
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 11 June 2020
Sec. Plant Breeding
Volume 11 - 2020 | https://doi.org/10.3389/fpls.2020.00686
 Sen Li1,2*†
Sen Li1,2*† Fangfang Ji1,2†Feifan Hou1,2Huliang Cui1,2Qingqing Shi1,2Guoming Xing1,2
Fangfang Ji1,2†Feifan Hou1,2Huliang Cui1,2Qingqing Shi1,2Guoming Xing1,2 Yiqun Weng3*Xiuping Kang1,2*
Yiqun Weng3*Xiuping Kang1,2*Hemerocallis spp. commonly known as daylilies and night lilies, are among the most popular ornamental crops worldwide. In Eastern Asia, H. citrina is also widely cultivated as both a vegetable crop and for medicinal use. However, limited genetic and genomic resources are available in Hemerocallis. Knowledge on the genetic diversity and population structure of this species-rich genus is very limited. In this study, we reported transcriptome sequencing of H. citrina cv. ‘Datonghuanghua’ which is a popular, high-yielding variety in China. We mined the transcriptome data, identified and characterized the microsatellite or simple sequence repeat (SSR) sequences in the expressed genome. From ∼14.15 Gbp clean reads, we assembled 92,107 unigenes, of which 41,796 were annotated for possible functions. From 41,796 unigenes, we identified and characterized 3,430 SSRs with varying motifs. Forty-three SSRs were used to fingerprint 155 Hemerocallis accessions. Clustering and population structure analyses with the genotypic data among the 155 accessions reveal broader genetic variation of daylilies than the night lily accessions which form a subgroup in the phylogenetic tree. The night lily group included accessions from H. citrina, H. lilioasphodelus, and H. minor, the majority of which bloom in the evening/night, whereas the ∼100 daylily accessions bloomed in the early morning suggesting flowering time may be a major force in the selection of night lily. The utility of these SSRs was further exemplified in association analysis of blooming time among these accessions. Twelve SSRs were found to have significant associations with this horticulturally important trait.
Hemerocallis species are among the most popular ornamental crops worldwide because of the large, conspicuous flowers and their adaptation to a wide range of soils and climates. In Eastern Asian countries, some species, especially H. citrina (night lily, or long yellow daylily) are important vegetables or medicinal plants with a long history of cultivation. For example, the first reference of H. fulva (daylily) was found in a writing in the Chou Dynasty of China (ca. 112–255 BCE) (Hu, 1968; Kitchingnan, 1985). The unopened flower buds (fresh or dried) of the night lily are consumed as a special vegetable (also known as “golden needle vegetable”). The medicinal value of Hemerocallis species has received more attention in recent years. The flower buds seem to be enriched with antioxidants, such as stelladerol and caffeoylquinic acid derivatives (Cichewicz and Nair, 2002; Hsu et al., 2011; Lin et al., 2011; Jiao et al., 2016). These secondary metabolites are used to treat anxiety and swelling (Uezu, 1998; Cichewicz et al., 2002; Gu et al., 2012; Yi et al., 2012; Du et al., 2014; Liu et al., 2014), and for other applications in modern medicine and biology (Venugopalan and Srivastava, 2015). Thus, Hemerocallis flowers have considerable potentials as “nutraceutical” or “functional” foods (Rop et al., 2011).
In addition to their economic and medical values, some features of Hemerocallis species are of particular biological significance. For example, the day and night lilies provide an excellent model for understanding the genetic and molecular mechanisms of flower opening and flower senescing. All known species in this genus show strict circadian rhythm of flowering with the rapid opening and withering that lasts only a few hours indicting precise regulation of floral death probably by a programmed cell death system (Hasegawa et al., 2006; Nitta et al., 2010; Hirota et al., 2012, 2013). All Hemerocallis species exhibit self-incompatibility. As such, Hemerocallis was proposed as a future model system to study these biologically interesting phenomena (Rodriguez-Enriquez and Grant-Downton, 2012).
As an ornamental plant, extensive breeding efforts during the last century have resulted in varieties with different flower size, color, shape, scent and blooming time. In the US, over 83,000 daylily varieties have been registered in the American Hemerocallis Society online database1. Some morphological characteristics of daylily and night lily plants and flowers are exemplified in Figure 1. Despite the economic and biological importance as well as extensive breeding efforts on Hemerocallis species, not much has been accomplished on genetic studies in this genus. As demonstrated in numerous other horticulture crops, molecular markers are important tools for genetic research and breeding in Hemerocallis with many potential applications such as evaluation of genetic diversity and population structure of germplasm collection, development of linkage maps, gene and QTL (quantitative trait loci) mapping or cloning, as well as marker-assisted selection of horticulturally important traits in breeding. Nevertheless, limited work has been done in the development of molecular markers for Hemerocallis species. The natural range of Hemerocallis encompasses temperate and sub-tropical Asia, with the main center of diversity of the genus in China, Korea and Japan (Flora of China, 2000). However, reports on the genetic diversity and population genetic analysis of Hemerocallis especially for the collections in China are sporadic (e.g., Tomkins et al., 2001; Podwyszynska et al., 2009; Vendrame et al., 2009; Miyake and Yahara, 2010). In these early reports, molecular markers employed included random amplified polymorphic DNA (RAPD) and amplified fragment length polymorphism (AFLP), each of which has limitations in practical uses (Varshney et al., 2007). Despite the increased use of single nucleotide polymorphism (SNPs), SSR markers remain popular in plant genetic and breeding studies because of their many desirable attributes, including hyper variability, a multi allelic nature, co-dominant inheritance, reproducibility, relative abundance, extensive genome coverage (including organellar genomes), relatively low cost, and amenability to high throughput genotyping (Parida et al., 2009). However, very few SSRs have been reported for Hemerocallis (e.g., Zhu et al., 2009). While there are several methods to develop SSR markers, transcriptome sequencing (RNA-Seq) has been an efficient and affordable way for large-scale, and genome-wide SSR discovery and marker development for various marker-based studies in non-model plants (e.g., Hudson, 2010; Strickler et al., 2012; Yuan et al., 2013; Zhang et al., 2013; Guo et al., 2016; Smithunna et al., 2016). Thus, the objective of the present study is to develop EST (expressed sequence tag)-SSR markers through transcriptome sequencing and examine their utility in evaluation of genetic diversity and population structure of our Hemerocallis collection. We also conducted association analysis to identify potential association of molecular markers with horticulturally important traits.

Figure 1. Phenotypic diversity in flowers, petals, sepals, and buds of two typical Hemerocallis species: H. citrina (night lily; A), and H. fulva (daylily, B). (C) Flower buds and opened flowers from different accessions. C1–C5 are opened flowers. C1: Hemerocallis cv. ‘Stella de Oro 2’; C2: Hemerocallis cv. ‘Frans Hals’; C3: H. citrina cv. ‘Datonghuanghua’ C4: Hemerocallis cv. ‘Blazing sun’; C5: Hemerocallis cv. ‘H400’; C6-C9 are young flower buds. C6: Hemerocallis cv. ‘Beijing 7’; C7: Hemerocallis cv. ‘Blue Sheen 1’; C8: H. citrina cv. ‘Zaohuanghua’; C9: Hemerocallis cv. ‘Little Grapette’.
One hundred and fifty-five Hemerocallis accessions were employed for genetic diversity analysis with 43 EST-SSRs. These accessions belong to at least 13 species from different geographic regions around the world including commercial varieties, landraces, and collections from the wild. The details of these 155 accessions are presented in Supplementary Table S1, and their geographic distributions are illustrated in Figure 2. The taxonomic status of more than half of the 155 accessions was labeled uncertain during collection (Hemerocallis spp. in Supplementary Table S1). All accessions were grown in the Hemerocallis Germplasm Resource Nursery located on the campus of Shanxi Agricultural University (Taigu, Shanxi Province, China). All plants used for sample collection have been grown for 3–5 years in the nursery. At full flowering stage, the fresh roots, flower buds, and leaves of the H. citrina cv ‘Datonghuanghua’ were collected, flash frozen in liquid nitrogen, and used for total RNA extraction and RNA-Seq.
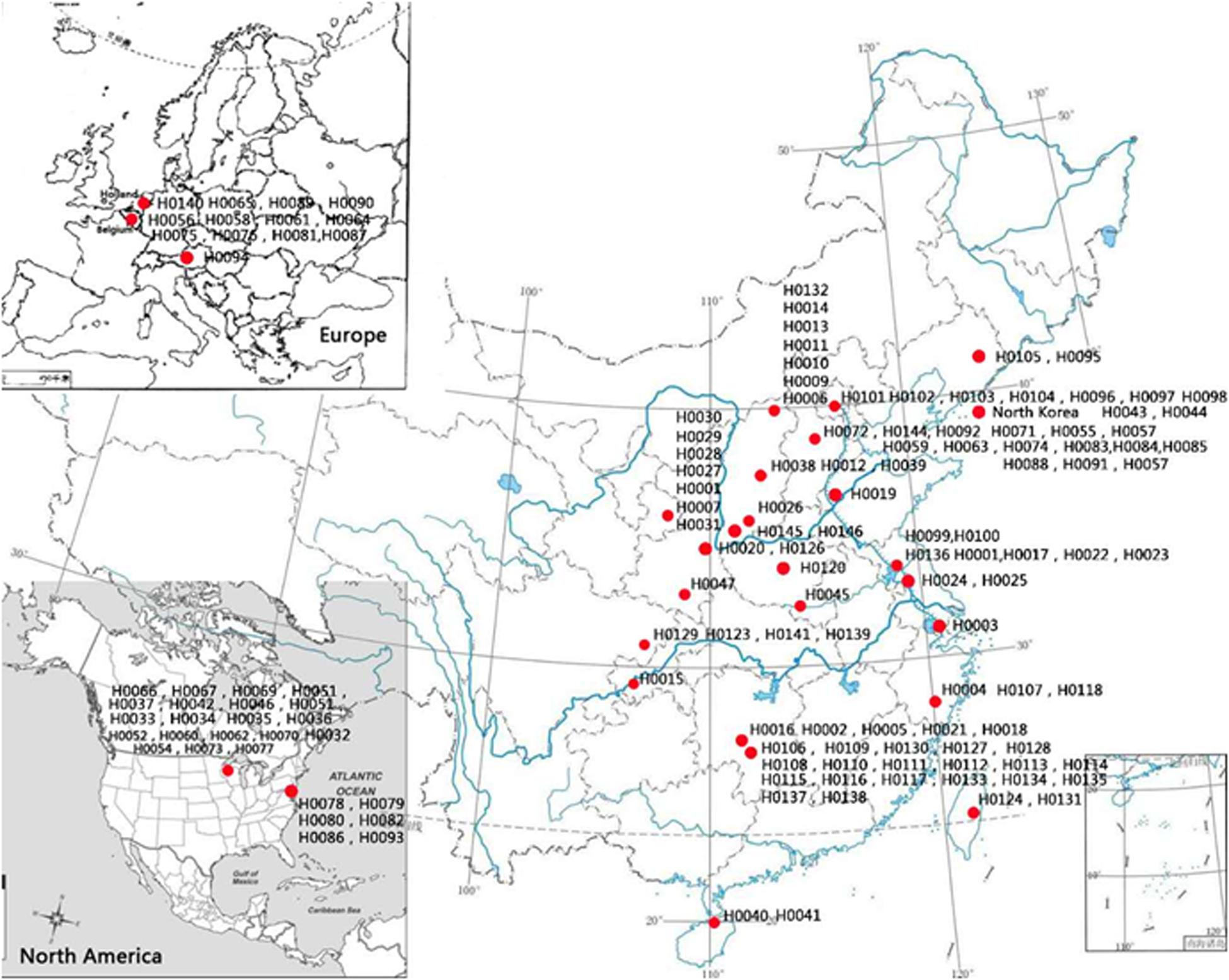
Figure 2. Geographic distribution 155 Hemerocallis accessions used in this study.
For RNA-Seq, total RNA was isolated from each sample with three plants pooled with a modified CTAB protocol (Tel-Zur et al., 1999). RNA quantity and quality were evaluated using the Agilent Bioanalyzer (Agilent Technologies, CA, United States). The integrity of the total RNA was also assessed through agarose gel electrophoresis. The cDNA library for RNA-Seq was prepared using Illumina TruseqTM RNA sample prep kit (Illumina, CA, United States) following manufacturer’s protocols. The concentration and insert size of the library were assessed using Qubit2.0 and Agilent 2100. Pair-end (125 PE) Illumina high-throughput sequencing was performed with a Hi-Seq 2500 sequencing machine. All samples were sequenced in the same instrument (HWI-7001455), same run (316), same follow cell (HH7VHADXX), and same lane (2).
After resequencing, the adaptors and low quality reads were filtered from the raw reads. The clean reads were de novo assembled into contigs with an optimized k-mer length = 25 and group pairs distance = 300 (Grabherr et al., 2011) using the Trinity program2. Unigene sequences were aligned against databases of the Clusters of Orthologous Groups (COG) (Tatusov et al., 2000), Gene Ontology (GO) (Michael et al., 2000), Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al., 2004), euKaryotic Orthologous Groups (KOG) (Eugene et al., 2004). The functions of unigenes were predicted by BLAST against the NCBI non-redundant protein (Nr), and Swiss-Prot databases at an E-value of 10–5. The resulting datasets were further aligned to the Protein family (PFAM) database with HMMER (E-value 10–10). For further quantitative assessment of the completeness of the assembly and annotation, the H. citrina cv ‘Datonghuanghua’ transcriptome was subjected to BUSCO (Benchmarking Universal Single-Copy Orthologs) analysis (Simão et al., 2015) against the Viridiplantae_odb10, embryophyta_odb10, and liliopsida_odb10 databases.
SSR identification among the unigene sequences (>1 kb) was performed with the MIcroSAtellite (MISA) program3. All microsatellites containing di-, tri-, tetra-, penta-, hexa-nucleotide, and compound motifs with more than five repeats were included. The Primer3 program4 was used to design primers for the identified SSRs. Each designed primer pair was first evaluated with in silico PCR procedure5 using unigene sequences as the template. Primers with multiple amplicons were filtered out.
Forty-three EST-SSR markers were selected for genotyping 155 Hemerocallis accessions. Genomic DNA was extracted from fresh leaves of each accession (pooled from five plants) using the CTAB method. A touch-down PCR procedure (Azhaguvel et al., 2009) was employed and the amplicons were separated using 9% non-denaturing polyacrylamide gel electrophoresis (PAGE) with 0.5 × TBE buffer for 1.5 h in 250V, and then visualized with silver staining.
The SSR genotypic data were organized in a matrix in which 0 and 1 representing absent and present alleles, respectively (9 = missing data). The genetic distances were calculated through the SM coefficient by the Sim Qual procedure in NTSYSpc 2.10 (Rohlf and Jensen, 1989). The dendrogram was constructed using the unweighted pair-group method with arithmetic mean (UPGMA) clustering and drawn by NTSYSpc 2.10. We also applied Neighbor Joining (NJ) clustering on the dataset, which was implemented in the MEGA5.05 software package.
A model-based Bayesian clustering was applied to infer genetic structure and define the number of clusters (gene pools) in the dataset using STUCTURE v.2.3.4 (Pritchard et al., 2000). No prior information was used to define the clusters. Independent runs were done by setting the number of clusters (K) from 1 to 15. Each run was comprised of a burn-in length of 10,000 followed by 100,000 MCMC (Monte Carlo Markov Chain); each replicate at a particular K-value was repeated 20 times. An ad hoc statistic ΔK based on the rate of changes in the log probability of data among successive K-values (Evanno et al., 2005) was calculated through Structure Harvester v.0.9.93 and used to estimate the most likely number of clusters (K). ΔK was calculated using ΔK = m[| L(K+1)-2L(K)+L(K-1)|]/s| L(K)|, where (L(K) is the logarithm of K, s is the standard deviation, and m is the mean.
We performed association analysis of blooming time with SSR markers among the 155 accessions of the Hemerocallis collection. During flowering season, the time of flower opening was continuously monitored real-time using a digital video camera (360 Smart Camera, D606, 360 China). The flowering time of each plant from each accession was recorded. The blooming time of at least five flowers per accession was collected. The blooming time of each accession is provided in Supplementary Table S1. In association analysis, the blooming time was treated a qualitative trait. Accessions with blooming time from 4:00 to 10:00 and 16:00 to 24:00 were assigned “0” and “1,” respectively.
Pairwise linkage disequilibrium (LD) and association analyses were performed using TASSEL4.3.6 (Bradbury et al., 2007; Dent et al., 2012), and calculation of LD and P-values. Association analysis was performed using both the general linear model (GLM, Q model) and the mixed linear model (MLM, Q+K model) in TASSEL. The comparison wise significance was computed using 1,000 permutations as implemented in GLM. The kinship matrix was generated by NTSYSpc 2.10 with option P3D for variance component estimation in MLM (Kang et al., 2008). Significance of marker-trait associations was determined at P ≤ 0.05 (Jaiswal et al., 2012). False discovery rate (FDR) was also used to detect true associations.
The expression level of blooming time-related gene c33464.graph_c0 in different tissues (fresh roots, flower buds and leaves) of H. citrina cv. ‘Datonghuanghua’ (H0006) was carried out with quantitative real-time PCR (RT-qPCR) on an ABI prism 7500 Fast Real-time PCR system 147 machine (Thermo Fisher Scientific Inc., Waltham, MA, United States). Sequences of the primers for c33464.graph_c0 were GGCGAATTAGTCTGGAAAGAACTAGG (forward primer) and TGTTATGTTCCTCGTCCGTCCAC (reverse primer). The H. citrina actin gene, HcACT, was used as the reference. The primers sequences for this gene were GAGCAAGGAAATCACGGCACT (forward primer) and GGAACCTCCAATCCAAACACTGTAC (reverse primer). RT-PCR procedure followed Hou et al. (2017). Each sample had three biological and three technical replications.
After filtering of the raw sequencing data, nearly 56 million clean reads (14.15 Gb) were obtained from root, bud and leaf transcriptomes. Main RNA-Seq statistics for the three tissues are presented in Supplementary Table S2. In all three transcriptomes, >90% reads had Q30 or higher quality scores. Using the Trinity de novo assembly program, the ∼14.15 Gb high-quality reads were assembled into contigs, transcripts and unigenes. Main statistics of the assemblies are shown in Table 1. From 6,390,477 contigs (>25 bp in length), 164,723 transcripts were assembled (mean length 838 bp) representing 92,107 unigenes. The length distribution of these unigenes is illustrated in Figure 3. The transcripts with >500 bp in length accounted for 47.06% of all transcripts. There were 13,724,749 (76.9%), 15,731,521 (79.2%), and 14,841,142 (80.4%) reads from the roots, buds and leaves mapped to the assembly transcripts and unigenes, respectively.
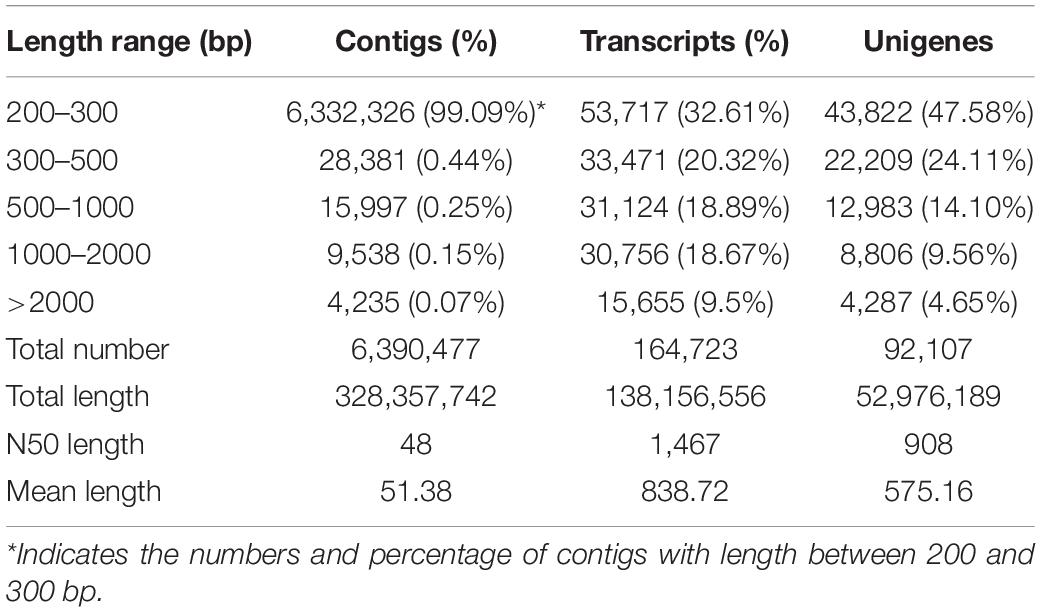
Table 1. Transcriptome assembly statistics of H. citrina cv. ‘Datonghuanghua’.
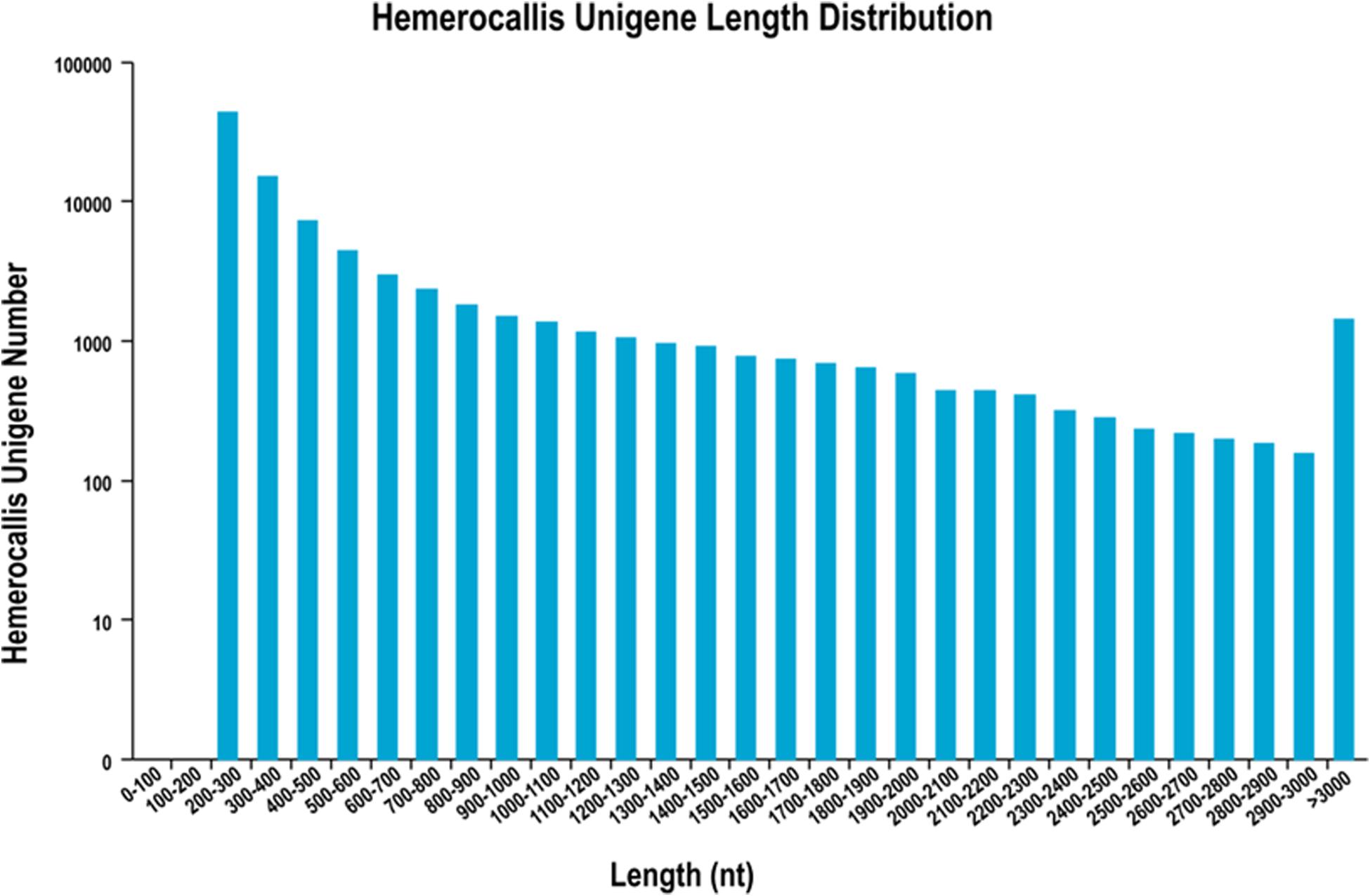
Figure 3. Length distribution of unigenes from leaf, root and flower bud transcriptomes of H. citrina cv. ‘Datonghuahua’.
We annotated the 92,107 unigenes by searches against the COG, GO, KEGG, KOG, Nr, and Swiss-Prot databases. Of them, 41,796 (45.4%) had hits in all 7 databases. The unigene hits in each database are shown in Table 2 with the number of annotated unigenes being the highest in the NR (41,053) and the lowest in KEGG (9,375). Further BLAST searches against other databases showed that 12,537, 23,716, 23,393, 28,711, and 24,125 unigenes had at least one match in the COG, GO, KOG, Swiss-Prot and PFAM databases, respectively (Table 2). In BLASTx homology searches (at cutoff E-value of 10–5), the ten top species with hits in the Nr database were: Elaeis guineensis (8,779; 21.38%), Phoenix dactylifera (7,194; 17.52%), Musa acuminate (2,912; 7.09%), Nelumbo nucifera (2,273; 5.54%), Vitis vinifera (2,204; 5.37%), Populus euphratica (761; 1.85%), Oryza sativa (650; 1.58%), Citrus sinensis (582; 1.42%), Populus trichocarpa (554; 1.35%), and Hordeum vulgare (474; 1.15%).
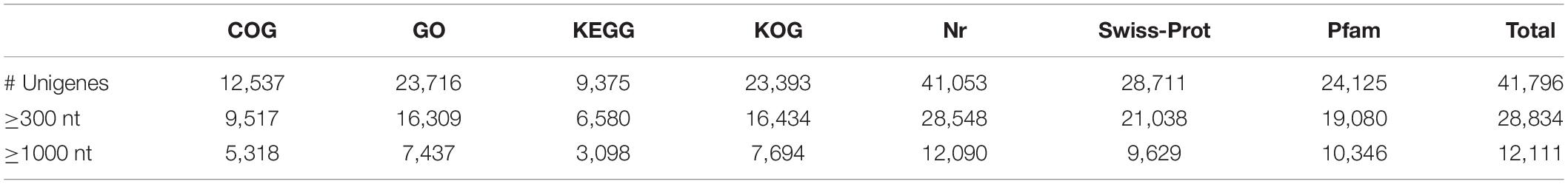
Table 2. Number of unigenes with annotations in six databases of H. citrina cv. ‘Datonghuanghua’.
To further evaluate the functions of unigenes, 12,537 of the assembled 41,796 unigenes were classified into 25 clusters based on the Cluster of Orthologous Groups (COG) analysis (Figure 4). Among the 25 COG categories, the cluster related to general function prediction (2,801 or 22.3%) was the largest, followed by the clusters of replication, recombination and repair (1,630 or 13.0%), transcription (1,397 or 11.1%), and secondary metabolites biosynthesis, transport and catabolism (519 or 4.1%). The clusters represented by the least number of unigenes were cell motility, nuclear structure, and extracellular structures (1, 0.008%).
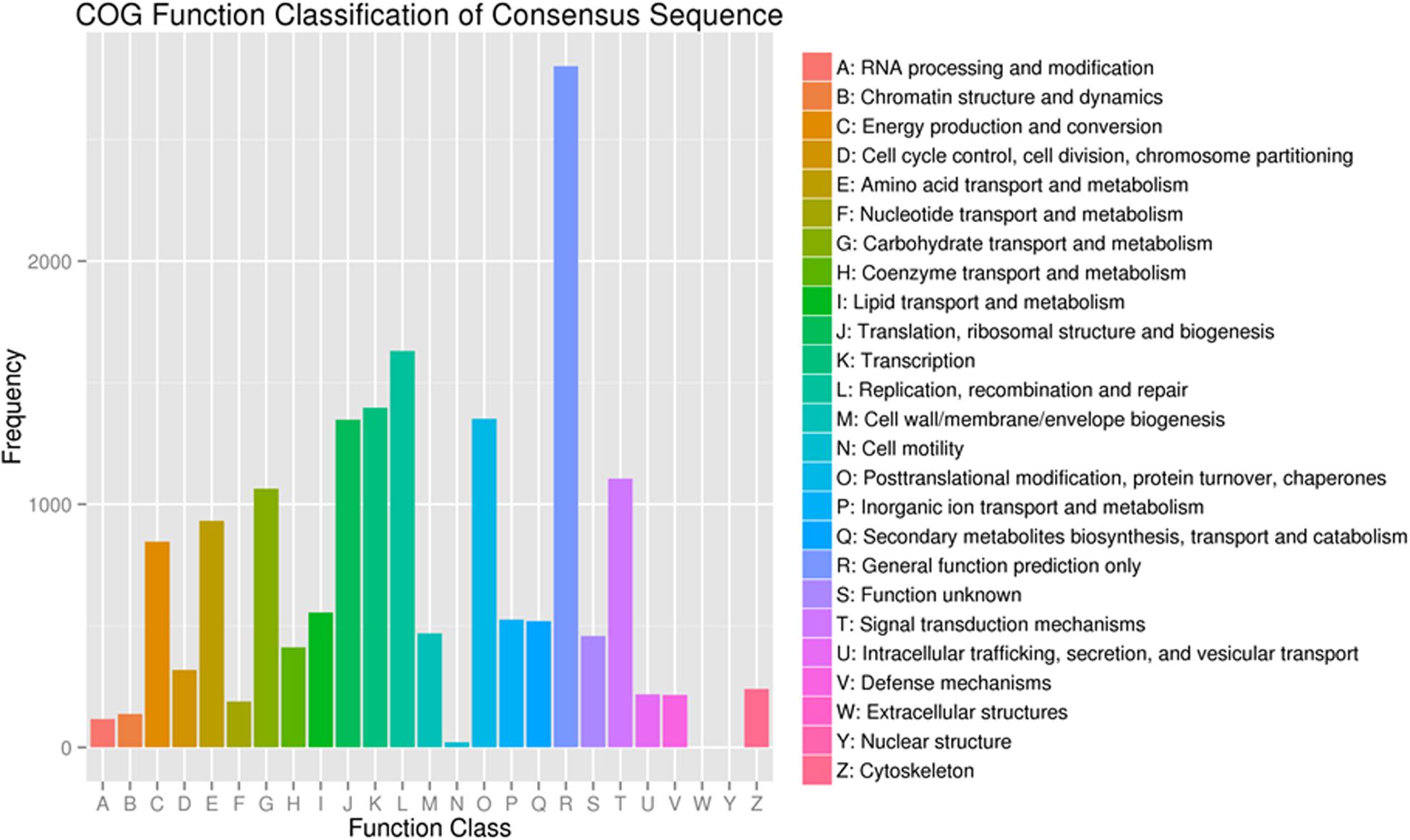
Figure 4. Functional classification of the assembled unigenes based on Clusters of Orthologous Groups (COG) categories.
In addition, 23,716 unigenes matched with the GO database were categorized into 51 functional sub-groups of the three main GO groups: cellular component, molecular function, and biological process (Figure 5). The majority of the unigenes was assigned to biological processes (63,089, 43.7%), followed by cellular components (35,677, 37.2%), and molecular functions (27,719, 19.2%). Under the category of biological processes, metabolic process (15,976, 25.3%) and cellular process (13,749, 21.8%) were predominant. In the cellular component category, cell part (12,837, 23.9%) and cell (12,746, 23.8%) were the most abundant classes. As for the molecular function, catalytic activities (12,157, 43.9%) and binding (11,677, 42.1%) were the top two categories in numbers.
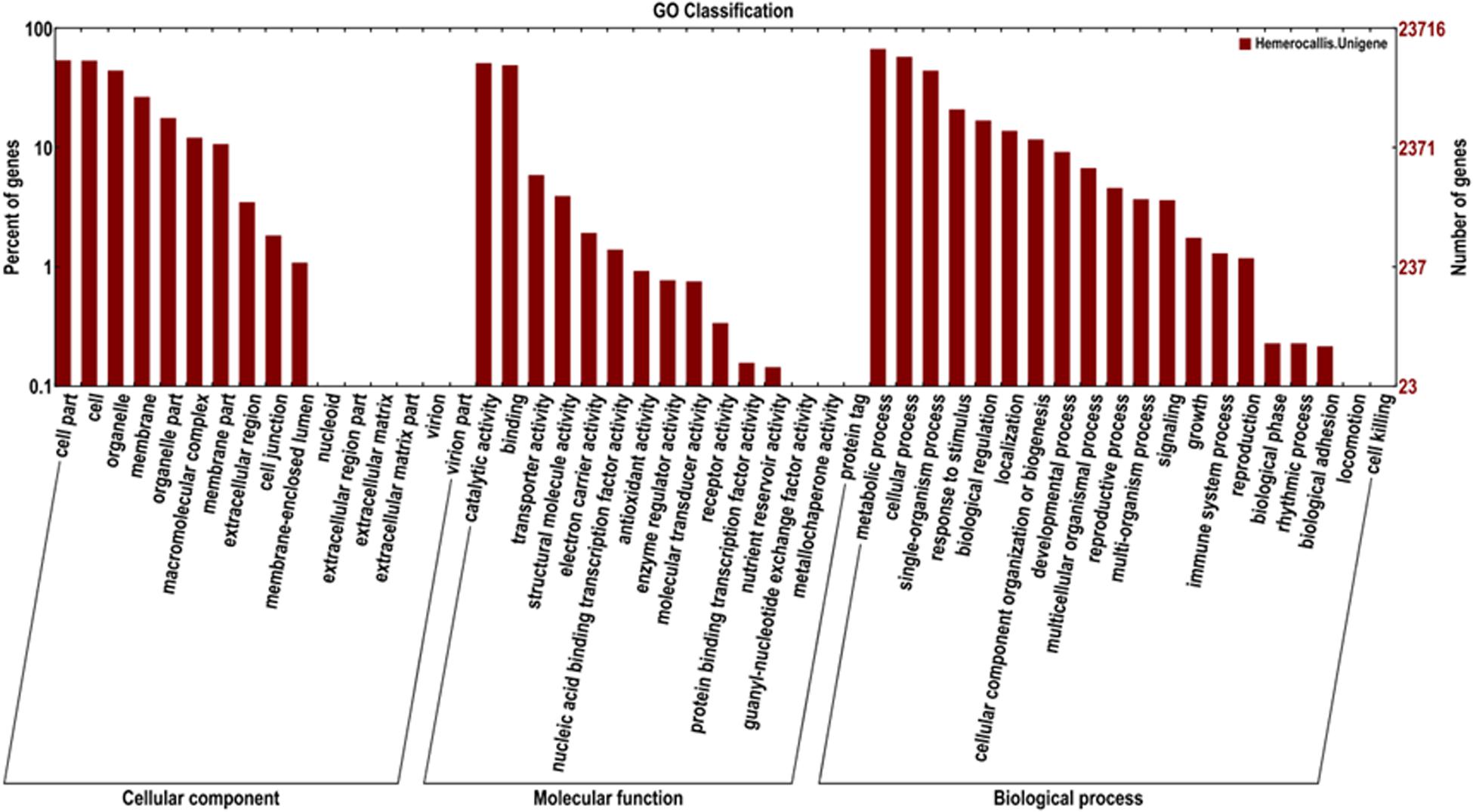
Figure 5. Functional classification of the assembled unigenes based on Gene Ontology (GO).
In KEGG pathway analysis of the 41,796 unigenes, 9,375 (22.4%) could be assigned to 117 pathways that belong to five main categories including metabolism (6,783), genetic information processing (3,013), cellular processes (495), environmental information processing (321), and organismal systems (305) (see details in Figure 6). The colchicine content is a very important trait for commercial production of night lily. Among annotated unigenes, 20 were involved in the isoquinoline alkaloids metabolic pathway that is related with colchicine biosynthesis. The category with the largest number of unigenes was metabolism, in which the most represented five pathways were oxidative phosphorylation (ko00190) (336, 4.9%), glycolysis/Gluconeogenesis (ko00010) (325, 4.8%), carbon fixation in photosynthetic organisms (ko00710) (306, 4.5%), purine metabolism (ko00230) (242, 3.6%), and starch and sucrose metabolism (ko00500) (228, 3.4%).
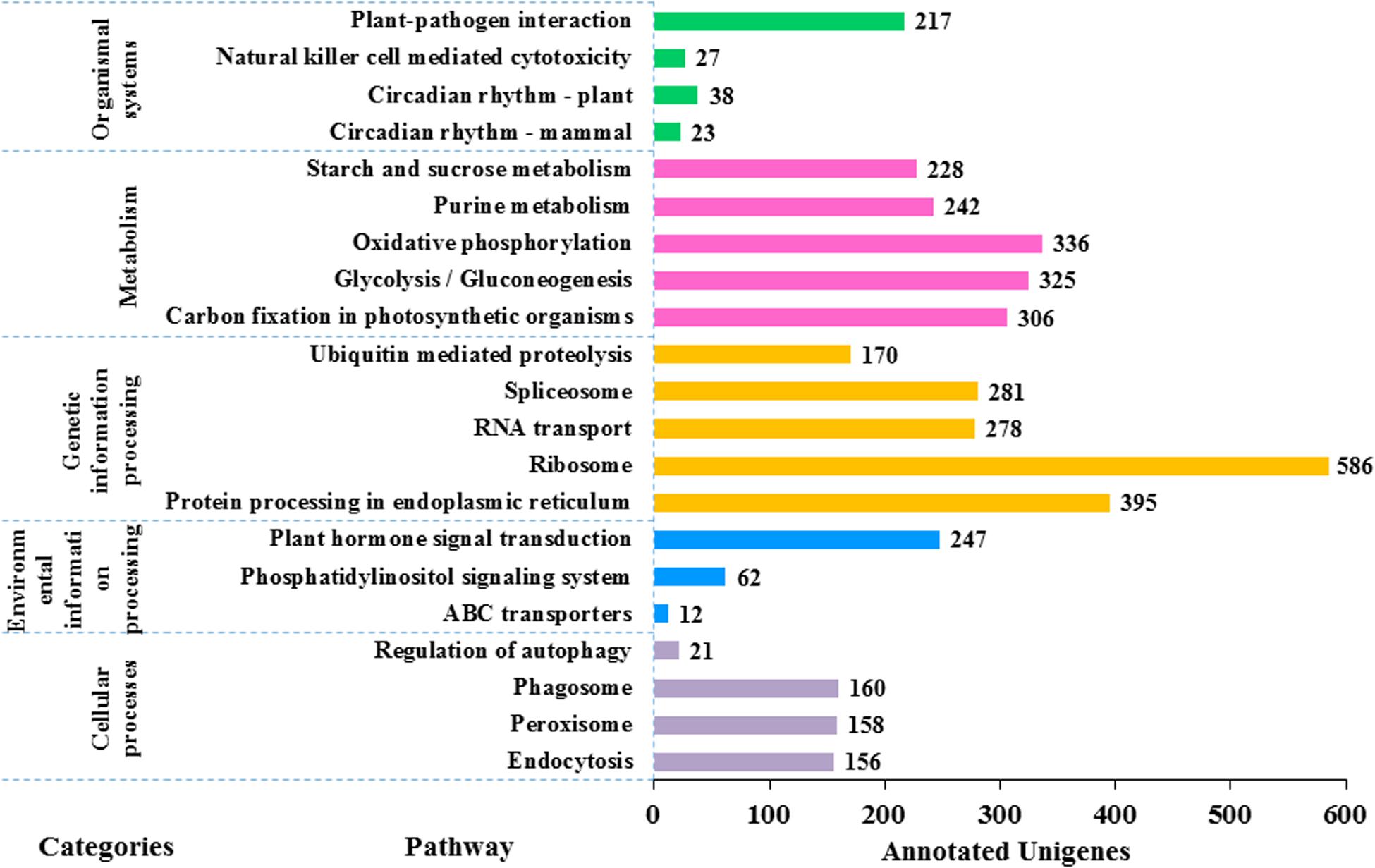
Figure 6. Functional classification of the assembled unigenes based on Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway analysis. 9,375 unigenes are annotated to five categories: metabolism, genetic information processing, cellular processes, environmental information processing and organic systems. There are a large number of pathways of metabolism and genetic information processing (in total 106 pathways) but the picture is limited, so the first five pathways with the largest number of each category are selected for plotting. The value to the right of each bar indicates the number of unigenes annotated in the pathway.
We conducted BUSCO analysis to evaluate completeness of the transcriptome using the near-universal orthologous gene groups. The details are presented in Supplementary Table S3 and illustrated in Supplementary Figure S1. For example, among the 430 BUSCOs in the viridiplantae_odb10 database, H. citrina cv. ‘Datonghuanghua’ transcriptome had 388 (90.2%) complete single or duplicated copies, 27 were fragmented, and 15 were missing, which indicated that this transcriptome assembly had adequate coverage and quality to represent the H. citrina transcriptome for various downstream analyses.
Using MISA, we mined microsatellite-containing sequences from 13,019 unigenes with >1,000 bp in length. As a result, 3,430 potential SSRs were identified from 2,977 unigenes, 453 of which contained more than one SSR (detailed in Supplementary Table S4). Details of the 2,977 unigenes, associated SSRs, and motif statistics are provided in Supplementary Tables S5, S6, respectively. The sequences of these unigenes in fasta format are provided in Supplementary Table S7. Among those SSRs, those with the tri-nucleotide repeat motifs were the most abundant (54.29%, 1,862), followed by SSRs with di-nucleotide (31.75%, 1,089), and tetra-nucleotide (3.24%, 111) motifs. Other types were rare. Among the di- and tri-nucleotide repeats, (GAA/TTC)n, and GA/TC)n or (AG/CT)n were the most common repeat motifs, respectively (Supplementary Table S6).
From the 2,977 SSR-containing unigene sequences, primers were designed for 3,577 SSRs. The primer sequences for each SSR and associated information are provided in Supplementary Table S8. From the list, we tested 192 experimentally in six accessions. Based on the preliminary data, we finally selected 43 SSRs that had clear, and unique amplicons with expected size. Information for the 43 EST-SSRs is provided in Table 3.
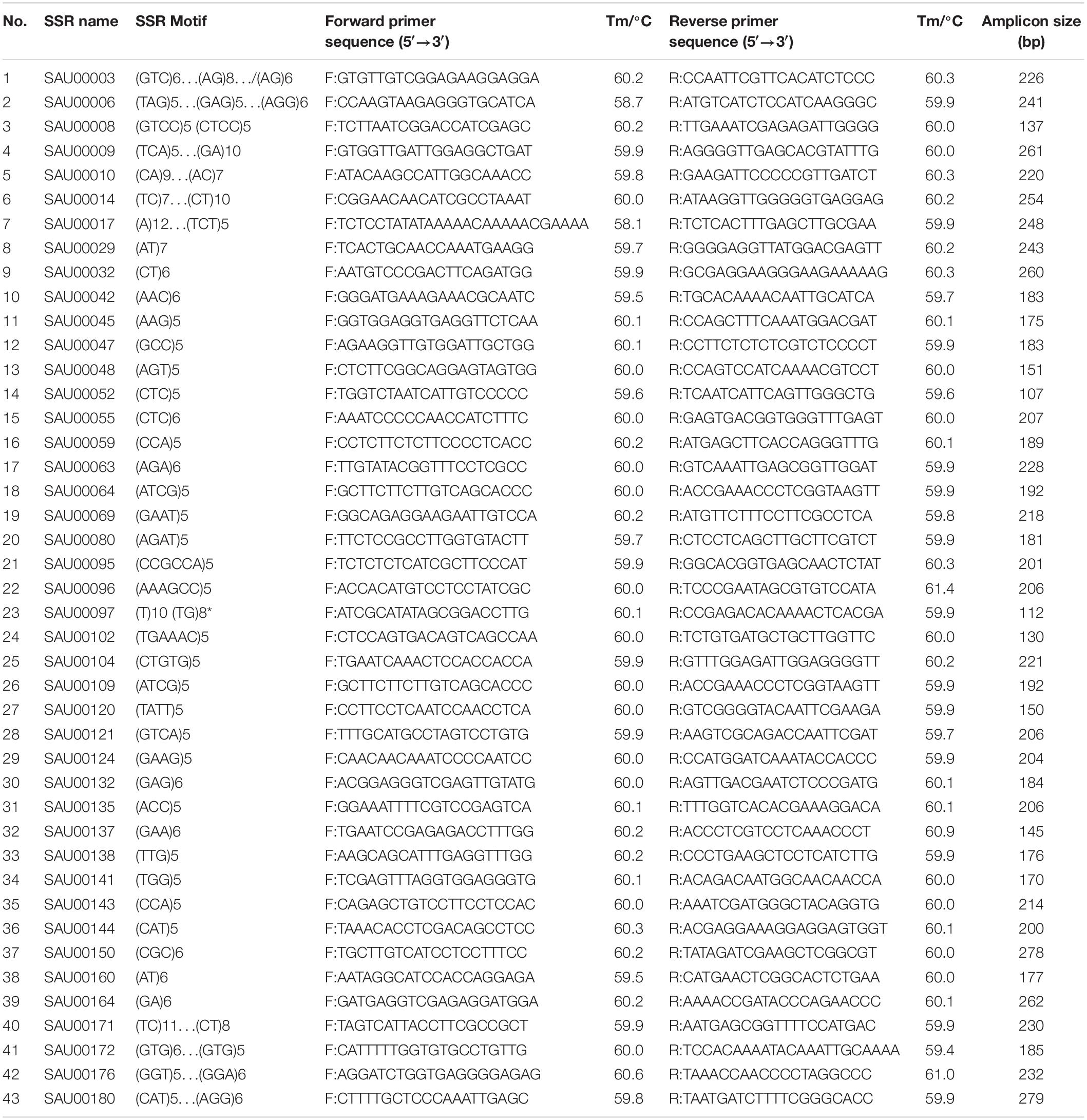
Table 3. Information of 43 SSRs used for assessment of genetic diversity among 155 Hemerocallis accessions.
The forty-three EST-SSRs were used to fingerprint 155 Hemeracallis accessions. A total of 396 alleles were detected at the 43 marker loci. The number of alleles detected at each locus varied from 2 to 25 with an average of 9.21 per locus. The polymorphism information content (PIC) ranged from 0.907 to 0.259 with an average of 0.606 (Table 4). Mean Shannon’s information index (I) was 1.294 (0.477–2.653). Mean observed heterozygosity (Ho) and mean expected heterozygosity (He) was 0.1757 (0.0000–0.5175) and 0.5935 (0.2104–0.9109). These indicated that the 43 SSRs provided rich genetic information (Table 4).
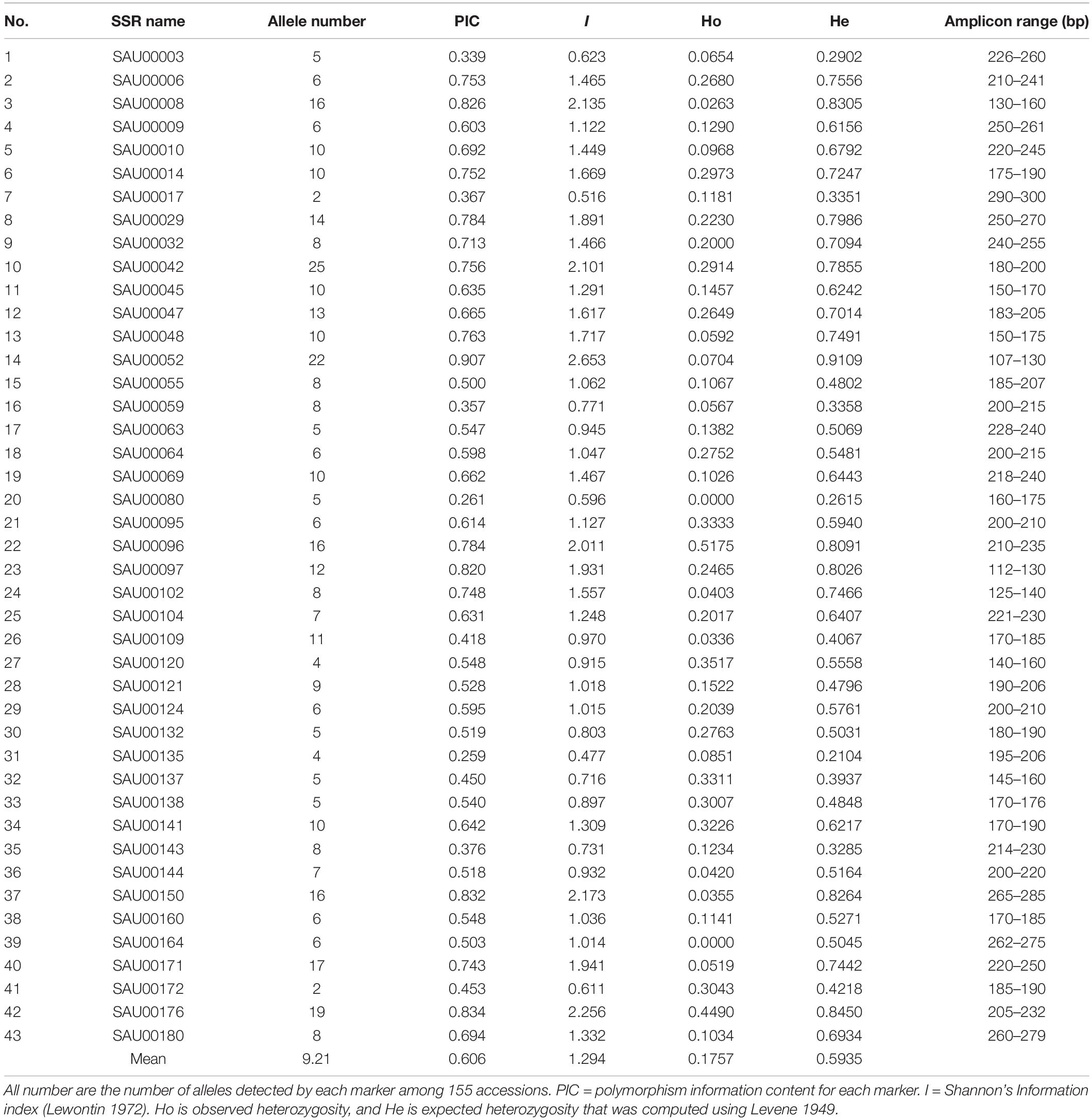
Table 4. Estimation of genetic diversity based on 43 SSRs among 155 Hemerocallis accessions.
Pairwise genetic similarity coefficients were calculated among all samples which are provided in Supplementary Table S9. The mean genetic similarity coefficient among these accessions was 0.8642 (range: 0.7828–1.000). The least genetic similarity coefficient was 0.7928 between “Suqian 1-C” and US4 suggesting they are genetically the most distant among these accessions. Meanwhile, the genetic similarity coefficient between ‘Datonghuanghua’ (H0006) and ‘Qiaotouhuanghua’ (H0010) was 1.000 indicating they may actually belong to the same accession.
A UPGMA dendrogram of the 155 accessions based on 43 EST-SSR markers was constructed (Supplementary Figure S2). A cophenetic correlation test (r = 0.8687) showed that the clustering results were credible. Among the 155 accessions, 55 from three species (H. citrina, H. lilioasphodelus, and H. minor) were clustered in one group (branches highlighted in red in Supplementary Figure S2). Fifty of the 55 accession in this group are known to be night lily accessions, which are represented by ‘Datonghuanghua’ (H0006) and ‘Panlonghua’ (H0004). Thus, this group could be called the “Night Lily Group.” Of the rest 100 accessions, 95 bloomed in the morning, and five [‘Guanglinghuanghua’ (H0011), ‘Stella de Oro 2’ (H0077), ‘Stella de Oro 3’ (H0139), ‘Xue Qiu Hong’ (H0092), and ‘Beijing 6’ (H0101)] open flowers at night (see flowering time data in Supplementary Table S1). These 100 accessions included multiple species such as H. fulva [var. sempervirens (H0050), and var. kwanso var. reasata (H0052)], H. aurantiaca (H0040), H. middendorffii (H0041), H. thunbergii (H0042), H. multiflora (H0045), H. altissima (H0046), and H. dumortieri (H0048) (Supplementary Table S1).
We also constructed a Neighbor-Joining (NJ) tree for the 155 accessions, which is presented in Figure 7. The groupings were largely the same as the UPGMA dendrogram (Supplementary Figure S2) with all night lily accession in one clade. These data suggested that night lily has narrower genetic base than daylily, and the night lily is likely the result of human selection from daylilies for human consumption during crop evolution.
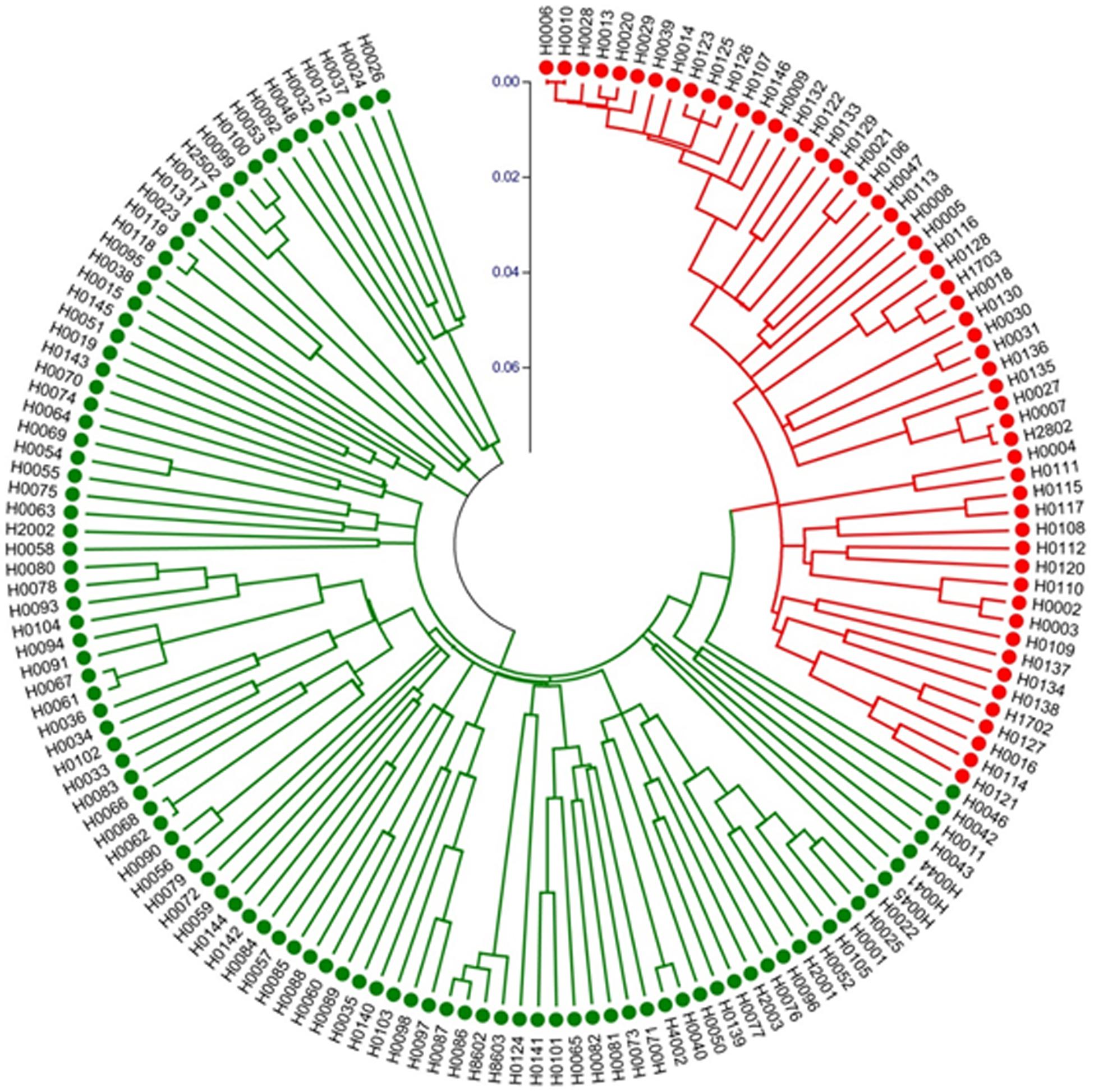
Figure 7. Neighbor-Joining tree of 155 Hemerocallis accessions based on 43 EST-SSR constructed using MEGA. Group I (branches in red) includes 55 accessions belonging mostly to the night lily group. Group II (branches in green) consisted of 100 accessions forming the daylily group.
A model-based Bayesian clustering approach was used to analyze 155 accessions by STRUCTURE. The logarithm of the likelihood [Ln P(D)] on average continued to increase with increasing numbers of assumed subpopulations (K) from 2 to 20 (Figure 8A). Difference between Ln P(D) values at two successive K-values became non-significant after K = 5. As the ad hoc statistic ΔK preferentially detects the uppermost level of structure and gave the highest value at K = 2 (Figure 8B; Lia et al., 2009; Emanuelli et al., 2013). Thus, K = 2 was considered as the most probable prediction for the number of subpopulations. Indeed, in the first round of structure analysis, the 155 accessions were split into two major clusters. Group I contained 53 accessions with an average Q value of 0.9867 (86.79% of them with Q > 0.98), and Group II contained 102 accessions with an average Q value of 0.9213 (78.43% of them with Q > 0.90) (Figure 8C and Supplementary Table S1).
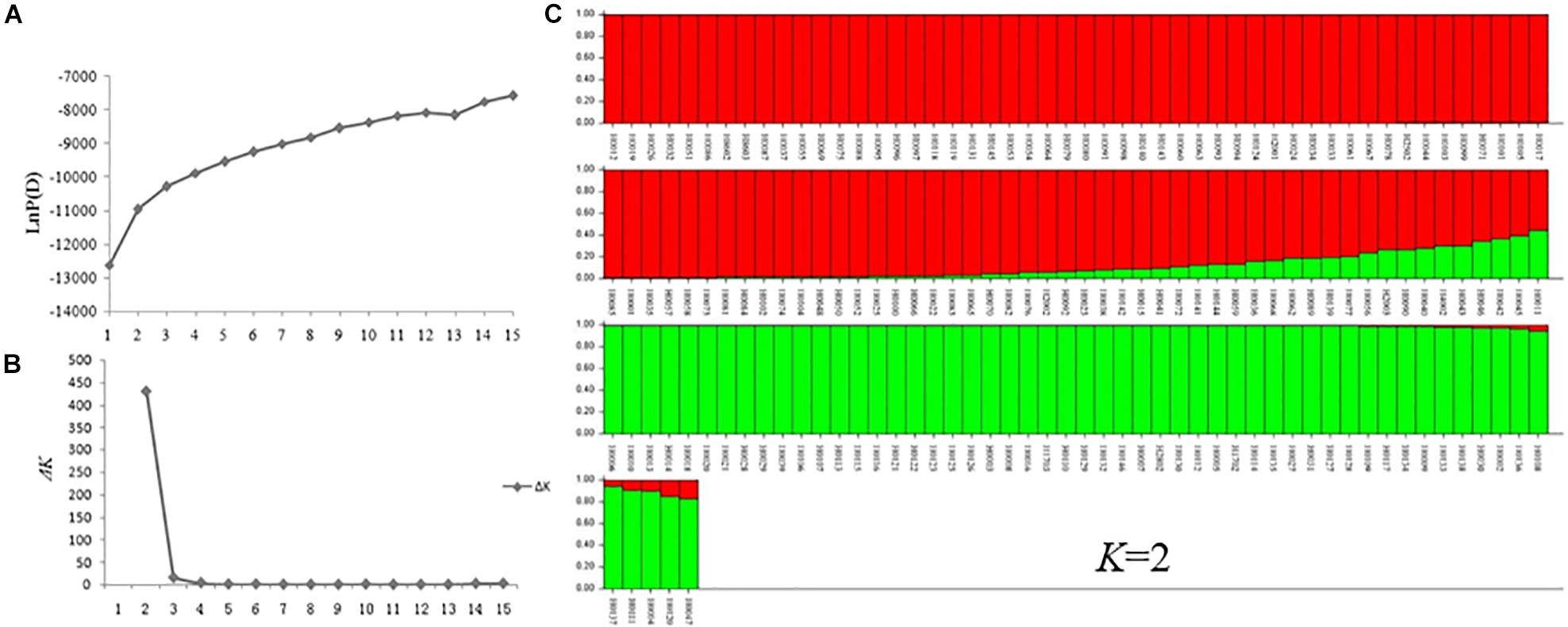
Figure 8. STRUCTURE analysis of 155 Hemerocallis accessions. (A) The relationship between K-value and LnP(D) value. (B) The relationship between K-value and ΔK value. (C) Clustering of 155 Hemerocallis accessions. The red is Groups II consisting of 102 accessions (daylily group), the green is Group I including 53 accessions (night lily group).
Group I consisted of 53 accessions, all of which were blooming at night (night lily group). Groups II consisted of 102 accessions, 97 accessions of them were blooming in the morning (daylily group) (Supplementary Table S1). Some accessions were admixtures of the two groups.
Blooming time is a very important horticultural trait for both daylily and night lily varieties. We recorded blooming time of all 155 accessions, and the data were presented in Supplementary Table S1. Among them, 96 bloomed (almost all daylily) in the morning (7–10 a.m.), and the rest (almost night lily) in the later afternoon or evening (4–11:30 p.m.). To confirm the utility of the EST-SSR markers developed in the present study, we conducted association analysis of blooming time using these markers. We first examined linkage disequilibrium (LD) in the Hemerocallis population. Of the 903 pairwise combinations generated for 43 the EST-SSR loci, 786 (87.0%) showed LD (Supplementary Table S10 and Supplementary Figure S3). At P ≤ 0.001, 74 pairs (8.2%) had mean r2 of 0.4266 indicating strong LD. At P ≤ 0.001, the standardized disequilibrium coefficients (D’) was 0.8435 which was close to 1.0 suggesting low chance of recombination between pairs of loci, which may be caused by the long-term asexual reproduction among the Hemerocallis collection.
Association analysis was performed using both the general linear model (GLM, Q model) and the mixed linear model (MLM, Q+K model) in TASSEL. The results are presented in Table 5, and illustrated in Figure 9. In the GLM model, 12 markers showed significant association with blooming time, 8 of which had extremely high significance (P ≤ 0.01; mean r2 = 0.0459) (Points above the blue line in Figure 9c). Similarly, under the MLM model, 10 markers were significantly associated with blooming time and 5 had extremely significant association (P ≤ 0.01; mean r2 = 0.0560) (Points above the blue line in Figure 9d). The 10 markers with significant associations with the blooming time in both GLM and MLM were SAU00008, SAU00045, SAU00047, SAU00045, SAU00063, SAU00064, SAU00109, SAU00121, SAU00143, and SAU00150. Among them, SAU00109 had the strongest association (P << 0.001 in MLM model) that could explain 8.8% phenotypic variance (8.0% in GLM) (Table 5). The predicted functions of the 12 unigenes associated with these SSRs are provided in Table 5. Both SAU00064 and SAU00109 were associated with the same unigenes c22731.graph_c0, which seems to encode a eukaryotic translation initiation factor 2D. Another unigene, c33464.graph_c0, associated with marker SAU00063 was annotated to encode a LHY-like protein, which is known to be regulated by circadian clock. The expression of c33464.graph_c0 in flower buds was significantly higher than that of roots and leaves in both RNA-Seq and RT-qPCR experiments (Figure 10). The unigene c47752.graph_c0 associated with SAU00008 was predicted to encode phosphatase 2C 35/65, and its expression level in roots was significantly different from that in flower buds and leaves.
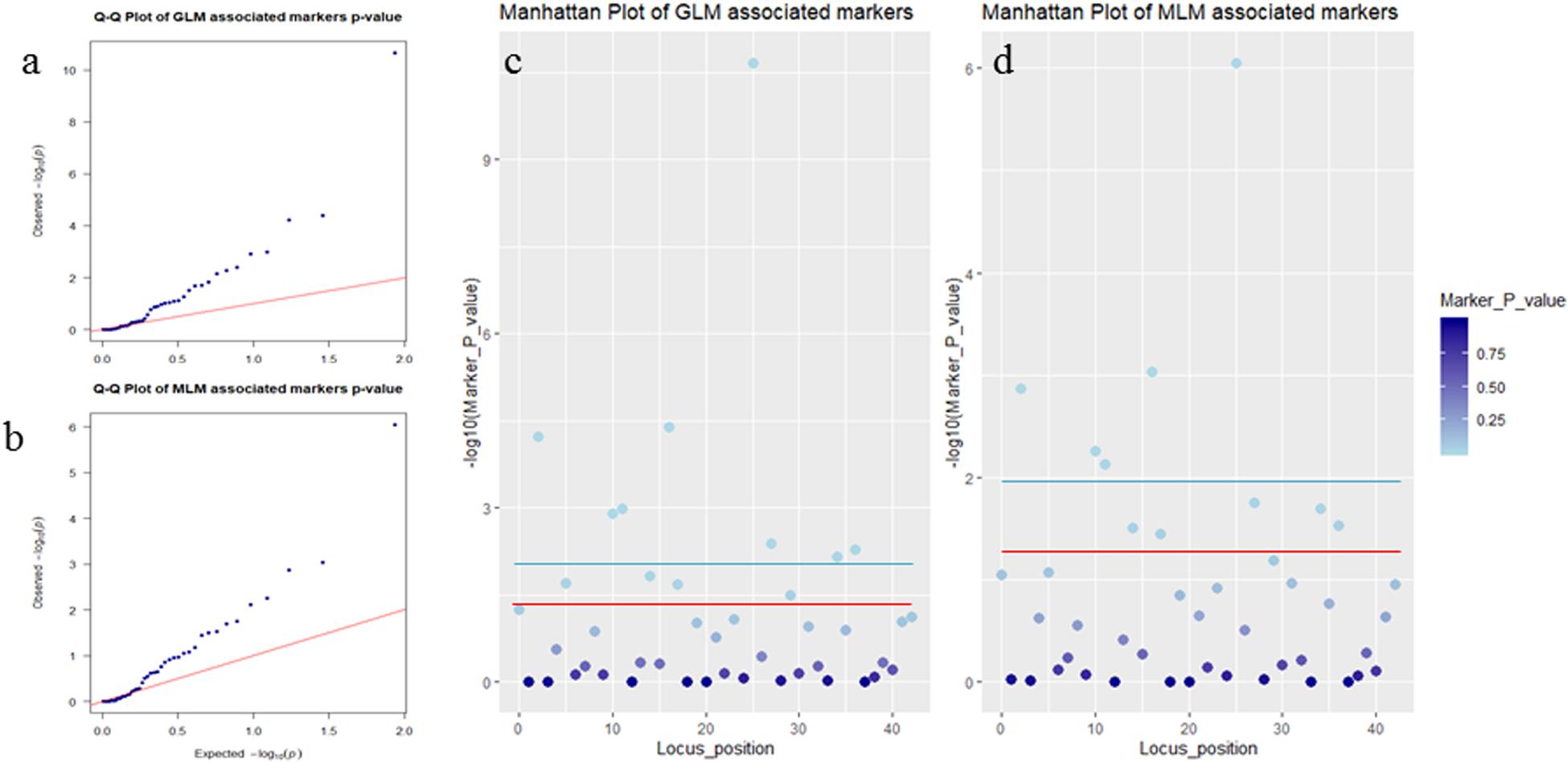
Figure 9. Association analysis of SSR markers with flower timing among 155 Hemerocallis accessions. In (a,b), QQ plots showing the P value of associated markers deviated from the expected P-value in GLM Model (a) and MLM Model (b). Blue dots and red lines indicate the P-value of the associated markers, and the expected P-value, respectively. In (c,d), Manhattan plots showed markers associated with flowering time in GLM Model (c) and MLM Model (d). Threshold: the red line is P = 0.05 and the blue line is P = 0.01.
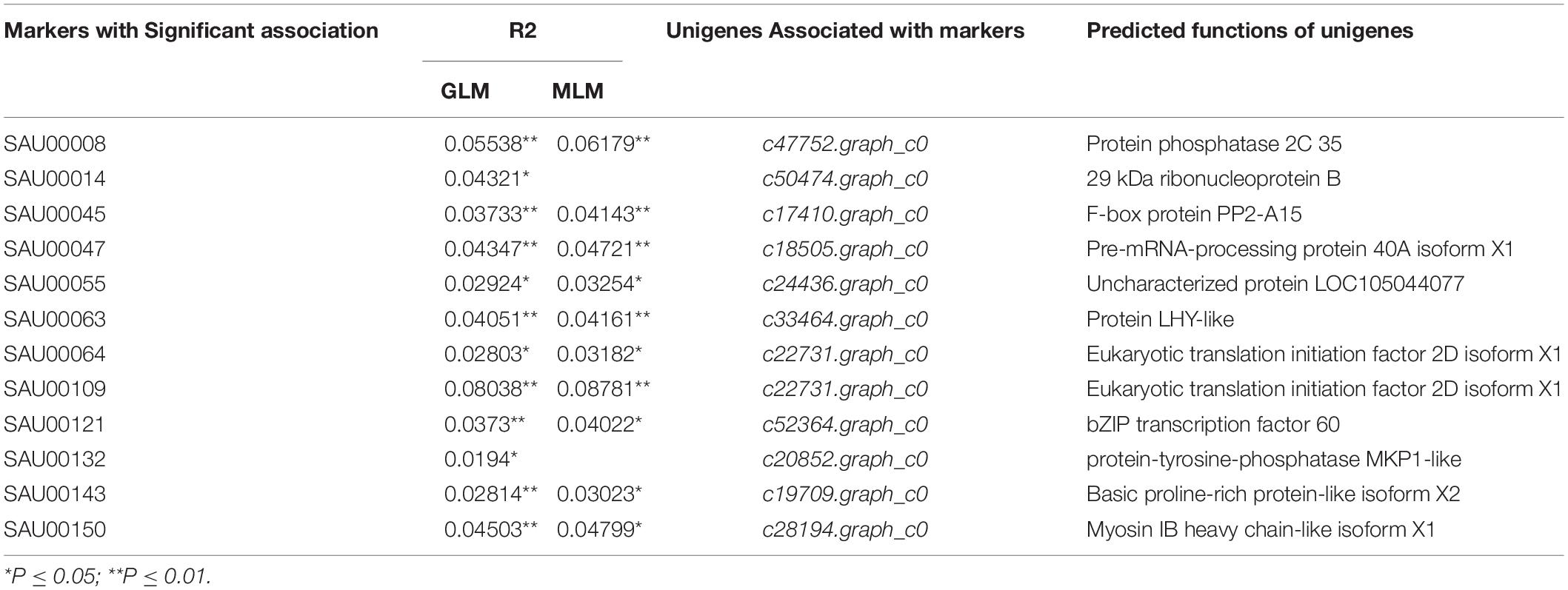
Table 5. Twelve SSR markers with significant association with blooming time in 155 Hemerocallis accessions*.
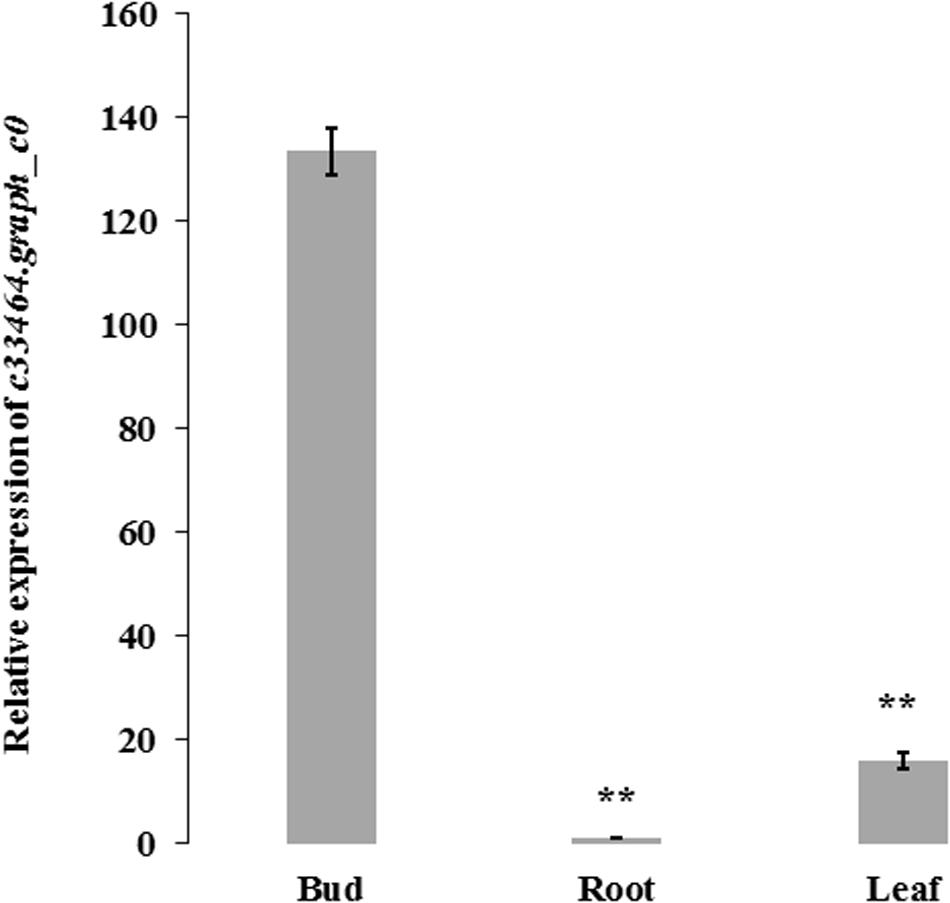
Figure 10. The expression of c33464.graph_c0 based on quantitative real-time PCR and RNA-Seq in bud, leaf and root of H. citrina cv. ‘Datonghuanghua’. The results show the mean ± SD (error bars) and were generated from three biological replicates. ** mean significantly difference, P ≤ 0.01. HcACT was used as an internal control for RT-qPCR.
In this study, 92,107 Hemerocallis transcriptomic unigenes were assembled using the Illumina Hi-Seq 2500 platform (Table 1). The N50 length of these unigenes was 908 bp, and the average length was 575 bp. These results were comparable to what obtained in other recent studies in species of the Liliaceae family, such as Lilium regale (N50 = 920 bp, average length = 682 bp) (Cui et al., 2018). The number of unigenes and contained SSRs obtained from the present study (92,107 unigenes with 3,430 SSRs) was also close the number identified in other three Liliaceae species (L. formolong, L. longiflorum, and L. longiflorum) (average unigenes = 72,256) (Biswas et al., 2018). These results suggested that closely related species share similar gene contents and distribution of SSR sequences in their genomes.
Fingerprinting is among the most popular uses of molecular markers. In Hemerocallis, hundreds of varieties have been released. Many varieties have the same names but may be morphologically different, or have the same appearances but with different names. Marker-based fingerprinting can help resolve these issues. In the study, the two landraces ‘Malinhuanghua (H0029)’ and ‘Malinhuanghua2 (H0125)’ had the same name (in Chinese), but have different geographic origins. Clustering analysis indicated that they were located in different clades (Figure 7 and Supplementary Figure S2). Therefore, these two accessions do not seem to share any common parent, but may have the same name by chance. The three accessions, ‘Taiguxuancao (H0038),’ ‘Stella de Oro 2 (H0077),’ and ‘Stella de Oro 3 (H0139)’ may have a similar situation. In addition, three accessions, H. minor Mill. (H0007) and H. minor Mill.2 (H2802) were all introduced from Qingyang, Gansu Province, China, but clustering analysis did not support their close relatedness. These apparent mis-identifications were probably because the original names of these foreign varieties were changed right after their introduction. This is also likely true for many accessions that were introduced from one place to another place inside China. Work in the present study presents a good example to show the power of molecular markers in variety identification and protection.
Both clustering analysis and STRUCTURE analysis place ∼50 accessions into one group (Figure 7 and Supplementary Figure S2) that bloom in the evening, while the rest of ∼100 accession all bloom in the morning (Supplementary Table S1). Marker-based phylogenetic analysis revealed that the night lily accessions were more genetically related, which seems to be consistent with their geographical distribution. Accessions in the daylily group were collected from more geographically diverse locations (Supplementary Table S1). Based on our multiple observations (Ji et al., 2018), accessions in the daylily group are also morphologically more diverse than those in the night lily group. For example, the colors of petals and sepals of daylilies may vary from yellow, yellow-orange, orange, orange-red, red, purple-red to purple. For flower size, the width variation coefficient was high in petals and sepals. Among them, the orange petal width variation coefficient was the highest of 42.15% and the average width was 35.90 mm (13.58–75.56 mm). Similarly, the variation coefficient of the orange sepal width was the highest at 38.74%, and the average width was 23.40 mm (13.20∼48.34 mm). The difference between stalk height and leaf width were highly significant. The inflorescences were rich in morphological variation ranging from mini inflorescence, extremely short branches, to large branched inflorescence, with varying amounts of blossoms. On the other hand, the 55 accessions in the night lily group (branches in red in Figure 7 and Supplementary Figure S2 were collected from nine provinces of China, all of which were diploids and are edible. For these accessions, both petals and sepals were yellow while the width of petals and sepals were very small, with the mean values 17.46 and 13.77 mm, respectively. As compared with accessions in the daylily group, the stalk height (main stem) of night lily group was in general higher (129.08 cm), the leaf was narrower (3.16 cm), and the average number of blossoms was 22.10. Fifty-three accessions were blooming at night, except for ‘Panlonghua’ (H0004) and ‘Panlonghua 2’ (H0111) that bloomed in the morning. The results from both marker analysis (Figure 7 and Supplementary Figure S2) and morphological observations indicate that the night lily group may have narrower genetic base than the daylily group accessions in this collection. This seems reasonable because the daylily group accessions had a more diverse geographic origin (Supplementary Table S1). Also, many of the night lily accessions may share common parents during their breeding and selection. From the crop evolution perspective, night lily was probably selected from the genetically more diverse daylily gene pool for vegetable use. During this process, blooming at night may be a main target of selection which is critical for vegetable use to collect the tender unopened flower bud during day time.
A few accessions were placed in different clades when different programs were used in clustering analysis, for example, ‘Panlonghua’ and ‘Panlonghua 2’ were clustered into night lily group by NTSYS and MEGA, which were in daylily group by STRUCTURE (Supplementary Table S1, Figures 7, 8, and Supplementary Figure S2). ‘Panlonghua’ and ‘Panlonghua 2’ were edible landraces, which were probably the hybrids between the night lily and the daylily. They displayed characteristics of both parents including morning blooming and the narrow perianth traits. ‘Stella de Oro 2’ (H0077), ‘Xue Qiu Hong’ (H0092), ‘Beijing 6’ (H0101), and ‘Little Bumble Bee’ (H0141) were daylily varieties that blossom at night, but they were clustered together with other daylily accessions that bloomed during the day. These observations suggest gene flow between the two cultivated groups during the long-term breeding and selection. Hermerocallis varieties are often recognized as either daylily or night lily based on their blooming time. Probably only a few genes with strong phenotypic effects are underlying the blooming time (Masahiro et al., 2006). In this study, we identified 12 SSRs with strong association with blooming time (Table 5). The SSR marker SAU00063 with the strongest association is in the unigene (c33464.graph_c0) that was annotated to encode a LHY-like protein (Table 5). The LHY (Late Elongated Hypocotyl) gene has been shown to regulate flower timing in a number of plants (e.g., Ramos-Sanchez et al., 2019; Zhang et al., 2019). Our work in the present study provides a foundation for gene discovery of flower timing in Hermerocallis. However, additional work is needed to validate this result.
This is the first report on the Hemerocallis transcriptome, and a large number of EST-SSR markers have been developed from this transcriptome, which will provide an excellent resource for researchers and breeders focusing on Hemerocallis. More importantly, our results showed that this strategy using high-throughput sequencing technology is feasible and extremely convenient and efficient in the genus Hemerocallis. The genes associated with circadian rhythm provide germplasm resources and genes basis for further study on flowering rhythm of this genus.
The datasets presented in this study can be found in NCBI under accession numbers SRR11610941, SRR11610942, and SRR11610943 (Submission ID: SUB7327030; BioProject ID: PRJNA628147).
SL designed and conducted the whole research. FJ collected the phenotype data and conducted the GWAS analysis. FH collected some accessions and developed the EST-SSR. HC analyzed the transcriptome data. QS ran the PAGE gel. GX and XK kept the Hemerocallis germplasm. YW advised the project and revised the manuscript.
This work was supported by the Innovative Talents Plan of Shanxi Agricultural University (BJRC201601), Key Research and Development Project of Shanxi Province (201703D211001-04-05, 201903D211011), the Shanxi Scholarship Council (2015-065), and the Collaborative Innovation Center for Improving Quality and Profits of Protected Vegetables in Shanxi.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2020.00686/full#supplementary-material
Azhaguvel, P., Li, W. L., Rudd, J. C., Gill, B. S., Michels, G. J. J., and Weng, Y. Q. (2009). Aphid feeding response and microsatellite-based genetic diversity among diploid Brachypodium distachyon (L.) Beauv accessions. Plant Genet. Resour. 7, 72–79. doi: 10.1017/s1479262108994235
Biswas, M. K., Nath, U. K., Howlader, J., Bagchi, M., Natarajan, S., Kayum, M. A., et al. (2018). Exploration and exploitation of novel SSR markers for candidate transcription factor genes in Lilium Species. Genes Basel 9:97. doi: 10.3390/genes9020097
Bradbury, P. J., Zhang, Z., Kroon, D. E., Casstevens, T. M., and Buckler, E. S. (2007). Tassel: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635. doi: 10.1093/bioinformatics/btm308
Cichewicz, R. H., Lim, K., Mckerrow, J. H., and Nair, M. G. (2002). Kwanzoquinones A-G and other constituents of Hemerocallis fulva ‘kwanzo’ roots and their activity against the human pathogenic trematode Schistosoma mansoni. Tetrahedron 58, 8597–8606. doi: 10.1016/s0040-4020(02)00802-5
Cichewicz, R. H., and Nair, M. G. (2002). Isolation and characterization of stelladerol, a new antioxidant naphthalene glycoside, and other antioxidant glycosides from edible daylily (Hemerocallis) flowers. J. Agr. Food Chem. 50, 87–91. doi: 10.1021/jf010914k
Cui, Q., Liu, Q., Gao, X., Yan, X., and Jia, G. X. (2018). Transcriptome-based identification of genes related to resistance against Botrytis elliptica in Lilium regale. Can. J. Plant Sci. 98, 1058–1071. doi: 10.1139/cjps-2017-0254
Dent, A., Earl, B., and vonHoldt, M. (2012). Structure harvester: a website and program for visualizing structure output and implementing the evanno method. Conserv. Genet. Resour. 4, 359–361. doi: 10.1007/s12686-011-9548-7
Du, B., Tang, X., Liu, F., Zhang, C., Zhao, G., and Ren, F. (2014). Antidepressant-like effects of the hydroalcoholic extracts of Hemerocallis citrina, and its potential active components. BMC Complem. Altern. 14:326. doi: 10.1186/1472-6882-14-326
Emanuelli, F., Lorenzi, S., Grzeskowiak, L., Catalano, V., Stefanini, M., Troggio, M., et al. (2013). Genetic diversity and population structure assessed by SSR and SNP markers in a large germplasm collection of grape. BMC Plant Biol. 13:39. doi: 10.1186/1471-2229-13-39
Eugene, V. K., Natalie, D. F., John, D. J., Aviva, R. J., Dmitri, M. K., and Kira, S. M. (2004). A comprehensive evolutionary classification of proteins encoded in complete eukaryotic genomes. Genome Biol. 5, R7–R7.
Evanno, G., Regnaut, S., and Goudet, J. (2005). Detecting the number of clusters of individuals using the software structure: a simulation study. Mol. Ecol. 14, 2611–2620. doi: 10.1111/j.1365-294x.2005.02553.x
Flora of China (2000). International Symposium of Plant Diversity and Conservation in China. Available online at: http://foc.eflora.cn/content.aspx?TaxonID=114981 (accessed April 6, 2019).
Grabherr, M. G., Haas, B. J., Yassour, M., Levin, J. Z., Thompson, D. A., and Amit, I. (2011). Full-length transcriptome assembly from RNA-Seq data without a reference genome. Nat. Biotechnol. 29:644. doi: 10.1038/nbt.1883
Gu, L., Liu, Y. J., Wang, Y. B., and Yi, L. T. (2012). Role for monoaminergic systems in the antidepressant-like effect of ethanol extracts from Hemerocallis citrina. J. Ethnopharmacol. 139, 780–787. doi: 10.1016/j.jep.2011.11.059
Guo, L. N., Zhao, X. L., and Gao, X. F. (2016). De novo assembly and characterization of leaf transcriptome for the development of EST-SSR markers of the non-model species Indigofera szechuensis. Biol. Chem. Syst. Ecol. 68, 36–43. doi: 10.1016/j.bse.2016.06.010
Hasegawa, M., Yahara, T., Yasumoto, A., and Hotta, M. (2006). Bimodal distribution of flowering time in a natural hybrid population of daylily (Hemerocallis fulva) and nightlily (Hemerocallis citrina). J. Plant Res. 119, 63–68. doi: 10.1007/s10265-005-0241-3
Hirota, S. K., Kozue, N., Yuni, K., Aya, K., Nobumitsu, K., and Yasumoto, A. A. (2012). Relative role of flower color and scent on pollinator attraction: experimental tests using F1 and F2 hybrids of daylily and nightlily. PLoS One 7:e39010. doi: 10.1371/journal.pone.0039010
Hirota, S. K., Nitta, K., Suyama, Y., Kawakubo, N., and Yasumoto, A. A. (2013). Pollinator-mediated selection on flower color, flower scent and flower morphology of Hemerocallis: evidence from genotyping individual pollen grains on the stigma. PLoS One 8:e85601. doi: 10.1371/journal.pone.0085601
Hou, F., Li, S., Wang, J., Kang, X., Weng, Y., and Xing, G. (2017). Identification and validation of reference genes for quantitative real-time PCR Studies in long yellow daylily Hemerocallis citrina Borani. PLoS One 12:e0174933. doi: 10.1371/journal.pone.00174933
Hsu, Y. W., Tsai, C. F., Chen, W.-K., Ho, Y.-C., and Lu, F. J. (2011). Determination of lutein and zeaxanthin and antioxidant capacity of supercritical carbon dioxide extract from daylily (Hemerocallis disticha). Food Chem. 129, 1813–1818. doi: 10.1016/j.foodchem.2011.05.116
Hudson, M. E. (2010). Sequencing breakthroughs for genomic ecology and evolutionary biology. Mol. Ecol. Resour. 8, 3–17. doi: 10.1111/j.1471-8286.2007.02019.x
Jaiswal, V., Mir, R. R., Mohan, A., Balyan, H. S., and Gupta, P. K. (2012). Association mapping for pre-harvest sprouting tolerance in common wheat (Triticumaestivum L.). Euphytica 188, 89–102. doi: 10.1007/s10681-012-0713-1
Ji, F., Li, S., Shi, Q., Hou, F., and Kang, X. (2018). Genetic diversity analysis of Hemerocallis and constructing the primary core collection. J. Hebei Agric. Univer. 41, 55–61.
Jiao, F., Liu, Q., Sun, G. F., Li, X. D., and Zhang, J. Z. (2016). Floral fragrances of Hemerocallis (daylily) evaluated by headspace solid-phase microextraction with gas chromatography-mass spectrometry. J. Pomol. Horti. Sci. 91, 573–581. doi: 10.1080/14620316.2016.1193427
Kanehisa, M., Goto, S., Kawashima, S., Okuno, Y., and Hattori, M. (2004). The KEGG resource for deciphering the genome. Nucleic Acids Res. 32:D277.
Kang, H. M., Zaitlen, N. A., Wade, C. M., Kirby, A., Heckermann, D., Daly, M. J., et al. (2008). Efficient control of population structure in model organism association mapping. Genetics 178, 1709–1723. doi: 10.1534/genetics.107.080101
Lia, V. V., Poggio, L., and Confalonieri, V. A. (2009). Microsatellite variation in maize landraces from Northwestern Argentina: genetic diversity, population structure and racial affiliations. Theor. Appl. Genet. 119, 1053–1067. doi: 10.1007/s00122-009-1108-0
Lin, Y. L., Lu, C. K., Huang, Y. J., and Chen, H. J. (2011). Antioxidative caffeoylquinic acids and flavonoids from Hemerocallis fulva flowers. J. Agric. Food Chem. 59, 8789–8795. doi: 10.1021/jf201166b
Liu, X. L., Luo, L., Liu, B. B., Li, J., Geng, D., and Liu, Q. (2014). Ethanol extracts from Hemerocallis citrina attenuate the upregulation of proinflammatory cytokines and indoleamine 2,3-dioxygenase in rats. J. Ethnopharmacol. 153, 484–490. doi: 10.1016/j.jep.2014.03.001
Masahiro, H., Tetsukazu, Y., Akiko, Y., and Mitsuru, H. (2006). Bimodal distribution of flowering time in a natural hybrid population of daylily (Hemerocallis fulva) and nightlily (Hemerocallis citrina). J. Plant Res. 119, 63–68. doi: 10.1007/s10265-005-0241-3
Michael, A., Catherine, A. B., Judith, A. B., David, B., Heather, B., and Michael, C. J. (2000). Gene ontology: tool for the unification of biology. Nat. Genet. 25, 25–29.
Miyake, T., and Yahara, T. (2010). Isolation of polymorphic microsatellite loci in Hemerocallis fulva, and Hemerocallis citrina, (Hemerocallidaceae). Mol. Ecol. Notes 6, 909–911. doi: 10.1111/j.1471-8286.2006.01396.x
Nitta, K., Yasumoto, A. A., and Yahara, T. (2010). Variation of flower opening and closing times in F1 and F2 hybrids of daylily (Hemerocallis fulva; Hemerocallidaceae) and nightlily (H. citrina). Am. J. Bot. 97, 261–267. doi: 10.3732/ajb.0900001
Parida, S. K., Kalia, S. K., Kaul, S., Dalal, V., Hemaprabha, G., and Selvi, A. (2009). Informative genomic microsatellite markers for efficient genotyping applications in sugarcane. Theor. Appl. Genet. 118, 327–338. doi: 10.1007/s00122-008-0902-4
Podwyszynska, M., Gabryszewska, E., Korbin, M., and Jasinski, A. (2009). Evaluation of somaclonal variation in micropropagated Hemerocallis.spp plants using phenotype and ISSR markers. Acta Biol. Cracov. 51, 58–58.
Pritchard, J. K., Stephens, M., and Donnelly, P. (2000). Inference of population structure using multi-locus genotype data. Genetics 155, 945–959.
Ramos-Sanchez, J., Triozzi, P., Alique, D., Geng, F., Gao, M., and Jaeger, E. (2019). LHY2 integrates night-length information to determine timing of Popular photoperiodic growth. Curr. Biol. 29, 2402–2406.
Rodriguez-Enriquez, M. J., and Grant-Downton, R. T. (2012). A new day dawning: Hemerocallis (daylily) as a future model organism. AOB Plants 5:ls055.
Rop, O., ŘEzníček, V., Mlček, J., Juríková, T., Balík, J., and Sochor, J. (2011). Antioxidant and radical oxygen species scavenging activities of 12 cultivars of blue honeysuckle fruit. Horticul. Sci. 38, 63–70. doi: 10.17221/99/2010-hortsci
Simão, F. A., Waterhouse, R. M., Ioannidis, P., Kriventseva, E. V., and Zdobnov, E. M. (2015). BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31, 3210–3212. doi: 10.1093/bioinformatics/btv351
Smithunna, R., Boursnell, C., Patro, R., Hibberd, J. M., and Kelly, S. (2016). Transrate: reference-free quality assessment of de novo transcriptome assemblies. Genome Res. 26:1134. doi: 10.1101/gr.196469.115
Strickler, S. R., Bombarely, A., and Mueller, L. A. (2012). Designing a transcriptome nextgeneration sequencing project for a nonmodel plant species. Am. J. Bot. 99, 257–266. doi: 10.3732/ajb.1100292
Tatusov, R. L., Galperin, M. Y., Natale, D. A., and Koonin, E. V. (2000). The cog database: a tool for genome-scale analysis of protein functions and evolution. Nucleic Acids Res. 28, 33–36. doi: 10.1093/nar/28.1.33
Tel-Zur, N., Abbo, S., Myslabodski, D., and Mizrahi, Y. (1999). Modified CTAB procedure for DNA isolation from epiphytic cacti of the genera Hylocereus and Selenicereus (Cactaceae). Plant Mol. Biol. Report. 17, 249–254.
Tomkins, J. P., Wood, T. C., Barnes, L. S., Westman, A., and Wing, R. A. (2001). Evaluation of genetic variation in the daylily (Hemerocallis spp.) using AFLP markers. Theor. Appl. Genet. 102, 489–496. doi: 10.1007/s001220051672
Uezu, E. (1998). Effects of Hemerocallis on sleep in mice. Psych. Clin. Neurosci. 52, 136–137. doi: 10.1111/j.1440-1819.1998.tb00992.x
Varshney, R. K., Mahendar, T., Aggarwal, R. K., and Börner, A. (2007). “Genic molecular markers in plants: development and applications,” in Genomics-Assisted Crop Improvement, eds R. K. Varshney and R. Tuberosa (Dordrecht: Springer).
Vendrame, W. A., Fogaa, L. A., Pinares, A., Cuquel, F. L., and Filho, J. C. B. (2009). Assessment of Ploidy Level and Genetic Relationships Among Selected Hemerocallis Hybrids. ASHS Annual Conference.
Venugopalan, A., and Srivastava, S. (2015). Endophytes as in vitro production platforms of high value plant secondary metabolites. Biotechnol. Adv. 33, 873–887. doi: 10.1016/j.biotechadv.2015.07.004
Yi, L. T., Li, J., Li, H. C., Zhou, Y., Su, B. F., and Yang, K. F. (2012). Ethanol extracts from Hemerocallis citrina, attenuate the decreases of brain-derived neurotrophic factor, TrkB levels in rat induced by corticosterone administration. J. Ethnopharmacol. 144, 328–334. doi: 10.1016/j.jep.2012.09.016
Yuan, S., Ge, L., Liu, C., and Ming, J. (2013). The development of EST-SSR markers in Lilium regale and their cross-amplification in related species. Euphytica 189, 393–419. doi: 10.1007/s10681-012-0788-8
Zhang, L., Yan, H. F., Wei, W., Hui, Y., and Ge, X. J. (2013). Comparative transcriptome analysis and marker development of two closely related primrose species (Primula poissonii, and Primula wilsonii). BMC Genomics 14:329. doi: 10.1186/1472-6882-14-329
Zhang, Z., Fu, Y., Zhou, Y., Wang, C., Lan, T., and Chen, G. (2019). Nitrogen and nitric oxide regulate Arabidopsis flowering differently. Plant Sci. 284, 177–184. doi: 10.1016/j.plantsci.2019.04.015
Keywords: daylily, genetic diversity, Hemerocallis, night lily, population structure, SSR, transcriptome
Citation: Li S, Ji F, Hou F, Cui H, Shi Q, Xing G, Weng Y and Kang X (2020) Characterization of Hemerocallis citrina Transcriptome and Development of EST-SSR Markers for Evaluation of Genetic Diversity and Population Structure of Hemerocallis Collection. Front. Plant Sci. 11:686. doi: 10.3389/fpls.2020.00686
Received: 16 November 2019; Accepted: 30 April 2020;
Published: 11 June 2020.
Edited by:
Jose Luis Gonzalez Hernandez, South Dakota State University, United StatesReviewed by:
Claudio Benicio Cardoso-Silva, Center for Molecular Biology and Genetic Engineering, State University of Campinas, BrazilCopyright © 2020 Li, Ji, Hou, Cui, Shi, Xing, Weng and Kang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Sen Li, c2F1bGlzZW5AMTYzLmNvbQ==; Yiqun Weng, eWlxdW4ud2VuZ0B3aXNjLmVkdQ==; Xiuping Kang, c3huZGt4cEAxNjMuY29t
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.