 Xiao Du
Xiao Du Xiao Zhang
Xiao Zhang Haidong Bu
Haidong Bu Ticao Zhang
Ticao Zhang Yongchun Lao1
Yongchun Lao1 Wenxuan Dong
Wenxuan Dong
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 09 April 2019
Sec. Plant Systematics and Evolution
Volume 10 - 2019 | https://doi.org/10.3389/fpls.2019.00443
Hawthorn is of high economic value owing to its medicinal properties and health benefits. Crataegus is a member of the Rosaceae family; the genus has a complicated taxonomic history, and several theories on its origin have been proposed. In this study, 53 accessions from seven Crataegus taxa native to China and accessions of exotic Crataegus species (two from Europe and one from North America) were analyzed by specific locus amplified fragment sequencing (SLAF-seq). In total, 933,450 single-nucleotide polymorphisms were identified after filtering and used to investigate the species’ genomic evolution. Phylogenetic trees derived from nuclear simple sequence repeats (SSRs) and SLAF-seq data showed the same topology, in which Crataegus maximowiczii and Crataegus sanguineae formed a closely related cluster that was clearly separated from the cluster composed of Crataegus hupehensis, Crataegus pinnatifida, Crataegus pinnatifida var. major, Crataegus bretschneideri and Crataegus scabrifolia. Phylogenetic and structure analysis indicated that the seven Chinese Crataegus taxa had two separate speciation events. Plants that evolved the southwestern route shared the genepool with the European species, whereas plants along the northeastern route shared the genepool with the North American species. TreeMix genetic analysis revealed that C. bretschneideri may have a hybrid origin. This study provides valuable information on the origins of Chinese Crataegus and suggests an evolutionary model for the main Crataegus species that native to China.
The genus Crataegus (hawthorn), a member of the Rosaceae family, ranges from small shrubs to trees distributed in Eurasia and America (Phipps et al., 1990). Hawthorns are among the most economically important plant species in China, owing to their pleasant flavor, attractive color, and nutrient-rich fruit (Xu et al., 2016). Hawthorn use in preventive medicine dates to the late 1800s. Hawthorns contain biologically active compounds, such as flavonoids, phenols, and oligomeric procyanidins, which have therapeutic benefits (Dickinson et al., 2014; Dahmer and Scott, 2018). Previous laboratory tests and clinical trials have demonstrated the efficacy of hawthorn in the treatment and prevention of cardiovascular disease (Edwards et al., 2012).
On the basis of cladistics analyses of morphological data, Phipps (1990) suggested southwest China and Mexico were ancestral areas for the genus and that trans-Beringian migration of Asian and American Crataegus had occurred. Crataegus was postulated to have migrated westward from southwest China to Europe, and eastward from East Asia to North America. However, other authors hold conflicting opinions. Evans and Campbell (2002) treated Crataegus as subtribe Pyrinae, and based on molecular and non-molecular characters, suggested that Crataegus originated in North America. Lo et al. (2009) used sequences for the internal transcribed spacer region, chloroplast DNA regions, LEAFY intron2 to infer relationships among species from eastern Asia, western North America, eastern North America, and Europe; their findings indicated that eastern North America and Europe are probably the most recent common areas of origin for Crataegus.
China is the center of Crataegus cultivation, and the place of origin of both cultivated and some wild Crataegus species. Based on morphological characters, some researchers have proposed that 18 species and six varieties of Crataegus are widely distributed across China (Zhao and Feng, 1996; Xin and Zhang, 1997), other authors recognize 20 species of Chinese Crataegus, with seven varieties (Dong and Li, 2015). Among these species, C. hupehensis, C. pinnatifida var. major, C. bretschneideri, and C. scabrifolia are cultivated. Previous efforts to understand the phylogenetic and biogeographic history of Crataegus have included representatives of Chinese Crataegus. Phipps (1990) suggested that C. scabrifolia evolved into European Crataegus and other Chinese Crataegus species (C. pinnatifida, C. hupehensis, and C. sanguineae). Based on sequence data from 14 plastid loci, Zarrei et al. (2015) treated C. maximowiczii as section Sanguineae and suggested that the origin of the section involved east-to-west trans-Beringian migration from western North America into eastern Asia. Lo et al. (2009) suggested that ancestors of C. hupehensis, C. songorica, and C. pinnatifida dispersed from Europe into Asia. However, a consensus is lacking on the migration direction of Chinese and European Crataegus.
Recent studies have examined intraspecific (Wu et al., 2008; Zhang et al., 2008; Sheng et al., 2017) and interspecific relationships (Su et al., 2015; Ma and Lu, 2016) of Chinese Crataegus. Previous investigations have used morphological data analyses and limited molecular data, but no study has explored the origin and evolution of cultivated Crataegus and related species that are native to China at the genomic level. Plant DNA contains abundant genetic information, and an increasing number of researchers have explored interspecific relationships and diversification of plants using molecular marker information. Molecular markers are used to determine genetic relationships within plant populations with almost 100% reliability (Güney et al., 2018), and random amplified repeats (Erfani-Moghadam et al., 2016; Zarei et al., 2017), inter-simple sequence repeats (Sheng et al., 2017; Emami et al., 2018), and SSRs (Lo et al., 2009; Khiari et al., 2015; Brown et al., 2016) have been widely used for genetic characterization of Crataegus, and to analyze genetic diversity among and within accessions of Crataegus. Of these molecular markers types, SSR markers have attained considerable popularity in genetic research because they are highly polymorphic, convenient, and codominant. Based on double barcode genotyping systems and sequencing, specific-locus amplified fragment sequencing (SLAF-seq) was developed for de novo single nucleotide polymorphism (SNP) discovery and genotyping using reduced representation library sequencing. SLAF-seq is a high-throughput, high-accuracy, and low-cost method with short cycles, and has been used for molecular breeding and analysis of germplasm resources (Huang et al., 2016). SLAF-seq does not depend on a reference genome sequence and is particularly useful for species the lack an assembled reference genome (Sun et al., 2013), because it is possible to perform polymorphism analysis and develop molecular markers directly from the sequence data provided by SLAF-seq (Zheng et al., 2016). Given these advantages, SLAF-seq has been used for rapid mass discovery of SNP markers for polymorphism analysis, system evolution, and germplasm resource identification (Chen et al., 2013; Zhang et al., 2013; Xu et al., 2015).
In the present study, we used SLAF-seq to gain insight into evolutionary relationships among seven Crataegus taxa native to China, namely C. maximowiczii, C. sanguineae, C. hupehensis, C. pinnatifida, C. pinnatifida var. major, C. bretschneideri and C. scabrifolia. These taxa are widely distributed in China and cover most of the different climatic regions in the country. The sampled taxa include the four taxa cultivated in China (C. hupehensis, C. pinnatifida var. major, C. bretschneideri, and C. scabrifolia) and three species distributed in close proximity to cultivated Crataegus. C. maximowiczii and C. sanguineae are typically distributed in northeastern China. Of the cultivated taxa, C. pinnatifida var. major is endemic to China and has the longest history in cultivation, and C. scabrifolia is considered to be the ancestral Crataegus species. C. pinnatifida is a species that is widespread throughout China. The seven taxa were selected by other researchers as representatives of Chinese Crataegus in previous phylogenetic studies (e.g., C. pinnatifida, C. hupehensis, and C. sanguineae: Phipps, 1990; C. hupehensis, C. sanguineae, C. maximowiczii, and C. pinnatifida: Lo et al., 2009; C. maximowiczii and C. sanguineae: Zarrei et al., 2015). Previous studies provided useful information that partially resolved the phylogenetic history of cultivated Crataegus in China. However, the interspecific relationships and evolution of cultivated Crataegus in China remain unclear. In the present study, SLAF-seq was used to analyze phylogenetic relationships among 53 accessions of cultivated Crataegus and three related species in China based on SSRs and SNPs.
In total, 53 accessions of Chinese Crataegus were sampled, which consisted of C. maximowiczii (5 accessions), C. sanguineae (4 accessions), C. hupehensis (6 accessions), C. pinnatifida (14 accessions), C. pinnatifida var. major (12 accessions), C. bretschneideri (10 accessions) and C. scabrifolia (2 accessions). The outgroup comprised accessions of three exotic taxa collected from abroad (C. monogyna and C. laevigata from Europe, and C. cruss-galli from North America). The 53 Chinese accessions (Table 1) were broadly distributed across a range of geographic and climatic conditions, and thus were considered to be representative of Chinese hawthorn diversity and to encompass all possible introductory sources in China.
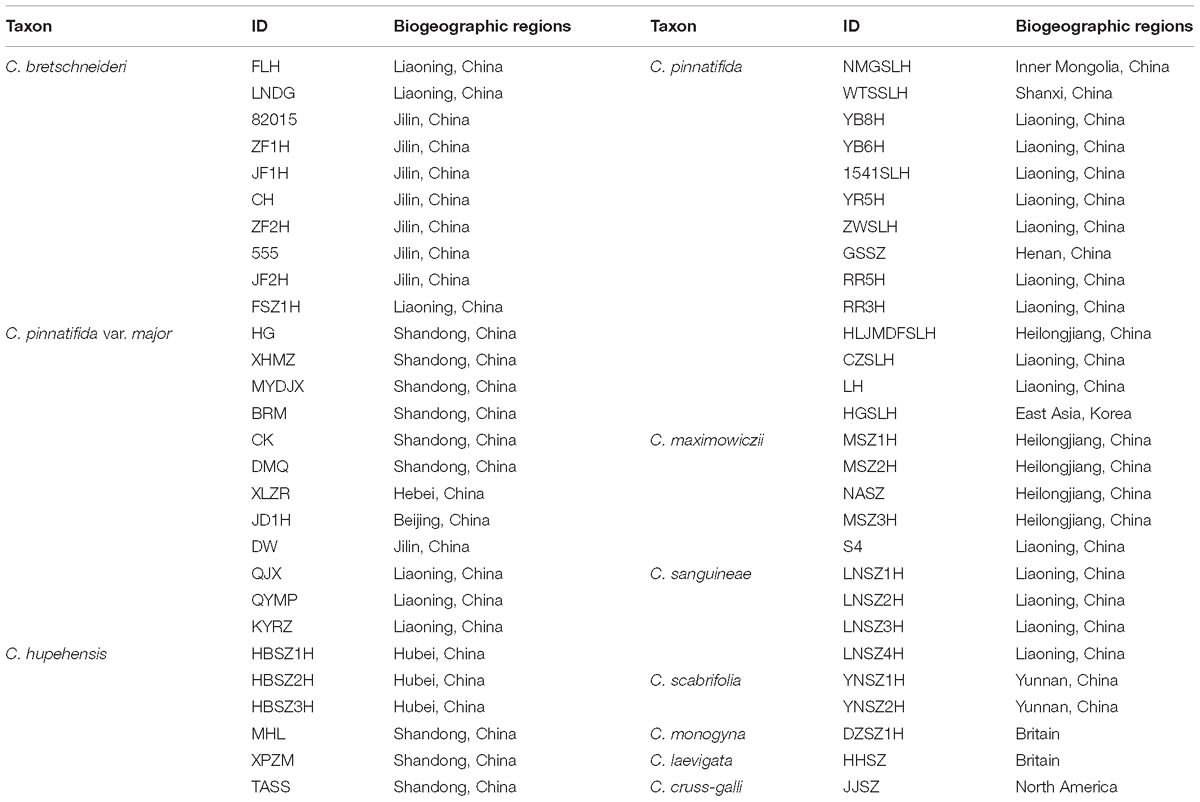
Table 1. Details of geographic and sampling information for Crataegus investigated in this study.
All sampled trees were maintained at the National Hawthorn Germplasm Repository at Shenyang Agricultural University, China (Supplementary Table S1).
Fresh leaves and fruits were sampled for morphological measurements when mature (from September to early October). Twenty-five leaves and fruits per accession were sampled. The measurement methods followed the technical code for evaluation crop germplasm resources for hawthorn (Crataegus L.) (Dong, 2013). Twenty-one characters, including leaf shape, leaf blade lobes, leaf blade margin, and leaf color, were measured. The qualitative trait characteristics and classifications are summarized in Supplementary Table S2.
Samples of young leaves were collected, labeled, frozen with liquid nitrogen, and stored at −80°C until DNA extraction. One gram of frozen leaf material was ground for genomic DNA extraction using cetyl-tri-methylammonium bromide in accordance with the protocol of Doyle and Doyle (1990). The DNA quality was checked using a Nanodrop-2000 spectrophotometer (Thermo Fisher Scientific, Wilmington, DE, United States).
Fifty-six samples were analyzed using nuclear SSR (nSSR) markers. PCR amplification was performed in 20 μl reaction mixture consisting of 1 μl template DNA (20–30 ng), 7 μl of 2× Es MasterMix buffer (CWBIO, Beijing, China), 2 μl primers (20 ng/μl), and 10 μl sterile nuclease-free distilled H2O. The PCR protocol was as follows: initial denaturation at 94°C for 5 min; 30 cycles at 94°C for 1 min, 56–59°C for 1 min, and 72°C for 1 min; and a final extension at 72°C for 7 min. PCR amplification was carried out in a Thermal Cycler (Applied Biosystems, Foster City, CA, United States). Genotyping was performed using 20 SSR primers (Supplementary Table S3) that were arbitrarily selected from hawthorn transcriptional data (Xu et al., 2016).
The PCR products were separated on a 6% polyacrylamide gel in 0.5× TBE buffer. After electrophoresis, the gel was stained as previously described (Cairns and Murray, 1994). The resulting bands were recorded in a presence–absence matrix in which values represented the presence (1) or absence (0) of a band. Genetic similarities based on Jaccard’s coefficients were calculated using the SIMQUAL program with the Numerical Taxonomy Multivariate Analysis System (NTSYS-PC) v.2.0. The matrix of genetic similarity coefficients was used to generate a dendrogram with the unweighted pair group method with arithmetic mean (UPGMA) method implemented in NTSYS-PC v.2.0 (Rohlf, 1997).
Library preparation and sequencing of SLAF-markers from genomic DNA were performed by the Biomarker Technology Company (Beijing, China). Owing to the lack of a reference genome, the Malus genome1 (GCA_ 002114115.1) was used for enzyme cutting site prediction. The following criteria were applied to determine the enzyme cutting scheme: (i) the smallest fragments in the repeat sequence, (ii) the fragments are distributed even in the genome, (iii) the length of fragments must be consistent with the scheme, and (iv) the number of SLAFs must meet expectations. After filtering, we used RsaI + HaeIII (NEB, Ipswich, MA, United States) for digestion, and the predicted target fragment length was 314–344 bp. Sequence data for the control, Oryza sativa subsp. japonica, was obtained from the Rice Annotation Project Database2 for quality control and to ensure the effectiveness of the enzyme cutting scheme. In accordance with the enzyme cutting scheme, the restriction enzymes digested the qualified DNA to obtain the SLAFs; then, the adenine nucleotide (A) was added to the 3′ end of the SLAFs, the dual-index adapter was ligated, and the extract was purified and submitted for pair-end sequencing using an Illumina HiseqTM 2500 platform (Illumina, Inc., San Diego, CA, United States). Dual-indexing was used to identify the original data from the sequence and obtain reads for the 56 samples. SNPs were called using the Burrows-Wheeler Alignment tool (BWA; Li and Durbin, 2009). Population polymorphism analysis was conducted using high-quality SNPs. Reads with clear index information were clustered based on sequence similarity.
A phylogenetic tree was constructed based on the SNPs using maximum likelihood (ML) analyses (Kobert et al., 2016). The ML analyses were performed using RaxML v.8.2.0 (Stamatakis, 2014). Principal component analysis (PCA) was performed using EIGENSOFT (Price et al., 2006). Population structure was investigated using STRUCTURE (Hubisz et al., 2009). To determine the most likely number of ancestral kinships (K) in the population, STRUCTURE was run 20 times for each K value from 3 to 10. We calculated ΔK, which indicates the change in likelihood of different numbers of clusters, and determined the cluster number with the highest ΔK value, which represents the most likely number of clusters in the population. We inferred admixture graphs using TreeMix v.1.12 (Pickrell and Pritchard, 2012). Divergence time estimation was conducted using PAML (Stadler and Yang, 2013) based on the SNPs data and fossil age. Fossils for calibration were selected from the TimeTree database3. The divergence times between species from the TimeTree database were extracted from all peer-reviewed publications in molecular evolution and phylogenetic that reported estimates of the time of divergence among species. A hierarchical average linkage method was used to estimate divergence times (Ts) of clade pairs to build a Super Time tree, together with a procedure for testing and updating topological partitions to ensure the highest degree of consistency with individual time trees in every study (Hedges et al., 2015).
The split time between C. pinnatifida-1541SLH and C. monogyna-DZ1H was searched in the database. The estimated divergence data (14.7 Ma) was set as the ancestral node time, which was then used to estimate species divergence times.
The leaf and fruit characteristics recorded are shown in Supplementary Table S2. The most obvious differences between species were in the leaf blade lobes and fruit size. Based on morphological characteristics (Figure 1), the seven Chinese Crataegus taxa were separated into the following four groups: (i) plants with shallowly dissected leaves and small fruit, which contained C. maximowiczii and C. sanguineae; (ii) plants with moderately lobed leaves and medium-sized fruit, which comprised C. pinnatifida; (iii) plants with deeply dissected leaves and large fruit, which consisted of C. pinnatifida var. major and C. bretschneideri; and (iv) plants with non-dissected or shallowly lobed leaves and large fruit, which comprised C. hupehensis and C. scabrifolia. The C. maximowiczii is the only species with have pubescence on the leaf.

Figure 1. Morphological illustration of the seven Crataegus species investigated. (A) C. bretschneideri – 82015, (B) C. hupehensis – HB1H, (C) C. maximowiczii – NASZ, (D) C. pinnatifida var. major – JD1H, (E) C. sanguineae – LN1H, (F) C. pinnatifida – HGSLH, (G) C. scabrifolia – YN1H.
The genetic relationships among the Crataegus taxa were determined based on a cluster analysis that used loci generated by the nSSR markers. Twenty microsatellite primer pairs were used for the nSSR analysis. Similarity values ranged from 0.49 to 0.96. Genotyping data for 48 alleles were obtained with the 20 primer pairs and used to construct a dendrogram with the UPGMA method (Supplementary Figure S1). The 56 accessions were well separated using the SSR markers.
The nSSR phylogenetic tree showed that C. monogyna and C. laevigata, which are from Europe, were entirely distinct from the other Crataegus species, and C. cruss-galli, which is from North America, formed a sister group with Crataegus from northeastern China. The seven Chinese Crataegus taxa were grouped into two major clusters; one cluster contained C. maximowiczii and C. sanguineae, whereas the other contained C. hupehensis, C. pinnatifida, C. pinnatifida var. major, C. bretschneideri, and C. scabrifolia. These two clusters were separated at a similarity value of approximately 0.65. Four accessions (ZWSLH, GSSZ, RR5H, and RR3H) were considered to belong to C. pinnatifida based on their morphological characteristics; however, in the nSSR dendrogram, these accessions showed higher similarity to C. maximowiczii and C. sanguineae.
In this study, all 56 samples were sequenced and 803,196 SLAF tags were developed in total. The average sequencing depth of the SLAF tag was 12.31-fold, and 164,397 SLAF tags were polymorphic. Fifty-six DNA libraries were sequenced using the SLAF-seq technique, which generated 157.75 M reads, with a mean Q30 of 91.83% and GC content of approximately 41.04% (Table 2).
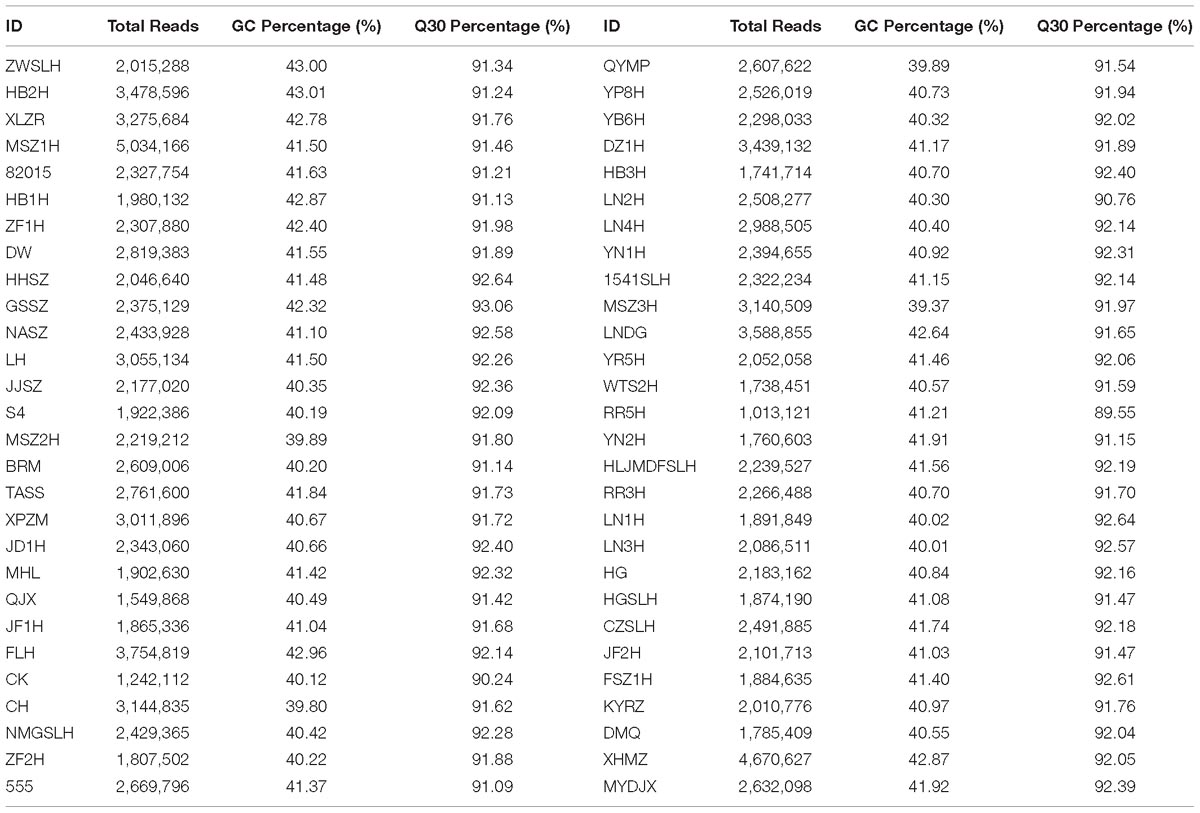
Table 2. Summary statistics for specific-locus amplified fragment sequence data.
The maximum depth sequence of every SLAF tag was accounted for as a reference; using BWA (Li and Durbin, 2009), the reads were aligned to the Malus genome, and SNPs were identified with GATK (McKenna et al., 2010) and SAMtools methods (Li et al., 2009). When both methods identified the same SNP, SNP identification was considered to be reliable. Thus, we identified 933,450 SNPs after filtering and used them to investigate genomic evolution among the 56 accessions.
We first constructed a ML phylogenetic tree based on the SNPs data. Given that the morphological characters divided the Chinese Crataegus taxa into four groups, based on the molecular data these four groups were predicted to form two clusters. The ML tree (Figure 2A) showed strong evidence for two clusters: cluster A was a northeastern group (C. maximowiczii and C. sanguineae) that was sister to all other Crataegus accessions; cluster B was a widely distributed group, with species ranging from southwestern to central, northern, eastern, and northeastern China (C. scabrifolia, C. hupehensis, C. pinnatifida, C. pinnatifida var. major, and C. bretschneideri). In cluster B, accessions of the southwestern Chinese species C. scabrifolia was indicated to be an early divergence in the cluster. The central Chinese, species C. hupehensis was indicated to be paraphyletic with the northern, eastern, and northeastern species nested within it. These two subclusters were well separated and therefore distantly related. The PCA (Figure 2B) suggested relationships in ordination space that were consistent with topology the A of the phylogenetic tree. C. maximowiczii and C. sanguineae accessions formed a closely related cluster that was distinctly separated from the cluster comprising the accessions of C. scabrifolia, C. hupehensis, C. pinnatifida, C. pinnatifida var. major, and C. bretschneideri.
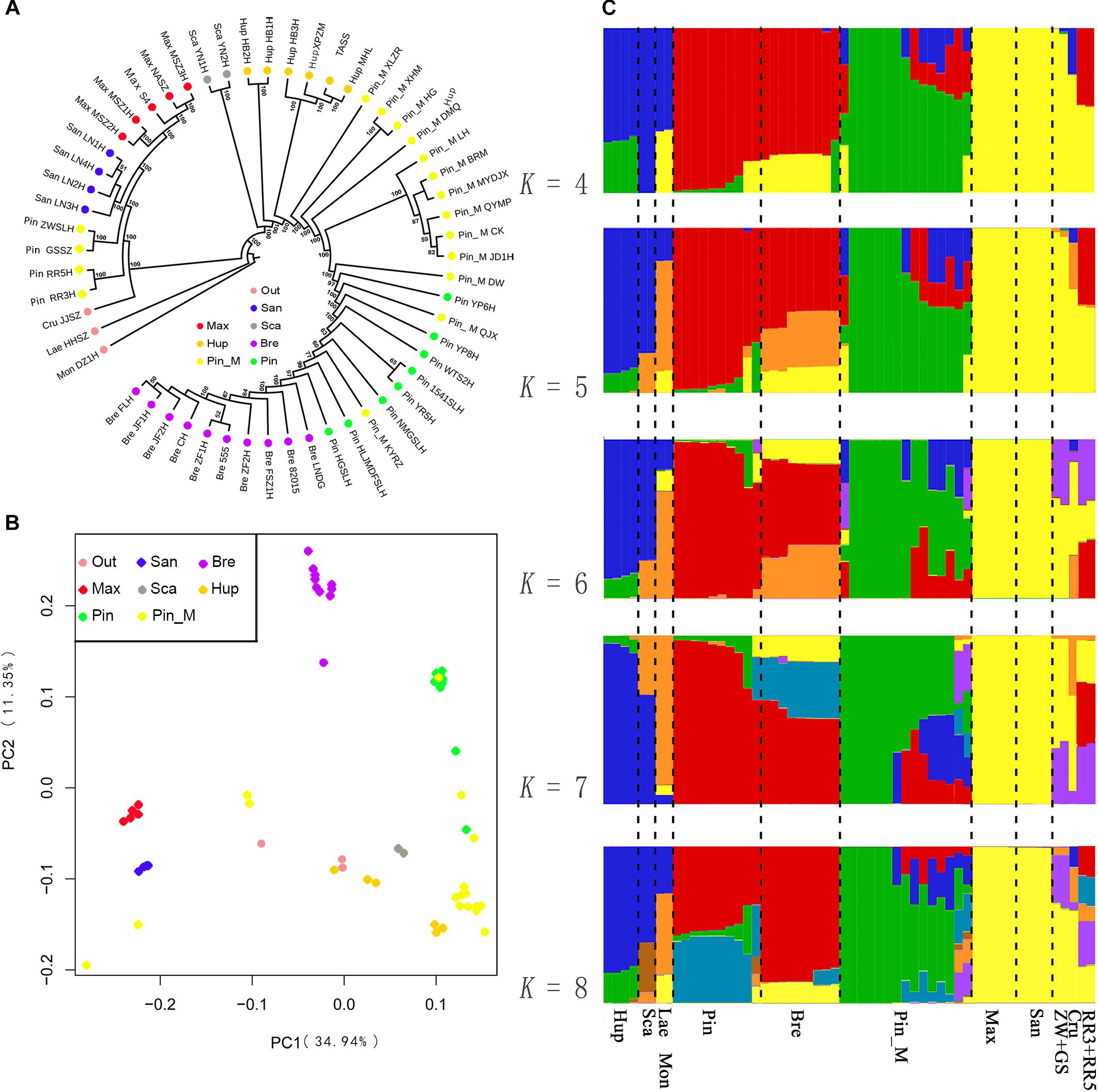
Figure 2. Population structure of 56 samples. (A) ML phylogenetic tree constructed using SNPs. Each species group is color coded. (B) Principal component analysis (PCA) of the 56 samples. (C) Bayesian model-based clustering of 56 samples with the number of ancestry kinship (K) from 4 to 8. Each vertical bar represents one hawthorn sample. Each color represents one putative ancestral background.
To further understand the evolutionary history of Chinese Crataegus, we used a Bayesian clustering algorithm with admixed models (Hubisz et al., 2009) to estimate the ancestral proportions for each sample (Figure 2C). The ΔK analysis revealed that five populations (K = 5) represented the best model for these 56 samples (Supplementary Figure S2). When K = 4, the C. bretschneideri genepool was indicated to be derived from C. maximowiczii (yellow) and C. pinnatifida (red); when K = 5 to 8, the C. bretschneideri genepool predominantly (more than half) was suggested to be derived from C. maximowiczii and C. pinnatifida. The STRUCTURE analysis (Figure 2C) showed that the southwestern Chinese species C. scabrifolia harbored had the genepool from the European species C. monogyna and C. laevigata, and showed evidence for introgression from C. scabrifolia and the two European species. The genepool of the North American species C. cruss-galli was similar to that of the northeastern Chinese species C. sanguineae. Similar to the SSR dendrogram, the genepool of the accessions ZWSLH, GSSZ, RR5H, and RR3H shared with C. maximowiczii and C. sanguineae; these findings demonstrated that four accessions did not belong to C. pinnatifida. When K = 4 to 8, the accessions RR3H and RR5H shared the genepool of C. pinnatifida and C. bretschneideri. To explore the true identity of these four accessions, we considered them to be “unknown” accessions in the following analysis.
To understand the history of divergence and admixtures, we applied TreeMix to the 10 groups, six species and one variety, and the groups unknown1 (GSSZ and ZWSLH) and unknown2 (RR3H and RR5H). C. cruss-galli, C. monogyna, and C. laevigata were used as the outgroup taxa. TreeMix uses genome-wide allele frequency data and a Gaussian approximation to genetic drift to analyze whether the migration events between species is a pattern of population splits and mixtures in multiple populations. The result contains the population splits and gene flow between species. The arrow in the figure corresponds to the migration events, the darkness of the arrow color indicates the migration edge weight. In the TreeMix result (Figure 3), introgression occurred among C. hupehensis, C. pinnatifida var. major, and C. pinnatifida, which indicated that extensive gene flow had occurred in central, northern, and northeastern China. The C. maximowiczii, C. sanguineae, and two unknown groups formed a cluster that differed markedly from the other groups. Between the two clusters, gene flow from C. maximowiczii to C. bretschneideri was observed, with an arrow weight of 0.43, which indicated that C. bretschneideri is the result of admixture between C. maximowiczii and C. pinnatifida, with about 43% of the genome derived from C. maximowiczii. We did not observe gene flow between the four unknown accessions and other species. Based on the STRUCTURE and TreeMix results, we propose that C. bretschneideri may have been of hybrid origin, and shared the genepool with C. pinnatifida and C. maximowiczii.
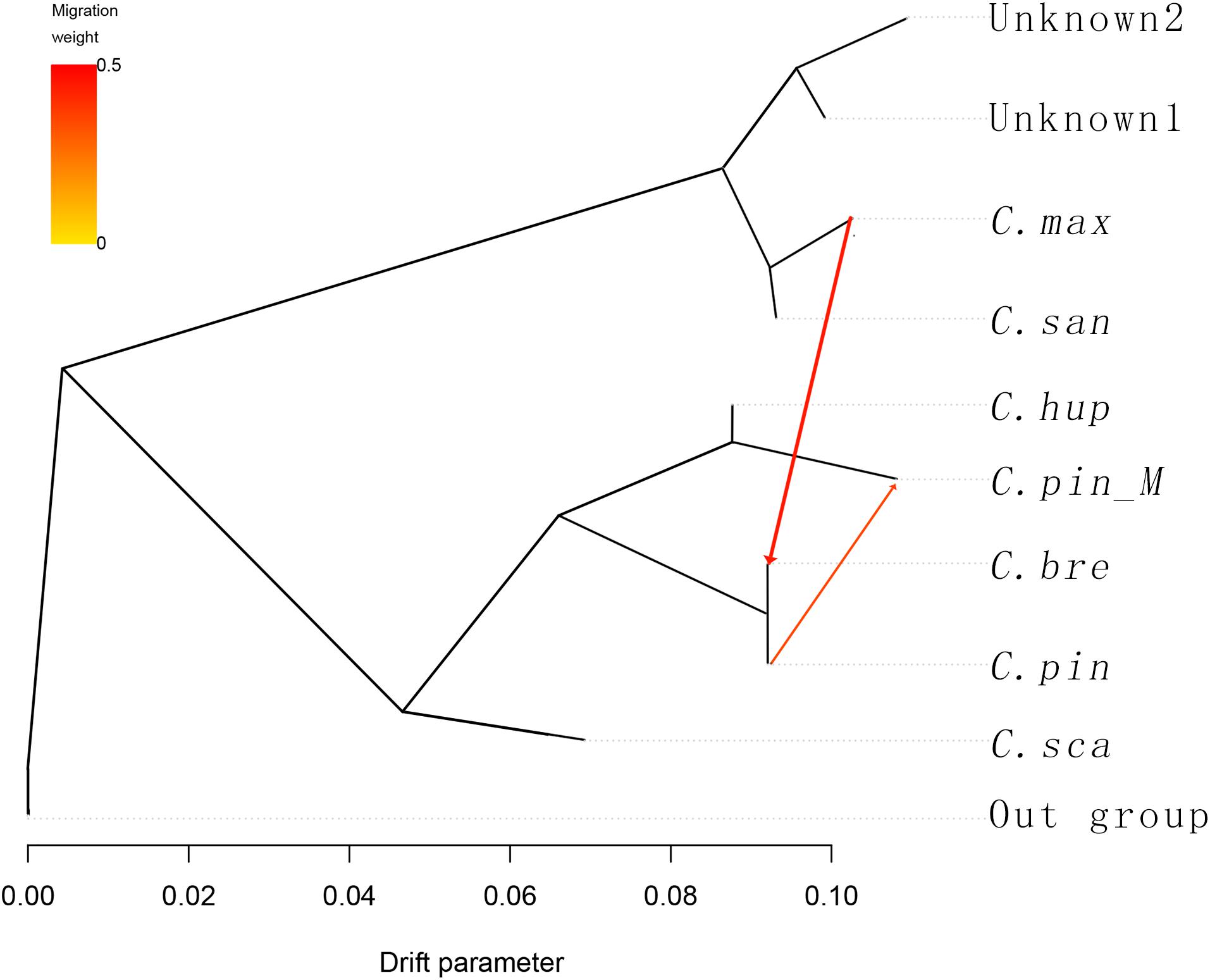
Figure 3. TreeMix analysis of 53 hawthorn samples divided into 9 groups. With C. cruss-galli, C. monogyna and C. laevigata serving as the outgroup population, the arrow corresponds to the direction of migration.
Based on the SLAF-seq data and fossil calibration, the major diversification events of Chinese Crataegus species were estimated to have occurred in the late Miocene and Pliocene (Figure 4). The split between clusters A and B was estimated to be ∼10.8 Ma. The earliest Chinese Crataegus to diverge was the southwestern species C. scabrifolia, which split at ∼8.81 Ma, followed by the central species C. hupehensis, which diverged at ∼6.85 Ma. Subsequent diversification events occurred during the Pliocene. The northeastern species C. sanguineae and C. maximowiczii split at ∼4.12 Ma; C. pinnatifida var. major, which is distributed in northern, eastern, and northeastern China, diverged at ∼3.25 Ma; the northern and northeastern species C. pinnatifida was diverged at ∼1.23 Ma; and the northeastern species C. bretschneideri was the most recent species to diverge and arose at ∼0.22 Ma.
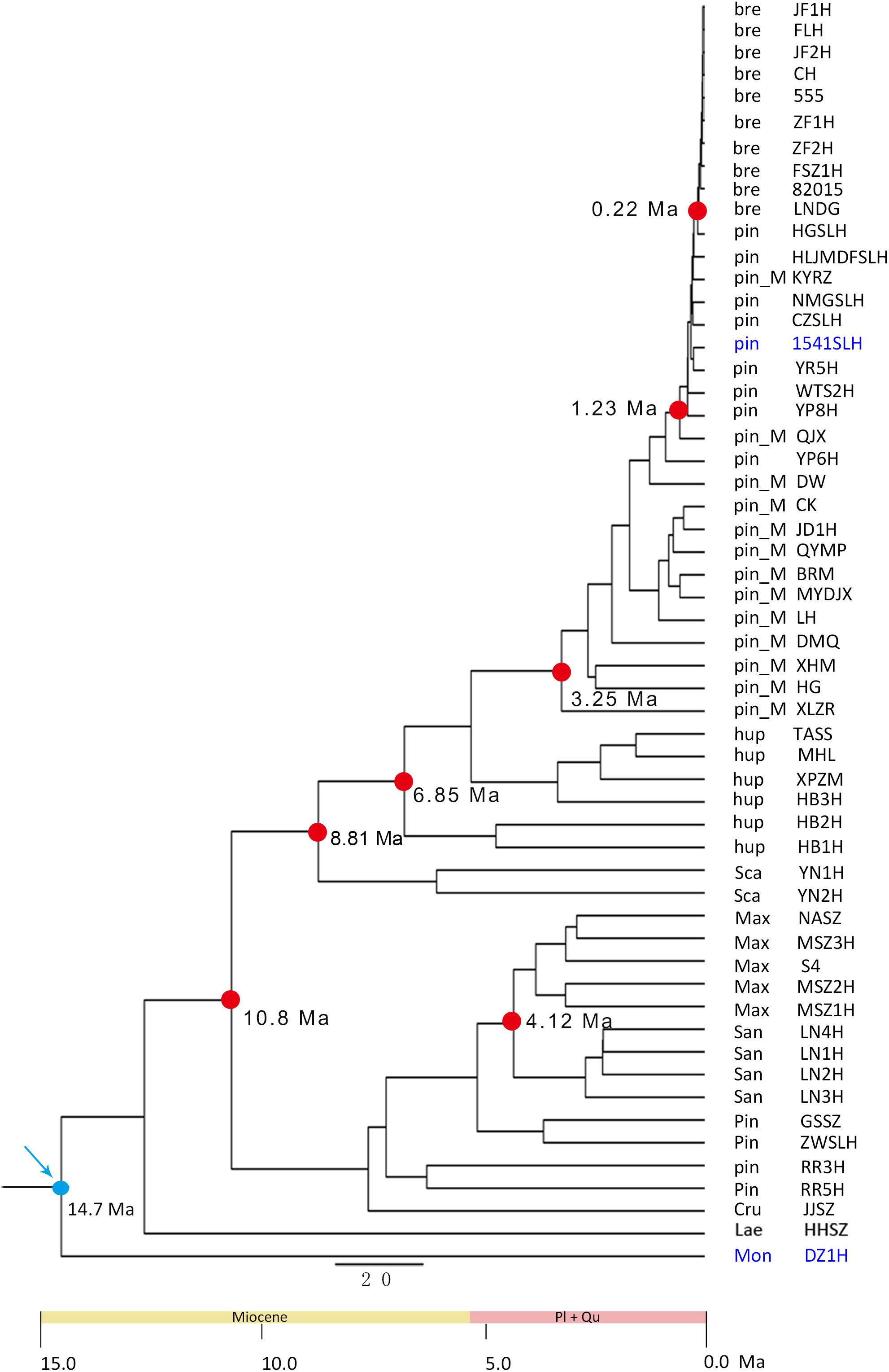
Figure 4. Time-calibrated phylogeny inferred from SLAF-seq data in BEAST V.1.7.5 using node age based on fossil data. Blue arrows designate calibrated node (14.7 Ma). The blue words represent the fossils for calibration. The dots represents the split time.
We presented a robust phylogenetic construction for seven Chinese Crataegus species based on SLAF-seq data. The ML phylogenetic tree showed evidence for two main clusters. Cluster A includes C. maximowiczii and C. sanguineae, whereas cluster B comprised C. hupehensis, C. pinnatifida, C. pinnatifida var. major, C. bretschneideri, and C. scabrifolia. From a geographically perspective, cluster A consists of the species in northeastern China, whereas cluster B includes the widespread species that extend from southwestern to northeastern China. Combined the molecular data and the morphological characters, fruit size is an important character for Crataegus classification. Consistent with the morphological classification, molecular data showed strong support for the grouping of the two small-fruited species, namely C. maximowiczii and C. sanguineae. These two species are closely related, and the only differentiating character is pubescence on the leaf of C. maximowiczii. In addition to fruit size, leaf dissection is also an important character for discrimination of Crataegus species. C. sanguineae and C. maximowiczii are sympatric with C. bretschneideri, C. pinnatifida, and C. pinnatifida var. major, which are distributed in northeastern China. These five species from the northeast were divided into two groups, of which the group that contained C. bretschneideri, C. pinnatifida, and C. pinnatifida var. major was more closely related to the southwestern species. The STRUCTURE analysis showed that C. scabrifolia, which indicated to be the earliest species to diverge in cluster A, belonged to the same lineage as the European species C. laevigata and the North American species C. cruss-galli. When K = 4 to 7, all C. scabrifolia accessions clustered with C. laevigata and C. cruss-galli.
There are two contrasting perspectives on the migration direction of Chinese and European Crataegus. Based on the nSSR dendrogram and STRUCTURE analysis (Figure 2C), we suggest that the seven Chinese hawthorn species may have experienced two different speciation events. The first speciation event occurred in northeastern China, the northeastern species shared the genepool with the North American species. The second speciation event originated in southwestern China and then progressed northward and eastward to northeastern China. The taxa in southwestern route shared genepool with Europe Crataegus. The southwestern routed is consistent with Wang’s (1992) description of the plants of other genus and families stretched from the Hengduan Mountains toward the northeast through the Qinling Range, the eastern fringe of the Loess Plateau including the Taihang Range, the Yinshan Range, the Changbai Mountains, and the Xiao Hinggan Mountains of Siberia or the adjacent regions.
Population structure is the result of both present and historical processes, and many factors may change the geographical distributions of plant species (Comes and Kadereit, 1998). Similar to the Quaternary climatic events that caused vegetation changes between southern and northern China (Sun et al., 2000), geological events are critical to the formation and development of the regional flora (Zhe and Jian, 2017; Zhe et al., 2017). In the late Miocene, interspecific divergence events occurred within Chinese Crataegus species. Since the late Pliocene, numerous intraspecific differentiation events have occurred. According to previous studies, the third intense uplift of the Qinghai–Tibet Plateau (QTP) and formation of the Hengduan Mountains began at this time (Li and Fang, 1998, 1999; Shi, 1998). The QTP uplift is one of the most important geological events of the Cenozoic; it changed the geography and climate in Asia and led to the Asian monsoon system (Li, 2006; Liu and Dong, 2013). The Asian monsoon system resulted in uneven distribution of precipitation in Yunnan Province and caused the migration of plants (Jacques et al., 2011; Su et al., 2013). Consequently, these events also may have caused the Crataegus species in southwestern China to migrate toward the northeast.
In this study, we observed that four accessions were misidentified based on morphological characteristics. The molecular data support the conclusion that these accessions may belong to species other than C. pinnatifida; however, additional research is needed to elucidate the true identity of these accessions.
Hybridization is recognized to be an important driving force in plant evolution (Mallet, 2007; Paun et al., 2009) that creates new species, or ecotypes, and results in reticulate evolution (Whitham et al., 1994; Linder and Rieseberg, 2004). Changes in geographical distribution provide opportunities for speciation through hybridization. Hybrids are commonly found in regions where different species overlap. Where species’ ranges overlap, hawthorns show introgressive hybridization, which results in a variety of morphological variants (Talent and Dickinson, 2007). Radford et al. (1968) and Phipps (2015) posited that hybridization is a potential explanatory factor for speciation in Crataegus. Furthermore, molecular data have provided evidence for hybridization in Crataegus (Lo et al., 2010a,b). C. bretschneideri is morphologically very similar to C. pinnatifida, and some researchers consider the former to be a variant of the latter species (Dai, 2007). Based on peroxidase isozymograms, Guo and Jiao (1995) suggested that C. bretschneideri is closely related to C. pinnatifida. On the basis of the present results, we propose that C. bretschneideri has a hybrid origin. The STRUCTURE analysis (Figure 2C) indicated that C. bretschneideri shared a genepool with C. pinnatifida and C. maximowiczii; when K = 4, the genepool of C. bretschneideri was derived from C. maximowiczii (yellow) and C. pinnatifida (red); when K = 5 to 8, the C. bretschneideri genepool dominated (more than half) was derived from C. maximowiczii and C. pinnatifida. The TreeMix results (Figure 3) indicated that gene flow had occurred from C. maximowiczii to C. bretschneideri. C. bretschneideri is the most recently divergent species, and C. pinnatifida was diverged before C. bretschneideri. Geographically, C. bretschneideri is distributed at the border of C. maximowiczii and C. pinnatifida. Based on these results, we hypothesize that C. bretschneideri arose through hybridization between C. pinnatifida and C. maximowiczii. The northern species migrated northeast, and hybridization with native species gave rise to new species.
This study is the first to use SLAF-seq to investigate the evolution and phylogenetic relationships of Chinese Crataegus. We hypothesize that the seven Crataegus species analyzed in this study experienced two speciation events. The southwestern species, C. scabrifolia, was indicated to be the earliest-diverging Chinese species, and shares a genepool with European Crataegus species. The northeastern Crataegus species share a genepool with the North American species C. cruss-galli. Overall, the present results provide valuable information on the origin of Crataegus in China.
The datasets generated for this study can be found in NCBI, SRP151024.
XD and WD conceived this project and designed the work. XD, XZ, HB, and YL performed the research. XD and TZ analyzed the data. XD, WD, and XZ wrote the manuscript. All authors contributed critically to the drafts and gave final approval for publication.
This work was supported by the Program (project) of “The Conservation and Utilization of Crop Germplasm Resource-Hawthorn (2015–2017).”
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Dr. Zhang Qijing and Dr. Hou Yali for assisting with the experiments and commenting on the manuscript.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.00443/full#supplementary-material
Brown, J. A., Beatty, G. E., Finlay, C., Montgomery, I., Tosh, D. G., and Provan, J. (2016). Genetic analyses reveal high levels of seed and pollen flow in hawthorn (Crataegus monogyna, Jacq.), a key component of hedgerows. Tree Genet. Genomes 12:58. doi: 10.1007/s11295-016-1020-0
Cairns, M. J., and Murray, V. (1994). Rapid silver staining and recovery of PCR products separated on polyacrylamide gels. BioTechniques 17,915–919.
Chen, S., Huang, Z., Dai, Y., Qin, S., Gao, Y., Zhang, L. (2013). The development of 7E chromosome-specific molecular markers for Thinopyrum elongatum based on Slaf-seq technology. PLoS One 8:e65122. doi: 10.1371/journal.pone.0065122
Comes, H. P., and Kadereit, J. W. (1998). The effect of quaternary climatic changes on plant distribution and evolution. Trends Plant Sci. 3, 432–438. doi: 10.1016/s1360-1385(98)01327-2
Dahmer, S., and Scott, E. (2018). Health effects of hawthorn (Complementary and Alternative Medicine). Am. Fam. Physician 81,465–469.
Dai, H. Y. (2007). Molecular Identification and Enhancement of Germplasms in Hawthorn. Ph.D. Thesis, Shenyang Agricultural University, Shenyang.
Dickinson, T. A., Proctor, S., Shipley, P. R., Lee, J., Coughlan, J., and Zarrei, M. (2014). “Natural health products and Crataegus of the Pacific Northwest,” in Proceedings of the Annual Meeting, Canadian Botanical Association, (Montréal, QC),
Dong, W., and Li, Z. X. (2015). The Science and Practice of Chinese Fruit Tree: Hawthorn. Shanxi: Science Press, 23–52.
Dong, W. X. (2013). Technical Specifications for Crop Germplasm Characterization and Evaluation of Hawthorn (NY/T 2325-2013). Beijing: China Agriculture Publishin House.
Edwards, J. E., Brown, P. N., Talent, N., Dickinson, T. A., and Shipley, P. R. (2012). A review of the chemistry of the genus Crataegus. Phytochemistry 79, 5–26. doi: 10.1016/j.phytochem.2012.04.006
Emami, A., Shabanian, N., Rahmani, M. S., Khadivi, A., and Mohammad-Panah, N. (2018). Genetic characterization of the Crataegus genus: implications for in situ conservation. Sci.Hortic. 231, 56–65. doi: 10.1016/j.scienta.2017.12.014
Erfani-Moghadam, J., Mozafari, M., and Fazeli, A. (2016). Genetic variation of some hawthorn species based on phenotypic characteristics and RAPD marker. Biotechnol. Biotechnol. Equip. 30, 247–253. doi: 10.1080/13102818.2015.1121790
Evans, R. C., and Campbell, C. S. (2002). The origin of the apple subfamily (Maloidea; Rosaceae) is clarified by DNA sequence data from duplicated GBSSI genes. Am. J. Bot. 89, 1478–1484. doi: 10.3732/ajb.89.9.1478
Güney, M., Kafkas, S., Keles, H., Aras, S., and Ercişli, S. (2018). Characterization of hawthorn (Crataegus spp.) genotypes by SSR markers. Physiol. Mol. Biol. Plants 24, 1221–1230. doi: 10.1007/s12298-018-0604-6
Guo, T. J., and Jiao, P. J. (1995). Hawthorn (Crataegus) resources in China. Hortscience. 30, 1132–1134. doi: 10.21273/HORTSCI.30.6.1132
Hedges, S. B., Marin, J., Suleski, M., Paymer, M., and Kumar, S. (2015). Tree of life reveals clock-like speciation and diversification. Mol. Biol. Evol. 32, 835–845. doi: 10.1093/molbev/msv037
Huang, H. R., Wu, W., Zhang, J. X., Wang, L. J., Yuan, Y. M., and Ge, X. J. (2016). A genetic delineation of Patchouli (Pogostemon cablin) revealed by specific-locus amplified fragment sequencing. J. Syst. Evol. 54, 491–501. doi: 10.1111/jse.12195
Hubisz, M. J., Falush, D., Stephens, M., and Pritchard, J. K. (2009). Inferring weak population structure with the assistance of sample group information. Mol. Ecol. Resour. 9, 1322–1332. doi: 10.1111/j.1755-0998.2009.02591.x
Jacques, F. M. B., Guo, S. X., Su, T., Xing, Y. W., and Huang, Y. J. (2011). Quantitative reconstruction of the late miocene monsoon climates of Southwest China: a case study of the Lincang flora from Yunnan Province. Palaeoge. Palaeocli. Palaeoe. 304, 318–327. doi: 10.1016/j.palaeo.2010.04.014
Khiari, S., Boussaid, M., and Messaoud, C. (2015). Genetic diversity and population structure in natural populations of Tunisian Azarole (Crataegus azarolus, L. var. aronia L.) assessed by microsatellite markers. Biochem. Syst. Ecol. 59, 264–270. doi: 10.1016/j.bse.2015.01.025
Kobert, K., Stamatakis, A., and Flouri, T. (2016). Efficient detection of repeating sites to accelerate phylogenetic likelihood calculations. Syst. Biol. 66, 205–217. doi: 10.1093/sysbio/syw075
Li, H., and Durbin, R. (2009). Fast and accurate short read alignment with burrows–wheeler transform. Bioinformatics. 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Li, H., Handsaker, B., Wysoker, A., Fennell, T., Ruan, J., Homer, N., et al. (2009). The sequence alignment/map format and SAMtools The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. doi: 10.1093/bioinformatics/btp352
Li, J., and Fang, X. (1998). Uplift of the Tibetan Plateau and environmental changes. Chin. Sci. Bull. 43, 1569–1574.
Li, J., and Fang, X. (1999). Uplift of the Tibetan Plateau and environmental changes. Chin. Sci. Bull. 44, 2117–2124. doi: 10.1007/BF03182692
Li, J. J. (2006). The Qinghai - Tibet Plateau Uplifting and Environmental Evolution in Asia: Article Collection of Academician Li Ji-Jun. Beijing: Science Press.
Linder, C. R., and Rieseberg, L. H. (2004). Reconstructing patterns of reticulate evolution in plants. Am. J. Bot. 91, 1700–1708. doi: 10.3732/ajb.91.10.1700
Liu, X. D., and Dong, B. W. (2013). Influence of the Tibetan Plateau uplift on the Asian monsoon-arid environment evolution. Chin. Sci. Bull. 58, 2906–2919. doi: 10.1186/s12862-017-1058-0
Lo, E. Y., Stefanovic, S., Dickinson, T. A., and Christensen, K. I. (2009). Evidence for genetic association between East Asian and western North American Crataegus L. (Rosaceae) and rapid divergence of the eastern North American lineage based on multiple DNA sequences. Mol. Phylogenet. Evol. 51, 157–168. doi: 10.1016/j.ympev.2009.01.018
Lo, E. Y., Stefanovic’, S., and Dickinson, T. A. (2010a). Reconstructing reticulation history in a phylogenetic framework and the potential of allopatric speciation driven by polyploidy in an agami complex in Crataegus (Rosaceae). Evolution 64, 3593–3608. doi: 10.1111/j.1558-5646.2010.01063.x
Lo, E. Y. Y., Stefanoviæ, S., Ritland, K., and Dickinson, T. A. (2010b). Fine-scale comparisons of genetic variability in seed families of asexually and sexually reproducing Crataegus (Hawthorn; Rosaceae). Am. J. Bot. 97, 1014–1024. doi: 10.3732/ajb.0900091
Ma, S. L. Y., and Lu, Y. M. (2016). Classification and phylogenetic analysis of Chinese hawthorn assessed by plant and pollen morphology. Gen. Mol. Res. 15, gmr.15038739. doi: 10.4238/gmr.15038739
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–1303. doi: 10.1101/gr.107524.110
Paun, O., Forest, F., Fay, M. F., and Chae, M. W. (2009). Hybrid speciation in angiosperms: parental divergence drives ploid. New Phytol. 182, 507–518. doi: 10.1111/j.1469-8137.2009.02767.x
Phipps, J. B. (1990). Mespilus canescens, a new Rosaceous endemic from Arkansas. Syst. Bot. 15, 26–32. doi: 10.2307/2419013
Phipps, J. B. (2015). “Crataegus,” in Flora of North America Editorial Committee, Vol. 9, ed. Flora of North America North of Mexico (New York, NY: Oxford University Press), 491–643.
Phipps, J. B., Robertson, K. R., Smith, P. G., and Roher, J. R. (1990). A checklist of the subfamily Maloideae (Rosaceae). Can. J. Bot. 68, 303–332. doi: 10.1139/b90-288
Pickrell, J. K., and Pritchard, J. K. (2012). Inference of population splits and mixtures from genome-wide allele frequency data. PLoS Genet. 8:e1002967. doi: 10.1371/journal.pgen.1002967
Price, A. L., Patterson, N. J., Plenge, R. M., Weinblatt, M. E., Shadick, N. A., and Reich, D. (2006). Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909. doi: 10.1038/ng1847
Radford, A. E., Ahles, H. E., and Bell, C. R. (1968). Manual of the Vascular Flora of the Carolinas. Chapel Hill, NC: University of North Carolina Press.
Rohlf, F. J. (1997). NTSYSpc Numerical Taxonomy and Multivariate Analysis System,v.2.2. Setauket, NY: Exeter Software.
Sheng, F., Chen, S. Y., Tian, J., Tian, J., Peng, L., Xue, Q., et al. (2017). Morphological and ISSR molecular markers reveal genetic diversity of wild hawthorns (Crataegus songorica K. Koch.) in xinjiang, China. J. Integr. Agric. 10, 2402–2406. doi: 10.1016/S2095-3119(17)61688-5
Shi, Y. (1998). Uplift and Environmental Changes of Qinghai-Xizang (Tibetan) Plateau in the Late Cenozoic. Guangzhou: Guangdong Scirence & Technology Press.
Stadler, T., and Yang, Z. (2013). Dating phylogenies with sequentially sampled tips. Syst. Biol. 62, 674–688. doi: 10.1093/sysbio/syt030
Stamatakis, A. (2014). RaxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Su, K., Guo, Y. S., Wang, G., Zhao, Y. H., and Dong, W. X. (2015). Genetic diversity analysis of fruit characteristics of hawthorn germplasm. Genet. Mol. Res. 14, 16012–16017. doi: 10.4238/2015.December.7.14
Su, T., Liu, Y. S., Jacques, F. M. B., Huang, Y. J., Xing, Y. W., and Zhou, Z. K. (2013). The intensification of the East Asian winter monsoon contributed to the disappearance of Cedrus (Pinaceae) in southwestern China. Quat. Res. 80, 316–325. doi: 10.1016/j.yqres.2013.07.001
Sun, X., Liu, D., Zhang, X., Li, W., Liu, H., and Hong, W. (2013). SLAF-seq: an efficient method of large-scale de novo SNP discovery and genotyping using highthroughput sequencing. PloS One 8:e58700. doi: 10.1371/journal.pone.0058700
Sun, X. J., Chen, X. D., and Liu, P. M. (2000). Pollen-based biome reconstruction at middle holocene (6ka BP) and last glacial maximum (18ka BP) in China. Acta Bot. Sin. 42, 1201–1209.
Talent, N., and Dickinson, T. A. (2007). Ploidy level increase and decrease in seeds from crosses between sexual diploids and asexueal triploids and tetraploids in Crataegus L. (Rosaceae, Spiraeoideae, Pyreae). Can. J. Bot. 85, 570–584. doi: 10.1139/B07-028
Wang, W. T. (1992). On some distribution patterns and some migration routes found in the eastern Asiatic region. Acta Phy. Sin. 30, 1–24.
Whitham, T. G., Morrow, P. A., and Potts, B. M. (1994). Plant hybrid zones as centers of biodiversity: the herbivore community of two endemic Tasmanian eucalypts. Oecologia 97, 481–490. doi: 10.1007/BF00325886
Wu, F. F., Zhang, Z. H., Dai, H. Y., Zhang, Y., and Zhang, L. L. (2008). Genetic relationship of some Crataegus spp. (Hawthorn) revealed by chloroplast DNA PCR-RFLP. J. Biotechnol. 136:S103. doi: 10.1016/j.jbiotec.2008.07.235
Xin, X., and Zhang, Y. M. (1997). Chinese Hawthorn Germplasm Resources and Utilization. Beijing: China Agricultural Press.
Xu, X., Xu, R., Zhu, B., Yu, T., Qu, W., Lu, L., et al. (2015). A high density genetic map of cucumber derived from specific length amplified fragment sequencing (SLAF-seq). Front. Plant Sci. 5, 768. doi: 10.3389/fpls.2014.00768
Xu, J. Y., Zhao, Y. H., Zhang, X., Zhang, L. J., and Dong, W. X. (2016). Transcriptome Analysis and ultrastructure observation reveal that hawthorn fruit softening is due to cellulose/hemicellulose degradation. Front. Plant Sci. 7:1524. doi: 10.3389/fpls.2016.01524
Zarei, A., Erfanimoghadam, J., and Mozaffari, M. (2017). Phylogenetic analysis among some pome fruit trees of Rosaceae family using RAPD markers. Biotechnol. Biotechnol. Equip. 31:10. doi: 10.1080/13102818.2016.1276414
Zarrei, M., Talent, N., Kuzmina, M., Lee, J., Lund, J., Shipley, P. R., et al. (2015). DNA barcodes from four loci provide poor resolution of taxonomic groups in the genus Crataegus. AoB Plants 7:lv045. doi: 10.1093/aobpla/plv045
Zhang, Y., Dai, H. Y., Zhang, Q. J., Li, H., and Zhang, Z. H. (2008). Assessment of genetic relationship in Crataegus genus by the apple SSR primers. J. Fruit Sci. 25, 521–525.
Zhang, Y., Wang, L., Xin, H., Li, D., Ma, C., Ding, X., et al. (2013). Construction of a high-density genetic map for sesame based on large scale marker development by specific length amplified fragment (SLAF) sequencing. BMC Plant Biol. 13, 141. doi: 10.1186/1471-2229-13-141
Zhao, H. C., and Feng, B. T. (1996). China Fruit-Plant Monograph of Hawthorn (Crataegus) Flora. Beijing: Zhongguo Linye Press.
Zhe, K. Z., Huang, J., and Ding, W. N. (2017). The impact of major geological events on Chinese flora. Biodivers. Sci. 25, 123–125. doi: 10.17520/biods.2016120
Zhe, K. Z., and Jian, H. W. (2017). The impact of major geological events on Chinese flora. Biodivers. Sci. 25, 123–125. doi: 10.17520/biods.2016120
Keywords: hawthorn, Crataegus, SLAF-seq, nSSR marker, molecular evolution
Citation: Du X, Zhang X, Bu H, Zhang T, Lao Y and Dong W (2019) Molecular Analysis of Evolution and Origins of Cultivated Hawthorn (Crataegus spp.) and Related Species in China. Front. Plant Sci. 10:443. doi: 10.3389/fpls.2019.00443
Received: 08 November 2018; Accepted: 25 March 2019;
Published: 09 April 2019.
Edited by:
Marcial Escudero, Universidad de Sevilla, SpainReviewed by:
Kyong-Sook Chung, Jungwon University, South KoreaCopyright © 2019 Du, Zhang, Bu, Zhang, Lao and Dong. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Wenxuan Dong, d3hkb25nNjNAMTI2LmNvbQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.