 Chuanmei Zhu
Chuanmei Zhu Jiani Yang
Jiani Yang Mathew S. Box
Mathew S. Box Elizabeth A. Kellogg
Elizabeth A. Kellogg Andrea L. Eveland
Andrea L. Eveland
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 12 September 2018
Sec. Plant Breeding
Volume 9 - 2018 | https://doi.org/10.3389/fpls.2018.01309
This article is part of the Research Topic Setaria as a Model Genetic System to Accelerate Yield Increases in Cereals, Forage Crops, and Bioenergy Grasses View all 12 articles
The morphological and functional diversity of plant form is governed by dynamic gene regulatory networks. In cereal crops, grain and/or pollen-bearing inflorescences exhibit vast architectural diversity and developmental complexity, yet the underlying genetic framework is only partly known. Setaria viridis is a small, rapidly growing grass species in the subfamily Panicoideae, a group that includes economically important cereal crops such as maize and sorghum. The S. viridis inflorescence displays complex branching patterns, but its early development is similar to that of other panicoid grasses, and thus is an ideal model for studying inflorescence architecture. Here we report a detailed transcriptional resource that captures dynamic transitions across six sequential stages of S. viridis inflorescence development, from reproductive onset to floral organ differentiation. Co-expression analyses identified stage-specific signatures of development, which include homologs of previously known developmental genes from maize and rice, suites of transcription factors and gene family members, and genes of unknown function. This spatiotemporal co-expression map and associated analyses provide a foundation for gene discovery in S. viridis inflorescence development, and a comparative model for exploring related architectural features in agronomically important cereals.
Extensive morphological diversity is exhibited by inflorescences across grass species, family Poaceae. Since the inflorescences bear grain, understanding the genetic and molecular bases for this variation can accelerate the generation of higher yielding crops through breeding or precision engineering. Grasses encompass the world’s important cereal crops; e.g., maize, sorghum, and millets in the subfamily Panicoideae, wheat, oats, and barley in subfamily Pooideae, and rice in subfamily Oryzoideae (Soreng et al., 2017). Many genes that regulate aspects of inflorescence architecture in grasses have been discovered and characterized, largely in maize and rice (Zhang and Yuan, 2014; Bommert and Whipple, 2017). In many cases gene function is conserved across species (Whipple et al., 2010; Bommert and Whipple, 2017), whereas in others, it varies between species or clades (McSteen, 2006; Whipple, 2017; Bommert and Whipple, 2017). Relatively little is known about how these genes interact in the larger context of a developmental network to control inflorescence form and how these networks are rewired across species.
All inflorescence structures are ultimately derived from a group of pluripotent cells called the inflorescence meristem (IM). The IM is indeterminate and transitions from the shoot apical meristem (SAM) during the shift from vegetative to reproductive growth (Bartlett and Thompson, 2014; Kyozuka et al., 2014). In grasses, the terminal reproductive structure is the spikelet, which bears one or more flowers that produce the seeds. The different routes that the IM takes to form a determinate spikelet meristem (SM) largely determine the morphology of the mature inflorescence (Kyozuka et al., 2014; Whipple, 2017). For example, the IM can produce SMs directly on its flanks, as in wheat, or it can produce few to many indeterminate branch meristems (BMs), which is typical for most grasses. Like the IM, BMs can produce SMs directly, as in finger millet, or can initiate higher order branches, as in rice. The IM and BM may ultimately convert to a terminal SM or simply cease development. Eventually, SMs initiate sterile bracts (glumes) and floral meristems (FMs), which produce lateral organs that differentiate into floral structures including lemma, palea, anthers, and ovary.
In species of the Andropogoneae tribe, which includes maize and sorghum, spikelets are borne in pairs where two SMs arise from a spikelet pair meristem (SPM). Paired spikelets also arose independently in some other closely related groups. In the “bristle clade” of grasses, which includes Setaria species and other members of the subtribe Cenchrinae (see Doust and Kellogg, 2002 and references therein), BMs can alternatively differentiate into sterile branches called bristles. While spikelets are not paired in these species, morphological analyses suggest that spikelets may be paired with bristles (Doust and Kellogg, 2002). Recent work in Setaria viridis showed that BMs poised to form bristles first initiate an SM identity program before the homeotic shift to bristle formation. This developmental switch is dependent on proper spatiotemporal synthesis of growth-promoting brassinosteroids (BRs) during SM development (Yang et al., 2017).
Despite this variation in inflorescence morphology among grasses, the core underlying developmental processes are shared, and often leverage common regulatory modules. Pathways that regulate IM size (Somssich et al., 2016), SM identity (Bommert and Whipple, 2017), and flower development (Hirano et al., 2014), for instance, are largely conserved across those grass species studied. However, these processes can vary by the spatiotemporal expression of certain factors, functional divergence of gene family members, and/or co-option of novel factors all together. For example, the RAMOSA1 (RA1) transcription factor (TF) is expressed at the base of SPMs in Andropogoneae species and acts to suppress branching by conferring determinacy on the SPM. The timing and degree of RA1 expression dictates inflorescence branching patterns; i.e., during maize tassel development ra1 is induced early and suppresses higher order branching whereas in sorghum and Miscanthus, delayed ra1 expression leads to highly branched inflorescences (Vollbrecht et al., 2005). By leveraging comparative transcriptome analyses across grasses, we can gain invaluable insight into the core components that regulate these processes, and what variations are associated with species-specific morphologies.
Transcriptome studies of early inflorescence development have been reported for a few important cereal crops, including maize (Eveland et al., 2014), rice (Furutani et al., 2006; Harrop et al., 2016), barley (Digel et al., 2015), and wheat (Feng et al., 2017), leading to systematic discovery of underlying regulatory modules. To our knowledge, maize is the only panicoid grass with existing transcriptome data across early inflorescence development (Eveland et al., 2014). Given the variation in architectures among important cereals in the Panicoideae, comparative expression maps from additional species will help determine conserved and species-specific components. Here, we present a comprehensive developmental and transcriptomics analysis of inflorescence development in S. viridis, a model panicoid grass with a rapid life cycle, sequenced genome, and emerging genetics and genomics toolkit (Doust et al., 2009; Brutnell et al., 2010; Bennetzen et al., 2012; Huang et al., 2016; Zhu et al., 2017). We profiled six sequential stages of inflorescence development that captured key events from the reproductive transition to floral organ differentiation and identified stage-specific co-expression signatures. Our detailed characterization of S. viridis inflorescence development at both morphological and molecular levels provides an invaluable resource for the community as a foundation for gene discovery and a comparative platform for studying diverse architectures in agronomically important cereal crops.
The reference S. viridis genotype A10.1 was used for this study. A10.1 seeds from 2-year-old stocks were used to ensure full loss of dormancy and promote synchronized germination. Seeds were planted in MetroMix 360 soil (Sun Gro Horticulture company) and grown in a controlled growth chamber with the following conditions: temperature of 31°C/23°C (day/night), light intensity of 200 μmol/sq.meter/s for 12 h (6am–6pm) and 50% relative humidity, at the Donald Danforth Plant Science Center’s Integrated Plant Growth Facility. Plants were fertilized with Jack’s 15-16-17 (Hummert International) twice a week.
In our controlled conditions, the transition from a vegetative to IM occurred after 8 days after sowing (DAS) and before 10 DAS. Accordingly, vegetative SAMs were hand-dissected from plants at 8 DAS, and IMs from plants at 10, 11, 12, 13, 14, 15, 16, and 18 DAS. Samples were fixed immediately in FAA solution (3.75% formaldehyde, 50% ethanol, and 5% glacial acetic acid) and left overnight. They were then dehydrated using 50% ethanol for a minimum of 1 h and then shifted to 70% ethanol. For critical point drying, samples were dehydrated in an ethanol series (80, 95, 100, 100% ethanol) for at least 1 h for each step; they were then left in 100% ethanol overnight and then moved to fresh 100% ethanol from a newly opened bottle the next day. Samples were then critical point dried using a Tousimis Samdri-780a, mounted on stubs and sputter coated using a Tousimis Samsputter-2a. Images were taken with a Hitachi S2600 SEM at 20 kV at Washington University’s Central Institute for the Deaf.
Inflorescences were hand-dissected into fresh 100% acetone on dry ice from S. viridis seedlings at 10, 12, 14, 15, 16, and 18 DAS. Depending on the representative size at a given stage, 10–30 individual inflorescence primordia were pooled per biological replicate; three to four biological replicates were collected for each stage. All sampling was performed within a 2-h window in the morning to control for circadian effects. Acetone was removed and samples were flash-frozen in liquid nitrogen and ground into a fine powder using 3 mm tungsten-carbide beads (Qiagen) in a Tissue Lyser-II. Total RNA was isolated using the PicoPure RNA Isolation Kit (Thermo Fisher Scientific) with in-column DNase I treatment following manufacturer’s protocols. RNA-seq libraries were generated from 1 μg total RNA using the NEBNext Ultra Directional RNA Library Prep Kit for Illumina (New England BioLabs Inc.) and size-selected for 200 bp inserts. Libraries were quantified on a Bioanalyzer (Agilent 2100) using a DNA 1000 chip to confirm the insert size and quantified again on a Qubit 3.0 Fluorometer (Life Technologies) to ensure different libraries were equally loaded, and then sequenced using standard Illumina protocols (Illumina, Inc.) for either paired-end (PE) or single-end (SE) sequencing at 100 bp on the Illumina HiSeq 2500 platform at the University of Illinois at Urbana-Champaign W.M. Keck Center.
Sequenced reads were quality-checked using fastQC with Phred scores all above 30. For read mapping and transcript quantification, we used Salmon (v0.8.1) (Patro et al., 2017) in a quasi-mapping mode for indexing reference transcripts from the S. viridis primary transcript file (Sviridis_311_v1; Phytozome1 v12.1) (Supplementary Table S1). Transcripts were quantified using the option numBootstraps 30. Parameters not specified were run as default. Salmon outputs were imported into R with the Bioconductor package tximport (v1.0.3). Raw counts and transcripts per million (TPM) were extracted for 35,214 S. viridis primary transcripts (Supplementary Table S2). All subsequent analyses were conducted using TPM (Soneson et al., 2015).
To reduce noise from genes with ubiquitously low expression or that do not change expression during development, we applied a set of criteria to filter genes that: (1) showed more than twofold change in expression between at least two of the stages, and (2) collectively had an expression value of at least 1 TPM across all stages. This resulted in 11,425 dynamically expressed genes during inflorescence development (Supplementary Table S3). To determine the optimal number of clusters to use for fuzzy c-means (FCM) cluster analysis, we used two functions in the R Bioconductor package, Mfuzz (Kumar and Futschik, 2007): (1) Dmin, which calculates the minimum centroid distance for a range of cluster numbers and (2) cselection, which reports the cluster number where empty clusters are detected in the repeated soft clustering. Cluster number was chosen for FCM analysis based on the centroids of each cluster being well separated based on Dmin and there were no empty clusters based on cselection. FCM was performed using Mfuzz. The 11,425 genes were clustered into groups based on their standardized mean expression profiles (for each transcript, average expression is 0 and the standard deviation across 6 stages is 1). Each gene was assigned to the cluster with its highest membership score. Heatmaps and hierarchical clustering using Euclidean distances and complete linkage clusters were generated using MeV2 (version 4.8) (Saeed et al., 2003).
The S. viridis genome (v1.1) and annotation files were downloaded from Phytozome v12.1. Functional annotations were assigned to S. viridis genes based on these files and information extracted from EnsemblPlants Biomart3. To determine homologs of S. viridis genes in other grass species [maize (v3 and v4 genomes), sorghum (v3.1.1), Brachypodium (v3.1), and Setaria italica (v2.2)], blastp4 was used with two as the maximum target sequence for each gene. All genome sequence data were downloaded from Phytozome v12.1, except for maize v4, which was downloaded from MaizeGDB5.
An HMM (Hidden Markov Model6, HMMER 3.1b2) was used to classify genes encoding MADS-Box proteins in the proteome database of S. viridis (v1.1) and coding sequences of the primary transcripts were retrieved from Phytozome1. Previously identified MIKC type MADS-box genes from Arabidopsis, rice and maize genomes (Parenicová et al., 2003; Arora et al., 2007; Zhao et al., 2011) were included in building the phylogenetic tree. All protein coding sequences were manually checked for the presence of a MADS domain and were aligned using MUSCLE (Edgar, 2004) to construct a maximum likelihood tree by RAxML (Edgar, 2004; Stamatakis, 2014), with 1,000 bootstrap replicates. The final trees were drawn with the R Bioconductor package, phytools (Revell, 2011).
The R Bioconductor package wgcna was used to perform weighted gene co-expression network analysis (WGCNA) on the same set of 11,425 genes used in FCM analysis (Langfelder and Horvath, 2008). A matrix of all genes with their TPM values across 23 samples (including individual biological replicates) was used as input. An adjacency matrix was generated to determine similarity between genes (i.e., correlation for every gene pair across the 23 samples) and transformed through a soft-thresholding procedure using the function “pickSoftThreshold,” where a soft power of 18 was chosen for module detection. A topological overlap measure (TOM) was then calculated from the adjacency matrix to estimate network interconnectedness. The dissimilarity of TOM (1-TOM) was used as the input for average linkage hierarchical clustering to identify co-expression modules. Module eigengenes (MEs), the first principal component of a given module, was used as a representative gene expression profile for that module. Modules were further merged based on their MEs (using cutHeight = 0.25) and the module membership (MM) for each gene indicated the degree of similarity between its expression profile and each ME. The entire network including WGCNA-calculated weights for each edge between genes (nodes) is available on NCBI GEO. Gene-to-gene connections were filtered if the weight of interaction was <0.02. Sub-networks were visualized using Cytoscape7 v3.4.10.
To characterize the developmental progression of the S. viridis inflorescence, we used scanning electron microscopy (SEM) to examine sequential stages from floral transition to floral organ development. At 8 DAS, the SAM had not yet transitioned, but had emerged from surrounding leaf primordia (Figure 1A). By 10 DAS, the SAM had transitioned to the IM and BMs were initiated but barely visible (Figure 1B). By 11 DAS, primary BMs had initiated at the base of the developing inflorescence and continued to form in a helical pattern at 12 DAS (Figures 1C,D). From 12 to 14 DAS, secondary and tertiary BMs developed sequentially in a distichous pattern (Figures 1D–F). By 15 DAS, the IM had ceased to produce new BMs and those at the inflorescence tip were the first to differentiate into SMs or bristles (Figures 1G,J). At 16 DAS, differentiation of SMs and bristles continued basipetally and the two developing structures became morphologically distinguishable. SMs then initiated glumes and FMs, while bristles started to elongate and form an indented ring below the meristem tip (Figures 1H,K), which often later detached (Doust and Kellogg, 2002; Yang et al., 2017). By 18 DAS, floral organs such as lemma, palea, and anther primordia formed in the spikelets, and bristles further elongated and produced prickle hairs (Doust and Kellogg, 2002; Figures 1I,L).
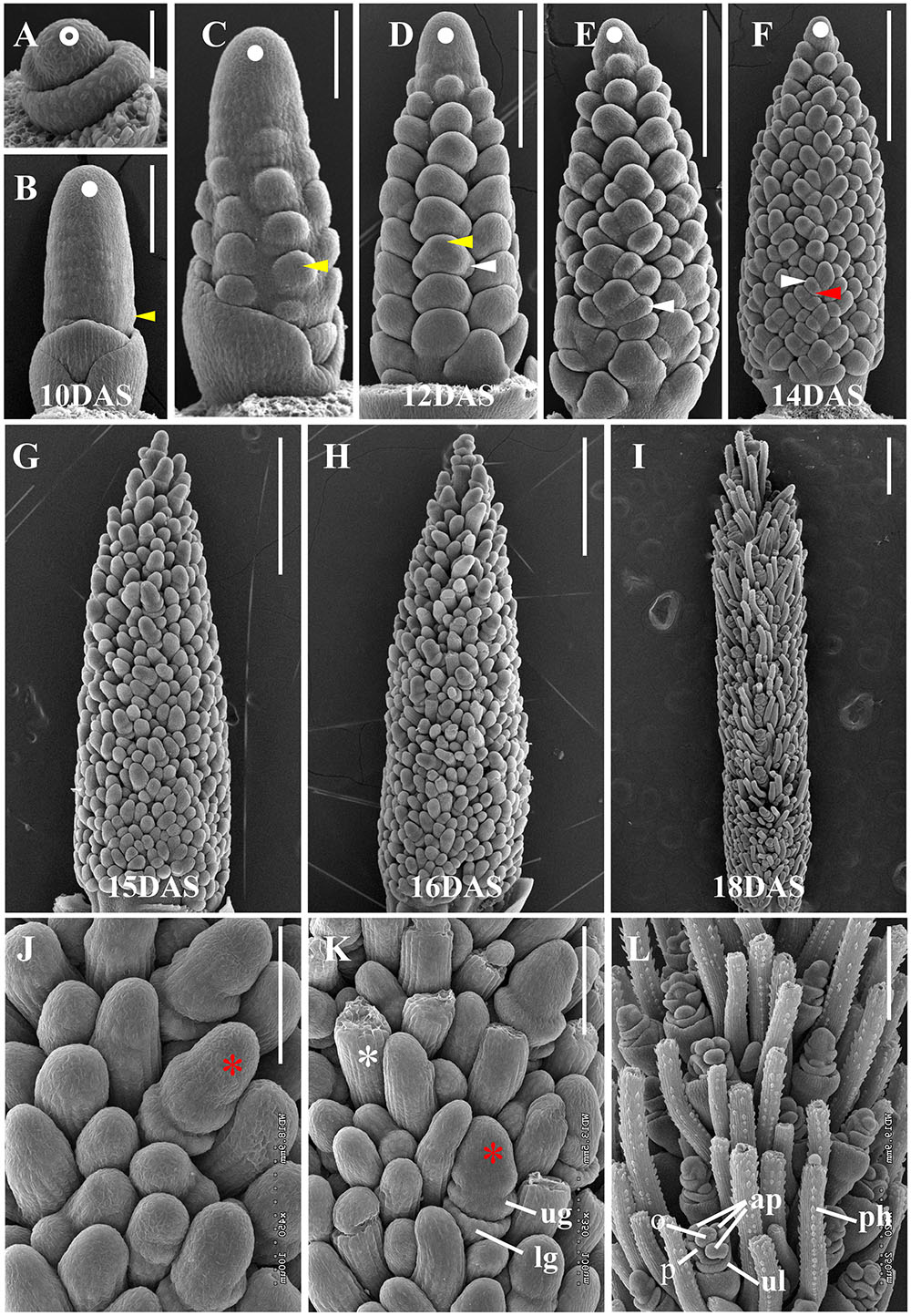
FIGURE 1. Key developmental stages of Setaria viridis inflorescence development by SEM. (A) At 8 DAS, the S. viridis SAM (white open circle) had not yet transitioned to reproductive development. (B) By 10 DAS, the SAM had transitioned to an indeterminate IM (white dot) and primary BMs (yellow arrow) were initiated. As development proceeds through (C) 11 DAS, (D) 12 DAS, (E) 13 DAS, and (F) 14 DAS stages, primary, secondary, and higher order branching was progressively initiated (white dot, IM; yellow arrow, primary BM; white arrow, secondary BM; red arrow, tertiary BM). (G, J) At 15 DAS, SMs (red asterisk) had begun to differentiate and by 16 DAS (H, K), were distinguishable from developing bristles (white asterisk) and initiated glume primordia (lg, lower glume; ug, upper glume). (I, L) At 18 DAS, bristles were elongated and developed prickle hairs (ph) and SMs further differentiated floral organs (ul, upper lemma; p, palea; ap, anther primordium; o, ovary). Developmental stages that were used for RNAseq-based transcript profiling are labeled. Scale bars = 50 μm in (A), 100 μm in (B, C, J, and K), 250 μm in (D–F and L), and 500 μm in (G–I).
To establish a dynamic transcriptome map of S. viridis inflorescence development, we used RNA-seq to link global changes in gene expression with developmental transitions. Based on our detailed morphological characterization, we selected six stages that captured key events in inflorescence development for transcriptome profiling by RNA-seq: the initiation of the IM (10 DAS), primary and secondary (12 DAS), and higher order (14 DAS) branching events, transition to SMs (15 DAS), differentiation of spikelets and bristles (16 DAS), and development of floral organs (18 DAS) (Figure 1). For each of these six stages, inflorescence primordia were hand-dissected and RNA-seq libraries prepared for three to four biological replicates as described in the Section “Materials and Methods” (Supplementary Table S1). Transcript abundance was quantified in TPM at each of the six stages; these analyses showed strong correlations among biological replicates and dynamic patterns of gene expression across developmental stages (Supplementary Figures S1, S2 and Supplementary Table S2). Based on our filtering criteria described in the section “Materials and Methods,” all analyses in this manuscript were performed using a subset of 11,425 dynamically expressed genes (Supplementary Table S3).
We explored developmental dynamics of TF families across the six stages. Spatiotemporal expression and combinatorial action of TF family members fine-tune developmental decisions, and knowledge of their individual expression profiles can provide insight into underlying regulatory mechanisms. We annotated several TF families in S. viridis based on annotations of functional TF domains from various sources. Specifically, we focused on families with multiple members previously implicated in plant development and plotted relative expression of family members across the six stages: MADS-box, TCP, SBP (Figure 2), AP2/ERF, bHLH, C2H2_Zinc, bZIP, LOB, and NAC families (Supplementary Figure S3; see abbreviations in Box 1). Individual TF family members showed dynamic expression profiles during early inflorescence development in S. viridis (Figures 2, Supplementary Figures S3, S4 and Supplementary Table S4). These included orthologs of previously characterized developmental regulators from maize and rice, as well as many uncharacterized family members with potential functions in inflorescence development based on their co-expression patterns. For example, we identified a set of 21 candidate TFs that were highly expressed and showed large fold changes in expression that peaked at specific stages of inflorescence development (Supplementary Figure S4 and Supplementary Table S4).
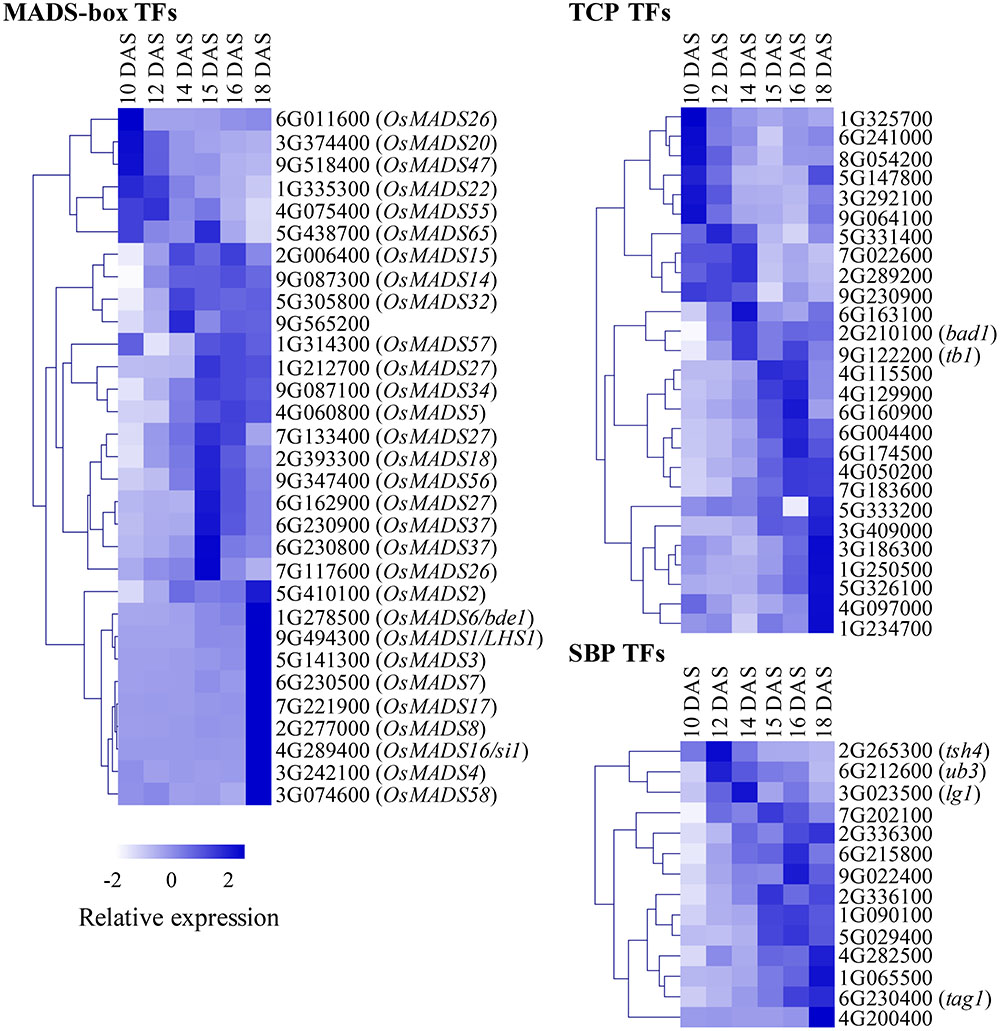
FIGURE 2. Transcription factor (TF) family members show dynamic expression profiles across inflorescence development. Individual members of MADS-box, TCP and SBP TF families were annotated, TPM values for each were normalized based on a Z-scale and plotted as a heatmap across the six stages of Setaria viridis inflorescence development. For MADS-box genes, names of orthologous genes from rice are included. For TCP and SBP families, orthologs of classical maize genes are indicated. White-to-dark shading represents low-to-high relative expression level.

BOX 1. Abbreviations and names of TF families annotated in this study.
In addition, we performed a detailed phylogenetic analysis for the MIKC type MADS-box family based on these annotations to determine homology between the S. viridis genes and those in maize, rice and/or Arabidopsis (Supplementary Figure S5). This phylogenetic framework enables comparative analyses among MADS-box family members that have been shown in multiple systems to work in a combinatorial manner to regulate development (Theißen et al., 2016; Bartlett, 2017).
To define signatures of gene expression across this developmental trajectory, we used an FCM clustering approach (Kumar and Futschik, 2007) and our filtered set of 11,425 dynamically expressed genes. Based on simulations and selection criteria as described in the Section “Materials and Methods,” we determined 25 as an optimal number of clusters to use for the FCM analyses (Supplementary Figure S6A). FCM was used to assign a membership score for each gene to each cluster, and based on the highest score for each gene, 25 clusters with unique expression profiles were defined ranging from 290 to 608 genes per cluster (Supplementary Figure S6B and Supplementary Table S3).
The mean relative expression value across the six developmental stages was determined for each of the 25 clusters and hierarchical clustering on these values revealed four groups (Figure 3A), each representing a distinct developmental expression pattern: (1) floral transition and IM identity, (2) BM initiation and determinacy, (3) SM differentiation and FM initiation, and (4) floral organ development. We first located orthologs of known developmental genes; expression profiles of representative genes within each group were comparable to those of their orthologs in maize and rice, consistent with their predicted roles in development (Figure 3B and Supplementary Table S3). In the following four subsections, we describe expression of these and other key developmental regulators during S. viridis inflorescence development compared to other grasses and highlight some key findings from each of the major expression groups. For simplicity, we listed full names and abbreviations of genes discussed in Table 1.
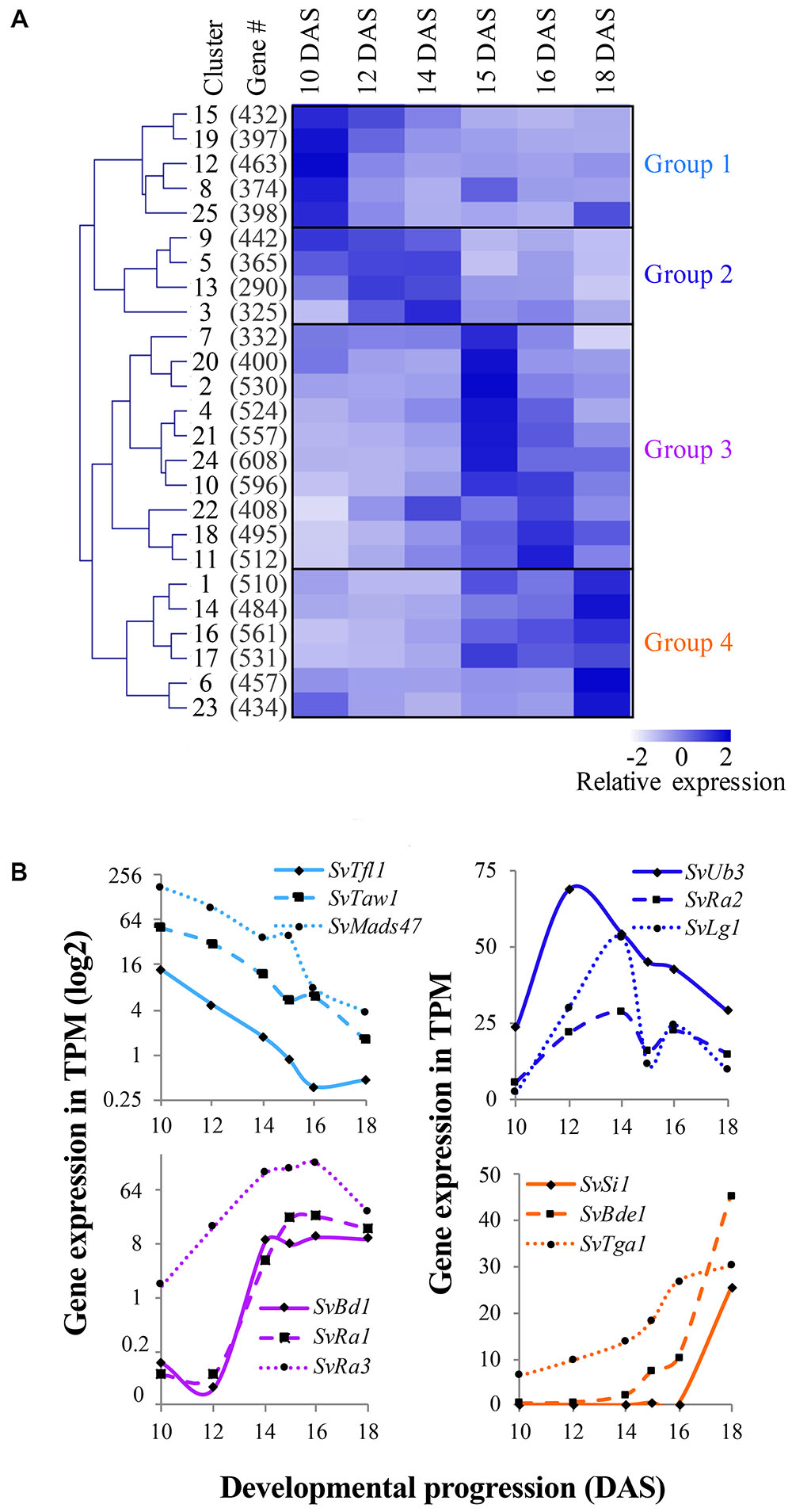
FIGURE 3. Co-expression analysis revealed distinct gene expression signatures during Setaria viridis inflorescence development. (A) FCM clustering of 11,425 dynamically expressed genes identified 25 co-expression clusters. The mean relative expression value of genes in each cluster across development was used for hierarchical clustering and is displayed by the heatmap. Overall clusters organized into four general groups of expression associated with key developmental transitions. White-to-dark shading represents low-to-high relative expression level. (B) Within each of the four expression groups, homologs of known developmental genes from other species showed expected expression patterns.
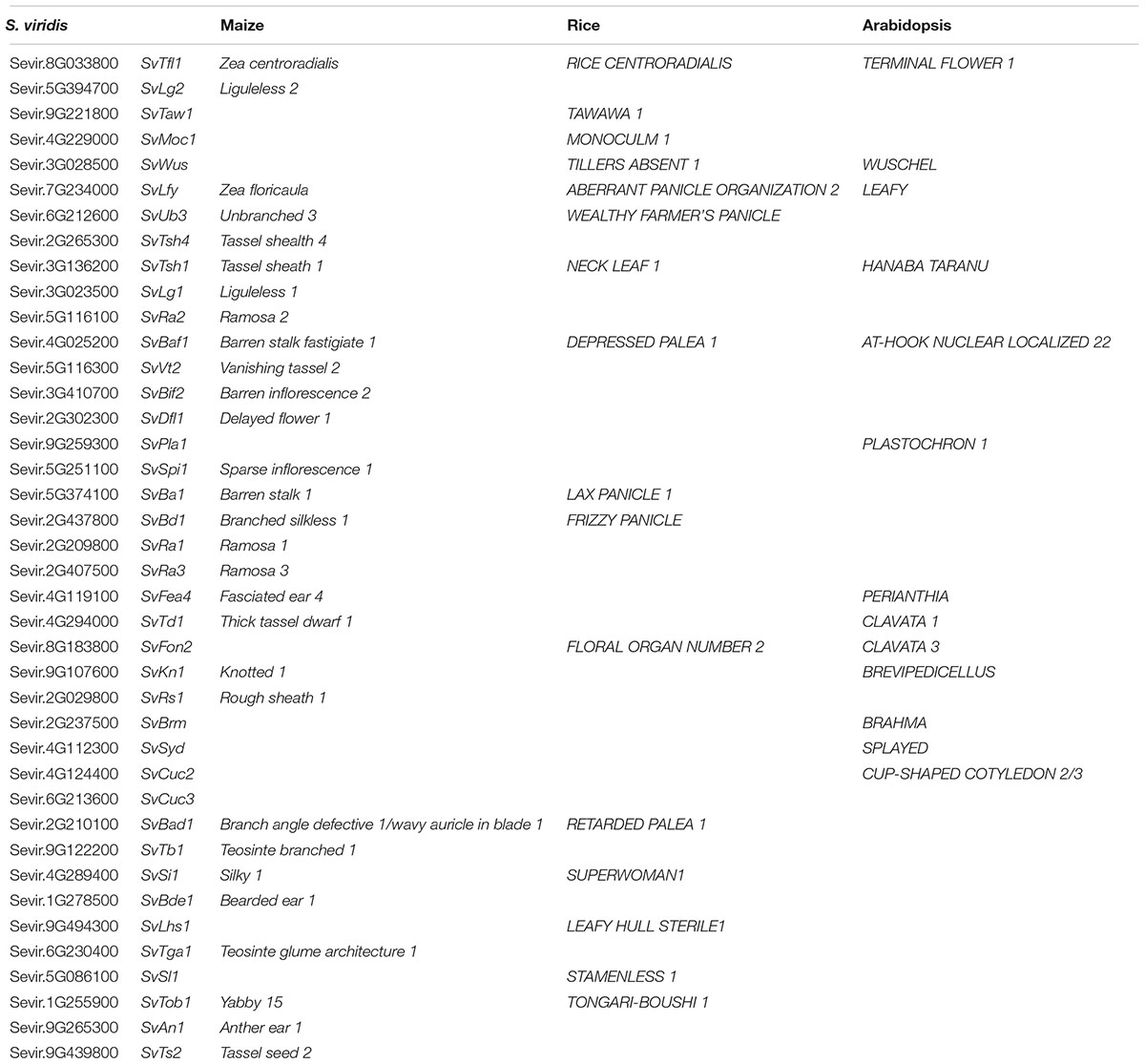
TABLE 1. Setaria viridis genes, short name descriptions and homologs in maize, rice, and Arabidopsis.
Genes in Group 1 (including clusters 8, 12, 15, 19, and 25) showed highest expression at 10 DAS as the IM was initiated and decreased at later stages of development (Figure 3A, Supplementary Figure S6 and Supplementary Table S3). We inferred that genes involved in regulating the transition to reproductive growth, IM identity, and/or primary branch initiation would be included in these clusters. For example, SvTfl1 (Sevir.8G033800), an ortholog of TERMINAL FLOWER1, a general repressor of flowering in many species including maize, rice, and Arabidopsis (Nakagawa et al., 2002; Danilevskaya et al., 2010; Hanano and Goto, 2011), was grouped in cluster 19 (Figure 3B). Three other TFL1-like homologs clustered in Group 1; Sevir.7G334800, a closely related paralog of SvTfl1, Sevir.7G097700 and Sevir.1G183200. The latter gene, which we named SvTfl1-like, was expressed markedly higher than the other two at 10–12 DAS in cluster 15 (Supplementary Table S3). Co-expressed in cluster 15 was SvLg2 (Sevir.5G394700), the ortholog of liguleless 2 (lg2) from maize, which encodes a bZIP TF responsible for early establishment of lateral organ boundary positioning (Harper and Freeling, 1996; Walsh et al., 1998). Loss-of-function lg2 mutants in maize produce few to no tassel branches. Consistent with its expression profile in S. viridis (Supplementary Figure S4), lg2 in maize has been implicated in the transition from vegetative to reproductive growth (Walsh and Freeling, 1999).
In rice, TAWAWA1 (TAW1) encodes a nuclear protein belonging to the ALOG (ARABIDOPSIS LSH1 and ORYZA G1) gene family that is highly expressed in the IM and BMs early in inflorescence development, and decreases during the phase change to SM identity (Yoshida et al., 2013). TAW1 promotes indeterminate IM and BM activity by suppressing acquisition of SM identity, and thus is a major regulator of inflorescence architecture (Yoshida et al., 2013). The S. viridis ortholog, SvTaw1 (Sevir.9G221800), was found in cluster 19 (Figures 3A,B), which shows a very similar expression profile to cluster 15, consistent with TAW1 expression in rice. Five other ALOG genes were found in Group 1 and shared similar expression profiles (Table S3); Sevir.7G17600 and Sevir.1G244200, which we named SvTaw1-like 1 and SvTaw1-like 2, respectively, shared highest homology with SvTaw1 and were co-expressed in cluster 15 (Figure 4). Functional studies of other ALOG genes in various species indicate that they generally share a role in developmental phase changes and organ identity, and their spatiotemporal expression patterns determine their specificity (Yoshida et al., 2009; Cho and Zambryski, 2011; Takeda et al., 2011). Notably, three genes belonging to a subfamily of MADS-box TFs called SHORT VEGETATIVE PHASE (SVP), were also co-expressed in Group 1 (Figures 3B, 4 and Supplementary Table S3). In rice, homologs of these SVP genes (OsMADS47, OsMADS55, and OsMADS22) suppress FM initiation and were positively regulated by TAW1 to promote indeterminacy (Yoshida et al., 2013; Kyozuka et al., 2014).
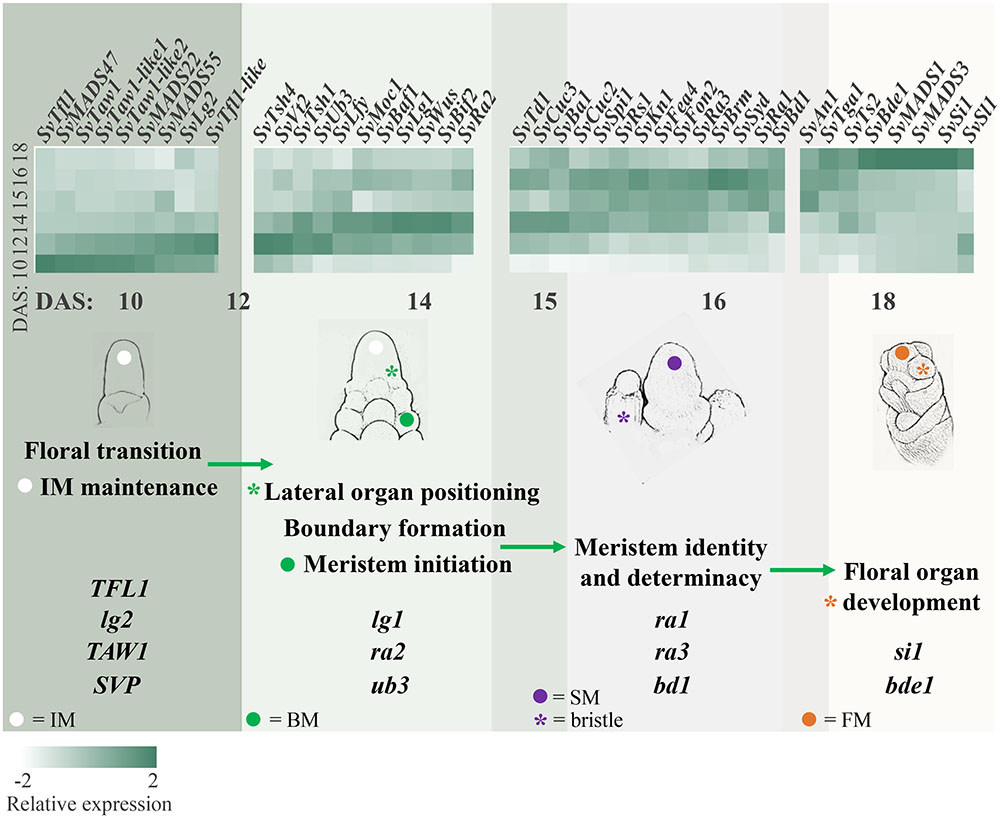
FIGURE 4. Orthologs of genes known to regulate inflorescence architecture in maize and rice showed expected expression patterns during Setaria viridis inflorescence development and transitions between meristem types. The schematic diagrams sequential developmental transitions captured by the RNA-seq data and shows relative expression of genes predicted to control these transitions. For example, consistent with the initiation of BMs from the IM between 10 and 14 DAS, positive regulators of lateral meristem formation (e.g., SvBif2 and SvUb3) showed increased expression between 10 and 14 DAS, and negative regulators of axillary meristem formation (e.g., SvTd1 and SvFea4) increased later at 14–16 DAS. Genes displayed in the heatmap are divided by group and developmental processes can transition across groups. Light-to-dark color indicates low-to-high relative expression.
Genes in Group 2 (clusters 3, 5, 9, and 13) tended to show highest expression at 12 and 14 DAS, decreasing by 15 DAS and into later stages (Figure 3A and Supplementary Figure S6). This expression pattern coincides with the initiation, maintenance and patterning of BMs, which occurs mostly between 10 and 14 DAS, and is largely completed by 15 DAS. Among genes in this group were those with putative roles in axillary meristem initiation and maintenance. For example, SvMoc1 (Sevir.4G229000), a GRAS family protein homologous to MONOCULM 1 (MOC1) from rice, was found in cluster 3. This expression pattern is consistent with a possible role for SvMoc1 in axillary meristem initiation, analogous to how it controls tiller outgrowth in rice (Li et al., 2003). The rice WUSCHEL (WUS) ortholog, TILLERS ABSENT 1 (TAB1/OsWUS), was also shown to play a role in initiating axillary meristems (Tanaka et al., 2015). Consistent with this, the S. viridis ortholog, SvWus (Sevir.3G028500), was co-expressed in cluster 3 (Supplementary Table S3). Also co-expressed with these developmental regulators was SvLfy, the S. viridis ortholog of zea floricaula1/2/ABERRANT PANICLE ORGANIZATION 2 (APO2)/LEAFY (SvLfy, Sevir.7G234000). These genes play universal roles in regulation of meristem determinacy, but with some species-specific differences; LFY and zfl1/2 promote meristem determinacy and FM identity in Arabidopsis and maize, respectively (Schultz and Haughn, 1991; Bomblies et al., 2003), but APO2 promotes meristem indeterminacy as well as BM initiation in rice (Kyozuka et al., 1998; Ikeda-Kawakatsu et al., 2012).
Among the SBP TFs annotated in developing S. viridis inflorescences (Figure 2), three were associated with Group 2. SvUb3 (Sevir.6G212600), the ortholog of unbranched 3 (ub3) from maize and WEALTHY FARMERS PANICLE (WFP) from rice, was expressed in cluster 3. The function of this gene is generally conserved in both species in controlling partitioning of cells between lateral organ formation and meristem maintenance (Jiao et al., 2010; Miura et al., 2010; Chuck et al., 2014; Li et al., 2016). In maize, ub3 shares redundant roles with tassel sheath 4 (tsh4), another SPB TF that functions also to suppress formation of leaves in the inflorescence (Chuck et al., 2010, 2014). SvTsh4 (Sevir.2G265300) was also found in Group 2, and was co-expressed in cluster 13 with SvTsh1 (Sevir.3G136200), which encodes a GATA TF that functions in the same pathway (Wang et al., 2009; Whipple et al., 2010; Figures 2, 3A).
Initiation of lateral organs involves the formation of boundaries, regions of decreased cell division that separate the growing organ primordia from meristematic signals to promote proper organ identity. Genes implicated in setting up lateral organ boundaries were identified in Group 2. SvLg1 (Sevir.3G023500), the third SBP TF in this group and orthologous to liguleless 1 (lg1) from maize (Moreno et al., 1997; Lewis et al., 2014) and OsLG1 from rice (Lee et al., 2007), was found in cluster 3 (Figures 2, 3B). In maize, lg1 is classically known for its role in setting up the proximal-distal boundary that separates leaf blade from sheath and, along with lg2, is required for ligule and auricle development, thus controlling leaf angle. Recently, lg1 was also shown to regulate inflorescence architecture by controlling tassel branch number in maize (Lewis et al., 2014) and branch angle in rice (Ishii et al., 2013; Zhu et al., 2013). Consistent with our data from S. viridis, lg2 was proposed to act upstream of lg1 in maize, with lg2 expression initiated earlier in development to specify boundary position (Harper and Freeling, 1996; Lewis and Hake, 2016).
Also co-expressed with lg1 in cluster 3 was SvRa2 (Sevir.5G116100), ortholog of the maize ramosa 2 (ra2) gene, which encodes a LATERAL ORGAN BOUNDARY (LOB) domain TF that is expressed transiently in positions where axillary meristems will form on the inflorescence (Moreno et al., 1997; Bortiri et al., 2006; Figure 3B and Supplementary Figure S3). In maize, ra2 and lg1 were shown to act in parallel pathways to control branch number and angle during early inflorescence development (Bai et al., 2012). The S. viridis ortholog of maize barren stalk fastigiate 1 (baf1), SvBaf1 (Sevir.4G025200), was also co-expressed in cluster 3 (Figure 4 and Supplementary Table S3). In maize, baf1 encodes an AT-hook protein that is involved in demarcating the boundary region of axillary meristems and plays a role in their initiation (Gallavotti et al., 2011).
The positioning of axillary meristems is strongly influenced by transport and function of the phytohormone auxin. As auxin flows in and out of emerging primordia, localized auxin response maxima arise at places of lateral meristem initiation, and this mechanism is generally conserved in maize inflorescence development (Barazesh and McSteen, 2008; Gallavotti, 2013; Eveland et al., 2014). Therefore, we would expect suites of auxin-related genes to be dynamically expressed between 12 and 15 DAS in S. viridis while branching occurs; i.e., in Groups 2 and 3. We annotated genes in these groups based on homology to those implicated in synthesis, transport, signaling and/or response of auxin, and plotted their expression profiles (Supplementary Figure S7 and Supplementary Table S5). The expression of SvVt2 (Sevir.5G116300), the ortholog of maize vanishing tassel 2 (vt2), was initiated early in development and associated with cluster 13, while expression of SvBif2 (Sevir.3G410700), the ortholog of barren inflorescence 2 (bif2), peaked slightly later and was in cluster 3. Vt2 encodes a tryptophan aminotransferase that regulates an early step of auxin biosynthesis (Phillips et al., 2011), and bif2 encodes a PINOID serine/threonine kinase that regulates auxin transport (McSteen et al., 2007; Supplementary Figure S7).
Other key genes implicated in auxin-mediated lateral branching in maize and rice inflorescence development were expressed slightly later and in Group 3 (Figure 3A and Supplementary Figure S7). For example, genes in cluster 22 increased expression between 10 and 14 DAS, but expression was stable after 15 DAS unlike Group 2 clusters that decreased expression between 15 and 18 DAS (Figure 3A and Supplementary Figure S6). These included S. viridis orthologs of maize sparse inflorescence 1 (spi1; SvSpi1; Sevir.5G251100), encoding a YUCCA-like gene involved in a late step of auxin biosynthesis (Gallavotti et al., 2008), and barren stalk 1 (ba1)/LAX PANICLE 1 (LAX1) (SvBa1; Sevir.5G374100), a bHLH protein involved in auxin signaling (Komatsu et al., 2003a; Gallavotti et al., 2004; Supplementary Figure S7). In maize, functional BA1 is required for creation of auxin maxima at the meristem anlagen to promote lateral meristem initiation. Our expression data from S. viridis are consistent with expression and genetics analyses in maize that show ba1 acts downstream of baf1 (Gallavotti et al., 2011).
Clusters 10, 11, 18, and 22 in Group 3 have similar expression signatures that show a progressive increase during early stages of development, peaking around 16 DAS, and decreasing slightly by 18 DAS (Figure 3A and Supplementary Figure S6). Among these clusters were a number of genes related to axillary meristem identity and determinacy. For example, SvBd1 (Sevir.2G437800), a marker of SM identity in S. viridis (Yang et al., 2017), was found in cluster 18 (Figure 3B and Supplementary Table S3). Orthologs of this AP2/ERF TF across grass species, e.g., branched silkless 1 (bd1) in maize, FRIZZY PANICLE (FZP) in rice, and MORE SPIKELETS 1 (MOS1) in Brachypodium distachyon, share a conserved function in specifying SM identity (Chuck et al., 2002; Komatsu et al., 2003b; Derbyshire and Byrne, 2013).
Genes in the maize RAMOSA pathway regulate meristem determinacy prior to SM identity (Vollbrecht et al., 2005). Among these genes, ra2 is widely conserved across grasses and its expression comes on early during axillary meristem initiation. Consistent with genetics and expression data from maize (Bortiri et al., 2006; Satoh-Nagasawa et al., 2006), the S. viridis orthologs of ra1 (SvRa1; Sevir.2G209800) and ramosa 3 (ra3) (SvRa3; Sevir.2G407500), which encodes a trehalose-phosphate phosphatase (Satoh-Nagasawa et al., 2006), were expressed after initiation of SvRa2 and were found in clusters 10 and 11, respectively, their expression peaking at 15–16 DAS (Figures 3A,B).
Genetic interactions among genes controlling meristem determinacy and meristem size pathways in maize indicated these processes interface generally (Bommert and Whipple, 2017). Interestingly, many genes orthologous to known players in meristem size pathways were also co-expressed in Group 3. For example, SvFea4 (Sevir.4G119100), the ortholog of fasciated ear 4 (fea4) from maize and PERIANTHIA (PAN) from Arabidopsis encoding a bZIP TF that negatively regulates meristem size (Maier et al., 2009; Pautler et al., 2015), was found in cluster 22 (Figure 3A and Supplementary Figure S3). Regulatory components of the CLAVATA-WUSCHEL negative feedback signaling pathway central to meristem maintenance (Somssich et al., 2016) were also co-expressed in cluster 22. Among these were SvTd1 (Sevir.4G294000) and SvFon2 (Sevir.8G183800), orthologs of maize thick tassel dwarf 1 (td1)/CLAVATA1, encoding a leucine-rich repeat receptor-like kinase (Bommert et al., 2005), and rice FLORAL ORGAN NUMBER 2/CLV3, encoding a CLV3/ESR related peptide (Chu et al., 2006; Goad et al., 2017), respectively (Supplementary Figure S2 and Supplementary Table S3). Both td1/CLV1 and FON2/CLV3 negatively control meristem size.
Certain Class I KNOX homeodomain TFs also function in pathways to maintain meristem cell identity and size. Orthologs of the maize knotted 1 (kn1) and rough sheath 1 (rs1) genes, SvKn1 (Sevir.9G107600) and SvRs1 (Sevir.2G029800), respectively, both homologs of the Arabidopsis BREVIPEDICELLUS (BP) gene, were co-expressed in cluster 11 (Figure 3A and Supplementary Figure S6). In maize, kn1 is a key marker of meristem maintenance (Kerstetter et al., 1997; Bolduc et al., 2012), and the localized expression of rs1 predicted and subtended the initiation of axillary meristems on the flanks of the IM (Schneeberger et al., 1995). Interestingly, the S. viridis homologs of the BP interaction partner, BRAHMA (BRM), and a closely related gene, SPLAYED (SYD), both encoding SWI/SNF chromatin remodeling ATPases in Arabidopsis (Wagner and Meyerowitz, 2002; Zhao et al., 2015), Sevir.2G237500 and Sevir.4G112300, respectively, were also found cluster 11 (Supplementary Table S3). In Arabidopsis, BRM upregulates CUC genes (Kwon et al., 2006), which are essential players in lateral meristem establishment and proper boundary formation; SvCuc2 (Sevir.4G124400) and SvCuc3 (Sevir.6G213600) were co-expressed in cluster 22. In addition, two class II TCP TFs orthologous to branch angle defective 1 (bad1)/wavy auricle blade 1 (wab1) (Bai et al., 2012; Lewis et al., 2014) and teosinte branched 1 (tb1) (Doebley et al., 1997) were co-expressed in cluster 22 [SvBad1 (Sevir.2G210100) and SvTb1 (Sevir.9G122200), respectively]. Consistent with their expression profiles during S. viridis inflorescence development (Figure 2), their localized expression overlaps in axillary meristems of young maize tassels and both genes contribute to the regulation of branch outgrowth (Bai et al., 2012).
Genes in Group 4 (clusters 1, 6, 14, 16, 17, and 23) progressively increased in expression after 14 DAS and peaked largely at 18 DAS, coinciding with floral organ formation and differentiation (Figure 3A and Supplementary Figure S6). A number of genes encoding MIKC-type MADS-box TFs were found in this group, which was expected given their conserved roles as master regulators of floral organ identity across species (Theißen et al., 2016; Figures 2 and Supplementary Figure S5). Among them were two B-class function TFs known to regulate lodicule identity and stamen development and co-expressed in cluster 6; an ortholog of the maize silky1 (si1) gene (SvSi1; Sevir.4G289400) and homolog of OsMADS4 from rice (SvMADS4; Sevir.3G242100) (Ambrose et al., 2000; Nagasawa et al., 2003; Yao et al., 2008). A number of C-class, AGAMOUS-like genes were also co-expressed in cluster 6, including SvBde1 (Sevir.1G278500), orthologous to the maize bearded ear 1 (bde1) gene (Thompson et al., 2009), and homologs of rice OsMADS17, OsMADS3, and OsMADS58, Sevir.7G221900, Sevir.5G141300, and Sevir.3G074600, respectively (Yamaguchi et al., 2006). Also, three SEPALLATA-like E-class genes were expressed in cluster 6 including homologs of rice OsMADS1/LEAFY HULL STERILE 1 (LHS1), OsMADS7, and OsMADS8, Sevir.9G494300, Sevir.6G230500, and Sevir.2G277000, respectively, which all presumably function in specifying floral organ identity (Jeon, 2000; Zahn et al., 2005; Cui et al., 2010).
Additional genes that function in floral organ specification were also found in Group 4. Among these, SvTga1 (Sevir.6G230400), the ortholog of teosinte glume architecture 1 (tga1) encoding an SBP TF that regulates glume development and was a key domestication locus in maize (Dorweiler and Doebley, 1997; Preston et al., 2012), was found in cluster 16 (Figure 3B and Supplementary Figure S3). In addition, orthologs of the C2H2 TF STAMENLESS 1 from rice (Sevir.5G086100; SvSl1) (Xiao et al., 2009) and maize yabby 15/rice TONGARI-BOUSHI 1 (TOB1) (Sevir.1G255900; SvTob1) (Tanaka et al., 2012), were expressed in clusters 6 and 14, respectively (Tanaka et al., 2012). TOB1 and close homologs regulate maintenance and fate of reproductive meristems whereas OsSL1 regulates floral organ identity. Sex determination genes from maize were also associated with Group 4, including orthologs of maize anther ear 1 (an1), a terpenoid synthase involved in the gibberellic acid biosynthesis (Sevir.9G265300; SvAn1) (Bensen et al., 1995) and tassel seed 2 (ts2) (Sevir.9G439800; SvTs2) (DeLong et al., 1993) in cluster 17 and 16, respectively (Figure 3A and Supplementary Table S3).
To extend our co-expression analyses, we also performed a WGCNA (Langfelder and Horvath, 2008). We used WGCNA to independently define modules of co-expressed genes and to construct a co-expression network for early S. viridis inflorescence development (Supplementary Table S6). WGCNA produced 14 co-expression modules based on parameters detailed in the Section “Materials and Methods,” which were highly consistent with our FCM results; i.e., each WGCNA module largely corresponded to the sum of several FCM clusters (Supplementary Figure S8). In addition to grouping genes into co-expression modules, WGCNA was used to determine the interconnectedness of genes within and between modules. A strength value (i.e., weight) was assigned to the connection (edge) between each gene (node) and every other gene in the network. An integrated co-expression network was generated after filtering gene-to-gene connections with very low connectivity (weight >0.02; available in NCBI GEO).
To define sub-networks related to specific developmental processes and potentially identify new regulatory factors, we filtered the co-expression network for genes with strong connections to known genes of interest within a module. For example, we explored a co-expression module that included SvLg1, SvBaf1, and SvBif2, which are known players in boundary formation and axillary meristem initiation (“Plum” module; Supplementary Figure S8 and Supplementary Table S6). To reduce complexity in visualization and interpretation of this module, we included only genes with strong interactions (edges between genes having a weight >0.2) to a set of ten predicted developmental genes (Figure 5 and Supplementary Table S7). One sub-network was defined that included strong co-expression “interactions” among SvDfl1 (Sevir.2G302300), a homolog of maize delayed flower 1 (Muszynski et al., 2006), SvVt2 and a GH3 IAA-amido synthase (Sevir.9G364900) (Supplementary Figure S7), homologous to Arabidopsis YADOKARI 1 (YDK1) and predicted to regulate auxin levels (Staswick et al., 2005), via a common set of genes. In another sub-network, SvPla1 (Sevir.9G259300), a cytochrome P450 homologous to Arabidopsis PLASTOCHRON1 (Miyoshi et al., 2004; Sun et al., 2017), was identified as a hub gene directly or indirectly connected to SvBaf1, SvLg1, and SvBif2, consistent with its conserved role in lateral organ initiation (Figure 5 and Supplementary Table S7). Within these subnetworks, some developmental regulators showed high connectivity to many other genes, while others showed relatively low connectivity (Figure 5).
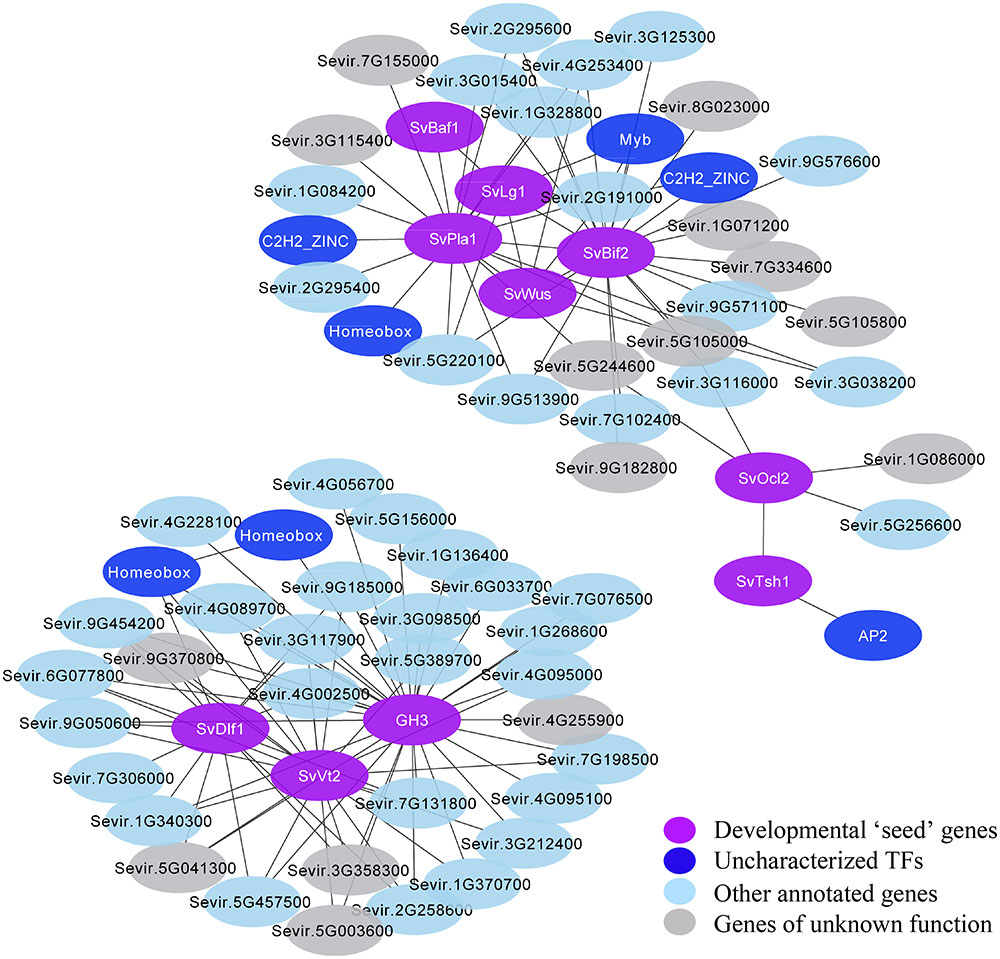
FIGURE 5. Two sub-networks extracted from the WGCNA were defined based on gene connectivity to a set of co-expressed regulators of inflorescence development (shown in purple). Genes (or nodes) were included if they were connected to these developmental genes by edges with a weight >0.2. Developmental regulators within these sub-networks either show high connectivity (e.g., SvVt2, SvBif2) or low connectivity (e.g., SvTsh1, SvBaf1). Uncharacterized TFs are indicated in dark blue, other genes with assigned functional annotations in light blue, and genes of unknown function in gray.
Our transcriptomics analysis in S. viridis identified homologs of known developmental regulators as well as uncharacterized genes with putative roles in panicoid grass inflorescence development. Based on spatiotemporal expression profiles, we showed that genes controlling transitions in meristem types during inflorescence development are largely conserved between S. viridis and maize (Figure 4). Since the diversity of inflorescence architectures found across grasses are largely determined by subtle regulatory variation on common developmental processes, it is not surprising that suites of genes have conserved functions across grasses, and often analogous functions in more distantly related species. For example, tsh1/NL1 genes share conserved functions in boundary establishment and bract suppression in maize and rice (Wang et al., 2009; Whipple et al., 2010), whereas the close Arabidopsis homolog, HANABA TARANU, functions in floral organ development (Zhao et al., 2004). However, in some cases orthologous genes can take on context-specific functional roles. The rice ortholog of baf1, DEPRESSED PALEA 1 (DP1), regulates palea formation, and floral organ number (Jin et al., 2011), while its close homolog in Arabidopsis, AHL22, functions in the floral transition (Yun et al., 2012).
We anticipate that species-specific expression differences underlie the variation in inflorescence form in Setaria spp. compared to maize and other closely related grasses. While we are beginning to elucidate some of the genes responsible for these morphological differences through mutant screens and experimental analyses (Yang et al., 2017), identifying subtle variation in gene regulation at the network level will require parallel transcriptomics resources from other species. In addition, using co-expression analyses to prioritize novel candidate genes as having previously undefined functions in development will also be most powerful when evaluated in the context of comparable datasets. Thus, a detailed knowledgebase of how the expression of individual genes shift during development and how co-expressed gene modules are rewired across grass species, is invaluable to understanding the genetic basis for morphological diversity in grass inflorescence architecture.
The apparent similarity between maize and S. viridis at the transcriptome level supports its use as a functional model for panicoid cereals, especially when paired with rapid advances in gene editing and transformation technologies (Zhu et al., 2017). Our transcriptomics analyses provide a platform for gene discovery in S. viridis inflorescence development and a comparative model for studying diverse architectures of agronomically important cereal crops. With respect to the former, it is anticipated that these data will help uncover novel expression patterns associated with the unique features of S. viridis inflorescence morphology, which can be translated to other millets (Huang et al., 2016), including subsistence crops in many developing countries that remain largely untapped for genetic improvement.
RNA-seq data (raw sequence reads and processed data files) generated in this study and the WGCNA gene co-expression matrix are available from the NCBI Gene Expression Omnibus (GEO; www.ncbi.nlm.nih.gov/geo/) under accession number GSE118673.
AE and EK conceptualized and supervised the work. MB established the developmental sampling scheme. JY and MB performed SEM and RNA-seq experiments. JY and CZ performed bioinformatics analyses and data visualization. CZ, JY, EK, and AE wrote the paper. All authors read and approved the final manuscript.
CZ and MB were supported by National Science Foundation (NSF) grant IOS-1413824 to EK. JY was supported by PGRP-NSF grant IOS-1733606 to AE.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Drs. Edoardo Bertolini, Michael McKain, and Indrajit Kumar at the Donald Danforth Plant Science Center (DDPSC) for advice on bioinformatics analyses, and Kevin Reilly and his team at the DDPSC Integrated Growth Facility for growth chamber maintenance. We also thank the U.S. Department of Energy (DOE) Joint Genome Institute for use of the S. viridis genome prior to publication, which is supported by the Office of Science of the U.S. DOE under Contract No. DE-AC02-05CH11231.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2018.01309/full#supplementary-material
FIGURE S1 | Correlations of global gene expression profiles across all samples.
FIGURE S2 | Expression profiles of orthologs of known developmental genes across six stages of Setaria viridis inflorescence development showing standard deviation among biological replicates.
FIGURE S3 | Dynamic expression profiles of TF families during inflorescence development in Setaria viridis: LOB, NAC, AP2/ERF, C2H2, bZIP, and bHLH families.
FIGURE S4 | Expression profiles of uncharacterized TFs with highly dynamic changes during inflorescence development.
FIGURE S5 | Phylogenetic relationship of Setaria viridis MIKC-type MADS-box TFs with homologous genes in rice, maize, and Arabidopsis.
FIGURE S6 | Cluster number selection for FCM and developmental trajectories of 25 clusters.
FIGURE S7 | Expression profiles of auxin-related genes from Groups 2 and 3.
FIGURE S8 | Assignment of genes to co-expression modules using WGCNA and comparison to FCM analyses.
TABLE S1 | RNA-seq library sequencing and mapping statistics.
TABLE S2 | Gene expression values for all S. viridis genes, their closest orthologs in other species, and available functional annotations.
TABLE S3 | Cluster assignment of 11,425 dynamically expressed genes and associated functional annotation.
TABLE S4 | Expression values and annotations of selected transcriptional factor (TF) families.
TABLE S5 | Expression values and functional annotations of auxin-related genes from Groups 2 and 3.
TABLE S6 | Output from WGCNA using 11,425 dynamically expressed genes showing the relative connectivity of each gene to each of 14 modules.
TABLE S7 | Genes (or nodes) and their connections (edges) to each other within the two sub-networks depicted in Figure 5.
Ambrose, B. A., Lerner, D. R., Ciceri, P., Padilla, C. M., Yanofsky, M. F., and Schmidt, R. J. (2000). Molecular and genetic analyses of the silky1 gene reveal conservation in floral organ specification between eudicots and monocots. Mol. Cell 5, 569–579. doi: 10.1016/S1097-2765(00)80450-5
Arora, R., Agarwal, P., Ray, S., Singh, A. K., Singh, V. P., Tyagi, A. K., et al. (2007). MADS-box gene family in rice: genome-wide identification, organization and expression profiling during reproductive development and stress. BMC Genomics 8:242. doi: 10.1186/1471-2164-8-242
Bai, F., Reinheimer, R., Durantini, D., Kellogg, E. A., and Schmidt, R. J. (2012). TCP transcription factor, BRANCH ANGLE DEFECTIVE 1 (BAD1), is required for normal tassel branch angle formation in maize. Proc. Natl. Acad. Sci. U.S.A. 109, 12225–12230. doi: 10.1073/pnas.1202439109
Barazesh, S., and McSteen, P. (2008). Hormonal control of grass inflorescence development. Trends Plant Sci. 13, 656–662. doi: 10.1016/j.tplants.2008.09.007
Bartlett, M. E. (2017). Changing MADS-box transcription factor protein–protein interactions as a mechanism for generating floral morphological diversity. Integr. Comp. Biol. 57, 1312–1321. doi: 10.1093/icb/icx067
Bartlett, M. E., and Thompson, B. (2014). Meristem identity and phyllotaxis in inflorescence development. Front. Plant Sci. 5:508. doi: 10.3389/fpls.2014.00508
Bennetzen, J. L., Schmutz, J., Wang, H., Percifield, R., Hawkins, J., Pontaroli, A. C., et al. (2012). Reference genome sequence of the model plant Setaria. Nat. Biotechnol. 30, 555–561. doi: 10.1038/nbt.2196
Bensen, R. J., Johal, G. S., Crane, V. C., Tossberg, J. T., Schnable, P. S., Meeley, R. B., et al. (1995). Cloning and characterization of the maize An1 gene. Plant Cell 7, 75–84. doi: 10.1105/tpc.7.1.75
Bolduc, N., Yilmaz, A., Mejia-Guerra, M. K., Morohashi, K., O’Connor, D., Grotewold, E., et al. (2012). Unraveling the KNOTTED1 regulatory network in maize meristems. Genes Dev. 26, 1685–1690. doi: 10.1101/gad.193433.112
Bomblies, K., Wang, R.-L., Ambrose, B. A., Schmidt, R. J., Meeley, R. B., and Doebley, J. (2003). Duplicate FLORICAULA/LEAFY homologs zfl1 and zfl2 control inflorescence architecture and flower patterning in maize. Development 130, 2385–2395. doi: 10.1242/dev.00457
Bommert, P., Lunde, C., Nardmann, J., Vollbrecht, E., Running, M., Jackson, D., et al. (2005). thick tassel dwarf1 encodes a putative maize ortholog of the Arabidopsis CLAVATA1 leucine-rich repeat receptor-like kinase. Development 132, 1235–1245. doi: 10.1242/dev.01671
Bommert, P., and Whipple, C. (2017). Grass inflorescence architecture and meristem determinacy. Semin. Cell Dev. Biol. 79, 37–47. doi: 10.1016/j.semcdb.2017.10.004
Bortiri, E., Chuck, G., Vollbrecht, E., Rocheford, T., Martienssen, R., and Hake, S. (2006). ramosa2 encodes a LATERAL ORGAN BOUNDARY domain protein that determines the fate of stem cells in branch meristems of maize. Plant Cell 18, 574–585. doi: 10.1105/tpc.105.039032
Brutnell, T. P., Wang, L., Swartwood, K., Goldschmidt, A., Jackson, D., Zhu, X.-G., et al. (2010). Setaria viridis: a model for C4 photosynthesis. Plant Cell 22, 2537–2544. doi: 10.1105/tpc.110.075309
Cho, E., and Zambryski, P. C. (2011). ORGAN BOUNDARY1 defines a gene expressed at the junction between the shoot apical meristem and lateral organs. Proc. Natl. Acad. Sci. U.S.A. 108, 2154–2159. doi: 10.1073/pnas.1018542108
Chu, H., Qian, Q., Liang, W., Yin, C., Tan, H., Yao, X., et al. (2006). The FLORAL ORGAN NUMBER4 gene encoding a putative ortholog of Arabidopsis CLAVATA3 regulates apical meristem size in rice. Plant Physiol. 142, 1039–1052. doi: 10.1104/pp.106.086736
Chuck, G., Muszynski, M., Kellogg, E., Hake, S., and Schmidt, R. J. (2002). The control of spikelet meristem identity by the branched silkless1 gene in maize. Science 298, 1238–1241. doi: 10.1126/science.1076920
Chuck, G., Whipple, C., Jackson, D., and Hake, S. (2010). The maize SBP-box transcription factor encoded by tasselsheath4 regulates bract development and the establishment of meristem boundaries. Development 137, 1243–1250. doi: 10.1242/dev.048348
Chuck, G. S., Brown, P. J., Meeley, R., and Hake, S. (2014). Maize SBP-box transcription factors unbranched2 and unbranched3 affect yield traits by regulating the rate of lateral primordia initiation. Proc. Natl. Acad. Sci. U.S.A. 111, 18775–18780. doi: 10.1073/pnas.1407401112
Cui, R., Han, J., Zhao, S., Su, K., Wu, F., Du, X., et al. (2010). Functional conservation and diversification of class E floral homeotic genes in rice (Oryza sativa). Plant J. 61, 767–781. doi: 10.1111/j.1365-313X.2009.04101.x
Danilevskaya, O. N., Meng, X., and Ananiev, E. V. (2010). Concerted modification of flowering time and inflorescence architecture by ectopic expression of TFL1-like genes in maize. Plant Physiol. 153, 238–251. doi: 10.1104/pp.110.154211
DeLong, A., Calderon-Urrea, A., and Dellaporta, S. L. (1993). Sex determination gene TASSELSEED2 of maize encodes a short-chain alcohol dehydrogenase required for stage-specific floral organ abortion. Cell 74, 757–768. doi: 10.1016/0092-8674(93)90522-R
Derbyshire, P., and Byrne, M. E. (2013). MORE SPIKELETS1 is required for spikelet fate in the inflorescence of Brachypodium. Plant Physiol. 161, 1291–1302. doi: 10.1104/pp.112.212340
Digel, B., Pankin, A., and von Korff, M. (2015). Global transcriptome profiling of developing leaf and shoot apices reveals distinct genetic and environmental control of floral transition and inflorescence development in barley. Plant Cell 27, 2318–2334. doi: 10.1105/tpc.15.00203
Doebley, J., Stec, A., and Hubbard, L. (1997). The evolution of apical dominance in maize. Nature 386, 485–488. doi: 10.1038/386485a0
Dorweiler, J. E., and Doebley, J. (1997). Developmental analysis of Teosinte glume architecture1: a key locus in the evolution of maize (Poaceae). Am. J. Bot. 84:1313. doi: 10.2307/2446130
Doust, A. N., and Kellogg, E. A. (2002). Inflorescence diversification in the panicoid “bristle grass” clade (Paniceae, Poaceae): evidence from molecular phylogenies and developmental morphology. Am. J. Bot. 89, 1203–1222. doi: 10.3732/ajb.89.8.1203
Doust, A. N., Kellogg, E. A., Devos, K. M., and Bennetzen, J. L. (2009). Foxtail millet: a sequence-driven grass model system. Plant Physiol. 149, 137–141. doi: 10.1104/pp.108.129627
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Eveland, A. L., Goldshmidt, A., Pautler, M., Morohashi, K., Liseron-Monfils, C., Lewis, M. W., et al. (2014). Regulatory modules controlling maize inflorescence architecture. Genome Res. 24, 431–443. doi: 10.1101/gr.166397.113
Feng, N., Song, G., Guan, J., Chen, K., Jia, M., Huang, D., et al. (2017). Transcriptome profiling of wheat inflorescence development from spikelet initiation to floral patterning identified stage-specific regulatory genes. Plant Physiol. 174, 1779–1794. doi: 10.1104/pp.17.00310
Furutani, I., Sukegawa, S., and Kyozuka, J. (2006). Genome-wide analysis of spatial and temporal gene expression in rice panicle development. Plant J. 46, 503–511. doi: 10.1111/j.1365-313X.2006.02703.x
Gallavotti, A. (2013). The role of auxin in shaping shoot architecture. J. Exp. Bot. 64, 2593–2608. doi: 10.1093/jxb/ert141
Gallavotti, A., Barazesh, S., Malcomber, S., Hall, D., Jackson, D., Schmidt, R. J., et al. (2008). sparse inflorescence1 encodes a monocot-specific YUCCA-like gene required for vegetative and reproductive development in maize. Proc. Natl. Acad. Sci. U.S.A. 105, 15196–201. doi: 10.1073/pnas.0805596105
Gallavotti, A., Malcomber, S., Gaines, C., Stanfield, S., Whipple, C., Kellogg, E., et al. (2011). BARREN STALK FASTIGIATE1 is an AT-hook protein required for the formation of maize ears. Plant Cell 23, 1756–1771. doi: 10.1105/tpc.111.084590
Gallavotti, A., Zhao, Q., Kyozuka, J., Meeley, R. B., Ritter, M. K., Doebley, J. F., et al. (2004). The role of barren stalk1 in the architecture of maize. Nature 432, 630–635. doi: 10.1038/nature03148
Goad, D. M., Zhu, C., and Kellogg, E. A. (2017). Comprehensive identification and clustering of CLV3/ESR-related (CLE) genes in plants finds groups with potentially shared function. New Phytol. 216, 605–616. doi: 10.1111/nph.14348
Hanano, S., and Goto, K. (2011). Arabidopsis TERMINAL FLOWER1 is involved in the regulation of flowering time and inflorescence development through transcriptional repression. The Plant Cell 23, 3172–3184. doi: 10.1105/tpc.111.088641
Harper, L., and Freeling, M. (1996). Interactions of liguleless1 and liguleless2 function during ligule induction in maize. Genetics 144, 1871–1882.
Harrop, T. W. R., Ud Din, I., Gregis, V., Osnato, M., Jouannic, S., Adam, H., et al. (2016). Gene expression profiling of reproductive meristem types in early rice inflorescences by laser microdissection. Plant J. 86, 75–88. doi: 10.1111/tpj.13147
Hirano, H.-Y., Tanaka, W., and Toriba, T. (2014). Grass flower development. Methods Mol. Biol. 1110, 57–84. doi: 10.1007/978-1-4614-9408-9_3
Huang, P., Shyu, C., Coelho, C. P., Cao, Y., and Brutnell, T. P. (2016). Setaria viridis as a model system to advance millet genetics and genomics. Front. Plant Sci. 7:1781. doi: 10.3389/fpls.2016.01781
Ikeda-Kawakatsu, K., Maekawa, M., Izawa, T., Itoh, J.-I., and Nagato, Y. (2012). ABERRANT PANICLE ORGANIZATION 2/RFL, the rice ortholog of Arabidopsis LEAFY, suppresses the transition from inflorescence meristem to floral meristem through interaction with APO1. Plant J. 69, 168–180. doi: 10.1111/j.1365-313X.2011.04781.x
Ishii, T., Numaguchi, K., Miura, K., Yoshida, K., Thanh, P. T., Htun, T. M., et al. (2013). OsLG1 regulates a closed panicle trait in domesticated rice. Nat. Genet. 45, 465e1–462e1. doi: 10.1038/ng.2567
Jeon, J.-S. (2000). leafy hull sterile1 is a homeotic mutation in a rice MADS box gene affecting rice flower development. Plant Cell 12, 871–884. doi: 10.1105/tpc.12.6.871
Jiao, Y., Wang, Y., Xue, D., Wang, J., Yan, M., Liu, G., et al. (2010). Regulation of OsSPL14 by OsmiR156 defines ideal plant architecture in rice. Nat. Genet. 42, 541–544. doi: 10.1038/ng.591
Jin, Y., Luo, Q., Tong, H., Wang, A., Cheng, Z., Tang, J., et al. (2011). An AT-hook gene is required for palea formation and floral organ number control in rice. Dev. Biol. 359, 277–288. doi: 10.1016/j.ydbio.2011.08.023
Gallavotti, A., Barazesh, S., Malcomber, S., Hall, D., Jackson, D., Schmidt, R. J., et al. (2008). sparse inflorescence1 encodes a monocot-specific YUCCA-like gene required for vegetative and reproductive development in maize. Proc. Natl. Acad. Sci. U.S.A. 105, 15196–15201. doi: 10.1073/pnas.0805596105
Kerstetter, R. A., Laudencia-Chingcuanco, D., Smith, L. G., and Hake, S. (1997). Loss-of-function mutations in the maize homeobox gene, knotted1, are defective in shoot meristem maintenance. Development 124, 3045–3054.
Komatsu, K., Maekawa, M., Ujiie, S., Satake, Y., Furutani, I., Okamoto, H., et al. (2003a). LAX and SPA: major regulators of shoot branching in rice. Proc. Natl. Acad. Sci. U.S.A. 100, 11765–11770.
Komatsu, M., Chujo, A., Nagato, Y., Shimamoto, K., and Kyozuka, J. (2003b). FRIZZY PANICLE is required to prevent the formation of axillary meristems and to establish floral meristem identity in rice spikelets. Development 130, 3841–3850.
Kumar, L., and Futschik, M. E. (2007). Mfuzz: a software package for soft clustering of microarray data. Bioinformation 2, 5–7. doi: 10.6026/97320630002005
Kwon, C. S., Hibara, K.-I., Pfluger, J., Bezhani, S., Metha, H., Aida, M., et al. (2006). A role for chromatin remodeling in regulation of CUC gene expression in the Arabidopsis cotyledon boundary. Development 133, 3223–3230. doi: 10.1242/dev.02508
Kyozuka, J., Konishi, S., Nemoto, K., Izawa, T., and Shimamoto, K. (1998). Down-regulation of RFL, the FLO/LFY homolog of rice, accompanied with panicle branch initiation. Proc. Natl. Acad. Sci. U.S.A. 95, 1979–1982. doi: 10.1073/pnas.95.5.1979
Kyozuka, J., Tokunaga, H., and Yoshida, A. (2014). Control of grass inflorescence form by the fine-tuning of meristem phase change. Curr. Opin. Plant Biol. 17, 110–115. doi: 10.1016/j.pbi.2013.11.010
Langfelder, P., and Horvath, S. (2008). WGCNA: an R package for weighted correlation network analysis. BMC Bioinformatics 9:559. doi: 10.1186/1471-2105-9-559
Lee, J., Park, J.-J., Kim, S. L., Yim, J., and An, G. (2007). Mutations in the rice liguleless gene result in a complete loss of the auricle, ligule, and laminar joint. Plant Mol. Biol. 65, 487–499. doi: 10.1007/s11103-007-9196-1
Lewis, M. W., Bolduc, N., Hake, K., Htike, Y., Hay, A., Candela, H., et al. (2014). Gene regulatory interactions at lateral organ boundaries in maize. Development 141, 4590–4597. doi: 10.1242/dev.111955
Lewis, M. W., and Hake, S. (2016). Keep on growing: building and patterning leaves in the grasses. Curr. Opin. Plant Biol. 29, 80–86. doi: 10.1016/j.pbi.2015.11.008
Li, M., Li, X., Zhou, Z., Wu, P., Fang, M., Pan, X., et al. (2016). Reassessment of the four yield-related genes Gn1a, DEP1, GS3, and IPA1 in rice using a CRISPR/Cas9 system. Front. Plant Sci. 7:377. doi: 10.3389/fpls.2016.00377
Li, X., Qian, Q., Fu, Z., Wang, Y., Xiong, G., Zeng, D., et al. (2003). Control of tillering in rice. Nature 422, 618–621. doi: 10.1038/nature01518
Maier, A. T., Stehling-Sun, S., Wollmann, H., Demar, M., Hong, R. L., Haubeiss, S., et al. (2009). Dual roles of the bZIP transcription factor PERIANTHIA in the control of floral architecture and homeotic gene expression. Development 136, 1613–1620. doi: 10.1242/dev.033647
McSteen, P. (2006). Branching out: the ramosa pathway and the evolution of grass inflorescence morphology. Plant Cell 18, 518–522. doi: 10.1105/tpc.105.040196
McSteen, P., Malcomber, S., Skirpan, A., Lunde, C., Wu, X., Kellogg, E., et al. (2007). barren inflorescence2 encodes a co-ortholog of the PINOID serine/threonine kinase and is required for organogenesis during inflorescence and vegetative development in maize. Plant Physiol. 144, 1000–1011. doi: 10.1104/pp.107.098558
Miura, K., Ikeda, M., Matsubara, A., Song, X.-J., Ito, M., Asano, K., et al. (2010). OsSPL14 promotes panicle branching and higher grain productivity in rice. Nat. Genet. 42, 545–549. doi: 10.1038/ng.592
Miyoshi, K., Ahn, B.-O., Kawakatsu, T., Ito, Y., Itoh, J.-I., Nagato, Y., et al. (2004). PLASTOCHRON1, a timekeeper of leaf initiation in rice, encodes cytochrome P450. Proc. Natl. Acad. Sci. U.S.A. 101, 875–880. doi: 10.1073/pnas.2636936100
Moreno, M. A., Harper, L. C., Krueger, R. W., Dellaporta, S. L., and Freeling, M. (1997). liguleless1 encodes a nuclear-localized protein required for induction of ligules and auricles during maize leaf organogenesis. Genes Dev. 11, 616–628. doi: 10.1101/gad.11.5.616
Muszynski, M. G., Dam, T., Li, B., Shirbroun, D. M., Hou, Z., Bruggemann, E., et al. (2006). delayed flowering1 encodes a basic leucine zipper protein that mediates floral inductive signals at the shoot apex in maize. Plant Physiol. 142, 1523–1536. doi: 10.1104/pp.106.088815
Nagasawa, N., Miyoshi, M., Sano, Y., Satoh, H., Hirano, H., Sakai, H., et al. (2003). SUPERWOMAN1 and DROOPING LEAF genes control floral organ identity in rice. Development 130, 705–718. doi: 10.1242/dev.00294
Nakagawa, M., Shimamoto, K., and Kyozuka, J. (2002). Overexpression of RCN1 and RCN2, rice TERMINAL FLOWER 1/CENTRORADIALIS homologs, confers delay of phase transition and altered panicle morphology in rice. Plant J. 29, 743–750. doi: 10.1046/j.1365-313X.2002.01255.x
Parenicová, L., de Folter, S., Kieffer, M., Horner, D. S., Favalli, C., Busscher, J., et al. (2003). Molecular and phylogenetic analyses of the complete MADS-box transcription factor family in Arabidopsis: new openings to the MADS world. Plant Cell 15, 1538–1551. doi: 10.1105/tpc.011544
Patro, R., Duggal, G., Love, M. I., Irizarry, R. A., and Kingsford, C. (2017). Salmon provides fast and bias-aware quantification of transcript expression. Nat. Methods 14, 417–419. doi: 10.1038/nmeth.4197
Pautler, M., Eveland, A. L., LaRue, T., Yang, F., Weeks, R., Lunde, C., et al. (2015). FASCIATED EAR4 encodes a bZIP transcription factor that regulates shoot meristem size in maize. Plant Cell 27, 104–120. doi: 10.1105/tpc.114.132506
Phillips, K. A., Skirpan, A. L., Liu, X., Christensen, A., Slewinski, T. L., Hudson, C., et al. (2011). vanishing tassel2 encodes a grass-specific tryptophan aminotransferase required for vegetative and reproductive development in maize. Plant Cell 23, 550–566. doi: 10.1105/tpc.110.075267
Preston, J. C., Wang, H., Kursel, L., Doebley, J., and Kellogg, E. A. (2012). The role of teosinte glume architecture (tga1) in coordinated regulation and evolution of grass glumes and inflorescence axes. New Phytol. 193, 204–215. doi: 10.1111/j.1469-8137.2011.03908.x
Revell, L. J. (2011). phytools: an R package for phylogenetic comparative biology (and other things). Methods Ecol. Evol. 3, 217–223. doi: 10.1111/j.2041-210X.2011.00169.x
Saeed, A. I., Sharov, V., White, J., Li, J., Liang, W., Bhagabati, N., et al. (2003). TM4: a free, open-source system for microarray data management and analysis. Biotechniques 34, 374–378.
Satoh-Nagasawa, N., Nagasawa, N., Malcomber, S., Sakai, H., and Jackson, D. (2006). A trehalose metabolic enzyme controls inflorescence architecture in maize. Nature 441, 227–230. doi: 10.1038/nature04725
Schneeberger, R. G., Becraft, P. W., Hake, S., and Freeling, M. (1995). Ectopic expression of the knox homeo box gene rough sheath1 alters cell fate in the maize leaf. Genes Dev. 9, 2292–2304. doi: 10.1101/gad.9.18.2292
Schultz, E. A., and Haughn, G. W. (1991). LEAFY, a homeotic gene that regulates inflorescence development in Arabidopsis. Plant Cell 3, 771–781. doi: 10.1105/tpc.3.8.771
Somssich, M., Je, B. I., Simon, R., and Jackson, D. (2016). CLAVATA-WUSCHEL signaling in the shoot meristem. Development 143, 3238–3248. doi: 10.1242/dev.133645
Soneson, C., Love, M. I., and Robinson, M. D. (2015). Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res. 4:1521. doi: 10.12688/f1000research.7563.2
Soreng, R. J., Peterson, P. M., Romaschenko, K., Davidse, G., Teisher, J. K., Clark, L. G., et al. (2017). A worldwide phylogenetic classification of the Poaceae (Gramineae) II: an update and a comparison of two 2015 classifications. J. Syst. Evol. 55, 259–290. doi: 10.1111/jse.12262
Stamatakis, A. (2014). RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30, 1312–1313. doi: 10.1093/bioinformatics/btu033
Staswick, P. E., Serban, B., Rowe, M., Tiryaki, I., Maldonado, M. T., Maldonado, M. C., et al. (2005). Characterization of an Arabidopsis enzyme family that conjugates amino acids to indole-3-acetic acid. Plant Cell 17, 616–627. doi: 10.1105/tpc.104.026690
Sun, X., Cahill, J., Van Hautegem, T., Feys, K., Whipple, C., Novák, O., et al. (2017). Altered expression of maize PLASTOCHRON1 enhances biomass and seed yield by extending cell division duration. Nat. Commun. 8:14752. doi: 10.1038/ncomms14752
Takeda, S., Hanano, K., Kariya, A., Shimizu, S., Zhao, L., Matsui, M., et al. (2011). CUP-SHAPED COTYLEDON1 transcription factor activates the expression of LSH4 and LSH3, two members of the ALOG gene family, in shoot organ boundary cells. Plant J. 66, 1066–1077. doi: 10.1111/j.1365-313X.2011.04571.x
Tanaka, W., Ohmori, Y., Ushijima, T., Matsusaka, H., Matsushita, T., Kumamaru, T., et al. (2015). Axillary meristem formation in rice requires the WUSCHEL ortholog TILLERS ABSENT1. Plant Cell 27, 1173–1184. doi: 10.1105/tpc.15.00074
Tanaka, W., Toriba, T., Ohmori, Y., Yoshida, A., Kawai, A., Mayama-Tsuchida, T., et al. (2012). The YABBY gene TONGARI-BOUSHI1 is involved in lateral organ development and maintenance of meristem organization in the rice spikelet. Plant Cell 24, 80–95. doi: 10.1105/tpc.111.094797
Theißen, G., Melzer, R., and Rümpler, F. (2016). MADS-domain transcription factors and the floral quartet model of flower development: linking plant development and evolution. Development 143, 3259–3271. doi: 10.1242/dev.134080
Thompson, B. E., Bartling, L., Whipple, C., Hall, D. H., Sakai, H., Schmidt, R., et al. (2009). bearded-ear encodes a MADS box transcription factor critical for maize floral development. Plant Cell 21, 2578–2590. doi: 10.1105/tpc.109.067751
Vollbrecht, E., Springer, P. S., Goh, L., Buckler, E. S. IV, and Martienssen, R. (2005). Architecture of floral branch systems in maize and related grasses. Nature 436, 1119–1126. doi: 10.1038/nature03892
Wagner, D., and Meyerowitz, E. M. (2002). SPLAYED, a novel SWI/SNF ATPase homolog, controls reproductive development in Arabidopsis. Curr. Biol. 12, 85–94. doi: 10.1016/S0960-9822(01)00651-0
Walsh, J., and Freeling, M. (1999). The liguleless2 gene of maize functions during the transition from the vegetative to the reproductive shoot apex. Plant J. 19, 489–495. doi: 10.1046/j.1365-313X.1999.00541.x
Walsh, J., Waters, C. A., and Freeling, M. (1998). The maize gene liguleless2 encodes a basic leucine zipper protein involved in the establishment of the leaf blade-sheath boundary. Genes Dev. 12, 208–218. doi: 10.1101/gad.12.2.208
Wang, L., Yin, H., Qian, Q., Yang, J., Huang, C., Hu, X., et al. (2009). NECK LEAF 1, a GATA type transcription factor, modulates organogenesis by regulating the expression of multiple regulatory genes during reproductive development in rice. Cell Res. 19, 598–611. doi: 10.1038/cr.2009.36
Whipple, C. J. (2017). Grass inflorescence architecture and evolution: the origin of novel signaling centers. New Phytol. 216, 367–372. doi: 10.1111/nph.14538
Whipple, C. J., Hall, D. H., DeBlasio, S., Taguchi-Shiobara, F., Schmidt, R. J., and Jackson, D. P. (2010). A conserved mechanism of bract suppression in the grass family. Plant Cell 22, 565–578. doi: 10.1105/tpc.109.073536
Xiao, H., Tang, J., Li, Y., Wang, W., Li, X., Jin, L., et al. (2009). STAMENLESS 1, encoding a single C2H2 zinc finger protein, regulates floral organ identity in rice. Plant J. 59, 789–801. doi: 10.1111/j.1365-313X.2009.03913.x
Yamaguchi, T., Lee, D. Y., Miyao, A., Hirochika, H., An, G., and Hirano, H.-Y. (2006). Functional diversification of the two C-class MADS box genes OSMADS3 and OSMADS58 in Oryza sativa. Plant Cell 18, 15–28. doi: 10.1105/tpc.105.037200
Yang, J., Thames, S., Best, N. B., Jiang, H., Huang, P., Dilkes, B. P., et al. (2017). Brassinosteroids modulate meristem fate and differentiation of unique inflorescence morphology in Setaria viridis. Plant Cell 30, 48–66. doi: 10.1105/tpc.17.00816
Yao, S.-G., Ohmori, S., Kimizu, M., and Yoshida, H. (2008). Unequal genetic redundancy of rice PISTILLATA orthologs, OsMADS2 and OsMADS4, in lodicule and stamen development. Plant Cell Physiol. 49, 853–857. doi: 10.1093/pcp/pcn050
Yoshida, A., Sasao, M., Yasuno, N., Takagi, K., Daimon, Y., Chen, R., et al. (2013). TAWAWA1, a regulator of rice inflorescence architecture, functions through the suppression of meristem phase transition. Proc. Natl. Acad. Sci. U.S.A. 110, 767–772. doi: 10.1073/pnas.1216151110
Yoshida, A., Suzaki, T., Tanaka, W., and Hirano, H.-Y. (2009). The homeotic gene long sterile lemma (G1) specifies sterile lemma identity in the rice spikelet. Proc. Natl. Acad. Sci. U.S.A. 106, 20103–20108. doi: 10.1073/pnas.0907896106
Yun, J., Kim, Y.-S., Jung, J.-H., Seo, P. J., and Park, C.-M. (2012). The AT-hook motif-containing protein AHL22 regulates flowering initiation by modifying flowering LOCUS T chromatin in Arabidopsis. J. Biol. Chem. 287, 15307–15316. doi: 10.1074/jbc.M111.318477
Zahn, L. M., Kong, H., Leebens-Mack, J. H., Kim, S., Soltis, P. S., Landherr, L. L., et al. (2005). The evolution of the SEPALLATA subfamily of MADS-box genes: a preangiosperm origin with multiple duplications throughout angiosperm history. Genetics 169, 2209–2223. doi: 10.1534/genetics.104.037770
Zhang, D., and Yuan, Z. (2014). Molecular control of grass inflorescence development. Annu. Rev. Plant Biol. 65, 553–578. doi: 10.1146/annurev-arplant-050213-040104
Zhao, M., Yang, S., Chen, C.-Y., Li, C., Shan, W., Lu, W., et al. (2015). Arabidopsis BREVIPEDICELLUS interacts with the SWI2/SNF2 chromatin remodeling ATPase BRAHMA to regulate KNAT2 and KNAT6 expression in control of inflorescence architecture. PLoS Genet. 11:e1005125. doi: 10.1371/journal.pgen.1005125
Zhao, Q., Weber, A. L., McMullen, M. D., Guill, K., and Doebley, J. (2011). MADS-box genes of maize: frequent targets of selection during domestication. Genet. Res. 93, 65–75. doi: 10.1017/S0016672310000509
Zhao, Y., Medrano, L., Ohashi, K., Fletcher, J. C., Yu, H., Sakai, H., et al. (2004). HANABA TARANU is a GATA transcription factor that regulates shoot apical meristem and flower development in Arabidopsis. Plant Cell 16, 2586–2600. doi: 10.1105/tpc.104.024869
Zhu, C., Yang, J., and Shyu, C. (2017). Setaria comes of age: meeting report on the Second International Setaria Genetics Conference. Front. Plant Sci. 8:1562. doi: 10.3389/fpls.2017.01562
Keywords: inflorescence development, Setaria, panicoid grasses, transcriptome profiling, meristem, spikelet
Citation: Zhu C, Yang J, Box MS, Kellogg EA and Eveland AL (2018) A Dynamic Co-expression Map of Early Inflorescence Development in Setaria viridis Provides a Resource for Gene Discovery and Comparative Genomics. Front. Plant Sci. 9:1309. doi: 10.3389/fpls.2018.01309
Received: 01 May 2018; Accepted: 20 August 2018;
Published: 12 September 2018.
Edited by:
Prem Bhalla, The University of Melbourne, AustraliaReviewed by:
Michael J. Scanlon, Cornell University, United StatesCopyright © 2018 Zhu, Yang, Box, Kellogg and Eveland. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Andrea L. Eveland, YWV2ZWxhbmRAZGFuZm9ydGhjZW50ZXIub3Jn
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.