 Jing Sun†
Jing Sun† Xuean Cui
Xuean Cui Jinxia Wu
Jinxia Wu Zhiguo Zhang
Zhiguo Zhang- Biotechnology Research Institute, National Key Facility for Genetic Resources and Gene Improvement, the Chinese Academy of Agricultural Sciences, Beijing, China
Foxtail millet (Setaria italica) is attractive to plant scientists as a model plant because of several distinct characteristics, such as its short stature, rapid life cycle, sufficient seed production per plant, self-compatibility, true diploid nature, high photosynthetic efficiency, small genome size, and tolerance to abiotic and biotic stress. However, the study on the genetic resources of foxtail millet largely lag behind those of the other model plants such as Arabidopsis, rice and maize. Mutagenized populations cannot only create new germplasm resources, but also provide materials for gene function research. In this manuscript, an ethyl methanesulfonate (EMS)-induced foxtail millet population comprising ∼15,000 individual M1 lines was established. Total 1353 independent lines with diverse abnormal phenotypes of leaf color, plant morphologies and panicle shapes were identified in M2. Resequencing of sixteen randomly selected M2 plants showed an average estimated mutation density of 1 loci/213 kb. Moreover, we provided an example for rapid cloning of the WP1 gene by a map-based cloning method. A white panicle mutant, named as wp1.a, exhibited significantly reduced chlorophyll (Chl) and carotenoid contents in leaf and panicle. Map-based cloning results showed an eight-base pair deletion located at the sixth exon of wp1.a in LOC101786849, which caused the premature termination. WP1 encoded phytoene synthase. Moreover, the sequencing analysis and cross test verified that a white panicle mutant wp1.b was an allelic mutant of wp1.a. The filed phenotypic observation and gene cloning example showed that our foxtail millet EMS-induced mutant population would be used as an important resource for functional genomics studies of foxtail millet.
Introduction
Foxtail millet is a member of the Paniceae tribe (subfamily Panicoideae of the Poaceae) and came from green millet domestication in northern China about for 8000 years ago (Barton et al., 2009). Foxtail millet is extensively cultivated in the developing countries in semiarid and arid regions of Africa, Americas, Asia (Lata et al., 2013) because of its health benefits (a particular balance of nutrients, e.g., starch, protein, dietary fibers, fat, vitamins, and low-glycemic and hypolipidemic effects), good yield with minimal agricultural inputs, and adaptation to different biotic and abiotic stresses such as salinity (Lata et al., 2011), drought (Lata et al., 2011; Feldman et al., 2017; Tang et al., 2017), and fungal diseases (Xu et al., 2011). A healthy and environmentally friendly small crop of foxtail is an increasingly attractive alternative for crop production, and China have planted 70 M ha more foxtail millet in 2018 than in 2017, increasing to 130 M ha1.
In recent years, foxtail millet and its ancestor green foxtail (Setaria viridis) became more attractive to plant scientists as an alternative model plant because of several distinct characteristics, such as their short stature, rapid life cycle, sufficient seed production per plant, self-compatibility, true diploid nature (2n = 18), and small genome size (515 and 395 Mb, respectively) (Doust et al., 2009; Li and Brutnell, 2011; Huang et al., 2016; Pant et al., 2016). More importantly, foxtail millet and green foxtail are typical C4 plants, similar to maize, sorghum and sugarcane, and therefore can be a valuable model plant for studies of C4 photosynthesis (Brutnell et al., 2010; Lata et al., 2013; Pant et al., 2016; Huang et al., 2017; Yang et al., 2018). These plants can also be used as model systems for panicoid grasses such as switchgrass (Panicum virgatum), and napier grass (Pennisetum purpureum) for biofuel studies (Li and Brutnell, 2011; Lata et al., 2013; Pant et al., 2016). The distinct drought tolerant characteristic is very beneficial to dissect the molecular mechanisms for drought tolerance.
Genetic and genomic resources are necessary for studies of gene function. Reference genome sequences and resequencing of the core germplasm of foxtail millet are available to the public (Bennetzen et al., 2012; Zhang et al., 2012; Jia et al., 2013a) and several high density molecular marker maps and integrated databases were developed based on public data (Jia et al., 2013b; Pandey et al., 2013; Muthamilarasan et al., 2014; Yadav et al., 2015; Zhang et al., 2014; Muthamilarasan and Prasad, 2015). Genetic variation resources have been collected, and high-density genetic maps have been constructed (Wang et al., 2012, 2017; Jia et al., 2013b; Muthamilarasan and Prasad, 2015; Fang et al., 2016). Genes controlling agronomically important traits such as branching (Doust et al., 2004) and panicle shape (Xiang et al., 2017) have been cloned using those resources. However, compared with Setaria viridis, much less attention has been paid to Setaria italica; for example, mutant resources are plentiful for Setaria viridis (Brutnell et al., 2010; Huang et al., 2016), but only occasional reports are found for Setaria italica (Gupta and Yashvir, 1976). Thus, the construction of a Setaria italica mutant library is urgently needed to accelerate functional research. In this study, we described the generation and field phenotype characterization of an ethyl methyl sulfonate-induced foxtail millet mutant population. We also reported the mutation frequency of this population by resequencing and identified a gene that controls carotenoid biosynthesis, namely, WP1.
Materials and Methods
Mutagenesis and Plant Growth
Newly harvested mature seeds of an elite inbred line Yugu No. 1(the most influential variety in North China because of its strong disease resistance, drought and waterlogging resistance, insensitivity to light and temperature, and wide adaptability) were stored at 4°C for 30 days to break the dormancy, and seeds with at least a 90% germination rate were utilized for chemical mutagenesis. After immersing foxtail millet seeds for 8 h, the soaked seeds were then incubated in a hood with different concentrations of ethyl methyl sulfonate (Sigma-Aldrich, United States) of 0.5, 1.0, 1.5, or 2% in 0.1 M sodium phosphate buffer (pH 7.0) for different times (8, 16, or 24 h). After determining the appropriate concentration and time (1% EMS concentration and 16 h treatment time) based on the germination rate (20–25%), approximately one hundred thousand seeds were treated in EMS solution on a shaker at 80 rpm in dark conditions at room temperature. After incubation, seeds were washed with running water for 4 h, germinated on wet filter paper in a 25 × 30 × 10 cm plastic box covered with transparent cling film at 28°C for 3 days in darkness and then transferred to a greenhouse at 26–30°C with natural light conditions (14 h light/10 h dark). Germinated seedlings of approximately 0.3–0.5 cm in height were then transferred to a greenhouse and grown in soil. The spikelet of each plant was paper-bagged before flowering to prevent pollen contamination from other plants. Golden spikelets with mature seeds were harvested, air-dried completely and kept at 4°C.
Mutant Characterization
Approximately 1000 seeds for each line were sown in an experimental field in June. All the seedlings were first characterized by the leaf color, i.e., albino or yellow, at 15 days after germination, and then excess seedlings were thinned to ensure the remaining seedlings were grown at 15 cm for a line and 30 cm to a row, with approximately 30 M2 plants for each line grown in the field. All plants were managed according to standard protocols for watering, fertilizing and other practices. The remaining M2 plants grown in the field were subjected to carefully recorded observation and characterization every 2 weeks.
Chlorophyll and Carotenoid Contents Measurement
The mutant wp1 and wild type chlorophyll contents were measured by using spectrophotometrical method (Arnon, 1949). In total, 400 mg leaves and panicles were ground into powder in liquid nitrogen and then shifted to a 10 ml tube. Five milliliters acetone (80% V/V) was put into the tube and mixed thoroughly, and the tube was kept overnight in darkness. Centrifugation was carried out for 15 min (∼750 g) at 4°C. The supernatant was washed three times with the same amount of hexane. The chlorophyll contents were determinated with spectrophotometer (Beckman Coulter DU 800 UV/Vis).
Mutant Frequency Evaluation
Sixteen foxtail millet mutant samples were randomly selected and grown in an incubator with 10 h of light at 28°C per day for a month. To construct the library for resequencing, total genomic DNAs of 4th-leaf-stage leaf tissue were isolated by CTAB method. 1.5 μg DNA each sample was used for construct the sequencing libraries.
Sequencing libraries were created based on a TruSeq Nano DNA HT Sample Preparation Kit (Illumina, United States) as followed. Firstly, sonication treatment made the DNA sample fragment to a size of 350 bp, secondly, DNA fragments were end-blunted, A-tailed, and ligated with the full-length adapter for Illumina sequencing, thirdly, the PCR product was amplified and purified. The size was decided by an Agilent 2100 Bioanalyzer and quantity was analyzed using Q-PCR.
The libraries constructed of these sixteen lines were sequenced by an Illumina HiSeq platform, and 150 bp paired-end reads were set up with ∼ 350 bp size. Average 38x sequencing depth per sample was produced and 277.39 Gb of clean bases were produced in total. The alignment of reads was used to build a consensus genome sequence for foxtail millet2. The GATK tools package (McKenna et al., 2010) was used to detect SNP sites and small INDEL s (insertion and deletion), which included local realignment, and the duplicates were marked by Picard3 to ensure the accuracy. According to standard procedure, if less than 60% of the loci appear in samples, the loci is regarded as a SNP or INDEL site, otherwise or not.
Map-Based Cloning and Candidate Gene Identification
To map WP1 gene, a wp1.a × “SSR41” F2 population was created. In F2 progeny, a total 246 white panicle individuals were collected. Their DNA samples were extracted and used to map. Ten samples with equal DNA concentration were mixed as a pool. Three pools were used for rough mapping. A set of high resolution SSR/ INDEL s molecular markers were designed based on the published primer sequences (Zhang et al., 2014; Supplementary Table S2) and 30× genome resequence result both Yugu No.1 and SSR41. Using this set of SSR/ INDEL markers, WP1 gene was mapped on chromosome 4. By further screen the 246 F2 recessive individuals, we narrowed the interval to 80 kb using newly developed primers (Supplementary Table S1).
Allelic Analysis
To determine whether wp1.b was allelic with wp1.a, a cross was conducted between white panicle mutant wp1.a and wp1.b. In F1 progeny, the phenotype was investigated. If wp1.b was not allelic with the wp1.a, the panicle phenotype would restore to the normal level in F1 progeny.
Results
EMS Induced Foxtail Millet Population Construction
To determine the appropriate concentration of EMS mutagenesis, four EMS solution concentrations (v/v) of 0.5, 1.0, 1.5, or 2% were used to treat seeds of Foxtail millet cv. Yugu No.1 overnight. At the 0.5% concentration, at least 50% of Yu1 seeds germinated, whereas at the 2% concentration, the seed germination rate was only 2.5%. We selected the 1% concentration as an appropriate concentration with a 25% seeds germination rate (Supplementary Figure S1A). The optimal treatment time was determined by testing three times: 8, 16, and 24 h. At the 1% concentration, the 16 h treatment resulted in a 20% seed germination rate. Thus, we selected the 1% EMS concentration and 16 h treatment time for further trials (Supplementary Figure S1B). Approximately 100,000 seeds of foxtail millet cv. Yugu No.1 were mutagenized with EMS. Of these seeds, 20,000 seeds normally germinated as M0 progeny. Approximately 5000 seeds resulted in M0 progeny plants that did not produce seeds owing to albino leaf, sterility or severe dwarfing (Figure 1).
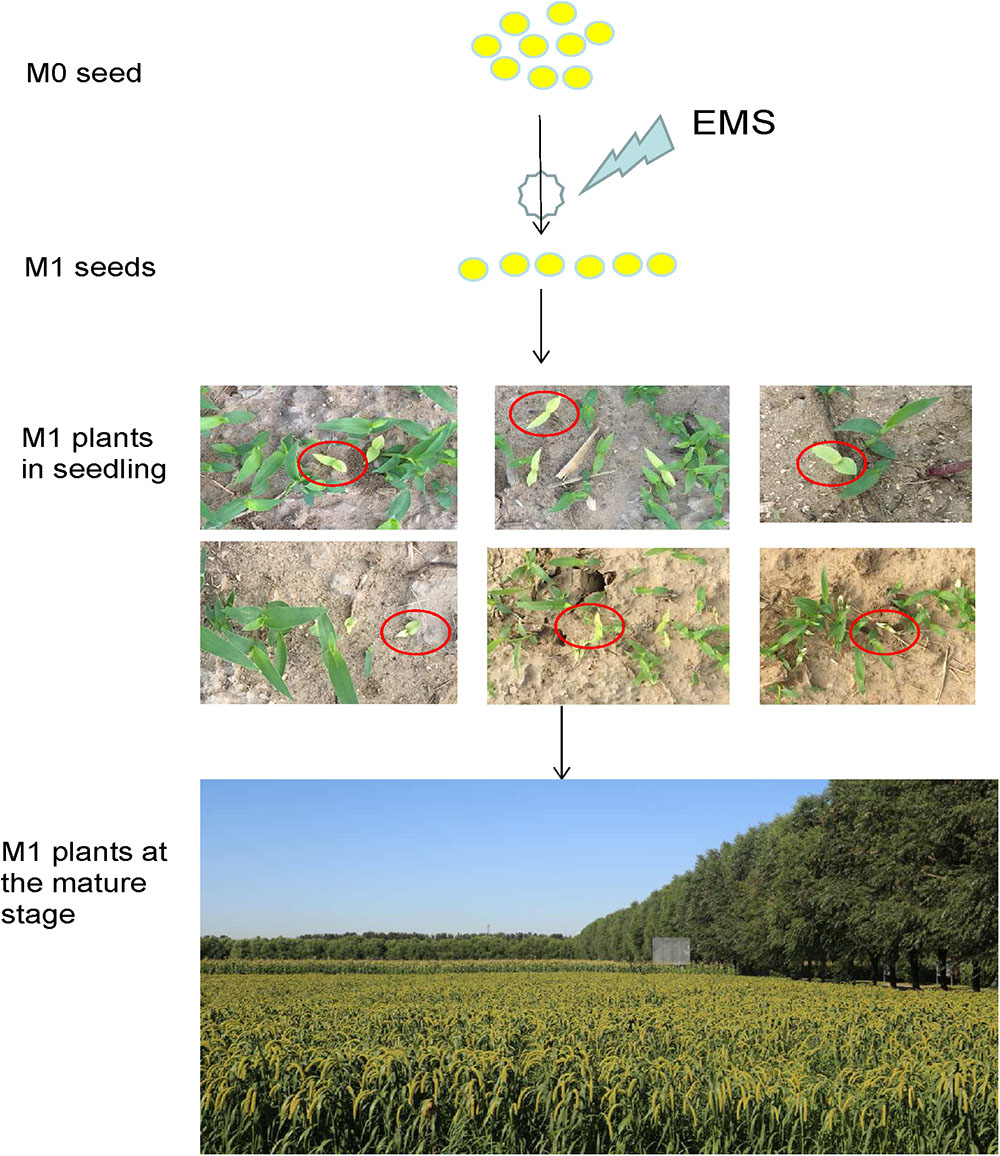
Figure 1. Development of a foxtail millet ethyl methanesulfonate (EMS)-induced mutant population. M0 seeds were mutated and propagated, and a single M2 seed was selected from each chimeric M1 plant.
Phenotypic Screening in M2 Plants
Seeds of approximately 15,000 M1 plants were harvested. All M1 seeds (minimum 20 plants per line) were planted to screen for plant morphological mutants. According to the phenotypic variation at the seedling and mature stages, mutants were classified into six types based on leaf color, heading date, plant height, senescence, leaf shape, and panicle development (Table 1 and Figure 2). A total of 1353 independent mutants with a phenotype visible to the naked eye were obtained. The mutation frequency reached 9.02% in the M2 plants (Table 1). The largest mutation type was that of leaf color mutant, wherein the albino mutant numbers reached 425, with the highest mutant frequency of 2.83%. Notably, panicle phenotypic variation was rich in our foxtail millet EMS mutant library, which reached 348 with the high mutant frequency of 2.32%. Morphological variation of various types of panicles included the dense panicle, loose panicle, degenerated panicle, white panicle, small panicle, large panicle, red panicle, and long awn (Figure 2), which indicated that the foxtail millet panicle morphological variation mutants could be a valuable resource for studying panicle development.
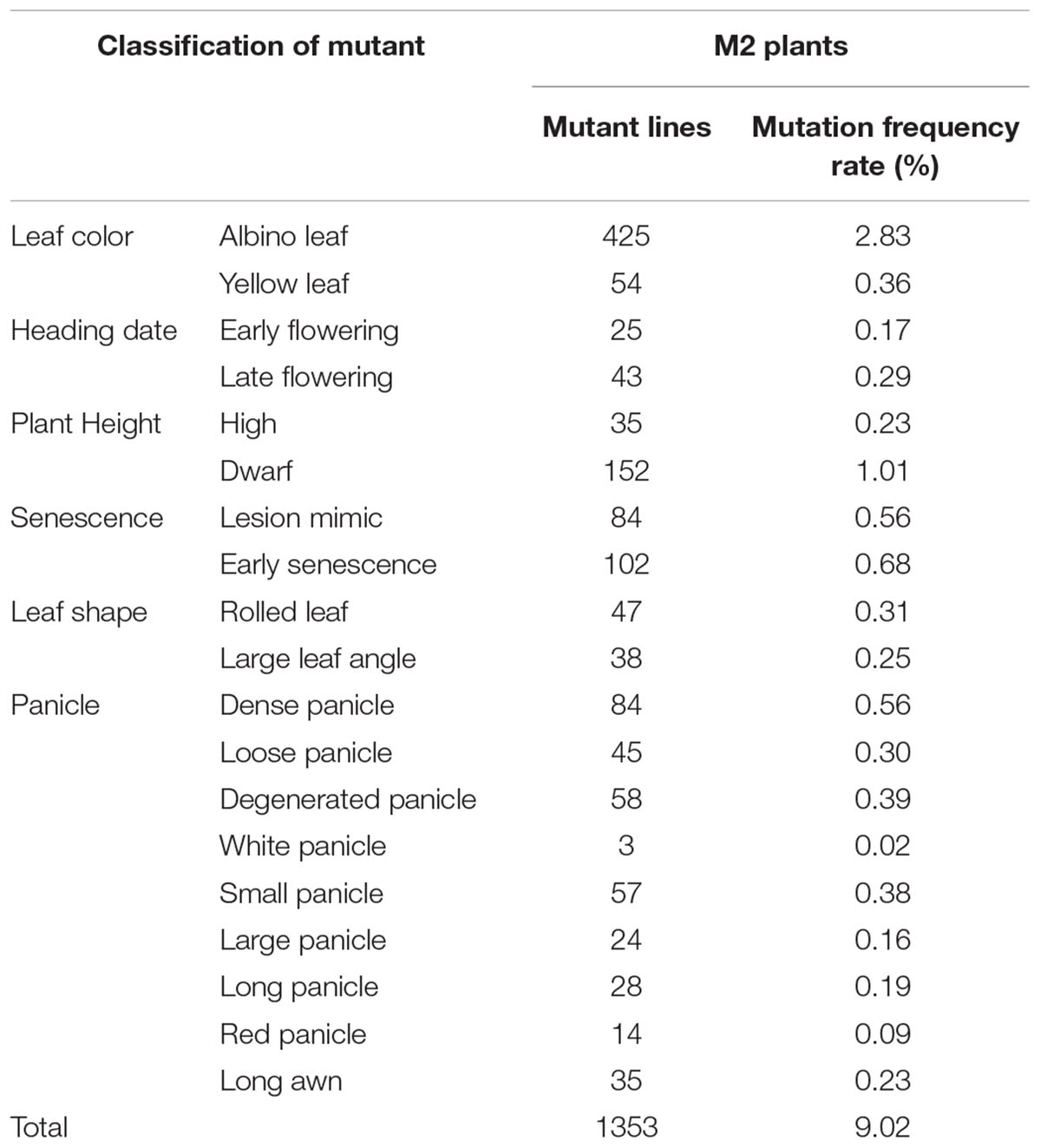
Table 1. Characterization of different mutant types of foxtail millet in the M2 plants [15,000 EMS lines (minimum 20 plants per line)] from the EMS library.

Figure 2. Pictures of mutants of foxtail millet in the M2 progeny from the EMS library. (A) T1, Height mutant. (B) T2, Dwarf mutant. (C) T3, Large panicle mutant. (D) T4, Yellow leaf mutant. (E) T5, Rolling leaf mutant. (F) T6, Large leaf angle mutant (Bar = 5 cm). (G) Different leaf phenotype mutants. L1–L11, Yellow leaf mutants with different chlorophyll contents. (H) Different panicle variation mutants. P1–P2, Loose panicle mutants; P3, Red awn mutant; P4, Small panicle mutant; P5, Large panicle mutant; P6, Red panicle mutant; P7, Long awn mutant; P8, White panicle mutant; P9, Degenerated panicle mutant; P10, Dense panicle mutant; CK, Yugu No. 1 (Bar = 0.5cm).
Mutation Density in the EMS Induced Population
Total > 38 × coverage depth was generated and over 98% of the reads were uniquely mapped to the Yugu No.1 reference genome. Based on comparison on the Yugu No.1 reference genome, the EMS-induced SNP/INDEL results are shown in Table 2. A total number of INDEL/SNPs were 36711, with 2295.44 average were identified compared with Yugu No.1, corresponding to an average estimated mutation density of 1/213 kb. Among the 16 resequenced mutant lines, intergenic variations were the maximum type and occupied at least 45.75% of the total variation. The regulatory region variation including upstream region, downstream region, 5′-untranslated region (5′-UTR), and 3′-untranslated region (3′-UTR) summed to 885.13 loci, with the proportion 38.56% of the total variation, as shown in Table 2. The variation that occurred within open reading frames (ORFs) containing intron, stop gained, splice variation, frame shift, codon insertion/deletion, synonymous coding, and nonsynonymous coding reached 359.68 loci, with the proportion 15.66% of the total variation. Further, SNP or INDEL loci were analyzed. The SNP results are shown in Table 3, and the INDEL results are shown in Table 4. On average, each line had 1100.94 loci for SNP variation and 1194.50 loci for INDEL variation. Average intergenic variation had 565.56 loci for SNPs and 1050.63 loci for INDELs, with proportions of 51.37 and 45.77% of the total variation, respectively (Figure 3 and Tables 3, 4).
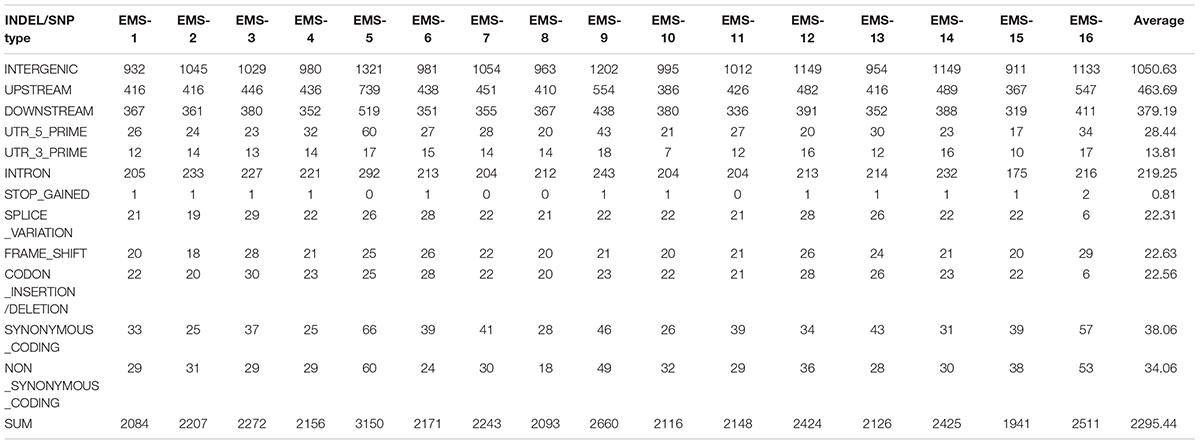
Table 2. Overview of all variation data containing INDELs/SNPs generated analyzing the 16 resequenced EMS induced mutant lines.
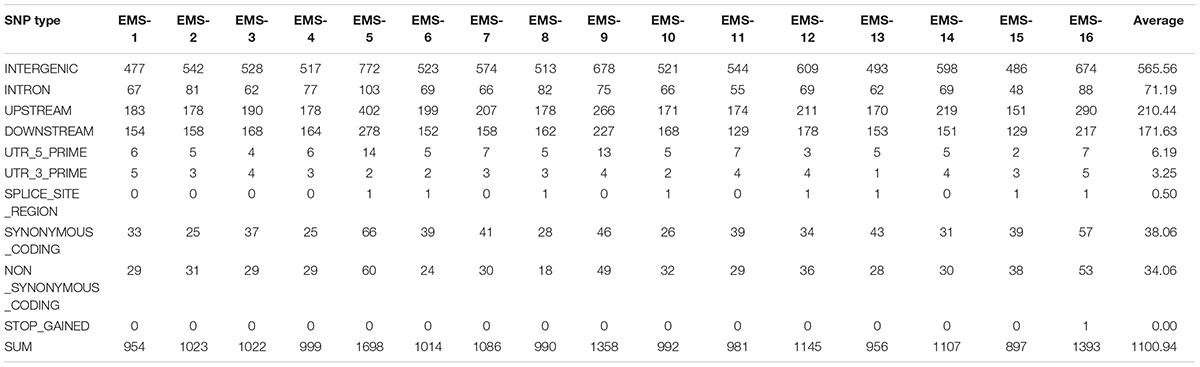
Table 3. Overview of single nucleotide polymorphism (SNP) data generated analyzing the 16 resequenced EMS induced mutant lines.
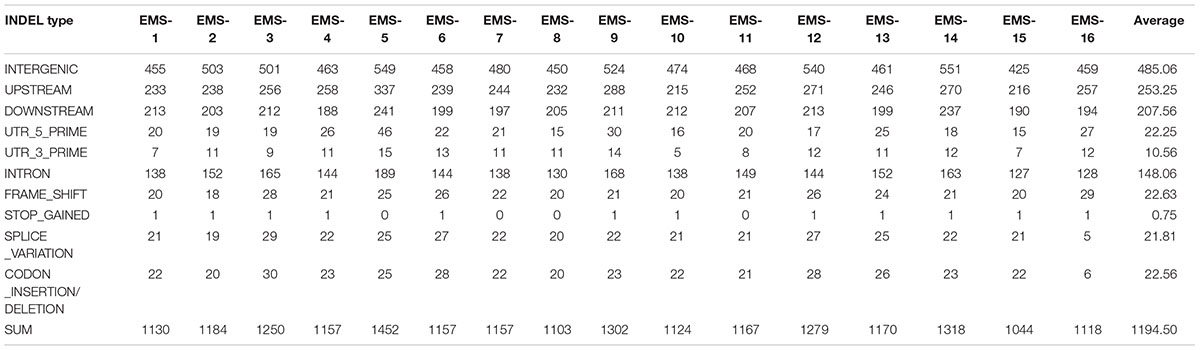
Table 4. Overview of insertion/deletion nucleotide polymorphism (INDEL) data generated analyzing the 16 resequenced EMS induced mutant lines.
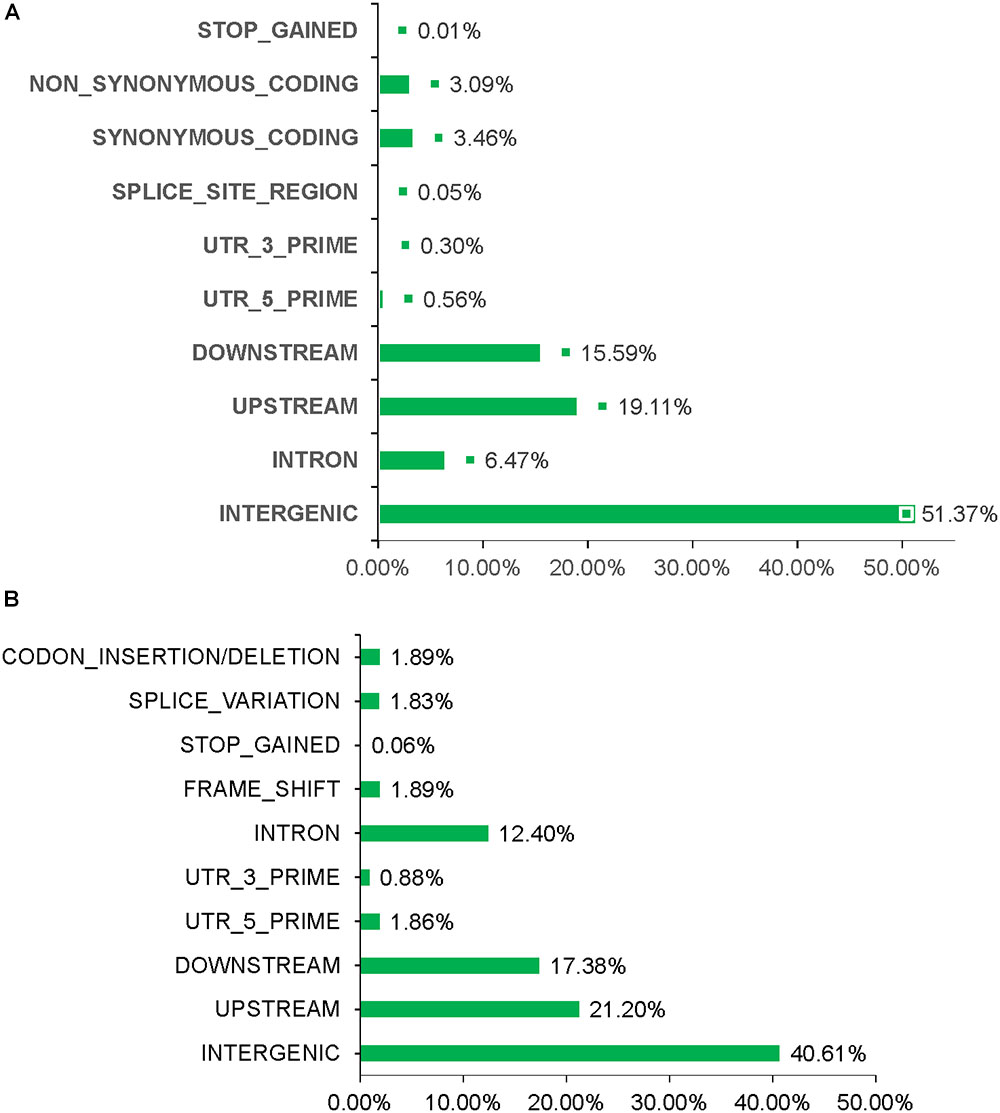
Figure 3. Overview of INDEL/SNP polymorphism data generated analyzing the sixteen resequenced mutant lines. (A) Different variation types for SNPs. (B) Different variation types for INDEL.
Map-Based Cloning Identified the Causal Gene WP1
To evaluate whether our foxtail millet EMS induced population was beneficial to clone a gene, a white panicle mutant (wp1.a) was selected to identify the causal gene (Figure 4A). The wp1.a plant was crossed with another landrace “SSR41.”
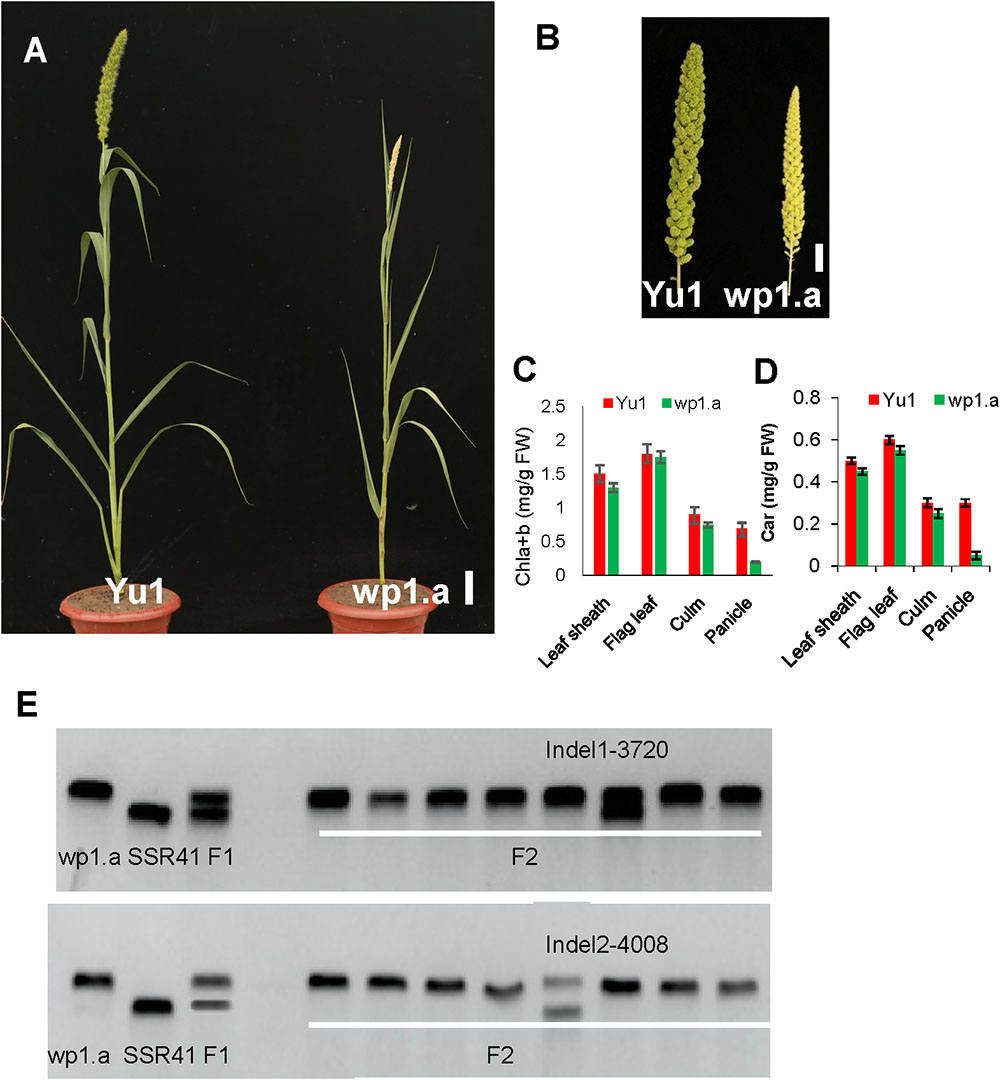
Figure 4. Phenotypic analysis of the white panicle mutant (wp1.a) and the wild type. (A) Phenotype comparison between a white panicle mutant (wp1.a) and the wild type (Yu1) at the mature stage (Bar = 5cm). (B) Panicle comparison between a white panicle mutant (wp1.a) and the wild type (Yu1) at the mature stage (Bar = 0.5cm). (C,D) Comparison of the pigment contents in different tissues between wp1.a mutants and the wild type; Chla+b, total chlorophyll; Car, carotenoid. Bars represent the sd of three measurements. Student’s t-test was performed on the raw data; asterisk indicates statistical significance at P < 0.01. (E) Rough mapping analysis for the WP1 gene using the F2 progeny. The rough mapping indicated that WP1 was located between Indel1 and Indel2 of chromosome 4.
In F1 progeny, all the plants displayed the normal panicle phenotype. In F2 progeny, the panicle phenotype segregated in normal phenotype plants (156) and white panicle phenotype plants (45), which accord with 3:1 ratio (χ2 = 1.21 < χ20.05,1), indicating that the wp1.a white phenotype was controlled by a single recessive mutation.
Notably, no great differences occurred except for panicle color and plant height between the wp1.a plant and the wild type (Figures 4A,B). The wp1.a plant had reduced height. Pigment assays showed that the chlorophyll (Chl) contents of leaf sheath, flag leaf and culm in wp1.a only were slightly lower than those of the wild type (Figure 4C). The content of chlorophyll in wp1.a panicle only reached 30% of that in wild type. (Figure 4D).
For rough mapping cloning, molecular markers were selected equally distributed on the 9 chromosomes with an average physical interval of approximately 5 Mb. A bulked pool analysis revealed that the molecular markers Indel1 -3720 and Indel2-4008 closely linked to WP1 gene ranging from 37.25 to 40.08 cM (Figure 4E), implying that WP1 is positioned at the end of chromosome 4. For fine mapping, a total 246 F2 homozygous recessive individuals were used, and INDEL markers (Indel3-4000, Indel4-3857) were developed (Figure 5A). Finally, WP1 was mapped to an 80 kb region between markers lndel3-4000 and lndel2-4008 (Figure 5A), which contained five candidate genes. We sequenced the five open reading frames in the wp1.a plant and found only an eight-base pair deletion within LOC101786849 (Figure 5B). The eight-base pair deletion located at the sixth exon in wp1.a caused the LOC101786849 transcript led to premature termination (Figure 5C).
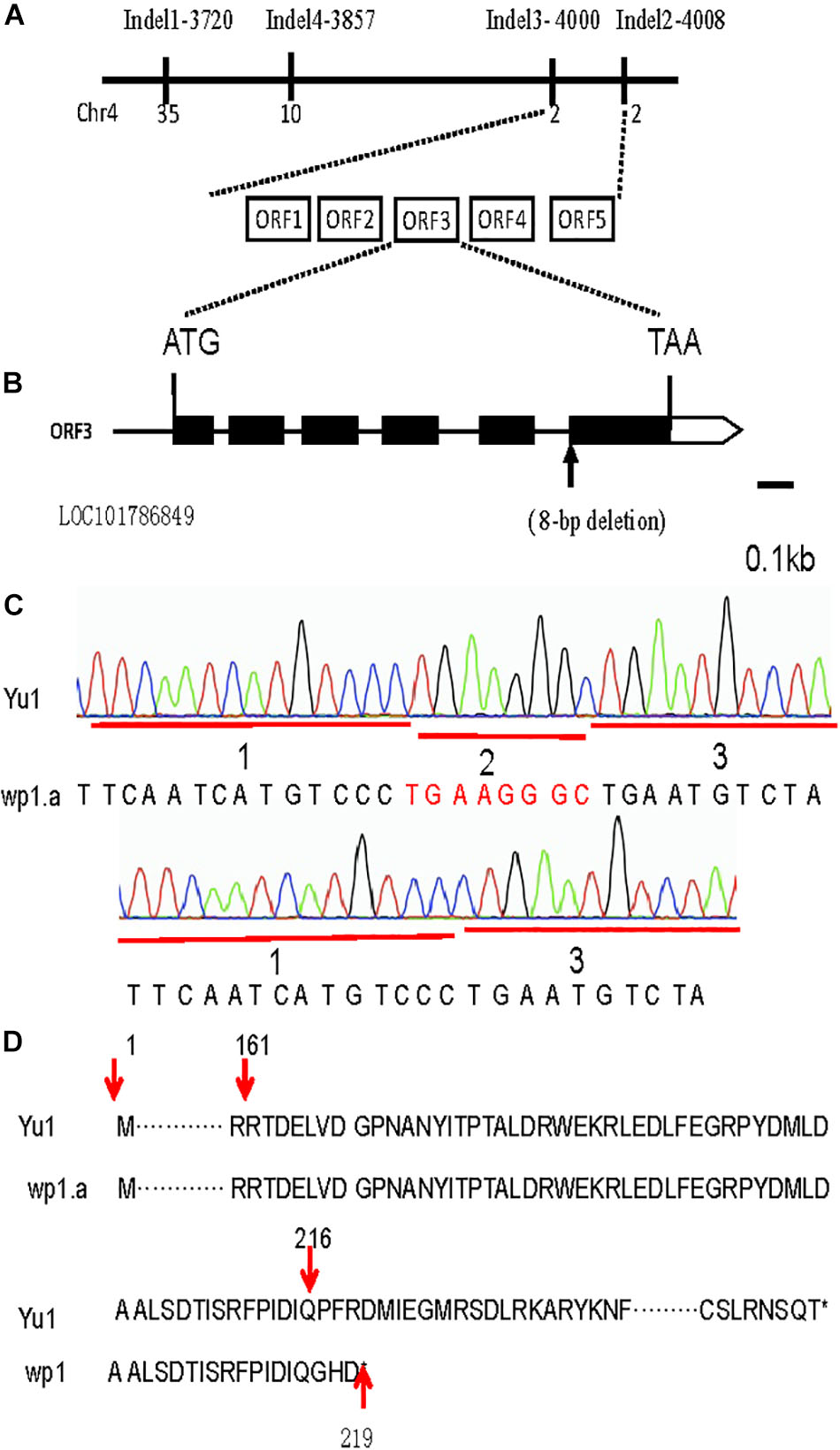
Figure 5. Map-based cloning of the WP1 gene. (A) The WP1 locus was narrowed down to an 80 kb region between InDel markers 4000 and 4008 of chromosome 4 using 246 F2 homozygous white panicle plants. Five open reading frames (ORF1-5) were predicted in the mapped region. (B) An eight-base pair deletion located at the junction of the fifth intron and sixth exon of LOC101786849 in wp1. ATG and TAA were the start and stop codons, respectively. (C) The mutant site comparison of LOC101786849 between white panicle (wp1) and Yu1. (D) The eight-base pair deletion caused the LOC101786849 transcript to not be normally cut and led to premature termination.
In our EMS library, there are five white panicle mutants. We sequenced the LOC101786849 for the five white panicle mutants. One of the white panicle mutants, named white panicle (wp1.b), was found to have a base pair change located at the junction of fifth intron and sixth exon of LOC101786849. WP1 transcript was checked between the wp1.b and Yu1. The PCR product was amplified by forward primer fp1 and reverse primer fp2 approaching the splicing site. The result showed that PCR product in wp1.b had a higher molecular weight (∼820bp) than in Yu1 (∼ 750 bp), which indicated that mutant splicing site (G-A) affected the splicing of LOC101786849 transcript products in wp1.b (Supplementary Figures S2A–D).
Cross between wp1.a and wp1.b was proceeded. The 21 seeds were gained successfully in F0. All the 21 plants showed the white panicle phenotype in F1 progeny. The analysis strongly verified that a white panicle mutant wp1.b was an allelic mutant of wp1.a. This result demonstrated that the methods could be used to quickly clone a foxtail millet gene using our EMS induced mutant library.
Discussion
Foxtail millet, as the oldest cultivated millet crops, needs warm weather and minimal water to ripen rapidly in dry and hot months of each year. In 2012, two departments both the Joint Genome Institute (JGI) of the Department of Energy, United States, and BGI (Beijing Genome Initiative), China, announced and published the foxtail millet genome sequence (Zhang et al., 2012). The foxtail millet genome sequence is closely related to several bioenergy crops such as pearl millet, napier grass (Pennisetum purpureum), switchgrass (Panicum virgatum). Compared with several bioenergy crops, foxtail millet has the advantage of being selected as a model system due to low amount of repetitive DNA, its small genome, short life cycle, more seeds, rich genetic diversity and inbreeding nature (Doust et al., 2009). At present, foxtail millet has been selected as a C4 model crop to explore crop architectural, evolutionary genomics from C3 to C4, and physiological characteristics of the C4 grass crops (Doust et al., 2009; Li and Brutnell, 2011). S. viridis is ancestor of S. italica. S. viridis widely distributed in the earth, whereas S. italica is one of the most earliest domesticated crops in China. S. viridis and S. italica have rich natural diversity, which provided a rich resource to unearth novel gene/allele for the important agronomical traits.
Jia went on the population genetics analysis of S. italica. He resequenced the 916 diverse accessions of S. italica and associated analysis using GWAS in S. italica. 47 agronomically important traits were associated with 512 loci including flowering time, plant height and inflorescence architecture (Jia et al., 2013a). China’s foxtail millet resources account for more than 80% of the world’s stock, but for a long time, due to the lack of reliable and efficient molecular marker information, researchers still lack of understanding of the population structure of these genetic resources, thus limiting the efficient exploration and deep utilization of millet genetic resources. Jia’s work provided a large amount of basic data information for genetic improvement and gene discovery of foxtail millet, greatly enrich the research of comparative genetics and functional genomics of cereal crops, and would have a profound impact on the improvement of cereal crops and genetic analysis of energy crops. In addition, S. viridis diversity samples have also been collected across the United States (Huang et al., 2016). Together, these collections provide an unprecedented opportunity for evolutionary research and domestication studies. However, compared with other staple cereal crops, not much research has been conducted on S. italica mutant resources for the development of genetic and functional resources. At present, the S. viridis mutant populations (NMU) and S. italica mutant populations (EMS) have also been constructed in the Brutnell and Diao labs (DDPSC and Chinese Academy of Agricultural Sciences, respectively), offering a lot of useful mutants resources for genetic studies in Setaria. In this study, we constructed a large capacity foxtail millet cv. Yugu No. 1 mutant library. Various mutant phenotypes including those for leaf color, plant stature, and panicle shape were obtained at a high (9%) mutation percentage. The mutation frequency was similar as those of other previous rice (Suzuki et al., 2008), soybean (Li et al., 2017), and maize mutant libraries (Till et al., 2004). Notably, panicle phenotypic variation was rich in our foxtail millet EMS mutant library, which reached up to 2.32% mutant frequency. Morphological variation of various panicle phenotypes indicated the foxtail millet mutant library would provide a valuable resource for studying panicle development. This foxtail millet mutant library enriched the foxtail millet germplasm and will accelerate functional genomics research.
EMS at a high concentration induced a high frequency of loci variation. Since multiple mutations may mask the mutant phenotype of interest, functional mutations need to be identified by cross analysis experiments with wild-type plants. By selecting an optimal EMS concentration, we avoided higher frequency variation, which was a result verified by random sequencing for sixteen M2 mutant materials. Based on random sequencing, the mutation density in our mutant population reached 1 mutation per 213 kb. EMS is an alkylating agent causing mainly G/C to A/T transitions. SNP variations accord with expectations, but there are many indel variation in each line after EMS treatment. We analysis the reason of so many indels. From the InDels length distribution (Figure S3 in the new version), we found that a large number of INDELs mainly center on insertion and deletion only 1 bp in length. Frankly speaking, there must be a lot of ineffective INDELs results that lead to overnumber. Other reason is, our SNP/INDELs analysis procedure adopted is looser standard (that less than 60% of the loci appears in samples), If the strict standards was adopted such as less than 40% of the loci appears in samples, the loci (SNP or INDELs) would become more few. LOC10178684 mutation in wp1.b happens from G to A, which conforms to this mutagenesis rule. Compared with the rice mutation frequency (1 mutant site/265 kb) (Suzuki et al., 2008), Arabidopsis mutation frequency (1 mutant site/89 kb) (Martín et al., 2009), tomato mutation frequency (1 mutant site/737 kb) (Okabe et al., 2011), sorghum mutation frequency (1 mutant site/526 kb) (Xin et al., 2008), maize (1 mutant site/485 kb) (Till et al., 2004), the mutation frequency in our foxtail mutant library was basically similar as the these species, sometimes even lower, which indicated that one test cross or two could reduce the background and reduce the complexity of confirmation of a functional gene.
Protocols for map-based cloning and bulked segregantanalysis (BSA) are now widely used for the discovery of causal genes in a time-efficient and cost-effective manner. Using these methods, AUX1which controlled the inflorescence architecture, was cloned in S. viridis (Huang et al., 2017). Similarly, Xiang (Chinese Academy of Agricultural Sciences, China) reported using this method to clone genes in S. italica that control grain size (Xiang et al., 2017). A transposon tagging population in S. viridis had been constructed in the Brutnell lab at DDPSC (Personal communication), strengthening resources for both forward and reverse genetic studies. Using the BSA cloning method, we provided an example for rapid cloning with the WP1 gene using F2 cross progeny. An eight-base pair deletion located at the sixth exon of wp1.a in LOC101786849 caused the premature termination in wp1. An allele wp1.b verified that WP1 was responsible for white panicle phenotype. The WP1 gene encoded a member of the phytoene synthase family. Phytoene synthase (PSY) is the first step in the synthesis of carotenoids. It makes the C20-geranyl diphosphate (GGPP) molecule head-to-head condensation to produce a C40 carotenoid phytoene molecule. Subsequently, phytoene undergoes four desaturation reactions and produce lycopene (Cunningham and Gantt, 1998). Due to the lack of carotenoids, mutants with deficiencies in carotenoid precursor synthesis exhibit a variety of phenotypes, such as albino or pale seedlings that are not viable in light, which support the conclusion that defects in a carotenoid precursor are related with chloroplast development/synthesis (Fang et al., 2008).
Because the wp1.a mutant had a white panicle phenotype; the indication was that the chloroplast development/synthesis was indirectly influenced in the wp1.a mutant. This example verified that our EMS mutant library was beneficial for cloning a gene quickly by map-based cloning methods. In the future, our foxtail millet mutants could be primarily abiotic stress tolerant, particularly to drought and salinity, with high photosynthetic efficiency; therefore, manipulating these agronomic traits can improve its water use efficiency in genetic engineering for abiotic stress tolerance and breeding for high photosynthetic rate.
Author Contributions
ZZ constructed the foxtail millet mutant library. ZZ, NL, JW, and XC conceived the original screening and cross experiments. ZZ, ZC, JS, NL, and BC analyzed the phenotypes and cloned WP1. ZZ and TL supervised and contributed to the writing.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We thank National Key Research and Development Program of China (2018YFD1000706), Project973 (2015CB150103), CAAS Innovation Project (CAAS-XTCX2016002), National Natural Science Foundation Project (31861143006), and Fundamental Research Funds for Central Non-profit Scientific Institution for support.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2019.00369/full#supplementary-material
Footnotes
- ^http://www.fao.org/faostat/zh/#data/QC
- ^https://www.ncbi.nlm.nih.gov/genome/10982
- ^http://sourceforge.net/projects/picard/
References
Arnon, D. I. (1949). Copper enzymes in isolated chloroplasts. Polyphenoloxidase in Beta vulgaris. Plant Physiol. 24, 1–15. doi: 10.1104/pp.24.1.1
Barton, L., Newsome, S. D., Chen, F. H., Wang, H., and Guilderson, T. P. (2009). Agricultural origins and the isotopic identity of domestication in northern China. Proc. Natl. Acad. Sci. U.S.A. 106, 5523–5528. doi: 10.1073/pnas.0809960106
Bennetzen, J. L., Schmutz, J., Wang, H., Percifield, R., and Hawkins, J. (2012). Reference genome sequence of the model plant setaria. Nat. Biotechnol. 30, 555–561. doi: 10.1038/nbt.2196
Brutnell, T. P., Wang, L., Swartwood, K., Goldschmidt, A., and Jackson, D. (2010). Setaria viridis: a model for C4 photosynthesis. Plant Cell 22, 2537–2544. doi: 10.1105/tpc.110.075309
Cunningham, J. A., and Gantt, E. (1998). Genes and enzymes of carotenoid biosynthesis in plants. Annu. Rev. Plant Biol. 49, 557–583. doi: 10.1146/annurev.arplant.49.1.557
Doust, A. N., Devos, K. M., Gadberry, M. D., Gale, M. D., and Kellogg, E. A. (2004). Genetic control of branching in foxtail millet. Proc. Natl. Acad. Sci. U.S.A. 101, 9045–9050. doi: 10.1073/pnas.0402892101
Doust, A. N., Kellogg, E. A., Devos, K. M., and Bennetzen, J. L. (2009). Foxtail millet: a sequence-driven grass model system. Plant Physiol. 149, 137–141. doi: 10.1104/pp.108.129627
Fang, J., Chai, C., Qian, Q., Li, C., and Tang, J. (2008). Mutations of genes in synthesis of the carotenoid precursors of ABA lead to pre-harvest sprouting and photo-oxidation in rice. Plant J. 54, 177–189. doi: 10.1111/j.1365-313X.2008.03411.x
Fang, X., Dong, K., Wang, X., Liu, T., and He, J. (2016). A high density genetic map and QTL for agronomic and yield traits in Foxtail millet [Setaria italica (L.) P. Beauv.]. BMC Genomics 17:336. doi: 10.1186/s12864-016-2628-z
Feldman, M. J., Paul, R. E., Banan, D., Barrett, J. F., and Sebastian, J. (2017). Time dependent genetic analysis links field and controlled environment phenotypes in the model C4 grass setaria. PLoS Genet. 13:e1006841. doi: 10.1371/journal.pgen.1006841
Gupta, P. K., and Yashvir (1976). Induced mutations in foxtail millet (Setaria italica Beauv.) : II. viable mutations in ear characters induced by gamma rays, EMS and dES. Theor. Appl. Genet. 48, 131–136. doi: 10.1007/BF00281655
Huang, P., Jiang, H., Zhu, C., Barry, K., and Jenkins, J. (2017). Sparse panicle1 is required for inflorescence development in Setaria viridis and maize. Nat. Plants 3:17054. doi: 10.1038/nplants.2017.54
Huang, P., Shyu, C., Coelho, C. P., Cao, Y., and Brutnell, T. P. (2016). Setaria viridis as a model system to advance millet genetics and genomics. Front. Plant Sci. 7:1781. doi: 10.3389/fpls.2016.01781
Jia, G., Huang, X., Zhi, H., Zhao, Y., and Zhao, Q. (2013a). A haplotype map of genomic variations and genome-wide association studies of agronomic traits in foxtail millet (Setaria italica). Nat. Genet. 45, 957–961. doi: 10.1038/ng.2673
Jia, G., Shi, S., Wang, C., Niu, Z., and Chai, Y. (2013b). Molecular diversity and population structure of chinese green foxtail [Setaria viridis (L.) Beauv.] revealed by microsatellite analysis. J. Exp. Bot. 64, 3645–3656. doi: 10.1093/jxb/ert198
Lata, C., Gupta, S., and Prasad, M. (2013). Foxtail millet: a model crop for genetic and genomic studies in bioenergy grasses. Crit. Rev. Biotechnol. 33, 328–343. doi: 10.3109/07388551.2012.716809
Lata, C., Jha, S., Dixit, V., Sreenivasulu, N., and Prasad, M. (2011). Differential antioxidative responses to dehydration-induced oxidative stress in core set of foxtail millet cultivars [Setaria italica (L.)]. Protoplasma 248, 817–828. doi: 10.1007/s00709-010-0257-y
Li, P., and Brutnell, T. P. (2011). Setaria viridis and Setaria italica, model genetic systems for the panicoid grasses. J. Exp. Bot. 62, 3031–3037. doi: 10.1093/jxb/err096
Li, Z., Jiang, L., Ma, Y., Wei, Z., and Hong, H. (2017). Development and utilization of a new chemically-induced soybean library with a high mutation density? J. Integr. Plant Biol. 59, 60–74. doi: 10.1111/jipb.12505
Martín, B., Ramiro, M., Martínez-Zapater, J., and Alonso-Blanco, C. (2009). A high-density collection of EMS-induced mutations for TILLING in Landsberg erecta genetic background of arabidopsis. BMC Plant Biol. 9:147. doi: 10.1186/1471-2229-9-147
McKenna, A., Hanna, M., Banks, E., Sivachenko, A., Cibulskis, K., Kernytsky, A., et al. (2010). The genome analysis toolkit: a mapreduce framework for analyzing next-generation DNA sequencing data. Genome Res. 20, 1297–303. doi: 10.1101/gr.107524.110
Muthamilarasan, M., and Prasad, M. (2015). Advances in setaria genomics for genetic improvement of cereals and bioenergy grasses. Theor. Appl. Genet. 128, 1–14. doi: 10.1007/s00122-014-2399-3
Muthamilarasan, M., Venkata Suresh, B., Pandey, G., Kumari, K., and Parida, S. K. (2014). Development of 5123 intron-length polymorphic markers for large-scale genotyping applications in foxtail millet. DNA Res. 21, 41–52. doi: 10.1093/dnares/dst039
Okabe, Y., Asamizu, E., Saito, T., Matsukura C., and Ariizumi, T. (2011). Tomato TILLING technology: development of a reverse genetics tool for the efficient isolation of mutants from micro-tom mutant libraries. Plant Cell Physiol. 52, 1994–2005. doi: 10.1093/pcp/pcr134
Pandey, G., Misra, G., Kumari, K., Gupta, S., and Parida, S. K. (2013). Genome-wide development and use of microsatellite markers for large-scale genotyping applications in foxtail millet [Setaria italica (L.)]. DNA Res. 20, 197–207. doi: 10.1093/dnares/dst002
Pant, S. R., Irigoyen, S., Doust, A. N., Scholthof, K. B., and Mandadi, K. K. (2016). Setaria: a food crop and translational research model for c4 grasses. Front. Plant Sci. 7:1885. doi: 10.3389/fpls.2016.01885
Suzuki, T., Eiguchi, M., Kumamaru, T., Satoh, H., and Matsusaka, H. (2008). MNU-induced mutant pools and high performance TILLING enable finding of any gene mutation in rice. Mol. Genet. Genomics 279, 213–223. doi: 10.1007/s00438-007-0293-2
Tang, S., Li, L., Wang, Y., Chen, Q., and Zhang, W. (2017). Genotype-specific physiological and transcriptomic responses to drought stress in Setaria italica (an emerging model for Panicoideae grasses). Sci. Rep. 7:10009. doi: 10.1038/s41598-017-08854-6
Till, B. J., Reynolds, S., Weil, C., Springer, N., and Burtner, C. (2004). Discovery of induced point mutations in maize genes by TILLING. BMC Plant Biol. 4:12. doi: 10.1186/1471-2229-4-12
Wang, C., Jia, G., Zhi, H., Niu, Z., and Chai, Y. (2012). Genetic diversity and population structure of chinese foxtail millet [Setaria italica (L.) Beauv.] landraces. G3 2, 769–777. doi: 10.1534/g3.112.002907
Wang, J., Wang, Z., Du, X., Yang, H., and Han, F. (2017). A high-density genetic map and QTL analysis of agronomic traits in foxtail millet [Setaria italica (L.) P. Beauv.] using RAD-seq. PLoS One 12:e0179717. doi: 10.1371/journal.pone.0179717
Xiang, J., Tang, S., Zhi, H., Jia, G., and Wang, H. (2017). Loose panicle1 encoding a novel WRKY transcription factor, regulates panicle development, stem elongation, and seed size in foxtail millet [Setaria italica (L.) P. Beauv.]. PLoS One 12:e0178730. doi: 10.1371/journal.pone.0178730
Xin, Z., Wang, M., Barkley, N., Burow, G., and Franks, C. (2008). Applying genotyping (TILLING) and phenotyping analyses to elucidate gene function in a chemically induced sorghum mutant population. BMC Plant Biol. 8:103. doi: 10.1186/1471-2229-8-103
Xu, W., Wei, L., Qu, W., Liang, Z., and Wang, J. (2011). A novel antifungal peptide from foxtail millet seeds. J. Sci. Food Agric. 91, 1630–1637. doi: 10.1002/jsfa.4359
Yadav, C. B., Bonthala, V. S., Muthamilarasan, M., Pandey, G., and Khan, Y. (2015). Genome-wide development of transposable elements-based markers in foxtail millet and construction of an integrated database. DNA Res. 22, 79–90. doi: 10.1093/dnares/dsu039
Yang, J., Thames, S., Best, N. B., Jiang, H., and Huang, P. (2018). Brassinosteroids modulate meristem fate and differentiation of unique inflorescence morphology in Setaria viridis. Plant Cell 30, 48–66. doi: 10.1105/tpc.17.00816
Zhang, G., Liu, X., Quan, Z., Cheng, S., and Xu, X. (2012). Genome sequence of foxtail millet (Setaria italica) provides insights into grass evolution and biofuel potential. Nat. Biotechnol. 30, 549–554. doi: 10.1038/nbt.2195
Keywords: foxtail millet, EMS, mutant, mutation frequency, white panicle
Citation: Sun J, Luu NS, Chen Z, Chen B, Cui X, Wu J, Zhang Z and Lu T (2019) Generation and Characterization of a Foxtail Millet (Setaria italica) Mutant Library. Front. Plant Sci. 10:369. doi: 10.3389/fpls.2019.00369
Received: 13 December 2018; Accepted: 11 March 2019;
Published: 29 March 2019.
Edited by:
Goetz Hensel, Leibniz-Institut für Pflanzengenetik und Kulturpflanzenforschung (IPK), GermanyReviewed by:
Miriam Ewa Szurman-Zubrzycka, University of Silesia of Katowice, PolandJishan Niu, Henan Agricultural University, China
Copyright © 2019 Sun, Luu, Chen, Chen, Cui, Wu, Zhang and Lu. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Zhiguo Zhang, emhhbmd6aGlndW9AY2Fhcy5jbg== Tiegang Lu, bHV0aWVnYW5nQGNhYXMuY24=
†These authors have contributed equally to this work