists. Mol. Ecol. 27 (16), 3193–3206. doi: 10.1111/mec.14792
Ortego, J., Gugger, P. F., Sork, V. L. (2018). Genomic data reveal cryptic lineage diversification and introgression in Californian golden cup oaks (section Protobalanus). New Phytol. 218, 804–818. doi: 10.1111/nph.14951
Ouellette, L. A., Reid, R. W., Blanchard, J. S. G., Brouwer, C. R. (2017). LinkageMapView - Rendering High Resolution Linkage and QTL Maps. Bioinformatics 34 (2), 306–307. doi: 10.1093/bioinformatics/btx576
Perea Garcia-Calvo, R. (2006). Estudio de la estructura de masa de una dehesa de encina con alcornoque en “El Deheson del Encinar” (Toledo: [dissertation]: Universidad Politécnica de Madrid).
Peterson, B. K., Weber, J. N., Kay, E. H., Fisher, H. S., Hoekstra, H. E. (2012). Double digest RADseq: an inexpensive method for de novo SNP discovery and genotyping in model and non-model species. PLoS One 7 (5), e37135. doi: 10.1371/journal.pone.0037135
Petit, R. J., Bodénès, C., Ducousso, A., Roussel, G., Kremer, A. (2003). Hybridization as a mechanism of invasion in oaks. New Phytol. 161, 151–164. doi: 10.1046/j.1469-8137.2003.00944.x
Plomion, C., Aury, J. M., Amselem, J., Leroy, T., Murat, F., Duplessis, S., et al. (2018). Oak genome reveals facets of long lifespan. Nat. Plants 4, 440–452. doi: 10.1038/s41477-018-0172-3
Poland, J. A., Rife, T. W. (2012). Genotyping-by-Sequencing for Plant Breeding and Genetics. Plant Genome 5, 92–102. doi: 10.3835/plantgenome2012.05.0005
Pootakham, W., Ruang-Areerate, P., Jomchai, N., Sonthirod, C., Sangsrakru, D., Yoocha, T., et al. (2015). Construction of a high-density integrated genetic linkage map of rubber tree (Hevea brasiliensis) using genotyping-by-sequencing (GBS). Front. Plant Sci. 6, 367. doi: 10.3389/fpls.2015.00367
Pootakham, W., Sonthirod, C., Naktang, C., Jomchai, N., Sangsrakru, D., Tangphatsornruang, S. (2016). Effects of methylation-sensitive enzymes on the enrichment of genic SNPs and the degree of genome complexity reduction in a two-enzyme genotyping-by-sequencing (GBS) approach: a case study in oil palm (Elaeis guineensis). Mol. Breed. 36 (11), 154. doi: 10.1007/s11032-016-0572-x
Potts, B., Reid, J. B. (1988). Hybridization as a dispersal mechanism. Evolution 42, 1245–1255. doi: 10.1111/j.1558-5646.1988.tb04184.x
Pritchard, J. K., Stephens, M., Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genomics 155, 945–959. doi: 10.1111/j.1471-8286.2007.01758.x
Radosavljevic, I., Bogdanovic, S., Celep, F., Filipovic, M., Satovic, Z., Surina, B., et al. (2019). Morphological, genetic and epigenetic aspects of homoploid hybridization between Salvia officinalis L. and Salvia fruticose Mill. Sci. Rep. 9, 3276. doi: 10.1038/s41598-019-40080-0
Ramos, A., Usié, A., Barbosa, P., Barros, P. M., Capote, T., Chaves, I., et al. (2018). The draft genome sequence of cork oak. Sci. Data 5, 180069. doi: 10.1038/sdata.2018.69
Saintagne, C., Bodenes, C., Barreneche, T., Pot, D., Plomion, C., Kremer, A. (2004). Distribution of genomic regions differentiating oak species assessed by QTL detection. Heredity 92 (1), 20–30. doi: 10.1038/sj.hdy.6800358
Salmon, A., Ainouche, M. L., Wendel, J. F. (2005). Genetic and epigenetic consequences of recent hybridization and polyploidy in Spartina (Poaceae). Mol. Ecol. 14, 1163–1175. doi: 10.1111/j.1365-294X.2005.02488.x
Sexton, J. P., McIntyre, P. J., Angert, A. L., Rice, K. J. (2009). Evolution and ecology of species range limits. Annu. Rev. Ecol. Evol. Syst. 40, 415–436. doi: 10.1146/annurev.ecolsys.110308.120317
Simeone, M. C., Grimm, G. W., Papini, A., Vessella, F., Cardoni, S., Tordoni, E., et al. (2016). Plastome data reveal multiplegeographic origins of Quercus Group Ilex. PeerJ 4, e1897. doi: 10.7717/peerj.1897
Simeone, M. C., Cardoni, S., Piredda, R., Imperatori, F., Avishai, M., Grimm, G. W., et al. (2018). Comparative systematics and phylogeography of Quercus Section Cerris in western Eurasia: inferences from plastid and nuclear DNA variation. PeerJ 6, e5793. doi: 10.7717/peerj.5793
Soto, Á., Rodríguez-Martínez, D., López de Heredia, U. (2018). SimHyb: a simulation software for the study of the evolution of hybridizing populations. Application to Quercus ilex and Q. suber suggests hybridization could be underestimated. iForest 11, 99–103. doi: 10.3832/ifor2569-011
Suárez-González, A., Lexer, C., Cronk, Q. C. B. (2018). Adaptive introgression: a plant perspective. Biol. Lett. 14 (3), 20170688. doi: 10.1098/rsbl.2017.0688
Ueno, S., Le Provost, G., Leger, V., Klopp, C., Noirot, C., Frigerio, J. M., et al. (2010). Bioinformatics analysis of ESTs collected by Sanger and pyrosequencing methods for a keystone forest tree species: oak. BMC Genomics 11, 650. doi: 10.1186/1471-2164-11-650
Valbuena-Carabaña, M., González-Martínez, S. C., Sork, V. L., Collada, C., Soto, Á., Goicoechea, P. G., et al. (2005). Gene flow and hybridisation in a mixed oak forest (Quercus pyrenaica Willd. and Quercus petraea (Matts.) Liebl.) in central Spain. Heredity 95, 457–465. doi: 10.1038/sj.hdy.6800752
van Valen, L. (1976). Ecological species, multispecies and oaks. Taxon 25, 233–239. doi: 10.2307/1219444
Vitelli, M., Vessella, F., Cardoni, S., Pollegioni, P., Denk, T., Grimm, G. W., et al. (2017). Phylogeographic structuring of plastome diversity in Mediterranean oaks (Quercus Group Ilex, Fagaceae). Tree Genet. Genomes 13, 3. doi: 10.1007/s11295-016-1086-8
Wu, T. D., Reeder, J., Lawrence, M., Becker, G., Brauer, M. J. (2016). GMAP and GSNAP for Genomic Sequence Alignment: Enhancements to Speed, Accuracy, and Functionality. Methods Mol. Biol. 1418, 283–334. doi: 10.1007/978-1-4939-3578-9_15
Keywords: ddRADseq, Quercus, hybridization, introgression, genomic boundaries, SNPs
Citation: López de Heredia U, Mora-Márquez F, Goicoechea PG, Guillardín-Calvo L, Simeone MC and Soto Á (2020) ddRAD Sequencing-Based Identification of Genomic Boundaries and Permeability in Quercus ilex and Q. suber Hybrids. Front. Plant Sci. 11:564414. doi: 10.3389/fpls.2020.564414
Received: 21 May 2020; Accepted: 13 August 2020;
Published: 04 September 2020.
Edited by:
Gonzalo Nieto Feliner, Real Jardín Botánico (RJB, CSIC), SpainReviewed by:
John McVay, Florida Department of Agriculture and Consumer Services, United StatesCopyright © 2020 López de Heredia, Mora-Márquez, Goicoechea, Guillardín-Calvo, Simeone and Soto. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Álvaro Soto, YWx2YXJvLnNvdG8uZGV2aWFuYUB1cG0uZXM=
†Present address: Laura Guillardín-Calvo, Department of Plant Sciences, University of Oxford, Oxford, United Kingdom
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.