 Yucui Wu1,2†
Yucui Wu1,2† Xiaoyan Cao
Xiaoyan Cao Zhezhi Wang
Zhezhi Wang- 1National Engineering Laboratory for Resource Development of Endangered Crude Drugs in Northwest of China, Key Laboratory of the Ministry of Education for Medicinal Resources and Natural Pharmaceutical Chemistry, Shaanxi Normal University, Xi’an, China
- 2School of Landscape and Ecological Engineering, Hebei University of Engineering, Handan, China
- 3College of Chemistry, Biology and Materials Science, East China University of Technology, Nanchang, China
Phenolic acids from Salvia miltiorrhiza have drawn considerable attention in recent years because of their remarkable pharmacological activities. We previously reported that Arabidopsis thaliana transcription factor production of anthocyanin pigment 1 (AtPAP1) has strong capability to promote the production of phenolic acids in S. miltiorrhiza. However, the responsible molecular mechanism is unclear. Here, we analyzed the transcriptome of transgenic S. miltiorrhiza that over-expressed AtPAP1. Transcriptome analysis revealed 4,152 genes that were differentially expressed due to ectopic AtPAP1 overexpression. SmbHLH51, a novel bHLH gene significantly up-regulated by constitutive expression of AtPAP1, was isolated from S. miltiorrhiza for detailed functional characterization. SmbHLH51 localizes in the nuclei and interacts with AtPAP1, indicating that they probably comprise a regulatory transcription complex. Enhanced or reduced expression of SmbHLH51 was achieved in S. miltiorrhiza by gain- or loss-of-function assays, respectively, revealing that SmbHLH51 is a positive transcriptional regulator of the pathway for phenolic acid biosynthesis. We propose that applying this functional genomics approach through the transcriptomic analyses is an efficient means for identifying novel genes involved in plant secondary metabolism.
Introduction
Salvia miltiorrhiza Bunge, a well-known member of the Labiatae family, is emerging as a model plant system for research in regulation of secondary metabolites because of its significant medicinal values, high transformation efficiency and published genome data (Shao and Lu, 2013; Xu et al., 2015b). Its dry roots or rhizomes (called ‘danshen’ in Chinese) have been widely and successfully used in traditional Chinese medicine for thousands of years to treat cardiovascular disease, amenorrhea, and dysmenorrhea (Cheng, 2006). The bioactive components of S. miltiorrhiza are divided into two groups: lipid-soluble tanshinones and water-soluble phenolic acids. The latter group includes rosmarinic acid (RA) and salvianolic acid B (Sal B), which are attracting increased attention because of their marked pharmacological activities coupled with their traditional use as herbs steeped in boiling water in China (Petersen and Simmonds, 2003; Wu et al., 2012b). Because its active ingredients have such high economic value, studies are now focusing more on the pathway for phenolic acid biosynthesis (Hua et al., 2011; Wang et al., 2015). The phenolic acids distribute in different parts of Salvia miltiorrhiza and the content of total phenolic acids from leaves was significantly higher than those in roots and stems (Li et al., 2011).
Generally, overexpression of transcription factor (TF) that regulate the expression of genes involved in entire metabolic pathways is an effective tool for engineering high levels of metabolites (Butelli et al., 2008). Our research group was the first to show that the formation of health-promoting phenolic acids, especially Sal B, in S. miltiorrhiza, can be enriched by the ectopic overexpression of the Arabidopsis thaliana MYB TF production of anthocyanin pigment 1 (AtPAP1) (Zhang et al., 2010). However, the underlying molecular mechanism is unclear.
The phenylpropanoid pathway in different plant organs and tissues of higher plants is under the control of specific R2R3-MYB TFs and bHLH families (Xue et al., 2011). In S. miltiorrhiza, only a few endogenous MYBs have been functionally characterized to be involved in the regulation of phenolic acids biosynthesis (Zhang et al., 2013; Hao et al., 2016; Ding et al., 2017; Li et al., 2018). Among the 127 bHLHs identified in S. miltiorrhiza (Zhang et al., 2015), none has been verified to be involved in the regulation of phenolic acids by now. The bHLH genes are regulated by R2R3-MYB TFs. For example, constitutive expression of AcMYB110 that is highly activated in the anthocyanin pathway in tobacco up-regulates interacting endogenous NtAN1 bHLH (Montefiori et al., 2015). Ectopic expression of the MYB NtAN2 in tobacco promotes anthocyanin biosynthesis by up-regulating the expression of biosynthetic genes and bHLH NtAN1 (Bai et al., 2011). Activity of TT8 is strongly affected in AtPAP1 overexpressors (Baudry et al., 2006). Besides, it’s well documented that flavonoid biosynthesis is regulated by MYB-bHLH-WDR (MBW) complexes (Xu et al., 2015a). In Arabidopsis, AtPAP1 can interact with TT8, GL3, EGL3, or AtMYC1, together with TTG1 (WDR family), to form MBW complexes and activate flavonoid biosynthesis (Feller et al., 2011).
Recently, transcriptome analysis has been used to identify candidate genes on the biosynthesis pathways of secondary metabolites and explore the potential mechanisms associated with important traits in S. miltiorrhiza (Yang et al., 2013; Song et al., 2017; Wei et al., 2017; Pei et al., 2018). Manipulating a single TF can reprogram the transcriptome profiling and is an effective way to regulate some specific secondary metabolites. Since ectopic expression of AtPAP1 in S. miltiorrhiza greatly improved the level of Sal B (Zhang et al., 2010), the expression levels of many genes associated with Sal B biosynthesis pathway must be changed. Comparison of the transcriptomes between transgenic S. miltiorrhiza over-expressing AtPAP1 and the wild-type (WT) plants will help to identify not only novel genes associated with Sal B biosynthesis but also specific targets of AtPAP1, which will improve our insights into the Sal B accumulation mechanisms in S. miltiorrhiza.
Here, we report an examination of the transcriptome of AtPAP1-overexpressing S. miltiorrhiza. Our objectives were to investigate (1) the role of a single TF in global gene expression, and (2) the specific target/mechanism that results in the enrichment of phenolic acid biosynthesis in S. miltiorrhiza. To answer the first question, we studied transcriptomics using the Solexa/Illumina digital gene expression (DGE) system. From this we determined that a set of genes involved in phenolics accumulations are up-regulated in conjunction with the production of phenolic acids, and we identified the genes that function in that process. SmbHLH51, significantly up-regulated by constitutive expression of AtPAP1, was identified as a positive regulator for the phenolic acids biosynthesis by overexpressing and suppressing SmbHLH51 in S. miltiorrhiza. Our results indicate that AtPAP1 stimulates phenolic acid biosynthesis in S. miltiorrhiza by activating the expression of SmbHLH51 and probably forming a potential transcription complex with SmbHLH51.
Materials and Methods
Experimental Materials
Plant materials were cultured under the conditions described in our previous study (Hua et al., 2012). Transgenic line AtPAP1-14 (Zhang et al., 2010) and wild-type (WT) plants of S. miltiorrhiza were sampled on day 180 (D180) of growth for use in transcriptome and metabolome analysis. All chemicals were purchased from Sigma Chemical Co. (St. Louis, MO, United States). Solvents were HPLC grade. Standards of RA and Sal B were obtained from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). All standards were prepared as stock solutions in methanol and were stored in the dark at -18°C. Primer pairs are listed in Supplementary Table S1.
Transcriptome Analysis
We performed DGE profile analysis of AtPAP1-14 and WT plants by Solexa sequencing, as described before (Ge et al., 2015). Briefly, all clean tags obtained by filtering out any adaptor-only tags and low-quality tags were annotated based on S. miltiorrhiza reference genes (Hua et al., 2011). We identified DEGs based on false discovery rates ≤ 0.001, P-values < 0.005, and an estimated absolute log2 fold-change of ≥1 in sequence counts across libraries. Significantly enriched metabolic pathways were functionally classified into several categories with Kyoto Encyclopedia of Genes and Genomes (KEGG1) by comparing those DEGs with the control genome background. Pathways with Q-values < 0.05 were considered significantly enriched in DEGs.
Isolation and Sequence Analysis of SmbHLH51
We designed primers bHLH51-F/bHLH51-R to clone SmbHLH51 (GenBank Accession Number KT215166) with LA DNA polymerase (Takara, Japan) at both the genomic DNA level and the transcriptome cDNA level. Total RNA was isolated from 3-month-old plantlets of S. miltiorrhiza with a Plant RNA kit (OMEGA, United States), according to the manufacturer’s protocol, and was converted into cDNA using a primeScript RT reagent kit (TaKaRa, Dalian, China). Sequencing-verified SmbHLH51 cDNA cloned in pMD19-T (pMD19-cSmbHLH51) was used as template in some of our PCR reactions.
Combining the DNA and cDNA sequences, we obtained a diagram of exon/intron structures using public online software from the Gene Structure Display Server2. For our homology search we used BLAST3. The MW and pI of each corresponding protein were predicted with the ProParam tool4. Homologous bHLH protein sequences were aligned by CLUSTAL W (version 1.83) (Thompson et al., 1994) and the phylogenetic tree was constructed with MEGA 6.0 software (Tamura et al., 2013) and the Neighbor-Joining method (Gascuel and Steel, 2006).
Subcellular Localization Assay of SmbHLH51
The subcellular localization assay was performed by transiently expressing the SmbHLH51-GFP fusion protein in onion epidermal cells. The entire CDS region of SmbHLH51 was amplified from pMD19-cSmbHLH51 with primers 207-SmbHLH51-F/R, which contain the attB1/attB2 sites. The PCR products were cloned into entry vector pDONR207, using the BP recombination reaction, and then transferred into destination vector pEarleyGate103 (Earley et al., 2006) through an LR reaction, according to the protocol from the Gateway technology manufacturer (Invitrogen, United States). The pEarleyGate103-SmbHLH51 vector was transformed into onion epidermal cells by particle bombardment at a helium pressure of 1100 psi with the PDS-1000/He system (Bio-Rad, United States) and observed using a Leica DM6000B microscope (Leica, Germany) as described before (Li et al., 2018). The pEarleyGate103 plasmid was transformed into onion epidermal cells as a positive control.
Monitoring Transient Expression in Tobacco for Assaying Transcriptional Activation
We used AtPAP1-overexpressing plasmid pAtPAP1-OE, which we had constructed in our laboratory as the effector plasmid (Zhang et al., 2010). Fragments of the SmbHLH51 promoters were cloned using primers Pro-SmbHLH51-F/R and fused with the GUS reporter gene in pCAMBIA1391Z to obtain the reporter plasmid pProbHLH51-1391Z. The resulting constructs were transformed into the A. tumefaciens GV3101 strain. Tobacco plants were grown in a light incubator to the four- to six-leaf stage before being infiltrated with A. tumefaciens (Yin et al., 2010). After they were then grown in the dark for 24 h, they were exposed to a 16-h/8-h light/dark cycle for 48 h at 23°C. The infiltrated tobacco leaves were used for GUS staining (Jefferson et al., 1987).
Yeast One-Hybrid (Y1H) Assays
The ORF of AtPAP1 (GenBank Accession Number NM_104541.3) was amplified by PCR using primers containing BamHI and XhoI restriction sites and then fused to the GAL4 activation domain in vector pGADT7-Rec2 (Clontech) to create the fusion proteins pGADT7-AtPAP1. A trimer of MBSII element (AAAAGTTAGTTA) and A trimer mutant sequence (AAAAGTAAGGTA) were synthesized and cloned into pHIS2 (Clontech) to obtain pHIS2-MBSII and pMutant-MBSII, respectively. These recombinant vectors were co-transformed into yeast strain Y187 according to the reported protocol (Huang et al., 2013). The transformed cells were cultured on an SD/-Leu/-Trp medium and then selected on an SD/-Leu/-Trp/-His medium supplemented with 60 mM 3-amino-1,2,4-triazole to examine any protein-DNA interactions.
Yeast Two-Hybrid (Y2H) Assays
For Y2H assays, the ORFs of AtPAP1 and SmbHLH51 were cloned into the entry vector pDONR207 and then, respectively, transferred into Gateway vectors pGADT7 and pGBKT7 according to the manufacturer’s protocol. The resulting pGADT7-AtPAP1 and pGBKT7-SmbHLH51 were transformed into yeast strain AH109. Primers 207-AtPAP1-F/R and 207-SmbHLH51-F/R used for vector construction are listed in Supplementary Table S1. The interaction assays were performed according to the Matchmaker Gold Yeast Two-Hybrid System manufacturer’s protocol (Clontech, United States), and Y2H images were taken on day 5 of incubation.
Firefly Luciferase Complementation Imaging Assay
AtPAP1 was fused to the N-terminal domain of firefly luciferase (Nluc) of p1301-NLuc to produce destination vector pAtPAP1-NLuc. SmbHLH51 and AtTT8 were fused to the C-terminal domain of firefly luciferase (Cluc) of p1301-CLuc to obtain pSmbHLH51-CLuc and pAtTT8-CLuc. The pAtPAP1-NLuc and pAtTT8-CLuc served as positive controls. Primers AtPAP1-NLF/R, AtTT8-CLF/R, and SmbHLH51-CLF/R (Supplementary Table S1) were used for constructing the vectors, which were transformed into the A. tumefaciens GV3101 strain. Cultures harboring each of the CLuc and NLuc constructs were co-infiltrated into fully expanded tobacco leaves before Luc activity was visualized and pictured with IVIS Spectrum (Xenogen, United States) (Wu et al., 2012a).
Construction of Plant Expression Vectors and Plant Transformation
To construct the SmbHLH51-overexpressing vectors, we designed gene-specific primers OEbHLH51-F and OEbHLH51-R (Supplementary Table S1) to amplify SmbHLH51. The PCR products were digested with KpnI/BamHI (Takara) and ligated into the pKANNIBAL vector (Wesley et al., 2001) and sub-cloned into the pART27 vector to generate overexpression plasmid pAK–SmbHLH51 as described previously (Wang et al., 2017).
To generate the RNAi plasmid, a 260-bp fragment of SmbHLH51 cDNA (located from coding sequence positions 23–283 bp) was amplified with primers bHLH37i-F/R (Supplementary Table S1) and used to construct RNAi vector pSmbHLH51–RNAi according to previously published protocols (Wang et al., 2017).
Transgenic plants were generated through Agrobacterium-mediated transformation (Yan and Wang, 2007). In parallel, plasmid pAK, constructed in our laboratory (Wu et al., 2014), was introduced into WT S. miltiorrhiza as the empty-vector control PDK.
Transformant Selection and Transcript Analysis
To evaluate whether the overexpressing and interfering box had been integrated into the transgenic plant genome, we amplified the 35S promoter from isolated genomic DNA, using previously published protocols (Wu et al., 2014).
Transcript abundances for SmbHLH51 were analyzed by real-time quantitative PCR (RT-qPCR), using Actin as the internal reference gene (Yang et al., 2010). Relative gene expression level was performed using the 2-ΔΔC(T) method (Livak and Schmittgen, 2001). Based on the transcript levels of SmbHLH51, two SmbHLH51-OE lines (OE1 and OE35) and two SmbHLH51-RNAi lines (in4 and in10) were selected to detect the expression levels of 19 genes for enzymes involved in pathways for phenolic acid, flavonoid, and lignin biosynthesis. All experiments were performed on three independent biological samples, with each including three technical replicates. All primers are listed in Supplementary Table S1.
HPLC Analysis of Phenolic Acids
Roots were collected from 2-month-old SmbHLH51-overexpressing, SmbHLH51-RNAi and control tube plantlets. Extractions and HPLC analysis of phenolic acids were performed as described before except that the LC-2010A HPLC system was substituted by LC-2010 CHT (Zhang et al., 2010).
Determination of Total Phenolic, Total Flavonoid, and Anthocyanin Concentrations
The levels of total phenolics, total flavonoids, and anthocyanins in 2-month-old transgenic and control tube plantlets were determined by our laboratory procedures (Zhang et al., 2010).
Results
Gene Expression Profile of AtPAP1-Overexpressing (AtPAP1-14) and WT Salvia miltiorrhiza
To investigate the underlying mechanism of AtPAP1 in S. miltiorrhiza, we compared the global expression profiles of AtPAP1-14 versus the WT. Two cDNA libraries were sequenced by Illumina deep sequencing to obtain 3,567,545 and 3,679,557 clean tags for AtPAP1-14 and WT, respectively (Supplementary Table S2). These sequences were mapped to the S. miltiorrhiza reference transcriptome database (Hua et al., 2011). Tags with a complete match or a mismatch of only one base pair received further consideration. A comparable proportion of tags (44.24% for PAP1-14 and 50.96% for the control) matched the S. miltiorrhiza transcriptome (Supplementary Table S2). Transcriptome analysis identified 4152 differentially expressed genes (DEGs), with 1,601 unigenes being up-regulated and 2551 down-regulated in AtPAP1-14 when compared with expression in the WT (Figure 1A). Our KEGG analysis identified 23 significantly enriched metabolic pathways for those DEGs (Supplementary Table S3). Their functions included photosynthesis, as well as the biosynthesis of plant hormones, terpenoids, steroids, and phenylpropanoids. The pathway for phenylpropanoids enriched the most DEGs, 114, among the pathways involved in biosynthesis of secondary metabolites. Expression of four genes (PAL2, C4H, 4CL, and RAS) in the phenylpropanoid pathway and the tyrosine pathway gene TAT1 was increased more than twofold (Supplementary Table S4 and Figure 1B), a finding in accord with our earlier report (Zhang et al., 2010). The DEGs related to secondary metabolites, including those in the shikimate and flavonoid pathways and genes active in terpenoid backbone biosynthesis, are listed in Supplementary Table S4. Among the 10 DEGs in the terpenoid pathway, only one (encoding DXS) was up-regulated, which is consistent with the result from our metabolome analysis that showed no obvious changes in the level of tanshinones due to overexpression.
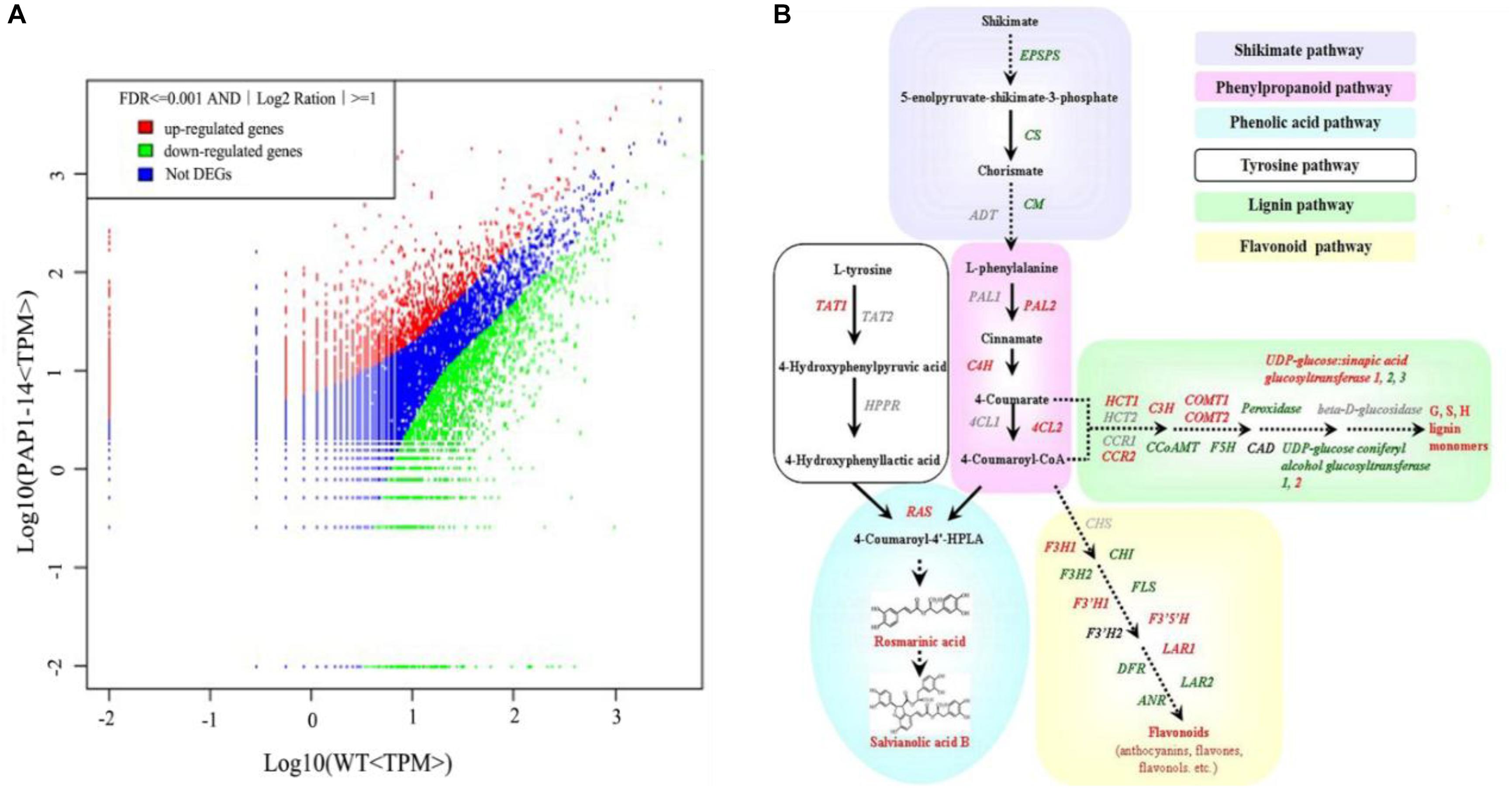
FIGURE 1. Transcriptome response of Salvia miltiorrhiza to overexpression of AtPAP1. (A) Comparison of gene expression between two libraries, with each normalized to one million tags. Red dots represent transcripts more prevalent in AtPAP1-14 library; green dots, those present at lower frequency in transgenic plants; and blue dots, transcript levels with no significant change. Parameters “FDR < 0.001” and “log2Ratio ≥ 1” were used as threshold to judge significance of differences in expression. (B) Proposed biosynthetic pathway for phenolics, beginning with core phenylpropanoid and tyrosine pathways, and leading to three major branch pathways: phenolic acid, flavonoid, and lignin (red, upregulated genes; green, downregulated). TAT, tyrosine aminotransferase; HPPR, hydroxyl phenylpyruvate reductase; PAL, phenylalanine ammonia lyase; C4H, cinnamate 4-hydroxylase; 4CL, hydroxycinnamate-CoA ligase; RAS, rosmarinic acid synthase; CYP, cytochrome P450 enzymes; CHS, chalcone synthase; CHI, chalcone isomerase; FLS, flavonol synthase; F3H, flavanone 3-hydroxylase; F3’H, flavonoid 3’-hydroxylase; F3′5′H, flavonoid 3′,5′-hydroxylase; DFR, dihydroflavonol 4-reductase; ANS, anthocyanin synthase; HCT, hydroxyl cinnamoyl transferase; CCR, cinnamoyl-CoA reductase; C3’ H, coumarate 3’-hydroxylase; COMT, caffeic acid O-methyltransferase; CCoAMT, caffeoyl-CoA O-methyltransferase; F5H, ferulate 5-hydroxylase; GT, glycosyltransferase; CAD, cinnamyl alcohol dehydrogenase.
In addition to analyzing the abundance of genes for enzymes in the biosynthesis pathway for secondary metabolites, we analyzed any TFs for which expression was changed by at least twofold between OE and WT plants. The 42 identified DEGs included those within the bHLH superfamily (8 members), WRKY superfamily (8), myeloblastosis (MYB) superfamily (6), and NAC superfamily (5) (Supplementary Table S5). We then focused on the bHLH genes because that family helps regulate the phenylpropanoid pathway. Among those eight bHLHs, the coding sequence of unigene37696 shows higher amino acid identity with MYC-RP (BAA75513, 79%), MYC-GP (BAA75514, 79%), AmDEL2 (AEM63394, 57%) and AmDEL (AAA32663, 56%), which are known to modulate flavonoids accumulation (Gong et al., 1999; Wang et al., 2016). This indicated that it has a critical role in phenolics biosynthesis.
Sequence Analysis and Subcellular Localization of SmbHLH51
Using unigene37696 and currently available transcriptome data, we experimentally isolated cDNA from this gene and initially named it SmMYC (GenBank Accession Number: KT215166). This gene contains a 1,911-bp open reading frame (ORF) that is predicted to encode a 636-amino acid (aa) protein with a molecular weight (MW) of 69.7 kDa and an isoelectric point (pI) of 5.70. We then used PCR to amplify a 3,420-bp genomic sequence from its genomic DNA. Comparison of the genomic DNA and cDNA sequences revealed that SmMYC harbors eight exons. Sequence analysis indicated that SmMYC contains not only a typical bHLH domain (440–489 aa) but also a bHLH-MYC_N pfam (13–196 aa). We compared the gene sequence of SmMYC with that of 127 bHLHs identified based on the entire genome sequence of S. miltiorrhiza (Zhang et al., 2015). The sequence of SmMYC is almost completely consistent with that of SmbHLH51 except that the latter is missing 12 bp, i.e., the ORF length for SmbHLH51 is 1,899 bp. We speculate that the missing 12 bp is probably an error caused by de novo assembly. To keep the consistence of gene name, SmMYC was renamed SmbHLH51 in this study.
Alignment of protein sequences between SmbHLH51 and homologous bHLH TFs revealed the presence of a typical bHLH domain and N-terminal conserved domains, including BOX11, BOX18, and BOX13 (Supplementary Figure S1), which interact with R2R3-MYB TFs (Heim et al., 2003; Pires and Dolan, 2010). We then conducted a phylogenetic analysis of bHLH IIIf proteins from various plant species, and found that SmbHLH51 had a close relationship with MYC-RP/MYC-GP from P. frutescens, AmDEL and AmDEL2 from Antirrhinum majus (Figure 2A). Subcellular localization assay showed that SmbHLH51 was localized to the nucleus (Figure 2B).
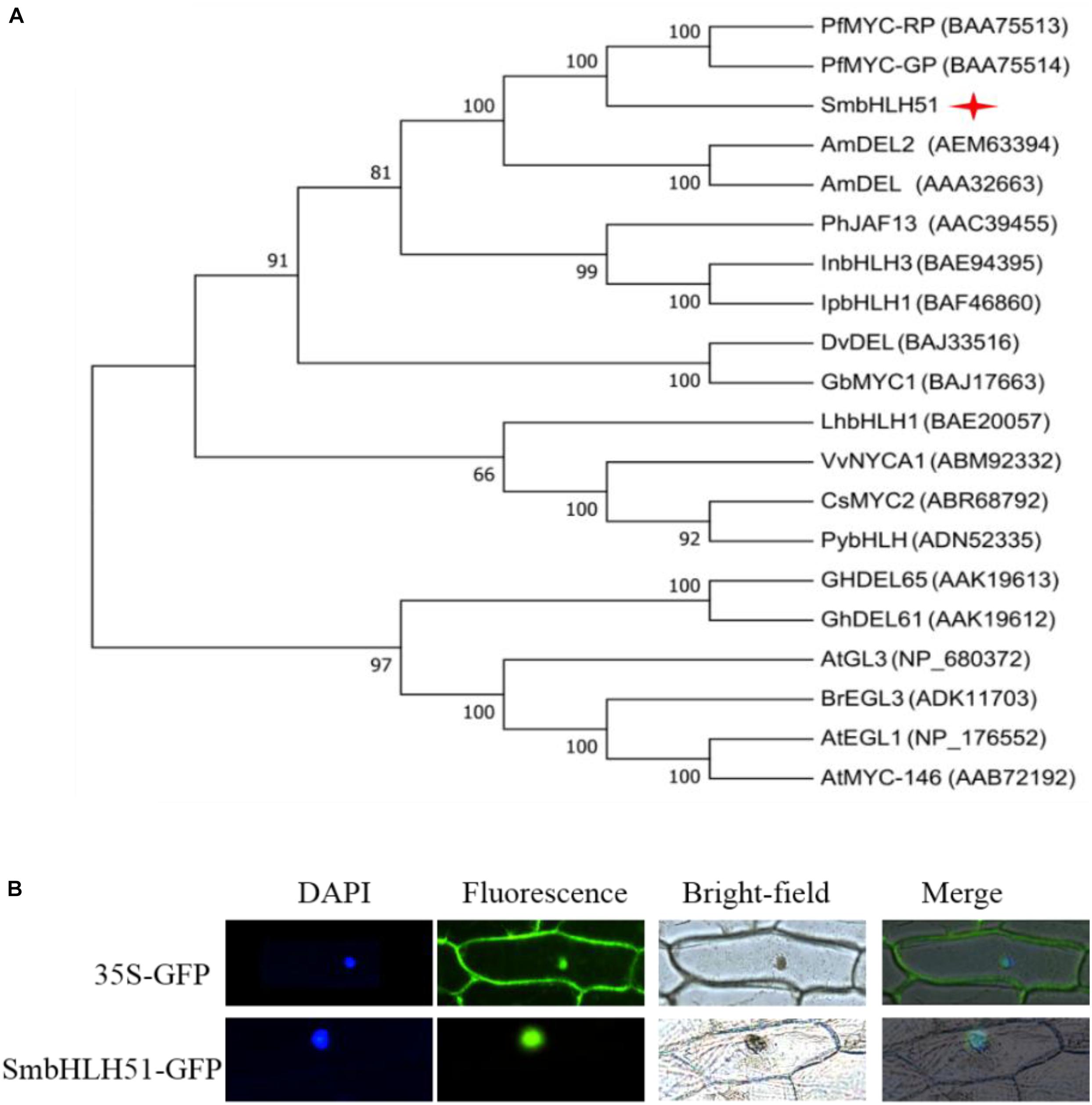
FIGURE 2. Sequence analysis of SmbHLH51 and subcellular localization. (A) Phylogenetic tree of SmbHLH51 and bHLH protein sequences from other species. (B) Subcellular localization of SmbHLH51 in onion epidermal cells.
SmbHLH51 Is Directly Activated by AtPAP1
Results from RT-qPCR further validated that the expression level of SmbHLH51 significantly increased in PAP1-14 when compared with the WT (Figure 3A), which is consistent with our transcriptome data. Analysis of the promoter region indicated that MBSII elements (AAAAGTTAGTTA) were located -1,435 bp upstream of the translation initiation codon ATG, a MYB binding site involved in regulating genes for flavonoid biosynthesis (Supplementary Figure S2). Our Y1H results showed that AtPAP1 could specifically bind to the MBSII elements in vivo (Figure 3B).
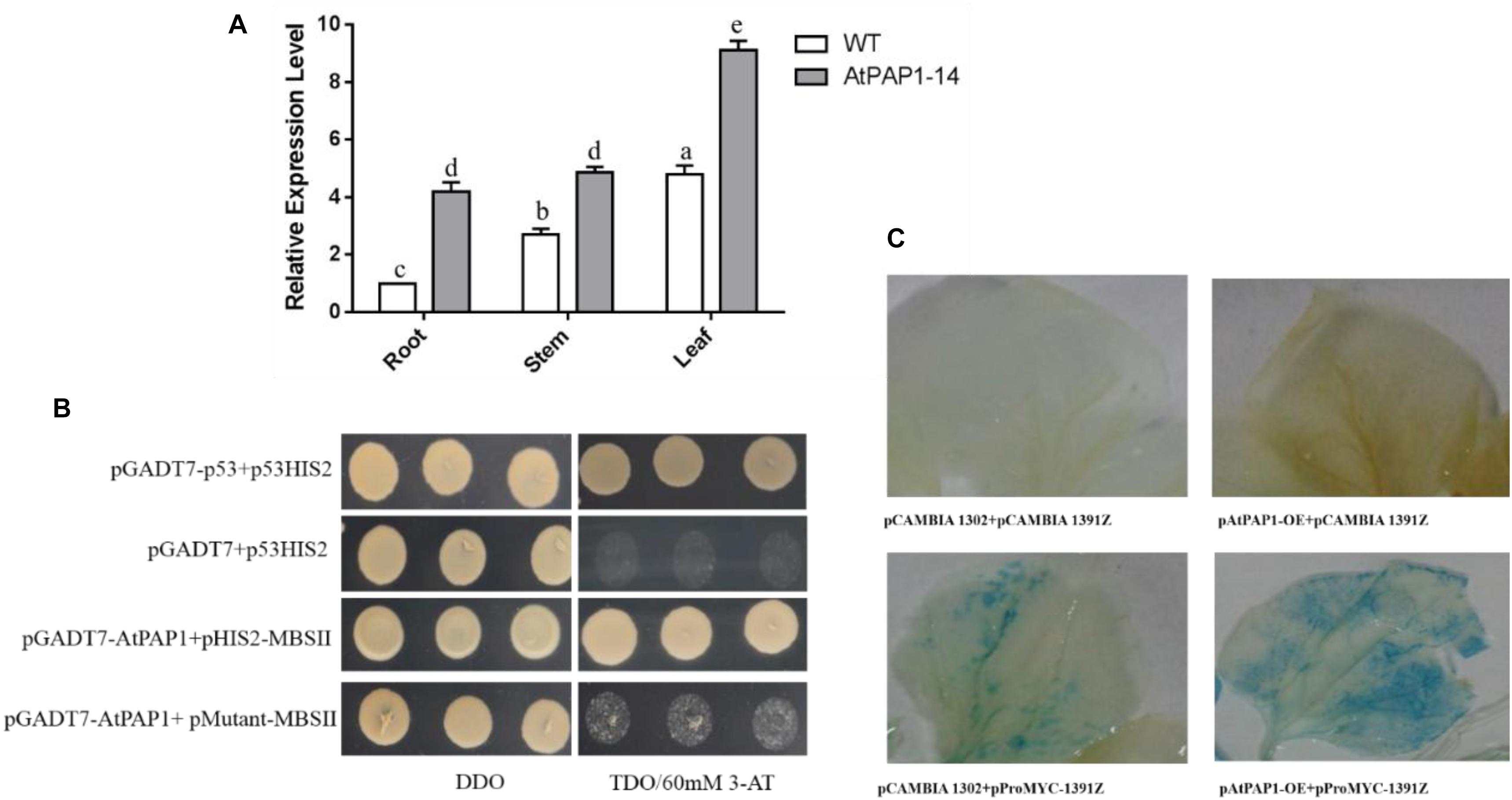
FIGURE 3. The relationship between AtPAP1 and the SmbHLH51 gene. (A) Relative expression level of SmbHLH51 in AtPAP1-14 and wild type (WT) roots. The results were analyzed using the comparative Ct method and presented as fold changes compared with the WT. The data represent means ± SD of three independent experiments. One-way ANOVA (followed by a Turkey comparison) tested for significant differences among the means (indicated by different letters at p < 0.01). (B) Interaction of AtPAP1 with the MBSII element as revealed by yeast one-hybrid assays. AtPAP1 was fused to GAL4 activation domain (AD). The core sequence of SmbHLH51 promoter MBSII elements were synthesized and cloned into pHIS2 to construct pHIS2-MBSII. Recombinant vectors were co-transformed into yeast strain Y187 and transformed cells were cultured on SD/-Leu/-Trp medium (DDO), then selected on SD/-Leu/-Trp/-His medium (TDO) supplemented with 60 mM 3-amino-1,2,4-triazole (3-AT) to examine protein-DNA interaction. The p53HIS2/pGADT7-p53 and p53HIS2/pGADT7 served as positive control and negative control, respectively. (C) AtPAP1 activates SmbHLH51 promoter via transient expression in tobacco leaves. The effector plasmid pAtPAP1-OE contains AtPAP1 driven by the CaMV 35S promoter. The reporter plasmid pProbHLH51-1391Z contains the GUS reporter gene driven by SmbHLH51 promoter.
We isolated the promoter of SmbHLH51 and used it to examine whether AtPAP1 could directly activate the expression of SmbHLH51 by Agrobacterium-mediated transient expression in tobacco (Nicotiana benthamiana). Results from GUS (β-glucuronidase) histochemical staining suggested that, indeed, AtPAP1 directly induced SmbHLH51 (Figure 3C). Taken together, our results based on RT-qPCR, Y1H and GUS histochemical staining indicated that SmbHLH51 is directly activated by AtPAP1.
AtPAP1 Interacts With SmbHLH51
The bHLH proteins within the IIIf subgroup (i.e., TT8, GL3, EGL3, and AtMYC1) can interact with various R2R3-MYBs, such as TT2 and PAP1–4 (Xu et al., 2015a). This implies that AtPAP1 has the ability to form a protein complex with IIIf bHLH factor SmbHLH51. Yeast cells co-transformed with vectors harboring AtPAP1-AD/SmbHLH51-BD and the pGBKT7/pGADT7 empty vector could not grow on plates holding a synthetic dextrose/-Ade/-His/-Leu/-Trp medium. However, the yeast cells transformed with AtPAP1-AD and SmbHLH51-BD grew normally, suggesting that AtPAP1 interacts with SmbHLH51 in yeast (Figure 4A).
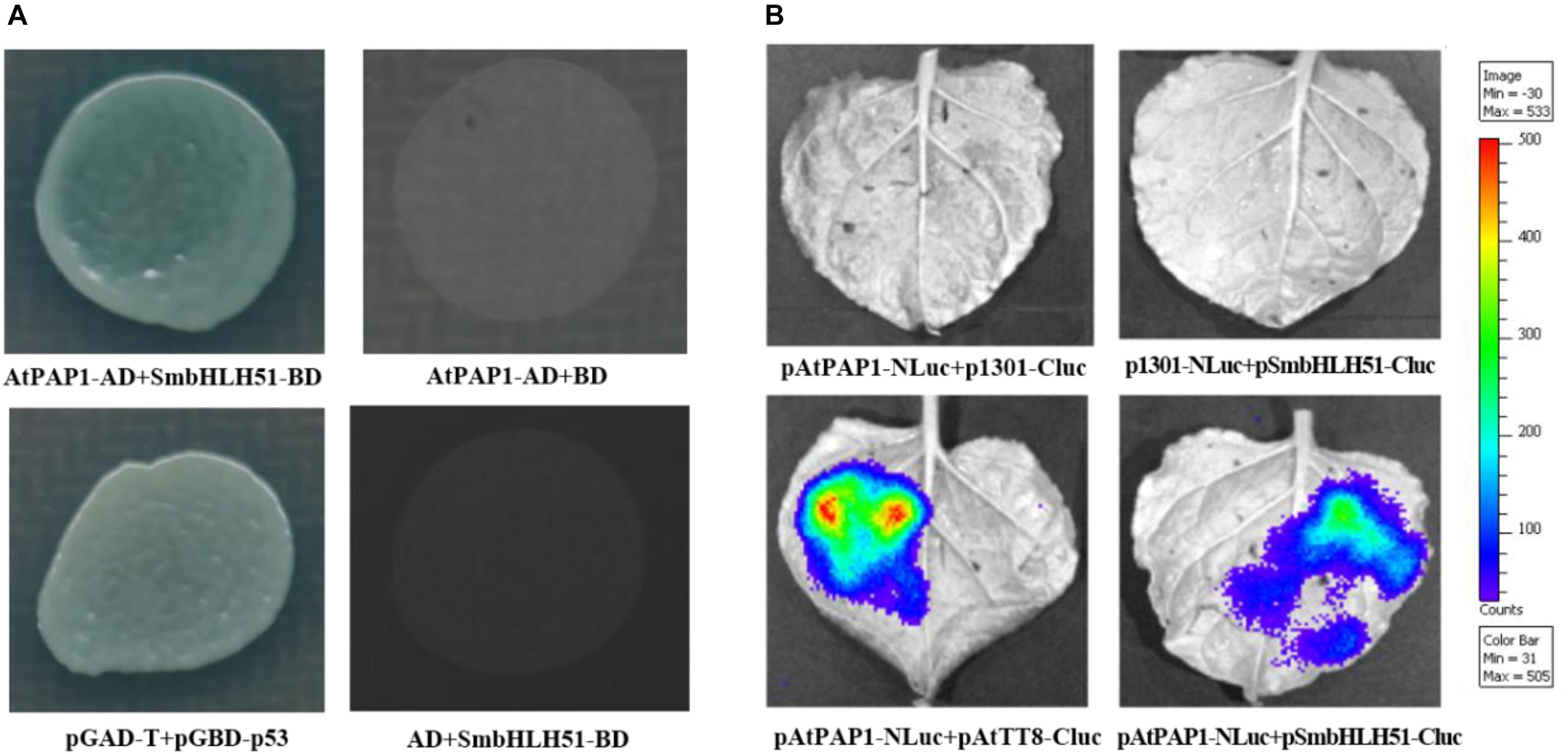
FIGURE 4. AtPAP1 physically interacts with SmbHLH51. (A) Y2H assay to test interactions of AtPAP1 with SmbHLH51. AtPAP1 was fused with activation domain while SmbHLH51 was fused with DNA-binding domain. (B) LCI assay to detect interactions of AtPAP1 with SmbHLH51. AtPAP1was fused with N-terminal of luciferase to produce pAtPAP1-NLuc. AtTT8 (GenBank Accession Number NM_117050) and SmbHLH51 were fused with C-terminal of luciferase to produce pAtTT8-Cluc and pSmbHLH51-Cluc, respectively.
To confirm those Y2H results, we performed a firefly luciferase complementation imaging (LCI) assay to detect the interaction between AtPAP1 and SmbHLH51. Whereas AtPAP1 was fused with the N-terminal to the NLuc fragment, SmbHLH51 was fused with its C-terminal to the CLuc fragment. As expected, co-injection of the vector constructs into the tobacco leaves presented the same interaction features of the proteins (Figure 4B) that had been observed in the Y2H test. Therefore, all of our Y2H and LCI results demonstrated that AtPAP1 interacts with SmbHLH51 in vivo.
Generation of Transgenic S. miltiorrhiza Plants
Explants were transformed via Agrobacterium mediation with overexpression plasmid pAK-SmbHLH51, RNAi vector pSmbHLH51-RNAi (Figure 5A), and the empty control pAK. After selective culturing on a kanamycin medium, PCR-amplification was conducted with genomic DNA as templates. An expected 901-bp fragment of the CaMV 35S promoter was amplified in the transgenic S. miltiorrhiza plants but not in the WT (Figure 5B). Our PCR analysis showed that the positive transformation rate of SmbHLH51-overexpressing (SmbHLH51-OE) and SmbHLH51-RNAi lines was 70% and 95%, respectively. We then conducted RT-qPCR analysis of those transgenics (Figure 5C). From this we selected for further examination two SmbHLH51-OE lines (OE1 and OE35) with higher SmbHLH51 transcription levels and two SmbHLH51-RNAi lines (in4 and in10) with lower SmbHLH51 transcription levels. Our controls were the WT and those plants that had been transformed with the corresponding empty vector (PDK).
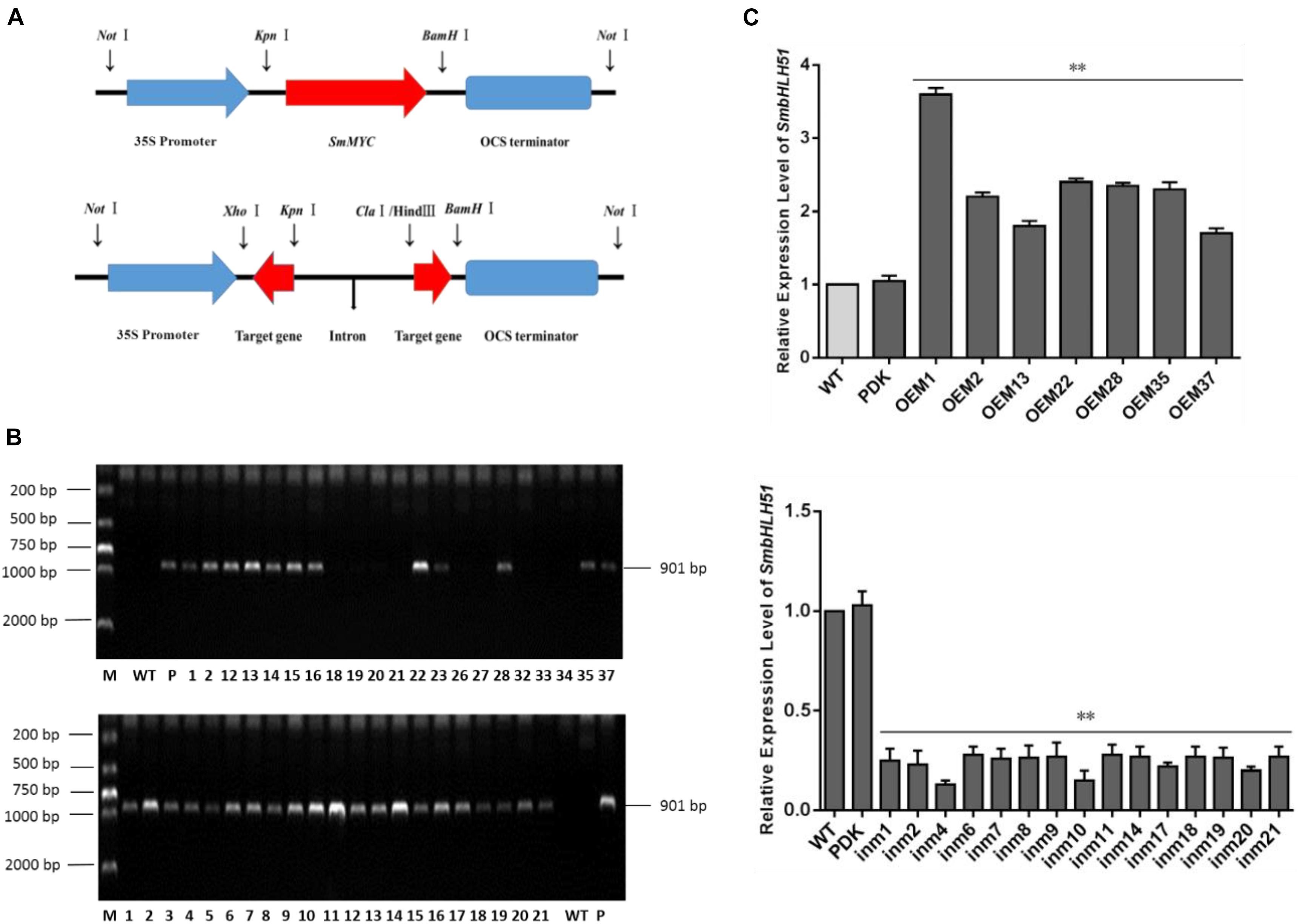
FIGURE 5. Detection of SmbHLH51-overexpressing and SmbHLH51-RNAi transgenic lines. (A) Sketch map of overexpression and transcription box for RNAi vectors. (B) PCR amplification product of 35S promoter from transgenic Salvia miltiorrhiza gDNA. Lanes: M, DL2000 DNA marker; P, plasmids as positive control; WT, wild-type plants as negative control. (C) Relative expression levels of SmbHLH51 in SmbHLH51-overexpressing transgenic lines (OE) and SmbHLH51-RNAi transgenic lines (in). The data represent means ± SD of three independent experiments. Level of SmbHLH51 in the wild type was set as 1. ∗∗p < 0.01 compared with the wild type (Student’s t-test).
Phenotypes of SmbHLH51-Overexpressing and SmbHLH51-RNAi S. miltiorrhiza
To investigate whether phenolic acids metabolism was modified in our SmbHLH51-transgenic plants, we extracted their phenolic acids and separated them via HPLC. The results showed that levels of RA and Sal B did not differ significantly between the WT and PDK. However, when compared with the WT, the concentrations of both RA and Sal B were increased significantly (P < 0.01) in OE1 and OE35 (Figure 6A). For example, the amount of RA was 1.54-fold and 2.77-fold higher in OE1 and OE35, respectively, than in the WT, while Sal B concentrations were approximately 1.28-fold and 2.54-fold higher, respectively, than in the WT. In contrast, the levels of RA and Sal B were significantly lower (P < 0.01) in the SmbHLH51-RNAi lines in4 and in10 (Figure 6A), with those reductions being 17.99 % and 12.32%, respectively, for RA and 50.00% and 45.20%, respectively, for Sal B.
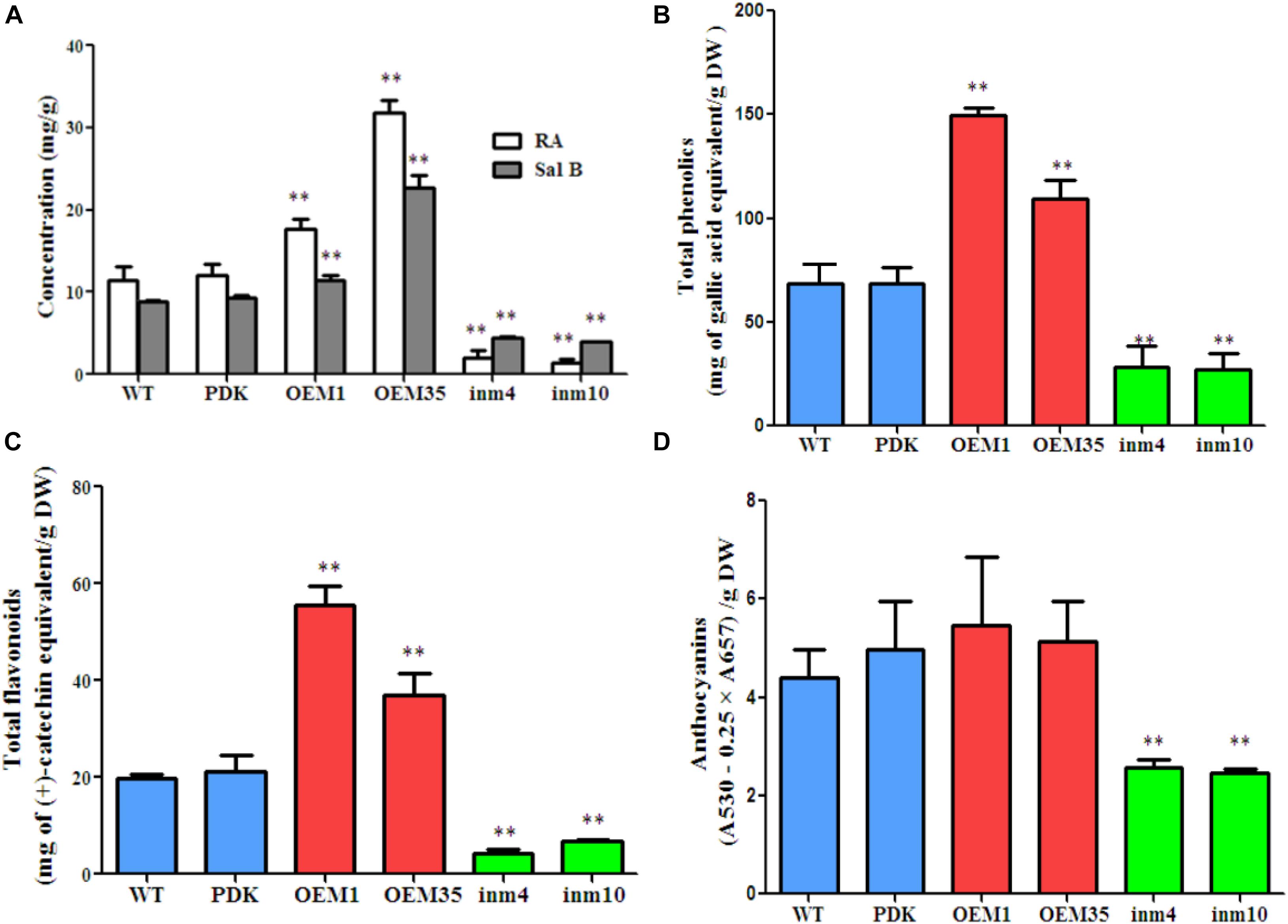
FIGURE 6. Comparison of secondary metabolite concentrations between transgenic lines and controls. (A) Levels of RA and Sal B in root extracts. (B–D) Levels of total phenolics (B), total flavonoids (C), and anthocyanins (D) from root extracts. The data represent means ± SD of three independent experiments. ∗∗p < 0.01 compared with the wild type (Student’s t-test).
We also performed global assays for phenolics, flavonoids, and anthocyanins because they share an upstream core phenylpropanoid metabolism with Sal B. Total phenolics and total flavonoids accumulated at higher levels in the roots of the two SmbHLH51-OE lines than in the WT. Differences were significant and corresponded to 2.19-fold and 1.59-fold increases in total phenolics concentrations for OE1 and OE35, respectively, and 2.81-fold and 1.87-fold increases in total flavonoids (Figures 6B,C). No significant changes in anthocyanin concentrations among genotypes were observed (Figure 6D). In contrast, when compared with the WT, the levels of total phenolics, total flavonoids, and anthocyanins were dramatically lower in both SmbHLH51-RNAi lines, with decreases being 41% (in4) and 40% (in10) for total phenolics, 22% (in4) and 34% (in10) for total flavonoids, and 56% (in4) and 54% (in10) for anthocyanins (Figures 6B–D). These results implied that SmbHLH51 is an important regulator of biosynthesis for phenolic acids and flavonoids (including anthocyanins).
Effect of SmbHLH51 on the Expression of Genes in the Phenolics Pathway
To evaluate whether these substantial changes in the levels of phenolics in the transgenics were a result of significant alterations in gene expression relative to the control, we examined the transcript levels of 19 genes for enzymes in the phenolics biosynthesis pathway. Relative to expression in the WT, expression in the SmbHLH51-OE or SmbHLH51-RNAi transgenic plants was markedly different for genes in the core phenylpropanoid pathway and specific branch pathways (Figure 7). Of those 19 genes, 11 were significantly up-regulated in the SmbHLH51-OE lines while nine were dramatically down-regulated in the SmbHLH51-RNAi lines. Expression of SmCAD was unexpectedly increased in the SmbHLH51-RNAi lines. Six genes (SmTAT1, SmHPPR, SmRAS1, SmRAS5, SmDFR, and SmANS) were significantly induced in SmbHLH51-OE lines but their expression was dramatically reduced in SmbHLH51-RNAi lines. Another five genes (SmPAL2, Sm4CL2, SmCYP98A14, SmCHS, and SmF3’5’H) were significantly up-regulated in SmbHLH51-OE lines but their expression was not significantly changed in SmbHLH51-RNAi lines. Three genes (SmPAL1, SmPAL3, and SmC4H) were markedly down-regulated in SmbHLH51-RNAi lines but not significantly altered in SmbHLH51-OE lines. Finally, the expression of four genes – Sm4CL1, SmHCT, SmCCR, and SmCOMT – was not influenced significantly in either SmbHLH51-OE or SmbHLH51-RNAi lines. All of these findings demonstrated that SmbHLH51 positively regulates many genes that encode enzymes in the pathways for phenolic acid and flavonoid biosynthesis.
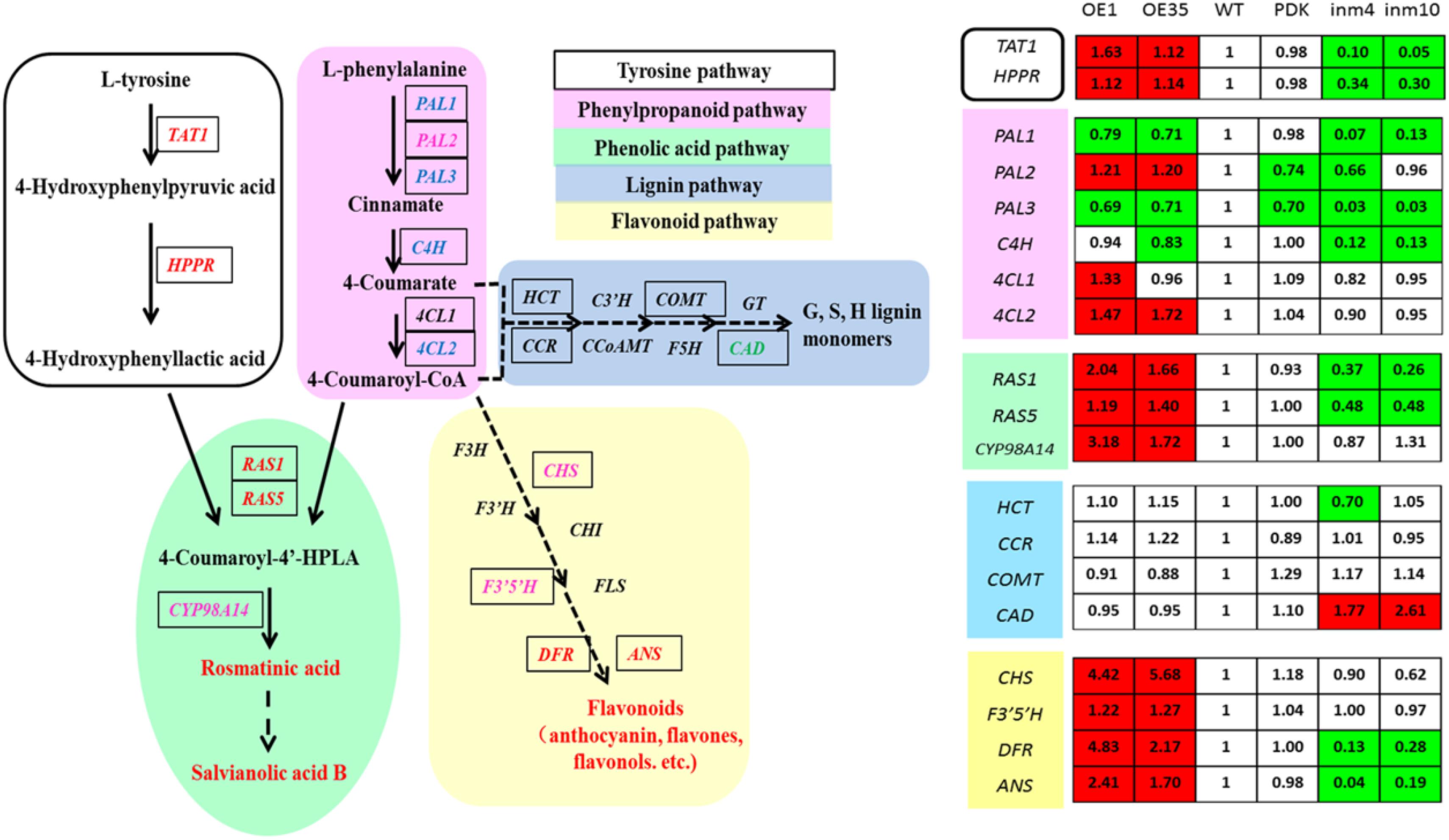
FIGURE 7. Relative expression levels of 19 phenolics-pathway genes in SmbHLH51-overexpressing and SmbHLH51-RNAi Salvia miltiorrhiza were analyzed by RT-qPCR. The data represent fold changes of phenolics-pathway genes compared with the wild type (WT). Red box indicates the data is significantly upregulated compared with the WT (p < 0.05, Student’s t-test); green box indicates the data is significantly downregulated compared with the WT (p < 0.05, Student’s t-test). The following genes were retrieved from GenBank databases: TAT1 (DQ334606.1), HPPR (DQ099741.1), PAL1 (EF462460.1), PAL2 (GQ249111.1), PAL3 (KF220569.1), C4H (DQ355979.1), 4CL1 (AY237163.1), 4CL2 (AY237164.1), RAS1 (FJ906696.1), RAS5 (KF220573.1), CYP98A14 (HQ316179.1), CAD (HQ162287.1), CHS (KF255832.1), F3’5’H (MH447665.1), DFR (MH447664.1), ANS (MH447663.1). HCT (Smil-00001226), CCR (Smil-00000329) and COMT (Smil-00026281) were retrieved from the web portal at http://www.ndctcm.org/shujukujieshao/2015-04-23/27.html.
The bHLH TFs can form homodimers or heterodimers to bind both canonical and non-canonical E-box sites (CANNTG) (Earley et al., 2006). We analyzed the promoter sequences of the 15 phenolics-pathway genes that were most significantly regulated in SmbHLH51-OE and SmbHLH51-RNAi transgenic lines and found that most, except PAL2 and CAD, contained E/G-box sequences in their promoters (Supplementary Table S6). We also determined that the promoter regions of 10 genes (TAT1, HPPR, PAL3, C4H, 4CL2, RAS5, CYP98A14, CHS, DFR, and ANS) contained not only E-box sites but also MBS elements. Whether these genes are directly activated by SmbHLH51 or AtPAP1-SmbHLH51 complex requires further study.
Discussion
Global Changes in Transcriptome Caused by Ectopic AtPAP1 Expression in Salvia miltiorrhiza
Altering the expression of TFs can be an effective means for coordinately modulating entire metabolic pathways in plants (Massari and Murre, 2000). AtPAP1 is a well studied TF which could be used to stimulate not only anthocyanin accumulation but also the biosynthesis of some valuable phenolic compounds in medicinal plants. For instance, Overexpression of AtPAP1 enhances chlorogenic acid production in hairy roots of Platycodon grandiflorum (Tuan et al., 2014). We previously reported that overexpression of AtPAP1 in S. miltiorrhiza significantly enhanced the levels of not only Sal B, but also total flavonoids, anthocyanins, and lignin (Zhang et al., 2010). However, the responsible mechanism has not been well understood. To improve our knowledge about the regulatory mechanisms by which AtPAP1 promotes phenolic acid biosynthesis, we used transcriptomic analyses to characterize transgenic S. miltiorrhiza plants that over-expressed that gene.
In this study, we compared the transcriptomes between AtPAP1-overexpressing S. miltiorrhiza and the WT. Transcriptome analysis demonstrated that a novel bHLH TF, SmbHLH51, is activated by AtPAP1 in S. miltiorrhiza. Usually the bHLH genes are positively regulated by R2R3-MYB TF (Baudry et al., 2006; Bai et al., 2011; Montefiori et al., 2015). Based on previous reports, we propose that bHLHs, which is activated by AtPAP1 and can interact with AtPAP1 in S. miltiorrhiza, is potentially involved in the regulation of phenolic acids biosynthesis. The results from our RT-qPCR, Y1H and Agrobacterium-mediated transient expression in tobacco indicated that AtPAP1 positively regulates SmbHLH51 by directly binding to the MBSII element of promoter region (Figure 3). Therefore, the latter gene is functionally identified as a positive regulator of phenolic acid biosynthesis. In addition to its direct transcriptional regulation activities, AtPAP1 interacts with SmbHLH51 and functions as a protein complex to promote the accumulation of phenolics.
The Integrated Strategy Reveals That SmbHLH51 Is a Putative Regulator of Phenolics Accumulations in S. miltiorrhiza
We employed this integrated approach to identify regulatory factors that influence phenolics accumulations because that process has been poorly understood in S. miltiorrhiza. The bHLH proteins, comprising one of the largest TF families, regulate various physiological or morphological events, including those in different branches of the flavonoid pathway (Hichri et al., 2011). The bHLHs Booster1 (B) and Red1 (R), which control the anthocyanin biosynthesis pathway in seeds, were first found in Zea mays (Chandler and Turks, 1989). Since then, several other bHLH proteins regulating the flavonoid pathway have been identified, including Delila (DEL) in Antirrhinum majus (Martin et al., 1991); MYC-RP in P. frutescens (Gong et al., 1999); AN1 and JAF13 in Petunia hybrid (Quattrocchio et al., 1998; Spelt et al., 2000). AtMYC-146, GLABRA3 (GL3), and TT8 in Arabidopsis (Nesi et al., 2000; Ramsay et al., 2003); and VvMYC1 and VvMYCA1 in Vitis vinifera (Hichri et al., 2010). Usually the bHLH genes are positively regulated by R2R3-MYB TF (Baudry et al., 2006; Bai et al., 2011; Montefiori et al., 2015). Based on these reports, we propose that bHLHs, which is activated by AtPAP1 and can interact with AtPAP1 in S. miltiorrhiza, is potentially involved in the regulation of phenolic acids biosynthesis.
To date, at least 43 bHLH TFs have been identified as regulators of secondary metabolism in at least 21 plant species, with those active components including flavonoids, alkaloids, and terpenoids (Zhang et al., 2015). The bHLHs have been divided into 26 subgroups (Pires and Dolan, 2010), those related to flavonoids are in subgroup IIIf (Montefiori et al., 2015). We also were able to assign SmbHLH51 to the IIIf subfamily based on the bHLH group nomenclature presented above. Members of the same plant bHLH subfamily are frequently involved in the same biological process.
SmbHLH51 shows high similarity with MYC-RP from P. frutescens (Figure 2C), which regulates anthocyanin biosynthesis by controlling the transcription of structural genes PAL, DFR, and ANS in that pathway (Gong et al., 1999). We found here that SmbHLH51 positively regulates not only DFR, ANS, CHS, and F3’5’H in the flavonoid biosynthesis pathway, but also PAL and C4H in the core phenylpropanoid pathway, TAT and HPPR in the tyrosine pathway, and RAS1 and RAS5 in the phenolic acid pathway (Figure 6). This might explain the significant changes in phenylpropanoid-derived end products in our transgenic lines. We also showed that SmbHLH51 positively regulates different branches of the phenylpropanoid pathway, altering the accumulations of phenolic acids and flavonoids, which is consistent with the function of AtPAP1 in S. miltiorrhiza.
Interactions Between AtPAP1 and SmbHLH51
Although MYB and bHLH can individually regulate phenylpropanoid metabolism and enhance the production of secondary metabolites, many studies have confirmed that they could play more prominent roles when in the form of a MYB/bHLH/WD40 (MBW) protein complex (Gonzalez et al., 2008; Qi et al., 2011; Xu et al., 2015a). The interactions between R2R3-MYB TFs and the bHLHs from subgroup IIIf are among the best-described examples of cooperation between plant transcription regulators (Xu et al., 2015a). In Arabidopsis, AtPAP1 can interact with TT8, GL3, EGL3, or AtMYC1, together with TTG1 (WDR family), to form MBW complexes and activate flavonoid biosynthesis (Feller et al., 2011). Our Y2H and LCI results showed that AtPAP1 interacts with SmbHLH51 (Figure 3), indicating that AtPAP1 elicits strong induction of phenolic acids in S. miltiorrhiza through an efficient interaction with the endogenous bHLH factor SmbHLH51, so that they are functionally formed as a regulatory transcription complex.
AtPAP1 has a critical role in controlling the production of soluble phenolic compounds in S. miltiorrhiza (Zhang et al., 2010) By now only a few endogenous R2R3-MYBs in S. miltiorrhiza have been functionally characterized through genetic screening, including SmMYB39 and SmMYB36, two repressors of the phenolic acids pathway (Zhang et al., 2013; Ding et al., 2017). Although SmPAP1, an endogenous R2R3-MYB TF from S. miltiorrhiza, was reported to be a positive regulator for the phenolic acid biosynthetic pathway (Hao et al., 2016), its ability in controlling the production of phenolic acids is far from that of AtPAP1 (Zhang et al., 2010). Therefore, it would be very beneficial to identify more endogenous TFs related to phenolic acid biosynthesis in S. miltiorrhiza. In the present study, SmbHLH51 was identified as a positive regulator for phenolic acid biosynthesis. We speculate that the endogenous R2R3-MYB, interacting with SmbHLH51, has the same role.
Conclusion
We have characterized AtPAP1-overexpressing plants of S. miltiorrhiza through transcriptomic and metabolomic analyses. Our data provide a molecular phenotype for these transgenics in which AtPAP1 strongly promotes the accumulation of phenolics, consistent with the upregulation of genes in the pathway for phenylpropanoid biosynthesis. Through integrative analyses of the metabolome and transcriptome of OE plants, we discovered SmbHLH51, a novel bHLH gene that we functionally identified as a positive transcriptional regulator of the phenolics pathway. Our findings demonstrate that AtPAP1 stimulates the production of phenolic acids in S. miltiorrhiza by activating the expression of SmbHLH51 and forming a transcriptional complex with SmbHLH51. This improves our knowledge about the regulatory mechanism of AtPAP1. Further experiments are needed to elucidate the precise molecular mechanism by which AtPAP1/SmbHLH51 regulates the biosynthesis of phenolic acids in S. miltiorrhiza.
Author Contributions
ZW and XC designed and coordinated the study. YW, YZ, LL, XG and BW performed the experiments and analyzed the data. YZ and XC wrote the manuscript.
Funding
This work was funded by the National Natural Science Foundation of China (Grant Nos. 31670299 and 31870276), National Key Technologies R&D Program for Modernization of Traditional Chinese Medicine (Grant No. 2017YFC1701300), Major Project of Shaanxi Province, China (Grant No. 2017ZDXM-SF-005), Natural Science Foundation of Shaanxi Province, China (Grant No. 2018JZ3001), Fundamental Research Funds for the Central Universities (Grant No. GK201706004) and Natural Science Foundation of Hebei Province, China (Grant No. C2018402145).
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2018.01687/full#supplementary-material
Footnotes
- ^http://www.genome.jp/kegg/
- ^http://gsds.cbi.pku.edu.cn
- ^www.ncbi.nlm.nih.gov/BLAST/
- ^http://www.expasy.ch/tools/pi_tool.html
References
Bai, Y., Pattanaik, S., Patra, B., Werkman, J. R., Xie, C. H., and Yuan, L. (2011). Flavonoid-related basic helix-loop-helix regulators, NtAn1a and NtAn1b, of tobacco have originated from two ancestors and are functionally active. Planta 234, 363–375. doi: 10.1007/s00425-011-1407-y
Baudry, A., Caboche, M., and Lepiniec, L. (2006). TT8 controls its own expression in a feedback regulation involving TTG1 and homologous MYB and bHLH factors, allowing a strong and cell-specific accumulation of flavonoids in Arabidopsis thaliana. Plant J. 46, 768–779. doi: 10.1111/j.1365-313X.2006.02733.x
Butelli, E., Titta, L., Giorgio, M., Mock, H., Matros, A., Peterek, S., et al. (2008). Enrichment of tomato fruit with health-promoting anthocyanins by expression of select transcription factors. Nat. Biotechnol. 26, 1301–1308. doi: 10.1038/nbt.1506
Chandler, V. L., and Turks, D. (1989). Two regulatory genes of the maize anthocyanin pathway are homologous: isolation of B utilizing R genomic sequences. Plant Cell 1, 1175–1183. doi: 10.1105/tpc.1.12.1175
Cheng, T. O. (2006). Danshen: a popular chinese cardiac herbal drug. J. Am. Coll. Cardiol. 47, 1499–1500. doi: 10.1016/j.jacc.2006.01.001
Ding, K., Pei, T., Bai, Z., Jia, Y., Ma, P., and Liang, Z. (2017). SmMYB36, a novel R2R3-MYB transcription factor, enhances tanshinone accumulation and decreases phenolic acid content in Salvia miltiorrhiza hairy roots. Sci. Rep. 7:5104. doi: 10.1038/s41598-017-04909-w
Earley, K. W., Haag, J. R., Pontes, O., Opper, K., Juehne, T., Song, K., et al. (2006). Gateway-compatible vectors for plant functional genomics and proteomics. Plant J. 45, 616–629. doi: 10.1111/j.1365-313X.2005.02617.x
Feller, A., Machemer, K., Braun, E., and Grotewold, E. (2011). Evolutionary and comparative analysis of MYB and bHLH plant transcription factors. Plant J. 66, 94–116. doi: 10.1111/j.1365-313X.2010.04459.x
Gascuel, O., and Steel, M. (2006). Neighbor-joining revealed. Mol. Biol. Evol. 23, 1997–2000. doi: 10.1093/molbev/msl072
Ge, Q., Zhang, Y., Hua, W. P., Wu, Y. C., Jin, X. X., Song, S. H., et al. (2015). Combination of transcriptomic and metabolomic analyses reveals a JAZ repressor in the jasmonate signaling pathway of Salvia miltiorrhiza. Sci. Rep. 5:14048. doi: 10.1038/srep14048
Gong, Z. Z., Yamagishi, E., Yamazaki, M., and Saito, K. (1999). A constitutively expressed Myc-like gene involved in anthocyanin biosynthesis from Perilla frutescens: molecular characterization, heterologous expression in transgenic plants and transactivation in yeast cells. Plant Mol. Biol. 41, 33–44. doi: 10.1023/A:1006237529040
Gonzalez, A., Zhao, M., Leavitt, J. M., and Lloyd, A. M. (2008). Regulation of the anthocyanin biosynthetic pathway by the TTG1/bHLH/Myb transcriptional complex in Arabidopsis seedlings. Plant J. 53, 814–827. doi: 10.1111/j.1365-313X.2007.03373.x
Hao, G., Jiang, X., Feng, L., Tao, R., Li, Y., and Huang, L. (2016). Cloning, molecular characterization and functional analysis of a putative R2R3-MYB transcription factor of the phenolic acid biosynthetic pathway in S. miltiorrhiza Bge. f. alba. Plant Cell Tissue Organ Cult. 124, 151–168. doi: 10.1007/s11240-015-0883-3
Heim, M. A., Jakoby, M., Werber, M., Martin, C., Weisshaar, B., and Bailey, P. C. (2003). The basic helix–loop–helix transcription factor family in plants: a genome-wide study of protein structure and functional diversity. Mol. Biol. Evol. 20, 735–747. doi: 10.1093/molbev/msg088
Hichri, I., Barrieu, F., Bogs, J., Kappel, C., Delrot, S., and Lauvergeat, V. (2011). Recent advances in the transcriptional regulation of the flavonoid biosynthetic pathway. J. Exp. Bot. 62, 2465–2483. doi: 10.1093/jxb/erq442
Hichri, I., Heppel, S. C., Pillet, J., Léon, C., Czemmel, S., Delrot, S., et al. (2010). The basic helix-loop-helix transcription factor MYC1 is involved in the regulation of the flavonoid biosynthesis pathway in grapevine. Mol. Plant 3, 509–523. doi: 10.1093/mp/ssp118
Hua, W., Song, J., Li, C., and Wang, Z. (2012). Molecular cloning and characterization of the promoter of SmGGPPs and its expression pattern in Salvia miltiorrhiza. Mol. Biol. Rep. 39, 5775–5783. doi: 10.1007/s11033-011-1388-8
Hua, W., Zhang, Y., Song, J., Zhao, L., and Wang, Z. (2011). De novo transcriptome sequencing in Salvia miltiorrhiza to identify genes involved in the biosynthesis of active ingredients. Genomics 98, 272–279. doi: 10.1016/j.ygeno.2011.03.012
Huang, X. S., Wang, W., Zhang, Q., and Liu, J. H. (2013). A basic helix-loop-helix transcription factor, PtrbHLH, of Poncirus trifoliata confers cold tolerance and modulates peroxidase-mediated scavenging of hydrogen peroxide. Plant Physiol. 162, 1178–1194. doi: 10.1104/pp.112.210740
Jefferson, R. A., Kavanagh, T. A., and Bevan, M. W. (1987). GUS fusions: beta-glucuronidase as a sensitive and versatile gene fusion marker in higher plants. EMBO J. 6, 3901–3907. doi: 10.1002/j.1460-2075.1987.tb02730.x
Li, S., Wu, Y., Kuang, J., Wang, H., Du, T., Huang, Y., et al. (2018). SmMYB111 is a key factor to phenolic acid biosynthesis and interacts with both SmTTG1 and SmbHLH51 in Salvia miltiorrhiza. J. Agric. Food Chem. 66, 8068–8078. doi: 10.1021/acs.jafc.8b02548
Li, X., Xue, Z., and Zhu, W. (2011). Antioxidant activities and contents of total flavonoids and phenols from different parts of Salvia miltiorrhiza bunge. Food Sci. 32, 108–111.
Livak, K. J., and Schmittgen, T. D. (2001). Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 25, 402–408. doi: 10.1006/meth.2001.1262
Martin, C., Prescott, A., Mackay, S., Bartlett, J., and Vrijlandt, E. (1991). Control of anthocyanin biosynthesis in flowers of Antirrhinum majus. Plant J. 1, 37–49. doi: 10.1111/j.1365-313X.1991.00037.x
Massari, M. E., and Murre, C. (2000). Helix-loop-helix proteins: regulators of transcription in eucaryotic organisms. Mol. Cell. Biol. 20, 429–440. doi: 10.1128/mcb.20.2.429-440.2000
Montefiori, M., Brendolise, C., Dare, A. P., Linwang, K., Davies, K. M., Hellens, R. P., et al. (2015). In the solanaceae, a hierarchy of bHLHs confer distinct target specificity to the anthocyanin regulatory complex. J. Exp. Bot. 66, 1427–1436. doi: 10.1093/jxb/eru494
Nesi, N., Debeaujon, I., Jond, C., Pelletier, G., Caboche, M., and Lepiniec, L. (2000). The TT8 gene encodes a basic helix-loop-helix domain protein required for expression of DFR and BAN genes in Arabidopsis siliques. Plant Cell 12, 1863–1878. doi: 10.1105/tpc.12.10.1863
Pei, T., Ma, P., Ding, K., Liu, S., Jia, Y., Ru, M., et al. (2018). SmJAZ8 acts as a core repressor regulating JA-induced biosynthesis of salvianolic acids and tanshinones in Salvia miltiorrhiza hairy roots. J. Exp. Bot. 69, 1663–1678. doi: 10.1093/jxb/erx484
Petersen, M., and Simmonds, M. S. (2003). Rosmarinic acid. Phytochemistry 62:121. doi: 10.1007/978-3-540-71095-0_8989
Pires, N., and Dolan, L. (2010). Origin and diversification of basic-helix-loop-helix proteins in plants. Mol. Biol. Evol. 27, 862–874. doi: 10.1093/molbev/msp288
Qi, T., Song, S., Ren, Q., Wu, D., Huang, H., Chen, Y., et al. (2011). The jasmonate-ZIM-domain proteins interact with the WD-repeat/bHLH/MYB complexes to regulate jasmonate-mediated anthocyanin accumulation and trichome initiation in Arabidopsis thaliana. Plant Cell 23, 1795–1814. doi: 10.1105/tpc.111.083261
Quattrocchio, F., Wing, J. F., Van, D. W. K., Mol, J. N., and Koes, R. (1998). Analysis of bHLH and MYB domain proteins: species-specific regulatory differences are caused by divergent evolution of target anthocyanin genes. Plant J. 13, 475–488. doi: 10.1046/j.1365-313X.1998.00046.x
Ramsay, N. A., Walker, A. R., Mooney, M., and Gray, J. C. (2003). Two basic-helix-loop-helix genes (MYC-146 and GL3) from Arabidopsis can activate anthocyanin biosynthesis in a white-flowered Matthiola incana mutant. Plant Mol. Biol. 52, 679–688. doi: 10.1023/A:1024852021124
Shao, F., and Lu, S. (2013). Genome-wide identification, molecular cloning, expression profiling and posttranscriptional regulation analysis of the Argonaute gene family in Salvia miltiorrhiza, an emerging model medicinal plant. BMC Genomics 14:512. doi: 10.1186/1471-2164-14-512
Song, Z., Guo, L., Liu, T., Lin, C., Wang, J., and Li, X. (2017). Comparative RNA-sequence transcriptome analysis of phenolic acid metabolism in Salvia miltiorrhiza, a traditional Chinese medicine model plant. Int. J. Genomics 2017:9364594. doi: 10.1155/2017/9364594
Spelt, C., Quattrocchio, F., Mol, J. N., and Koes, R. (2000). Anthocyanin1 of petunia encodes a basic helix-loop-helix protein that directly activates transcription of structural anthocyanin genes. Plant Cell 12, 1619–1632. doi: 10.1105/tpc.12.9.1619
Tamura, K., Stecher, G., Peterson, D., Filipski, A., and Kumar, S. (2013). MEGA6: molecular evolutionary genetics analysis version 6.0. Mol. Biol. Evol. 30, 2725–2729. doi: 10.1093/molbev/mst197
Thompson, J. D., Higgins, D. G., and Gibson, T. J. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res. 22, 4673–4680. doi: 10.1007/978-1-4020-6754-9_3188
Tuan, P., Kwon, D., Lee, S., Arasu, M., Al-Dhabi, N., Park, N., et al. (2014). Enhancement of chlorogenic acid production in hairy roots of Platycodon grandiflorum by over-expression of an Arabidopsis thaliana transcription factor AtPAP1. Int. J. Mol. Sci. 15:14743. doi: 10.3390/ijms150814743
Wang, B., Sun, W., Li, Q., Li, Y., Luo, H., Song, J., et al. (2015). Genome-wide identification of phenolic acid biosynthetic genes in Salvia miltiorrhiza. Planta 241, 711–725. doi: 10.1007/s00425-014-2212-1
Wang, F., Zhu, H., Kong, W., Peng, R., and Liu, Q. (2016). The Antirrhinum AmDEL gene enhances flavonoids accumulation and salt and drought tolerance in transgenic Arabidopsis. Planta 244, 59–73. doi: 10.1007/s00425-016-2489-3
Wang, H., Wu, Y., Yang, X., Guo, X., and Cao, X. (2017). SmLEA2, a gene for late embryogenesis abundant protein isolated from Salvia miltiorrhiza, confers tolerance to drought and salt stress in Escherichia coli and S. miltiorrhiza. Protoplasma 254, 685–696. doi: 10.1007/s00709-016-0981-z
Wei, T., Deng, K., Zhang, Q., Gao, Y., Liu, Y., Yang, M., et al. (2017). Modulating AtDREB1C expression improves drought tolerance in Salvia miltiorrhiza. Front. Plant Sci. 8:52. doi: 10.3389/fpls.2017.00052
Wesley, S. V., Helliwell, C. A., Smith, N. A., Wang, M. B., Rouse, D. T., Liu, Q., et al. (2001). Construct design for efficient, effective and high-throughput gene silencing in plants. Plant J. 27, 581–590. doi: 10.1046/j.1365-313X.2001.01105.x
Wu, X. M., Yu, Y., Han, L. B., Li, C. L., Wang, H. Y., Zhong, N. Q., et al. (2012a). The tobacco BLADE-ON-PETIOLE2 gene mediates differentiation of the corolla abscission zone by controlling longitudinal cell expansion. Plant Physiol. 159, 835–850. doi: 10.1104/pp.112.193482
Wu, Y. B., Ni, Z. Y., Shi, Q. W., Dong, M., Kiyota, H., Gu, Y. C., et al. (2012b). Constituents from Salvia species and their biological activities. Chem. Rev. 112, 5967–6026. doi: 10.1021/cr200058f
Wu, Y., Liu, C., Kuang, J., Ge, Q., Zhang, Y., and Wang, Z. (2014). Overexpression of SmLEA enhances salt and drought tolerance in Escherichia coli and Salvia miltiorrhiza. Protoplasma 251, 1191–1199. doi: 10.1007/s00709-014-0626-z
Xu, W., Dubos, C., and Lepinie, L. (2015a). Transcriptional control of flavonoid biosynthesis by MYB-bHLH-WDR complexes. Trends Plant Sci. 20, 176–185. doi: 10.1016/j.tplants.2014.12.001
Xu, Z., Peters, R. J., Weirather, J., Luo, H., Liao, B., Zhang, X., et al. (2015b). Full-length transcriptome sequences and splice variants obtained by a combination of sequencing platforms applied to different root tissues of Salvia miltiorrhiza and tanshinone biosynthesis. Plant J. 82, 951–961. doi: 10.1111/tpj.12865
Xue, S., Valdez, D., Collman, P. I., and Diamant, N. E. (2011). Recent advances on the regulation of anthocyanin synthesis in reproductive organs. Plant Sci. 181, 219–229. doi: 10.1016/j.plantsci.2011.05.009
Yan, Y., and Wang, Z. (2007). Genetic transformation of the medicinal plant Salvia miltiorrhiza by Agrobacterium tumefaciens-mediated method. Plant Cell Tissue Organ Cult. 88, 175–184. doi: 10.1007/s11240-006-9187-y
Yang, L., Ding, G., Lin, H., Cheng, H., Kong, Y., Wei, Y., et al. (2013). Transcriptome analysis of medicinal plant Salvia miltiorrhiza and identification of genes related to tanshinone biosynthesis. PLoS One 8:e80464. doi: 10.1371/journal.pone.0080464
Yang, Y., Hou, S., Cui, G., Chen, S., Wei, J., and Huang, L. (2010). Characterization of reference genes for quantitative real-time PCR analysis in various tissues of Salvia miltiorrhiza. Mol. Biol. Rep. 37, 507–513. doi: 10.1007/s11033-009-9703-3
Yin, X., Allan, A. C., Chen, K., and Ferguson, I. B. (2010). Kiwifruit EIL and ERF genes involved in regulating fruit ripening. Plant Physiol. 153, 1280–1292. doi: 10.1104/pp.110.157081
Zhang, S., Ma, P., Yang, D., Li, W., Liang, Z., Liu, Y., et al. (2013). Cloning and characterization of a putative R2R3 MYB transcriptional repressor of the rosmarinic acid biosynthetic pathway from Salvia miltiorrhiza. PLoS One 8:e73259. doi: 10.1371/journal.pone.0073259
Zhang, X., Luo, H., Xu, Z., Zhu, Y., Ji, A., Song, J., et al. (2015). Genome-wide characterisation and analysis of bHLH transcription factors related to tanshinone biosynthesis in Salvia miltiorrhiza. Sci. Rep. 5:11244. doi: 10.1038/srep11244
Keywords: AtPAP1, phenolic acids, salvianolic acid B, Salvia miltiorrhiza, SmbHLH51
Citation: Wu Y, Zhang Y, Li L, Guo X, Wang B, Cao X and Wang Z (2018) AtPAP1 Interacts With and Activates SmbHLH51, a Positive Regulator to Phenolic Acids Biosynthesis in Salvia miltiorrhiza. Front. Plant Sci. 9:1687. doi: 10.3389/fpls.2018.01687
Received: 22 July 2018; Accepted: 30 October 2018;
Published: 20 November 2018.
Edited by:
Agnieszka Ludwików, Adam Mickiewicz University in Poznań, PolandReviewed by:
Toshihiro Obata, University of Nebraska-Lincoln, United StatesAgnieszka Kiełbowicz-Matuk, Institute of Plant Genetics (PAN), Poland
Copyright © 2018 Wu, Zhang, Li, Guo, Wang, Cao and Wang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiaoyan Cao, Y2FveGlhb3lhbkBzbm51LmVkdS5jbg== Zhezhi Wang, enp3YW5nQHNubnUuZWR1LmNu
†These authors have contributed equally to this work