 Tsubasa Shoji
Tsubasa Shoji- Department of Biological Science, Nara Institute of Science and Technology (NAIST), Ikoma, Japan
Plants produce a vast array of structurally diverse specialized metabolites with various biological activities, including medicinal alkaloids and terpenoids, from relatively simple precursors through a series of enzymatic steps. Massive metabolic flow through these pathways usually depends on the transcriptional coordination of a large set of metabolic, transport, and regulatory genes known as a regulon. The coexpression of genes involved in certain metabolic pathways in a wide range of developmental and environmental contexts has been investigated through transcriptomic analysis, which has been successfully exploited to mine the genes involved in various metabolic processes. Transcription factors are DNA-binding proteins that recognize relatively short sequences known as cis-regulatory elements residing in the promoter regions of target genes. Transcription factors have positive or negative effects on gene transcription mediated by RNA polymerase II. Evolutionarily conserved transcription factors of the APETALA2/ETHYLENE RESPONSE FACTOR (AP2/ERF) and basic helix-loop-helix (bHLH) families have been identified as jasmonate (JA)-responsive transcriptional regulators of unrelated specialized pathways in distinct plant lineages. Here, I review the current knowledge and propose a conceptual model for the evolution of metabolic pathways, termed “recruitment model of metabolic evolution.” According to this model, structural genes are repeatedly recruited into regulons under the control of conserved transcription factors through the generation of cognate cis-regulatory elements in the promoters of these genes. This leads to the adjustment of catalytic activities that improve metabolic flow through newly established passages.
Introduction
Biological processes generally depend on the coordinated expression of multiple genes. Transcription factors play a central role in controlling the RNA polymerase-mediated transcription of downstream genes. These genes form gene networks, or regulons, with transcription factors recognizing specific cis-regulatory elements in the promoter regions of these groups of target genes. Complex, often long, metabolic pathways rely on the proper functioning of a large series of metabolic enzymes, membrane transporters, and other proteins. The activity of transcription factors often underlies the coordination of gene expression during various metabolic processes (Patra et al., 2013; Chezem and Clay, 2016; Zhou and Memelink, 2016).
A diverse range of specialized metabolites, such as bioactive alkaloids and terpenoids, are produced and accumulate in various plant species. These metabolites contribute to plant defense and reproduction in a changing environment. Due to their useful attributes, many natural products derived from plants or phytochemicals are utilized as medicines, drugs, dyes, perfumes, or other industrial materials. In contrast to universally present primary metabolites, the occurrence of specialized (or so-called secondary) metabolites is usually restricted to certain taxonomic groups. Metabolite levels are often highly variable, even within a single species or individual plant, reflecting the temporal and spatial dynamics of their production. Genomic and molecular approaches, often involving coexpression network analysis to select candidate genes (Yonekura-Sakakibara and Saito, 2013), have greatly facilitated the identification of structural genes involved in metabolic pathways. By contrast, the regulatory aspects of these genes, such as regulatory mechanisms at the transcriptional and other levels, have remained unexplored, representing a promising area of research in the coming years.
The central roles of MYB and basic helix-loop-helix (bHLH) family transcription factors in regulating the anthocyanin and related flavonoid pathways of many plant species have been a cornerstone example of the persistence of metabolic regulons comprising master transcriptional regulators with their downstream structural genes (Patra et al., 2013; Chezem and Clay, 2016). Well-studied instances include the regulation of the glucosinolate pathway by MYB family factors in Arabidopsis (Chezem and Clay, 2016). In notable contrast to the regulators that target a specific metabolic pathway (or set of related pathways), jasmonate (JA)-responsive factors in certain subgroups of the APETALA2/ETHYLENE RESPONSE FACTOR (AP2/ERF) and bHLH families have been found to be master regulators for a diverse range of specialized pathways, mostly for important alkaloids and terpenoids, in distinct plant linages (Zhou and Memelink, 2016).
JAs are phytohormones derived from the octadecanoid pathway that play central roles as signaling molecules during biotic and abiotic stress responses in plants (Goossens et al., 2016a). Many specialized pathways are readily elicited by JA treatment (Goossens et al., 2016a; Zhou and Memelink, 2016). Indeed, many phytochemicals are thought to be involved in plant defense responses against pathogens and herbivores based on the JA-dependent elicitation of their biosynthetic pathways, along with their toxicity to biological agents. The perception of JA signals and the resulting cascades leading to gene regulation primarily occur via proteasome-dependent degradation of JAZ repressor proteins and the subsequent liberation of a few key transcription factors, including bHLH family member MYC2, from JAZ-mediated repression (Thines et al., 2007; Sheard et al., 2010; Zhang et al., 2015; Goossens et al., 2016a). It is important to address how the upstream JA signaling circuit is anchored to downstream defense metabolism. A handful of the JA-responsive transcription factors of AP2/ERF and bHLH families, have been identified as missing links between the highly conserved JA signaling module and more divergent downstream pathways (Zhou and Memelink, 2016).
In this article, I provide an overview of the JA-responsive factors and their target metabolic pathways, which encompass a substantial portion of the specialized pathways for which transcriptional regulators have been defined (Patra et al., 2013; Zhou and Memelink, 2016). The identification of such evolutionarily conserved regulators targeting divergent pathways prompt me to contemplate how these metabolic regulons have been established during the evolution. This evolutionary issue is discussed and a conceptual model is proposed, mainly focusing on the JA-responsive factors and their regulons.
Clade II, Subgroup IXa ERF Transcription Factors
Transcription factors of the AP2/ERF family are widespread in plants. The GCC-box (5′-AGCCGCC-3′) element is a typical sequence recognized by AP2/ERF transcription factors. The DNA-binding AP2/ERF domain contains a three-stranded β-sheet followed by an α-helix, which form a unique interface required for DNA binding (Allen et al., 1998).
A group of AP2/ERF family transcription factors, including Octadecanoid-derivative Responsive Catharanthus AP2-domain (ORCA) proteins from Catharanthus roseus (Van der Fits and Memelink, 2000; Paul et al., 2017), OpERF2 from Ophiorrhiza pumila (Udomsom et al., 2016), ERF189 and ORC1 from tobacco (Nicotiana tabacum; Shoji et al., 2010; De Boer et al., 2011), JASMONATE RESPONSIVE ERF4 (JRE4)/GLYCOALKALOID METABOLISM9 (GAME9) from tomato and potato (Cárdenas et al., 2016; Thagun et al., 2016; Nakayasu et al., 2018), and AaORA from Artemisia annua (Lu et al., 2013), are classified into clade II of subgroup IXa (Nakano et al., 2006; Shoji et al., 2010, 2013). These transcription factors are involved in regulating JA-mediated defense metabolism in various plants. The JA-responsive ERF genes are present in a wide range of eudicots, usually as multicopy genes (Figure 1A). Multiple ERF genes are tandemly clustered on chromosomes in some plant genomes (Figure 1A). The phylogenetic relationships of ERFs from different species (Figure 1A) imply that independent generations of these gene clusters in distinct plant families through tandem gene duplication.
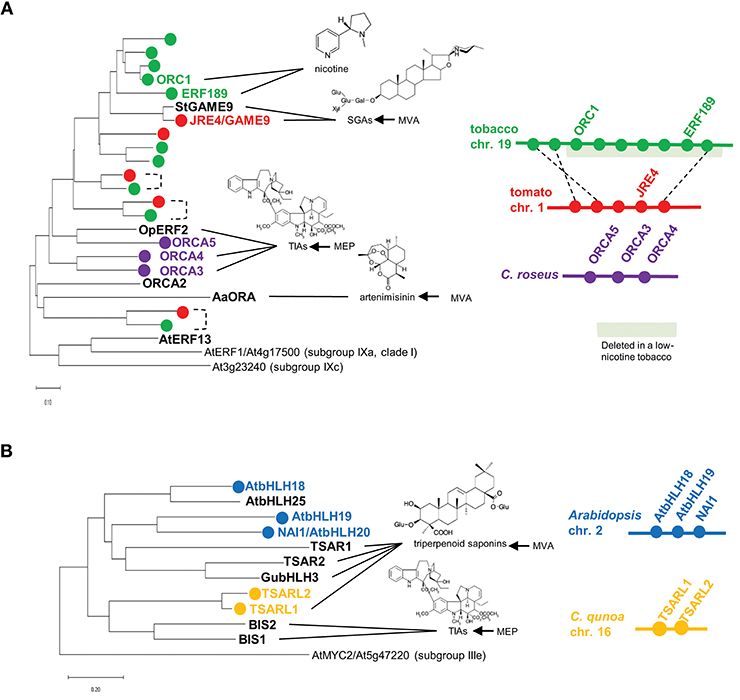
Figure 1. Phylogenetic relationships and gene clustering of JA-responsive ERF (A) and bHLH (B) transcription factors of clade II, subgroup IXa and IVa, respectively. Full-length amino acid sequences were aligned using ClustalW (Thompson et al., 1994), and the trees were generated with the neighbor-joining algorithm using MEGA-X (Kumar et al., 2018). The Arabidopsis genes are included in the trees as an outgroup. The scale bars indicate the number of amino acid substitutions per residue. For isoprenoid-derived metabolites, the contribution of the mevalonate (MVA) or methylerythritol phosphate (MEP) pathway to their production is indicated on the right. Apparent orthologs from Solanaceae plants are connected by dotted lines. Gene clusters are depicted schematically. The region deleted in a low-nicotine tobacco cultivar (Kajikawa et al., 2017) is highlighted in light green.
ORCAs in C. roseus
Terpenoid indole alkaloids (TIAs) are a large group of specialized products, including the valuable chemotherapy drugs vinblastine and vincristine. A variety of TIAs are derived from the key intermediate strictosidine, which is formed by condensation between tryptamine (a product of the shikimate pathway) and the seco-iridoid compound secologanin. TIA biosynthesis and its regulation have been intensively studied in the medicinally important species C. roseus (Apocynaceae) (Zhu et al., 2014). In C. roseus, ORCA2 and ORCA3 function as transcriptional regulators that induce the expression of TIA biosynthesis genes, including strictosidine synthase and tryptophan decarboxylase, encoding key enzymes in this pathway (Menke et al., 1999; Van der Fits and Memelink, 2000; Li et al., 2013). ORCA3 is physically linked to ORCA4 and ORCA5, forming a gene cluster in the genome (Figure 1A, Paul et al., 2017). ORCA4 shares an overlapping function with ORCA2 and ORCA3, but ORCA4 also targets additional TIA genes (Paul et al., 2017). Unlike ORCA2 (Li et al., 2013) and ORCA3 (Van der Fits and Memelink, 2000), the overexpression of ORCA4 causes a drastic increase in TIA accumulation (Paul et al., 2017). C. roseus MYC2 (CrMYC2) directly upregulates the expression of ORCA3 by recognizing a G-box-like element in its promoter (Zhang et al., 2011), and it also coregulates TIA structural genes with ORCA3 (Paul et al., 2017). In addition to their role in transcriptional regulation, ORCAs and CrMYC2 are activated by phosphorylation by a kinase involved in a JA-activated MAP kinase cascade (Paul et al., 2017).
OpERF2 in Ophiorrhiza pumila
Camptothecin is an antitumor TIA that functions by inhibiting DNA topoisomerase I activity. This clinically important TIA is produced by various angiosperms from taxonomically distant families, including Ophiorrhiza pumila (Rubiaceae) (Sirikantaramas et al., 2007). OpERF2 was originally isolated from O. pumila hairy roots. The suppressed expression of this gene resulted in the reduced expression of genes involved in seco-iridoid and upstream methylerythritol phosphate (MEP) pathways, which supply secologanin for downstream camptothecin production, although this did not have a significant impact on TIA accumulation (Udomsom et al., 2016).
ERF189 and ORC1 in Tobacco
Nicotine is composed with two heterocyclic rings, the ornithine-derived pyrrolidine ring and the nicotinate-derived pyridine ring. In tobacco (Nicotiana tabacum, Solanaceae), this toxic alkaloid is produced in roots and primarily accumulates in leaves, functioning as a defense compound against herbivores (Dewey and Xie, 2013). Tobacco ERF189, ORC1, and related ERF genes are clustered together in the tobacco genome (Figure 1A, Shoji et al., 2010; De Boer et al., 2011; Kajikawa et al., 2017). A cluster of ERFs including ERF189 and ORC1 were found to be deleted to a large extent in a tobacco cultivar with low nicotine content (Figure 1A, Shoji et al., 2010; Kajikawa et al., 2017). Although not yet proven, ERF189 is considered to be a primary transcriptional regulator of nicotine biosynthesis, given that its expression profiles are similar to those of the downstream biosynthesis genes: strong expression in roots (Kajikawa et al., 2017), no induction in response to NaCl (Shoji and Hashimoto, 2015; Kajikawa et al., 2017), and the suppression of JA-dependent induction by ethylene (Shoji et al., 2000, 2010). A large series of metabolic and transport genes in this pathway are upregulated by ERF189, which recognizes P-box elements, but not the typical GCC-box elements, in their promoters (Shoji et al., 2010, 2013; Shoji and Hashimoto, 2011a). Tobacco MYC2 regulates the expression of ERF189 and directly activates the transcription of nicotine biosynthesis genes together with ERF189 (Shoji and Hashimoto, 2011b; Zhang et al., 2012).
JRE4/GAME9 in Tomato and Potato
Steroidal glycoalkaloids (SGAs) are cholesterol-derived, nitrogen-containing metabolites found in the inedible parts of Solanaceae plants such as tomato (Solanum lycopersicum) and potato (S. tuberosum) (Cárdenas et al., 2015). In the tomato and potato genomes, the JRE4/GAME9 gene is present in a cluster with related ERF genes (Cárdenas et al., 2016; Thagun et al., 2016). JRE4/GAME9 regulates nearly an entire series of SGA metabolic steps, including those in the upstream isoprenoid-producing mevalonate (MVA) pathway (Cárdenas et al., 2016; Thagun et al., 2016; Nakayasu et al., 2018). A loss of JRE4/GAME9 function drastically reduced SGA accumulation and resistance to chewing insects in tomato, demonstrating the major role of this transcription factor in defense-related SGA formation (Nakayasu et al., 2018). Tomato MYC2 and JRE4/GAME9 synergistically activated the promoters of SGA genes in tobacco protoplasts (Cárdenas et al., 2016). In agreement with the results of promoter binding studies, cognate cis-regulatory elements are significantly enriched in the proximal promoter regions of SGA biosynthesis genes, supporting the direct regulation of these genes by JRE4/GAME9 (Thagun et al., 2016). A comparison of the genomes of ancestral and cultivated species of the Solanum genus pointed to the possible selection of certain alleles of JRE4/GAME9 during domestication, which might have contributed to the decrease in antinutritional SGA levels in cultivated Solanum species (Hardigan et al., 2017; Zhu et al., 2018).
AaORA in Artemisia annua
Artemisinin, a sesquiterpene lactone produced by the traditional Chinese herb Artemisia annua (Asteraceae), has been exploited as an effective anti-malaria agent (Tang et al., 2014). A. annua ORA (AaORA) is a transcriptional regulator of artemisinin biosynthesis that upregulates the expression of genes involved in this pathway, including amorpha-4,11-diene synthase, CYP71AV1, and double bond reductase 2 (Lu et al., 2013). AaORA is specifically expressed in the trichomes of aerial organs, including artemisinin-producing glandular trichomes (Olofsson et al., 2011; Lu et al., 2013). Since numerous transcription factors from various families (e.g., bHLH, ERF, bZIP, and WRKY), in addition to AaORA, were shown to regulate artemisinin biosynthesis (Tang et al., 2014; Lv et al., 2017), the relative importance of each transcription factor and their functional relationships in this process should be addressed.
Arabidopsis thaliana ERF13
AtERF13 is the only member of clade II, subgroup IXa in Arabidopsis thaliana (Brassicaceae). In contrast to the ERFs mentioned above, AtERF13 was not yet shown to be involved in a specific metabolic pathway. AtERF13 is induced in response to a range of biotic and abiotic stresses, such as JA, wounding, insect feeding, colonization of beneficial bacteria, high osmolality, and NaCl (Lee et al., 2010; Sogabe et al., 2011; Srivastava et al., 2012; Schweizer et al., 2013). AtERF13 binds to COUPLING ELEMENT1 (CE1), a cis-regulatory element required for abscisic acid (ABA)-responsive gene expression, and the overexpression of AtERF13 confers increased sensitivity to ABA in Arabidopsis, suggesting this gene functions in abiotic stress resistance (Lee et al., 2010). AtERF13 is also involved in resistance to insect herbivores, acting downstream of MYC2 (a central player in JA signaling) and mediating the expression of a subset of MYC2-regulated defense genes (Schweizer et al., 2013). AtERF13 is phosphorylated at its tyrosine residues, as revealed by phosphoproteomic analysis, suggesting that its activity is regulated via post-translational modification (Nemoto et al., 2015).
Subgroup IVa bHLH Transcription Factors
Another group of JA-responsive transcription factors is attracting attention as regulators of metabolic pathways in diverse plants. These transcription factors include bHLH IRIDOID SYNTHESIS1 (BIS1) and BIS2 from C. roseus (Van Moerkercke et al., 2015, 2016), TRITERPENE SAPONIN BIOSYNTHESIS ACTIVATING REGULATOR1 (TSAR1) and TSAR2 from Medicago truncatula (Mertens et al., 2016a), TSAR-LIKE1 (TSARL1) from Chenopodium quinoa (Jarvis et al., 2017), GubHLH3 from Glycyrrhiza uralensis (Tamura et al., 2018), and bHLH18, bHLH19, bHLH20/NAI1, and bHLH25 from Arabidopsis (Matsushima et al., 2004), which all belong to subgroup IVa of the bHLH family (Figure 1B, Heim et al., 2003; Goossens et al., 2016b).
Unlike AP2/ERF family members, which are specific to plants, the bHLH transcription factor family is widely present in eukaryotic organisms and has expanded, especially in land plants (Feller et al., 2011). The signature bHLH domain is composed of an N-terminal basic region that binds to negatively charged DNA and a helix-loop-helix motif responsible for protein dimerization. bHLH transcription factors form homo- or heterodimers that typically bind to E-box (5′-CANNTG-3′) elements, such as G-box (5′-CACGTG-3′) and N-box (5′-CACGAG-3′) elements, in the promoter regions of their target genes.
BISs in C. roseus
In addition to ORCAs and CrMYC2, BIS1 and BIS2, a pair of homologous JA-responsive bHLH transcription factors, are involved in regulating TIA formation in C. roseus. BISs specifically act on a branch of the TIA pathway that supplies the seco-iridoid intermediate, secologanin, for incorporation into TIAs. Overexpression of BIS1 or BIS2 results in the upregulation of genes involved in seco-iridoid and upstream MEP pathways, thereby increasing the accumulation of downstream TIAs (Van Moerkercke et al., 2015, 2016). The finding that BIS2 is induced by BIS1 or BIS2 overexpression points to the existence of a positive feedback loop (Van Moerkercke et al., 2016). In contrast to subgroup IIIe MYC2 transcription factors, BISs cannot interact with JAZ proteins and are thus not direct targets of the repressors integrated into the JA signaling module (Van Moerkercke et al., 2016).
TSARs in Medicago truncatula
The model legume plant Medicago truncatula (Fabaceae) produces oleanane-type triterpenoid saponins. These amphipathic glycosides, containing triterpenoid aglycones, exhibit a diverse range of biological activities (Osbourn et al., 2011). In M. truncatula, TSAR1 and TSAR2, two homologous bHLH transcription factors, are JA-responsive transcriptional regulators of triterpenoid saponin biosynthesis (Mertens et al., 2016a). While the isoprenoid-producing MVA pathway is commonly targeted by both TSARs, TSAR1 and TSAR2 specifically regulate two distinct downstream branches of this pathway, producing nonhemolytic and hemolytic saponins, respectively (Mertens et al., 2016a). TSARs activate the gene encoding 3-Hydroxy-3-Methylglutaryl-CoA Reductase, a rate-limiting enzyme in the MVA pathway, by directly recognizing the N-box element in its promoter (Mertens et al., 2016a).
TSARL1 in Chenopodium qunoa
Chenopodium quinoa (Chenopodiaceae), or quinoa, is a staple food crop in Andean countries. Quinoa seeds have high nutritional value, but bitterness of the seeds due to the accumulation of triperpenoid saponins (oleanane-type) is disadvantageous (Kuljanabhagavad et al., 2008). In C. quinoa, TSARL1 and TSARL2 are clustered together (Figure 1B) and are expressed in seeds and roots, respectively (Jarvis et al., 2017). In sweet quinoa strains, loss-of-function mutations of TSARL1, including one that appears to cause alternative splicing, allowed the down-regulation of genes involved in the production of the antinutritional saponins (Jarvis et al., 2017).
GubHLH3 in Glycyrrhiza uralensis
The medicinal legume Glycyrrhiza uralensis (Fabaceae) is rich in oleanane-type triterpenoid saponins, such as glycyrrhizin, which is used as a pharmaceutical compound and sweetener, as well as soyasaponins (Hayashi and Sudo, 2009). G. uralensis bHLH3 (GubHLH3), a JA-responsive bHLH transcription factor, upregulates the expression of soyasaponin biosynthesis genes, such as those encoding CYP93E3 and CYP72A566, which are involved in oxidative modifications of the triterpenoid backbone (Tamura et al., 2018). Consistently, the overexpression of GubHLH3 increased the levels of soyasapogenol B and other intermediates of the soyasaponin pathway in G. uralensis hairy roots (Tamura et al., 2018).
bHLH18, bHLH19, bHLH20/NAI1, and bHLH25 in Arabidopsis
In Arabidopsis, four genes, bHLH18, bHLH19, bHLH20/NAI1, and bHLH25, encode subgroup IVa bHLH transcription factors; three of them, except bHLH25, form a gene cluster (Figure 1B). NAI1, which resides in this three-gene cluster, is indispensable for the formation of the ER body, an ER-derived rod-shape organelle found in plants of the Brassicales order (Matsushima et al., 2004). ER bodies are constitutively present in Arabidopsis seedlings and roots. By contrast, in rosette leaves, wounding and JA treatment induce the formation of this defense-related organelle, which accumulates large amounts of β-glucosidases, whose activities increase when the compartment is disrupted (Nakano et al., 2014). NAI1 regulates the expression of genes encoding proteins required for ER body formation and activity, including PYK10, a major β-glucosidase in this organelle. PYK10 functions as a myrosinase that hydrolyzes indole glucosinolates, a group of important defense compounds in Arabidopsis and related species (Nakano et al., 2017). The phylogenetic co-occurrence of ER bodies and indole glucosinolates and the co-expression of the associated genes also support the functional coordination between this organelle and glucosinolate metabolism (Nakano et al., 2017).
A previous study suggested the involvement of bHLH18, bHLH19, bHLH20/NAI1, and bHLH25 in JA-mediated inhibition of iron uptake in Arabidopsis (Cui et al., 2018). JA represses iron uptake by promoting the degradation of FIT/bHLH29, a central transcriptional regulator of iron-uptake genes critical to metal homeostasis (Cui et al., 2018). The four subgroup IVa bHLHs interact with FIT protein and promote its JA-stimulated removal through proteasome-dependent degradation (Cui et al., 2018).
The Gain of cis-Regulatory Elements
The recruitment of metabolic genes into regulons likely requires the gain of transcription factor-binding cis-regulatory elements in the appropriate promoter regions. Such a process is fairly likely, considering the relatively frequent, simple generation of short sequence elements in noncoding promoter regions that can have a degree of redundancy and acquire functions through mutational changes, such as point mutations and transpositions (Wray, 2007; Swinnen et al., 2016).
A case study of a tobacco gene involved in nicotine biosynthesis supports such a scenario. Quinolinate phosphoribosyltransferase (QPT) is a primary metabolic enzyme involved in NAD biosynthesis in all organisms. However, in tobacco, QPT also supplies a significant amount of intermediates required for downstream nicotine biosynthesis (Figure 2A). To satisfy such a metabolic demand, tandem duplication of QPT has occurred in the Nicotiana lineage, generating a cluster of QPT1 and QPT2 genes (Figure 2, Shoji and Hashimoto, 2019). These genes are thought to be involved in NAD and nicotine biosynthesis, respectively, based on their distinct expression patterns (Shoji and Hashimoto, 2011a). QPT2 harbors multiple ERF189-binding P-box elements in its promoter required for its transcriptional activation by ERF189 (Shoji and Hashimoto, 2011a). The progressive acquisition of these elements after gene duplication has ensured the involvement of QPT2 in the ERF189-controlled regulon of the nicotine pathway (Figure 2B). The frequent occurrence of JRE4/GAME9 binding elements in the proximal promoter regions of SGA biosynthesis genes in tomato implies that such a notion is also applicable to these genes (Thagun et al., 2016).
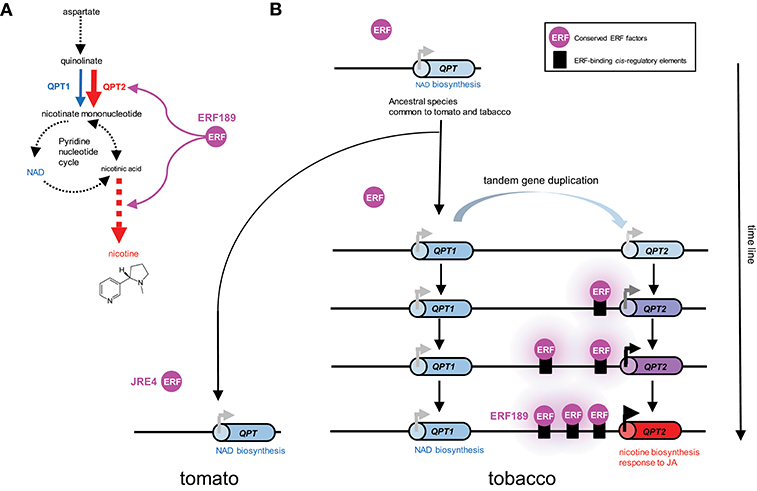
Figure 2. Recruitment of metabolic genes into regulons under the control of conserved transcription factors through the generation of cognate cis-regulatory elements in their promoter regions; a case study of the evolution of QPT2 in the ERF189-controlled nicotine biosynthesis regulon in the tobacco lineage. (A) Quinolinate phosphoribosyltransferase (QPT) catalyzes a reaction at the entry point in NAD and nicotine biosynthesis. In tobacco, QPT is encoded by distinct QPT1 (blue) and QPT2 (red) genes, which are thought to contribute to NAD and nicotine formation, respectively (Shoji and Hashimoto, 2011a). QPT2 and downstream steps specific to nicotine formation (red arrows) are regulated by ERF189 in tobacco. Steps including multiple enzymes and undefined reactions are represented by broken arrows. (B) Schematic depiction of the evolution of QPT genes in the tomato and tobacco lineages. In the tobacco genome, QPT1 and QPT2, which are thought to have arisen through tandem duplication, are located ~75 kb apart on the chromosome. Tomato contains one QPT gene copy in a genomic region syntenic to the tobacco cluster (Shoji and Hashimoto, 2019). One of the duplicates, QPT2, has become regulated by an evolutionarily conserved ERF transcription factor by gaining ERF-binding cis-regulatory elements in its promoter. Three functional P-box elements bound by ERF189 are present in the proximal promoter region of QPT2 in extant tobacco (Shoji and Hashimoto, 2011a).
Evolutionary Changes in Transcription Factors
In contrast to the gains (and losses) of cis-regulatory elements that strongly contribute to the rewiring of gene regulatory networks, mutational changes in transcription factors, which have profound, pleiotropic effects on numerous downstream genes, are relatively constrained. Nevertheless, there are also examples of the modification of the functionalities and expression patterns of trans-acting factors (Maerkl and Quake, 2009).
A series of subgroup IXa ERFs have divergent DNA-binding specificities to GCC-box elements and to related but distinct P-box and CS1-box elements. Such distinct binding specificities can be accounted for by a few amino acid differences in a small stretch of the DNA-binding domain (Figure 3, Shoji et al., 2013). It appears that a progressive evolutionary trajectory has led from transcription factors that recognize only a canonical GCC-box to Nicotiana-specific ERF189-type transcription factors that bind to P-box but not GCC-box elements via functional intermediates, such as ORCA3-type transcription factors, which bind to multiple elements, including both GCC-box and P-box elements (Figure 3, Shoji et al., 2013). The development of unique combinations of cis-elements and trans-factors may have been indispensable for avoiding missed connections among unrelated regulatory circuits and, thus, the establishment of lineage-specific specialized pathways. This process appears to have occurred independently of the development of a broad range of ERFs targeting GCC-box elements involved in general defense responses.
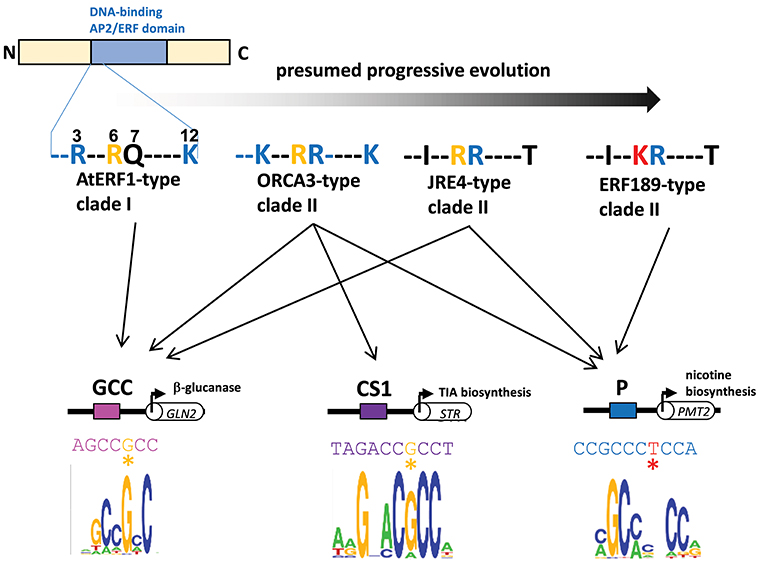
Figure 3. Divergent DNA-binding specificities of subgroup IXa ERF transcription factors. Amino acid residues at positions 3, 6, 7, and 12 in an N-terminal portion of the DNA-binding AP2/ERF domain are shown (counting from the end of the domain). Basic residues R and K at positions 3, 7, and 12 (blue) increase the nonspecific affinity of the proteins for DNA by interacting with negatively charged phosphate backbones (Shoji et al., 2013). The substitution of one residue at position 6 from R (yellow) to K (red) in ERF189-type transcription factors alters the recognition of nucleotide bases (G to T) at a specific position (marked with asterisks) in the cis-regulatory elements (Shoji et al., 2013). Perhaps progressive evolution has occurred from ancestral AtERF1-type transcription factors to more specialized ERF189-type transcription factors via functional intermediates with amino acid substitutions at a few positions. The in vitro binding specificities of recombinant ORCA3 are represented by sequence logos (Shoji et al., 2013) GLN, β-1,3-glucanase; STR, strictosidine synthase; PMT, putrescine N-methyltransferase.
Nicotine and SGA biosynthesis pathways in distinct lineages of the same Solanaceae family, which are regulated by orthologous ERFs, share many properties, such as JA-dependent induction and suppression by ethylene (Shoji et al., 2010; Nakayasu et al., 2018). By contrast, the site of their biosynthesis differs between the two lineages: nicotine is synthesized exclusively in tobacco roots, whereas SGAs are produced in nearly all inedible parts of tomato and potato, including leaves and roots. This difference depends on the differential expression patterns of the transcriptional regulators ERF189 and JRE4/GAME9 (Cárdenas et al., 2016; Thagun et al., 2016; Kajikawa et al., 2017). To guarantee the function of each group of metabolites in plant defense, the tissue-specific expression patterns of these regulators may have developed independently after the separation of the two lineages, whereas their responses to JA and other features have been conserved between lineages. These ideas point to the elastic evolution of sets of a particular transcription factor and its downstream metabolic genes as independent units with specialized roles in chemical defense in a lineage-specific manner.
Despite the functional differences noted above, the JA-responsive transcription factors are considered components of conserved regulatory mechanisms present in various species. For instance, in transgenic tomato plants, a promoter reporter of tobacco QPT2 regulated by ERF189 was expressed in a JA-responsive and cell type-specific manner, as in tobacco, and this expression was mediated by JRE4 (Shoji and Hashimoto, 2019). TSARs and BISs were shown to be functionally exchangeable, as the orthologous bHLH factors regulate each other's target genes in C. roseus and M. truncatula (Mertens et al., 2016b). Both of these examples clearly point to the interchangeable nature of these factors among species with entirely different pathways (e.g., ornithine-derived nicotine vs. MVA pathway-derived SGAs for ERF189 and JRE4, and MVA pathway-derived saponins vs. MEP pathway-derived TIAs for TSARs and BISs), supporting the functional conservation of these factors.
Recruitment Model of Metabolic Evolution
Metabolism is a fundamental requirement of all living organisms. Primeval metabolism, or the simple conversion of substances, is thought to rely on a small number of proteinaceous or other catalysts with low reaction specificities and efficiencies (Figure 4i). Metabolic systems have evolved toward increasing order and efficiency (Weng et al., 2012). Contemporary primary metabolism, which was established early and has been maintained, is carried out by robust systems mediated by enzymes with high specificities and efficiencies (Figure 4ii).
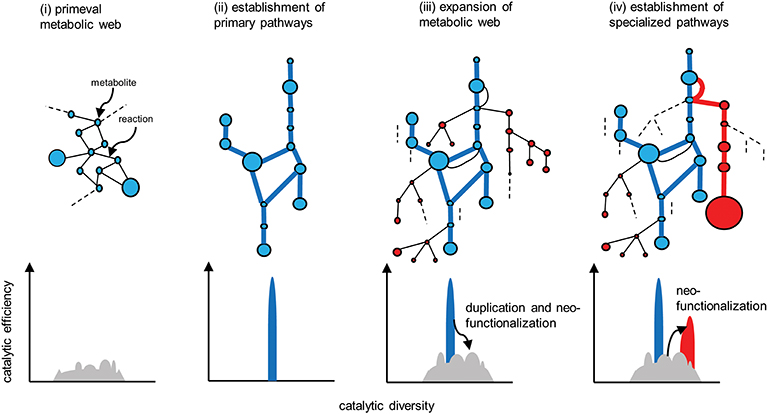
Figure 4. Evolution of metabolic pathways (upper) and the associated changes in the catalytic functionalities of the enzymes (lower). Catalytic efficiency (or the activity of a given reaction) and diversity (or the reaction specificity) are represented on the vertical and horizontal axes, respectively. (i) Primeval metabolism depends on low catalytic activities in terms of reaction specificities and efficiencies. (ii) Improved catalytic specificities and efficiencies allow for the emergence of highly efficient primary metabolic pathways (blue lines). (iii) Expansion of the metabolic web occurs via the duplication of metabolic genes, and the subsequent mutational changes in the genes contribute to neofunctionalization of the encoded enzymatic proteins. (iv) A certain series of enzymes are selected and subjected to catalytic improvement (neofunctionalization). Such processes have enabled the emergence of specialized pathways (red lines) that contribute to the substantial accumulation of the metabolites (red circles). Reactions that do not contribute to metabolic flow (and are therefore hidden) are indicated by dotted lines.
Enzymes involved in specialized metabolism are thought to have emerged through duplication, beginning with sophisticated primary enzymes as progenitors, followed by mutational changes in the duplicates and leading to neofunctionalization of the enzymes (Figure 4iii). Relaxed constraints on the specificities and efficiencies of newly generated duplicates allow them to explore a wide range of catalytic possibilities. The promiscuity of multifunctional enzymes, with broader specificities emerging through this process, contributes to the expansion of the metabolic web (Figure 4iii). This web even includes the virtual activities of hidden enzymes (dotted lines in Figure 4) that do not contribute to actual metabolic flow due to limited substrate availability or marginal enzymatic activity; such hidden activities are not readily eliminated by (and are more tolerant to) selection. If these changes are not deleterious or neutral and are thus not eliminated by purifying selection, the metabolic grids continue to build through neutral evolutionary processes such as genetic drift. It seems reasonable that autotrophic plants accumulate low-molecular-weight metabolites derived from photosynthetic assimilates, which usually have antioxidant properties to some extent and are often sequestered in cellular compartments such as vacuoles. These natural products, including those that accumulate in trace amounts, do not necessarily have adaptive significance (Koonnin, 2016).
The emergence of specialized pathways allowing for the efficient production and accumulation of substantial amounts of metabolites requires the selection of specific flows from the expanded metabolic web, again increasing order and efficiency (Figure 4iv). This process largely relies on positive natural selection rather than neutral evolution, which is dependent on randomness. I propose a conceptual model, recruitment model of metabolic evolution, describing this process. According to this model, structural genes are repeatedly recruited into regulons under the control of evolutionarily conserved transcription factors (which should be activators rather than repressors), such as the JA-responsive ERFs and bHLHs (Figure 5). When a gene in the metabolic web becomes regulated by a transcription factor, obtaining cognate cis-regulatory elements, metabolic flows are generated or altered accordingly. Although such events readily occur at high frequency, most of these mutational changes are immediately eliminated and are not maintained in the population. On the other hand, when the newly generated flows result in the accumulation of beneficial products, such as defense compounds, conferring adaptive advantages to the plant, the probability that such changes will be maintained and eventually fixed in the population increases tremendously. Once the beneficial flows occur, the likelihood that mutational events that enhance these flows (such as the transcriptional activation of other metabolic genes and the optimization of catalytic specificities and efficiencies associated with the flows) is expected to rise markedly as well. An initial, mostly accidental, event creating new metabolic flows may trigger cascading mutational changes associated with and improving the flows, eventually leading to the establishment of metabolic regulons and pathways, perhaps within a relatively short evolutionary timescale. The bioinformatic and mathematical bases of the model remain to be explored.
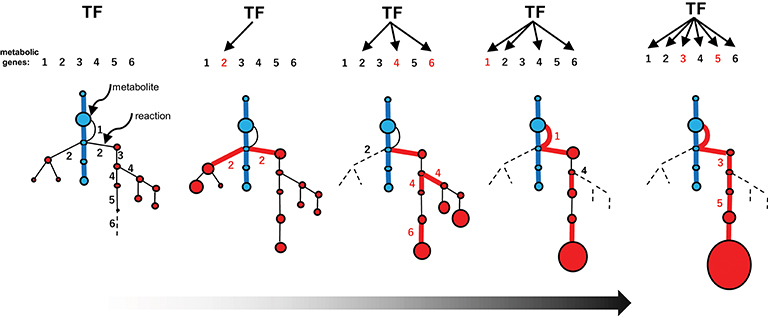
Figure 5. Recruitment model of metabolic evolution. The emergence of an efficient specialized pathway (red lines) from an expanded metabolic web, including the promiscuous steps (part of the web shown in Figure 4iii), is depicted schematically. Metabolic genes and the corresponding steps are numbered (1–6). The recruitment of metabolic genes and enzymes into the regulon under the control of a conserved transcription factor (TF) occurred repeatedly (red numbers). The catalytic specificity of promiscuous enzymes 2 and 4 was optimized (black numbers in the web). The recruitment of genes affecting metabolic flows in the web (which increase the possibility of recruiting other genes into the regulon) and catalytic optimization further enhance favorable flows once established.
The extensive rewiring of transcriptional circuits alters metabolic regulons that were once established under the original transcription factors (Figure 6). During such processes, takeover of the regulons by new transcription factors (including those derived from the original transcription factors by duplication) and the associated changes in the connections in the circuits occur frequently through changes in cis-regulatory elements and transcription factors (Figure 6, Johnson, 2017). Contemporary regulatory networks are often complex and include multiple transcription factors, which act as either activators or repressors, and in some cases regulate only specific parts of pathways (e.g., ORCA3 regulates some but not all TIA genes). Extensive rewiring of circuits may contribute to the advent of the complicated regulatory organization found in extant metabolic pathways, which also could account for the fact that few metabolic pathways have simple regulons comprising only a single master regulator and its downstream structural genes.
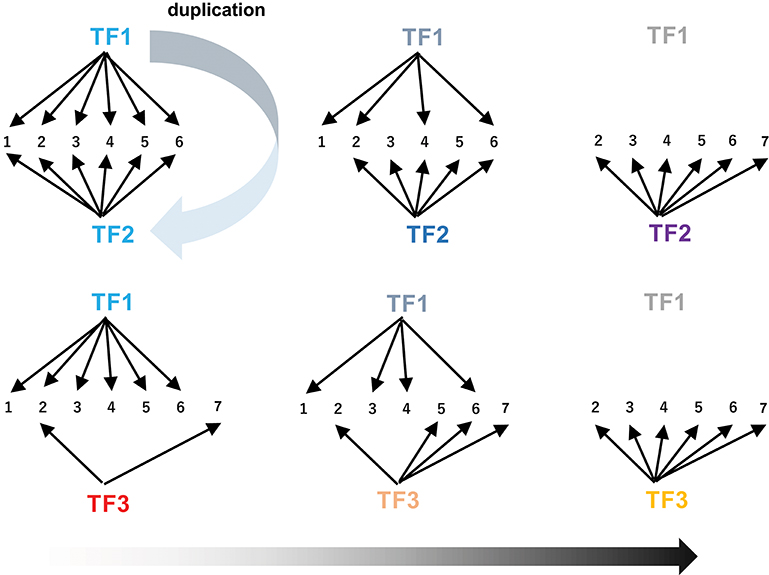
Figure 6. Rewiring of transcriptional circuits. An original regulon (including genes 1 to 6) controlled by TF1 is hijacked by other transcription factors (TF2 and TF3) and eventually modified (removal of 1 and addition of 7). TF2 arose from TF1 through gene duplication. The colors of the TFs reflect functional changes in these proteins during evolution.
Analyses of metabolic evolution have emphasized mutational changes to catalytic enzymes (Weng et al., 2012; Moghe and Last, 2015). If metabolic genes are not functionally expressed and no flows are associated with these genes, they are not subjected to positive or purifying selection. The recruitment model, which presumes that transcription factors activate genes prior to changes in catalytic activity, adequately addresses this point as well.
Metabolic evolution is driven by functional changes in catalytic enzymes and changes in the expression patterns of metabolic genes. There appear to be limits to the changes made to the catalytic functionalities of enzymes belonging to a limited number of protein families without hampering the structural and functional stability of protein frames. Therefore, changes in the combinations of metabolic genes with specific temporal and spatial expression patterns might also have significantly contributed to the rise in chemodiversity found in plants. Plants have exploited their limited repertoire of enzymes in a combinatorial manner to produce these diverse compounds.
Perspectives of “Evo-Meta” Biology
“Nothing in biology makes sense except in the light of evolution” is a famous quotation by Dr. Theodosius Dobzhansky, a Ukrainian-American evolutionary geneticist (Dobzhansky, 1973). The discovery of homeotic genes encoding a group of transcription factors that direct the organization of the body plans of vertebrates and invertebrates has helped elucidate the evolutionarily conserved mechanisms governing development, leading to the rise of Evolutionary Developmental (Evo-Devo) biology. Classic anatomy and embryology, as well as modern developmental biology, share some affinity with evolutionary biology. Paleontology based on fossil evidence is one of the main areas of focus in evolutionary biology.
Chemodiversity of specialized products in plants has been shaped through (and is a product of) evolution. Unfortunately, it is difficult to predict the hues and fragrances of ancient flowers from extinct plants. Nevertheless, the chemodiversity found in extant species and the diverse series of plant genomes, including those yet to be explored, is highly informative (Nakamura et al., 2014). A long period of collection of natural products with divergent chemical structures and biological activities, along with a better understanding of the grouping of biosynthetic pathways and associated enzymes, has led to an awareness of some sort of order behind this chemodiversity. Elucidating the molecular biology behind regulatory factors, such as master transcription factors, that orchestrate these metabolic processes is expected to reveal the universal principles ruling the metabolic processes that produce a diverse range of specialized products. The challenges of “Evo-Meta” (Evolutionary Metabolic) biology aimed at uncovering the origins of this chemodiversity are just beginning.
Author Contributions
The author confirms being the sole contributor of this work and has approved it for publication.
Funding
This work was supported in part by the Japan Society for the Promotion of Science (Grants-in-Aids for Scientific Research number 17K07447 to TS).
Conflict of Interest Statement
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
I thank Drs. Takashi Hashimoto and Yasuyuki Yamada (NAIST) for their long-term collaboration and continuous encouragement. I appreciate the encouraging and constructive remarks by Dr. Shigetada Nakanishi (SUNBOR) on our research related to this article.
References
Allen, M. D., Yamasaki, K., Ohme-Takagi, M., Tateno, M., and Suzuki, M. (1998). A novel mode of DNA recognition by a β-sheet revealed by the solution structure of the GCC-box binding domain in complex with DNA. EMBO J. 18, 5484–5496. doi: 10.1093/emboj/17.18.5484
Cárdenas, P. D., Sonawane, P. D., Heinig, U., Bocobza, S. E., Burdman, S., and Aharoni, A. (2015). The bitter side of the nightshades: genomics drives discovery in Solanaceae steroidal alkaloid metabolism. Phytochemistry 113, 24–32. doi: 10.1016/j.phytochem.2014.12.010
Cárdenas, P. D., Sonawane, P. D., Pollier, J., Bossche, R. V., Dewangan, V., Weithorn, E., et al. (2016). GAME9 regulates the biosynthesis of steroidal alkaloids and upstream isoprenoids in the plant mevalonate pathway. Nat. Commun. 7:10657. doi: 10.1038/ncomms10654
Chezem, W. R., and Clay, N. K. (2016). Regulation of plant secondary metabolism and associated specialized cell development by MYBs and bHLHs. Phytochemistry 131, 26–43. doi: 10.1016/j.phytochem.2016.08.006
Cui, Y., Chen, C. L., Cui, M., Zhou, W. J., Wu, H. L., and Ling, H. Q. (2018). Four IVa bHLH transcription factors are novel interactors of FIT and mediate JA inhibition of iron uptake in Arabidopsis. Mol. Plant 11, 1166–1183. doi: 10.1016/j.molp.2018.06.005
De Boer, K., Tillemann, S., Pauwels, L., Vanden Bossches, R., De Sutter, V., Vanderhaeghen, R., et al. (2011). APETALA2/ETHYENE RESPONSE FACTOR and basic helix-loop-helix tobacco transcription factors cooperatively mediates jasmonate-elicited nicotine biosynthesis. Plant J. 66, 1053–1065. doi: 10.1111/j.1365-313X.2011.04566.x
Dewey, R. E., and Xie, J. (2013). Molecular genetics of alkaloid biosynthesis in Nicotiana tabacum. Phytochemistry 94, 10–27. doi: 10.1016/j.phytochem.2013.06.002
Dobzhansky, T. (1973). Nothing in biology makes sense except in the light of evolution. Am. Biol. Teach. 35, 125–129. doi: 10.2307/4444260
Feller, A., Machemer, K., Braun, E. L., and Grotewold, E. (2011). Evolutionary and comparative analysis of MYB and bHLH plant transcription factors. Plant J. 66, 94–116. doi: 10.1111/j.1365-313X.2010.04459.x
Goossens, J., Fernández-Calvo, P., Schweizer, F., and Goossens, A. (2016a). Jasmonates: signal transduction components and their roles in environmental stress responses. Plant Mol. Biol. 91, 673–689. doi: 10.1007/s11103-016-0480-9
Goossens, J., Mertens, J., and Goossens, A. (2016b). Role and functioning of bHLH transcription factors in jasmonate signaling. J. Exp. Bot. 68, 1333–1347. doi: 10.1093/jxb/erw440
Hardigan, M. A., Laimbeer, F. P. E., Newton, L., Crisovan, E., Hamilton, J. P., Vaillancourt, B., et al. (2017). Genome diversity of tuber-bearing Solanum uncovers complex evolutionary history and targets of domestication in the cultivated potato. Proc. Natl. Acad. Sci. U.S.A. 114, E9999–E10008. doi: 10.1073/pnas.1714380114
Hayashi, H., and Sudo, H. (2009). Economic importance of licorice. Plant Biotechnol. 26, 101–104. doi: 10.5511/plantbiotechnology.26.101
Heim, M. A., Jakoby, M., Werber, M., Martin, C., Weisshaar, B., and Bailey, P. C. (2003). The basic helix-loop-helix transcription factor family in plants: a genome-wide study of protein structure and functional diversity. Mol. Biol. Evo. 20, 735–747. doi: 10.1093/molbev/msg088
Jarvis, D. E., Ho, Y. S., Lightfoot, D. J., Schöckel, S. M., Li, B., Borm, T. J. A., et al. (2017). The genome of Chenopodium quinoa. Nature 542, 307–312. doi: 10.1038/nature21370
Johnson, A. D. (2017). The rewiring of transcription circuits in evolution. Curr. Opin. Genet. Dev. 47, 121–127. doi: 10.1016/j.gde.2017.09.004
Kajikawa, M., Sierro, N., Kawaguchi, H., Bakaher, N., Ivanov, N. V., Hashimoto, T., et al. (2017). Genomic insights into the evolution of the nicotine biosynthesis pathway in tobacco. Plant Physiol. 174, 999–1011. doi: 10.1104/pp.17.00070
Koonnin, E. V. (2016). Splendor and misery of adaptation, or the importance of neutral null for understanding evolution. BMC Biol. 14:114. doi: 10.1186/s12915-016-0338-2
Kuljanabhagavad, T., Thongphasuk, P., Chamulitrat, W., and Wink, M. (2008). Triperpene saponins from Chenopodium qunoa Willd. Phytochemistry 69, 1919–1926. doi: 10.1016/j.phytochem.2008.03.001
Kumar, S., Stecher, G., Li, M., Knyaz, C., and Tamura, K. (2018). MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 35, 1547–1549. doi: 10.1093/molbev/msy096
Lee, S. J., Park, J. H., Lee, M. H., Yu, J. H., and Kim, S. Y. (2010). Isolation and functional characterization of CE1 binding proteins. BMC Plant Biol. 10:277. doi: 10.1186/1471-2229-10-277
Li, C. Y., Leopold, A. L., Sander, G. W., Shanks, J. V., Zhao, L., and Gibson, S. I. (2013). The ORCA2 transcription factor plays a key role in regulation of the terpenoid indole alkaloid pathway. BMC Plant Biol. 13:155. doi: 10.1186/1471-2229-13-155
Lu, X., Zhang, L., Zhang, F., Jiang, W., Shen, Q., Zhang, L., et al. (2013). AaORC, a trichome-specific AP2/ERF transcription factor of Artemisia annua, is a positive regulator in the artemisinin biosynthetic pathway and in disease resistance to Botrytis cinerea. New Phytol. 198, 1191–1202. doi: 10.1111/nph.12207
Lv, Z., Zhang, L., and Tang, K. (2017). New insights into artemisinin regulation. Plant Signal. Behav. 12:e1366398. doi: 10.1080/15592324.2017.1366398
Maerkl, S. J., and Quake, S. R. (2009). Experimantal determination of the evolvability of a transscription factor. Proc. Natl. Acad. Sci. U.S.A. 106, 18650–18655. doi: 10.1073/pnas.0907688106
Matsushima, R., Fukao, Y., Nishimura, M., and Hara-Nishimura, I. (2004). NAI1 gene encodes a basic helix-loop-helix-type putative transcription factor that regulates the formation of an endoplasmic reticulum-derived structure, the ER body. Plant Cell 16, 1536–1549. doi: 10.1105/tpc.021154
Menke, F. L. H., Champion, A., Kijne, J. W., and Memelinkm, J. (1999). A novel jasmonate- and elicitor-responsive element in the periwinkle secondary metabolite biosynthetic gene Str interacts with a jasmonate- and elicitor-inducible AP2-domain transcription factor, ORCA2. EMBO J. 16, 4455–4463. doi: 10.1093/emboj/18.16.4455
Mertens, J., Pollier, J., Vanden Bosshe, R., Lopez-Vidriero, I., Franco-Zorrilla, J. M., and Goossens, A. (2016a). The bHLH transcription factors TSAR1 and TSAR2 regulate triterpene saponin biosynthesis in Medicago truncatula. Plant Physiol. 170, 194–210. doi: 10.1104/pp.15.01645
Mertens, J., Van Moerkercke, A., Vanden Bosshe, R., Pollier, J., and Goossens, A. (2016b). Clade IVa basic helix-loop-helix transcription factors form part of a conserved jasmonate signaling circuit for the regulation of bioactive plant terpenoid biosynthesis. Plant Cell Physiol. 57, 2564–2575. doi: 10.1093/pcp/pcw168
Moghe, G. D., and Last, R. L. (2015). Something old, something new: conserved enzymes and the evolution of novelity in plant specialized metabolism. Plant Physiol. 169, 1512–1523. doi: 10.1104/pp.15.00994
Nakamura, Y., Afendi, F. M., Parvin, A. K., Ono, N., Tanaka, K., Hirai, A., et al. (2014). KNApSAcK metabolite activity database for retrieing the relationships between metabolites and biological activities. Plant Cell Physiol. 55:e7. doi: 10.1093/pcp/pct176
Nakano, R. T., Piślewska-Bednarek, M., Yamada, K., Edger, P. P., Miyahara, M., Kondo, M., et al. (2017). PYK10 myrosinase reveals a functional coordination between endoplasmic reticulum bodies and glucosinolates in Arabidopsis thaliana. Plant J. 89, 204–210. doi: 10.1111/tpj.13377
Nakano, R. T., Yamada, K., Bednarek, P., Nishimura, M., and Hara-Nishimura, I. (2014). ER bodies in plants of the Brassicales order: biogenesis and association with innate immunity. Front. Plant Sci. 5:73. doi: 10.3389/fpls.2014.00073
Nakano, T., Suzuki, K., Fujimura, T., and Shinshi, H. (2006). Genome-wide analysis of ERF gene family in Arabidopsis and rice. Plant Physiol. 140, 411–432. doi: 10.1104/pp.105.073783
Nakayasu, M., Shioya, N., Shikata, M., Thagun, C., Abdelkareem, A., Okabe, Y., et al. (2018). JRE4 is a master transcriptional regulator of defense-related steroidal glycoalkaloids in tomato. Plant J. 94, 975–990. doi: 10.1111/tpj.13911
Nemoto, K., Takemori, N., Seki, M., Shinozaki, K., and Sawasaki, T. (2015). Members of the plant CRK superfamily are capable of trans- and autophosphorylation of tyrosine residues. J. Biol. Chem. 290, 16665–16677. doi: 10.1074/jbc.M114.617274
Olofsson, L., Engstrom, A., Lundgren, A., and Brodelius, P. E. (2011). Relative expression of genes of terpene metabolism in different tissues of Artemisia annua L. BMC Plant Biol. 11:45. doi: 10.1186/1471-2229-11-45
Osbourn, A., Goss, R. J., and Field, R. A. (2011). The saponins: polar isoprenoids with important and diverse biological activities. Nat. Prod. Rep. 28, 1261–1268. doi: 10.1039/c1np00015b
Patra, B., Schluttenhofer, C., Wu, Y., Pattanaik, S., and Yuan, L. (2013). Transcriptional regulation of secondary metabolite biosynthesis in plants. Biochim. Biophys. Acta 1929, 1236–1247. doi: 10.1016/j.bbagrm.2013.09.006
Paul, P., Singh, S. K., Patra, B., Sui, X., Pattanaik, S., and Yuan, L. (2017). A differentially regulated AP2/ERF transcription factor gene cluster acts downstream of a MAP kinase cascade to modulate terpenoid indole alkaloid biosynthesis in Catharanthus roseus. New Phytol. 213, 1107–1123. doi: 10.1111/nph.14252
Schweizer, F., Bodenhausen, N., Lassueur, S., Masclaux, F. G., and Reymond, F. (2013). Differential contribution of transcription factors to Arabidopsis thaliana defense against Spodoptera littoralis. Front. Plant Sci. 4:13. doi: 10.3389/fpls.2013.00013
Sheard, L. B., Tan, X., Mao, H., Withers, J., Ben-Nissan, G., Hinds, T. R., et al. (2010). Jasmonate perception by inositol-phosphate-potentiated COI1-JAZ co-receptor. Nature 468, 400–405. doi: 10.1038/nature09430
Shoji, T., and Hashimoto, T. (2011a). Recruitment of a duplicated primary metabolism genes into the nicotine biosynthesis regulon in tobacco. Plant J. 67, 949–959. doi: 10.1111/j.1365-313X.2011.04647.x
Shoji, T., and Hashimoto, T. (2011b). Tobacco MYC2 regulates jasmonate-inducible nicotine biosynthesis genes directly and by the way of the NIC2-locus ERF genes. Plant Cell Physiol. 52, 1117–1130. doi: 10.1093/pcp/pcr063
Shoji, T., and Hashimoto, T. (2015). Stress-induced expression of NICOTINE2-locus genes and their homologs encoding Ethylene Response Factor transcription factors in tobacco. Phytochemistry 113, 41–49. doi: 10.1016/j.phytochem.2014.05.017
Shoji, T., and Hashimoto, T. (2019). Expression of a tobacco nicotine biosynthesis gene depends on the JRE4 transcription factor in heterogeneous tomato. J. Plant Res. 132, 173–180. doi: 10.1007/s10265-018-1075-0
Shoji, T., Kajikawa, M., and Hashimoto, T. (2010). Clustered transcription factor genes regulate nicotine biosynthesis in tobacco. Plant Cell 22, 3390–3409. doi: 10.1105/tpc.110.078543
Shoji, T., Mishima, M., and Hashimoto, T. (2013). Divergent DNA-binding specificities of a group of Ethylene Response Factor transcription factors involved in plant defense. Plant Physiol. 162, 977–990. doi: 10.1104/pp.113.217455
Shoji, T., Nakajima, K., and Hashimoto, T. (2000). Ethylene suppresses jasmonate-induced gene expression in nicotine biosynthesis. Plant Cell Physiol. 41:1072–1076. doi: 10.1093/pcp/pcd027
Sirikantaramas, S., Asano, T., Sudo, H., Yamazaki, M., and Saito, K. (2007). Camptothecin: therapeutic potential and biotechnology. Curr. Pharm. Biotechnol. 8, 196–202. doi: 10.2174/138920107781387447
Sogabe, Y., Nakamura, H., Nakagawa, T., Hasegawa, S., Asano, T., Ohta, H., et al. (2011). Plant Signal. Behaviour 6, 1037–1039. doi: 10.4161/psb.6.7.15602
Srivastava, S., Chaudhry, V., Mishra, A., Chauhan, P. S., Rehman, A., Yadav, A., et al. (2012). Gene expression profiling through microarray analysis in Arabidopsis thaliana colonized by Pseudomonas putida MTC5279, a plant growth promoting rhizobacterium. Plant Signal. Behav. 7, 235–245. doi: 10.4161/psb.18957
Swinnen, G., Goossens, A., and Pauwels, L. (2016). Lessons from domestication: targeting cis-regulatory elements for crop improvement. Trends Plant Sci. 21, 506–515. doi: 10.1016/j.tplants.2016.01.014
Tamura, K., Yoshida, K., Hiraoka, Y., Sakaguchi, D., Chikugo, A., Mochida, K., et al. (2018). The basic helix-loop-helix transcription factor GubHLH3 positively regulates soyasaponin biosynthetic genes in Glycyrrhiza uralensis. Plant Cell Physiol. 59, 783–796. doi: 10.1093/pcp/pcy046
Tang, K., Shen, Q., Yan, T., and Fu, X. (2014). Transgenic approach to increase artemisinin content in Artemisia annua L. Plant Cell Rep. 33, 605–615. doi: 10.1007/s00299-014-1566-y
Thagun, C., Imanishi, S., Kudo, T., Nakabayashi, R., Ohyama, K., Mori, T., et al. (2016). Jasmonate-responsive ERF transcription factors regulate steroidal glycoalkaloid biosynthesis in tomato. Plant Cell Physiol. 57, 961–975. doi: 10.1093/pcp/pcw067
Thines, B., Katsir, L., Melotto, M., Niu, Y., Mandaokar, A., Liu, G., et al. (2007). JAZ repressor proteins are targets of the SCF(COI1). complex during jasmonate signalling. Nature 448, 661–665. doi: 10.1038/nature05960
Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680. doi: 10.1093/nar/22.22.4673
Udomsom, N., Rai, A., Suzuki, H., Okuyama, J., Imai, R., Mori, T., et al. (2016). Function of AP2/ERF transcription factors involved in the regulation of specialized metabolism in Ophiorrhiza pumila revealed by transcriptomics and metabolomics. Front Plant Sci. 7:1861. doi: 10.3389/fpls.2016.01861
Van der Fits, L., and Memelink, J. (2000). ORCA3, a jasmonate-responsive transcriptional regulator of plant primary and secondary metabolism. Science 289, 295–297. doi: 10.1126/science.289.5477.295
Van Moerkercke, A., Steensma, P., Gariboldi, I., Espoz, J., Purnama, P. C., Schweizer, F., et al. (2016). The basic helix-loop-helix transcription factor BIS2 is essential for monoterpenoid indole alkaloid production in medicinal plant Catharanthus roseus. Plant J. 88, 3–12. doi: 10.1111/tpj.13230
Van Moerkercke, A., Steensma, P., Schweizer, F., Pollier, J., Gariboldi, I., Payne, R., et al. (2015). The bHLH transcription factor BIS1 controls the iridoid branch of the monoterpenoid indole alkaloid pathway in Catharanthus roseus. Proc. Natl. Acad. Sci. U.S.A. 112, 8130–8135. doi: 10.1073/pnas.1504951112
Weng, J. K., Philippe, R. N., and Noel, J. P. (2012). The rise of chemodiversity in plants. Science 336, 1667–1670. doi: 10.1126/science.1217411
Wray, G. A. (2007). The evolutionary significance of cis-regulatory elements. Nat. Rev. Genet. 8, 206–216. doi: 10.1038/nrg2063
Yonekura-Sakakibara, K., and Saito, K. (2013). Transcriptome coexpression analysis using ATTED-II for integrated transcriptomic/metabolomics analysis. Methods Mol. Biol. 1011, 317–326. doi: 10.1007/978-1-62703-414-2_25
Zhang, F., Yao, J., Ke, J., Zhang, L., Lam, V. Q., Xin, X. F., et al. (2015). Structural basis of JAZ repression of MYC transcription factors in jasmonate signalling. Nature 525, 269–273. doi: 10.1038/nature14661
Zhang, H., Hedhill, S., Montiel, G., Zhang, Y., Chatel, G., Pré, M., et al. (2011). The basic helix-loop-helix transcription factor CrMYC2 controls the jasmonate-responsive expression of the ORCA genes that regulate alkaloid biosynthesis in Catharanthus roseus. Plant J. 67, 61–71. doi: 10.1111/j.1365-313X.2011.04575.x
Zhang, H. B., Bokowiec, M. T., Rushton, P. J., Han, S. C., and Timko, M. P. (2012). Tobacco transcription factors NtMYC2a and NtMYC2b form nuclear complexes with the NtJAZ1 repressor and regulate multiple jasmonate-inducible steps in nicotine biosynthesis. Mol. Plant 5, 73–84. doi: 10.1093/mp/ssr056
Zhou, M., and Memelink, J. (2016). Jasmonate-responsive transcription factors regulating plant secondary metabolism. Biotechnol. Adv. 34, 441–449. doi: 10.1016/j.biotechadv.2016.02.004
Zhu, G., Wang, S., Huang, Z., Zhang, S., Liao, Q., Zhang, C., et al. (2018). Rewiring of the fruit metabolome in tomato breeding. Cell 172, 249–261. doi: 10.1016/j.cell.2017.12.019
Keywords: alkaloids, cis-regulatory element, jasmonates, recruitment model of metabolic evolution, regulon, specialized metabolism, terpenoids, transcription factor
Citation: Shoji T (2019) The Recruitment Model of Metabolic Evolution: Jasmonate-Responsive Transcription Factors and a Conceptual Model for the Evolution of Metabolic Pathways. Front. Plant Sci. 10:560. doi: 10.3389/fpls.2019.00560
Received: 21 November 2018; Accepted: 12 April 2019;
Published: 14 May 2019.
Edited by:
Philipp Zerbe, University of California, Davis, United StatesReviewed by:
Justin William Walley, Iowa State University, United StatesNobutaka Mitsuda, National Institute of Advanced Industrial Science and Technology (AIST), Japan
Jacob Pollier, Flanders Institute for Biotechnology, Belgium
Copyright © 2019 Shoji. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Tsubasa Shoji, dC1zaG91amlAYnMubmFpc3QuanA=