 Azman H. Farah
Azman H. Farah Shiou Yih Lee
Shiou Yih Lee Zhihui Gao
Zhihui Gao Tze Leong Yao
Tze Leong Yao Maria Madon
Maria Madon Rozi Mohamed
Rozi Mohamed
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 29 May 2018
Sec. Evolutionary and Population Genetics
Volume 9 - 2018 | https://doi.org/10.3389/fpls.2018.00712
The tribe Aquilarieae of the family Thymelaeaceae consists of two genera, Aquilaria and Gyrinops, with a total of 30 species, distributed from northeast India, through southeast Asia and the south of China, to Papua New Guinea. They are an important botanical resource for fragrant agarwood, a prized product derived from injured or infected stems of these species. The aim of this study was to estimate the genome size of selected Aquilaria species and comprehend the evolutionary history of Aquilarieae speciation through molecular phylogeny. Five non-coding chloroplast DNA regions and a nuclear region were sequenced from 12 Aquilaria and three Gyrinops species. Phylogenetic trees constructed using combined chloroplast DNA sequences revealed relationships of the studied 15 members in Aquilarieae, while nuclear ribosomal DNA internal transcribed spacer (ITS) sequences showed a paraphyletic relationship between Aquilaria species from Indochina and Malesian. We exposed, for the first time, the estimated divergence time for Aquilarieae speciation, which was speculated to happen during the Miocene Epoch. The ancestral split and biogeographic pattern of studied species were discussed. Results showed no large variation in the 2C-values for the five Aquilaria species (1.35–2.23 pg). Further investigation into the genome size may provide additional information regarding ancestral traits and its evolution history.
Accommodating about 800 species worldwide, the family Thymelaeaceae was given its name by the French botanist Michel Adanson in 1763 (Adanson, 1763). At present, the family is divided into subfamilies Octolepidoidae and Thymelaeoideae, with the former having two groups, Gonystylus and Octolepis, and the latter having three tribes, Aquilarieae, Daphneae, and Synandrodaphneae (Herber, 2003). In general, Thymelaeaceae is a cosmopolitan family consisting of 45 genera, including two well-known agarwood-producing genera, Aquilaria Lam. and Gyrinops Gaertn., both in the tribe Aquilarieae. There are 21 accepted species in the genus Aquilaria and nine in the genus Gyrinops (The Plant List, 2013), of which 13 from Aquilaria (A. baillonii, A. beccariana, A. crassna, A. filaria, A. hirta, A. khasiana, A. malaccensis, A. microcarpa, A. rostrata, A. rugosa, A. sinensis, A. subintegra, and A. yunnanensis.) and five from Gyrinops (G. caudata, G. ledermannii, G. salicifolia, G. versteegii, and G. walla), reportedly produce agarwood (Mulyaningsih and Yamada, 2007; Lee and Mohamed, 2016b). In the natural environment, it takes several years for a wild tree to form agarwood after damage from a natural catastrophe, pest, or insect disturbance and microorganism colonization (Rasool and Mohamed, 2016). Some of the major uses of agarwood include its incorporation into perfumes, incenses, and traditional medicines. Although agarwood has been cultivated for at least two decades, the high demand for natural agarwood in the market continues to threaten the survival of wild populations. Due to overwhelming illegal harvesting, these species are currently protected and listed as endangered in Appendix II of the Convention on International Trade in Endangered Species of Wild Fauna and Flora (CITES, 2013).
Of the many agarwood species, only a few have been cultivated intensively, including A. crassna, A. filaria, A. malaccensis, A. sinensis, A. subintegra, and G. versteegii. However, basic information such as genome size and chromosome count, which are important for breeding efforts, are lacking for these non-model plant species. Generally, the chromosome number for the family Thymelaeaceae is reported as x = 9 (Herber, 2003), however, variations exist depending on the species. The chromosome count for A. agallocha (synonym A. malaccensis) has been determined as x = 8 (2n = 16) and the karyotype formula as 2n = 2x = 16 = 10m + 6sm (Debnath et al., 1995), where ‘n’ is chromosome count, ‘x’ is the basic number, ‘m’ is metacentric, and ‘sm’ is submetacentric. There are two karyotypes for A. sinensis from China; the 2A type (2n = 2x = 16 = 10m +4sm + 2st) (Huang, 2009) is considered more primitive than the 2B type (2n = 16 = 6m + 6sm + 4st) (Shen et al., 2009), where ‘st’ is subtelocentric. From karyotype analysis, A. agallocha appears more primitive than A. sinensis (Zou et al., 2013). Aquilaria species are generally diploid, however, polyploids have been shown as possible through colchicine treatment in vitro. Naturally, A. malaccensis is 2x = 14, but induced tetraploids had double the chromosome number (4x = 28) (Siti Suhaila et al., 2015). There are also variations in genome size. For example, the genome size estimation for A. agallocha of Myanmar origin is 2C = 1.51 pg (Chen C.H. et al., 2014) while the estimation for A. malaccensis of Peninsular Malaysia is 2C = 1.86 ± 0.02 pg (Siti Suhaila et al., 2013). The estimation of C-value data for a certain plant family could contribute toward understanding the biological diversity significance and evolution of the family (Ng et al., 2017). Although the plant genome size database has been constantly increasing, information on genome size for tropical woody angiosperms is still lacking.
Several molecular phylogenetic studies have been carried out to investigate the broad scale genetic relationships in Thymelaeaceae. Molecular data from combined chloroplast locus rbcL and intergenic spacer region trnL-trnF are found insufficient to clarify relatedness of all the genera (Van der Bank et al., 2002). However, by adding the nuclear internal transcribed spacer (ITS) and multiplying the sampling size, Beaumont et al. (2009) increased the molecular data; the results support the taxonomy classification proposed by Herber (2003; Van der Bank et al., 2002). For its constituent genera in Thymelaeaceae, several molecular phylogenies have been reported: Aquilaria (Eurlings and Gravendeel, 2005; Lee and Mohamed, 2016a), Dirca (Schrader and Graves, 2004; Floden et al., 2009), Gnidia (Beaumont et al., 2009), Gyrinops (Eurlings and Gravendeel, 2005), Lachnaea (Robinson, 2008), Passerina (Van Niekerk, 2008), Pimelea (Motsi et al., 2010; Squire, 2016), Thecanthes (Motsi et al., 2010), and Thymelaeae (Galicia, 2006). Generally, Gyrinops is regarded as a sister to Aquilaria, and both genera are under the tribe Aquilarieae (Herber, 2003), nonetheless, its taxonomy category is still under debate. Some studies have suggested that Gyrinops should be synonymized with Aquilaria, because the sole delineating characteristic – the number of stamens – is not a strong taxonomic character (Eurlings and Gravendeel, 2005; Lee and Mohamed, 2016b).
Here, we constructed two phylogenetic trees from 15 different species in the tribe Aquilarieae, consisting of two genera, Aquilaria and Gyrinops, based on a combination of chloroplast DNA (cpDNA) matK, rbcL, trnL intron, trnL-trnF and psbC-trnS sequences, and the nuclear ribosomal DNA (nrDNA) ITS region, and reported the genome size of five commonly available Aquilaria species using flow cytometry. Our analysis is the first that provides estimation on the time when genetic divergence occurred in the tribe Aquilarieae.
Fresh young leaves from 5-year-old Aquilaria species: A. hirta, A. malaccensis, A. microcarpa, A. sinensis, and A. subintegra, were collected from lateral branches and immediately processed. Trees were grown in polybags in the plant nursery of the Faculty of Forestry, Universiti Putra Malaysia (UPM), Serdang, Malaysia. Identities of these species were authenticated in our previous studies (Lee and Mohamed, 2016a; Lee et al., 2016). A total of five pieces of leaves were collected from three individual plants to represent biological replicates of each species. For use as an external reference standard, oil palm (Elaeis guineensis, Arecaceae, 2C = 3.98 pg) (Madon et al., 2008) leaf sample was collected from trees growing in the arboretum of the Malaysian Palm Oil Board, Bandar Baru Bangi, Kajang, Malaysia. A 1 cm2 fragment was aseptically excised from the leaf using a sterile scalpel and directly transferred to prepare the nuclei for flow cytometry.
Using a sharp razor blade, approximately 1 cm2 of the leaf fragment was finely chopped in a Petri dish containing 1 mL of Otto I buffer (Dolezel et al., 2007). Subsequently, 500 μL of the same buffer was added and the suspension was filtered through a 40 μm nylon mesh and transferred into a Falcon tube. The nuclei suspension was mixed with 2 mL of Otto II buffer containing 1 μL of 50 μg mL-1 RNase A and 50 μm mL-1 propidium iodide (Dolezel et al., 2007). The stained nuclei suspension was then filtered through a 50 μm nylon mesh and incubated for 24 h at 4°C. The fluorescent nuclei were analyzed using a flow cytometer (FACSCalibur, Becton Dickinson) equipped with a 488 nm argon ion laser and 1,088 channels. The software CellQuest was used to measure nuclei images and the data was analyzed in the form of histogram peaks with a coefficient of variation (CV) generated from approximately 5,000 nuclei for each technical replicate. There were five technical replicates and three biological replicates for each species. Data collected included genome size values (2C) and the means were analyzed using an analysis of variance (ANOVA) and tested with Tukey HSD post hoc tests. P < 0.05 indicated significant differences.
For sequence amplification, 15 species (Table 3) were selected based on prior knowledge of their identities. A. beccariana, A. hirta, A. malaccensis, and A. rostrata were part of our own field collection (Lee and Mohamed, 2016a; Lee et al., 2016, 2017), while other samples were donated as follows: A. sinensis and A. yunnanensis from Prof. Jianhe Wei [Institute of Medicinal Plant Development (IMPLAD), China], A. crassna and A. subintegra from Dr. Mohd Noor Mahat [Forest Research Institute Malaysia (FRIM), Malaysia], A. rugosa from Prof. Arunrat Chaveerach [Khon Kaen University (KKU), Thailand], A. cumingiana, A. microcarpa, G. caudate, and G. versteegii from Dr. Maman Turjaman [Forestry and Environment Research, Development and Innovation Agency (FOERDIA), Indonesia]. A. agallocha and G. walla were obtained from planted sources by traders.
Other plant materials were acquired as follows: Gonystylus bancanus and Phaleria macrocarpa were collected from a planted tree in the arboretum of the Faculty of Forestry, UPM. Wikstroemia ridleyi was collected as a plant specimen during a field expedition in Terengganu, Malaysia. Aquilaria agallocha is synonym to A. malaccensis at present, with a few morphological differences which were exempted. However, in order to aid our discussion, we treated A. agallocha and A. malaccensis separately, with the former confined to the northeast of the Indian continent and the latter to the Malesian region. Whenever possible, fresh leaves were used for genomic DNA extraction, while for donated samples, leaves were oven-dried at 60°C overnight, prior to transport. Voucher specimens were prepared and kept in the Forest Biotechnology Lab, Faculty of Forestry, UPM.
Genomic DNA was extracted using the FavorprepTM Plant Genomic DNA Extraction Mini Kit (Favorgen, Taiwan) according the manufacturer’s suggested protocol. The isolated DNA was quantified using a Nanophotometer (Implen, Germany). A total of five chloroplast DNA (cpDNA) regions—two coding cpDNA loci, matK and rbcL; the trnL intron; and two non-coding cpDNA intergenic spacer loci, trnL-trnF and psbC-trnS—and an nrDNA ITS region were amplified (Table 2). PCR was conducted in a final reaction volume of 25 μL, containing 12.5 μL of 2x PCRBIO Taq Mix Red (PCRBiosystems, United Kingdom), 10 mM of each primer, and 20 ng of genomic DNA as a template. PCR amplification was conducted in a MyCyclerTM thermal cycler system (Bio-Rad, United States). PCR conditions (Lee et al., 2016) and annealing temperatures for each primer set are shown in Supplementary Table S1. PCR products were visualized in 1% agarose gel prior to direct DNA sequencing (ABI PRISM 3730xl Genetic Analyzer, Applied Biosystems, United States), performed by the First Base Laboratory Sdn. Bhd., Malaysia. In the case where targeted sequences were available in the GenBank database through previous studies of the same plant specimen, the records were included in this study without performing additional PCR and sequencing (Table 3).
Five cpDNA and one nrDNA sequences from each species were edited to remove primer sequences, and then the cpDNA sequences were combined into a contiguous sequence in the order of matK, rbcL, trnL intron, trnL-trnF, and psbC-trnS, resulting in a sequence 3,285–3,304 bp long; while the ITS sequence was 786–792 bp long. The length of the alignment generated using MUSCLE was 3,323 bp for the combined cpDNA sequences and 795 bp for the ITS sequence (Edgar, 2004). The number of indels and substitutions were calculated using MEGA 7 software (Kumar et al., 2016). A DNA substitution model, the jModelTest version 2.1.4 (Guindon and Gascuel, 2003; Darriba et al., 2012) was selected to assess our data set by implementing the hierarchical likelihood ratio test. According to the Akaike Information Criterion (AIC), the model best fitting the observed data for the combined cpDNA sequence was the general time reversible (GTR) model and invariable site (+I) (=GTR+I); while the nrDNA ITS sequence correlated best to the Tamura and Nei (TN93) model and gamma distributed (+G) (=TN93+G). Maximum likelihood (ML) trees were constructed using default parameters in MEGA 7, with 1,000 bootstrap replicates for each individual clade. Gaps and missing data were treated as complete deletions in this analysis. Gonystylus bancanus, P. macrocarpa, and W. ridleyi were used as an outgroup to root both trees before divergence times were estimated.
The evolution tree was constructed using the phylogenetic tree derived from the combined cpDNA sequences. There are no useful reports in the fossil evidence about Aquilarieae, therefore pairwise divergence time was conducted using fossil records from Cistaceae, a sister family that was recorded closest to Thymelaeaceae, using the TimeTree program (Hedges et al., 2006). The divergence time estimation was conducted using the Bayesian method implemented in the MCMCTREE v4.0 program in the PAML package (Yang, 2007). A Markov Chain Monte Carlo (MCMC) analysis was carried out based on the predicted divergence time calculated from the TimeTree program using the parameter: –rootage 102 –clock 2 –alpha 0.020000.
The isolation of nuclei suspensions from five Aquilaria species was successful following the method delineated by Dolezel et al. (2007). The histogram of the fluorescence intensity outputs (Figure 1) had sharp peaks at the G1 stage of the cell cycle and the CV values were less than 3%. A CV of <5% is adequate, while a CV above this threshold suggests the presence of polyphenols, dried-silica, or debris contamination, making the sample unacceptable (Dolezel et al., 2007; Pellicer and Leitch, 2014). Generally, Aquilaria species are diploid (Siti Suhaila et al., 2013; Chen C.H. et al., 2014). The 2C genome size values for the selected Aquilaria spp. were in the range of 1.35–2.23 pg (Table 1). There was only a 1.65-fold difference between the largest genome size (A. microcarpa) and the smallest (A. subintegra). Aquilaria malaccensis and A. sinensis had significantly larger genome sizes than did A. subintegra (Tukey test, P < 0.05), while A. microcarpa and A. hirta had similar sized genomes. Based on this result, the selection of the tips of growing shoots in the self-renewing region of Aquilaria plants is suitable for flow cytometry analysis. It would be ideal to estimate the genome size for other species in the genus to have a better idea of the genome size variation, however, it was impossible to get specimens in the form of fresh young shoots for many of the species for this purpose, as many of the trees are not accessible or have unknown locations.
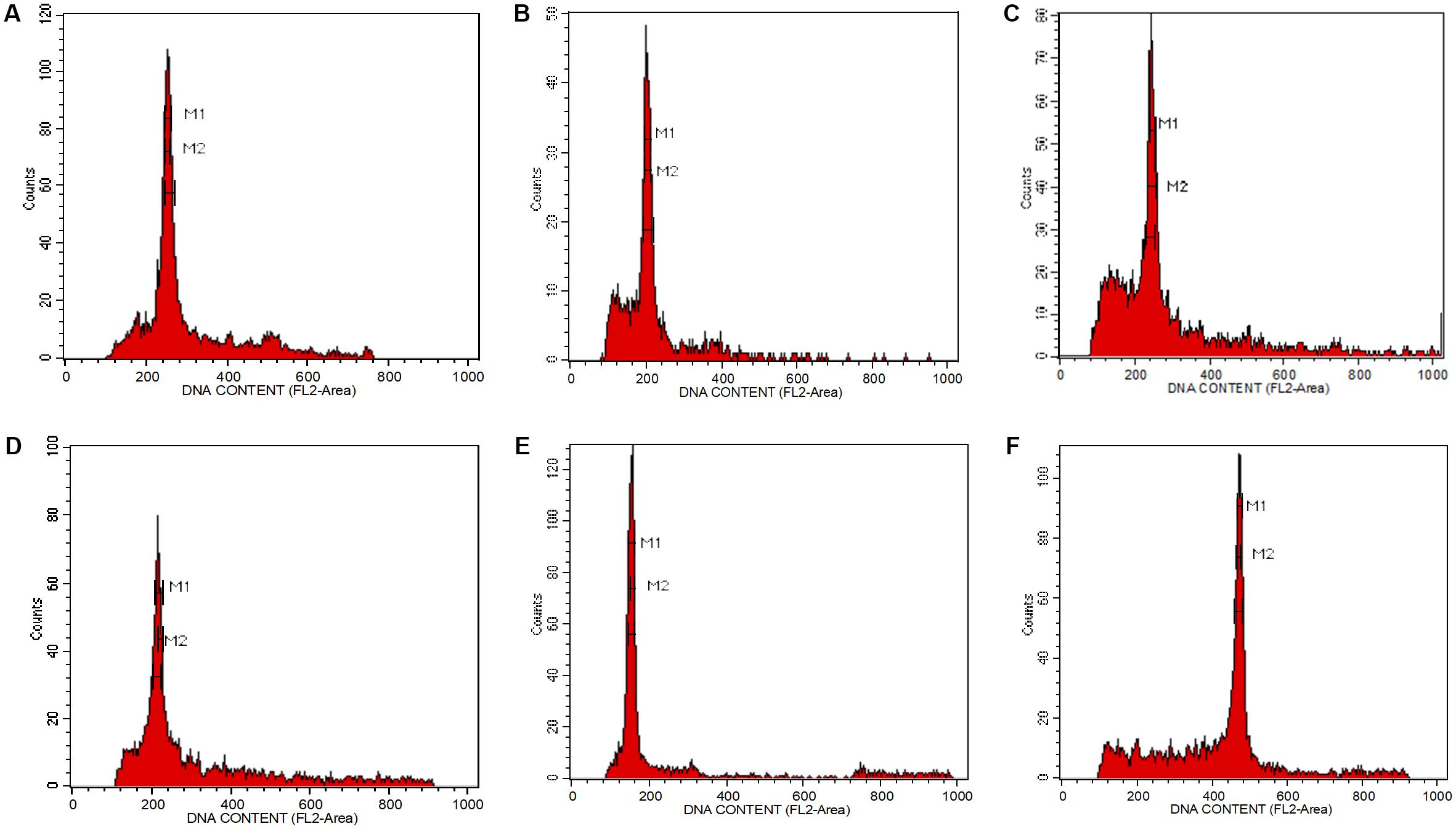
FIGURE 1. Fluorescence intensity histogram peaks showing the number of nuclei per channel as a function of relative fluorescence obtained after flow cytometric analysis of propidium iodide-stained nuclei. (A) Aquilaria hirta, (B) Aquilaria malaccensis, (C) Aquilaria microcarpa, (D) Aquilaria sinensis, (E) Aquilaria subintegra, and (F) Elaeis guineensis, which acted as an external reference standard.
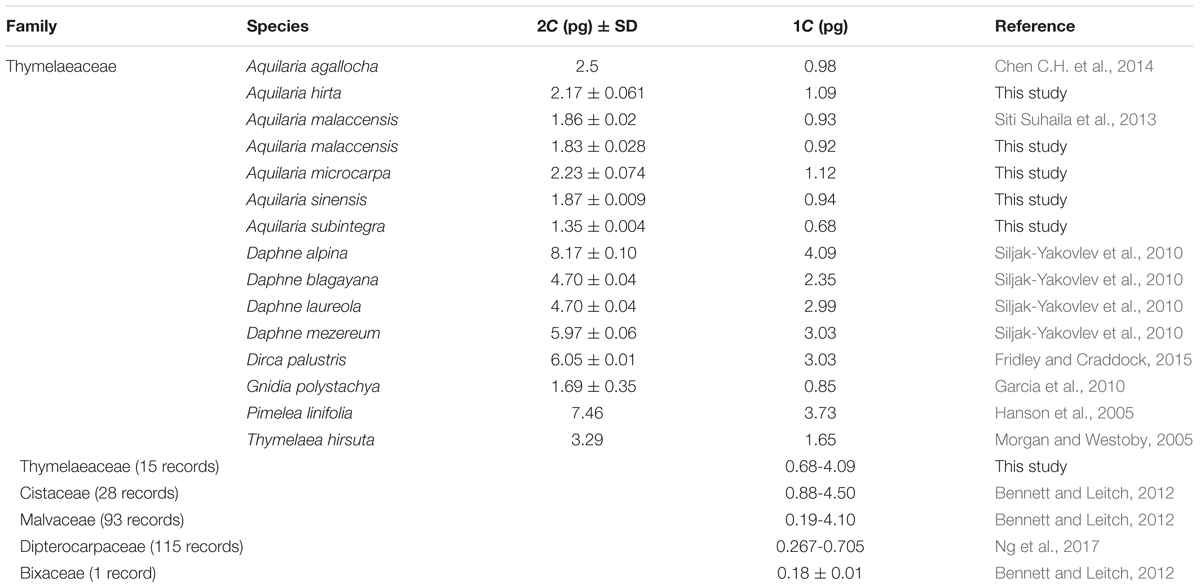
TABLE 1. Genome size of species used in this study and their closely related genera and families.
The entire alignment length for the 12 Aquilaria and three Gyrinops species contains 3,323 nucleotides of combined cpDNA sequences from matK, rbcL, trnL intron, trnL-trnF and psbC-trnS fragments and 795 nucleotides from the ITS region. A total of 55 and 88 characters (1.66% and 11.07%) were variable informative; while 19 and 37 characters (0.57% and 4.65%) were parsimony informative, for the combined cpDNA and ITS region, respectively (Table 2). All sequences were deposited in the GenBank (Table 3). Occurrences of poly A/T sequence repetition were detected in the aligned trnL intron, trnL-trnF, and psbC-trnS sequences.
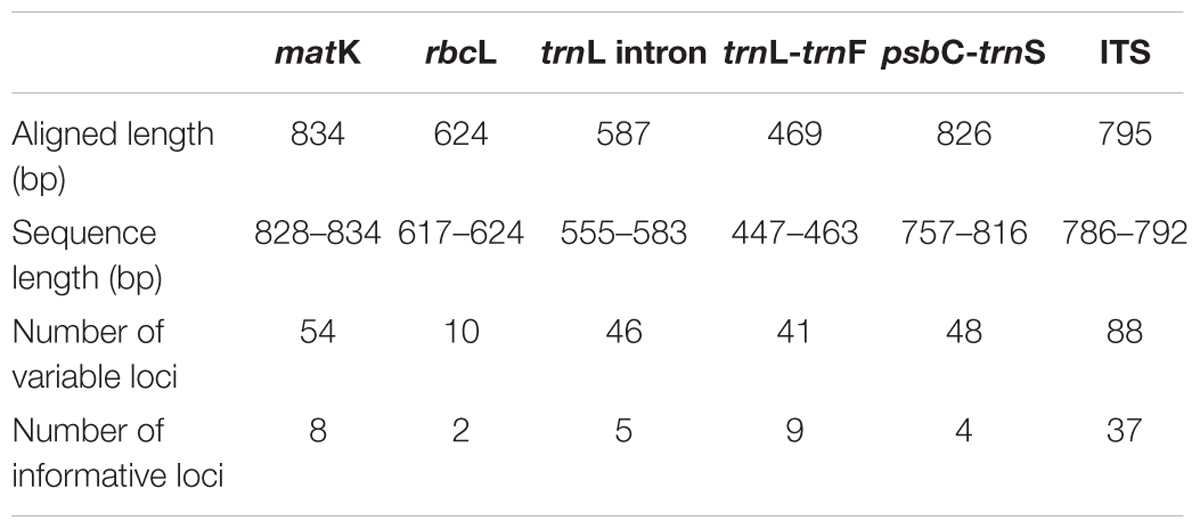
TABLE 2. Sequence attributes of the five loci used in this study.
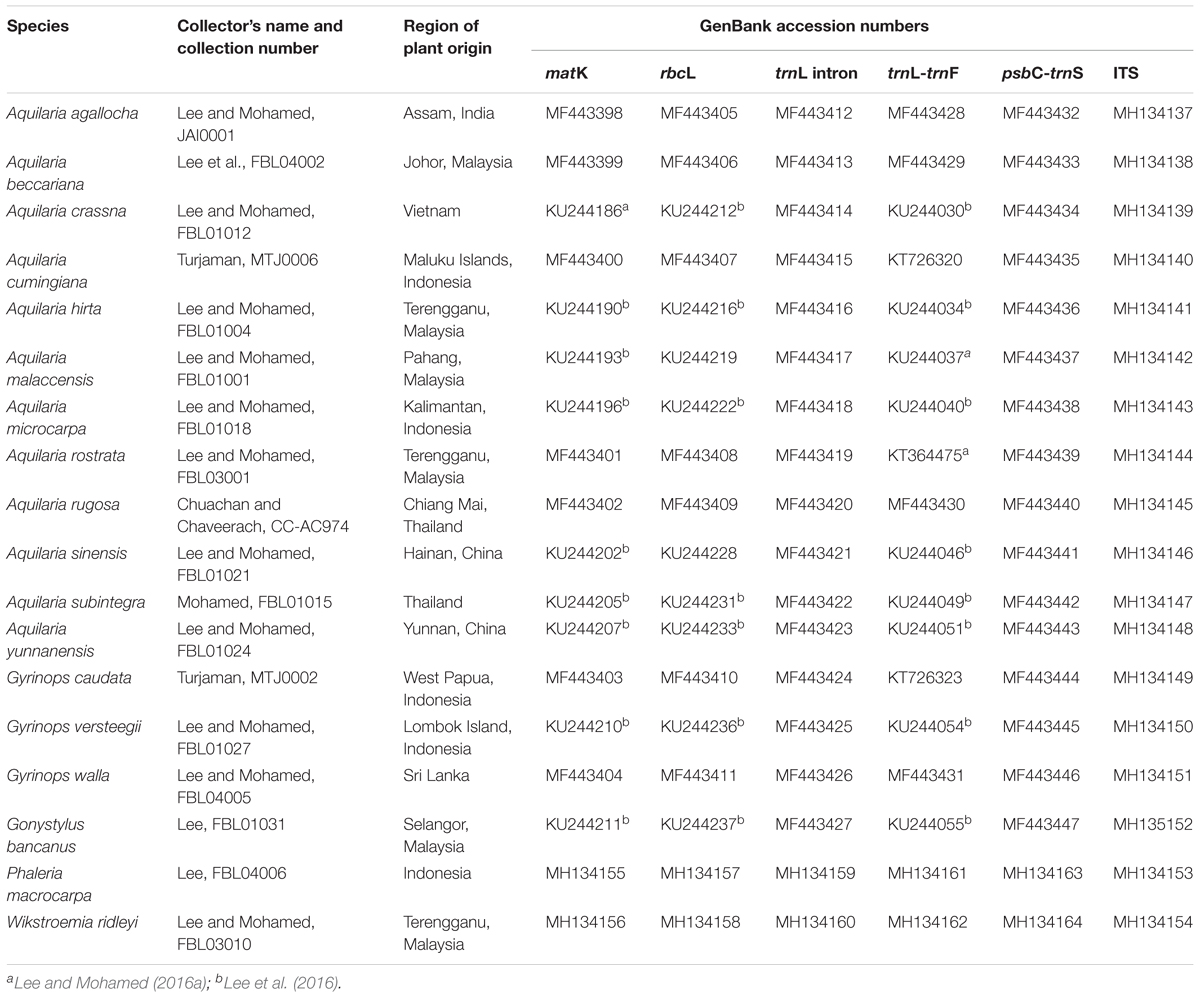
TABLE 3. Localities, voucher details, and GenBank accession numbers of the selected species generated from this study, unless stated otherwise (refer to footnote).
Combined sequences of the five chloroplast loci and the nrDNA ITS region were used to create the phylogenetic tree. Two methods, ML and Bayesian, were employed to acquire divergence time estimates for these species. For the combined cpDNA sequences, the jModeltest was used to identify the best substitution model for the mutation rate required for this analysis, which would further infer divergence times. The ML tree was constructed using MEGA 7, with 1,000 bootstrap replicates and a GTR+I model of substitution for the combined cpDNA sequences (Figure 2A), and a TN93+G model of substitution for the ITS region (Figure 2B). With the exception of A. cumingiana, the Aquilaria species were separated into two clusters in the ITS tree, in which clade A consisted of A. agallocha, A. crassna, A. rugosa, A. sinensis, A. subintegra, and A. yunnanensis, from the Indochina region; while clade B consisted of A. beccariana, A. hirta, A. malaccensis, A. microcarpa, and A. rostrata, from the Malesian region (Figure 2B). The branching between the Indochina and Malesian Aquilaria species showed moderate (67%) bootstrap support.
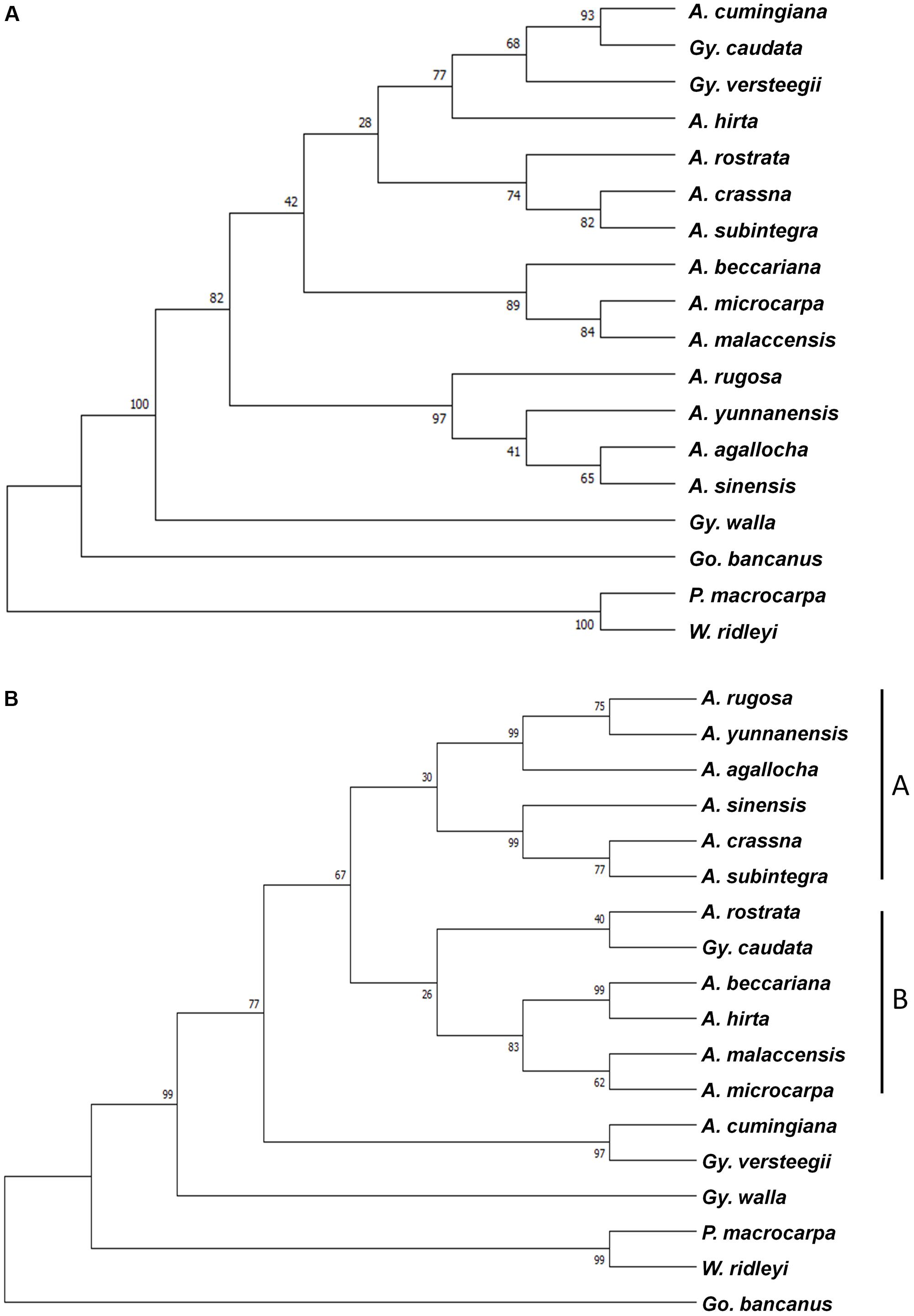
FIGURE 2. Maximum likelihood tree of 15 species from the Aquilarieae tribe based on (A) a combined data set of five chloroplast loci (matK, rbcL, trnL intron, trnL-trnF, and psbC-trnS), and (B) the nuclear region DNA internal transcribed spacer (ITS) region. Gonystylus bancanus, Phaleria macrocarpa, and Wikstroemia ridleyi served as outgroups. Bootstrap values for a majority of divergences are >50%, as indicated on the corresponding nodes.
To estimate the divergence times of the branching of the phylogenetic tree, we used the estimated confidence interval (CI) provided by the TimeTree program (Hedges et al., 2006), between Cistaceae and Thymelaeaceae. Cistaceae served as the reference for the divergence time calibration. Ancestors of the three target species, G. bancanus, A. malaccensis and G. walla, had diverged at approximately the same time, 71 million years ago (Ma), with CI = 40–102 Ma and a median time of 80 Ma. The maximum value of the CI (102 Ma) was used as the anchor point for the calculations of the divergence times. Figure 3 shows the estimated divergence time of the 15 studied species in Aquilarieae having a total of 11 clades. The first clade shows P. macrocarpa and W. ridleyii had split at 60.7 Ma (CI: 38.4–99.4 Ma). The two species then split to their respective clades at 44.4 Ma (CI: 22.6–79.4 Ma); while G. bancanus further split to form the fourth clade from other species of tribe Aquilarieae at 52.3 Ma (CI: 29.5–89.6 Ma). The fifth clade comprising A. agallocha, A. rugosa, A. sinensis, and A. yunnanensis, was formed at 9.9 Ma (CI: 3.8–24.8 Ma). Further speciation happened at 8.2 Ma (CI: 3.1–20.7 Ma) when A. beccariana, A. malaccensis, and A. microcarpa diverged from the fifth clade. The seventh clade (A. crassna, A. rostrata, and A. subintegra) diverged at 7.0 Ma (CI: 2.6–17.5 Ma), followed by the eight clade (A. hirta) at 5.4 Ma (CI: 1.9–13.9 Ma), and the ninth clade (G. versteegii) at 4.4 Ma (CI: 1.4–11.6 Ma). The last divergence for the tenth (G. caudata) and eleventh (A. cumingiana) clades happened quite recently at 1.7 Ma (CI: 0.1–5.7 Ma). The latest divergence that happened in Aquilarieae as found in this study was between A. malaccensis and A. microcarpa at 1.0 Ma (CI: 0.0–3.8 Ma), and between A. crassna and A. subintegra at 0.6 Ma (CI: 0.0–2.7 Ma).
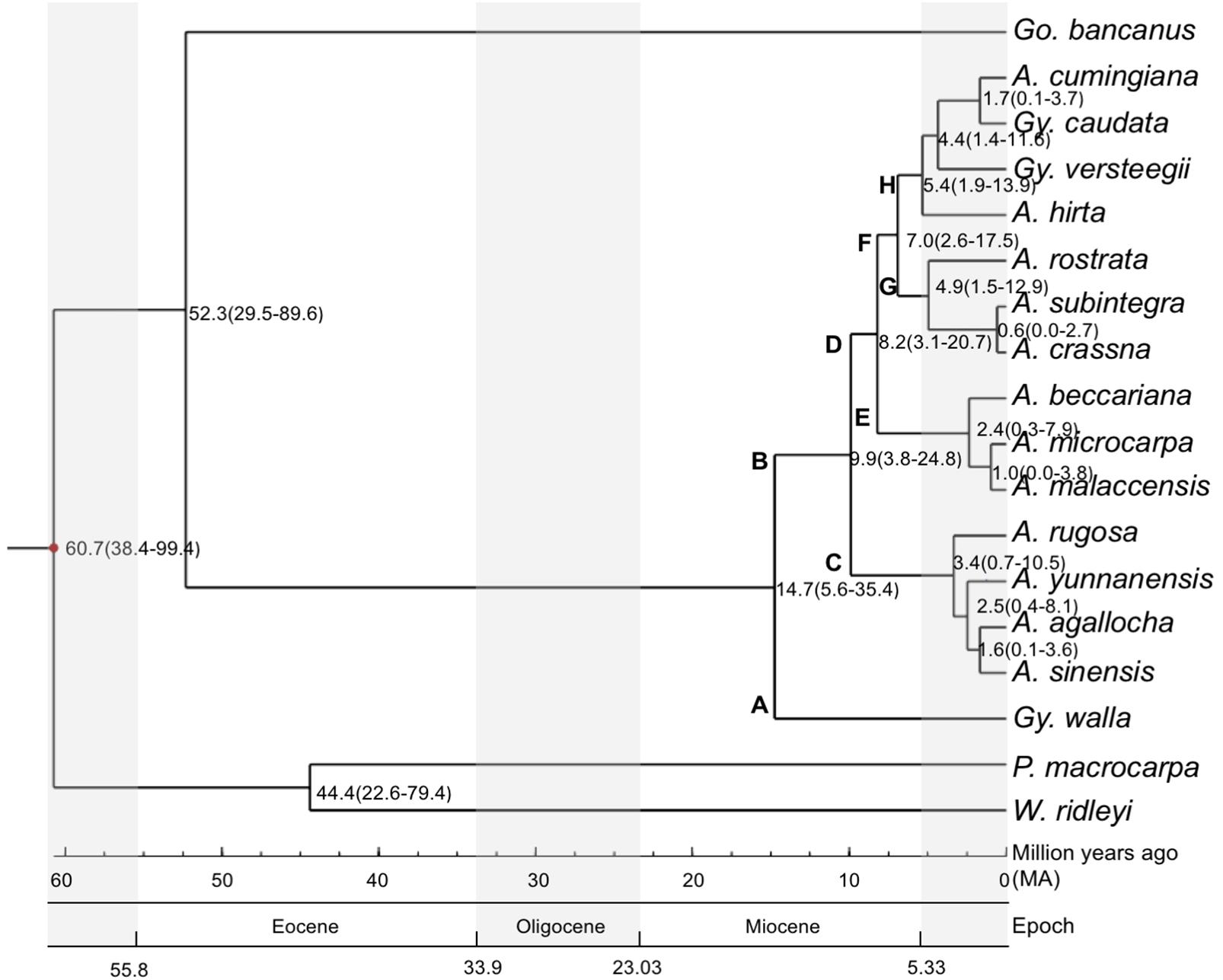
FIGURE 3. Divergence time estimation for species in the tribe Aquilarieae. Bayesian consensus tree inferred from the combined sequences of chloroplast non-coding genes matK and rbcL, chloroplast gene intron trnL, and chloroplast intergenic spacer region trnL-trnF and psbC-trnS from 15 species in the Aquilarieae tribe. Gonystylus bancanus, Phaleria macrocarpa, and Wikstroemia ridleyi were used as outgroups. The red dot on the node represent estimated divergence time used to calibrate the analysis. The scales below the phylogenetic tree indicate the estimated divergence time in million years ago (Ma), which happened over several epochs. (A, Aquilaria; Gy, Gyrinops; Go, Gonystylus; P, Phaleria; W, Wikstroemia).
As information about the Aquilaria genome size is still lacking, we conducted genome size estimations for selected species commonly used in plantation to provide ground base information for this genus. Genome size or C-value refers to the amount of DNA in a cell nucleus whether in an unreplicated monoploid chromosome set (n) or in a polyploid nucleus (Leitch and Leitch, 2013). Genome size has been shown to have a significant influence on plant ecophysiology and biodiversity (Greilhuber and Leitch, 2013). The genome sizes of several species in Thymelaeaceae, with the exception of Aquilaria, such as from the genera Daphne, Dirca, Gnidia, Pimelea, and Thymelaea, have been reported (Table 1). Generally, Aquilaria species have a genome size in the range of 2C = 1.35–2.5 pg, while other species from the same family, namely Daphne alpina, D. blagayana, D. laureola, D. mezereum, Dirca palustris, Gnidia polystachya, Pimelea linifolia, and Thymelaea hirsuta, have genome sizes between 1.69 and 8.17 pg (Hanson et al., 2005; Morgan and Westoby, 2005; Garcia et al., 2010; Siljak-Yakovlev et al., 2010; Fridley and Craddock, 2015). Thus, Aquilaria species have a smaller genome size than other group members like Daphne, Dirca, Pimelea, and Thymelaea, but no clear pattern was found through this alignment within the same family. Genome size in plants could be an important indicator for their growth behavior; for example, plants with a smaller genome size exhibit faster canopy growth when compared to plants with a larger genome size (Siljak-Yakovlev et al., 2010), suggesting that the smaller genome might allow for faster growth due to faster cell division in the plant itself. From our personal observations in the field and through a review of the literature, A. subintegra can be regarded as a fast-growing species while A. hirta is slow-growing, relative to other commonly cultivated Aquilaria species (Lee and Mohamed, 2016b). In contrast, when comparing genome sizes of close families within the same order Malvales, the genome size of Thymelaeaceae is smaller (median 1C = 1.73 pg; 0.68–4.09, 15 records) than that of Cistaceae (median 1C = 2.40 pg; 0.88–4.50 pg, 28 records) and Malvaceae (median 1C = 1.39 pg; 0.19–4.10 pg, 93 records), but greater than that of Dipterocarpaceae (median 1C = 0.417; 0.267–0.705 pg, 115 records) (Bennett and Leitch, 2012; Ng et al., 2017). Based on this information, our findings were consistent with those of Ohri (2005) and Chen S.C. et al. (2014), at least at the family level, suggesting that woody angiosperms may possess smaller genome sizes and rarely exhibit polyploidism.
To obtain first-hand preliminary information on the evolution pattern of the Aquilarieae tribe, we selected five different chloroplast loci and combined their sequences to construct a phylogenetic tree consisting of 15 species from the tribe (Figure 2). Although it is advisable to include more loci in the tree, based on our experiences, we found that some of the chloroplast loci may display little or no nucleotide variations between different species within a genus. This is especially true for Aquilaria, when using gene markers such as rpoB, rpoC1, and psbA-trnH (Lee et al., 2016). In phylogeny studies, a high percentage of polymorphic sites is requisite to obtain useful information on the genetic distance between different species. The sequences amplified by the five selected primer sets in this study contain a number of polymorphic sites, which were helpful for the phylogenetic analysis. However, as these species are closely related via taxonomy, the percentage of conserved sites may be high relative to taxonomically distant species. For a better resolution and thorough information on the evolution analysis of a studied genus, the full chloroplast genome sequences is commonly utilized to calculate the divergence times of its taxa and to draw comprehensive conclusions and inferences regarding taxonomical aspects, genetic diversity, and the pattern of evolution of the studied species (Yang et al., 2013; Daniell et al., 2016). The only two published full chloroplast genome sequences of Aquilarieae species is of A. sinensis (Wang et al., 2016) and A. yunnanensis (Zhang et al., 2018); while a complete chloroplast genome record of Daphne kiusiana (Cho et al., 2017) and an unpublished partial chloroplast genome of G. bancanus (EU849490) in the NCBI GenBank database are the third and fourth references available for the family Thymelaeaceae. Without a concerted effort from various parties to sequence as many species of diverse origins as possible, the ability to sequence a complete genome by one research group requires unobtainable resources. Thus, we resorted to sequencing multiple chloroplast loci.
Taxonomy of the Aquilarieae tribe is still inconclusive. An attempt to resolve its taxonomical standing by relying on phylogenetic observations is beyond the scope of this study. Yet some suggestions for changes are worth highlighting in order to clarify relationships during future revisions. Although the cpDNA ML tree in general indicates a less powerful discrimination, out of the 13 nodes, two had bootstrap support values ≥95%, four have bootstrap support values between 75% and 94%, and seven have <75% (Figure 2). From general observation, the branching of the cpDNA ML tree does not reveal a clear pattern of the grouping. Eurlings and Gravendeel (2005) reported the paraphyletic placement of Aquilaria and Gyrinops using a single chloroplast locus, trnL-trnF. In this study, with the addition of four chloroplast loci sequences, the two genera in the Aquilarieae tribe appeared to show a different relationship. The type species A. malaccensis and G. walla, instead of forming two separate clusters comprising all species of the same genus, were, instead, traced to a common ancestral origin of both genera. The suggestion to reduce Gyrinops to Aquilaria synonym was again brought forward to reflect the natural resemblances among agarwood-producing species (Eurlings and Gravendeel, 2005; Lee and Mohamed, 2016b). This controversy had been raised since 1922, when the unification of the two genera was advocated as they share many similarities in morphological characteristics (Hallier, 1922; Hou, 1960).
The use of the nrDNA ITS region to gather useful genetic information has revolutionized plant phylogenetic studies at the species level for the last two decades due to its biparental inheritance that can be used to assess phylogeny at family and higher levels (Baldwin, 1992). The ITS region has a tendency to be homogenized for its sequence variation in concerted evolution and is easily amplified in the majority of plant species. These properties have caused the ITS region to gain wide popularity in modern plant phylogenetic studies (Neves and Forrest, 2011). The first report to utilize ITS to evaluate phylogeny relationships at the nuclear level among agarwood-producing species was carried out by Lee and Mohamed (2016a), and comprised seven Aquilaria species of Malaysia and Thailand origins, followed by a phylogeny study on the genetic diversity of eight different species from Aquilaria and Gyrinops from Indonesia (Lee et al., 2018). In this study, the ITS analysis was less congruent with the combined cpDNA analysis, however, both had similar amounts of strong bootstrap supports (≥75%, 9/15 in combined cpDNA analysis; 10/15 in ITS analysis) although the informative sequence variation was likely to be higher in the ITS region than in the combined cpDNA sequences. However, studies have shown that data from the ITS region could indicate that biogeographical information of a large genus is separated into clades (Fritsch, 2001; Li et al., 2010). Based on our own hypothesis, at least in the case of the genus Aquilaria (excluding A. cumingiana), the non-monophyletic relationship may be related to biogeographical factors, because separation of the two clades is in agreement with their regions of origin (Indochina and Malesia). The latest taxonomy revision for Thymelaeaceae proposed two major sub-families: Octolepidoidae and Thymelaeoideae (Herber, 2002), supported by molecular phylogenetic analysis using combined cpDNA loci, rbcL and trnL-trnF, and the nrDNA ITS (Beaumont et al., 2009). The phylogenetic relationship shows the sub-family Octolepidoidae (which includes Gonystylus) of having ancestral ties to Thymelaeoideae, while the tribe Synandrodaphneae has ancestral ties to Aquilarieae and Daphneae (which includes Phaleria and Wikstroemia). The same displacement was observed in our ITS tree (Figure 2B), but not in the combined cpDNA tree (Figure 2A). This could be explained with the first molecular phylogeny on Thymelaeaceae carried out only with the combined cpDNA of the rbcL gene and trnL-trnF, returned with an inaccurate classification at tribe level in Thymelaeaceae (Van der Bank et al., 2002). The inclusion of an additional ITS region with the former two cpDNA loci had fully resolved the latest taxonomy displacement proposed for Thymelaeaceae. Explanations based on the genetic information from the ITS region in this family plays a major role in constructing a matching result for the phylogenetic tree to the proposed classical taxonomical system. Considering that molecular phylogenetics in Thymelaeaceae is well-resolved at the tribe level, our study analyzed the two nucleotide loci separately, as they are from two different inheritance systems.
Another interesting finding is the dispersing clade of A. agallocha that originated from India and its synonym A. malaccensis from the Malay Peninsula (Figures 2A,B) (The Plant List, 2013). While identification of these species is primarily based on conventional techniques relying mostly on their morphological characteristics, the sequences of the same species from different geographical regions may contain variations. Phenotypic changes due to geographical boundaries, which function as buffer zones that prevent out-crossings between natural populations, may be one of the few possible factors leading to genetic variation in the natural population of A. malaccensis, since their intra-specific genetic variation can be larger than inter-specific genetic variation (Lee et al., 2017). To synonym A. agallocha with A. malaccensis was first proposed in 1836 (Society for the Diffusion of Useful Knowledge, 1838). However, Ridley (1901) observed that A. agallocha, when compared to A. malaccensis, had: (1) bigger size, (2) leaves with more veins, (3) a twofold to fourfold higher number of flowers per umbel (A. malaccensis has 10 flowers per umbel), (4) solitary umbels (A. malaccensis has panicled umbels), (5) larger flowers, and several other differences. Both tree species can be differentiated based on their morphological characteristics, even if their fruits may look alike. In contrast, Hou (1964) was inclined to synonym the two species, categorically classifying the morphological differences as exemptions. To aid our discussion, we treated A. agallocha and A. malaccensis separately, with the former confined to the northeastern section of the Indian continent and the latter to the Malesian region. We acknowledge that a detailed taxonomic revision should be undertaken by plant taxonomists to resolve these species names.
In common practice, useful references for divergence time calibrations are based on fossil evidence. Our searches for recorded fossil evidence for Thymelaeaceae in the Paleobiology database1 returned 10 records, of which only one was old, dated during the Miocene period (23.0–2.6 Ma), while the remaining nine specimens were quite recent, dated in Early Pleistocene (2.6–0.8 Ma). Based on the review by Herber (2003), the oldest fossil evidence for Thymelaeaceae was recorded from the Eocene (Venkatachala and Kar, 1969), which was closely related to Gonystylus (Venkatachala et al., 1988). Additional fossil records in pollen form, which dated to the Oligocene and Miocene (Muller, 1972; Anderson and Muller, 1975), also resemble Gonystylus. Unfortunately, we could not locate fossil evidence for specimens in the tribe Aquilarieae. As an alternative to fossil evidence, we used the estimated confidence interval (CI) provided by the TimeTree program to estimate the divergence times of the species, as indicated on the branching nodes of the phylogenetic tree (Figure 3). We selected the sister family Cistaceae as the reference specimen to conduct pairwise divergence time analyses targeting the tribe Aquilarieae since the paleobotany of this species is well documented (Guzman and Vargas, 2009). Although there are many possible molecular clock estimations of branching times errors (Sanderson et al., 2004), we obtained branching times through integrative methods, which utilized all present information from published literatures and compiled databases. We concluded that the estimated divergence time tree is considerably reliable, as supported by splitting of G. bancanus at 52.3 Ma, during the Eocene (55.8–33.9 Ma) age, and this complements the oldest fossil recorded from India, which resembles Gonystylus (Venkatachala and Kar, 1969).
We deduced the biogeographic pattern in the speciation of the tribe Aquilarieae by observing the estimated divergence time tree (Figure 3). It can be postulated that Aquilarieae had diverged during the Miocene (23.03–5.33 Ma) age. This assumption is based on the earliest estimated origin of Aquilarieae at 14.7 Ma, from which the first lineage A gave rise to G. walla, currently confined to Sri Lanka and the extremity of southwest India (Subasinghe et al., 2012), and to lineage B, presently distributed in northern India, Indochina, and Malesia. Lineage C diverged into the extant species now primarily confined to mainland Asia, ranging from northeast India (A. agallocha), southern China and Indochina (A. sinensis, A. yunnanensis, and A. rugosa). Lineage D gave rise to the Southeast Asian species. It further diversified into lineages E and F, with the extant species of lineage E confined to west Malesia and lineage F diversified into lineages G and H. Most of the species included in the current study had diversified during the Pliocene (5.33–2.58 Ma) and Pleistocene (2.58–0.01 Ma), when intensification of icehouse condition was present and much of it persisted through Pleistocene (Corner, 1960). Extant species of lineage G are confined to Indochina, Thailand and northern Malay Peninsula. Apparently, the narrowly distributed endemics, A. subintegra and A. rostrata had arose from allopatric speciation. Lineage H diversified in the Malay Archipelago (A. hirta), and nearby regions such as Celebes, Lesser Sunda Islands (G. versteegii), Moluccas Archipelago (A. cumingiana) and the Sahul Shelf (G. caudata).
Aquilaria beccariana and A. hirta display a disjunctive distribution across the South China Sea. Aquilaria beccariana is predominant north and east of Borneo Island, with a small population found in the southeastern Malay Peninsula. Aquilaria hirta is a swamp species widely distributed in the eastern and southern Malay Peninsula, western Kalimantan, and larger islands of the Riau Archipelago. These two species were once probably more widely distributed but retracted into the humid forest refuge known as Riau Pocket (Corner, 1960; Ashton, 1992, 1995, 2014) during the dry Last Glacial Maximum that affected Sundaland (Cannon et al., 2009).
This is the first account attempting to explain the biogeographic pattern of Aquilarieae using time-calibrated phylogeny. Fossil calibration, if available, would increase the accuracy of our postulation. We demonstrated the use of flow cytometry techniques to estimate the genome size of five Aquilaria species, which are found to be significantly smaller than those of other genera in the same family. Future investigation into the correlation between genome sizes in the tribe Aquilarieae based on the two different genera may provide further information into ancestral traits and association in their evolutionary history. Moreover, we used ITS analysis to show that Aquilaria is non-monophyletic. Using a chloroplast gene-based phylogenetic tree, we described the relationship and divergence times between the species in the tribe Aquilarieae. It will be highly interesting to re-analyze this data using chloroplast genome phylogeny, to recalculate the divergence times, and to address the differences, if any, in order to test the robustness of what we put forth here as our postulation in speciation and biogeographic of these species. We expect that information obtained from this work can help clarify the genetic relationship between the two genera in the tribe Aquilarieae, as well as to provide a preliminary view of the ancestral time divergence pattern within the tribe.
The datasets generated or analyzed during this study can be found in the GenBank, accession codes MF443398–MF44344 and MH134137–MH134164, and in the Supplementary Information for this article.
RM and SL designed the study. AF and SL performed the experiments, led the writing with contributions from ZG and TY, and analyzed the data with help from ZG and TY. ZG provided the analyses for phylogeny and divergence time. MM aided in the cytometry experiment. RM reviewed and edited the manuscript. All authors reviewed and approved the final manuscript.
This work received financial support from the Ministry of Science, Technology and Innovation (Science Fund Project No. 02-01-04-SF2102) and the Ministry of Higher Education (FRGS/1/2016/WAB07/UPM/02/3), both of Malaysia.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The reviewer QF and handling Editor declared their shared affiliation.
We thank Ir. Dr. Maman Turjaman from FOERDIA, Prof. Jianhe Wei from IMPLAD, Prof. Dr. Arunrat Chaveerach from KKU, and Dr. Mohd Noor Mahat from FRIM, for providing valuable plant specimens for this study.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2018.00712/full#supplementary-material
Anderson, J. A. R., and Muller, J. (1975). Palynological study of a holocene peat and a Miocene coal deposit from NW Borneo. Rev. Palaeobot. Palynol. 19, 291–351. doi: 10.1016/0034-6667(75)90049-4
Ashton, P. S. (1992). “Plant conservation in the Malaysian region,” in Proceedings of the International Conference in Tropical Biodiversity –In Harmony with Nature, eds S. K. Yap and S. W. Lee (Kuala Lumpur: Malayan Nature Society), 86–93.
Ashton, P. S. (1995). “Biogeography and ecology,” in Tree Flora of Sabah and Sarawak, Vol. 1, eds E. Soepadmo and K. M. Wong (Kuala Lumpur: Forest Research Institute Malaysia).
Ashton, P. S. (2014). On the Forests of Tropical Asia, Lest the Memory Fade. Kew: Royal Botanic Garden.
Baldwin, B. G. (1992). Phylogenetic utility of the internal transcribed spacers of nuclear ribosomal DNA in plants: an example from the compositae. Mol. Phylogenet. Evol. 1, 3–16. doi: 10.1016/1055-7903(92)90030-K
Beaumont, A. J., Edwards, T. J., Manning, J., Maurin, O., Rautenbach, M., Motsi, M. C., et al. (2009). Gnidia (Thymelaeaceae) is not monophyletic: taxonomic implications for Thymelaeoideae and a partial new generic taxonomy for Gnidia. Bot. J. Linn. Soc. 160, 402–417. doi: 10.1111/j.1095-8339.2009.00988.x
Bennett, M. D., and Leitch, I. J. (2012). Angiosperm DNA C-values Database. Release 8.0 Dec. 2012. http://www.kew.org/cvalues/ [accessed 20 March, 2017]
Cannon, C. H., Morley, R. J., and Bush, A. B. (2009). The current refugial rainforests of Sundaland are unrepresentative of their biogeographic past and highly vulnerable to disturbance. Proc. Natl. Acad. Sci. U.S.A. 106, 11188–11193. doi: 10.1073/pnas.0809865106
Chen, C. H., Kuo, T. C. Y., Yang, M. H., Chien, T. Y., Chu, M. J., Huang, L. C., et al. (2014). Identification of cucurbitacins and assembly of a draft genome for Aquilaria agallocha. BMC Genomics 15:578. doi: 10.1186/1471-2164-15-578
Chen, S. C., Cannon, C. H., Kua, C. S., Lui, J. J., and Galbraith, D. W. (2014). Genome size variation in the Fagaceae and its implications for trees. Tree Genet. Genomes 10, 977–988. doi: 10.3390/molecules16064884
Cho, W. B., Han, E. K., Choi, G., and Lee, J. H. (2017). The complete chloroplast genome of Daphne kiusiana, an evergreen broad-leaved shrub on Jeju Island. Conserv. Genet. Resour. 10, 103–106. doi: 10.1007/s12686-017-0774-5
CITES (2013). Sixteenth Meeting of the Conference of the Parties Regarding of Amendment of Appendices and Inclusion of Species in Appendix II. Geneva: CITES Secretariat.
Corner, E. J. H. (1960). “The Malayan flora,” in Proceedings of the Centenary and Bicentenary Congress of Biology, ed. R. D. Purchon (Kuala Lumpur: Malaya University Press), 21–24.
Daniell, H., Lin, C. S., Yu, M., and Chang, W. J. (2016). Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biol. 17:134. doi: 10.1186/s13059-016-1004-2
Darriba, D., Taboada, G. L., Doallo, R., and Posada, D. (2012). jModelTest 2: more models, new heuristics and parallel computing. Nat. Method 9:772. doi: 10.1038/nmeth.2109
Debnath, B., Sil, S., Sinha, R. K., and Sinha, S. (1995). Chromosome number and karyotype of Aquilaria agallocha Roxb. (Thymelaeaceae). Cytologia 60, 407–409. doi: 10.1508/cytologia.60.407
Dolezel, J., Greilhuber, J., and Suda, J. (2007). Estimation of nuclear DNA content in plants using flow cytometry. Nat. Protoc. 2, 2233–2244. doi: 10.1038/nprot.2007.310
Edgar, R. C. (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 32, 1792–1797. doi: 10.1093/nar/gkh340
Eurlings, M. C. M., and Gravendeel, B. (2005). TrnL-trnF sequence data imply paraphyly of Aquilaria and Gyrinops (Thymelaeaceae) and provide new perspectives for agarwood identification. Plant Syst. Evol. 254, 1–12. doi: 10.1007/s00606-005-0312-x
Floden, A. J., Mayfield, M. H., and Ferguson, C. J. (2009). A new narrowly endemic species of Dirca (Thymelaeaceae) from Kansas and Arkansas, with a phylogenetic overview and taxonomic synopsis of the genus. J. Bot. Res. Inst. Tex. 3, 485–499.
Fridley, J. D., and Craddock, A. (2015). Contrasting growth phenology of native and invasive forest shrubs mediated by genome size. New Phytol. 207, 659–668. doi: 10.1111/nph.13384
Fritsch, P. W. (2001). Phylogeny and biogeography of the flowering plant genus Styrax (Styracaceae) based on chloroplast DNA restriction sites and DNA sequences of the internal transcribed spacer region. Mol. Phylogenet. Evol. 19, 387–408. doi: 10.1006/mpev.2001.0933
Galicia, H. D. (2006). Origin and diversification of Thymelaea (Thymelaeaceae): inferences from a phylogenetic study based on ITS (rDNA) sequences. Plant Syst. Evol. 257, 159–187. doi: 10.1007/s00606-005-0371-z
Garcia, S., Garnatje, T., Hidalgo, O., Mas, G., Pellicer, J., Sanchez, I., et al. (2010). First genome size estimations for some Eudicot families and genera. Collect. Bot. 29, 7–16. doi: 10.3989/collectbot.2010.v29.001
Greilhuber, J., and Leitch, I. J. (2013). “Genome size and the phenotype,” in Plant Genome Diversity, Vol. 2, eds J. Greilhuber, J. Dolezel, and J. F. Wendel (Vienna: Springer), 323–344. doi: 10.1007/978-3-7091-1160-4_20
Guindon, S., and Gascuel, O. (2003). A simple, fast and accurate method to estimate large phylogenies by maximum-likelihood. Syst. Biol. 52, 696–704. doi: 10.1080/10635150390235520
Guzman, B., and Vargas, P. (2009). Historical biogeography and character evolution of Cistaceae (Malvales) based on analysis of plastid rbcL and trnL-trnF sequences. Org. Divers. Evol. 9, 83–99. doi: 10.1016/j.ode.2009.01.001
Hallier, H. (1922). Beiträge zur Kenntnis der Thymelaeaceen und ihrer natürlichen Umgrenzung. Meded. Rijks Herb. Leiden. 44, 1–31.
Hanson, L., Boyd, A., Johnson, M. A., and Bennett, M. D. (2005). First nuclear DNA C-values for 18 Eudicot families. Ann. Bot. 96, 1315–1320. doi: 10.1093/aob/mci283
Hedges, S. B., Dudley, J., and Kumar, S. (2006). TimeTree: a public knowledge-base of divergence times among organisms. Bioinformatics 22, 2971–2972. doi: 10.1093/bioinformatics/btl505
Herber, B. (2003). “Thymelaeaceae,” in The Families and Genera of Vascular Plants. IV. Flowering Plants. Dicotyledons. Malvales, Capparales and Non-betalain Caryophyllales, ed. K. Kubitzki (Berlin: Springer),373–396.
Herber, B. E. (2002). Pollen morphology of the Thymelaeaceae in relation to its taxonomy. Plant Syst. Evol. 232, 107–121. doi: 10.1007/s006060200030
Huang, C. C. (2009). Study on Morphology and Cytology of Aquilaria Sinensis (Lour.) Gilg. Master thesis, Guangzhou University, Guangzhou.
Kumar, S., Stecher, G., and Tamura, K. (2016). MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 33, 1870–1874. doi: 10.1093/molbev/msw054
Lee, S. Y., and Mohamed, R. (2016a). Rediscovery of Aquilaria rostrata (Thymelaeaceae), a species thought to be extinct, and notes on Aquilaria conservation in Peninsular Malaysia. Blumea 61, 13–19. doi: 10.3767/000651916X691196
Lee, S. Y., and Mohamed, R. (2016b). “The origin and domestication of Aquilaria, an important agarwood-producing genus,” in Agarwood: Science Behind the Fragrance, eds S. Y. Lee and R. Mohamed (Singapore: Springer), 1–20. doi: 10.1007/978-981-10-0833-7_1
Lee, S. Y., Mohamed, R., Faridah-Hanum, I., and Lamasudin, D. U. (2017). Utilization of the internal transcribed spacer (ITS) DNA sequence to trace the geographical sources of Aquilaria malaccensis Lam. Populations. Plant Genet. Resour. 16, 103–111. doi: 10.1017/S1479262117000016
Lee, S. Y., Ng, W. L., Mahat, M. N., Nazre, M., and Mohamed, R. (2016). DNA Barcoding of the endangered Aquilaria (Thymelaeaceae) and its application in species authentication of agarwood products traded in the market. PLoS One 11:e0154631. doi: 10.1371/journal.pone.0154631
Lee, S. Y., Turjaman, M., and Mohamed, R. (2018). Phylogenetic relatedness of several agarwood-producing taxa (Thymelaeaceae) from Indonesia. Trop. Life Sci. Res. (in press).
Leitch, I. J., and Leitch, A. R. (2013). “Genome size diversity and evolution in land plants,” in Plant Genome Diversity, Vol. 2, eds I. J. Leitch, J. Greilhuber, J. Dolezel, and J. Wendel (Verlag Wien: Springer), 307–322. doi: 10.1007/978-3-7091-1160-4_19
Li, Q. Q., Zhou, S. D., He, X. J., Yu, Y., Zhang, Y. C., and Wei, X. Q. (2010). Phylogeny and biogeography of Allium (Amaryllidaceae: Allieae) based on nuclear ribosomal internal transcribed spacer and chloroplast rps16 sequences, focusing on the inclusion of species endemic to China. Ann. Bot. 106, 709–733. doi: 10.1093/aob/mcq177
Madon, M., Phoon, L. Q., Clyde, M. M., and Mohd, A. (2008). Application of flow cytometry for estimation of nuclear DNA content in Elaeis. J. Oil Palm Res 20, 447–452.
Morgan, H. D., and Westoby, M. (2005). The relationship between nuclear DNA content and leaf strategy in seed plants. Ann. Bot. 96, 1321–1330. doi: 10.1093/aob/mci284
Motsi, M. C., Moteetee, A. N., Beaumont, A. J., Rye, B. L., Powell, M. P., Savolainen, V., et al. (2010). A phylogenetic study of Pimelea and Thecanthes (Thymelaeaceae): evidence from plastid and nuclear ribosomal DNA sequence data. Aust. Syst. Bot. 23, 270–284. doi: 10.1071/SB09002
Muller, J. (1972). “Palynological evidence for change in germorpholgy, climate and vegetation in the Mio-Pliocene of Malesia,” in The Quaternary Era in Malesia, Miscellaneous Series 13, eds P. S. Ashton and M. Ashton (Hull: University of Hull), 6–34.
Mulyaningsih, T., and Yamada, I. (2007). “Notes on some species of agarwood in Nusa Tenggara, Celebes and West Papua,” in Natural Resource Management and Socio-Economic Transformation Under the Decentralization in Indonesia: Toward Sulawesi Area Studies, ed. K. Tanaka (Kyoto: Center of Integrated Area Studies Kyoto University), 365–371.
Neves, S. S., and Forrest, L. L. (2011). Plant DNA sequencing for phylogenetic analyses: from plants to sequences. Methods Mol. Biol. 781, 183–235. doi: 10.1007/978-1-61779-276-2_10
Ng, C. H., Lee, S. L., Tnah, L. H., Ng, K. K. S., Lee, C. T., and Madon, M. (2017). Genome size variation and evolution in Dipterocarpaceae. Plant Ecol. Divers. 9, 437–446. doi: 10.1080/17550874.2016.1267274
Ohri, D. (2005). Climate and growth form: the consequences for genome size in plants. Plant Biol. 7, 449–458. doi: 10.1055/s-2005-865878
Pellicer, J., and Leitch, I. J. (2014). “The application of flow cytometry for estimating genome size and ploidy level in plants,” in Molecular Plant Taxonomy: Methods and Protocols, Vol. 1115, ed. P. Besse (New York, NY: Springer Science+Business Media), 279–307. doi: 10.1007/978-1-62703-767-9_14
Rasool, S., and Mohamed, R. (2016). “Understanding agarwood formation and its challenges,” in Agarwood, ed. R. Mohamed (Singapore: Springer), 39–56. doi: 10.1007/978-981-10-0833-7_3
Robinson, C. (2008). Molecular Phylogenetics of Lachnaea (Thymelaeaceae): Evidence from Plastid and Nuclear Sequence Data. Ph.D. dissertation, University of Johannesburg, Johannesburg.
Sanderson, M. J., Thorne, J. L., Wikström, N., and Bremer, K. (2004). Molecular evidence on plant divergence times. Am. J. Bot. 91, 1656–1665. doi: 10.3732/ajb.91.10.1656
Schrader, J. A., and Graves, W. R. (2004). Systematics of Dirca (Thymelaeaceae) based on ITS sequences and ISSR polymorphisms. SIDA Contrib. Bot. 21, 511–524.
Shen, Y., Jiao, X., and Zhao, S. (2009). Chromosomal studies on populations of Aquilaria sinensis. Guangxi Zhi wu 29, 192–197.
Siljak-Yakovlev, S., Pustahija, F., Šolić, E. M., Bogunić, F., Muratović, E., Bašić, N., et al. (2010). Towards a genome size and chromosome number database of Balkan flora: C-values in 343 taxa with novel values for 242. Adv. Sci. Lett. 3, 190–213. doi: 10.1166/asl.2010.1115
Siti Suhaila, A. R., Norihan, M. S., Norwati, M., Azah, M. N., Mahani, M. C., Parameswari, N., et al. (2015). Aquilaria malaccensis polyploids as improved planting materials. J. Trop. For. Sci. 27, 376–387.
Siti Suhaila, A. R., Norihan, M. S., Norwati, M., Mahani, M. C., Azah, M. N., Parameswari, N., et al. (2013). “Chromosome doubling in A. malaccensis through in-vitro polyploidisation,” in Proceeding of the 10th Malaysian International Genetics Congress, ed. Genetics Society of Malaysia, (Kuala Lumpur: Genetics Society of Malaysia), 247–250.
Society for the Diffusion of Useful Knowledge (1838). “Eaglewood,” in The Penny Cyclopaedia of the Society for the Diffusion of Useful Knowledge, ed. G. Long (London: Charles Knight and Company), 235–236.
Squire, T. S. (2016). Systematics of New Zealand Pimelea (Thymelaeaceae). Master thesis, University of Waikato, Hamilton.
Subasinghe, S. M. C. U. P., Hettiarachchi, D. S., and Rathnamalala, E. (2012). Agarwood-type resin from Gyrinops walla Gaertn: a new discovery. J. Trop. For. Environ. 2, 43–48.
The Plant List (2013). Version 1.1. http://www.theplantlist.org/ [accessed February 28, 2017].
Van der Bank, M., Fay, M. F., and Chase, M. W. (2002). Molecular phylogenetics of Thymelaeaceae with particular reference to African and Australian genera. Taxon 51, 329–339. doi: 10.2307/1554930
Van Niekerk, A. (2008). Phylogenetic Relationships and Speciation in the Genus Passerina L. (Thymelaeaceae) Inferred from Chloroplast and Nuclear Sequence Data. Ph.D. dissertation, University of Johannesburg, Johannesburg.
Venkatachala, B. S., Caratini, C., Tissot, C., and Kar, R. K. (1988). Palaeocene-Eocene marker pollen from India and tropical Africa. Palaeobotanist 37, 1–25.
Venkatachala, B. S., and Kar, R. K. (1969). Palynology of the tertiary sediments of Kutch. 1. Spores and pollen from bore-hole no. 14. Palaeobotanist 17, 157–178.
Wang, Y., Zhan, D. F., Jia, X., Mei, W. L., Dai, H. F., Chen, X. T., et al. (2016). Complete chloroplast genome sequence of Aquilaria sinensis (Lour.) Gilg and evolution analysis within the Malvales order. Front. Plant Sci. 7:280. doi: 10.3389/fpls.2016.00280
Yang, J. B., Tang, M., Li, H. T., Zhang, Z. R., and Li, D. Z. (2013). Complete chloroplast genome of the genus Cymbidium: lights into the species identification, phylogenetic implications and population genetic analyses. BMC Evol. Biol. 13:84. doi: 10.1186/1471-2148-13-84
Yang, Z. (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. doi: 10.1093/molbev/msm088
Keywords: Aquilaria, Gyrinops, flow cytometry, chloroplast genes, ITS gene
Citation: Farah AH, Lee SY, Gao Z, Yao TL, Madon M and Mohamed R (2018) Genome Size, Molecular Phylogeny, and Evolutionary History of the Tribe Aquilarieae (Thymelaeaceae), the Natural Source of Agarwood. Front. Plant Sci. 9:712. doi: 10.3389/fpls.2018.00712
Received: 27 November 2017; Accepted: 11 May 2018;
Published: 29 May 2018.
Edited by:
Renchao Zhou, Sun Yat-sen University, ChinaReviewed by:
Qiang Fan, Sun Yat-sen University, ChinaCopyright © 2018 Farah, Lee, Gao, Yao, Madon and Mohamed. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rozi Mohamed, cm96aW1vaGRAdXBtLmVkdS5teQ==
†These authors have contributed equally to this work.
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.