 Pietro Gramazio
Pietro Gramazio Haidong Yan
Haidong Yan Tomas Hasing
Tomas Hasing Santiago Vilanova
Santiago Vilanova Jaime Prohens
Jaime Prohens Aureliano Bombarely
Aureliano Bombarely
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 08 October 2019
Sec. Plant Breeding
Volume 10 - 2019 | https://doi.org/10.3389/fpls.2019.01220
Whole-genome resequencing provides information of great relevance for crop genetics, evolution, and breeding. Here, we present the first whole-genome resequencing study using seven eggplant (Solanum melongena) and one wild relative (Solanum incanum) accessions. These eight accessions were selected for displaying a high phenotypic and genetic diversity and for being the founder parents of an eggplant multiparent advanced generation intercrosses population. By resequencing at an average depth of 19.8× and comparing to the high-quality reference genome “67/3” over 10 million high-reliable polymorphisms were discovered, of which over 9 million (84.5%) were single nucleotide polymorphisms and more than 700,000 (6.5%) InDels. However, while for the S. melongena accessions, the variants identified ranged from 0.8 to 1.3 million, over 9 million were detected for the wild S. incanum. This confirms the narrow genetic diversity of the domesticated eggplant and suggests that introgression breeding using wild relatives can efficiently contribute to broadening the genetic basis of this crop. Differences were observed among accessions for the enrichment in genes regulating important biological processes. By analyzing the distribution of the variants, we identified the potential footprints of old introgressions from wild relatives that can help to unravel the unclear domestication and breeding history. The comprehensive annotation of these eight genomes and the information provided in this study represents a landmark in eggplant genomics and allows the development of tools for eggplant genetics and breeding.
Common eggplant (Solanum melongena L., 2n = 2x = 24) is the fifth economically most important vegetable crop, and in the last few years, it has undergone a remarkable increase in yield and total production (Faostat, 2017). Despite its importance in many tropical and subtropical areas, eggplant has lagged behind other major crops in the development of genetic and genomic tools (Gramazio et al., 2018). However, recent efforts have been done to close the gap between eggplant and other model species where more genomic information is available (Yang et al., 2014; Gramazio et al., 2016; Zhou et al., 2016). In this way, the significant improvements in next-generation sequencing (NGS) technologies and the associated substantial cost reductions have fostered genomic studies for crops like eggplant. In this respect, the release of reference whole-genome sequences represents an important landmark for undertaking other genomic studies, as it has occurred for the most economically important cultivated crops (Weigel and Mott, 2009; Xu et al., 2012; Aflitos et al., 2014; Zhou et al., 2015; Du et al., 2018).
In eggplant, the first draft of a reference genome was released in 2014, which was built on 33,873 scaffolds and covered 833.1 Mb (∼74% of the eggplant genome) (Hirakawa et al., 2014). A new high-quality eggplant genome sequence from accession “67/3,” assembled combining by Illumina sequencing and optical mapping, has recently been released (http://www.eggplantgenome.org/) (Barchi et al., 2019a). As result, the improved eggplant genome has a lower fragmentation, a higher contiguity, and a higher genome coverage (1.06 of 1.20 Gb estimated genome size) compared with the 2014 version. Together with the draft reference genome of Hirakawa et al. (2014), the “67/3” high-quality reference genome will facilitate resequencing studies in cultivated eggplant and its wild genepool. It will also contribute to accelerate the understanding of the genetic basis of many traits of interest in breeding for which there is a lack of specific molecular and genomic tools. To our knowledge, no resequencing studies have been reported in eggplant, while in model species and other economically important crops, such as Arabidopsis thaliana (Cao et al., 2011; Alonso-Blanco et al., 2016), tomato (Aflitos et al., 2014; Lin et al., 2014; Gao et al., 2019), or soybean (Lam et al., 2010; Zhou et al., 2015), dozens, hundreds, and even thousands accessions have been resequenced. Examples include. In fact, in these studies, the resequencing approach has allowed capturing the natural variation across the genepool through the identification of millions of robust polymorphisms among cultivated and wild relative accessions. The high-confidence sets of polymorphisms provided a valuable opportunity to perform forward genetics and genome-wide association studies and unravel the genetic base of complex traits of agronomic importance (Huang et al., 2010; Tranchida-Lombardo et al., 2017). In addition, whole-genome resequencing helps to gain insight on the evolution of crops by identifying genetic diversity bottlenecks that occurred during the domestication, as well as the genes involved in the process (Zhou et al., 2015; Gao et al., 2019). Furthermore, resequencing studies have allowed associating genes and traits with geographical areas revealing how populations and subpopulations have adapted to specific geographical regions (Qi et al., 2013). Ultimately, the whole-genome resequencing approach is a powerful breeding tool that can foster the development of a new generation of resilient varieties adapted to present and future challenges like climate change.
In this paper, we describe the first whole-genome resequencing study in the eggplant genepool including a comprehensive structural and functional characterization of seven S. melongena and one wild eggplant relative (S. incanum L.) accessions of interest for eggplant breeding (Prohens et al., 2013; Gramazio et al., 2017a). These eight accessions were chosen to maximize the phenotypic, genetic, and geographic variation and are the founder parents of a multiparent advanced generation intercrosses (MAGIC) population that is under development. Solanum incanum, which is part of the secondary genepool of the common eggplant (Syfert et al., 2016), has been reported as a powerful source of phenolic compounds, displaying contents several times higher than those of S. melongena (Stommel and Whitaker, 2003; Prohens et al., 2013), and is tolerant to some biotic and abiotic stresses, mainly drought (Knapp et al., 2013). The aim of this study was to interrogate the entire genome of these accessions to provide a large set of robust and high-confidence molecular markers in a very diverse eggplant germplasm set for breeding purposes, genetic and genomic studies, and to develop a high-throughput genotyping platform to efficiently assist the future characterization of the MAGIC population under development. In addition, this exploratory study intends to pave the way for future studies to shed light on eggplant evolutionary and breeding history through the characterization of introgressions, transposable, and genomic elements. The information and tools developed in our study will be of great relevance for eggplant genomics-assisted breeding.
Seven Solanum melongena accessions were chosen to maximize the representation of phenotypic and genetic diversity in the common eggplant (Table 1). The accessions have different origins and display substantial differences in agronomic and morphological traits, such as prickliness, fruit size, fruit shape, fruit color, or calyx size (Figure 1). In addition, one S. incanum accession (MM577) was selected as an outgroup because of its interest in eggplant breeding. This specific S. incanum accession has been characterized in multiple genetic and genomic studies for several traits (Stommel and Whitaker, 2003; Gisbert et al., 2011; Salas et al., 2011; Prohens et al., 2013; Meyer et al., 2015; Gramazio et al., 2016; Gramazio et al., 2017b) and has been used as a parent for developing an interspecific linkage genetic map and the first introgression line population in the eggplant genepool (Gramazio et al., 2014, Gramazio et al., 2017a). All the accessions are maintained at the Universitat Politècnica de València (UPV) germplasm bank. Seeds were germinated in Petri dishes, following the Ranil et al. (2015) protocol, and were subsequently transferred to seedling trays in a climatic chamber under a photoperiod and temperature regime of 16 h light (25°C)/8 h dark (18°C). The plantlets were then transplanted to a glasshouse situated in the campus of the UPV, Valencia, Spain (GPS coordinates: latitude, 39° 28′ 55″ N; longitude, 0° 20′ 11″ W; 7 m above sea level). Plants were grown in 15-L pots filled with coconut fiber, irrigated and fertilized using a drip irrigation system, and pruned and trained with vertical strings.

Table 1 Plant materials used in this study including their country of origin and main phenotypic characteristics.

Figure 1 Pictures of the seven Solanum melongena and one S. incanum (MM577) accessions resequenced in this study. Fruits are not depicted at the same scale; the size of the grid cells is 1 cm × 1 cm.
Total genomic DNA was isolated from an individual plantlet of each accession according to the cetyl trimethylammonium bromide method (Doyle and Doyle, 1990), with slight modifications. DNA integrity was evaluated with agarose electrophoresis, DNA quality through the 260/280 and 260/230 nm ratios from NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies, Wilmington, Delaware, USA) and concentration with a Qubit® 2.0 Fluorometer (Thermo Fisher Scientific, Waltham, MA, USA). High-quality DNA samples were shipped to the Duke Center for Genomic and Computational Biology (Durham, NC, USA) for libraries construction and sequencing. Paired-end libraries were prepared using the KAPA HyperPrep Kits (Roche, Basel, Switzerland) with an insert size of approximately 300 bp and sequenced on two lines of an Illumina HiSeq 4000 sequencer.
The raw sequences were processed with the Ea-utils package for barcode demultiplexing, adapter trimming, quality filtering, and stats (Aronesty, 2013). Raw reads shorter than 50 bp and with Phred score lower than 30 were removed with the fastq-mcf tool. The processed reads were then mapped to the “67/3” high-quality eggplant reference genome (Barchi et al., 2019a; http://www.eggplantgenome.org/) with the Bowtie2 aligner (version 2.2.4), which was selected for its speed and capability to give a lower score for suboptimal alignments (Li, 2014), using default parameters (Langmead and Salzberg, 2012). The unmapped reads were assembled with the short-read assembler Minia with default settings (Salikhov et al., 2013), and the contigs obtained were blasted against the nr database of the GenBank. The SAMtools package (version 1.3.1) was used to convert the mapping results into the BAM format and to filter the unmapped reads (Li et al., 2009). The coverage of sequence alignments was calculated using the utility genomecov of the BEDtools package (version 2.21.0; Quinlan and Hall, 2010), while the average coverage was determined with an in-house script. Variant calling was performed using the Bayesian-based FreeBayes software (v0.9.20, Garrison and Marth, 2012) with a minimum depth coverage of 5, a minimum base quality of 20, and a minimum mapping quality of 20. The vcftools package (version 0.1.12; Danecek et al., 2010) was used to split the main variant call format (VCF) output file into eight individual files and to remove the allele missing data. In order to identify similar patterns of polymorphisms distribution to associate them with potential common ancestral introgressions, the main VCF file was divided into homozygous (0/0) or heterozygous (0/1) variants and into segments of 10 Mb. The number of variants for each segment was calculated with the utility Vcf2CountingBins of the package GenoToolBox (https://github.com/aubombarely/GenoToolBox) and plotted by chromosomes in R (Ihaka and Gentleman, 1996). Using the whole set of single nucleotide polymorphisms (SNPs), after filtering out the other marker types [InDels, multiple-nucleotide polymorphisms (MNP), and complex] and the missing data, a genetic matrix distance was calculated among the different accessions using the TASSEL software (version 5.0 Standalone, Bradbury et al., 2007). A multivariate principal coordinates analysis (PCoA) was performed using the “prcomp” (https://stat.ethz.ch/R-manual/R-devel/library/stats/html/prcomp.html) and “factoextra” (https://cran.r-project.org/web/packages/factoextra/index.html) packages of the R software.
The variants annotation was performed according to the “67/3” eggplant reference genome gff3 annotation file using the Bedtools package (version 2.21.0; Quinlan and Hall, 2010) and Blast2GO software (Conesa et al., 2005). In addition, the Gene Ontology (GO) terms were added from the GO database (http://www.geneontology.org/). The variant effects were annotated based on their genomic position using the SnpEff software (version 4.2; Cingolani et al., 2012). Specifically, the effects of the allelic variants were classified by impact (high, low, moderate, or modifier), by functional class (missense, nonsense, or silent mutation), by type (start lost, stop gained, stop lost, and others), and region affected (intergenic, intron, exon, and others). In addition, the DNA substitution mutations (transitions and transversion) and amino acids changes were reported.
Transposons in the eggplant reference genome “67/3” were searched by combining a de novo and a homology-based approaches. For the de novo-based approach, the RepeatModeler software (Smit and Hubley, 2015) was used for identifying repeat element boundaries and family relationships from the reference genome sequence. For the homology-based approach, RepeatMasker program (Smit et al., 2004) was used to obtain a detailed annotation of the repeats from the screening for interspersed repeats and low complexity DNA sequences against the Repbase TE library (https://www.girinst.org/). Subsequently, the transposons found with the two approaches were combined, and the redundant hits were removed using CD-HIT software (Fu et al., 2012) with default parameters. Transposon insertions were detected with the TEMP software (Zhuang et al., 2014), and their quantity were calculated based on the output of the McClintock pipeline (Nelson et al., 2017). Transposon size was calculated based on bed files generated from the McClintock pipeline.
The sequence data have been deposited into NCBI Short Read Archive under submission identifier SUB2829594 with the Bioproject identifier PRJNA392603. Raw reads of each accession are deposited under the accession numbers from SRR5796636 to SRR5796643. VCF files with the corresponding variants identified are available upon request to the corresponding author.
The genome sequencing of the eight accessions generated over 1.4 billion paired-end raw reads (109 Gb of data), with a mean of 180.9 million reads per sample (Table 2). After the cleaning step, 97.6% of sequences, with an average length of 135 bp, were kept and were mapped onto the eggplant reference genome “67/3” (Barchi et al., 2019a). The mapping rate was quite similar across the accessions with an average of 85.4% and a range from 76.9% (ASI-S-1) to 88.7% (MM1597). The lower mapping percentage of the S. melongena accession ASI-S-1 was due to a higher proportion of mitochondrial reads that did not map to the reference genome since the full mitochondrial genome is not included in the reference genome “67/3.” The mean coverage depth varied from 16.5x in H15 to 24.0x in A0416, with a mean in the set of accessions of 19.8x. The mapping coverage of common eggplant (S. melongena) accessions encompassed practically the whole length of the reference genome, except for small regions (∼1%) of chromosomes 1, 11, and 12 in A0416, chromosomes 9 and 12 in AN-S-26, and chromosome 12 in DH_ECAVI, H15, and IVIA-371 (Supplementary Data S1). The S. incanum mapping coverage was lower, with a mean of 95.4%, and a quite even distribution along the chromosomes.
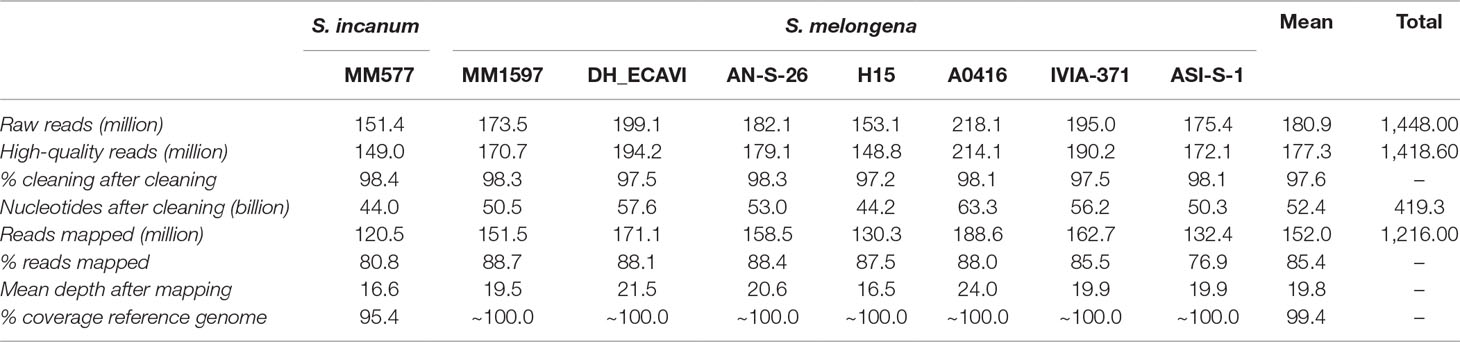
Table 2 Statistics of sequencing and mapping of the seven Solanum melongena and one S. incanum accessions resequenced in this study.
A total of 12,243,422 polymorphisms were detected among the eight resequenced accessions). To retain reliable variants for genotyping purposes and population studies, missing data were excluded when individual accessions were compared to the reference genome, yielding a final total of 10,916,466 high-quality variants (Table 3). Of those, 9,228,065 were SNPs (84.5%), 705,687 InDels (6.5%), 275,467 MNPs (2.5%), and 707,247 complex variations (6.5%). The total number of polymorphisms was homogeneously distributed among the S. melongena accessions and ranged from 832,366 of ASI-S-1 to 1,282,595 of IVIA-371. As expected, the wild S. incanum presented a much higher number of polymorphisms, with 9,343,703 variants when compared to the eggplant reference genome “67/3”.
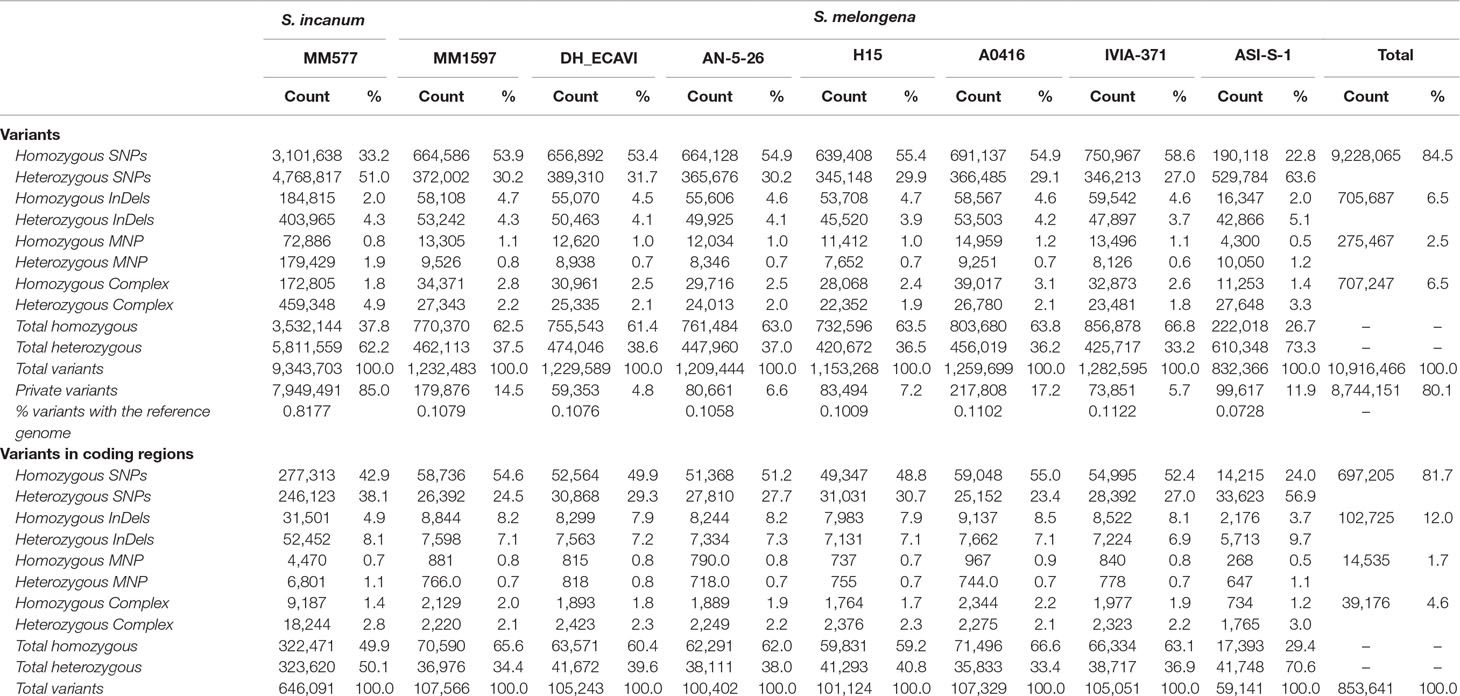
Table 3 Summary of the polymorphisms identified in the seven S. melongena and one S. incanum accessions of this study using the S. melongena accession 67/3 as a reference genome (Barchi et al., 2019a).
In all accessions, the total number of homozygous variants was higher (above 60%) than the heterozygous ones, except in the common eggplant ASI-S-1 and in the wild S. incanum MM577, where the total heterozygous variants represented 73.3% and 62.2% of the total variants, respectively, which may be due to the different degree of fixation among the eight accessions. The number of private variants (i.e., accession specific) was variable among the common eggplant accessions and ranged from 4.8% in DH_ECAVI with 59,353 polymorphisms to 17.2% in A0416 with 217,808 variants, while in the S. incanum MM577 accession, the percentage rose to 85.1% with over 8.7 million private variants (Table 3). Considering the variants between the accessions and the reference genome and its total length in base pairs, the percentages of variants with respect to the reference genome sequence ranged between 0.072% of ASI-S-1 and 0.112% of IVIA-371 in common eggplant and reached 0.817% with S. incanum accession MM577.
The variants identified in coding regions were much less abundant (853,641 and 7.8% of the total). Of these, 81.7% were SNPs, 12.0% InDels, 1.7% MNPs, and 4.6% complex variants. When considering only the seven S. melongena accessions, the total variants ranged from 59,141 in ASI-S-1 to 107,566 in MM1597, while in the S. incanum accession, there were 646,091 polymorphisms in coding regions. However, the gap in variants between the common eggplant accessions and the wild S. incanum decreased from almost 8-fold in the whole genome to around 6.5-fold in coding regions. Interestingly, while the rest of accessions maintained approximately their ratios of homozygous/heterozygous variants in the coding regions (from 71.4% in A0416 to 59.2% homozygous variants in H15), the S. incanum MM577 accession showed an almost equal proportion of them (322,471 homozygous vs. 323,620 heterozygous variants).
Substantial differences were observed in the average number of polymorphisms among the chromosomes, with differences over 4-fold between chromosome 5 (43,098) and chromosome 12 (181,603) among the seven common eggplant accessions and over 4.5-fold between chromosome 9 (257,781) and chromosome 7 (1,195,706) in the S. incanum accession (Supplementary Data S2). In addition, considerable variation within individual chromosome for the total number of polymorphisms was found among S. melongena accessions. In fact, in chromosome 12, the differences among the S. melongena lines was of up to fourfold; in chromosomes 1 and 2, it was around threefold; and in chromosomes 6, 7, and 10, over twofold. However, the most remarkable difference in concentration of polymorphisms was found in chromosome 1 for A0416 with 314,756 variants, while the mean for the rest of the lines was 107,979. The variants rate (physical length divided by the number of variants) was quite uniform for all the common eggplant accessions (on average 1 variant every 976 bp), ranging from one variant every 891 bp in IVIA-371 to one variant every 1,373 bp in ASI-S-1. For the S. incanum accession, the variation rate was considerably higher than for the S. melongena accessions (one variant every 122 bp), although for this calculation, we used the length of the S. melongena reference genome since the exact physical length of the S. incanum genome is still unknown. Regarding the average variation rate per chromosomes in the S. melongena accessions, the lowest value was found in chromosome 4 (one variant every 1,511 bp) and the highest in chromosome 12 (one variant every 553 bp). The range of variation for the S. incanum accession MM577 was narrower; chromosome 9 had the lowest rate with one variant every 140 bp, while chromosomes 2 and 8 had the highest rate with one variant every 117 bp. The number of variants was highly related to the physical length of the chromosomes since, for common eggplant accessions, the correlation was r = 0.7012 (p = 0.01105) and for S. incanum r = 0.9946 (p = 3.433 × 10-11).
The variants were separated in heterozygous and homozygous, and their distribution along the chromosomes was plotted (Figure 2; Supplementary Data S3). The peaks represent high concentrations of variants in a window size of 10 Mbp. Overall, the heterozygous variants were distributed more evenly than the homozygous ones, which were more abundant in distal parts compared to centromeric regions. Some accessions showed large regions of almost identical peaks pattern distribution for homozygous variants like DH_ECAVI and IVIA-371 in chromosomes 2, 6, and 11; H15 and IVIA-371 in chromosome 2; AN-S-26 and H15 and A0416 and ASI-S-1 in chromosome 6; AN-S-26, H15, and IVIA-371 in chromosome 9; and DH_ECAVI, AN-S-26, H15, and IVIA-371 in chromosome 12. For heterozygous variants, similar peak patterns were observed for DH_ECAVI and IVIA-371 in chromosomes 2 and 11; AN-S-26 and H15 in chromosome 6; AN-S-26, H15, and IVIA-371 in chromosome 9; and DH_ECAVI, AN-S-26, H15, and IVIA-371 in chromosome 12.
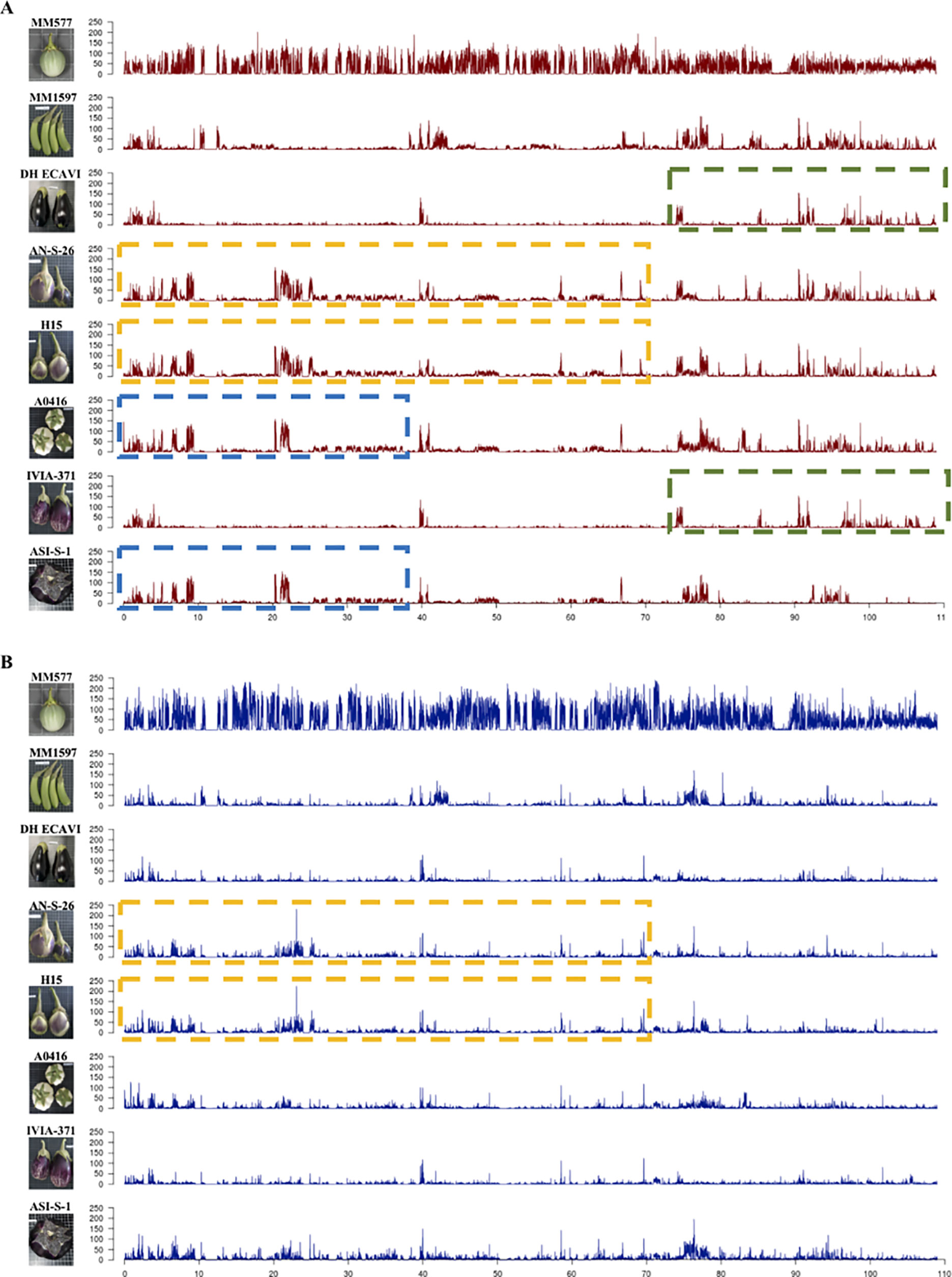
Figure 2 Distribution of homozygous (A) and heterozygous (B) polymorphisms along chromosome 6 for the seven S. melongena and one S. incanum (MM577) accessions resequenced identified using the S. melongena accession 67/3 as a reference genome (Barchi et al., 2019a). The peaks represent high frequencies of polymorphisms in a window size of 10 Mbp. The dashed lines of the same color indicate similar patterns of polymorphisms distribution.
According to the variant effect prediction software SnpEff, 98.72% of the variants were classified as “modifier” (i.e., the variants were located in intergenic or intronic regions, or affected noncoding genes), indicating that there is no evidence of impact or that their predictions are difficult to assess (Supplementary Data S4). The second most abundant variants impact class was “moderate” (0.77%) and ranged from 0.61% in MM577 to 0.78% in H15. These are nondisruptive variants, such as codon insertion/deletion or codon substitution, that might change protein effectiveness. The variant with “low” impact effects are those unlikely to change protein behavior, like start or stop codon mutated into another start or stop codon or into the same amino acid, represented an average of 0.46%, ranging from 0.43% in ASI-S-1 to 0.48% in H15. Finally, the less abundant impact class corresponded to the “high” variation effects (0.07%), which are considered to have a disruptive impact on the protein (like truncation or loss of function caused by exon deletion/deletion, start/stop codon mutation, or splice site modification), ranged from 0.05% in S. incanum MM577 to 0.09% from the total number of variants of each accession.
Among the variants that had been predicted to cause a high impact effect, the most abundant was the “frameshift variant,” caused by insertions or deletions that disrupt the translational reading frame, with a total mean of 0.037%. The other most abundant variants with high impact were “stop gained,” resulting in a premature stop codon and leading to a shortened polypeptide, with a mean of 0.024%; “stop lost,” corresponding to an elongated transcript due to the loss of stop codon, with a mean of 0.006%; “splice donor variant” and “splice acceptor variant,” causing the change of the two base pair regions at the 5′ end and at the 3′ of an intron, respectively, with total means of 0.006% and 0.005%; and finally, “start lost,” when a variant causes the mutation of a start codon into a nonstart codon, with a mean of 0.005%. Regarding the moderate impact, the “missense variant,” when a change in one or more bases causes a different amino acid sequence, was by far the most abundant effect with 0.717%, followed by “inframe insertion” (0.009%) and “inframe deletion” (0.007%) when one or many codons are inserted or deleted, respectively, and finally, “disruptive inframe insertion” (0.005%) and “disruptive inframe deletion” (0.005%) effects, which occur when one codon is changed and one or more codons are inserted or deleted. For the low impact effects, most polymorphisms were predicted as “synonymous variant” (0.377%), i.e., when a variant causes a codon that produces the same amino acid, followed by “splice region variant” (0.100%) when a variant occurs within the region of the splice site, and “5′ untranslated region (UTR) premature start codon gain variant” (0.012%) when a variant in 5′ UTR region produces a start codon. The modifier impact effects and the classification of variants effects by region indicated that over 93% of the variants were located outside the genes, in intergenic regions (68.4%), upstream (13.2%), and downstream (12.1%) of the genes, followed by intron (4.5%) and exon (1.1%), 3′ UTR (0.24%) and 5′ UTR (0.15%), splice site region (0.081%), transcript (0.03%), splice site donor (0.005%), and acceptor (0.005%). The details of amino acid changes are reported in Supplementary Data S5.
S. incanum had four times more genes affected by “high” impact effect variations (4,283) than other accessions (from 786 for ASI-S-1 to 1,130 for H15). An analysis of the biological processes (BP) GO enrichment associated to these genes showed a general enrichment in genes associated to the photosynthetic process. For example, “photosynthetic electron transport” (GO:0009772, GO:0009767), “photosynthesis” (GO:0015979), “carbon fixation” (GO:0015977), and/or “ATP synthesis coupled proton transport” (GO:0015986) were enriched for all the accessions (Supplementary Data S6). These genes were usually annotated as the different elements of the photosynthetic systems (e.g., SMEL_003g176830.1, “photosystem II D2 protein;” SMEL_002g157390.1, “ATP synthase subunit alpha, chloroplastic;” SMEL_002g159210.1, “Ycf2;” SMEL_005g233370.1, “ribulose bisphosphate carboxylase large chain”) (Supplementary Data 7). Nevertheless, there were some differences between accessions. S. incanum MM577 showed an enrichment in “telomere maintenance” (GO:0000723) and “DNA repair” (GO:0006281) (mostly helicases-like proteins) and RNA processing as “mRNA cleavage” (GO:0006379), “rRNA modification” (GO:0000154), “ncRNA processing” (GO:0034470), “RNA methylation” (GO:0001510), and “prolyl-tRNA aminoacylation” (GO:0006433) associated to genes such as RNA polymerases (e.g., SMEL_005g226240.1), ribonucleases (e.g., SMEL_009g322130.1), and tRNA-methyltransferases (e.g., SMEL_006g270090.1). In addition, S. incanum presented enrichment in genes associated to metabolic pathways such as “GDP-mannose biosynthetic process” (GO:0009298) (e.g., mannose-6-phosphate isomerase SMEL_006g255960.1) and “glutamine family amino acid biosynthetic process” (GO:0009084) (e.g., glutamine synthetase, SMEL_003g183980.1). Accession S. melongena MM1597 presented a differential enrichment in “nitrate transport” (GO:0015706) and “response to nitrate” (GO:0010167) associated to the gene SMEL_003g189360.1 (high-affinity nitrate transporter). DH_ECAVI presented a differential enrichment in “TOR signaling” (GO:0031929) linked to the genes SMEL_003g174880.1 and SMEL_010g353100.1 (regulatory-associated protein of TOR 1). Accession H15 had associated the enriched term “leaf formation” (GO:0010338) (gene SMEL_009g323460.1, an MYB transcription factor) and S. melongena A0416 genes associated to the term “recognition of pollen” (GO:0048544) (receptor-like protein kinases genes SMEL_001g143850.1, SMEL_004g218090.1, SMEL_006g251240.1, SMEL_007g292990.1, SMEL_007g293020.1, SMEL_007g293120.1, and SMEL_009g324070.1). Finally, S. melongena ASI-S-1 showed enrichment in the terms “cell volume homeostasis” (GO:0006884) and “chloride transport” (GO:0006821) associated to the genes SMEL_003g181350.1 (methylosome subunit pICln) and SMEL_009g330700.1 (unknown function).
Genetic identities among the accessions were calculated on 9,109,331 SNPs after removing all the missing data (Supplementary Data S8). High values of genetic identities (over 0.9) were obtained among common eggplant accessions, while lower values (below 0.5) were obtained when S. melongena accessions were compared with the S. incanum MM577. The higher genetic identity values were found between H15 and AN-S-26 (0.965) and between IVIA-371 and DH_ECAVI (0.963). Anyway, the pairwise comparisons of these four varieties resulted in the highest genetic identity values observed (0.951 between H15 and DH_ECAVI, 0.948 between AN-S-26 and DH_ECAVI, 0.947 between H15 and IVIA-371, and 0.946 between IVIA-371 and AN-S-26). Slightly lower genetic identity values were observed between ASI-S-1 and the cluster formed by H15 and AN-S-26 (0.938 and 0.936, respectively), A0416 (0.936), MM1597 (0.929), and the other cluster formed by DH_ECAVI (0.927) and IVIA-371 (0.921). Lower values, even though still quite high (the lowest value being 0.905), were obtained when comparing MM1597 and A0416 with the rest of accessions. As expected, the S. incanum accession displayed by far the lowest genetic identity values with a mean of 0.413. These values were also reflected in the multivariate PCoA analysis using the genetic matrix calculated by the TASSEL software (Figure 3). The first principal coordinate, which accounted for 98.6% of the variation, clearly separated the common eggplant accessions from the S. incanum accession (Figure 3A), while the second coordinate, which accounted for 0.6% of the variation, separated the Occidental (DH_ECAVI, AN-S-26, H15, and IVIA-371) from the Oriental accessions (MM1597, ASI-S-1) plus A0416 (of unknown origin). In order to gain a better landscape of the genetic relationships among the S. melongena accessions, a further analysis on partitioned data was performed by removing S. incanum (Figure 3B). The first axis, accounting for 50.0% of the intraspecific genetic variation, separated the S. melongena accessions of the Occidental group from the cluster formed by the Oriental group accessions plus A0416. On the other hand, the second axis, accounting for 20.7% of the genetic variation, separated the Indian accession MM1597 from the Chinese accession ASI-S-1 and the unknown origin accession A0416, as well as DH_ECAVI and IVIA-371 from H15 and AN-S-26 accessions.
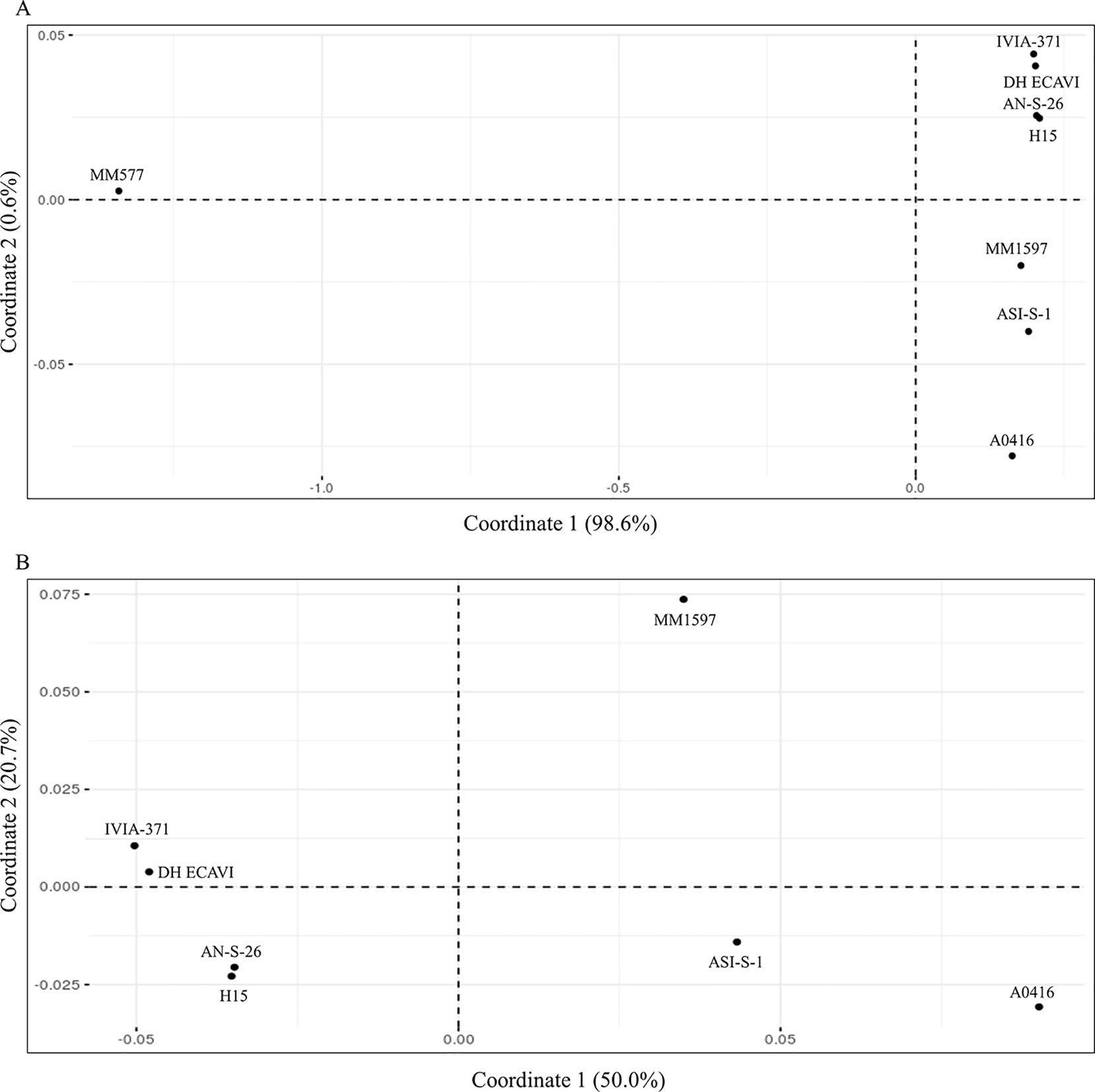
Figure 3 Principal coordinates analysis (PCoA) similarities based on the whole set of SNPs (9,228,065) for the seven S. melongena and one S. incanum (MM577) accessions (A) and on partitioned data without S. incanum (MM577) (B). The first and second principal coordinates are displayed.
In order to understand the dynamics of transposable elements (TEs) during domestication, the class and number of TEs were compared between the seven cultivated S. melongena accessions and the wild S. incanum (Table 4). The average TE copy number in the seven cultivated accessions (1,090,121) was similar than in S. incanum (1,093,637). The latter had higher number of TEs than DH_ECAVI, IVIA-371, A0416, MM1597, and AN-S-26 but lower than ASI-S-1 and H15. For most of the TE families, ASI-S-1 and H15 had higher number of TE copies than S. incanum (Supplementary Data S9). In contrast, for the TE class of long terminal repeat (LTR)/Caulimovirus and DNA_nMITE/Harbinger, the S. incanum accession MM577 displayed the highest copy number.
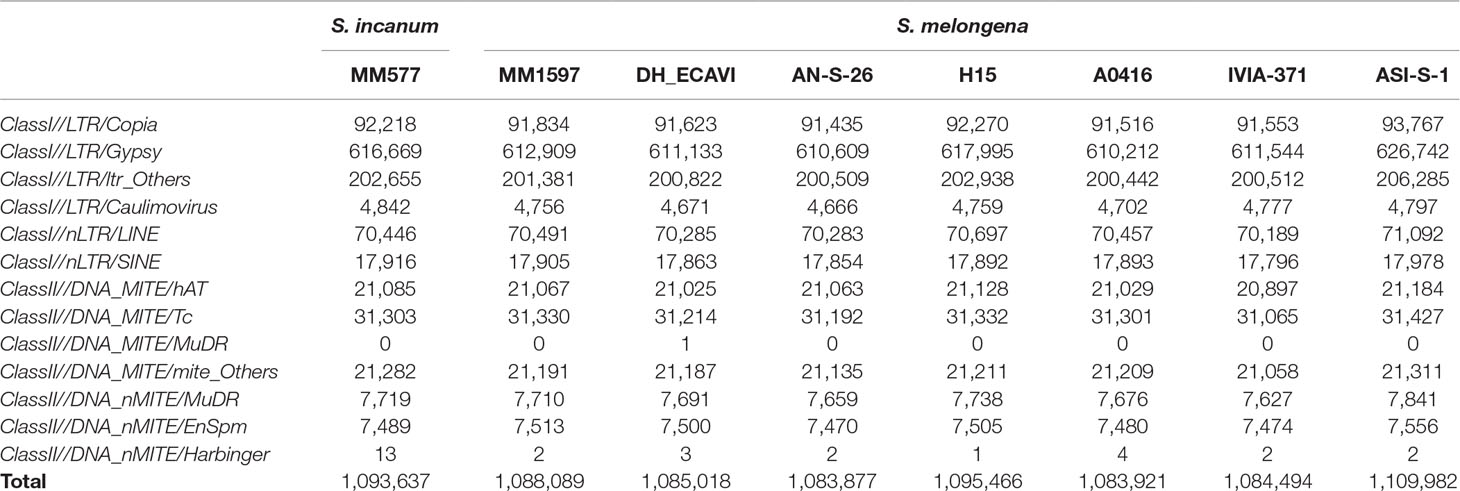
Table 4 Statistics of the transposable elements identified in seven Solanum melongena and one S. incanum accessions of this study.
The TE size proportion of the genome was compared among the eggplant accessions and other main crops. Eggplant had similar TE proportion (average of 60.4% for the eight accessions) as the two major Solanaceae crops, tomato (63.2%) (Tomato Genome Consortium, 2012) and potato (54.4%) (Potato Genome Sequencing Consortium, 2011), but lower than pepper (76.4%) (Kim et al., 2014) (Supplementary Data S10). By comparing with Poaceae crops, TE proportion in eggplant was lower than maize (84.2%) (Schnable et al., 2009), similar to Sorghum bicolor (62%) (Paterson et al., 2009), and higher than rice (39.5%) (Yu et al., 2002). Interestingly, nearly all TEs in eggplant belongs to LTR (97.3%), representing a superfamily in class I TE, which is consistent with the high LTR proportion in tomato, potato, and pepper, but much higher than the three Poaceae crops (rice, sorghum, and maize).
In this study, we performed the first whole-genome resequencing analysis of eggplant, in which seven phenotypically diverse eggplant materials from different geographic regions and a wild relative were included. This pioneering resequencing study has significant relevance for eggplant breeding. In this way, these eight accessions are the founders of an eight-way MAGIC population that is currently under development and which may make a significant impact in eggplant breeding (Cavanagh et al., 2008; Gramazio et al., 2018). Pascual et al. (2015) emphasized the importance of the information generated by the resequencing of the founder parents of a tomato MAGIC population in the mapping precision of quantitative trait loci, even if many tools and molecular markers were already available in this crop. In addition, in the eggplant genepool, only a few studies have generated a significant set of molecular markers (Barchi et al., 2011; Gramazio et al., 2016; Acquadro et al., 2017), although they are not sufficient for a deep molecular characterization of a multiparent population and for building the corresponding saturated genetic linkage map from the founders (Pascual et al., 2015). In addition, no arrays, chips, or other genotyping platforms, as those available in other Solanaceae crops like tomato (Sim et al., 2012a; Víquez-Zamora et al., 2013), potato (Felcher et al., 2012; Vos et al., 2015), or pepper (Hill et al., 2013; Hulse-Kemp et al., 2016), have been developed so far in eggplant for high-throughput genotyping. Thus, one of the main aims of this study was to develop a large genome variation data set of polymorphic markers among the accessions that can serve as an a source of markers, from a diverse germplasm, for the potential development of genotyping platforms, breeding purposes, and further eggplant genetic and genomic studies including the genotyping of our MAGIC population.
For this study, we have mapped the over 1.2 billion paired-end high-quality reads of the eight accessions against the new high-quality eggplant reference genome “67/3” (Barchi et al., 2019a). The high percentage of mapping rates of the eight lines (over 85%) confirms the high-quality of the “67/3” reference genome, although in other model species, the rates of unmapped reads were lower, such as the 3–5% in tomato (Causse et al., 2013; Aflitos et al., 2014) or 10–15% in rice (Subbaiyan et al., 2012; Xu et al., 2012). This might be due to different factors like the lower degree of the sequence assembly progress, the different level of repetitive elements, the genetic divergence between the sequenced accessions and the reference one, or the higher level of polymorphism, among others (Xu et al., 2012; Causse et al., 2013). However, the overall mapping coverage was around 20×, higher than the most common mapping coverage (around 10×) in resequencing studies of just a few years ago (Guo et al., 2013; Zhou et al., 2015), and encompassed practically the whole length of the reference genome for all the accessions except for S. incanum, in which the genome coverage onto the reference genome was on average 95%, evenly distributed for all the chromosomes. The S. incanum difference compared with the common eggplant accessions might be due to the phylogenetic distance between the two taxa, which is estimated in 1.56 MYA (Särkinen et al., 2013; Vorontsova et al., 2013; Acquadro et al., 2017) or to genome structural elements that are absent in S. melongena like repetitive sequences specific of S. incanum that difficult a sensitive mapping (Xu et al., 2012; Aflitos et al., 2014). This 5% of uncovered S. incanum genome may host important genic regions that have been lost during the domestication process, such as tolerance or resistance to stresses, as proposed by Aflitos et al. (2014) for tomato wild relatives. These authors also emphasized the need to focus efforts also in sequencing and assembly multiple reference genomes from crop wild relatives (CWRs) to avoid biased data interpretation and identify genome regions affected by genetic erosion. However, since the dynamics of gene gains and losses during domestication is not yet fully understood, other reasons may explain the lower genome coverage of S. incanum. In fact, among others, the 5% of uncovered regions could be also resulting from: i) regions gained by S. melongena during domestication by self-duplication, diversification or introgressions from other species, ii) regions present in the common ancestor of S. incanum and S. melongena that have been lost in S. incanum, and/or iii) hypervariable regions in S. incanum in which mapping using Burrows–Wheeler indexing may not be sensitive enough.
Our study has identified the largest set of polymorphisms to date in eggplant, consisting of almost 11 million of high-quality variants. Over 85% of these polymorphisms were SNPs, which are cheaper and easy to automate for high-throughput genotyping respect to other markers (Kim et al., 2016). In this respect, Yan et al. (2010) estimated that SNPs are on average 75% cheaper and 100-fold faster than simple sequence repeat gel-based methods to build a genetic map using a high-throughput genotyping platform. The whole-genome resequencing approach has been revealed as an excellent strategy to develop a large number of markers in many crops and mainly in their CWRs, where a general lack of information slows down their use in breeding programs and genetic studies (Aflitos et al., 2014; Brozynska et al., 2016). For example, 6 and 9 million SNPs were identified from wild and cultivated soybean accessions (Lam et al., 2010; Zhou et al., 2015), 6.5 million from wild rice accessions (Xu et al., 2012), and over 10 million from wild tomatoes (Aflitos et al., 2014; Gao et al., 2019). In these studies, the CWRs yielded up to 20-fold more variations compared to the heirloom and cultivated accessions, this without taking into account the genetic regions of CWRs that do not map onto the cultivated reference genome. Although in our study we just resequenced one wild relative (S. incanum), the results of the aforementioned studies are similar to ours, as S. incanum displayed a ninefold higher number of variations compared to cultivated common eggplants. The resequencing of more eggplant relatives will probably result in a greater difference in number of variants between the cultivated species and CWRs, particularly taking into account that many vegetables, including eggplant, have gone through severe genetic bottlenecks during domestication (Meyer et al., 2012; Meyer et al., 2015; Qi et al., 2013). Our study confirms that, as in other crops, the use of CWRs for introgression breeding can efficiently contribute to broadening the genetic basis of crops (Prohens et al., 2017). In this way, for several economically important crops like rice, tomato, or wheat, introgression breeding started several decades ago, and currently, many new varieties carry introgressed regions from CWRs (Dempewolf et al., 2017). For eggplant, the first efforts to harness the valuable large pool of genetic diversity of its wild relatives started just a few years ago thanks to the development of ad hoc breeding tools and strategies (Toppino et al., 2008; Liu et al., 2015; Kouassi et al., 2016; Plazas et al., 2016; Acquadro et al., 2017). The information produced in this study and the SNPs identified from S. incanum will accelerate the introgression of genomic regions of this drought-tolerant species (Knapp et al., 2013; Gramazio et al., 2017a) to develop new resilient eggplant varieties.
In addition, in our study, we have identified a large number of high-confidence sets of polymorphisms in common eggplant accessions, providing valuable information for performing high-throughput genotyping in cultivated or wild germplasm through the development of arrays, chips, or other high-throughput genotyping platforms. In this way, a first genotyping platform in eggplant, based on the single primer enrichment technology (SPET), was recently developed with the information produced in this study (Barchi et al., 2019b). The eggplant SPET platform, which consists of 5,093 probes designed from the SNPs identified throughout the genome of the eight eggplant accessions resequenced in our study, has been already used to genotype the diversity of a germplasm collection of 422 cultivated eggplant and CWR accessions (Barchi et al., 2019b). The results from the screening of a large eggplant germplasm dataset including eggplant CWRs using the polymorphisms identified in this study demonstrated their transferability in the assessment of closely related species from different genepools with high interest in breeding, as well as their potential a wide variety of studies like phylogenetic, domestication, or genome-wide association studies (Barchi et al., 2019b).
In this first eggplant resequencing study, we tried to maximize the phenotypic diversity for important breeding traits coupled with genetic diversity and the geographic origins. The genetic distance matrix and the PCoA obtained with the entire SNP dataset confirmed our selection criteria. In fact, the higher genetic identity values were between H15 and AN-S-26, which share some common traits like long calyx and being used for making pickles (Muñoz-Falcón et al., 2009a), and between IVIA-371 and DH_ECAVI, both having an élite background and with large fruit (Hurtado et al., 2014; Gramazio et al., 2017b). These four accessions clustered together since they were chosen to represent the Occidental diversity of eggplant (Vilanova et al., 2012). However, despite the phenotypic diversity, relatively low genetic diversity has been found among them, suggesting that the European accessions might have been diversified from a reduced genepool (Muñoz-Falcón et al., 2008; Muñoz-Falcón et al., 2009a; Muñoz-Falcón et al., 2009b; Meyer et al., 2012; Cericola et al., 2013). ASI-S-1 accession represented the Chinese domestication center and displays a great phenotypic similarity with the eggplant reference genome “67/3,” showing rounded and purple fruits and the recessive allele for the dominant pigment under calyx trait (Toppino et al., 2016). This similarity is confirmed by the lowest number of polymorphisms, compared to the rest of the accessions. Regarding accession A0416, the selection was made exclusively based on its peculiar phenotype, flattened ribbed white fruits, since its origin was unknown. Our results suggest that this accession might belong to the Oriental eggplant genepool (Vilanova et al., 2012) and, based on PCoA results, probably closer to the Chinese domestication center than to the Indian center (Meyer et al., 2012). Finally, the accession MM1597, representing the Indian domestication center, is closer to the European group than the Chinese accession. In this way, it has been suggested that Arab traders brought eggplants from southeast Asia into Europe in the 14th century (Lewicki et al., 1974; Daunay and Janick, 2007; Meyer et al., 2012).
An alternative way to visualize the relationships among the accessions could be done by analyzing the distribution of variants (Aflitos et al., 2014), which was uneven along the whole genome. More than 90% of the polymorphisms were found in intergenic and noncoding intragenic regions, being the variants in coding regions <9% on average. Similar ratios have been also observed in other crops (Xu et al., 2012; Causse et al., 2013; Aflitos et al., 2014; Zhou et al, 2015). Large differences were found among the 12 chromosomes in the number of polymorphisms, although the number of polymorphisms was highly related to the physical length of them. Differences were also observed among the accessions for a given chromosome. Interestingly, some accessions shared genomic regions with a similar large density of SNPs, which visually are like large blocks of peaks when the SNPs are divided and plotted into 10-Mbp sized bins. This similar SNPs distributions among some accessions have been interpreted as candidate introgressions and could help in reconstructing the breeding history of a crop (Sim et al., 2012b; Causse et al., 2013; Aflitos et al., 2014). In this study, footprints of old intraspecific and, more likely, interspecific hybridizations were identified in five chromosomes (2, 6, 9, 11, and 12) and more clearly in homozygous than in the heterozygous variants. The patterns of shared blocks of peaks in the Occidental accessions suggests that they share common ancestors, with many common candidate introgressions not only between IVIA-371 and DH_ECAVI, AN-S-26 and H15, but also among H15, IVIA-371, and AN-S-26. Furthermore, this alternative approach provides additional evidence to support that A0416 could have originated in the Chinese domestication center because of the general similarity in the pattern of SNPs databases and the similar distribution of SNPs in the first 70 Mbp of chromosome 6.
The analysis of the Gene Ontologies associated with genes with “high” impact variants, which were found in either homozygous and heterozygous state, shows a common trend in all the accessions related with variation on the photosynthetic pathway. Most of these genes, such as “ribulose bisphosphate carboxylase large chain” (e.g., SMEL_002g155400.1, SMEL_005g233370.1, and SMEL_012g386960.1) should be located in the chloroplast genome, but in these cases, there are located in the nuclear genome, in specific chromosomal locations. Chloroplast genes are commonly transferred to the nuclear genome by different mechanisms producing nuclear integrants of plastid DNAs (Michalovova et al., 2013). These chloroplast genomic fragments are under strong purifying selection, so it is not surprising that these genes present “high” impact variants that drive to a fast removal of the possible activity these genes. More intriguing is the high percentage of genes with “high” impact variants associated with the DNA maintenance and restoring after damage (mostly helicase-like proteins) machinery that can be found in S. incanum like “telomere maintenance” (GO:0000723, 25 out of 58 genes) and “DNA repair” (GO:0006281, 41 out of 195 genes). In plants, helicase-like proteins play a critical role in stress tolerance responses (Nidumukkala et al., 2019). Natural mutations and overexpression of DEAD box helicases confirmed the role of this subgroup of helicases in providing stress tolerance, like cold or salinity (Gong et al., 2002; Vashisht et al., 2005; Shivakumara et al., 2017; Mahesh and Udayakumar, 2018). As well, many RNA modification genes (mostly RNA methyltransferases and ribonucleases), related to transcriptional and posttranscriptional activities, presented “high” impact effect variants in S. incanum like “transcription by RNA polymerase III” (GO:0006383, five out of eight genes), “mRNA cleavage” (GO:0006379, two out of two genes), “rRNA modification” (GO:0000154, three out of five genes), “ncRNA processing” (GO:0034470, 20 out of 80 genes), “RNA methylation” (GO:0001510, 5 out of 15 genes), and “RNA 3’-end processing” (GO:0031123, 6 out of 18 genes). RNA methyltransferases and ribonucleases, along with many other epigenetic activities, and protein kinase (GO:0043549, 4 out of 17 genes) are reported to be involved in plant abiotic stress response (Deng et al., 2013; Wang et al., 2017, Hu et al., 2019). In addition, three out of four genes that encode for guanosine diphosphate mannose, which is a precursor for ascorbate, presented “high” impact variants. Ascorbic acid has important antioxidant and metabolic functions among which is environmental stress adaptation (Wheeler et al., 1998; Bao et al., 2016). The S. incanum accession MM577 was collected in a desertic area in Israel, where day and night temperatures differ considerably and plants experimented heat and cold stress, along with extreme drought conditions. It has already been reported that S. incanum is highly tolerant to drought and to some fungal diseases (Knapp et al., 2013), but no studies have been performed so far on its tolerance to other abiotic stresses. Based on these results, investigating the response of this wild species to other stresses, like heat, cold, and salinity, in order to introduce it in breeding pipelines, the advanced backcross materials with elite genetic background already developed using S. incanum as wild donor (Gramazio et al., 2017a).
Regarding the other S. melongena accessions, “high” impact effect variants were found in MM1597 for “response to nitrate” (GO:0010167 and GO:0015706, two out of two genes) affecting the gene SMEL_003g189360.1 “high-affinity nitrate transporter 3.1.” Nitrogen is one of the major limiting factors for plant growth and crop yield, and in the past decades, nitrogen fertilization has increase more than 20-fold with high economic and ecological costs (Marschner, 2011). At present, the improvement of nitrogen use efficiency is an agriculture challenge to maintain high crop yield under low nitrogen for more sustainable agriculture. Measuring the nitrogen uptake of this accession in order to associate these variants with a different absorption capacity may provide information relevant for improving nitrogen use efficiency. In DH_ECAVI, two out of four genes presented “high” impact variants in “TOR signaling” (GO:0031929), an evolutionarily conserved protein kinase that is involved in the growth processes of cotyledons, true leaves, petioles, and primary and secondary roots (Shi et al., 2018). In H15, a “high” impact effect variant in the Myb transcription factor “leaf formation” (GO:0010338) could be associated with the smaller leaves and fruit pedicel compared to its related Andalusian eggplants (Muñoz-Falcón et al., 2009a). Accession A0416 had 7 out of 54 genes related to “recognition of pollen” (GO:0048544) that could partially explain the difficulty of obtaining hybrids when it is used as a female parent. However, all the “high” impact effect variants of Supplementary Data 6 and 7 may be further investigated using our MAGIC population, which currently is under development, where the segregating recombinant lines will provide a more powerful resolution to dissect the genetics of many traits and associated variants with specific phenotypes.
TEs make up the vast majority of all investigated plant DNA, and TEs’ dynamic changes (activation or purification) in a genome frequently influence their insertions, duplications, and the genome size (Levin and Moran, 2011). This dynamic TE is commonly influenced during domestication. When comparing the copy number of S. melongena accessions with the wild accession of S. incanum, two of them had higher TE copy and the other five had lower TE copy number than the S. incanum accession, suggesting possible dynamic TE events occurring in the course of domestication. Two families including LTR/Caulimovirus and DNA_nMITE/Harbinger in the wild accession were identified to be purified potentially underlying selection influences. In rice, a miniature inverted-repeat TE (MITE/mPing) amplified from ∼50 to ∼1,000 copies in four rice strains, and 70% of the 280 inserted TEs were within 5 kb of the coding regions (Naito et al., 2006). Two MITE families in Medicago truncatula produced highly polymorphic insertion sites in 26 ecotypes. In this latter study, a subset of insertions was present only in a cultivar, compared to the other 25 wide ecotypes, indicating activations of these two families during M. truncatula domestication (Grzebelus et al., 2009). Some TE insertions underlying the dynamics altered plant phenotype simply by inducing the preexisting expression of genes selected during domestication. Overexpression of teosinte branched1 (tb1) in maize can induce a dramatic reduction in branch number relative to the progenitor species, resulting from a retrotransposon inserted in the promoter region of tb1 (Studer et al., 2011). These observations in eggplant suggest possible important roles of TEs during its domestication. However, in order to gain greater accuracy and sensitivity, we suggest resequencing further accessions of eggplant CWRs so as to identify TE copy number variations with statistical significance between the wild and cultivar accessions.
The progressive availability of high-quality reference genomes and the continuous improvements of high-throughput sequencing technologies are fostering resequencing studies even for no-model plant species, including eggplant. In this study, we performed the first resequencing of a set of eggplant accessions, identifying the largest set of polymorphisms so far in eggplant genepool using accessions phenotypically and genetically very diverse. The usefulness and transferability of these polymorphisms identified has already been demonstrated by developing the first high-throughput genotyping SPET platform in eggplant, which has been used to assess the genetic relationships among domesticated accessions and wild related from several species in a large eggplant germplasm dataset. All the variants were structurally and functionally annotated and their effects predicted, which will be very useful for a wide type of studies and approaches like genomic editing or the characterization of natural, segregating (like the MAGIC population that we are currently developing with the accessions resequenced in this study), and mutants populations. In addition, the distribution of the variants has revealed footprints of putative ancient introgressions that can help to shed light on the yet unclear eggplant domestication history. The analysis of the transposon composition across the different accessions reveals different trends among eggplant accessions with some of them having more and others less TE than the wild relative S. incanum. Further experiments increasing the number of accessions should be done in order to elucidate the impact of the TE in the eggplant domestication process.
This first resequencing study in eggplant has gathered relevant genomic data and information that will be extremely useful for the development of a new generation of improved eggplant varieties adapted to present and future challenges in eggplant production and with better fruit quality. The information obtained will also be highly relevant for answering a plethora of scientific and technical questions and needs, including allele and variants discovery, germplasm genomic characterization, marker-assisted mapping for dissecting agronomic-associated loci, or providing insight into underlying molecular mechanisms of eggplant genome evolution, among others. In this way, this first study paves the way and encourages other researchers and breeders to perform the resequencing of more eggplant accessions, especially CWRs, where many valuable alleles for eggplant breeding have still to be discovered and exploited.
The datasets generated for this study can be found in the NCBI Short Read Archive under submission identifier SUB2829594 with the Bioproject identifier PRJNA392603. Raw reads of each accession are deposited under the accession numbers from SRR5796636 to SRR5796643. VCF files with the corresponding variants identified are available upon request to the corresponding author
PG, SV, JP, and AB conceived and designed the research. PG, HY, and TH analyzed the data. PG wrote the manuscript. PG, HY, TH, SV, JP, and AB reviewed and edited the manuscript. All authors read and approved the manuscript.
This work has been funded by the European Union’s Horizon 2020 Research and Innovation Programme under grant agreement no. 677379 (G2P-SOL project: Linking genetic resources, genomes, and phenotypes of Solanaceous crops), by the Spanish Ministerio de Economía, Industria y Competitividad and Fondo Europeo de Desarrollo Regional/European Regional Development Fund (grant AGL2015-64755-R), and by the Spanish Ministerio de Ciencia, Innovación y Universidades (MCIU), Agencia Estatal de Investigación (AEI), and Fondo Europeo de Desarrollo Regional/European Regional Development Fund (grant RTI2018-09592-B-100). PG is grateful to Universitat Politècnica de València and to Japan Society for the Promotion of Science for their respective postdoctoral grants [PAID-10-18 and FY2019 JSPS Postdoctoral Fellowship for Research in Japan (Standard)].
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Authors also thank the Italian Eggplant Genome Sequencing Consortium for providing access to an improved version of the eggplant genome.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.01220/full#supplementary-material
Acquadro, A., Barchi, L., Gramazio, P., Portis, E., Vilanova, S., Comino, C., et al. (2017). Coding SNPs analysis highlights genetic relationships and evolution pattern in eggplant complexes. PLoS One 12, e0180774. doi: 10.1371/journal.pone.0180774
Aflitos, S., Schijlen, E., de Jong, H., de Ridder, D., Smit, S., Finkers, R., et al. (2014). Exploring genetic variation in the tomato (Solanum section Lycopersicon) clade by whole-genome sequencing. Plant J. 80, 136–148. doi: 10.1111/tpj.12616
Alonso-Blanco, C., Andrade, J., Becker, C., Bemm, F., Bergelson, J., Borgwardt, K. M., et al. (2016). 1,135 genomes reveal the global pattern of polymorphism in Arabidopsis thaliana. Cell 166, 481–491. doi: 10.1016/j.cell.2016.05.063
Aronesty, E. (2013). Comparison of sequencing utility programs. Open Bioinf. J. 7, 1–8. doi: 10.2174/1875036201307010001
Barchi, L., Lanteri, S., Portis, E., Acquadro, A., Valè, G., Toppino, L., et al. (2011). Identification of SNP and SSR markers in eggplant using RAD tag sequencing. BMC Genomics 12, 304. doi: 10.1186/1471-2164-12-304
Barchi, L., Pietrella, M., Venturini, L., Minio, A., Toppino, L., Acquadro, A., et al. (2019a). A chromosome-anchored eggplant genome sequence reveals key events in Solanaceae evolution. Sci. Rep. 9, 11769. doi: 10.1038/s41598-019-47985-w
Barchi, L., Acquadro, A., Alonso, D., Aprea, G., Bassolino, L., Demurtas, O., et al. (2019b). Single primer enrichment technology (SPET) for high-throughput genotyping in tomato and eggplant germplasm. Front. Plant Sci. 10, 1005. doi: 10.3389/fpls.2019.01005
Bao, G., Zhuo, C., Qian, C., Xiao, T., Guo, Z., Lu, S. (2016). Co-expression of NCED and ALO improves vitamin C level and tolerance to drought and chilling in transgenic tobacco and stylo plants. Plant Biotechnol. J. 14, 206–214. doi: 10.1111/pbi.12374
Bradbury, P. J., Zhang, Z., Kroon, D. E., Casstevens, R. M., Ramdoss, Y., Buckler, E. S. (2007). TASSELL. Software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635. doi: 10.1371/journal.pgen.0030004.4
Brozynska, M., Furtado, A., Henry, R. J. (2016). Genomics of crop wild relatives: expanding the gene pool for crop improvement. Plant Biotechnol. J. 14, 1070–1085. doi: 10.1111/pbi.12454
Cao, J., Schneeberger, K., Ossowski, S., Günther, T., Bender, S., Fitz, J., et al. (2011). Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat. Genet. 43, 956–963. doi: 10.1038/ng.911
Causse, M., Desplat, N., Pascual, L., Le Paslier, M.-C., Sauvage, C., Bauchet, G., et al. (2013). Whole genome resequencing in tomato reveals variation associated with introgression and breeding events. BMC Genomics 14, 791. doi: 10.1186/1471-2164-14-791
Cavanagh, C., Morell, M., Mackay, I., Powell, W. (2008). From mutations to MAGIC: resources for gene discovery, validation and delivery in crop plants. Curr. Opin. Plant Biol. 11, 215–221. doi: 10.1016/J.PBI.2008.01.002
Cericola, F., Portis, E., Toppino, L., Barchi, L., Acciarri, N., Ciriaci, T., et al. (2013). The population structure and diversity of eggplant from Asia and the Mediterranean basin. PLoS One 8, e73702. doi: 10.1371/journal.pone.0073702
Cingolani, P., Platts, A., Wang, L. L., Coon, M., Nguyen, T., Wang, L., et al. (2012). A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3. Fly 6, 80–92. doi: 10.4161/fly.19695
Conesa, A., Gotz, S., Garcia-Gomez, J. M., Terol, J., Talon, M., Robles, M. (2005). Blast2GO: a universal tool for annotation, visualization and analysis in functional genomics research. Bioinformatics 21, 3674–3676. doi: 10.1093/bioinformatics/bti610
Danecek, P., Auton, A., Abecasis, G., Albers, C. A., Banks, E., DePristo, M. A., et al. (2010). The variant call format and VCF tools. Bioinformatics 27, 2156–2158. doi: 10.1093/bioinformatics/btr330
Dempewolf, H., Baute, G., Anderson, J., Kilian, B., Smith, C., Guarino, L. (2017). Past and future use of wild relatives in crop breeding. Crop Sci. 57, 1070–1082. doi: 10.2135/cropsci2016.10.0885
Deng, Y., Srivastava, R., Howell, S. H. (2013). Protein kinase and ribonuclease domains of IRE1 confer stress tolerance, vegetative growth, and reproductive development in Arabidopsis. Proc. Natl. Acad. Sci. 110, 19633–19638. doi: 10.1073/pnas.1314749110
Doyle, J., Doyle, J. (1990). Isolation of Plant DNA from fresh tissue. Focus 12, 13–15. doi: 10.3923/rjmp.2012.65.73
Du, X., Huang, G., He, S., Yang, Z., Sun, G., Ma, X., et al. (2018). Resequencing of 243 diploid cotton accessions based on an updated A genome identifies the genetic basis of key agronomic traits. Nat. Genet. 50, 796. doi: 10.1038/s41588-018-0116-x
Felcher, K. J., Coombs, J. J., Massa, A. N., Hansey, C. N., Hamilton, J. P., Veilleux, R. E., et al. (2012). Integration of two diploid potato linkage maps with the potato genome sequence. PLoS One 7, e36347. doi: 10.1371/journal.pone.0036347
Fu, L., Niu, B., Zhu, Z., Wu, S., Li, W. (2012). CD-HIT: accelerated for clustering the next-generation sequencing data. Bioinformatics 28, 3150–3152. doi: 10.1093/bioinformatics/bts565
Gao, L., Gonda, I., Sun, H., Ma, Q., Bao, K., Tieman, D. M., et al. (2019). The tomato pan-genome uncovers new genes and a rare allele regulating fruit flavor. Nat. Genet. 1, 1546–1718. doi: 10.1038/s41588-019-0410-2
Garrison, E., Marth, G. (2012). Haplotype-based variant detection from short-read sequencing. arXiv Prepr. arXiv 1207, 3907
Gisbert, C., Prohens, J., Raigón, M. D., Stommel, J. R., Nuez, F. (2011). Eggplant relatives as sources of variation for developing new rootstocks: effects of grafting on eggplant yield and fruit apparent quality and composition. Sci. Hortic. 128, 14–22. doi: 10.1016/j.scienta.2010.12.007
Gong, Z., Lee, H., Xiong, L., Jagendorf, A., Stevenson, B., Zhu, J. K. (2002). RNA helicase-like protein as an early regulator of transcription factors for plant chilling and freezing tolerance. Proc. Natl. Acad. Sci. 99, 11507–11512. doi: 10.1073/pnas.172399299
Gramazio, P., Prohens, J., Plazas, M., Andújar, I., Herraiz, F., Castillo, E., et al. (2014). Location of chlorogenic acid biosynthesis pathway and polyphenol oxidase genes in a new interspecific anchored linkage map of eggplant. BMC Plant Biol. 14, 350. doi: 10.1186/s12870-014-0350-z
Gramazio, P., Blanca, J., Ziarsolo, P., Herraiz, F. J., Plazas, M., Prohens, J., et al. (2016). Transcriptome analysis and molecular marker discovery in Solanum incanum and S. aethiopicum, two close relatives of the common eggplant (Solanum melongena) with interest for breeding. BMC Genomics 17, 300. doi: 10.1186/s12864-016-2631-4
Gramazio, P., Prohens, J., Plazas, M., Mangino, G., Herraiz, F. J., Vilanova, S. (2017a). Development and genetic characterization of advanced backcross materials and an introgression line population of Solanum incanum in a S. melongena background. Front. Plant Sci. 8, 1477. doi: 10.3389/fpls.2017.01477
Gramazio, P., Prohens, J., Borràs, D., Plazas, M., Herraiz, F. J., Vilanova, S. (2017b). Comparison of transcriptome-derived simple sequence repeat (SSR) and single nucleotide polymorphism (SNP) markers for genetic fingerprinting, diversity evaluation, and establishment of relationships in eggplants. Euphytica 213, 264. doi: 10.1007/s10681-017-2057-3
Gramazio, P., Prohens, J., Plazas, M., Mangino, G., Herraiz, J., García-Fortea, E., et al. (2018). Genomic tools for the enhancement of vegetable crops: a case in eggplant. Not. Bot. Horti Agrobot. Cluj-Napoca 46, 1–13. doi: 10.15835/nbha46110936
Grzebelus, D., Gładysz, M., Macko-Podgórni, A., Gambin, T., Golis, B., Rakoczy, R., et al. (2009). Population dynamics of miniature inverted-repeat transposable elements (MITEs) in Medicago truncatula. Gene 448, 214–220. doi: 10.1016/j.gene.2009.06.004
Guo, S., Zhang, J., Sun, H., Salse, J., Lucas, W. J., Zhang, H., et al. (2013). The draft genome of watermelon (Citrullus lanatus) and resequencing of 20 diverse accessions. Nat. Genet. 45, 51–58. doi: 10.1038/ng.2470
Hill, T. A., Ashrafi, H., Reyes-Chin-Wo, S., Yao, J., Stoffel, K., Truco, M. J., et al. (2013). Characterization of Capsicum annuum genetic diversity and population structure based on parallel polymorphism discovery with a 30K unigene Pepper GeneChip. PLoS One 8, e56200. doi: 10.1371/journal.pone.0056200
Hirakawa, H., Shirasawa, K., Miyatake, K., Nunome, T., Negoro, S., Ohyama, A., et al. (2014). Draft genome sequence of eggplant (Solanum melongena L.): the representative Solanum species indigenous to the old world. DNA Res. 21, 649–660. doi: 10.1093/dnares/dsu027
Hu, J., Manduzio, S., Kang, H. (2019). Epitranscriptomic RNA methylation in plant development and abiotic stress responses. Front. Plant Sci. 10. doi: 10.3389/fpls.2019.00500
Huang, X., Wei, X., Sang, T., Zhao, Q., Feng, Q., Zhao, Y., et al. (2010). Genome-wide association studies of 14 agronomic traits in rice landraces. Nat. Genet. 42, 961. doi: 10.1038/ng.695
Hulse-Kemp, A. M., Ashrafi, H., Plieske, J., Lemm, J., Stoffel, K., Hill, T., et al. (2016). A HapMap leads to a Capsicum annuum SNP infinium array: a new tool for pepper breeding. Hortic. Res. 3, 16036. doi: 10.1038/hortres.2016.36
Hurtado, M., Vilanova, S., Plazas, M., Gramazio, P., Andújar, I., Herraiz, F. J., et al. (2014). Enhancing conservation and use of local vegetable landraces: the Almagro eggplant (Solanum melongena L.) case study. Genet. Resour. Crop Evol. 61, 787–795. doi: 10.1007/s10722-013-0073-2
Ihaka, R., Gentleman, R. (1996). R: a language for data analysis and graphics. J. Comput. Graph. Stat. 5, 299–314. doi: 10.1080/10618600.1996.10474713
Kim, C., Guo, H., Kong, W., Chandnani, R., Shuang, L., Paterson, A. H. (2016). Application of genotyping by sequencing technology to a variety of crop breeding programs. Plant Sci. 242, 14–22. doi: 10.1016/j.plantsci.2015.04.016
Kim, S., Park, M., Yeom, S., Kim, Y., Lee, J., Lee, H., et al. (2014). Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species. Nat. Genet. 46, 270. doi: 10.1038/ng.2877
Knapp, S., Vorontsova, M. S., Prohens, J. (2013). Wild relatives of the eggplant (Solanum melongena L.: Solanaceae): new understanding of species names in a complex group. PLoS One 8, e57039. doi: 10.1371/journal.pone.0057039
Kouassi, B., Prohens, J., Gramazio, P., Kouassi, A. B., Vilanova, S., Galán-Ávila, A., et al. (2016). Development of backcross generations and new interspecific hybrid combinations for introgression breeding in eggplant (Solanum melongena). Sci. Hortic. 213, 199–207. doi: 10.1016/J.SCIENTA.2016.10.039
Lam, H. M., Xu, X., Liu, X., Chen, W., Yang, G., Wong, F. L., et al. (2010). Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat. Genet. 42, 1053–1059. doi: 10.1038/ng.715
Langmead, B., Salzberg, S. L. (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. doi: 10.1038/nmeth.1923
Levin, H. L., Moran, J. V. (2011). Dynamic interactions between transposable elements and their hosts. Nat. Rev. Genet. 12, 615. doi: 10.1038/nrg3030
Lewicki, T., Johnson, M., Abrahamowicz, M. (1974). West African food in the Middle Ages: according to Arabic sources. Cambridge, UK: Cambridge University Press.
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, H. (2014). Toward better understanding of artifacts in variant calling from high-coverage samples. Bioinformatics 30, 2843–2851. doi: 10.1093/bioinformatics/btu356
Lin, T., Zhu, G., Zhang, J., Xu, X., Yu, Q., Zheng, Z., et al. (2014). Genomic analyses provide insights into the history of tomato breeding. Nat. Genet. 46, 1220.
Liu, J., Zheng, Z., Zhou, X., Feng, C., Zhuang, Y. (2015). Improving the resistance of eggplant (Solanum melongena) to Verticillium wilt using wild species Solanum linnaeanum. Euphytica 201, 463–469. doi: 10.1007/s10681-014-1234-x
Mahesh, S., Udayakumar, M. (2018). Peanut RNA helicase AhRH47 sustains protein synthesis under stress and improves stress adaptation in Arabidopsis. Plant Mol. Biol. Rep. 36, 58–70. doi: 10.1007/s11105-017-1056-9
Meyer, R. S., Karol, K. G., Little, D. P., Nee, M. H., Litt, A. (2012). Phylogeographic relationships among Asian eggplants and new perspectives on eggplant domestication. Mol. Phylogenet. Evol. 63, 685–701. doi: 10.1016/J.YMPEV.2012.02.006
Meyer, R. S., Whitaker, B. D., Little, D. P., Wu, S. B., Kennelly, E. J., Long, C. L., et al. (2015). Parallel reductions in phenolic constituents resulting from the domestication of eggplant. Phytochemistry 115, 194–206. doi: 10.1016/j.phytochem.2015.02.006
Michalovova, M., Vyskot, B., Kejnovsky, E. (2013). Analysis of plastid and mitochondrial DNA insertions in the nucleus (NUPTs and NUMTs) of six plant species: size, relative age and chromosomal localization. Heredity 111, 314–320. doi: 10.1038/hdy.2013.51
Muñoz-Falcón, J. E., Prohens, J., Vilanova, S., Nuez, F. (2008). Characterization, diversity, and relationships of the Spanish striped (Listada) eggplants: a model for the enhancement and protection of local heirlooms. Euphytica 164, 405–419. doi: 10.1007/s10681-008-9688-3
Muñoz-Falcón, J. E., Prohens, J., Vilanova, S., Ribas, F., Castro, A., Nuez, F. (2009a). Distinguishing a protected geographical indication vegetable (Almagro eggplant) from closely related varieties with selected morphological traits and molecular markers. J. Sci. Food Agric. 89, 320–328. doi: 10.1002/jsfa.3452
Muñeoz-Falcón, J. E., Prohens, J., Vilanova, S., Nuez, F. (2009b). Diversity in commercial varieties and landraces of black eggplants and implications for broadening the breeders’ gene pool. Ann. Appl. Biol. 154, 453–465. doi: 10.1111/j.1744-7348.2009.00314.x
Naito, K., Cho, E., Yang, G., Campbell, M. A., Yano, K., Okumoto, Y., et al. (2006). Dramatic amplification of a rice transposable element during recent domestication. Proc. Natl. Acad. Sci. U. S. A. 103, 17620–17625. doi: 10.1073/pnas.0605421103
Nelson, M., Linheiro, R., Bergman, C. M. (2017). McClintock: an integrated pipeline for detecting transposable element insertions in whole-genome shotgun sequencing data. G3: Genes, Genomes, Genet. 7, 2763–2778. doi: 10.1534/g3.117.043893
Nidumukkala, S., Tayi, L., Chittela, R. K., Vudem, D. R., Khareedu, V. R. (2019). DEAD box helicases as promising molecular tools for engineering abiotic stress tolerance in plants. Crit. Rev. Biotechnol. 39, 395–407. doi: 10.1080/07388551.2019.1566204
Pascual, L., Desplat, N., Huang, B. E., Desgroux, A., Bruguier, L., Bouchet, J. P., et al. (2015). Potential of a tomato MAGIC population to decipher the genetic control of quantitative traits and detect causal variants in the resequencing era. Plant Biotechnol. J. 13, 565–577. doi: 10.1111/pbi.12282
Paterson, A., Bowers, J., Bruggmann, R. (2009). The Sorghum bicolor genome and the diversification of grasses. Nature 4577229, 551. doi: 10.1038/nature07723
Plazas, M., Vilanova, S., Gramazio, P., Rodríguez-Burruezo, A., Fita, A., Herraiz, F. J., et al. (2016). Interspecific hybridization between eggplant and wild relatives from different genepools. J. Amer. Soc. Hort. Sci. 141, 34–44.
Potato Genome Sequencing Consortium (2011). Genome sequence and analysis of the tuber crop potato. Nature 475, 189–195 (7355). doi: 10.1038/nature10158
Prohens, J., Gramazio, P., Plazas, M., Dempewolf, H., Kilian, B., Díez, M. J., et al. (2017). Introgressiomics: a new approach for using crop wild relatives in breeding for adaptation to climate change. Euphytica 213, 158. doi: 10.1007/s10681-017-1938-9
Prohens, J., Whitaker, B. D., Plazas, M., Vilanova, S., Hurtado, M., Blasco, M., et al. (2013). Genetic diversity in morphological characters and phenolic acids content resulting from an interspecific cross between eggplant, Solanum melongena, and its wild ancestor (S. incanum). Ann. Appl. Biol. 162, 242–257. doi: 10.1111/aab.12017
Qi, J., Liu, X., Shen, D., Miao, H., Xie, B., Li, X., et al. (2013). A genomic variation map provides insights into the genetic basis of cucumber domestication and diversity. Nat. Genet. 45, 1510. doi: 10.1038/ng.2801
Quinlan, A., Hall, I. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
Ranil, R. H. G., Niran, H. M. L., Plazas, M., Fonseka, R. M., Fonseka, H. H., Vilanova, S., et al. (2015). Improving seed germination of the eggplant rootstock Solanum torvum by testing multiple factors using an orthogonal array design. Sci. Hortic. 193, 174–181. doi: 10.1016/j.scienta.2015.07.030
Salas, P., Prohens, J., Seguí-Simarro, J. M. (2011). Evaluation of androgenic competence through anther culture in common eggplant and related species. Euphytica 182, 261–274. doi: 10.1007/s10681-011-0490-2
Salikhov, K., Sacomoto, G., Kucherov, G. (2013). Using cascading bloom filters to improve the memory usage for de Brujin graphs. Algorithms Mol. Biol. 9, 2.
Särkinen, T., Bohs, L., Olmstead, R. G., Knapp, S. (2013). A phylogenetic framework for evolutionary study of the nightshades (Solanaceae): a dated 1000-tip tree. BMC Evol. Biol. 13, 214. doi: 10.1186/1471-2148-13-214
Scaglione, D., Pinosio, S., Marroni, F., Di Centa, E., Fornasiero, A., Magris, G., et al. (2019). Single primer enrichment technology as a tool for massive genotyping: a benchmark on black poplar and maize. Ann. Bot., mcz054.
Schnable, P. S., Pasternak, S., Liang, C., Zhang, J., Fulton, L., Graves, T. A., et al. (2009). The B73 maize genome: complexity, diversity, and dynamics. Science 326, 1112–1115.
Shi, L., Wu, Y., Sheen, J. (2018). TOR signaling in plants: conservation and innovation. Development 145, dev160887. doi: 10.1242/dev.160887
Shivakumara, T. N., Sreevathsa, R., Dash, P. K., Sheshshayee, M. S., Papolu, P. K., Rao, U., et al. (2017). Overexpression of Pea DNA Helicase 45 (PDH45) imparts tolerance to multiple abiotic stresses in chili (Capsicum annuum L.). Sci. Rep. 7. doi: 10.1038/s41598-017-02589-0
Sim, S. C., Durstewitz, G., Plieske, J., Wieseke, R., Ganal, M. W., van Deynze, A., et al. (2012a). Development of a large SNP genotyping array and generation of high-density genetic maps in tomato. PLoS One 7, e40563. doi: 10.1371/journal.pone.0040563
Sim, S. C., Van Deynze, A., Stoffel, K., Douches, D. S., Zarka, D., Ganal, M. W., et al. (2012b). High-density SNP genotyping of tomato (Solanum lycopersicum L.) reveals patterns of genetic variation due to breeding. PLoS One 7, e45520. doi: 10.1371/journal.pone.0045520
Smit, A. F. A., Hubley, R., Green, P. (2004). RepeatMasker Open-1.0. 2008-2015, Institute for Systems Biology.
Stommel, J. R., Whitaker, B. D. (2003). Phenolic acid content and composition of eggplant fruit in a germplasm core subset. J. Amer. Soc. Hort. Sci. 128, 704–710.
Studer, A., Zhao, Q., Ross-Ibarra, J., Doebley, J. (2011). Identification of a functional transposon insertion in the maize domestication gene tb1. Nat. Genet. 43, 1160.
Subbaiyan, G. K., Waters, D. L. E., Katiyar, S. K., Sadananda, A. R., Vaddadi, S., Henry, R. J. (2012). Genome-wide DNA polymorphisms in elite indica rice inbreds discovered by whole-genome sequencing. Plant Biotechnol. J. 10, 623–634. doi: 10.1111/j.1467-7652.2011.00676.x
Syfert, M. M., Castañeda-Álvarez, N. P., Khoury, C. K., Särkinen, T., Sosa, C. C., Achicanoy, H. A., et al. (2016). Crop wild relatives of the brinjal eggplant (Solanum melongena): poorly represented in genebanks and many species at risk of extinction. Am. J. Bot. 103, 635–651. doi: 10.3732/ajb.1500539
Tomato Genome Consortium (2012). The tomato genome sequence provides insights into fleshy fruit evolution. Nature 485-641, (7400) 635. doi: 10.1038/nature11119
Toppino, L., Valè, G., Rotino, G. L. (2008). Inheritance of Fusarium wilt resistance introgressed from Solanum aethiopicum Gilo and Aculeatum groups into cultivated eggplant (S. melongena) and development of associated PCR-based markers. Mol. Breed. 22, 237–250. doi: 10.1007/s11032-008-9170-x
Toppino, L., Barchi, L., Lo Scalzo, R., Palazzolo, E., Francese, G., Fibiani, M., et al. (2016). Mapping quantitative trait loci affecting biochemical and morphological fruit properties in eggplant (Solanum melongena L.). Front. Plant Sci. 7, 256. doi: 10.3389/fpls.2016.00256
Tranchida-Lombardo, V., Cigliano, R., Anzar, I., Landi, S., Palomberi, S., Colantuono, C., et al. (2017). Whole-genome re-sequencing of two Italian tomato landraces reveals sequence variations in genes associated with stress tolerance, fruit quality and long shelf-life. DNA Res. 25, 149–160. doi: 10.1093/dnares/dsx045
Vashisht, A. A., Pradhan, A., Tuteja, R., Tuteja, N. (2005). Cold- and salinity stress-induced bipolar pea DNA helicase 47 is involved in protein synthesis and stimulated by phosphorylation with protein kinase C. Plant J. 44, 76–87. doi: 10.1111/j.1365-313X.2005.02511.x
Vilanova, S., Manzur, J. P., Prohens, J. (2012). Development and characterization of genomic simple sequence repeat markers in eggplant and their application to the study of diversity and relationships in a collection of different cultivar types and origins. Mol. Breed. 30, 647–660. doi: 10.1007/s11032-011-9650-2
Víquez-Zamora, M., Vosman, B., de Geest, H., Bovy, A., Visser, R. G., Finkers, R., et al. (2013). Tomato breeding in the genomics era: insights from a SNP array. BMC Genomics 14, 354. doi: 10.1186/1471-2164-14-354
Vorontsova, M. S., Stern, S., Bohs, L., Knapp, S. (2013). African spiny Solanum (subgenus Leptostemonum, Solanaceae): a thorny phylogenetic tangle. Bot. J. Linn. Soc. 173, 176–193. doi: 10.1111/boj.12053
Vos, P. G., Uitdewilligen, J. G., Voorrips, R. E., Visser, R. G. F., van Eck, H. J. (2015). Development and analysis of a 20K SNP array for potato (Solanum tuberosum): an insight into the breeding history. Theor. Appl. Genet. 128, 2387–2401. doi: 10.1007/s00122-015-2593-y
Wang, Y., Pang, C., Li, X., Hu, Z., Lv, Z., Zheng, B., et al. (2017). Identification of tRNA nucleoside modification genes critical for stress response and development in rice and Arabidopsis. BMC Plant Biol. 17, 261. doi: 10.1186/s12870-017-1206-0
Weigel, D., Mott, R. (2009). The 1001 Genomes Project for Arabidopsis thaliana. Genome Biol. 10, 107. doi: 10.1186/gb-2009-10-5-107
Wheeler, G. L., Jones, M. A., Smirnoff, N. (1998). The biosynthetic pathway of vitamin C in higher plants. Nature 393, 365–369. doi: 10.1038/30728
Xu, X., Liu, X., Ge, S., Jensen, J., Hu, F., Li, X., et al. (2012). Resequencing 50 accessions of cultivated and wild rice yields markers for identifying agronomically important genes. Nat. Biotechnol. 30, 105. doi: 10.1038/nbt.2050
Yan, J., Yang, X., Shah, T., Sánchez-Villeda, H., Li, J., Warburton, M., et al. (2010). High-throughput SNP genotyping with the GoldenGate assay in maize. Mol. Breed. 25, 441–451. doi: 10.1007/s11032-009-9343-2
Yang, X., Cheng, Y. F., Deng, C., Ma, Y., Wang, Z. W., Chen, X. H., et al. (2014). Comparative transcriptome analysis of eggplant (Solanum melongena L.) and turkey berry (Solanum torvum Sw.): phylogenomics and disease resistance analysis. BMC Genomics 15, 412. doi: 10.1186/1471-2164-15-412
Yu, J., Hu, S., Wang, J., Wong, G. K. S., Li, S., Liu, B., et al. (2002). A draft sequence of the rice genome (Oryza sativa L. ssp. indica). Science 296, 79–92. doi: 10.1126/science.1068037
Zhou, X., Bao, S., Liu, J., Zhuang, Y. (2016). De novo sequencing and analysis of the transcriptome of the wild eggplant species Solanum aculeatissimum in response to Verticillium dahliae. Plant Mol. Biol. Rep. 34, 1193–1203. doi: 10.1007/s11105-016-0998-7
Zhuang, J., Wang, J., Theurkauf, W., Weng, Z. (2014). TEMP: a computational method for analyzing transposable element polymorphism in populations. Nucleic Acids Res. 42, 6826–6838
Keywords: whole-genome resequencing, Solanum melongena, S. incanum, polymorphisms, genome characterization
Citation: Gramazio P, Yan H, Hasing T, Vilanova S, Prohens J and Bombarely A (2019) Whole-Genome Resequencing of Seven Eggplant (Solanum melongena) and One Wild Relative (S. incanum) Accessions Provides New Insights and Breeding Tools for Eggplant Enhancement. Front. Plant Sci. 10:1220. doi: 10.3389/fpls.2019.01220
Received: 23 June 2019; Accepted: 04 September 2019;
Published: 08 October 2019.
Edited by:
Luigi Cattivelli, Council for Agricultural and Economics Research, ItalyCopyright © 2019 Gramazio, Yan, Hasing, Vilanova, Prohens and Bombarely. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Pietro Gramazio, cGllZ3JhQHVwdi5lcw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.