 Andrés J. Cortés
Andrés J. Cortés Luz N. Garzón2
Luz N. Garzón2 Jhon B. Valencia
Jhon B. Valencia Santiago Madriñán
Santiago Madriñán
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 03 December 2018
Sec. Evolutionary and Population Genetics
Volume 9 - 2018 | https://doi.org/10.3389/fpls.2018.01700
This article is part of the Research Topic Population Genetics and Evolutionary History of Plants in Mountainous Regions View all 29 articles
How diversity arises and what is the relative role of allopatric and ecological divergence are among the most persistent questions in evolution and ecology. Here, we assessed whether ecological divergence has enhanced the diversification of the Neotropical alpine plant complex Espeletia, also known as frailejones. This genus has one of the highest diversification rates ever reported and is distributed in the world’s fastest evolving biodiversity hotspot, the Páramo (Neotropical alpine grasslands at elevations of c. 2800–4700 m). Our goal was to determine whether ecology plays a role in divergence within the Espeletia complex by quantifying genome-wide patterns of ecological divergence. We characterized 162 samples of the three most common and contrasting ecotypes (distinct morphotypes occupying particular habitats) co-occurring in six localities in the northern Andes using Genotyping by Sequencing. Contrasting ecotypes were caulescent cloud forest populations, caulescent populations from wind-sheltered and well-irrigated depressions and acaulescent populations from wind-exposed drier slopes. We found high polymorphism with a total of 1,273 single nucleotide polymorphisms (SNPs) that defined the relationships among nine genetic clusters. We quantified allelic associations of these markers with localities and habitats using 18 different general and mixed-effects statistical models that accounted for phylogenetic distance. Despite that these models always yielded more SNPs associated with the localities, markers associated with the habitat types were recovered too. We found strong evidence for isolation-by-distance (IBD) across populations despite rampant gene flow, as expected for plant groups with limited seed dispersal. Contrasts between populations of different habitat types showed that an isolation-by-environment (IBE) trend emerged and masked the IBD signal. Maximum likelihood estimation of the number of migrants per generation (Nem) among ecotypes confirmed the IBE pattern. This result illustrates the importance of mountains’ environmental variation at a local scale in generating rapid morphological radiations and maintaining multiple adaptations in a fast-evolving ecosystem like the Páramo.
Diversification is recognized as an important process generating phenotypic and genetic variation in plants and animals. However, its relationship with ecological variation and genomic and morphological divergence is just starting to be understood (Nosil and Feder, 2011; Strasburg et al., 2011). For instance, despite ecological-driven diversification often being considered rare (Coyne and Orr, 2004), it has progressively been acknowledged as a main driver of diversification (Schluter, 2001, 2009; Rieseberg and Willis, 2007; Nosil and Harmon, 2009; Nosil, 2012). Yet, the consequences of ecological divergence at the genetic level remain poorly documented (Gompert et al., 2014). Rapidly evolving clades from highly heterogeneous ecosystems may bridge this gap since they offer a good set up to explore the genomic patterns associated with ecological variation during divergence with gene flow, ultimately leading to insights into how organisms adapt and diversify (Stinchcombe and Hoekstra, 2008; Hoffmann and Sgro, 2011; Savolainen et al., 2013).
The Páramo, the world’s fastest evolving biodiversity hotspot (Madriñán et al., 2013), is an alpine ecosystem dominated by species-rich grasslands above the treeline in the American tropics (at elevations of ca. 2800–4700), that despite its small surface area (35,000 km2), may contain over 3,000 plant species, many of which are found nowhere else on the planet (Luteyn, 1999; Hughes and Atchison, 2015). Plant groups that occupy these ‘sky islands’ (Sklenář et al., 2014) are therefore good candidates to explore ecological divergence because they are a likely product of unique adaptations to an extreme environment that evolved during the last five million years when the Andes reached an altitude that was capable of sustaining this type of vegetation (Antonelli et al., 2009; Hoorn et al., 2010; Madriñán et al., 2013). Three hypotheses have been proposed to explain the unparalleled diversification rate in the Páramo. First, Pleistocene glacial cycling coupled with the Andean uplift during the last 2.4 Myr led to repeated periods of connectivity and spatial isolation, which is thought to have generated many taxa in a short period – the ‘species pump hypothesis.’ Second, even though allopatric diversification may have been the main cause of isolation, the high levels of ultraviolet (UV) light of the high tropical mountains may have induced a rapid mutation rate (Davies et al., 2004; Willis et al., 2009) and in turn promoted morphological differentiation and even reproductive isolation. Third, it is also possible that environmental heterogeneity and ecological opportunity were the major factors driving rapid diversification. Mountains’ local scale environmental variation helps maintaining adaptation, which in turn can trigger the isolation needed for ecotypes to evolve into new species (Bridle and Vines, 2007; Cortés, 2013, 2015; Cortés et al., 2014; Cortés and Blair, 2018b; Cortés and Wheeler, 2018).
Here, we examine the iconic Espeletia complex, an appropriate model because it is one of the most rapidly evolving plant groups (Madriñán et al., 2013). Espeletia species (ca. 120) are ecologically abundant in the Páramo (Luteyn, 1999). Espeletia likely originated in the Venezuelan Andes (Pouchon et al., 2018) from where it spread southward through the Colombian Eastern Cordillera to the Ecuadorian Andes, followed by a northward colonization of the Colombian Central and Western Cordilleras (Cuatrecasas, 2013). The phylogeny of the genus is largely unresolved (Diazgranados and Barber, 2017) and represents a network due to weak species boundaries (Pouchon et al., 2018) and massive hybridization (Rauscher, 2002), likely consequences of its rapid diversification. The predominant pattern of genetic differentiation among Espeletia populations is isolation-by-distance (IBD; Diazgranados and Barber, 2017; Padilla-GonzáLez et al., 2017), indicating allopatric divergence reinforced by limited seed dispersal. Espeletia plants are ecologically heterogeneous, with populations adapted to grow in the wet depressions of high valleys, in the dry exposed slopes, or even within the forests at the tree line, therefore experiencing a wide range of climatic conditions within elevations and localities. Despite the heterogeneity in the habitat types, isolation-by-environment (IBE), in which genetic and environmental distances are positively correlated independent of geographic distance, has never been tested explicitly within this system, as has been partially envisioned for other Páramo genera such as Lupinus (Hughes and Eastwood, 2006; Contreras-Ortiz et al., 2018), Loricaria (Kolar et al., 2016) and Senecio (Duskova et al., 2017).
Genomic studies in plant populations have proven that genome–environment associations (GEA) are useful to identify genomic signatures of IBE, which are the genetic association with environmental variables. Mostly, these studies have associated single-nucleotide polymorphism (SNP) alleles and parameters from the environment of origin to infer genetic adaptive variants. For instance, Turner et al. (2010) and Fischer et al. (2013) predicted genetic adaptive variation to serpentine soils and to topo-climatic factors in Arabidopsis lyrata and A. halleri, respectively, Hancock et al. (2011) captured climate-adaptive loci in a set of geographically diverse A. thaliana, Yeaman et al. (2016) found convergent adaptation in two species of conifers, Pluess et al. (2016) detected local adaptation to climate at a regional scale in Fagus sylvatica, and Cortés and Blair (2018a) found evidence for disruptive selection on drought tolerance in wild Phaseolus vulgaris. Since the GEA approach is a robust strategy for characterizing the genomic landscape of IBE and discovering genetic sources of adaptive variation, in this study we couple GEA with an in-depth sampling of Espeletia ecotypes across the three contrasting habitats typically observed in the Páramo, which are: the upper limit of the wet cloud forest, the well-irrigated depressions of the high valleys, and the more wind-exposed and drier slopes. Abiotic stresses such as frost and flooding are known to vary among these habitats, as well as the plant traits (e.g., plant height, pubescence, presence of aerenchyma) that likely confer adaptation to this assortment of environmental conditions (Monasterio and Sarmiento, 1991; Sklenář et al., 2010a,b, 2012, 2016).
By combining a sampling spanning the three habitats where Espeletia is typically found across several representative localities of the genus’ distribution in the Central and Eastern Cordilleras of the northern Andes, with a whole-genome genotyping method such as Genotyping by Sequencing (GBS, Elshire et al., 2011), we aimed in this study to (1) quantify the genome-wide patterns of geographic and ecological divergence among Espeletia populations and ecotypes, and (2) compute SNP allelic associations with localities and habitats in order to identify sources for genetic isolation and adaptive variation. We hypothesized that if ecological differentiation contributed to genetic divergence in the Espeletia complex, then we should be able to recover for the first time more subtle and localized signals of IBE, besides a predominant pattern of IBD.
A total of 162 individual plants from the Espeletia complex were used in this study (Figure 1 and Supplementary Table S1). These individuals comprised 17 different taxonomic groups (Espeletia grandiflora, E. uribei, E. killipii, Espeletiopsis colombiana, E. lopezii, Espeletiopsis jimenez-quesadae, E. boyasensis, E. congestiflora, E. arbelaezii, E. discoidea, E. hartwegiana, E. estanislana, E. standleyana, E. conglomerata, E. argentea, E. cabrerensis, E. summapacis, from a total of ca. 120, according to Cuatrecasas (2013), although this taxonomy is likely to change, as proposed by Pouchon et al. (2018), because of weak species boundaries and reiterative paraphyly) and were chosen to be representative ecotypes of the three most common habitats: caulescent populations from the cloudy forest (F) and from wind-sheltered well-irrigated depressions (W), and acaulescent populations from wind-exposed drier slopes (D), co-occurring in six localities in the northern Andes (Ruíz in the Colombian Central Cordillera, and Santurbán, Cocuy, Guantiva, Chingaza and Sumapaz, from north to south, in the Colombian Eastern Cordillera). Two taxonomic groups were found in the wind-exposed drier slopes of Guantiva, so there was a total of 19, rather than 18 (6 × 3), locality–habitat combinations, hereinafter referred to as populations. This sampling is unique because the chosen populations merge properties associated with geographic as well as ecological isolation. Sampled leaves were shaved using a field knife in order to improve drying in silica gel. Genomic DNA was extracted using the QIAGEN DNeasy Plant Mini Kit (QIAGEN, Germany). DNA concentration was quantified in a Qubit® dsDNA HS Fluorometer (Life Technologies, Sweden). Two 96-plex genotyping-by-sequencing (GBS) libraries were performed with MsII digestions according to Elshire et al. (2011) and sequenced at LGC Genomics (Berlin, Germany).
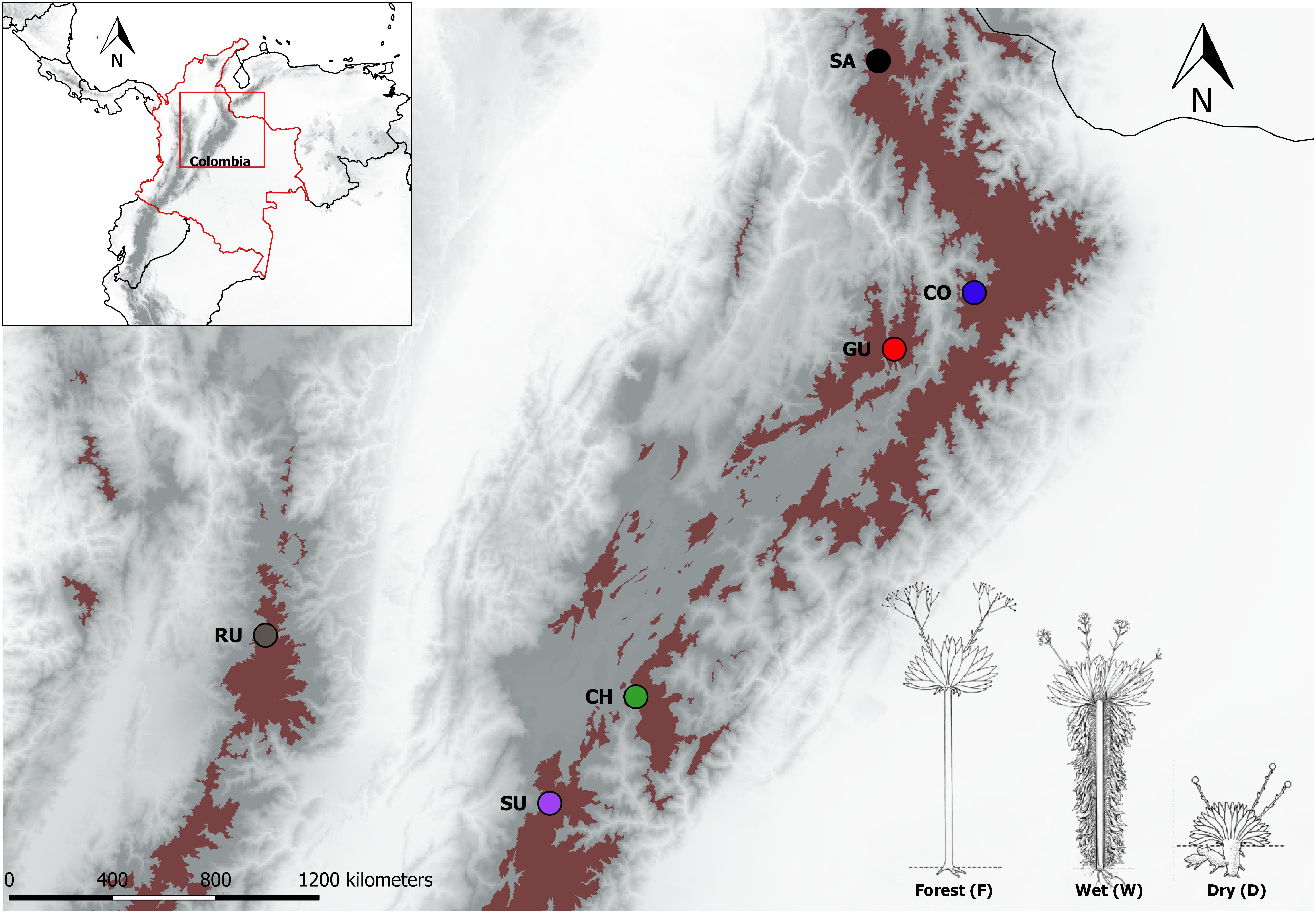
FIGURE 1. Páramo localities in the northern Andes sampled in this study. Red shaded areas mark the 2013 Páramo delimitation in the Colombian Andes as taken from http://paramo.uniandes.edu.co/. Localities are identified by different colors and abbreviated as follows: Chingaza (CH), Cocuy (CO), Guantiva (GU), Santurbán (SA), Sumapaz (SU), and Ruíz (RU). In the bottom right, a schematic drawing of the different ecotypes according to Cuatrecasas (2013), are coded as follows: caulescent populations from the cloud forest (F), caulescent populations from wind-sheltered well-irrigated depressions (W) and acaulescent populations from wind-exposed drier slopes (D).
Demultiplexing, cleaning, and filtering of Illumina reads was performed with the Illumina bcl2fastq v. 2.17.1.14 software (Illumina, San Diego, CA, United States). A total of 1 or 2 mismatches or Ns were allowed in the barcode read when the barcode distances between all libraries allowed for it. Demultiplexing of library groups into samples was done according to inline barcodes and verification of restriction site. No mismatches or Ns were allowed in the inline barcodes but Ns were allowed in the restriction site. Restriction enzyme site filtering of read 5′ ends was carried out so that reads with 5′ ends not matching the restriction enzyme site were discarded. Quality trimming of adapter-clipped reads was done by removing reads containing Ns, trimming reads at 3′-end to get a minimum average Phred quality score of 20 over a window of ten bases and discarding reads with final length < 20.
Alignment and clustering of combined reads was performed with CD-HIT-EST v. 11, allowing up to 5% difference. Alignment of subsampled quality-trimmed reads against all clusters was done using BWA (Li and Durbin, 2007) v. 0.7.122, resulting in a single combined alignment for all samples in a coordinate-sorted BAM format. Variant discovery and genotyping of samples was done with Freebayes v. 1.0.2-163, allowing for a minimum base quality and a minimum supporting allele qsum of 10, and a read mismatch limit of three. Filtering of variants was done using a GBS-specific rule set with >5 read count for a locus, >5% minimum allele frequency (MAF) across all samples, and an observation of genotype frequency of at least 72% (116 samples). The filtered dataset was inspected with TASSEL v. 3.0 (Glaubitz et al., 2014), resulting in a final set of 1,273 SNPs.
We examined broad patterns of population structure using principal coordinates analysis (PCoA) implemented in Trait Analysis by aSSociation, Evolution and Linkage v.5 (Bradbury et al., 2007). The construction of customized PCoA diagrams was carried out in R v.3.3.1 (R Core Team). Population structure was further examined by an AMOVA test according to Excoffier et al. (2005), and by allowing admixture through a Bayesian analysis using STRUCTURE (Pritchard et al., 2000) on LD-free sites (R2 = 0.0648 ± 0.0002, CI 95%: 0.003 – 0.625). Five independent runs for each K value from K = 2 to K = 19 (the total number of expected populations) used an admixture model with 100,000 burn-ins and 200,000 iterations in the MCMC analysis. A bar graph of the results was generated for each K value using CLUMPP (Jakobsson and Rosenberg, 2007). The optimal K value was determined based on the PCoA diagrams, cross-run cluster stability and likelihood of the graph model following Evanno et al. (2005). Finally, in order to compute phylogenetic distances a phylogenetic tree was inferred using the program SNAPP 1.3.0 (Bryant et al., 2012) included in the package BEAST 2.4.5 (Bouckaert et al., 2014). We used all 19 populations as a priori designated clusters. The analysis was run for 1,000,000 generations sampling every 1,000 generations. The log files were evaluated for convergence with the program Tracer 1.5 (Drummond and Rambaut, 2007) and trees in the 95% highest posterior density (HPD) set were analyzed with SNAPP-TreeSetAnalyser 2.4.5 using 10% of topologies as burn-in. Resulted tree files (cloudgrams) were visualized using DensiTree (Bouckaert, 2010). A consensus tree was rooted using the populations from Santurbán, following Diazgranados and Barber (2017).
In order to explore subtle divergence patterns among populations and ecotypes, the entire dataset was partitioned by habitats and localities, so that the habitat-based dataset included populations from the Cocuy and Guantiva localities, whereas the locality-based dataset included populations from the remaining localities. SNP markers were filtered for each of these new datasets using the same MAF and missing data rules than for the full dataset (see previous section), leading to 897 and 812 SNP markers, respectively. This partition led to three different datasets (entire, locality-based and habitat-based datasets) that were used in subsequent analyses. Four reiteratively misplaced samples led to an acceptable error rate (Glaubitz et al., 2014) of 2.5% and were excluded from further analyses.
In order to perform GEA analyses between the SNP markers and the localities, and between the SNP markers and the habitats, we used the software Trait Analysis by aSSociation Evolution and Linkage. For these GEA analyses the habitats were ranked according to the expected, hereinafter referred to as theoretical, exposure to frost and soil moisture content, as coded in Supplementary Table S1. Six generalized (GLM) and mixed (MLM) linear models were compared for each of the three datasets for a total of 18 models. Within each model family, three models were built as follows: (1) model with the locality as a fixed effect, (2) model with the theoretical exposure to frost as a fixed effect and (3) model with the theoretical soil moisture content as a fixed effect. All models included the phylogenetic distance computed from the phylogeny as a covariate. All MLMs used an IBS kinship matrix as a random effect to control for genomic background implementing the EMMA and P3D algorithms to reduce computing time (Zhang et al., 2010). Localities and habitats were never considered as random effects, as is tradition in the majority of association scans, because our main purpose was to detect allelic associations precisely with these factors. QQ-plots of the P-values were inspected to assess whether excessive numbers of false positives were generated and choose in this way the optimum model. Significant associations were determined using a strict Bonferroni correction of P-values at α = 0.05, leading to a significance threshold of 3.9 × 10-5, 6.2 × 10-5, and 5.6 × 10-5 (0.05 divided by the number of markers, 1,273, 812, and 897) or -log10 of 4.4, 4.2, and 4.6, for the full, locality-based and habitat-based datasets, respectively.
A single genome can exhibit heterogeneous patterns of geographic (IBD) and ecological (IBE) differentiation (Strasburg et al., 2011). Therefore, in order to understand the landscape of divergence in the Espeletia complex, the entire dataset and the contrasting datasets for localities and habitats were further subdivided according to the allelic associations found in the previous section, so that SNP markers were retained if they were significantly associated with the locality, the theoretical exposure to frost or the theoretical soil moisture content. Since the full marker set was also maintained in each case, this partition series led to a total of eleven datasets. The dataset that only included contrasting populations for localities and associated markers with the theoretical soil moisture content had just one polymorphic site and was excluded from the rest of the analyses. Pairwise FST values, according to Weir and Cockerham (1984), were computed in each of the eleven datasets using customized R scripts. Bidirectional gene flow among pairs of populations was then estimated as the number of migrants per generation (Nem) following Beerli and Felsenstein (1999), which is a highly robust maximum likelihood estimation coupled with a coalescent framework. Networks depicting pairwise bidirectional migration rates across all datasets were drawn using the R package “qgraph” (Epskamp et al., 2012). Even though more sophisticated methods have been developed to estimate gene flow, the conceptual clarity and robustness of FST-based methods and Beerli and Felsenstein (1999)’s approach is still undoubted.
Explicit correlations between genetic differentiation and geographic and ecological distances were obtained among populations across all eleven datasets. The genetic differentiation was computed as FST/(1 – FST) according to Rousset (1997). On the other hand, the geographic distance was taken from the sampling coordinates using the R package “geosphere”, whereas the ecological distance was calculated independently for the theoretical exposure to frost and the theoretical soil moisture content, as ranked in Supplementary Table S1, using an Euclidean distance measure implemented in the R function dist. Therefore, for each dataset three different correlations with the genetic differentiation were considered (against the geographic distance and the two ecological distances), leading to a total of 33 Mantel tests. These tests used 1,000 permutations implemented in R (R Core Team). Significant associations were determined based on a strict Bonferroni correction of P-values at α = 0.05 (as detailed in Table 1).
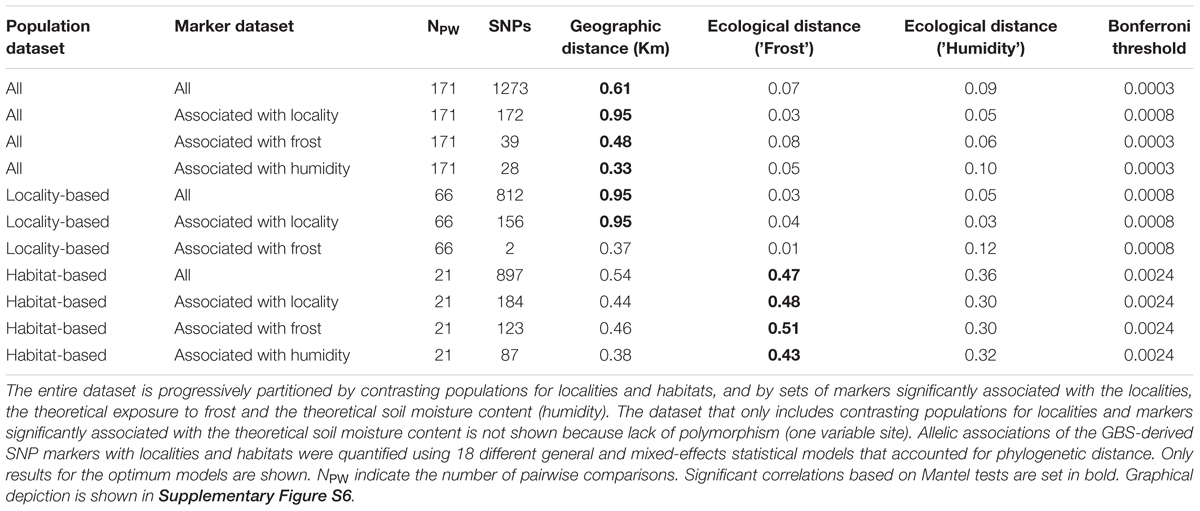
TABLE 1. Correlations between genetic differentiation, geographic distance, and ecological distance (exposure to frost and soil moisture content) among populations of Espeletia growing in 3 different habitats and 6 localities.
The principal components analysis of 1,273 GBS-derived SNP markers recovered nine genetic clusters (Figure 2) comprising the 17 taxonomic groups and 19 populations sampled. Individuals from the localities of Ruíz and Santurbán were both independent clusters, whereas individuals from the Chingaza and Sumapaz localities clustered with the caulescent individuals from the cloud forest and the wind-sheltered well-irrigated depressions of Guantiva. The acaulescent individuals from Guantiva formed two clusters and were situated between two clusters conformed by individuals from the Cocuy locality; one of them, close to the Santurbán cluster, clustered caulescent individuals from the cloud forest, while the other grouped the rest of the individuals from Cocuy. The cluster containing individuals from the localities of Chingaza and Sumapaz and caulescent individuals from the locality of Guantiva differentiated by locality but not by habitat (Supplementary Figure S1), suggesting that all 162 individuals were clustered in a maximum of nine different genetic groupings.
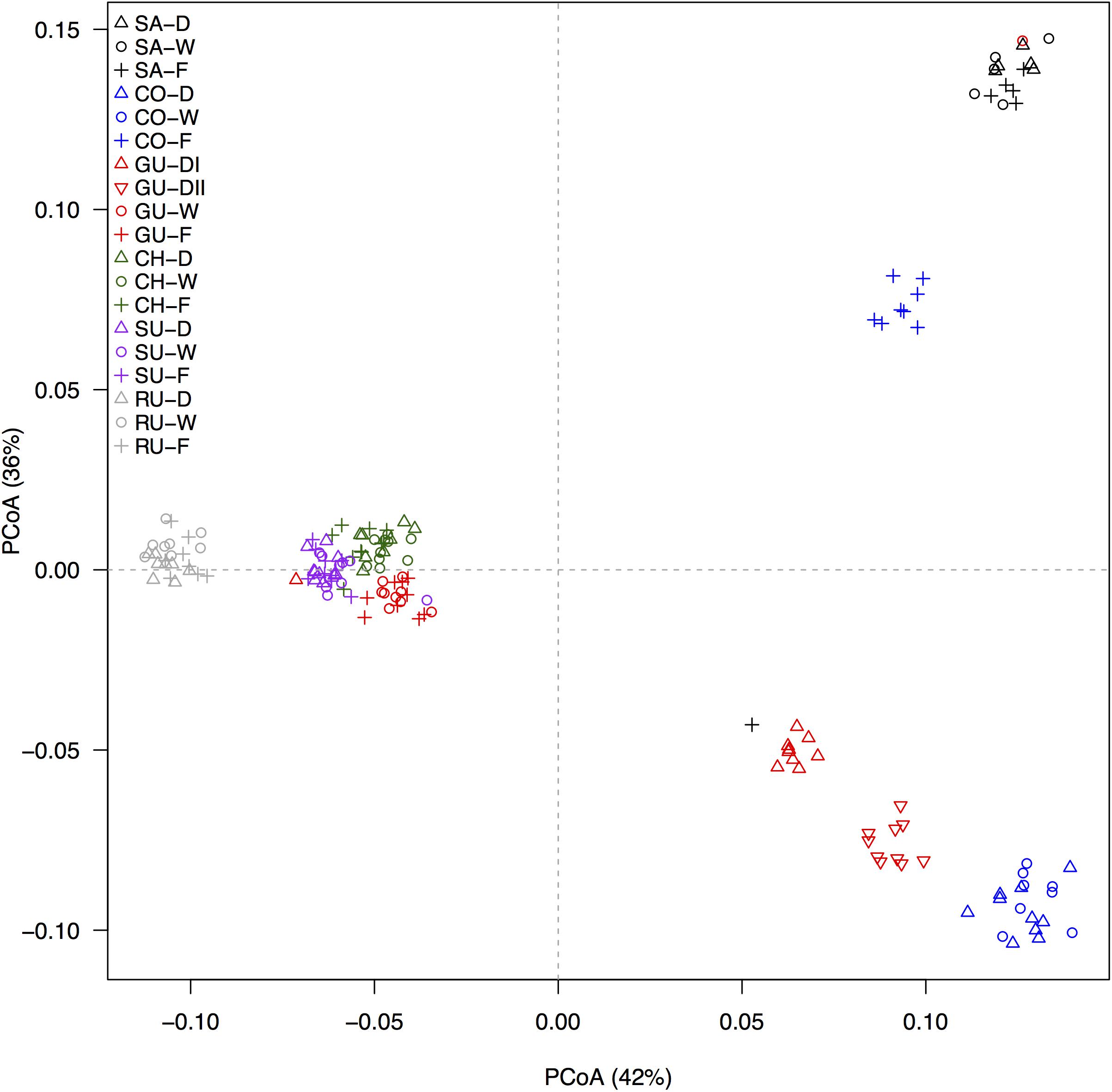
FIGURE 2. Differentiation, as revealed by principal coordinates analysis (PCoA), based on 1,273 GBS-derived SNP markers. Ecotypes are labeled by different symbols as follows: caulescent populations from cloud forest (+), caulescent populations from wind-sheltered well-irrigated depressions (o) and acaulescent populations from wind-exposed drier slopes ( ), and abbreviated in the figure legend according to the habitat as forest (F), wet (W) and dry (D), respectively. Localities are identified by different colors and coded as in Figure 1. The percentage of explained variation by each axis is shown within parenthesis in the label of the corresponding axis.
), and abbreviated in the figure legend according to the habitat as forest (F), wet (W) and dry (D), respectively. Localities are identified by different colors and coded as in Figure 1. The percentage of explained variation by each axis is shown within parenthesis in the label of the corresponding axis.
Evaluation of the population structure through unsupervised Bayesian genetic clustering allowing for admixture and implemented in STRUCTURE resulted in similar separations at K = 9 (Figure 3 and Supplementary Table S2), reinforcing the confirmatory observation (Rauscher, 2002; Diazgranados and Barber, 2017; Pouchon et al., 2018) of weak boundaries among taxonomic groups in the Espeletia complex (an AMOVA test indicated that 43.7% of the genetic variation could be explained by locality, 21.8% could be explained by habitat type within locality and 34.5% was found within ecotype). The most likely K-value of nine was selected based on the previous results and the increases in likelihood ratios between runs using Evanno et al.’s (2005) delta K statistic (Supplementary Figure S2). Separation of the populations at each K-value was informative. At the first level of population separation, K = 2, all individuals divided north–south with the acaulescent populations from Guantiva inside the northern pool. At K = 3 the individuals from Santurbán and the caulescent individuals from the cloud forest of Cocuy separated from the northern group. At K = 4 the individuals from Ruíz and the caulescent individuals from Guantiva detached from the southern group, and at K = 6 the latter split into an independent cluster. At K = 7 the population separation agreed with geographical distribution within the assembly of individuals from Chingaza and Sumapaz and the caulescent individuals from Guantiva. At K = 9 different levels of admixture differentiated the two acaulescent populations sampled in the wind-exposed drier slopes of Guantiva.
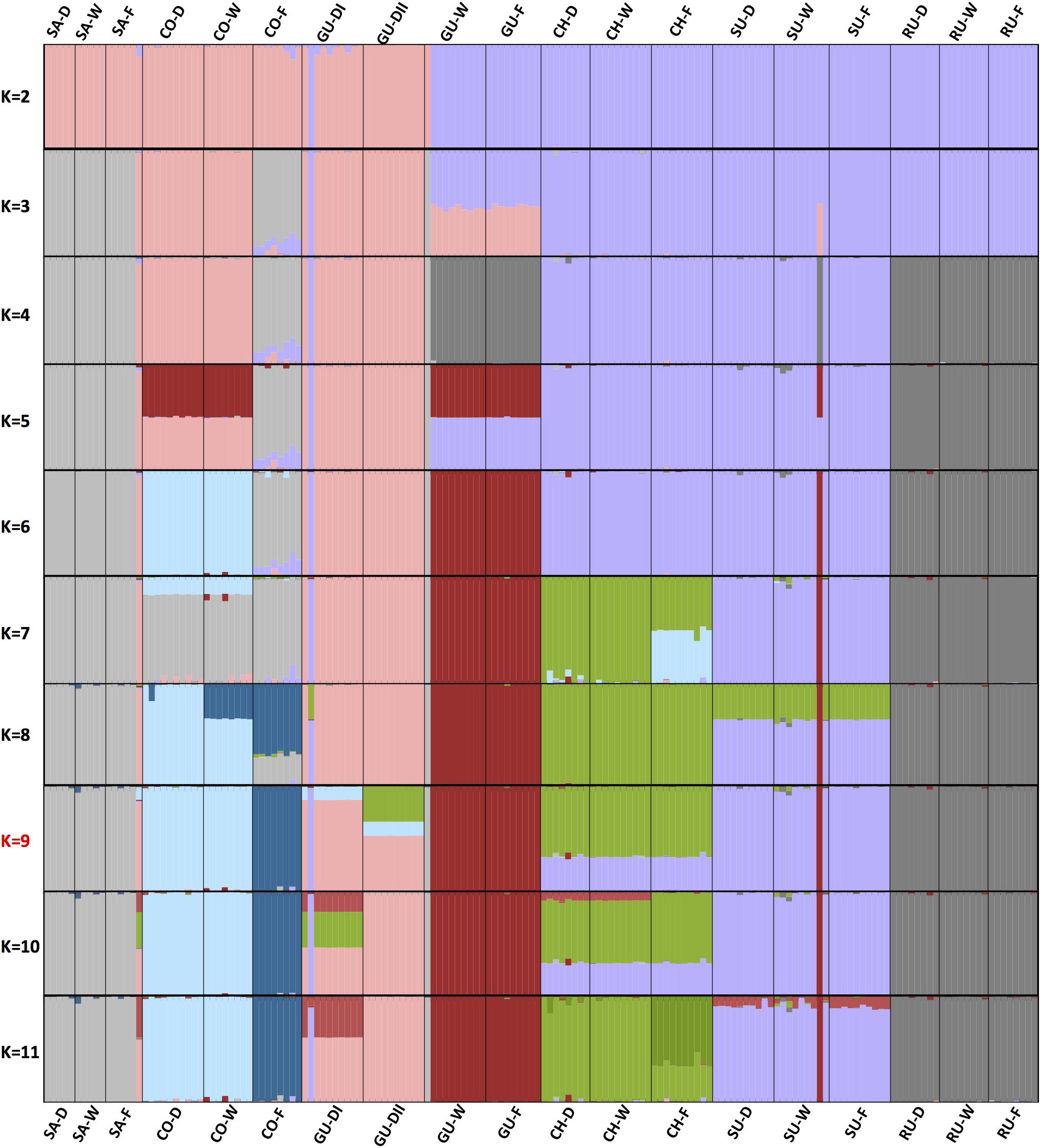
FIGURE 3. Population structure from assignment tests in STRUCTURE based on 1,273 variable SNP markers. K-values of 2–11 populations are shown. The optimum K-value (K = 9, Supplementary Figure S2) according to Evanno et al. (2005), the PCoA (Figure 2) and visual inspection is marked in red. Populations are sorted from north to south with the only locality from the Colombian Central Cordillera at the extreme right. The name of each population is given at the top of the bar plot and the localities and ecotypes are colored and coded as in Figure 1.
The consensus SNAPP-based phylogenetic tree (Figure 4) clarified the assignment of splitting events to populations defined in STRUCTURE. Populations from the localities of Sumapaz and Ruíz were monophyletic as well as the sister clade of the latter, which comprised all populations from Chingaza and Sumapaz. A sister monophyletic clade of all these populations comprised the caulescent populations from Guantiva so that all these linages were nested within the caulescent populations from Cocuy. Acaulescent populations from Cocuy and Guantiva were assembled in a monophyletic clade, which was a sister group of the other populations. The cloudgram depicting all SNAPP-based phylogenetic trees was fully consisted with the consensus tree (Supplementary Figure S3).
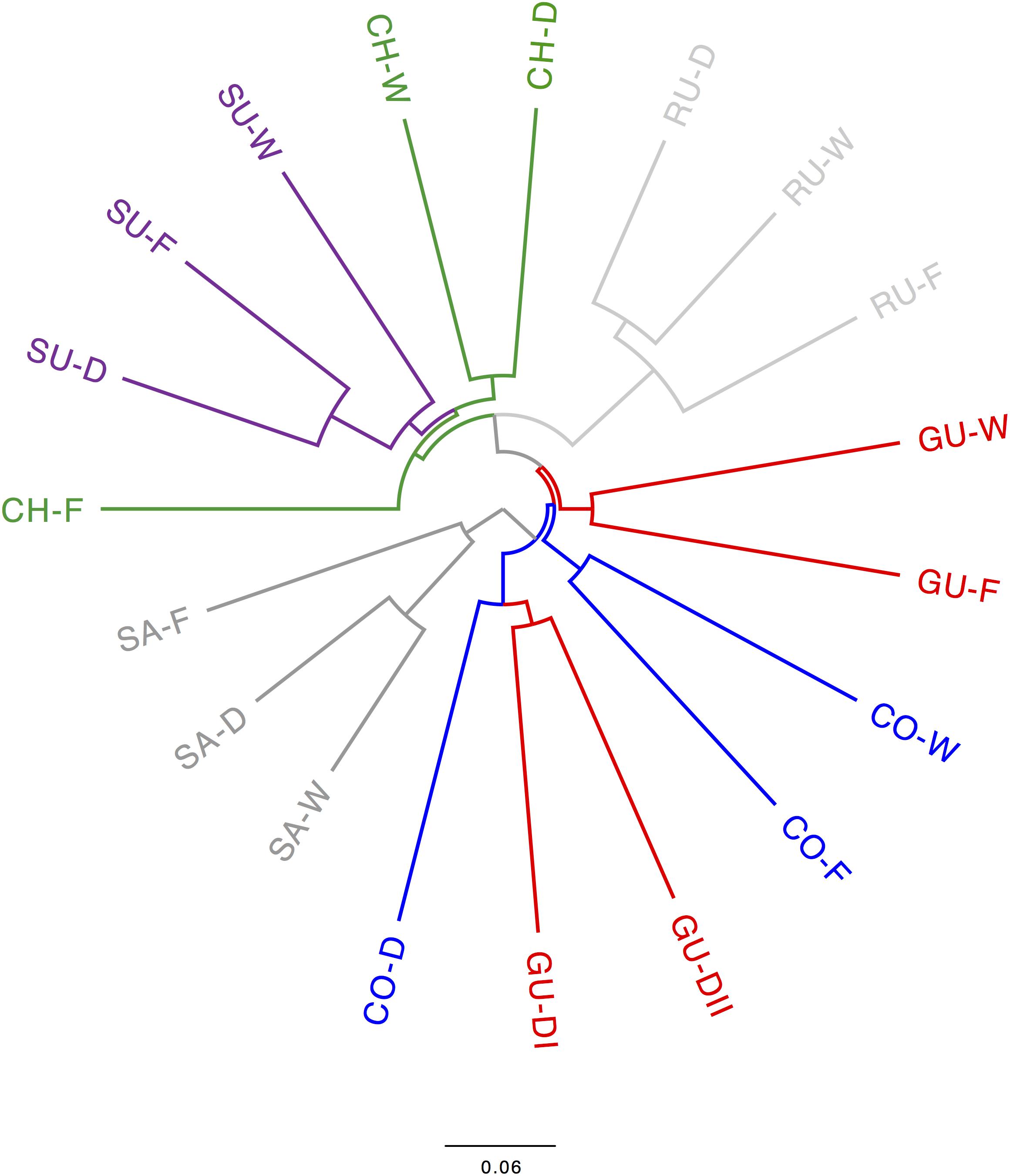
FIGURE 4. Consensus SNAPP-based phylogenetic tree of the Espeletia populations analyzed in the current study. Populations from Santurbán (SA) were used as outgroups for rooting the phylogenetic tree following Diazgranados and Barber (2017). Localities and ecotypes are colored and coded as in Figure 1.
QQ-plots from the association analyses between the SNP markers and the localities, and between the SNP markers and the habitats (ranked according to the theoretical exposure to frost and the theoretical soil moisture content) indicated that MLM models over-controlled for population structure by using a kinship matrix, whereas GLM analyses exhibited tolerable rates of false positives (Supplementary Figure S4). These last models yielded, at a Bonferroni-corrected significance threshold of 4.4 –log10 (P-value), a total of 172, 39 and 28 SNP markers associated with the localities and with the habitats when these were ranked according to the theoretical exposure to frost and the theoretical soil moisture content, respectively (as detailed in Table 1). When only contrasting populations for localities were considered, the GLM analyses respectively yielded a total of 156, 2 and 1 SNPs associated with the localities and with the habitats when these were ranked according to the theoretical exposure to frost and the theoretical soil moisture content, according to a Bonferroni-corrected significance threshold of 4.2 -log10 (P-value). The fact that few SNP markers were associated with the habitats in this contrasting dataset for localities was consistent with a low rate of false positives in a species complex well known for its IBD pattern. On the other hand, when only contrasting populations for the habitats were considered, the GLM analyses yielded, based on a Bonferroni-corrected significance threshold of 4.6 -log10 (P-value), a total of 184, 123 and 87 SNPs respectively associated with the localities and with the habitats when these were ranked according to the theoretical exposure to frost and the theoretical soil moisture content.
Estimates of migration rates (Nem) among populations were high and ranged from 0.3 to 7.5 for the entire dataset, and from 0.4 to 3.4 and from 0.3 to 7.5 when only contrasting populations for habitats and localities were considered, respectively. Estimated migration rates were asymmetric among these three types of datasets (Figure 5 and Table 2) with overall higher pairwise migration rates for the entire (Nem = 0.9 ± 0.1, CI 95%: 0.4 – 5.2, Figure 5A) and the locality-based (Nem = 1.2 ± 0.2, CI 95%: 0.4 – 5.8, Figure 5E) datasets than for the habitat-based dataset (Nem = 0.6 ± 0.1, CI 95%: 0.4 – 2.0, Figure 5H). Pairwise migration rates were higher among populations within the same locality than among populations from the same habitat from different localities for the entire (Nem = 3.4 ± 0.6, CI 95%: 2.9 – 4.6 vs. Nem = 0.7 ± 0.1, CI 95%: 0.5 – 1.5, P-value < 0.001, Figures 5A–D) and for the locality-based datasets (Nem = 3.8 ± 0.5, CI 95%: 3.3 – 4.6 vs. Nem = 0.6 ± 0.1, CI 95%: 0.4 – 1.0, P-value = 0.125, Figures 5E–G); whereas pairwise migration rates among contrasting populations for habitats did not exhibit this geographic clustering (Nem = 0.7 ± 0.3, CI 95%: 0.4 – 1.8 vs. Nem = 0.4 ± 0.1, CI 95%: 0.4 – 0.5, P-value = 0.002, Figures 5H–K). The datasets of alternative SNP clusters recovered the same results, except for the locality-based dataset that only included SNPs that were significantly associated with each habitat, ranked according to their theoretical exposure to frost, where the majority of pairwise migration rates were negligible except for the two Chingaza populations that were not in the cloud forest (Figure 5G). Results for theoretical soil moisture content were not possible because of a lack of polymorphism in the associated SNPs. These results were kept when considering only Nem estimates > 1 (Supplementary Figure S5), regarded as the minimum required for panmixia (Hartl and Clark, 2007).
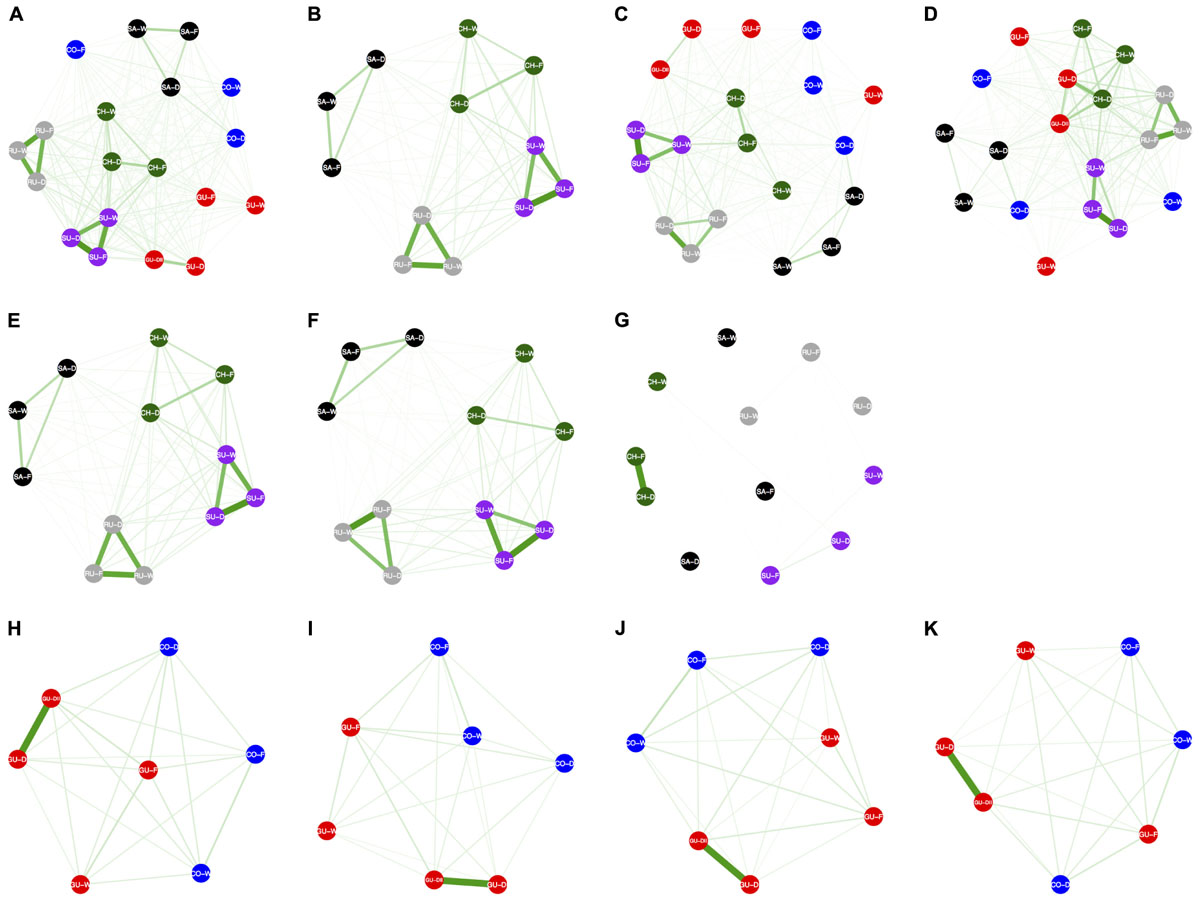
FIGURE 5. Networks depicting bidirectional gene flow patterns among populations of Espeletia. The width and color intensity of the green lines are proportional to the number of migrants per generation (Nem). The thinnest lines correspond to Nem values below 1. The first row of the diagrams are based on the entire SNP set including all populations (A–D), while second and third rows only include the datasets for contrasting populations for localities (E–G) and habitats (H–K), respectively. The first column of the diagrams are based on the entire SNP dataset (A,E,H), while second, third, and fourth columns only include markers significantly associated with the localities (B,F,I), the theoretical exposure to frost (C,G,J) and the theoretical soil moisture content (D,K), as coded in Supplementary Table S1. The dataset that only includes contrasting populations for localities and markers significantly associated with the theoretical soil moisture content is not shown because lack of polymorphism. Allelic associations of the GBS-derived SNP markers with localities and habitats were quantified using 18 different general and mixed-effects statistical models that accounted for phylogenetic distance. Only results for the optimal models are shown (see details in Supplementary Figure S4). Localities and ecotypes are colored and coded as in Figure 1.

TABLE 2. Average estimates of bidirectional migration rates among populations of Espeletia.
Mantel tests between genetic differentiation and geographic distance were significant when considering all populations and SNPs (average r = 0.59 ± 0.13, P-value < 0.001, Supplementary Figures S6A(i)–D(i) and Table 1). Mantel tests were also significant in the dataset enriched for contrasting localities when using the entire SNP panel (r = 0.95, P-value < 0.001, Figure 6A) and only those markers significantly associated with the localities (r = 0.95, P-value < 0.001, Figure 6D). In contrast, Mantel tests between genetic differentiation and ecological distance based on the theoretical exposure to frost were significant in the dataset with contrasting populations for habitats irrespective of the SNP set [average r = 0.47 ± 0.02, P-value < 0.001, Figures 6H,K and Supplementary Figures S6 H(ii)–K(ii)]. Mantel tests between genetic differentiation and the ecological distance based on the theoretical soil moisture content were not significant (average r = 0.16 ± 0.04, P-value > 0.05, third column in Figure 6 and Supplementary Figure S6).
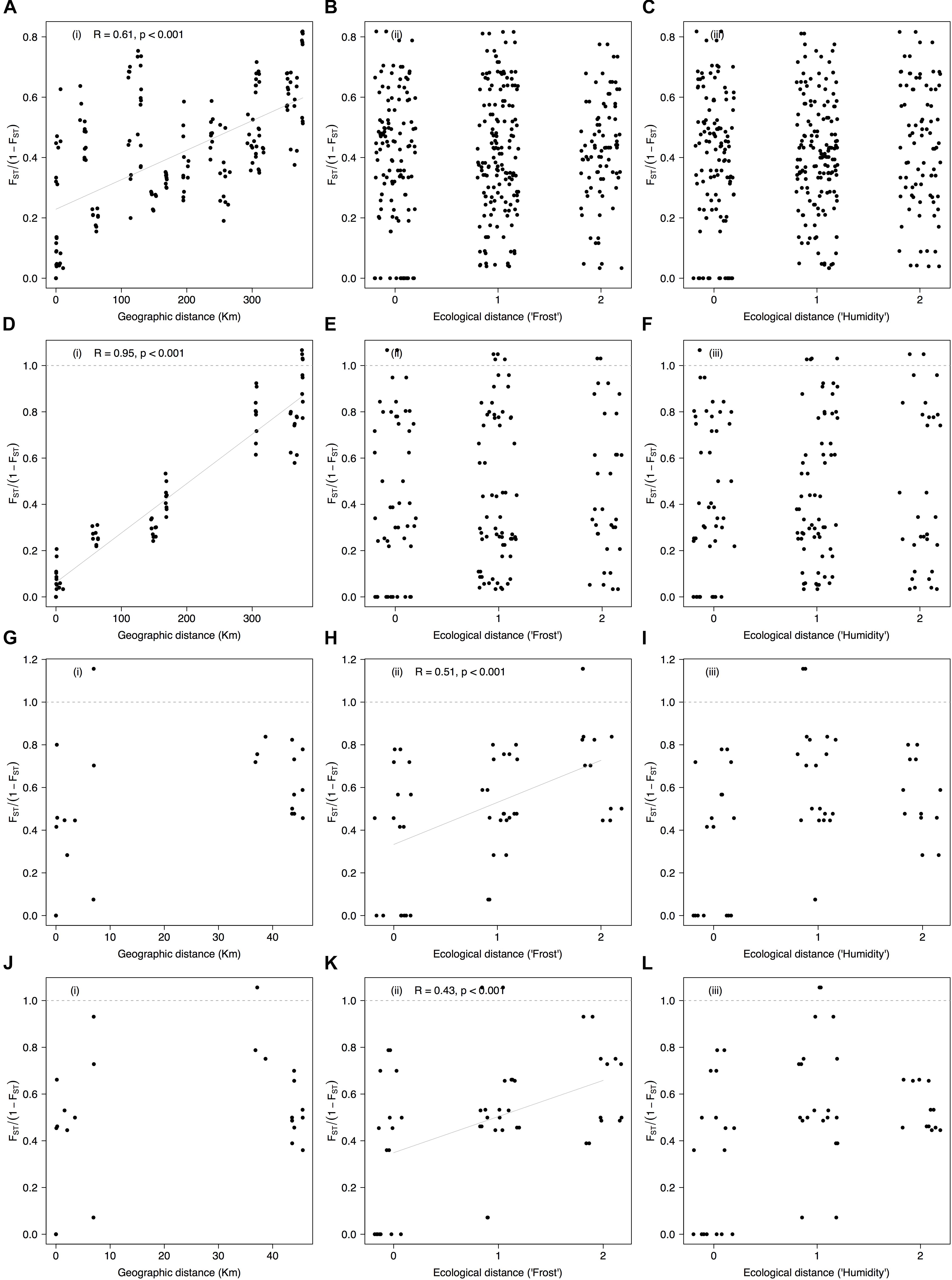
FIGURE 6. Correlations between genetic differentiation, geographic distance, and ecological distance (exposure to frost and soil moisture content) among populations of Espeletia growing in three different habitats and six localities. First column of diagrams show FST/(1 – FST) vs. geographic distance (A,D,G,J), following Rousset (1997), while second and third columns show FST/(1 – FST) vs. ecological distances based on the theoretical (as coded in Supplementary Table S1) exposure to frost (B,E,H,K) and soil moisture content (C,F,I,L), respectively. The first row of the diagrams are based on the entire dataset (A–C), second row only include contrasting populations and markers significantly associated with the localities (D–F), and third and fourth rows only include contrasting populations for habitats and markers significantly associated with the theoretical exposure to frost (G–I) and the theoretical soil moisture content (J–L), respectively. Allelic associations of the GBS-derived SNP markers with localities and habitats were quantified using 18 different general and mixed-effects statistical models that accounted for phylogenetic distance. Only results for the optimal models are shown. Lines are displayed where Mantel tests were significant according to Table 1.
If ecological differentiation contributed to genetic divergence in the Espeletia complex, then subtle and localized signals of IBE must be identifiable besides a predominant pattern of isolation-by-distance (IBD). We found strong evidence for IBD across populations despite rampant gene flow, as well as a subtle IBE trend that emerged and masked the IBD signal when we only considered contrasting populations for habitats. This finding is supported mainly by two significant results. First, migration rates were not higher among populations from the same habitat from different localities than among populations from different habitats within the same locality only for the SNP dataset enriched for habitat comparisons. Second, Mantel tests between genetic differentiation and ecological distance based on the theoretical exposure to frost, a proxy for adaptation to Páramo environments, were significant in the dataset with contrasting populations for habitats without exhibiting an, otherwise ubiquitous, relationship with the geographic distance. In other words, we have shown how IBD breaks down at the microenvironment level, allowing IBE to come into play. Mixed signatures of IBD and IBE indicate that geographical isolation and environmental heterogeneity both contribute to the spatial genetic patterns in the Espeletia complex. Since we were able to recover subtle and localized signals of IBE, besides a predominant pattern of IBD, there is evidence that ecological differentiation may have contributed to genetic divergence in the Espeletia complex. This illustrates the importance of local-scale environmental heterogeneity in keeping multiple putative adaptations and generating rapid morphological variation in a highly diverse ecosystem like the Páramo.
The migration estimates obtained in this study indicate rampant gene flow among populations from different localities and ecotypes (caulescent populations from the cloudy forest and from wind-sheltered well-irrigated depressions, and acaulescent populations from wind-exposed drier slopes). Judging by the result that maximum nine genetic clusters spanned 19 populations (locality–habitat combinations), we can conclude that there is little evidence pointing toward an assembly of genetically well-differentiated populations fully concordant with the ecological and morphological differentiation that is observed in the Espeletia complex. Results from Nem estimates >1 are often understood as an indication of panmixia (Hartl and Clark, 2007), and here were high in the pairwise comparisons among populations from different localities and habitats. However, we conjecture that this result does not preclude finding genetic differentiation at a narrower genomic scale, for which a close reference genome would be required. For instance, particular genomic regions may exhibit signatures of local adaptation despite the fact that most of the genome freely recombines (Andrew et al., 2012; Kremer et al., 2012). Mosaics of differentiation result from contrasting signatures of divergence and gene flow that simultaneously imprint the genomic landscape (Nosil and Feder, 2011; Ellegren and Galtier, 2016). Discerning among conflicting signatures is necessary in order to depict the causes of the genome-wide patterns of geographic and ecological divergence (Cortés and Blair, 2018b; Cortés et al., 2018).
There are several reasons why gene flow could be rampant in organisms with limited seed dispersal such as Espeletia. First, sporadic pollen dispersal may overcome the limited seed dispersal as the mechanism for gene flow. In Espeletia it has been hypothesized that pollen dispersal may not be as constrained as seed dispersal (Cuatrecasas, 2013). Second, intermediate ecotypes and hybrids may act as bridges for gene flow, particularly in the Espeletia complex that is well known for the high incidence of natural hybridization (Rauscher, 2002; Pouchon et al., 2018). Third, episodes of geographic and habitat connectivity as part of the glacial cycling could have offered a time window for gene flow to occur across barriers, that today appear stable. Because some Espeletia individuals may live for several centuries (Cuatrecasas, 2013), sporadic geographic connectivity may be frequent in Espeletia. Finally, the absence of strong population differentiation may be due to weak reproductive barriers resulting from recent divergence. The diversification of Espeletia occurred in recent geological time, after the final uplift of the Northern Andes about 2.3 Myr (Pouchon et al., 2018). Gene flow within the genus clearly introduces various caveats when it comes to the reconstruction of evolutionary history. Inferring a phylogenetic tree of Espeletia has proven to be challenging (Rauscher, 2002; Diazgranados and Barber, 2017), despite recent progress with next-generation sequencing (NGS) technologies (Pouchon et al., 2018), likely a consequence of divergence with gene flow. The uniqueness of our study is that we coupled a coalescent reconstruction with methods that allow for admixture and reticulate evolution in order to get a better representation of the porous meta-population structure instead of limiting our sampling to a collection of single voucher specimens. In any case, the most likely explanations and consequences for the widespread gene flow in the presence of morphological and ecological differentiation remain to be studied further within the Espeletia complex by targeting morphologically intermediate populations as well as their putative parental lineages.
Environmental effects play a role in shaping genetic diversity and generating morphological radiations in highly heterogeneous environments (Hughes and Atchison, 2015). Radiation has traditionally been described in the light of morphological traits and ecotypes. Yet, we have framed our study in an expanded view of radiation at the genetic level (Nosil and Feder, 2011; Nosil, 2012) by exploring the drivers that shape the continuum of genomic divergence in a rapidly evolving system. This expanded view is becoming increasingly common (Gompert et al., 2014; Ravinet et al., 2017) and an explosion of literature in this topic is expected in the years to come. We have concretely contributed evidence of ecological-driven divergence in the radiation of Andean Espeletia. Such divergence may be a consequence of local patterns of exposure to frost, being more extreme in the wind-exposed high valley slopes than in the wind-sheltered depressions of those valleys or in the upper boundary of the cloud forest. On the other hand, soil moisture content, higher in the well-irrigated high valley depressions and in the upper boundary of the cloud forest than in the drier slopes of the valleys, does not contribute to divergence. These predictions, as any outcome of a reverse genetics approach like the GEA, must be further validated throughout experimental approaches, such as growth chamber experiments and field reciprocal transplant assays of contrasting ecotypes.
In this study we also corroborated that geographical factors are associated with divergence in Espeletia. Since the role of geographic isolation has already been reported and discussed in extenso by recent works (Diazgranados and Barber, 2017; Padilla-GonzáLez et al., 2017; Pouchon et al., 2018), here we only point out that those reports made little reference to the consequences of habitat heterogeneity (Monasterio and Sarmiento, 1991). Furthermore, we also contributed evidence that adaptive genetic divergence can be recovered despite significant IBD, as has recently been discussed by Shih et al. (2018). Despite that our sampling was enriched for contrasting ecotypes in few localities, we were still capable of recovering a predominant IBD signal, likely the result of the strength of the IBD imprint (as revealed by the observation that there were more allelic associations with the localities than with the habitats). Even though a prevalent IBD pattern seems to obscure a subtler IBE trend, both geographic and ecological isolation are likely drivers of the rapid diversification in the Espeletia complex, acting as the major processes diversifying the genus.
One potential caveat of our results is the disadvantage of GBS due to missing data, for example from sequence divergence (Leaché et al., 2014). Nonetheless, we were able to identify, using stringent quality filters, 1,273 GBS-derived SNP markers spread throughout the genome and with contrasting signatures of geographic- and ecological-driven divergence. It is also worth to clarify that the power to detect marker associations with localities and habitats is unlikely limited by the number of markers. The information content of a given SNP set for this type of analyses is given by the linkage disequilibrium, due to high LD in selfing plants (Carlson et al., 2004), which is higher in selfing plants (Slatkin, 2008), like Espeletia. SNP redundancy due to LD (Carlson et al., 2004; Slate et al., 2009; Cortés et al., 2011; Blair et al., 2012, 2013, 2018; Kelleher et al., 2012) may therefore be sufficient and adequate to perform association analyses, as done here. On the other hand, the sample size used in the association analysis may overlook the majority of associations with low effect sizes and large-effect genes that segregate at low frequency (Maher, 2008). However, genes with major effects that segregate at moderate frequencies are still recognizable, particularly given the categorical nature of the fixed factors (i.e., localities and habitats). Furthermore, since our association models accounted for phylogenetic distance, the signatures displayed on the genetic variants that are associated with localities and habitats reflect true divergence rather that confounding processes (e.g., lineage sorting), even if not all associated markers may be causal, but rather linked with causal elements, and genetic distances may be intrinsically skewed because of gene flow. Then, our result about opposing signatures of geographic and ecological isolation is robust. Our research ultimately exemplifies that regions in the world that are experiencing extremely high diversification rates, such as the alpine Netropical Páramo ecosystem (Madriñán et al., 2013), are good candidates to serve as evolutionary playgrounds for today’s scientists, just as the Galápagos Islands. By accessing the genome-wide consequences of geographic and ecological differentiation in Espeletia, we were able to provide evidence of ecological divergence in this genus, as has already been suggested for other highly diverse genera in the Páramo like Loricaria (Kolar et al., 2016) and Lupinus (Hughes and Eastwood, 2006; Vásquez et al., 2016). However, similar genome-wide scans should still be undertaken by using a cross-taxonomic approach (Bacon et al., 2015; Antonelli et al., 2018) in other Páramo genera that exhibit well-described high diversification rates, such as Bartsia (Uribe-Convers and Tank, 2015), Diplostephium (Vargas et al., 2017), Hypericum (Nürk et al., 2013), Oreobolus (Chacón et al., 2006; Gómez-Gutiérrez et al., 2017) and Puya (Jabaily and Sytsma, 2013), in order to ultimately reveal the processes that make the Páramo the fastest evolving biodiversity hotspot on earth.
Besides allopatric and ecological diversification, hybridization, which was not explicitly addressed in the current sampling, is also regarded as a relevant process generating diversity in plants (Abbott et al., 2013). However, the genomic, morphological and ecological consequences of hybridization are generally not well documented across different taxa (Coyne and Orr, 2004; Payseur and Rieseberg, 2016). Espeletia is among the genera with a high incidence of natural hybridization and countless putative hybrids have been documented based on morphological characters (Rauscher, 2002; Cuatrecasas, 2013; Pouchon et al., 2018). Even though we have found ad hoc evidence of rampant gene flow, understanding the causes and consequences of hybridization in Espeletia and whether reticulate evolution has enhanced the diversification rate in this genus would ultimately require an in-depth ecological sampling design and an extended genotyping effort focused on hybrids and their putative parental populations.
Despite hybrids could occupy intermediate niches, persistence of populations in heterogeneous habitats that are facing changing environmental conditions is considered to be mostly mediated by phenotypic plasticity (Nicotra et al., 2010) or by adaptation from standing genetic variation by increasing the frequency of existing variants adapted to particular conditions (Bridle and Vines, 2007; Lascoux et al., 2016). Epigenetic mechanisms, those modulated by environmental factors that switch genes on and off and affect gene expression, may also alter responses to environmental variability in space and time (Bossdorf et al., 2008). Exploring the genetic architecture of ecologically relevant traits in natural surveys and field experiments, such as reciprocal transplant assays of ecotypes between habitats and space-by-time substitution trials, would help determining the relative role of plasticity and adaptation in generating morphological differentiation, as well as overall long-term adaptive potential (Barrett and Hoekstra, 2011; Valladares et al., 2014; Pacifici et al., 2017).
Some of the largest impacts of climate change are expected in highly heterogeneous alpine environments (Rumpf et al., 2018), where wind exposure and daytime vs. evening temperature fluctuations are the main drivers of vegetation composition (Körner, 2003). Temperature increases over the past decades have already led to upward migration of plants within mountains (Walther et al., 2002; Morueta-Holme et al., 2015; Steinbauer et al., 2018). However, these are mainly plant groups with short generation times (Lenoir et al., 2008) and the responses of long-living alpine plants with limited dispersal capacity are still unknown. Since Espeletia exhibits strong IBD and occurs in highly heterogeneous habitats, local scale variation can have important implications for their reaction to changing climatic conditions. For instance, environmental heterogeneity may provide new suitable locations for migrants within only a few meters of their current locations (Scherrer and Körner, 2011), while at the same time the populations adapted to a narrow range of conditions may respond poorly to future threats. Therefore, a key research line is to assess the evolutionary potential of Espeletia for coping with various types of stress that vary across habitats and that are expected to worsen at alpine ecosystems, such as frost (Wheeler et al., 2014, 2016), nutrient limitation (Sedlacek et al., 2014; Little et al., 2016), altered phenology (Cortés et al., 2014; Sedlacek et al., 2015, 2016), distorted biotic interactions (Wheeler et al., 2015), flooding and drought (well studied genetically, e.g., Cortés et al., 2012a,b, 2013; Galeano et al., 2012; Blair et al., 2016). Additionally, the evolution of the Páramo ecosystem during Andean uplift and glacial cycling presents similar conditions to current global warming and the contrasting habitats – depressions and colder and drier slopes, could be a proxy of current and future conditions. Understanding in more detail the adaptation of Espeletia, and other key plant groups in the Páramos, to local heterogeneous environments will thus assist with efforts to establish how future climate will impact plant populations in a highly diverse and threatened ecosystem (Vásquez et al., 2015; Pérez-Escobar et al., 2018). The contraction and expansion of populations in Páramos during past glacial fluctuations, moving up and down the mountains when it became either warmer or colder (Thuiller et al., 2008; Hazzi et al., 2018), is indicative of a general inability to adapt and a preponderant role of range shifts via migration (e.g., Bacon et al., 2018). It then remains to be seen whether the rapidly evolving and slowly dispersed Espeletia can keep pace with the quick rate of climate change and human expansion.
Finally, expanding our approach to other sky islands across the world’s mountains (Hoorn et al., 2018), especially in the African and Asian alpine regions, and generally to other island-like systems (Papadopoulou and Knowles, 2015; Lamichhaney et al., 2017), would help understanding how similar processes to those that that likely shaped the radiations in the neotropics have also influenced the diversity of unrelated plant groups in other regions (Condamine et al., 2018). Replicated island-like regions across the world configure an evolutionary playground to explore how radiations have taken place, as well as what are the chances for adaptation and migration to occur in response to environmental change.
The data analysis pipeline configuration files and cleaned dataset are archived at the Dryad Digital Repository under doi: 10.5061/dryad.23bd3ds.
AC and SM designed the study. AC, LG, and SM collected the samples. AC analyzed the data with contributions from JBV and SM. AC wrote up the results with editions from the other authors.
This research was funded by grants 4.1-2016-00418 from Vetenskapsrådet (VR) and BS2017-0036 from Kungliga Vetenskapsakademien (KVA) to AC as PI. Samples were collected under Permiso Marco of Uniandes for Proyecto Flora de Colombia to SM.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank A. Antonelli, C. D. Bacon, I. Calixto-Botía, N. Contreras-Ortiz, J. Mavarez and M. Pineda for discussion; A. Castillo, A. Correa, R. Cortés, J. L. Gómez-Diaz, D. Mojica, B. Moreno, G. Niño, J. D. Ramirez, B. V. Rodríguez-Cabeza, A. Rojas, A. M. Sequera, M. D. Sequera and M. T. Vera for field assistance; S. Arvidsson and R. Parol-Kryger for bioinformatics assistance; J. Bergman, P. Brandén, A. Perrigo, L. Sjöblom, S. Winkler and W. Zimmermann for administrative support, and the staff at Casa Grande, El Dorado, La Florida Casona, and Termales del Ruíz for logistical support and facilities.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2018.01700/full#supplementary-material
FIGURE S1 | Inset of the population structure for the Guantiva – Chingaza – Sumapaz complex as revealed by a principal coordinates analysis (PCoA) based on 1,273 GBS-derived SNP markers. Ecotypes are labeled by different symbols as follows: caulescent populations from the cloudy forest (+), caulescent populations from wind-sheltered well-irrigated depressions (o) and acaulescent populations from wind-exposed drier slopes (), and abbreviated in the figure legend according to the habitat as forest (F), wet (W) and dry (d), respectively. Localities are identified by different colors, as in Figure 1, and abbreviated in the figure legend as follows: Chingaza (CH), Guantiva (GU), and Sumapaz (SU). The percentage of explained variation by each axis is shown within parenthesis in the label of the correspond axis.
FIGURE S2 | Evanno’s delta K for the unsupervised Bayesian genetic clustering conducted in STRUCTURE with 1,273 GBS-derived SNP markers. K values ranged from K = 2 to K = 19. Transformed likelihoods of the graph model from Evanno et al. (2005) are shown in the vertical axis.
FIGURE S3 | Cloudgram depicting SNAPP-based phylogenetic trees of the Espeletia populations analyzed in the current study using 1,273 GBS-derived SNP markers. Populations from Santurbán (SA) were used as out-groups for rooting the phylogenetic trees following Diazgranados and Barber (2017). Populations’ names are given by the combination of codes for localities and ecotypes, coded as in Figure 1.
FIGURE S4 | Genome–environment association (GEA) analyses between the SNP markers and the localities, and between the SNP markers and the habitats. Habitats were ranked according to the theoretical exposure to frost and the theoretical soil moisture content, as coded in Supplementary Table S1. Given are eighteen QQ-plots of -log10 (P-value) for generalized (GLM, A–C,G–I,M–O) and mixed (MLM, D–F,J–L,P–R) linear models ran with the entire set of populations (A–F) or only with contrasting populations for localities (G–L) and for habitats (M–R). Models with the locality, the theoretical exposure to frost and the theoretical soil moisture content as fixed effects are respectively shown in the first, second, and third columns. Models include as covariate the phylogenetic distance computed in SNAPP (Figure 4). All MLMs use a centered IBS kinship matrix as a random effect. The gray dashed horizontal lines mark the P-value thresholds after Bonferroni-correction for multiple comparisons.
FIGURE S5 | Networks depicting bidirectional gene flow patterns leading to panmixia (Nem > 1) among populations of Espeletia. The width of the green lines is proportional to the number of migrants per generation (Nem) and are shown only if Nem > 1. First row of diagrams are based on the entire set of populations (A–D), whereas second and third rows only include contrasting populations for localities (E–G) and for habitats (H–K), respectively. First column of diagrams are based on the entire SNP dataset (A,E,H), while second, third and fourth columns only include markers significantly associated with the localities (B,F,I), the theoretical exposure to frost (C,G,J) and the theoretical soil moisture content (D,K), respectively. The dataset that only includes contrasting populations for localities and markers significantly associated with the theoretical soil moisture content is not shown because lack of polymorphism (one variable site). Allelic associations were quantified as described in the legend of Figure 5. Maximum Nem values are shown within parenthesis in the upper left corner. Localities are identified by different colors. Populations’ names within nodes are given by the combination of codes for localities and ecotypes, colored and coded as in Figure 1.
FIGURE S6 | Correlations between genetic differentiation, geographic distance and ecological distance (exposure to frost and soil moisture content) among populations of Espeletia growing in 3 different habitats and 6 localities. First column of diagrams show FST/(1 – FST) vs. geographic distance (i), following Rousset (1997), while second and third columns show FST/(1 – FST) vs. ecological distances based on theoretical (as coded in Supplementary Table S1) exposure to frost (ii) and soil moisture content (iii), respectively. First four rows of diagrams are based on the entire set of populations (A–D), whereas the following three and four rows only include contrasting populations for localities (E–G) and for habitats (H–K), respectively. First row of diagrams within these previous sets are based on the entire SNP dataset (A,E,H), whereas second, third and fourth rows only include markers significantly associated (based on the optimum model among 18 different statistical models that accounted for phylogenetic distance) with the localities (B,F,I), the theoretical exposure to frost (C,G,J) and the theoretical soil moisture content (D,K), respectively. Lines are displayed where Mantel tests were significant according to Table 1.
TABLE S1 | Identity of the 19 Espeletia populations used in this study and spanning 162 samples growing in 3 different habitats and 6 localities. Taxa names follow Cuatrecasas (2013). Populations’ abbreviations are given by the combination of codes for localities and ecotypes. Localities are coded as follows: Chingaza (CH), Cocuy (CO), Guantiva (GU), Santurbán (SA), Sumapaz (SU) and Ruíz (RU). Ecotypes are coded as follows: caulescent populations from the cloudy forest (F), caulescent populations from wind-sheltered well-irrigated depressions (W) and acaulescent populations from wind-exposed drier slopes (D). Under “Ecology” habitats are coded using an ordinal scale based on the theoretical exposure to frost and the theoretical soil moisture content.
TABLE S2 | Population structure from assignment tests in STRUCTURE based on 1,273 variable SNP markers. Probabilities are shown for the optimum K-value (K = 9, Supplementary Figure S2) according to Evanno et al. (2005), the PCoA (Figure 2) and visual inspection. Populations are abbreviated as in Supplementary Table S1.
Abbott, R., Albach, D., Ansell, S., Arntzen, J. W., Baird, S. J. E., Bierne, N., et al. (2013). Hybridization and speciation. J. Evol. Biol. 26, 229–246. doi: 10.1111/j.1420-9101.2012.02599.x
Andrew, R. L., Ostevik, K. L., Ebert, D. P., and Rieseberg, L. H. (2012). Adaptation with gene flow across the landscape in a dune sunflower. Mol. Ecol. 21, 2078–2091. doi: 10.1111/j.1365-294X.2012.05454.x
Antonelli, A., Nylander, J. A. A., Persson, C., and Sanmartin, I. (2009). Tracing the impact of the Andean uplift on neotropical plant evolution. Proc. Natl. Acad. Sci. U.S.A. 106, 9749–9754. doi: 10.1073/pnas.0811421106
Antonelli, A., Zizka, A., Carvalho, F. A., Scharn, R., Bacon, C. D., Silvestro, D., et al. (2018). Amazonia is the primary source of neotropical biodiversity. Proc. Natl. Acad. Sci. U.S.A. 115, 6034–6039. doi: 10.1073/pnas.1713819115
Bacon, C. D., Silvestro, D., Jaramillo, C., Smith, B. T., Chakrabarty, P., and Antonelli, A. (2015). Biological evidence supports an early and complex emergence of the isthmus of panama. Proc. Natl. Acad. Sci. U.S.A. 112:E3153. doi: 10.1073/pnas.1423853112
Bacon, C. D., Velasquez-Puentes, F. J., Hinojosa, L. F., Schwartz, T., Oxelman, B., Pfeil, B., et al. (2018). Evolutionary persistence in Gunnera and the contribution of southern plant groups to the tropical Andes biodiversity hotspot. PeerJ 6:e4388. doi: 10.7717/peerj.4388
Barrett, R. D. H., and Hoekstra, H. E. (2011). Molecular spandrels: tests of adaptation at the genetic level. Nat. Rev. Genet. 12, 767–780. doi: 10.1038/nrg3015
Beerli, P., and Felsenstein, J. (1999). Maximum likelihood estimation of migration rates and effective population numbers in two populations using a coalescent approach. Genetics 152, 763–773.
Blair, M. W., Cortes, A. J., Farmer, A. D., Huang, W., Ambachew, D., Penmetsa, R. V., et al. (2018). Uneven recombination rate and linkage disequilibrium across a reference snp map for common bean (Phaseolus vulgaris L.). PLoS One 13:e0189597. doi: 10.1371/journal.pone.0189597
Blair, M. W., Cortés, A. J., Penmetsa, R. V., Farmer, A., Carrasquilla-Garcia, N., and Cook, D. R. (2013). A high-throughput SNP marker system for parental polymorphism screening, and diversity analysis in common bean (Phaseolus vulgaris L.). Theor. Appl. Genetics 126, 535–548. doi: 10.1007/s00122-012-1999-z
Blair, M. W., Cortés, A. J., and This, D. (2016). Identification of an Erecta gene and its drought adaptation associations with wild and cultivated common bean. Plant Sci. 242, 250–259. doi: 10.1016/j.plantsci.2015.08.004
Blair, M. W., Soler, A,,and Cortés, A. J. (2012). Diversification and Population Structure in Common Beans (Phaseolus vulgaris L.). PLoS ONE 7:e49488. doi: 10.1371/journal.pone.0049488
Bossdorf, O., Richards, C. L., and Pigliucci, M. (2008). Epigenetics for ecologists. Ecol. Lett. 11, 106–115.
Bouckaert, R., Heled, J., Kuhnert, D., Vaughan, T., Wu, C. H., Xie, D., et al. (2014). Beast 2: a software platform for bayesian evolutionary analysis. PLoS Comput. Biol. 10:e1003537. doi: 10.1371/journal.pcbi.1003537
Bouckaert, R. R. (2010). Densitree: making sense of sets of phylogenetic trees. Bioinformatics 26, 1372–1373. doi: 10.1093/bioinformatics/btq110
Bradbury, P. J., Zhang, Z., Kroon, D. E., Casstevens, T. M., Ramdoss, Y., and Buckler, E. S. (2007). Tassel: software for association mapping of complex traits in diverse samples. Bioinformatics 23, 2633–2635. doi: 10.1093/bioinformatics/btm308
Bridle, J. R., and Vines, T. H. (2007). Limits to evolution at range margins: when and why does adaptation fail? Trends Ecol. Evol. 22, 140–147.
Bryant, D., Bouckaert, R., Felsenstein, J., Rosenberg, N. A., and Roychoudhury, A. (2012). Inferring species trees directly from biallelic genetic markers: bypassing gene trees in a full coalescent analysis. Mol. Biol. Evol. 29, 1917–1932. doi: 10.1093/molbev/mss086
Carlson, C. S., Eberle, M. A., Rieder, M. J., Yi, Q., Kruglyak, L., and Nickerson, D. A. (2004). Selecting a maximally informative set of single-nucleotide polymorphisms for association analyses using linkage disequilibrium. Am. J. Hum. Genet. 74, 106–120. doi: 10.1086/381000
Chacón, J., Madriñán, S., Chase, M. W., and Bruhl, J. J. (2006). Molecular phylogenetics of Oreobolus (Cyperaceae) and the origin and diversification of the American species. Taxon 55, 359–366. doi: 10.2307/25065583
Condamine, F. L., Antonelli, A., Lagomarsino, L. P., Hoorn, C., and Liow, L. H. (2018). “Teasing apart mountain uplift, climate change and biotic drivers of species diversification,” in Mountains, Climate, and Biodiversity, eds C. Hoorn, A. Perrigo, and A. Antonelli (New York, NY: Wiley).
Contreras-Ortiz, N., Atchison, G. W., Hughes, C. E., and Madriñán, S. (2018). Convergent evolution of high elevation plant growth forms and geographically structured variation in andean Lupinus (Fabaceae). Bot. J. Linn. Soc. 187, 118–136. doi: 10.1093/botlinnean/box095
Cortés, A. J. (2013). On the origin of the common bean (Phaseolus vulgaris L.). Am. J. Plant Sci. 4, 1998–2000. doi: 10.4236/ajps.2013.410248
Cortés, A. J. (2015). On the Big Challenges of a Small Shrub: Ecological Genetics of Salix herbacea L. Uppsala: Acta Universitatis Upsaliensis.
Cortés, A. J., and Blair, M. W. (2018a). Genotyping by sequencing and genome – environment associations in wild common bean predict widespread divergent adaptation to drought. Front. Plant Sci. 9:128. doi: 10.3389/fpls.2018.00128
Cortés, A. J., and Blair, M. W. (2018b). “Naturally available genetic adaptation in common bean and its response to climate change,” in Climate Resilient Agriculture – Strategies and Perspectives, eds C. Srinivasarao, A. K. Shanker, and C. Shanker (Rijeka: InTech).
Cortés, A. J., Chavarro, M. C., and Blair, M. W. (2011). Snp marker diversity in common bean (Phaseolus Vulgaris L.). Theor. Appl. Genet. 123, 827–845. doi: 10.1007/s00122-011-1630-8
Cortés, A. J., Chavarro, M. C., Madriñán, S., This, D., and Blair, M. W. (2012a). Molecular ecology and selection in the drought-related Asr gene polymorphisms in wild and cultivated common bean (Phaseolus vulgaris L.). BMC Genet. 13:58. doi: 10.1186/1471-2156-13-58
Cortés, A. J., Hurtado, P., Blair, M. W., and Chacón-Sánchez, M. I. (2018). Does the genomic landscape of species divergence in Phaseolus beans coerce parallel signatures of adaptation and domestication? Front. Plant Sci. 9:1816. doi: 10.3389/fpls.2018.01816
Cortés, A. J., This, D., Chavarro, C., Madriñán, S., and Blair, M. W. (2012b). Nucleotide diversity patterns at the drought-related Dreb2 encoding genes in wild and cultivated common bean (Phaseolus vulgaris L.). Theor. Appl. Genet. 125, 1069–1085. doi: 10.1007/s00122-012-1896-5
Cortés, A. J., Monserrate, F., Ramírez-Villegas, J., Madriñán, S., and Blair, M. W. (2013). Drought tolerance in wild plant populations: the case of common beans (Phaseolus vulgaris L.). PLoS One 8:e62898. doi: 10.1371/journal.pone.0062898
Cortés, A. J., Waeber, S., Lexer, C., Sedlacek, J., Wheeler, J. A., Van Kleunen, M., et al. (2014). Small-scale patterns in snowmelt timing affect gene flow and the distribution of genetic diversity in the alpine dwarf shrub Salix herbacea. Heredity 113, 233–239. doi: 10.1038/hdy.2014.19
Cortés, A. J., and Wheeler, J. A. (2018). “The environmental heterogeneity of mountains at a fine scale in a changing world,” in Mountains, Climate, and Biodiversity, eds C. Hoorn, A. Perrigo, and A. Antonelli (New York, NY: Wiley).
Cuatrecasas, J. (2013). A Systematic Study of the Subtribe Espeletiinae: Heliantheae, Asteraceae. New York, NY: The New York Botanical Garden.
Davies, T. J., Savolainen, V., Chase, M. W., Moat, J., and Barraclough, T. G. (2004). Environmental energy and evolutionary rates in flowering plants. Proc. R. Soc. Lond. B 271, 2195–2200. doi: 10.1098/rspb.2004.2849
Diazgranados, M., and Barber, J. C. (2017). Geography shapes the phylogeny of frailejones (Espeletiinae Cuatrec., Asteraceae): a remarkable example of recent rapid radiation in sky islands. PeerJ 5:e2968. doi: 10.7717/peerj.2968
Drummond, A. J., and Rambaut, A. (2007). Beast: bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7:214. doi: 10.1186/1471-2148-7-214
Duskova, E., Sklenář, P., Kolar, F., Vasquez, D. L. A., Romoleroux, K., Fer, T., et al. (2017). Growth form evolution and hybridization in Senecio (Asteraceae) from the high equatorial Andes. Ecol. Evol. 7, 6455–6468. doi: 10.1002/ece3.3206
Ellegren, H., and Galtier, N. (2016). Determinants of genetic diversity. Nat. Rev. Genet. 17, 422–433. doi: 10.1038/nrg.2016.58
Elshire, R. J., Glaubitz, J. C., Sun, Q., Poland, J. A., Kawamoto, K., Buckler, E. S., et al. (2011). A robust, simple genotyping-by-sequencing (Gbs) approach for high diversity species. PLoS One 6:e19379. doi: 10.1371/journal.pone.0019379
Epskamp, S., Cramer, A. O. J., Waldorp, L. J., Schmittmann, V. D., and Borsboom, D. (2012). Network visualizations of relationships in psychometric data. J. Stat. Softw. 48, 1–18. doi: 10.18637/jss.v048.i04
Evanno, G., Regnaut, S., and Goudet, J. (2005). Detecting the number of clusters of individuals using the software structure: a simulation study. Mol. Ecol. 14, 2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x
Excoffier, L., Laval, G., and Schneider, S. (2005). Arlequin (Version 3.0): an integrated software package for population genetics data analysis. Evol. Bioinform. Online 1, 47–50. doi: 10.1177/117693430500100003
Fischer, M. C., Rellstab, C., Tedder, A., Zoller, S., Gugerli, F., Shimizu, K. K., et al. (2013). Population genomic footprints of selection and associations with climate in natural populations of Arabidopsis halleri from the alps. Mol. Ecol. 22, 5594–5607. doi: 10.1111/mec.12521
Galeano, C. H., Cortés, A. J., Fernandez, A. C., Soler, A., Franco-Herrera, N., Makunde, G., et al. (2012). Gene-based single nucleotide polymorphism markers for genetic and association mapping in common bean. BMC Genet. 13:48. doi: 10.1186/1471-2156-13-48
Glaubitz, J. C., Casstevens, T. M., Harriman, J., Elshire, R. J., Sun, Q., and Buckler, E. S. (2014). Tassel-Gbs: a high capacity genotyping by sequencing analysis pipeline. PLoS One 9:e90346. doi: 10.1371/journal.pone.0090346
Gómez-Gutiérrez, M. C., Pennington, R. T., Neaves, L. E., Milne, R. I., Madriñán, S., and Richardson, J. E. (2017). Genetic diversity in the Andes: variation within and between the south American species of Oreobolus R. Br. (Cyperaceae). Alp. Bot. 127, 155–170. doi: 10.1007/s00035-017-0192-z
Gompert, Z., Comeault, A. A., Farkas, T. E., Feder, J. L., Parchman, T. L., Buerkle, C. A., et al. (2014). Experimental evidence for ecological selection on genome variation in the wild. Ecol. Lett. 17, 369–379. doi: 10.1111/ele.12238
Hancock, A. M., Brachi, B., Faure, N., Horton, M. W., Jarymowycz, L. B., Sperone, F. G., et al. (2011). Adaptation to climate across the Arabidopsis thaliana genome. Science 334, 83–86. doi: 10.1126/science.1209244
Hartl, D. L., and Clark, A. G. (2007). Principles of Population Genetics. Sunderland, MA: Sinauer Associates.
Hazzi, N. A., Moreno, J. S., Ortiz-Movliav, C., and Palacio, R. D. (2018). Biogeographic regions and events of isolation and diversification of the endemic biota of the tropical Andes. Proc. Natl. Acad. Sci. U.S.A. 115, 7985–7990. doi: 10.1073/pnas.1803908115
Hoffmann, A. A., and Sgro, C. M. (2011). Climate change and evolutionary adaptation. Nature 470, 479–485. doi: 10.1038/nature09670
Hoorn, C., Perrigo, A., and Antonelli, A. (2018). “Mountains, climate and biodiversity: an introduction,” in Mountains, Climate, and Biodiversity, eds C. Hoorn, A. Perrigo, and A. Antonelli (New York, NY: Wiley).
Hoorn, C., Wesselingh, F. P., Steege, H. T., Bermudez, M. A., Mora, A., Sevink, J., et al. (2010). Amazonia through time: andean uplift, climate change, landscape evolution, and biodiversity. Science 330, 927–931. doi: 10.1126/science.1194585
Hughes, C., and Eastwood, R. (2006). Island radiation on a continental scale: exceptional rates of plant diversification after uplift of the Andes. Proc. Natl. Acad. Sci. U.S.A. 103, 10334–10339. doi: 10.1073/pnas.0601928103
Hughes, C. E., and Atchison, G. W. (2015). The ubiquity of alpine plant radiations: from the Andes to the Hengduan Mountains. New Phytol. 207, 275–282. doi: 10.1111/nph.13230
Jabaily, R. S., and Sytsma, K. J. (2013). Historical biogeography and life-history evolution of andean Puya (Bromeliaceae). Bot. J. Linn. Soc. 171, 201–224. doi: 10.1111/j.1095-8339.2012.01307.x
Jakobsson, M., and Rosenberg, N. A. (2007). Clumpp: a cluster matching and permutation program for dealing with label switching and multimodality in analysis of population structure. Bioinformatics 23, 1801–1806. doi: 10.1093/bioinformatics/btm233
Kelleher, C. T., Wilkin, J., Zhuang, J., Cortés, A. J., Quintero,Á. L. P., Gallagher, T. F., et al. (2012). Snp discovery, gene diversity, and linkage disequilibrium in wild populations of Populus tremuloides. Tree Genet. Genomes 8, 821–829. doi: 10.1007/s11295-012-0467-x
Kolar, F., Duskova, E., and Sklenář, P. (2016). Niche shifts and range expansions along cordilleras drove diversification in a high-elevation endemic plant genus in the tropical Andes. Mol. Ecol. 25, 4593–4610. doi: 10.1111/mec.13788
Körner, C. (2003). Alpine Plant Life: Functional Plant Ecology of High Mountain Ecosystems. Berlin: Springer. doi: 10.1007/978-3-642-18970-8
Kremer, A., Ronce, O., Robledo-Arnuncio, J. J., Guillaume, F., Bohrer, G., Nathan, R., et al. (2012). Long-distance gene flow and adaptation of forest trees to rapid climate change. Ecol. Lett. 15, 378–392. doi: 10.1111/j.1461-0248.2012.01746.x
Lamichhaney, S., Han, F., Webster, M. T., Andersson, L., Grant, B. R., and Grant, P. R. (2017). Rapid hybrid speciation in Darwin’s finches. Science 359, 224–228. doi: 10.1126/science.aao4593
Lascoux, M., Glémin, S., and Savolainen, O. (2016). Local adaptation in plants. Encycl. Life Sci. 0025270, 1–7. doi: 10.1002/9780470015902.a0025270
Leaché, A. D., Fujita, M. K., Minin, V. N., and Bouckaert, R. R. (2014). Species delimitation using genome-wide snp data. Syst. Biol. 63, 534–542. doi: 10.1093/sysbio/syu018
Lenoir, J., Gegout, J. C., Marquet, P. A., De Ruffray, P., and Brisse, H. (2008). A significant upward shift in plant species optimum elevation during the 20th century. Science 320, 1768–1771. doi: 10.1126/science.1156831
Li, H., and Durbin, R. (2007). Fast and accurate short read alignment with burrows-wheeler transform. Bioinformatics 25, 1754–1760. doi: 10.1093/bioinformatics/btp324
Little, C. J., Wheeler, J. A., Sedlacek, J., Cortés, A. J., and Rixen, C. (2016). Small-scale drivers: the importance of nutrient availability and snowmelt timing on performance of the alpine shrub Salix herbacea. Oecologia 180, 1015–1024. doi: 10.1007/s00442-015-3394-3
Luteyn, J. L. (1999). PáRamos: A Checklist of Plant Diversity, Geographic Distribution and Botanical Literature. New York, NY: The New York Botanical Garden Press.
Madriñán, S., Cortés, A. J., and Richardson, J. E. (2013). Páramo is the world’s fastest evolving and coolest biodiversity hotspot. Front. Genet. 4:192. doi: 10.3389/fgene.2013.00192
Maher, B. (2008). Personal genomes: the case of the missing heritability. Nature 456, 18–21. doi: 10.1038/456018a
Monasterio, M., and Sarmiento, L. (1991). Adaptive radiation of Espeletia in the cold andean tropics. Trends Ecol. Evol. 6, 387–391. doi: 10.1016/0169-5347(91)90159-U
Morueta-Holme, N., Engemann, K., Sandoval-Acuna, P., Jonas, J. D., Segnitz, R. M., and Svenning, J. C. (2015). Strong upslope shifts in Chimborazo’s vegetation over two centuries since Humboldt. Proc. Natl. Acad. Sci. U.S.A. 112, 12741–12745. doi: 10.1073/pnas.1509938112
Nicotra, A. B., Atkin, O. K., Bonser, S. P., Davidson, A. M., Finnegan, E. J., Mathesius, U., et al. (2010). Plant phenotypic plasticity in a changing climate. Trends Plant Sci. 15, 684–692. doi: 10.1016/j.tplants.2010.09.008
Nosil, P. (2012). Ecological Speciation. Oxford: Oxford University Press. doi: 10.1093/acprof:osobl/9780199587100.001.0001
Nosil, P., and Feder, J. L. (2011). Genomic divergence during speciation: causes and consequences. Philos. Trans. R. Soc. B Biol. Sci. 367, 332–342. doi: 10.1098/rstb.2011.0263
Nosil, P., and Harmon, L. J. (2009). “Niche dimensionality and ecological speciation,” in Speciation and Patterns of Diversity, eds R. K. Butlin, J. Bridle, and D. Schluter (Cambridge: Cambridge University Press).
Nürk, N. M., Scheriau, C., and Madrinan, S. (2013). Explosive radiation in high andean Hypericum-rates of diversification among new world lineages. Front. Genet. 4:175. doi: 10.3389/fgene.2013.00175
Pacifici, M., Visconti, P., Butchart, S. H. M., Watson, J. E. M., Cassola, F. M., and Rondinini, C. (2017). Species’ traits influenced their response to recent climate change. Nat. Clim. Chang. 7, 205–208. doi: 10.1038/nclimate3223
Padilla-GonzáLez, G. F., Diazgranados, M., and Costa, F. B. D. (2017). Biogeography shaped the metabolome of the genus Espeletia: a phytochemical perspective on an andean adaptive radiation. Sci. Rep. 7:8835. doi: 10.1038/s41598-017-09431-7
Papadopoulou, A., and Knowles, L. L. (2015). Genomic tests of the species-pump hypothesis: recent island connectivity cycles drive population divergence but not speciation in caribbean crickets across the virgin islands. Evolution 69, 1501–1517. doi: 10.1111/evo.12667
Payseur, B. A., and Rieseberg, L. H. (2016). A genomic perspective on hybridization and speciation. Mol. Ecol. 25, 2337–2360. doi: 10.1111/mec.13557
Pérez-Escobar, O. A., Cámara-Leret, R., Antonelli, A., Bateman, R., Bellot, S., Chomicki, G., et al. (2018). Mining threatens Colombian ecosystems. Science 359:1475.
Pluess, A. R., Frank, A., Heiri, C., Lalague, H., Vendramin, G. G., and Oddou-Muratorio, S. (2016). Genome-environment association study suggests local adaptation to climate at the regional scale in Fagus sylvatica. New Phytol. 210, 589–601. doi: 10.1111/nph.13809
Pouchon, C., FernáNdez, A., Nassar, J. M., Boyer, F. D. R., Aubert, S., Lavergne, S. B., et al. (2018). Phylogenomic analysis of the explosive adaptive radiation of the Espeletia complex (Asteraceae) in the tropical Andes. Syst. Biol. 67, 1041–1060. doi: 10.1093/sysbio/syy022
Pritchard, J. K., Stephens, M., and Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959.
Rauscher, J. T. (2002). Molecular phylogenetics of the Espeletia complex (Asteraceae): evidence from nrDNA its sequences on the closest relatives of an andean adaptive radiation. Am. J. Bot. 89, 1074–1084. doi: 10.3732/ajb.89.7.1074
Ravinet, M., Faria, R., Butlin, R. K., Galindo, J., Bierne, N., Rafajlovic, M., et al. (2017). Interpreting the genomic landscape of speciation: a road map for finding barriers to gene flow. J. Evol. Biol. 30, 1450–1477. doi: 10.1111/jeb.13047
Rieseberg, L. H., and Willis, J. H. (2007). Plant speciation. Science 17, 910–914. doi: 10.1126/science.1137729
Rousset, F. (1997). Genetic differentiation and estimation of gene flow from F-statistics under isolation by distance. Genetics 145, 1219–1228.
Rumpf, S. B., Hulber, K., Klonner, G., Moser, D., Schutz, M., Wessely, J., et al. (2018). Range dynamics of mountain plants decrease with elevation. Proc. Natl. Acad. Sci. U.S.A. 115, 1848–1853. doi: 10.1073/pnas.1713936115
Savolainen, O., Lascoux, M., and Merilä, J. (2013). Ecological genomics of local adaptation. Nat. Rev. Genet. 14, 807–820. doi: 10.1038/nrg3522
Scherrer, D., and Körner, C. (2011). Topographically controlled thermal-habitat differentiation buffers alpine plant diversity against climate warming. J. Biogeogr. 38, 406–416. doi: 10.1111/j.1365-2699.2010.02407.x
Schluter, D. (2001). Ecology and the origin of species. Trends Ecol. Evol. 16, 372–380. doi: 10.1016/S0169-5347(01)02198-X
Schluter, D. (2009). Evidence for ecological speciation and its alternative. Science 323, 737–741. doi: 10.1126/science.1160006
Sedlacek, J., Bossdorf, O., Cortés, A. J., Wheeler, J. A., and Van-Kleunen, M. (2014). What role do plant-soil interactions play in the habitat suitability and potential range expansion of the alpine dwarf shrub Salix herbacea? Basic Appl. Ecol. 15, 305–315. doi: 10.1016/j.baae.2014.05.006
Sedlacek, J., Cortés, A. J., Wheeler, J. A., Bossdorf, O., Hoch, G., Klapste, J., et al. (2016). Evolutionary potential in the alpine: trait heritabilities and performance variation of the dwarf willow Salix herbacea from different elevations and microhabitats. Ecol. Evol. 6, 3940–3952. doi: 10.1002/ece3.2171
Sedlacek, J., Wheeler, J. A., Cortés, A. J., Bossdorf, O., Hoch, G., Lexer, C., et al. (2015). The response of the alpine dwarf shrub Salix herbacea to altered snowmelt timing: lessons from a multi-site transplant experiment. PLoS One 10:e0122395. doi: 10.1371/journal.pone.0122395
Shih, K. M., Chang, C. T., Chung, J. D., Chiang, Y. C., and Hwang, S. Y. (2018). Adaptive genetic divergence despite significant isolation-by-distance in populations of taiwan cow-tail fir (Keteleeria davidiana Var. formosana). Front. Plant Sci. 9:92. doi: 10.3389/fpls.2018.00092
Sklenář, P., Dušková, E., and Balslev, H. (2010a). Tropical and temperate: evolutionary history of páramo flora. Bot. Rev. 77, 71–108. doi: 10.1007/s12229-010-9061-9
Sklenář, P., Kucerová, A., Macek, P., and Macková, J. (2010b). Does plant height determine the freezing resistance in the páramo plants? Austral Ecol. 35, 929–934. doi: 10.1111/j.1442-9993.2009.02104.x
Sklenář, P., Hedberg, I., and Cleef, A. M. (2014). Island biogeography of tropical alpine floras. J. Biogeogr. 41, 287–297. doi: 10.3732/ajb.1600210
Sklenář, P., Kučerová, A., Macek, P., and Macková, J. (2012). The frost-resistance mechanism in páramo plants is related to geographic origin. N. Z. J. Bot. 50, 391–400. doi: 10.1080/0028825X.2012.706225
Sklenář, P., Kučerová, A., Macková, J., and Romoleroux, K. (2016). Temperature microclimates of plants in a tropical alpine environment: how much does growth form matter? Arct. Antarct. Alp. Res. 48, 61–78. doi: 10.1657/AAAR0014-084
Slate, J., Gratten, J., Beraldi, D., Stapley, J., Hale, M., and Pemberton, J. M. (2009). Gene mapping in the wild with Snps: guidelines and future directions. Genetica 136, 97–107. doi: 10.1007/s10709-008-9317-z
Slatkin, M. (2008). Linkage disequilibrium — understanding the evolutionary past and mapping the medical future. Nat. Rev. Genet. 9, 477–485. doi: 10.1038/nrg2361
Steinbauer, M. J., Grytnes, J. A., Jurasinski, G., Kulonen, A., Lenoir, J., Pauli, H., et al. (2018). Accelerated increase in plant species richness on mountain summits is linked to warming. Nature 556, 231–234. doi: 10.1038/s41586-018-0005-6
Stinchcombe, J. R., and Hoekstra, H. E. (2008). Combining population genomics and quantitative genetics: finding the genes underlying ecologically important traits. Heredity 100, 158–170. doi: 10.1038/sj.hdy.6800937
Strasburg, J. L., Sherman, N. A., Wright, K. M., Moyle, L. C., Willis, J. H., and Rieseberg, L. H. (2011). What can patterns of differentiation across plant genomes tell us about adaptation and speciation? Philos. Trans. R. Soc. B Biol. Sci. 367, 364–373. doi: 10.1098/rstb.2011.0199
Thuiller, W., Albert, C., Araújo, M. B., Berry, P. M., Cabeza, M., Guisan, A., et al. (2008). Predicting global change impacts on plant species’ distributions: future challenges. Perspect. Plant Ecol. Evol. Syst. 9, 137–152. doi: 10.1016/j.ppees.2007.09.004
Turner, T. L., Bourne, E. C., Von Wettberg, E. J., Hu, T. T., and Nuzhdin, S. V. (2010). Population resequencing reveals local adaptation of Arabidopsis lyrata to serpentine soils. Nat. Genet. 42, 260–263. doi: 10.1038/ng.515
Uribe-Convers, S., and Tank, D. C. (2015). Shifts in diversification rates linked to biogeographic movement into new areas: an example of a recent radiation in the Andes. Am. J. Bot. 102, 1854–1869. doi: 10.3732/ajb.1500229
Valladares, F., Matesanz, S., Guilhaumon, F., Araujo, M. B., Balaguer, L., Benito-Garzon, M., et al. (2014). The effects of phenotypic plasticity and local adaptation on forecasts of species range shifts under climate change. Ecol. Lett. 17, 1351–1364. doi: 10.1111/ele.12348
Vargas, O. M., Ortiz, E. M., and Simpson, B. B. (2017). Conflicting phylogenomic signals reveal a pattern of reticulate evolution in a recent high-andean diversification (Asteraceae: Astereae: Diplostephium). New Phytol. 214, 1736–1750. doi: 10.1111/nph.14530
Vásquez, D. L. A., Balslev, H., Hansen, M. M., Sklenář, P., and Romoleroux, K. (2016). Low genetic variation and high differentiation across sky island populations of Lupinus alopecuroides (Fabaceae) in the northern Andes. Alp. Bot. 126, 135–142. doi: 10.1007/s00035-016-0165-7
Vásquez, D. L. A., Balslev, H., and Sklenář, P. (2015). Human impact on tropical-alpine plant diversity in the northern Andes. Biodivers. Conserv. 24, 2673–2683. doi: 10.1007/s10531-015-0954-0
Walther, G. R., Post, E., Convey, P., Menzel, A., Parmesan, C., Beebee, T. J. C., et al. (2002). Ecological responses to recent climate change. Nature 416, 389–395. doi: 10.1038/416389a
Weir, B. S., and Cockerham, C. (1984). Estimating F-statistics for the analysis of population structure. Evolution 38, 1358–1370.
Wheeler, J. A., Cortés, A. J., Sedlacek, J., Karrenberg, S., Van Kleunen, M., Wipf, S., et al. (2016). The snow and the willows: accelerated spring snowmelt reduces performance in the low-lying alpine shrub Salix herbacea. J. Ecol. 104, 1041–1050. doi: 10.1111/1365-2745.12579
Wheeler, J. A., Hoch, G., Cortés, A. J., Sedlacek, J., Wipf, S., and Rixen, C. (2014). Increased spring freezing vulnerability for alpine shrubs under early snowmelt. Oecologia 175, 219–229. doi: 10.1007/s00442-013-2872-8
Wheeler, J. A., Schnider, F., Sedlacek, J., Cortés, A. J., Wipf, S., Hoch, G., et al. (2015). With a little help from my friends: community facilitation increases performance in the dwarf shrub Salix herbacea. Basic Appl. Ecol. 16, 202–209. doi: 10.1016/j.baae.2015.02.004
Willis, K. J., Bennett, K. D., and Birks, H. J. B. (2009). Variability in thermal and Uv-B energy fluxes through time and their influence on plant diversity and speciation. J. Biogeogr. 36, 1630–1644. doi: 10.1111/j.1365-2699.2009.02102.x
Yeaman, S., Hodgins, K. A., Lotterhos, K. E., Suren, H., Nadeau, S., Degner, J. C., et al. (2016). Convergent local adaptation to climate in distantly related conifers. Science 353, 1431–1433. doi: 10.1126/science.aaf7812
Keywords: isolation-by-distance (IBD), isolation-by-environment (IBE), genome–environment associations (GEA), genomic signatures of selection, GBS-derived SNP markers, neotropical alpine region
Citation: Cortés AJ, Garzón LN, Valencia JB and Madriñán S (2018) On the Causes of Rapid Diversification in the Páramos: Isolation by Ecology and Genomic Divergence in Espeletia. Front. Plant Sci. 9:1700. doi: 10.3389/fpls.2018.01700
Received: 19 June 2018; Accepted: 01 November 2018;
Published: 03 December 2018.
Edited by:
Kangshan Mao, Sichuan University, ChinaReviewed by:
Huafeng Wang, Hainan University, ChinaCopyright © 2018 Cortés, Garzón, Valencia and Madriñán. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrés J. Cortés, YWNvcnRlc0BhZ3Jvc2F2aWEuY28=
†Present address: Andrés J. Cortés, Corporación Colombiana de Investigación Agropecuaria (Agrosavia) - CI La Selva, Rionegro, Colombia; Universidad Nacional de Colombia - Sede Medellín, Facultad de Ciencias Agrarias - Departamento de Ciencias Forestales, Medellín, Colombia
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.