 Huijie Liu
Huijie Liu Jian He1†
Jian He1† Jin Cheng
Jin Cheng Lei Xie
Lei Xie
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci. , 27 July 2018
Sec. Plant Development and EvoDevo
Volume 9 - 2018 | https://doi.org/10.3389/fpls.2018.01097
Structural rearrangements of Anemone species' chloroplast genome has been reported based on genetic mapping of restriction sites but has never been confirmed by genomic studies. We used a next-generation sequencing method to characterize the complete chloroplast genomes of five species in the tribe Anemoneae. Plastid genomes were assembled using de novo assembling methods combined with conventional Sanger sequencing to fill the gaps. The gene order of the chloroplast genomes of tribe Anemoneae was compared with that of other Ranunculaceae species. Multiple inversions and transpositions were detected in tribe Anemoneae. Anemoclema, Anemone, Hepatica, and Pulsatilla shared the same gene order, which contained three inversions in the large single copy region (LSC) compared to other Ranunculaceae genera. Archiclematis, Clematis, and Naravelia shared the same gene order containing two inversions and one transposition in LSC. A roughly 4.4 kb expansion region in inverted repeat (IR) regions was detected in tribe Anemoneae, suggesting that this expansion event may be a synapomorphy for this group. Plastome phylogenomic analyses using parsimony and a Bayesian method with implementation of partitioned models generated a well resolved phylogeny of Ranunculaceae. These results suggest that evaluation of chloroplast genomes may result in improved resolution of family phylogenies. Samples of Anemone, Hepatica, and Pulsatilla were tested to form paraphyletic grades within tribe Anemoneae. Anemoclema was a sister clade to Clematis. Structual variation of the plastid genome within tribe Anemoneae provided strong phylogenetic information for Ranunculaceae.
Comparative analysis of chloroplast genomes can provide valuable information for phylogeny reconstruction and resolution of complex evolutionary relationships (Shaw et al., 2007; Mardanov et al., 2008; Moore et al., 2010; Park et al., 2017; Sun et al., 2017). In angiosperms, plastid genome gene number and order is conserved (Wolfe et al., 1987). This is because chloroplast sequences evolve at approximately half the speed of nuclear regions (Jansen et al., 2005; Walker et al., 2014). However, sequence rearrangements in chloroplast genomes have been reported from various kinds of plants (Doyle et al., 1996; Tangphatsornruang et al., 2011; Walker et al., 2015; Sun et al., 2017). These rearrangements include large inversions in large single copy region (LSC), and inverted repeats region (IR) expansions or contractions into single copy regions with inversions (Palmer et al., 1987; Tangphatsornruang et al., 2009). Intramolecular recombination may be the reason for large inversions in plastid genomes (Ogihara et al., 1988; Hiratsuka et al., 1989). These inversion events were probably triggered by tRNA activity (Hiratsuka et al., 1989) or intragenomic recombination in regions with variable G + C content (Fullerton et al., 2001; Smith et al., 2002; Walker et al., 2014). Gene rearrangements and inversions in plastid genomes are believed to have important value in phylogenetic analyses because they are rare, homology estimates are easy, and determination of inversion event polarity is easy (Johansson, 1999; Lee et al., 2007; Jansen et al., 2008; Walker et al., 2014; Yan et al., 2017). Comparison of whole plastid genomes provides the opportunity to explore sequence variation. These comparisons also permit examination of molecular evolutionary patterns associated with structural rearrangement and elucidation of the molecular mechanisms underlying those events.
Ranunculaceae, one of the earliest families that diverged from the eudicots (APG IV, 2016), is composed of more than 2000 mostly herbaceous species with a global distribution (Tamura, 1993, 1995; Ro et al., 1997). In recent years, molecular phylogenetics has provided deep insights and reassessment of Ranunculaceae taxonomy. Some genera have been reduced and a new genus (Gymnaconitum) has been proposed based on molecular phylogentic analysis results (Compton and Hedderson, 1997; Compton et al., 1998; Ro et al., 1997; Miikeda et al., 2006; Hoot et al., 2012; Wang et al., 2013; Falck and Lehtonen, 2014; Jiang et al., 2017a). All molecular studies to date were mainly based on tandemly repeated nrDNA and several commonly used plastid regions (Compton and Hedderson, 1997; Compton et al., 1998; Wang et al., 2005, 2010, 2013; Miikeda et al., 2006; Hörandl et al., 2009; Emadzade et al., 2010, 2011; Jabbour and Renner, 2012; Falck and Lehtonen, 2014; Cossard et al., 2016; Jiang et al., 2017a). There are only a few complete chloroplast genomes published and accessible from GenBank (http://www.ncbi.nlm.nih.gov). Phylogenetic inferences for Ranunculaceae taxa based on genomic data have yet to be conducted.
Chloroplast genome structural rearrangements and inversions in Anemone and other related genera have been previously reported based on genetic mapping by restriction enzyme site methods (Hoot and Palmer, 1994; Johansson, 1999). In recent years, several complete chloroplast genomes of Ranunculaceae have been published (Chen et al., 2015; Park et al., 2015, 2017; Li et al., 2016; Park and Park, 2016; Jiang et al., 2017b; Lim et al., 2017; Szczecinska et al., 2017; Liu et al., 2018). Recently, Jiang et al. (2017b) and Liu et al. (2018) published plastome sequences of Anemoclema and Clematis s.l. (including Archiclematis, Clematis, and Naravelia; Liu et al., 2018), and they also discovered striking structural rearrangements in plastome sequence of the reported genera comparing to that of other Ranunculaceae genera. However, structural variation of tribe Anemoneae plastomes were not discussed in detail by previous studies. The phylogenetic significance of the plastid genome structural variation in tribe Anemoneae and Ranunculaceae still needs to be assessed.
The tribe Anemoneae (as defined by Tamura, 1995) traditionally includes three subtribes (Kingdoniinae, Anemoninae, and Clematidinae), based on previous molecular phylogenetic studies that did not include the subtribe Kingdoniinae (Hoot et al., 2012; Jiang et al., 2017a). For subtribe Clematidinae, almost all satellite genera of Clematis (such as Naravelia and Archiclematis) were nested within Clematis by previous studies (Miikeda et al., 2006; Xie et al., 2011; Lehtonen et al., 2016; Jiang et al., 2017a; Liu et al., 2018). In subtribe Anemoninae, Hoot et al. (2012) reduced Hepatica, Pulsatilla, Oreithales, Knowltonia, and Barneoudia to the genus Anemone using molecular phylogenetic results inferred from nrITS and atpB-rbcL regions. This result was consistent with subsequent findings by Zhang et al. (2015). However, using six plastid DNA regions, Jiang et al. (2017a) disputed that Anemone s.l. (sensu Hoot et al., 2012) was confirmed to be a paraphyletic group and argues that Hepatica should not be included in Anemone s.l. These two competing phylogenetic hypotheses need to be reconciled using phylogenomic data.
In this study, we report on complete chloroplast genomes from five tribe Anemoneae species (Anemoclema, Anemone, Pulsatilla, and Hepatica). The plastomes of Anemone and Hepatica are reported for the first time. Together with the plastome sequence analyses of Clematis s.l., the aims of this study are to present whole chloroplast genome data for these species; to compare the plastid genomic structure and sequence variation within the tribe Anemoneae; to test two alternative hypotheses (tRNA activity or G + C content variation) that may cause gene rearrangement events; to test competing phylogenetic hypotheses within tribe Anemoneae using plastid phylogenomic data; and to clarify the phylogenetic significance of plastome structural variation of tribe Anemoneae. We also identified repeat sequences and SSRs inside these five plastomes. The data presented in this study should be useful for future phylogenetic studies of tribe Anemoneae and possibly the rest of the buttercup family.
We chose to sample five species, Anemone tomentosa, A. trullifolia, Hepatica henryi, Pulsatilla chinensis, and Anemoclema glaucifolium, for this study. Fresh young plant leaves were collected from the field for DNA extraction and were dried with silica-gel. Vouchers were deposited at the Beijing Forestry University (BJFC) Herbarium. In our previous study (Liu et al., 2018), complete chloroplast genome sequences of Clematis, Archiclematis, and Naravelia were reported. Thus, in this study, we included samples representing all of the major clades (Hepatica clade, Anemone s.l. clade, Anemoclema, and Clematis s.l. clade) of tribe Anemoneae (Hoot et al., 2012; Zhang et al., 2015; Jiang et al., 2017a) to check plastome structural variation. We also obtained whole chloroplast genomes of other Ranunculaceae species and outgroup species of Berberis available from Genbank for structural comparison and phylogenomic analysis (Table 1).
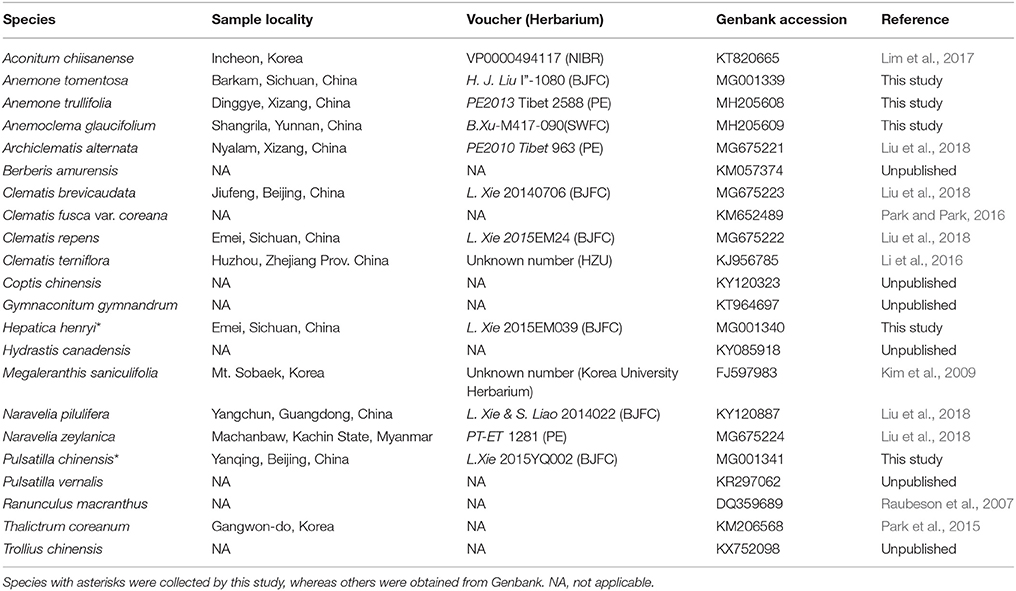
Table 1. Reference information for sequenced chloroplast genomes.
Approximately 50 mg dried leaves were ground for each species, and total DNAs were extracted using cetyl-trimethylammonium bromide (CTAB; Doyle and Doyle, 1987) with the quality of DNAs assessed by agarose gel electrophoresis. Total DNAs were subsequently sent to Novogene (http://www.novogene.com, China) for short insert (350 bp) library construction and next-generation sequencing. Pair end reads of 2 × 150 bp for all tested species were generated on an Illumina Hiseq 4000 genome analyzer platform. Original reads were filtered using the FASTX-Toolkit (http://hannonlab.cshl.edu/fastx_toolkit) to acquire high-quality data by deleting adaptors and low quality reads.
We used BLAT analysis with a Python script (Weitemier et al., 2014) to exclude nuclear and mitochondrial reads using published plastomes from Ranunculaceae as references. Putative chloroplast reads were used for de novo assembly to reconstruct the samples' complete plastid genomes using Geneious R11 (Kearse et al., 2012) with a low sensitivity setting. Contigs from de novo assembly were annotated in Geneious R11 and then were concatenated into larger contigs based on Pulsatilla vernalis (KR297062) plastomes. All gaps were bridged using conventional Sanger sequencing. The IR region was determined using the Repeat Finder function in Geneious R11. The IR region was subsequently manually inverted and copied to construct the complete plastome sequence. The IR and SC boundaries for all three species were checked using Sanger sequencing.
Complete plastid genomes were manually edited to remove ambiguous sites. The three plastomes were then annotated using the Unix program Plann (Huang and Cronk, 2015) and the annotations were verified using the online program DOGMA (http://dogma.ccbb.utexas.edu/; Wyman et al., 2004). If ambiguous annotations were present between Plann and DOGMA, we determined gene boundaries using the online program Blast (Gish and States, 1993). Illustrations of circular plastoms were generated using the Organellar Genome DRAW tool (Lohse et al., 2013). Final plastid genomes were deposited in GenBank (Table 1).
We obtained other complete chloroplast genomes of Ranunculaceae and an outgroup species Berberis from Genbank (Table 1) for comparative analyses. The IR/SC boundary regions of species from tribe Anemoneae were illustrated and compared to other Ranunculaceae species and Berberis. MAFFT was used to compare the similarity of plastid genome sequences (Katoh et al., 2005), and mVISTA was used to export visual results to evaluate similarity (Frazer et al., 2004). Visual results from mVISTA were further analyzed using two alignment programs: LAGAN, which produces true multiple alignments regardless of whether they contain inversions or not, and Shuffle-LAGAN, which can detect rearrangements and inversions (Brudno et al., 2003a,b). Detailed gene inversions and transpositions were identified by comparing the gene order of Ranunculaceae samples to Berberis with a whole plastome alignment method that used Mauve v2.3.1. (Darling et al., 2010).
The G + C content was calculated for the spacer regions boundary each of the major inversions and for transpositions of the tribe Anemoneae' plastomes. Flanking regions were defined as the noncoding sequence between the nearest coding genes on either side of the inversion/transposition boundary (Walker et al., 2014). We also conducted a sliding window analysis to identify the nucleotide variability (Pi) in the inversion/transposition regions using DnaSP version 5 (Librado and Rozas, 2009).
We used REPuter (Kurtz et al., 2001) to identify and locate the repeat sequences for the newly sampled species, including direct, reverse and palindromic repeats, within the plastid genome. For repeat identification, the following parameters were used: (1) 30 bp minimum repeat size and (2) 90% or greater sequence identity (Hamming distance = 3). In order to avoid redundancy, repeat sequence analysis was carried out with a single IR region. SSRs were determined using MISA (Thiel et al., 2003) and parameters were set to 10 repeat units ≥10 for mononucleotide SSRs, six repeat units ≥6 for dinucleotide, five repeat units ≥5 for trinucleotide, four repeat units ≥4 for tetranucleotide, and three repeat units ≥3 for pentanucleotide and hexanucleotide SSRs.
We used the plastome sequences of all the other genera of Ranunculaceae available from GenBank for phylogenomic analysis. The sampling covered 15 of the family's genera, seven of which belong to tribe Anemoneae. There is an accession of Actaea plastome (NC034704, unpublished) sequence available in GenBank. However, after extracting genes from this plastome and doing Blast in GenBank, we found that this sample belongs to Apiaceae and was mis-identified as Actaea. Thus, we did not include this sequence in the present analysis. Berberis amurensis plastome was chosen as the outgroup (Table 1).
In this study, we first separated the complete plastome sequences into coding regions (protein-coding genes, as well as tRNA genes and rRNA genes), intergenic spacer regions, and introns. Gene orders for all samples were tested using mVISTA, and then were shuffled in the same order with a Berberis amurensis plastome sequence whenever gene inversions/transpositions were found. All data sets were then aligned using MAFFT v6.833 (Katoh et al., 2005) and manually adjusted by MEGA 7.0 (Kumar et al., 2016). The ambiguous alignments were removed from the data sets for phylogeny reconstruction. Substitution models and data partitions for Bayesian analysis was determined by PartitionFinder v2.1.1 (Lanfear et al., 2016) and the best scheme selected by Akaike information criterion (AIC; Posada and Buckley, 2004).
Phylogeny reconstruction occurred using data sets of LSC, SSC, IR, and complete plastomes with Parsimony (MP) and Bayesian methods. Each data set was further separated into CDs, intron, intergenic spacer regions (Ma et al., 2014).
Parsimony (MP) analysis was conducted for all the separated data sets and the complete plastome data set using PAUP* 4.0b10 (Swofford, 2003). All characters were treated as unordered and equally weighted, and gaps were set as missing data. We used Branch-and-Bound or heuristic search (1000 replicates), simple addition, and tree bisection-reconnection branch swapping with MUL-trees to search the MP tree(s). Branch support values were assessed by performing 1000 bootstrap replicates using 1000 random taxon addition replicates with 10 trees held at each step and TBR swapping.
Bayesian inference (BI) was conducted with MrBayes v3.2.3 (Ronquist and Huelsenbeck, 2003) using partitioned substitution models tested by PartitionFinder. Two independent Markov chain Monte Carlo (MCMC) chains were run, each with three heated and one cold chain for 2,000,000 generations and sampling trees every 100 generations. The MCMC convergence in Bayesian inference was checked by AWTY (https://github.com/danlwarren/RWTY, Warren et al., 2017). The first 20% of trees were discarded as burn-in with the remaining trees being used for generating the consensus tree.
We obtained 2.7 Gb of average NGS clean data for each species, with minimums and maximums of 2.2 Gb (Pulsatila chinensis, 6,309,984 reads), and 3.3 Gb (Anemone tomentosa, 9,512,414 reads), respectively. Blat analysis selected out 542,906 putative plastid reads for Anemone tomentosa, 451,685 reads for A. trullifolia, 100,695 reads for Pulsatilla, 128,187 reads for Hepatica, and 335,675 reads for Anemoclema. Anemone tomentosa reads obtained three large contigs from de novo assembly (83,809 bp, 27,444 bp, and 20,496 bp). Three gaps were bridged using sanger sequencing. For A. trullifolia data, only one large contig (126,074 bp) was derived that included complete LSC, IR, and SSC regions and regions without gaps. For Pulsatilla chinensis plastid reads, eight large contigs ranging from 3,287 bp to 38,885 bp were obtained. We filled seven gaps for this sample. For Hepatica henryi, two large contigs (45,926 bp and 65,671 bp) were obtained and one large gap (ca. 1.5 kb) was filled by designing two pairs of primers. For Anemoclema data, three large contigs (92,619 bp, 33,225 bp, and 4,489 bp.) were concatenated. Two gaps were bridged using Sanger sequencing. Information for all of the gaps and primers for Sanger sequencing are presented in the Supplementary Materials.
The genome size of the five newly sequenced samples ranged from 157,096 bp (Anemone trullifolia) to 163,669 bp (Pulsatilla chinensis; Figure 1), and the overall GC content varied from 37.2% (Pulsatilla chinensis) to 37.9% (Anemoclema glaucifolium and Hepatica henryi; Table 2). All five plastid genomes consisted of a pair of IRs (31,022–31,490 bp) separated by the LSC (78,795–82,339 bp) and SSC (16,257–19,100 bp) regions, respectively (Table 2). The plastomes of the five samples encoded an identical set of 112 genes, including 78 protein-coding genes, 29 transfer RNAs, four ribosomal RNAs, and 25 genes are duplicated in IRs. There are 18 genes with introns in the plastomes of Anemoclema, Anemone tomentosa, and Pulsatilla chinensis, whereas, 17 genes with introns are present in Anemone trullifolia and Hepatica henryi plastomes (no introns in rps16).
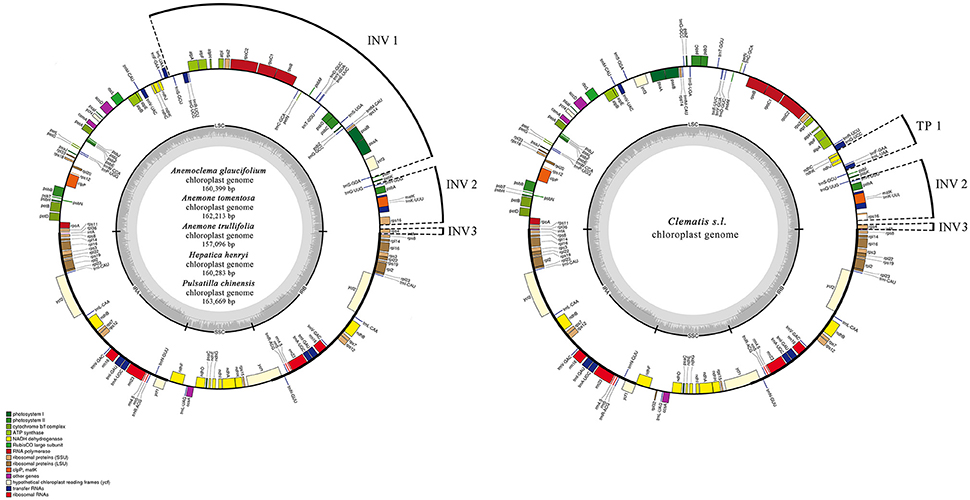
Figure 1. Chloroplast genome maps for Anemoclema, Anemone, Hepatica, and Pulsatilla samples (lefe), and Clematis s.l. (right). Thick lines on the outer complete circle identify the inverted repeat regions (IRa and IRb). The innermost track of the plastome shows the G + C content. Genes on the outside of the map are transcribed in a clockwise direction and genes on the inside of the map are transcribed in a counter clockwise direction. INV, inversion; TP, transposition; IR, inverted repeats; LSC, large single copy; SSC, small single copy; Pi, nucleotide variability.
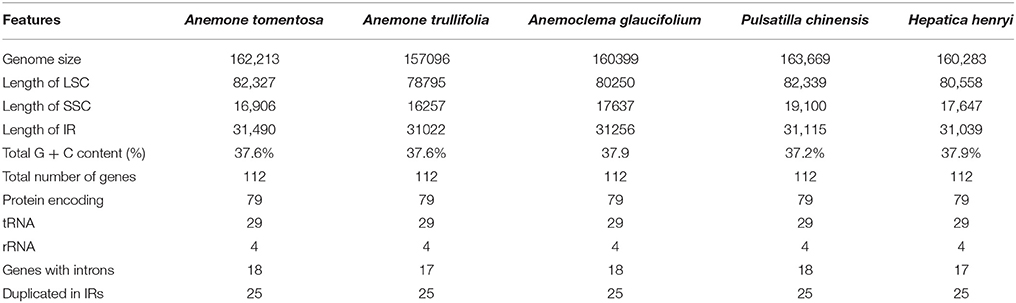
Table 2. Base compositions of five newly sequenced Anemoneae' plastomes.
We compared the IR/SC boundary regions of tribe Anemoneae to other Ranunculaceae species as well as an outgroup from Berberis. The junction positions are similar and conserved in tribe Anemoneae, yet differ from other Ranunculaceae species and Berberis (Figure 2). For example, rps19-infA (seven genes) is in the LSC region of Berberis, Hydrastis, Coptis, Thalictrum, Megaleranthis, Ranunculus, Aconitum, and Trollius plastomes, but is located in IR in Clematis (including Archiclematis and Naravelia), Anemoclema, Anomone, Pulsatilla, and Hepatica). This difference makes the IR region of tribe Anemoneae roughly 4.4 kb longer than in other genera of the family. The gene orders located within the IR-SSC and IR-LSC boundaries are similar among tribe Anemoneae samples but different from those in other Ranunculaceae genera (Figure 1).
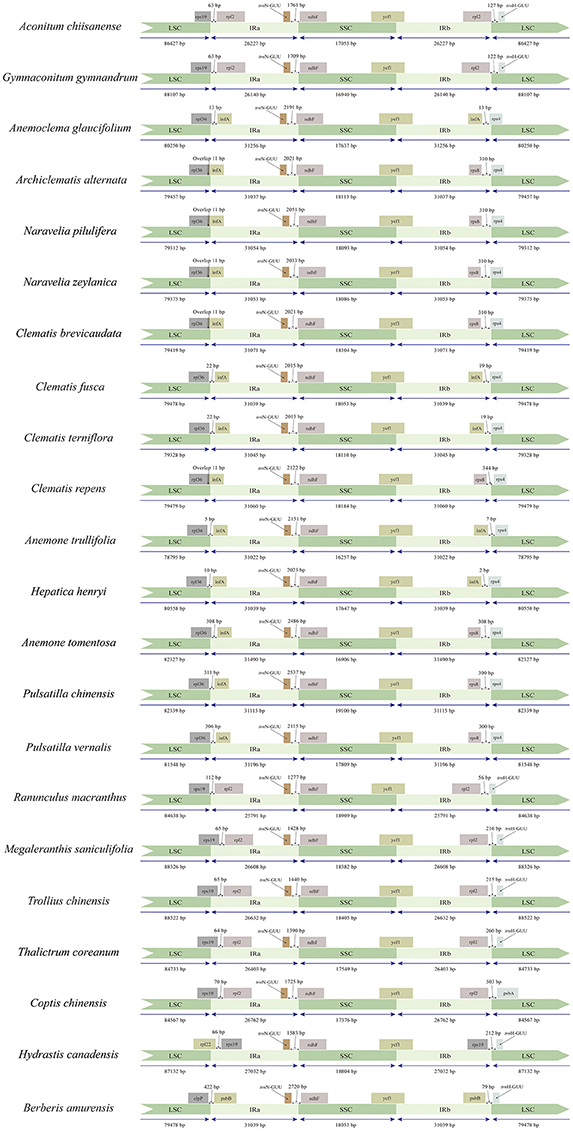
Figure 2. Comparison of the LSC, IRs and SSC boundary regions of tribe Anemoneae, published Ranunculaceae genera, and Berberis plastomes.
To investigate levels of genome divergence, multiple alignments of plastid genomes were performed (Figures 3A,B and Supplementary Materials). For mVISTA analysis, LAGAN, and Shuffle-LAGAN program results differed because of gene inversion/transposition occurring in tribe Anemoneae (Figures 3A,B). When using the LAGAN method (Figure 3A), plastomes of Anemoclema, Anemone, Pulsatilla, and Hepatica showed consistency in gene order, but carried large unalignable regions in LSC comparing to Clematis and other Ranunculaceae species. Plastomes of Clematis s.l. also showed some unalignable regions compared to other Ranunculaceae species. Conversely, all of the sequences aligned well when Shuffle-LAGAN methods were used (Figure 3B). This approach also revealed high sequence similarity across the coding region accompanied by more variability in non-coding regions.

Figure 3. Sequence alignment of tribe Anemoneae, published Ranunculaceae genera, and Berberis plastome using the mVISTA program. A cut-off of 70% similarity was used for the plot and the Y-scale represents the percent similarity ranging from 50-100%. Blue represents coding regions, and pink represents non-coding regions. (A) LAGAN method (as described in Materials and Methods); (B) Shuffle LAGAN method (as described in Materials and Methods).
tribe Anemoneae samples exhibited inversion and transposition regions that were detected by MAUVE (Supplementary Materials), compared to other Ranunculaceae plastomes. In Anemoclema, Anemone, Hepatica, and Pulsatilla, three inversions are present in the LSC region (Figure 1). The first inversion 1 (INV 1) is located between trnS-GGA and trnS-GCU (ca. 40 kb in length). The second inversion (INV 2) was between rps16 and trnH-GUG (ca. 5k in length). The third inversion region (INV3) was located in rps4 (ca. 600 bp in length). Two inversions and one transposition were detected in Clematis s.l. plastomes, with INV2, and INV3 being similar to those in Anemone s. l. plastomes. The transposition region 1 (TP1) was located between trnL UAA and ndhC (ca. 3k in length; Figure 1). No inversions or transpositions were found in published plastomes of other Ranunculaceae genera or in comparison to other plastomes of angiosperm species like Amborella trichopoda, Berberis amurensis, and Nicotiana tabacum.
The G + C content of the sequence in boundary regions was detected to be lower than the average G + C content of whole plastome and all the noncoding regions (Table 3). The sliding window analysis revealed that higher nucleotide variability (Pi) was exhibited at SC regions in comparison to IR regions (Figure 4). Genetic variation was particularly high at the boundary of INV1, which existed in Anemoclema, Anemone, Hepatica, and Pulsatilla plastomes. The INV2 and TP1, found in Clematis s.l. plastomes, has a high nucleotide varability at the borders as well. However, we did not find high genetic variation in INV3, which is shared among all the Anemoneae species.
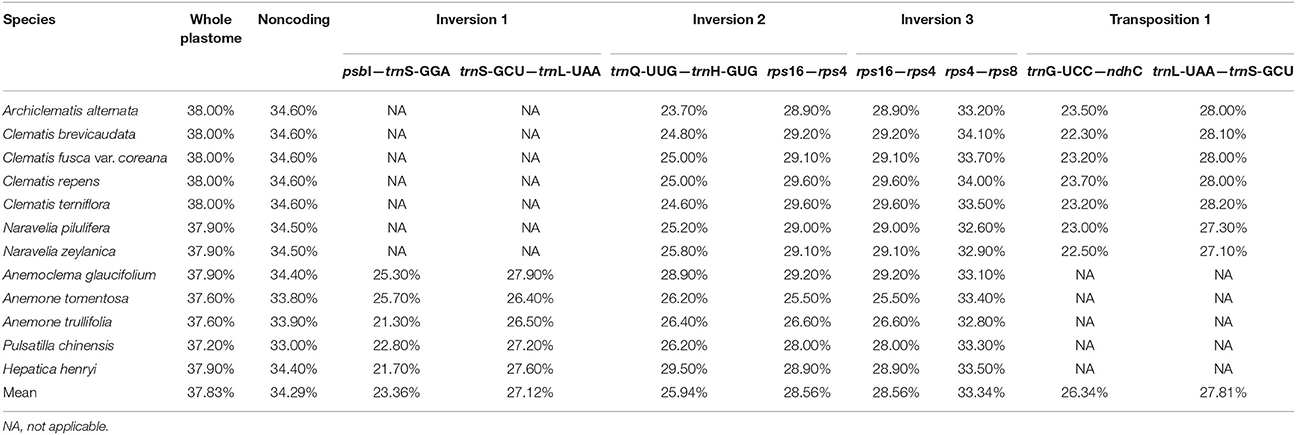
Table 3. Percent of G + C content of the whole plastomes, noncoding regions and the regions boundary INV 1, INV 2, INV3, and TP1 in Anemoneae samples.
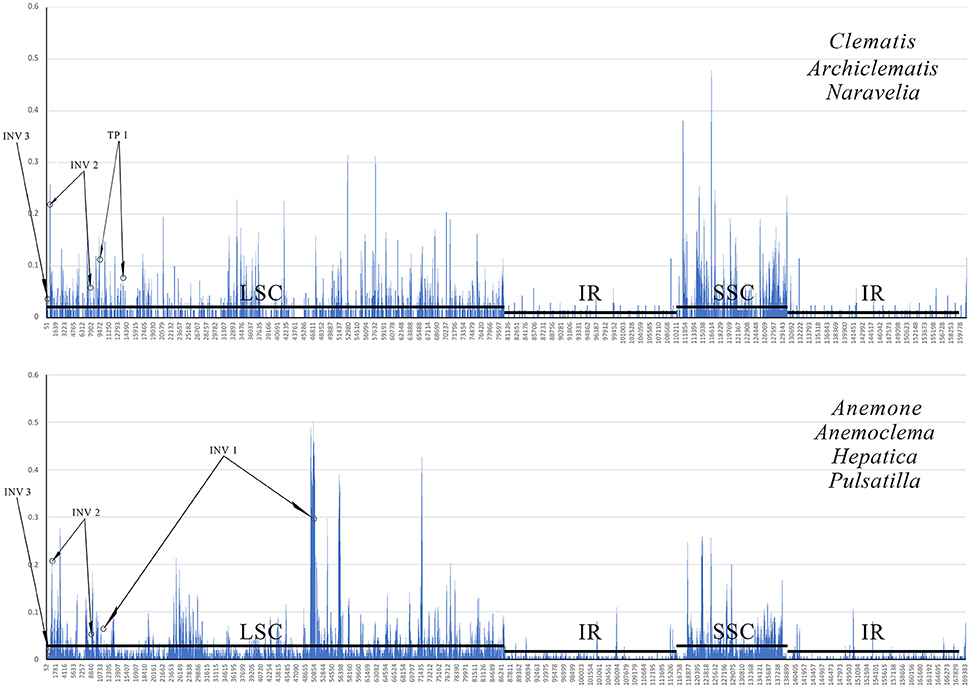
Figure 4. A sliding window analyses of the whole plastid genomes of Clematis s.l. (upper) and Anemoclema, Anemone, Hepatica, and Pulsatilla samples (lower). Circles identify the regions bordering inversion sites, and lines parallel to the x-axis identify the positions of the LSC, SSC and IR regions.
We detected a total of 173 repeats including direct, reverse, palindromic and complement repeats in the five newly sequenced plastomes (Figure 5). The most common repeat types are direct repeats, which account for 45% of the total repeats, followed by palindromic repeats (35%) and reverse repeats (16%). The only two complement repeats were found in Pulsatilla plastomes. Most of the repeats were short, ranging from 30–59 bp. However, a few much longer direct and reverse repeats (up to more than100 bp) were found in Hepatica and Anemone plastomes. The majority of repeats were located in noncoding regions (84%), among which only 3% were found in introns. There were 16% repeats detected in CDs.
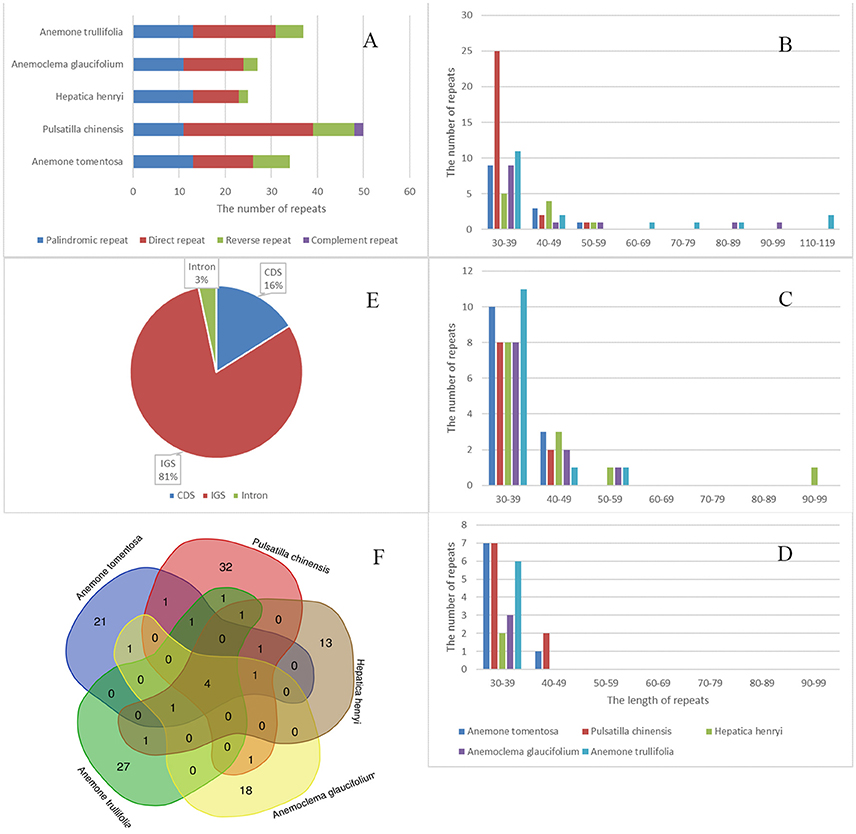
Figure 5. Analyses of repeated sequences in five newly sequenced plastomes. (A) number of four repeat types; (B) Frequency of direct repeats by length; (C) Frequency of reverse repeats by length; (D) Frequency of palindromic repeats by length; (E) Location of repeats; (F) Summary of shared repeats among the five plastomes.
We also investigated repeats that are shared among plastomes of the five samples by using strict criteria, i.e., repeats which are identical in length and located in homologus regions were defined as shared repeats. Under this criteria, there were four repeats shared by all five species. All tested plastomes possessed their own repeats, with the number varying from 13 (Hepatica henryi) to 32 (Pulsatilla chinesis; Figure 5). These repeats may serve as potential population genetic markers for further studies.
We identified 57, 57, 43, 51, and 61 SSRs in Anemone tomentosa, A. trullifolia, Anemoclema glaucifolium, H. henryi, and P. chinensis, respectively (Table 4). A mononucleotide repeat unit (A/T) was found to be the most abundant, accounting for 65–95% among the five species. This was followed by a dinucleotide repeat unit (AT/AT) with particular numbers of four, six, one, three and two SSRs, respectively. The trinucleotide repeat unit (AAT/ATT) was detected in Anemone trullifolia, Hepatica henryi, and Pulsatilla chinensis, and only one tetranucleotide repeat (AAGT) was found in Anemone tomentosa. Multiple pentanucleotide repeats were detected in Anemone tomentosa, Hepatica henryi, and Pulsatilla chinese. One hexanucleotide repeat was present in Pulsatilla chinensis plastome. The mononucleotide repeat unit C/G was also identified in all five species. Within the five plastomes, SSR loci were mainly located in IGS, followed by locations in CDS and introns. As expected, most SSRs were located in the LSC region, followed by SSC and IR regions.
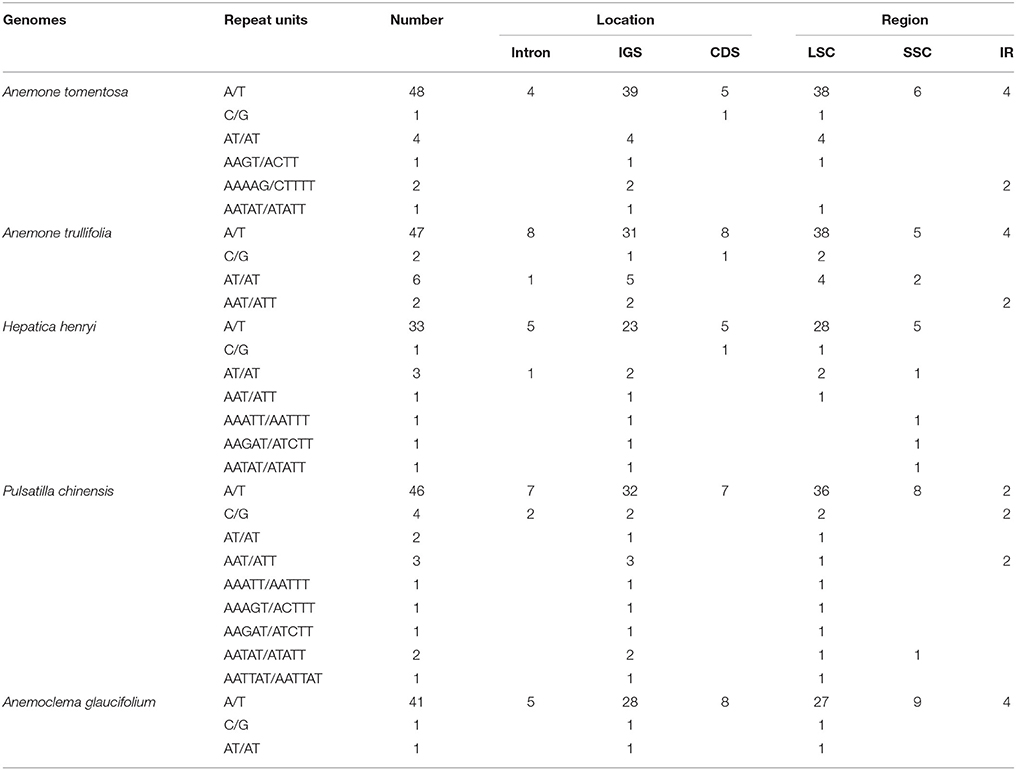
Table 4. Simple sequence repeats (SSRs) for five newly sequenced plastome samples.
Phylogenies, reconstructed with each data set and by both methods were consistent with each other and only differed for some nodes' supporting values. Because complete plastome sequence data provided the most robust phylogeny, we used this result (Figure 6). Excluding ambiguous aligments, the complete chloroplast genome data set alignment was 123,519 bp in length (including 16,916 informative characters). Only one parsimony tree with 58,536 steps was searched, along with a consistency index (CI) of 0.75, and a retention index (RI) of 0.75. The Bayesian analysis used partitioned substitution models checked by PartitionFinder. The length of each coding region, intron, intergenic spacer, and models of each partition subset tested by PartitionFinder are provided in the Supplementary Material.
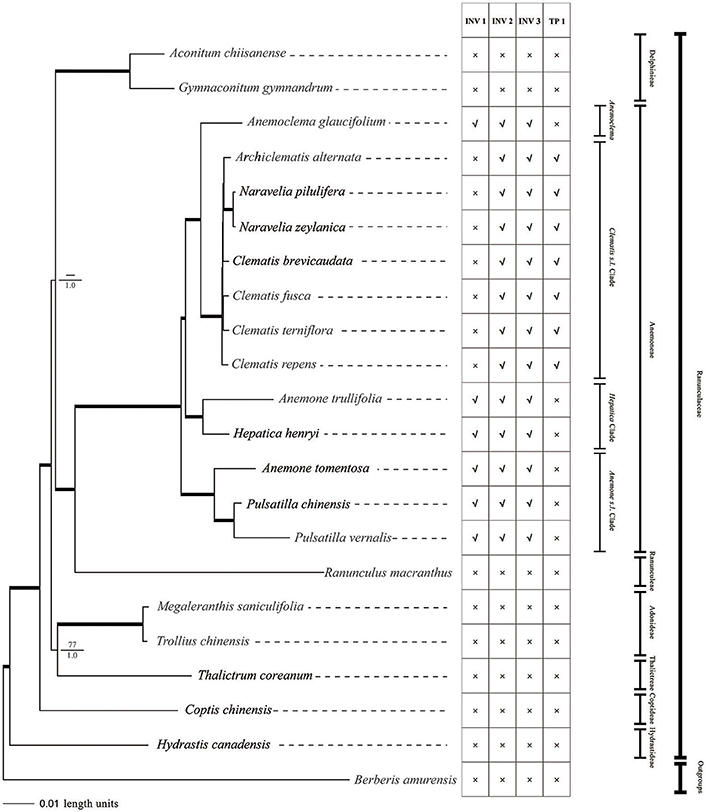
Figure 6. Phylogeny of Ranunculaceae species inferred from complete plastome sequences using MP and Bayesian methods. Bayesian phylograms are shown with MP bootstrap values/PP values for Bayesian analysis at each node. Internal branches which are fully supported by both analyses (with 100 bootstrap values and 1 Bayesian values) were thickened. Plastome Structural variations are also shown. INV, inversion; TP, transposition; ×, absent; √, present.
Parsimony analysis of the major Ranunculaceae clades did not resolve the sister relationship of Aconitum clade and Ranunculus + tribe Anemoneae clade. The Thalictrum, Trollius, and Megaleranthis clade was also not fully supported by MP analysis (Figure 6). The Bayesian analysis using partitioned models resolved all of the family's major clades. Except for the Clematis s.l. clade, all clades were fully supported (PP = 1) by the Bayesian method.
tribe Anemoneae was supported by our phylogenomic analyses and was closely related to Ranunculus. There was strong support for grouping Archiclematis, Clematis, and Naravelia as a sister clade to Anemoclema. Samples of Anemone, Pulsatilla, and Hepatica did not group as a clade but as a paraphyletic grade in tribe Anemoneae. The Hepatica henryi + Anemone trullifolia (sect. Omalocarpus) clade was a sister clade to Clematis + Anemoclema. Aneome tomentosa (sect. Rivularidium) + Pulsatilla clade was found to be the first diverged clade within the tribe.
We found two derived types of chloroplast genomes in tribe Anemoneae compared to other genera within Ranunculaceae, with one type (with two inversions and one transposition regions) in Clematis s.l. and the other (with three inversions) in the rest of the tribe's genera (Figures 1, 6). These gene rearrangements clearly bear important phylogenetic information. Two inversions (INV2 and INV3) in Clematis s.l. plastome were also present in Anemoclema, Anenome, Hepatica, and Pulsatilla plastomes. Thus, the presence of INV2 and INV3 could be considered as a synapomorphy of tribe Anemoneae. The largest inversion (INV 1) is present in Anemoclema, Anemone, Hepatica, and Pulsatilla plastomes, and these samples were paraphyletic to Clematis s.l. clade (Figure 6). This suggests that the presence of INV 1 may be a pleisiomorphy within the tribe. In contrast, the only transposition region (TP 1) present in the Clematis s.l. clade may represent a synapomorphy for Clematis, Archiclematis, and Naravelia. The phylogenomic results suggest that the ancestor of tribe Anemoneae may have a plastome sequence similar to that of Anemone, Anemoclema, Hepatica, and Pulsatilla which carried three inversions in its LSC regions. Two steps of gene rearrangements subsequently occurred in the plastome sequence of the Clematis s.l. ancestor. One of these was the loss of INV I, and the other was the addition of TP1.
Inversion and transposition events in the chloroplast genome may be triggered by tRNA activity (Hiratsuka et al., 1989; Walker et al., 2014) or intragenomic recombination at regions with variable G + C content (Fullerton et al., 2001; Smith et al., 2002). In this study, we evaluated the G + C content at the boundaries of each inversion and relocation (Table 3). The regions flanking inversions and relocations had lower G + C contents than non-coding regions of the whole plastome. In contrast, the flanks of all inversion/transposition regions had tRNA genes, as well as higher genetic variation (Figure 4). The tRNA activity, higher genetic variation, and lower G + C content present in flank regions could be the key factors promoting gene rearrangements in chloroplast genomes.
Although genome size and overall genomic structure are highly conserved in land plants, IR expansion/contraction is common in plastid genomes and is the main outcome of plastid gemone length variation in angiosperms (Kim and Lee, 2004). Gene duplications in plastid genomes are mainly caused by the expansion of the IR region to single copy regions (Goulding et al., 1996). Expansion events that result in the duplication of single genes, parts of genes, or several genes, have been documented in several plant taxa. This includes a 12-kb expansion in Nicotiana acuminata (Goulding et al., 1996), 4-kb expansion in Jasminum nudiflorum (Lee et al., 2007), and a remarkable 50-kb expansion in Pelargonium (Chumley et al., 2006). In Ranunculaceae, the termini of two genes, rps19 and trnH-GUG, were previously reported to have migrated into adjacent IRs (Park et al., 2015).
We observed an approximately 4.4 kb expansion of the IR toward the LSC region in tribe Anemoneae. This expansion caused duplicate copies of six ribosomal protein genes (rps8, rpl14, rpl16, rps3, rpl22, rps19). These are single genes in other genera of the family, such as Ranunculus, Thalictrum, Megaleranthis, Trollius, and Aconitum (Hoot and Palmer, 1994; Chen et al., 2015; Park et al., 2015), as well as most other angiosperms. For this reason, this IR expansion could also be considered as a synapomorphy of tribe Anemoneae.
Simple sequence repeats (SSRs), also known as microsatellites, are often used as genetic markers for population genetics studies. This is because they provide rich information for population genetics and evolutionary studies (Powell et al., 1995). However, plastid SSRs were rarely used in tribe Anemoneae. We identified 43 to 61 chloroplast SSRs in the five samples we evaluated (Table 4). Our results showed that Anemone tomentosa, Hepatica henryi, and Pulsatilla chinensis have pentanucleotide repeats in their plastomes, and that the Pulsatilla chinensis plastome has hexanucleotide repeats. The rich diversity of chloroplast SSR loci provides opportunities to survey the population genetic structure of those species.
All previous phylogenetic studies of Ranunculaceae were based on small numbers of DNA regions (Ro et al., 1997; Wang et al., 2009, 2016; Cossard et al., 2016), and there is a need for improved resolution of Ranunculaceae phylogeny needs to be further improved. In this study, phylogeny inferred from complete chloroplast genomic data was better resolved and more rigorous than previous studies (Figure 6), thereby demonstrating that plastome sequences may provide the ideal data sets for resolving family phylogenies. tribe Anemoneae was supported and tested to be sister to the genus Ranunculus, which is in agreement with morphological classifications (Tamura, 1995) and previous molecular phylogenetic studies (Wang et al., 2009, 2016; Cossard et al., 2016). Although the gene order of Anemoclema plastome was found to be identical with Anemone (Figure 1), this genus has a sister relationship to Clematis clade (Figure 6) as previously reported by molecular phylogenetic analyses (Zhang et al., 2015; Jiang et al., 2017a). Unlike results by Hoot et al. (2012), samples of Anemone, Hepatica, and Pulsatilla did not form a monophyletic group. Thus, our results did not supported classification by Hoot et al. (2012), which included Hepatica into Anemone s.l.
In this study, Hetapica henryi, and Anemone trullifolia (sect. Omalocarpus) are grouped together, whereas Pulsatilla chinensis and Anemone tomentosa (sect. Rivularidum) are grouped and these two clades were paraphyletic to the Anemoclema + Clematis clade (Figure 6). These results are somewhat similar to the phylogenetic analyses by Jiang et al. (2017a), but still different with their phylogenetic topology. In Jiang et al. (2017a), Pulsatilla + sect. Rivularidum clade was sister to Anemoclema + Clematis clade, and Hepatica + sect. Omalocarpus clade was outside. In the present study, Hepatica + sect. Omalocarpus clade was sisiter to Anemoclema + Clematis clade, whereas Pulsatilla + sect. Rivularidum clade was outside. Statistical support of sister relationshisp of Pulsatilla + sect. Rivularidum clade and Anemoclema + Clematis clade by Jiang et al. (2017a) was not very strong. However, the sister relationship of Hepatica + sect. Omalocarpus and Anemoclema + Clematis clade was fully supported by both MP and Bayesian analyses by plastome phylogenomic analyses in this study.
Within tribe Anemoneae, generic relationship within subtribe Clematidinae (sensu Tamura, 1995) was clear. All the small genera like Archiclematis and Naravelia should be included into Clematis s.l. (Miikeda et al., 2006; Xie et al., 2011; Zhang et al., 2015; Liu et al., 2018; Jiang et al., 2017a), and its sister relationship was supported by all the analyses (Zhang et al., 2015; Jiang et al., 2017a; and this study). However, subtribe Anemoninae (sensu Tamura, 1995) showed very complicated evolutionary patterns. Simply grouped together and treated them as Anemone s.l. should be reconsidered. According to this study, subtribe Anemoninae should be separated at least three genera (Anemoclema, Anemone s.l. including Pulsatilla and Pulsatilloides, and Hepatica including sect. Omalocarpus, sect. Anemonidium, and sect. Keiskea) as suggested by Jiang et al. (2017a), and Anemoclema is better to be treated as a member of subtribe Clematidinae.
HL and JH contributed equally. LX and JC initiated the project. LX, JH, HL, CD, and LP conceived and designed the experiments. HL, JH, and RL performed the experiments. JH and HL analyzed the data. HL, JH, and LX wrote the manuscript.
This study was supported by the Fundamental Research Funds for the Central Universities (2017PT05), the National Natural Science Foundation of China (grant no. 31670207), the Medium- and Long-term Scientific Study Projects for Young Teachers of Beijing Forestry University (grant no. 2015ZCQ-BH-03 to LX), and the Beijing Natural Science Foundation (grant no. 5182016).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We acknowledge Dr. Gregory Stull and Dr. Sun Miao for helping genomic assembling. We thank Dr. Xu Bo for kindly providing Anemoclema samples.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2018.01097/full#supplementary-material
Figure S1. MAUVE alignment of Ranunculaceae plastomes. Homologous regions are shown in the same color.
Table S1. Detailed informations of gaps between contigs in de novo assembly, and primers for Sanger sequencing that bridges the gaps.
Table S2. Results of substitution model and data partition by PartionFinder.
APG IV (2016). An update of the Angiosperm Phylogeny Group classification for the orders and families of flowering plants: APG IV. Bot. J. Linn. Soc. 181, 1–20. doi: 10.1111/boj.12385
Brudno, M., Do, C. B., Cooper, G. M., Kim, M. F., Davydov, E., Green, E. D., et al. (2003a). LAGAN and Multi-LAGAN: efficient tools for large-scale multiple alignment of genomic DNA outline of algorithms. Genome Res. 13, 721–731 doi: 10.1101/gr.926603
Brudno, M., Malde, S., Poliakov, A., Do, C. B., Couronne, O., Dubchak, I., et al. (2003b). Glocal alignment: finding rearrangements during alignment. Bioinformatics 19, i54–i62. doi: 10.1093/bioinformatics/btg1005
Chen, X., Li, Q., Li, Y., Qian, J., and Han, J. (2015). Chloroplast genome of Aconitum barbatum var. puberulum (Ranunculaceae) derived from CCS reads using the PacBio RS platform. Front. Plant Sci. 6:42. doi: 10.3389/fpls.2015.00042
Chumley, T. W., Palmer, J. D., Mower, J. P., Boore, J. L., Fourcade, H. M., Caile, P. J., et al. (2006). The completed chloroplast genome sequence of Pelargonium × hortorum: organization and evolution of the largest and most highly rearranged chloroplast genome of land plants. Mol. Biol. Evol. 23, 2175–2190. doi: 10.1093/molbev/msl089
Compton, J. A., Culham, A., and Jury, S. L. (1998). Reclassification of Actaea to include Cimicifuga and Souliea (Ranunculaceae): phylogeny inferred from morphology, nrDNA ITS, and cpDNA trnL-F sequence variation. Taxon 47, 593–634. doi: 10.2307/1223580
Compton, J. A., and Hedderson, T. A. (1997). A morphometric analysis of the Cimicifuga foetida L. complex (Ranunculaceae). Bot. J. Linn. Soc. 123, 1–23. doi: 10.1111/j.1095-8339.1997.tb01402.x
Cossard, G., Sannier, J., Sauquet, H., Damerval, C., de Craene, L. R., Jabbour, F., et al. (2016). Subfamilial and tribal relationships of Ranunculaceae: evidence from eight molecular markers. Plant Syst. Evol. 302, 419–431. doi: 10.1007/s00606-015-1270-6
Darling, A. E., Mau, B., and Perna, N. T. (2010). progressiveMauve: multiple genome alignment with gene gain, loss and rearrangement. PloS ONE 5:e11147. doi: 10.1371/journal.pone.0011147
Doyle, J. J., and Doyle, J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15.
Doyle, J. J., Doyle, J. L., Ballenger, J. A., and Palmer, J. D. (1996). The distribution and phylogenetic significance of a 50-kb chloroplast DNA inversion in the flowering plant family Leguminosae. Mol. Phylogenet. Evol. 5, 429–438. doi: 10.1006/mpev.1996.0038
Emadzade, K., Gehrke, B., Linder, H. P., and Hörandl, E. (2011). The biogeographical history of the cosmopolitan genus Ranunculus L.(Ranunculaceae) in the temperate to meridional zones. Mol. Phylogenet. Evol. 58, 4–21. doi: 10.1016/j.ympev.2010.11.002
Emadzade, K., Lehnebach, C., Lockhart, P., and Hörandl, E. (2010). A molecular phylogeny, morphology and classification of genera of Ranunculeae (Ranunculaceae). Taxon 59, 809–828. doi: 10.2307/25677670
Falck, D., and Lehtonen, S. (2014). Two new names in Clematis (Ranunculaceae). Phytotaxa 163, 58. doi: 10.11646/phytotaxa.163.1.7
Frazer, K. A., Pachter, L., Poliakov, A., Rubin, E. M., and Dubchak, I. (2004). VISTA: computational tools for comparative genomics. Nucleic Acids Res. 32(Suppl. 2), W273–W279. doi: 10.1093/nar/gkh458
Fullerton, S. M., Bernardo Carvalho, A., and Clark, A. G. (2001). Local rates of recombination are positively correlated with G + C content in the human genome. Mol. Biol. Evol. 18, 1139–1142. doi: 10.1093/oxfordjournals.molbev.a003886
Gish, W., and States, D. J. (1993). Identification of protein coding regions by database similarity search. Nat. Genet. 3, 266–272. doi: 10.1038/ng0393-266
Goulding, S. E., Olmstead, R. G., Morden, C. W., and Wolfe, K. H. (1996). Ebb and flow of the chloroplast inverted repeat. Mol. Gen. Genet. 252, 195–206. doi: 10.1007/BF02173220
Hiratsuka, J., Shimada, H., Whittier, R., Ishibashi, T., Sakamoto, M., Mori, M., et al. (1989). The complete sequence of the rice (Oryza sativa) chloroplast genome: intermolecular recombination between distinct tRNA genes accounts for a major plastid DNA inversion during the evolution of the cereals. Mol. Gen. Genet. 217, 185–194. doi: 10.1007/BF02464880
Hoot, S. B., Meyer, K. M., and Manning, J. C. (2012). Phylogeny and reclassification of Anemone (Ranunculaceae), with an emphasis on austral species. Syst. Bot. 37, 139–152. doi: 10.1600/036364412X616729
Hoot, S. B., and Palmer, J. D. (1994). Structural rearrangements, including parallel inversions, within the chloroplast genome of Anemone and related genera. Mol. Evol. 38, 274–281. doi: 10.1007/BF00176089
Hörandl, E., Greilhuber, J., Klímová, K., Paun, O., Temsch, E., and Emadzade, K. (2009). Reticulate evolution and taxonomic concepts in the Ranunculus auricomus complex (Ranunculaceae): insights from analysis of morphological, karyological and molecular data. Taxon, 58, 1194–1216.
Huang, D. I., and Cronk, Q. C. (2015). Plann: a command-line application for annotating plastome sequences. Appli. Plant Sci. 3:1500026. doi: 10.3732/apps.1500026
Jabbour, F., and Renner, S. S. (2012). A phylogeny of Delphinieae (Ranunculaceae) shows that Aconitum is nested within Delphinium and that Late Miocene transitions to long life cycles in the Himalayas and Southwest China coincide with bursts in diversification. Mol. Phylogenet. Evol. 62, 928–942. doi: 10.1016/j.ympev.2011.12.005
Jansen, R. K., Raubeson, L. A., Boore, J. L., Chumley, T. W., Haberle, R. C., Wyman, S. K., et al. (2005). Methods for obtaining and analyzing whole chloroplast genome sequences. Methods Enzymol. 395, 348–384. doi: 10.1016/S0076-6879(05)95020-9
Jansen, R. K., Wojciechowski, M. F., Sanniyasi, E., Lee, S. B., and Daniell, H. (2008). Complete plastid genome sequence of the chickpea (Cicer arietinum) and the phylogenetic distribution of rps12 and clpP intron losses among legumes (Leguminosae). Mol. Phylogenet. Evol. 48, 1204–1217. doi: 10.1016/j.ympev.2008.06.013
Jiang, N., Zhou, Z., Yang, J. B., and Yu, W. B. (2017b). Complete chloroplast genome of Anemoclema glaucifolium (Ranunculaceae), a vulnerable and threatened species endemic to the Hengduan Mountains. Conserv. Genet. Resour. 2017, 1–4. doi: 10.1007/s12686-017-0874-2
Jiang, N., Zhou, Z., Yang, J. B., Zhang, S. D., Guan, K. Y., Tan, Y. H., et al. (2017a). Phylogenetic reassessment of tribe Anemoneae (Ranunculaceae): non-monophyly of Anemone s.l. revealed by plastid datasets. PloS ONE 12:e0174792. doi: 10.1371/journal.pone.0174792
Johansson, J. T. (1999). There large inversions in the chloroplast genomes and one loss of the chloroplast generps16 suggest an early evolutionary split in the genus Adonis (Ranunculaceae). Plant Syst. Evol. 218, 133–143. doi: 10.1007/BF01087041
Katoh, K., Kuma, K., Toh, H., and Miyata, T. (2005). MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Res. 33, 511–518. doi: 10.1093/nar/gki198
Kearse, M., Moir, R., Wilson, A., Stones-Havas, S., Cheung, M., Sturrock, S., et al. (2012). Geneious Basic: an integrated and extendable desktop software platform for the organization and analysis of sequence data. Bioinformatics 28, 1647–1649. doi: 10.1093/bioinformatics/bts199
Kim, K. J., and Lee, H. L. (2004). Complete chloroplast genome sequences from Korean ginseng (Panax schinseng Nees) and comparative analysis of sequence evolution among 17 vascular plants. DNA Res. 11, 247–261. doi: 10.1093/dnares/11.4.247
Kim, Y. K., Park, C. W., and Kim, K. J. (2009). Complete chloroplast DNA sequence from a Korean endemic genus, Megaleranthis saniculifolia, and its evolutionary implications. Mol. cells 27, 365–381. doi: 10.1007/s10059-009-0047-6
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Kurtz, S., Choudhuri, J. V., Ohlebusch, E., Schleiermacher, C., Stoye, J., and Giegerich, R. (2001). REPuter: the manifold applications of repeat analysis on a genomic scale. Nucleic Acids Res. 29, 4633–4642. doi: 10.1093/nar/29.22.4633
Lanfear, R., Frandsen, P. B., Wright, A. M., Senfeld, T., and Calcott, B. (2016). PartitionFinder 2: new methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 34, 772–773. doi: 10.1093/molbev/msw260
Lee, H. L., Jansen, R. K., Chumley, T. W., and Kim, K. J. (2007). Gene relocations within chloroplast genomes of Jasminum and Menodora (Oleaceae) are due to multiple, overlapping inversions. Mol. Biol. Evol. 24, 1161–1180. doi: 10.1093/molbev/msm036
Lehtonen, S., Christenhusz, M. J., and Falck, D. (2016). Sensitive phylogenetics of Clematis and its position in Ranunculaceae. Bot. J. Linn. Soc. 182, 825–867. doi: 10.1111/boj.12477
Li, M., Yang, B., Chen, Q., Zhu, W., Ma, J., and Tian, J. (2016). The complete chloroplast genome sequence of Clematis terniflora DC.(Ranunculaceae). Mitochondrial DNA Part A 27, 2470–2472. doi: 10.3109/19401736.2015.1033702
Librado, P., and Rozas, J. (2009). DnaSP v5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Lim, C. E., Kim, G. B., Baek, S., Han, S. M., Yu, H. J., and Mun, J. H. (2017). The complete chloroplast genome of Aconitum chiisanense Nakai (Ranunculaceae). Mitochondrial DNA Part A 28, 75–76. doi: 10.3109/19401736.2015.1110805
Liu, H. J., Ding, C. H., He, J., Cheng, J., Pei, L. Y., and Xie, L. (2018). Complete chloroplast genomes of Archiclematis, Naravelia and Clematis (Ranunculaceae), and their phylogenetic implications. Phytotaxa, 343, 214–226. doi: 10.11646/phytotaxa.343.3.2
Lohse, M., Drechsel, O., Kahlau, S., and Bock, R. (2013). OrganellarGenomeDRAW-a suite of tools for generating physical maps of plastid and mitochondrial gemones and visualizing expression data sets. Nucleic Acids Res. 41, W575–W581. doi: 10.1093/nar/gkt289
Ma, P. F., Zhang, Y. X., Zeng, C. X., Guo, Z. H., and Li, D. Z. (2014). Chloroplast phylogenomic analyses resolve deep-level relationships of an intractable bamboo tribe Arundinarieae (Poaceae). Syst. Biol. 63, 933–950. doi: 10.1093/sysbio/syu054
Mardanov, A. V., Ravin, N. V., Kuznetsov, B. B., Samigullin, T. H., Antonov, A. S., Kolganova, T. V., et al. (2008). Complete sequence of the duckweed (Lemna minor) chloroplast genome: structural organization and phylogenetic relationships to other angiosperms. J. Mol. Evol. 66, 555–564. doi: 10.1007/s00239-008-9091-7
Miikeda, O., Kita, K., Handa, T., and Yukawa, T. (2006). Phylogenetic relationships of Clematis (Ranunculaceae) based on chloroplast and nuclear DNA sequences. Bot. J. Linn. Soc. 152, 153–168. doi: 10.1111/j.1095-8339.2006.00551.x
Moore, M. J., Soltis, P. S., Bell, C. D., Burleigh, J. G., and Soltis, D. E. (2010). Phylogenetic analysis of 83 plastid genes further resolves the early diversification of eudicots. Proc. Natl. Acad. Sci. U.S.A. 107, 4623–4628. doi: 10.1073/pnas.0907801107
Ogihara, Y., Terachi, T., and Sasakuma, T. (1988). Intramolecular recombination of chloroplast genome mediated by short direct-repeat sequences in wheat species. Proc. Natl. Acad. Sci. U.S.A. 85, 8573–8577. doi: 10.1073/pnas.85.22.8573
Palmer, J. D., Nugent, J. M., and Herbon, L. A. (1987). Unusual structure of Geranium chloroplast DNA: a triple-sized inverted repeat, extensive gene duplications, multiple inversions, and two repeat families. Proc. Natl. Acad. Sci. U.S.A. 84, 769–773. doi: 10.1073/pnas.84.3.769
Park, I., Kim, W. J., Yang, S., Yeo, S. M., Li, H., and Moon, B. C. (2017). The complete chloroplast genome sequence of Aconitum coreanum and Aconitum carmichaelii and comparative analysis with other Aconitum species. PloS ONE 12:e0184257. doi: 10.1371/journal.pone.0184257
Park, K. T., and Park, S. (2016). Complete chloroplast genome of Clematis fusca var. coreana (Ranunculaceae). Mitochondrial DNA Part A 27, 4056–4058. doi: 10.3109/19401736.2014.1003841
Park, S., Jansen, R. K., and Park, S. (2015). Complete plastome sequence of Thalictrum coreanum (Ranunculaceae) and transfer of the rpl32 gene to the nucleus in the ancestor of the subfamily Thalictroideae. BMC Plant Biol. 15:40. doi: 10.1186/s12870-015-0432-6
Posada, D., and Buckley, T. R. (2004). Model selection and model averaging in phylogenetics: advantages of Akaike information criterion and Bayesian approaches over likelihood ratio tests. Syst. Biol. 53, 793–808. doi: 10.1080/10635150490522304
Powell, W., Morgante, M., McDevitt, R., Vendramin, G. G., and Rafalski, J. A. (1995). Polymorphic simple sequence repeat regions in chloroplast genomes: applications to the population genetics of pines. Proc. Natl. Acad. Sci. U.S.A. 92, 7759–7763. doi: 10.1073/pnas.92.17.7759
Raubeson, L. A., Peery, R., Chumley, T. W., Dziubek, C., Fourcade, H. M., et al. (2007). Comparative chloroplast genomics: analyses including new sequences from the angiosperms Nuphar advena and Ranunculus macranthus. BMC Genomics 8:174. doi: 10.1186/1471-2164-8-174
Ro, K. E., Keener, C. S., and McPheron, B. A. (1997). Molecular phylogenetic study of the Ranunculaceae: utility of the nuclear 26S ribosomal DNA in inferring intrafamilial relationships. Mole. Phylogen. Evol. 8, 117–127. doi: 10.1006/mpev.1997.0413
Ronquist, F., and Huelsenbeck, J. P. (2003). MrBayes 3: bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Shaw, J., Lickey, E. B., Schilling, E. E., and Small, R. L. (2007). Comparison of whole chloroplast genome sequences to choose noncoding regions for phylogenetic studies in angiosperms: the tortoise and the hare III. Am. J. Bot. 94, 275–288. doi: 10.3732/ajb.94.3.275
Smith, N. G., Webster, M. T., and Ellegren, H. (2002). Deterministic mutation rate variation in the human genome. Genome Res. 12, 1350–1356. doi: 10.1101/gr.220502
Sun, M., Li, J., Li, D., and Shi, L. (2017). Complete chloroplast genome sequence of the medical fern Drynaria roosii and its phylogenetic analysis. Mitochondrial DNA Part B 2, 7–8. doi: 10.1080/23802359.2016.1275835
Szczecinska, M., Lazarski, G., Bilska, K., and Sawicki, J. (2017). The complete plastid genome and nuclear genome markers provide molecular evidence for the hybrid origin of Pulsatilla × hackelii Pohl. Turk. J. Bot. 41, 329–337. doi: 10.3906/bot-1610-28
Tamura, M. (1993). “Ranunculaceae,” in The Families and Genera of Vascular Plants II, eds K. Kubitzki, J. G. Rohwer, V, Bittrich (Berlin: Springer), 563–583. doi: 10.1007/978-3-662-02899-5_67
Tamura, M. (1995). “Clematis L,” in Engler's Die Natürlichen Pflanzenfamilien 2nd Edn, ed P. Heipko (Berlin: Duncker & Humblot), 368–387.
Tangphatsornruang, S., Sangsrakru, D., Chanprasert, J., Uthaipaisanwong, P., Yoocha, T., Jomchai, N., et al. (2009). The chloroplast genome sequence of mungbean (Vigna radiata) determined by high-throughput pyrosequencing: structural organization and phylogenetic relationships. DNA Res. 17, 11–22. doi: 10.1093/dnares/dsp025
Tangphatsornruang, S., Uthaipaisanwong, P., Sangsrakru, D., Chanprasert, J., Yoocha, T., Jomchai, N., et al. (2011). Characterization of the complete chloroplast genome of Hevea brasiliensis reveals genome rearrangement, RNA editing sites and phylogenetic relationships. Gene 475, 104–112. doi: 10.1016/j.gene.2011.01.002
Thiel, T., Michalek, W., Varshney, R. K., and Graner, A. (2003). Exploiting EST databases for the development and characterization of gene-derived SSR-markers in barley (Hordeum vulgare L.). Theor. Appl. Genet. 106, 411–422. doi: 10.1007/s00122-002-1031-0
Walker, J. F., Jansen, R. K., Zanis, M. J., and Emery, N. C. (2015). Sources of inversion variation in the small single copy (SSC) region of chloroplast genomes. Amer. J. Bot. 102, 1–2. doi: 10.3732/ajb.1500299
Walker, J. F., Zanis, M. J., and Emery, N. C. (2014). Comparative analysis of complete chloroplast genome sequence and inversion variation in Lasthenia burkei (Madieae, Asteraceae). Amer. J. Bot. 101, 722–729. doi: 10.3732/ajb.1400049
Wang, W., Dilcher, D. L., Sun, G., Wang, H. S., and Chen, Z. D. (2016). Accelerated evolution of early angiosperms: evidence from ranunculalean phylogeny by integrating living and fossil data. J. Syst. Evol. 54, 336–341. doi: 10.1111/jse.12090
Wang, W., Hu, H., Xiang, X. G., Yu, S. X., and Chen, Z. D. (2010). Phylogenetic placements of Calathodes and Megaleranthis (Ranunculaceae): Evidence from molecular and morphological data. Taxon 59, 1712–1720.
Wang, W., Li, R. Q., and Chen, Z. D. (2005). Systematic position of Asteropyrum (Ranunculaceae) inferred from chloroplast and nuclear sequences. Plant Syst. Evol. 255, 41–54. doi: 10.1007/s00606-005-0339-z
Wang, W., Liu, Y., Yu, S. X., Gao, T. G., and Chen, Z. D. (2013). Gymnaconitum, a new genus of Ranunculaceae endemic to the Qinghai-Tibetan Plateau. Taxon 62, 713–722. doi: 10.12705/624.10
Wang, W., Lu, A. M., Ren, Y., Endress, M. E., and Chen, Z. D. (2009). Phylogeny and classification of Ranunculales: evidence from four molecular loci and morphological data. Perspect. Plant Ecol. 11, 81–110. doi: 10.1016/j.ppees.2009.01.001
Warren, D. L., Geneva, A. J., and Lanfear, R. (2017). RWTY (R We There Yet): An R package for examining convergence of Bayesian phylogenetic analyses. Mol. Biol. Evol. 34, 1016–1020. doi: 10.1093/molbev/msw279
Weitemier, K., Straub, S. C., Cronn, R. C., Fishbein, M., Schmickl, R., McDonnell, A., et al. (2014). Hyb-Seq: combinging target enrichment and genome skimming for plant phylogenomics. Appl. Plant Sci. 2:1400042. doi: 10.3732/apps.1400042
Wolfe, K. H., Li, W. H., and Sharp, P. M. (1987). Rates of nucleotide substitution vary greatly among plant mitochondrial, chloroplast, and nuclear DNAs. Proc. Natl. Acad. Sci. U.S.A. 84, 9054–9058. doi: 10.1073/pnas.84.24.9054
Wyman, S. K., Jansen, R. K., and Boore, J. L. (2004). Automatic annotation of organellar genomes with DOGMA. Bioinformatics 20, 3252–3255. doi: 10.1093/bioinformatics/bth352
Xie, L., Wen, J., and Li, L. Q. (2011). Phylogenetic analyses of Clematis (Ranunculaceae) based on sequences of nuclear ribosomal ITS and three plastid regions. Syst. Bot. 36, 907–921. doi: 10.1600/036364411X604921
Yan, M., Moore, M. J., Meng, A., Yao, X., and Wang, H. (2017). The first complete plastome sequence of the basal asterid family Styracaceae (Ericales) reveals a large inversion. Plant Syst. Evol. 303, 61–70. doi: 10.1007/s00606-016-1352-0
Keywords: chloroplast genome, inversion, IR expansion, phylogenomics, Ranunculaceae, transposition, Tribe Anemoneae
Citation: Liu H, He J, Ding C, Lyu R, Pei L, Cheng J and Xie L (2018) Comparative Analysis of Complete Chloroplast Genomes of Anemoclema, Anemone, Pulsatilla, and Hepatica Revealing Structural Variations Among Genera in Tribe Anemoneae (Ranunculaceae). Front. Plant Sci. 9:1097. doi: 10.3389/fpls.2018.01097
Received: 03 January 2018; Accepted: 09 July 2018;
Published: 27 July 2018.
Edited by:
Elena M. Kramer, Harvard University, United StatesReviewed by:
Wen-Bin Yu, Xishuangbanna Tropical Botanical Garden (CAS), ChinaCopyright © 2018 Liu, He, Ding, Lyu, Pei, Cheng and Xie. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lei Xie, eGllbGVpX3NpQDEyNi5jb20=
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.