 Manish K. Vishwakarma1†
Manish K. Vishwakarma1† Sandip M. Kale1
Sandip M. Kale1 Manda Sriswathi1
Manda Sriswathi1 Talari Naresh1
Talari Naresh1 Yaduru Shasidhar1
Yaduru Shasidhar1 Vanika Garg1
Vanika Garg1 Manish K. Pandey1*
Manish K. Pandey1* Rajeev K. Varshney1,2*
Rajeev K. Varshney1,2*- 1International Crops Research Institute for the Semi-Arid Tropics, Hyderabad, India
- 2The University of Western Australia, Crawley, WA, Australia
Small insertions and deletions (InDels) are the second most prevalent and the most abundant structural variations in plant genomes. In order to deploy these genetic variations for genetic analysis in genus Arachis, we conducted comparative analysis of the draft genome assemblies of both the diploid progenitor species of cultivated tetraploid groundnut (Arachis hypogaea L.) i.e., Arachis duranensis (A subgenome) and Arachis ipaënsis (B subgenome) and identified 515,223 InDels. These InDels include 269,973 insertions identified in A. ipaënsis against A. duranensis while 245,250 deletions in A. duranensis against A. ipaënsis. The majority of the InDels were of single bp (43.7%) and 2–10 bp (39.9%) while the remaining were >10 bp (16.4%). Phylogenetic analysis using genotyping data for 86 (40.19%) polymorphic markers grouped 96 diverse Arachis accessions into eight clusters mostly by the affinity of their genome. This study also provided evidence for the existence of “K” genome, although distinct from both the “A” and “B” genomes, but more similar to “B” genome. The complete homology between A. monticola and A. hypogaea tetraploid taxa showed a very similar genome composition. The above analysis has provided greater insights into the phylogenetic relationship among accessions, genomes, sub species and sections. These InDel markers are very useful resource for groundnut research community for genetic analysis and breeding applications.
Introduction
The systematic development of different types of molecular markers over three decades through various approaches, such as storage proteins, isozymes, random amplified polymorphic DNA (RAPDs), restriction fragment length polymorphism (RFLP), amplified fragment length polymorphism (AFLP), cleaved amplified polymorphic sequence (CAPS), simple sequence repeat (SSR), start codon targeted polymorphism (SCoT), etc. helped in conducting genetic studies in several crops species including groundnut (Bechara et al., 2010; Pandey et al., 2012b; Varshney et al., 2013; Liu et al., 2015). Among different marker types, SSRs have dominated genetic studies in groundnut due to multiple advantages of SSR markers but lacked amenability for high throughput genotyping (Gupta and Varshney, 2000). Therefore, the current emphasis is now on developing single-nucleotide polymorphism (SNP) markers in groundnut due to their high preponderance throughout the genome and their amenability for high throughput genotyping for genome-wide breeding applications (Varshney et al., 2009; Pandey et al., 2016). Surprisingly, both SSRs and SNPs have their own drawbacks in spite of usefulness. For instance, the complicated and heterogenous mutation forms of SSRs can sometimes mislead the data analysis (Ellegren, 2004). Similarly, genotyping error may happen due to false bands and proficient artifacts (null or false alleles, allelic dropouts, size homoplasy) (Pompanon et al., 2005). Furthermore, regardless of many refined genotyping methods available (Syvänen, 2001), comparatively most of them are costly and needs special instruments for high throughput genotyping.
Insertions-deletions (InDels) are the second most prevalent and frequent structural variations detected across the plant genomes (Yang et al., 2014; Lu et al., 2015). InDels originate as a consequence of some cellular events like replication slippage, transposable elements, and crossing-over (Moghaddam et al., 2014). InDel process maintains beneficial as well as the deleterious effect on specific loci in the genome (Pearson et al., 2005). InDels are discernible as an efficient marker system for genetic studies in crops and a good addition to other sequence-based genetic markers mainly due to countless desirable inherent genetic ascribes as co-dominant inheritance and multi-allelic with genome-wide dispersion. In addition to easy detection in the genome, InDel markers can be selected by their anticipated fragment size and validated in genetic populations/germplasm using simple and cost-efficient agarose gel. These features make InDels an appropriate marker system for various translational genomics studies in crop plants.
Groundnut (Arachis hypogaea L.) is an economically important, nutritious and protein-rich oilseed crop grown in tropical and warm temperate regions of the world. The genus Arachis is indigenous to South America and its different species are widespread in >100 countries of the world and includes 80 species (Krapovickas and Gregory, 1994; Valls and Simpson, 2005). Based on the morphological variation, geographic distribution and ability to cross (cross compatibility), these species are grouped into nine sections namely Arachis, Caulorrhizae, Erectoides, Extranervosae, Heteranthae, Procumbentes, Rhizomatosae, Triseminatae, and Trierectoides. Of these sections, Arachis is the most diverse section with 30 different species including A. hypogaea, the cultivated groundnut species. Arachis section has much diversity at ploidy level i.e., two tetraploids (A. hypogaea and A. monticola with 2n = 4x = 40), three aneuploids (2n = 2x = 18; A. decora, A. palustris, and A. praecox) and remaining diploids (2n = 2x = 20; Valls and Simpson, 2005). The cultivated tetraploid, A. hypogaea, is considered to have originated from two diploid species namely Arachis duranensis (AA) and Arachis ipaënsis (BB) (Seijo et al., 2004). All the cultivated genotypes can be further grouped into subspecies (hypogaea and fastigiata), varieties (aequatoriana, fastigiata, hirsuta, hypogaea, peruviana, and vulgaris) and agronomic types (Spanish, Southeast-runner, Valencia, and Virginia) (Krapovickas and Gregory, 1994, 2007).
Albeit, with the boastful variation in phenotype, the species display less genetic diversity, as earlier reported with the analyses using RFLPs, SSRs, and SNPs (Pandey et al., 2012b; Varshney et al., 2013). Availability of genome sequences for both the diploid ancestors of tetraploid groundnut (Bertioli et al., 2016; Chen et al., 2016) fostered the process of genome resequencing of diverse cultivars. Therefore, this study reports in silico discovery of large-scale informative genome-wide InDels, development of InDel markers, validation and their deployment for conducting phylogenetic analysis of Arachis genus to understand the genetic complexity at the genome and species level.
Materials and Methods
Plant Materials and DNA Isolation
A panel of 96 groundnut genotypes comprising of cultivated groundnut (A. hypogaea L.) and its wild relatives were used to evaluate InDel polymorphisms and their discriminatory power. The panel of 96 accessions had representation from 41 different Arachis species of seven sections and two synthetics viz. ISATGR278-18 (A. duranensis ICG 8138 × A. batizocoi ICG 13160) and ISATGR184 (A. duranensis ICG 8128 × A. ipaënsis ICG 8206) (Supplementary Table 1). Genomic DNA isolation using the modified CTAB method (Doyle and Doyle, 1987) from freshly collected unopened leaves followed by DNA quantification was performed following the protocol explained in Mace et al. (2003).
Accessing Genome Sequences and Genome-Wide Indels Discovery
Considering the similarities between two diploid subgenomes of tetraploid groundnut (Bertioli et al., 2016), the genome sequences of A. duranensis (accession V14167 for A subgenome,) and A. ipaënsis (accession K30076 for B subgenome,) were used for mining the InDels using the methodology depicted by Yang et al. (2014). Briefly, the fasta files for A and B subgenomes were downloaded from PeanutBase site (https://www.peanutbase.org/) and InDels were identified using MUGSY software (Angiuoli and Salzberg, 2011). The alignment results were mined for InDel polymorphism using the Perl scripts provided by Dr. Wencai Yang (China Agricultural University, Beijing, China). Only the reads mapped on respective pseudomolecules of A and B subgenomes were considered for InDel discovery. The 100 bp (base pair) sequences flanking the candidate InDels were considered from A genome for insertion and from B genome for deletion. The low similarity sequences were removed by searching the extracted sequences against respective genomes using BLASTN program (Altschul et al., 1990) with an E-value of e-20. In house Perl script was used to extract (insertion or deletion) types, chromosomal position, and length of InDels (Supplementary Table 2). The Primers with desired features such as 100–200 bp product size and 20 bp optimum primer length were designed from the extracted sequences by using Primer3 software (Untergasser et al., 2012). The circular plots were constructed using standalone version of circos software (http://circos.ca/) developed at the Genome Sciences Centre in Vancouver, Canada (Krzywinski et al., 2009). The insertion and deletion densities are plotted at a window size of 100 Kb.
InDel Polymorphism and Genotyping
Primer pairs were first used on four accessions/genotypes namely ICG 8138 (A. duranensis), ICG 8206 (A. ipaënsis), TAG 24 (A. hypogaea), and GPBD 4 (A. hypogaea). Subsequently, polymorphic InDels were used for the genotyping set of 96 diverse accessions. Each PCR reaction mixture of total volume 5 μl, contained ~5 ng of template DNA with 2 picomoles of each primer (forward and reverse), 2 mM of each dNTP, 2 mM MgCl2, 1X buffer (Kapa Biosystems) and 0.1 U of Taq DNA polymerase (Kapa Biosystems). PCR (ABI 9700 thermal cycler, Applied Biosystems, USA) reaction was performed following touchdown PCR program explained in Pandey et al. (2012a) and Varshney et al. (2014). The amplified PCR products of all accessions were electrophoresed on 1.5% agarose gels for 2 h at 80–100 V and visualized under gel documentation unit (Syngene). The amplified products were scored on the basis of presence (1) or absence (0) of alleles and the fragment sizes were determined by comparing the 100 bp DNA ladder band.
Population Structure and Phylogenetic Analysis
The population structure of the 96 Arachis accessions was analyzed utilizing genotyping data for polymorphic InDel markers using the program STRUCTURE 2.2 (Pritchard et al., 2000). The presumed number of k (the number of groups) was set from 1 to 10 with admixture and related frequency models; five independent simulations were executed for each range of values. Admixture and concerned frequency models including standard parameters such as 10,000 iterations before a burn-in length of 10,000 MCMC (Markov Chain Monte Carlo) replications for each simulation were used for population structure analysis. The optimal k-value was estimated by posterior probability [LnP(D)] and accessions were assigned to a representing group, as elucidated by Remington et al. (2001). Shannon–Weaver diversity index was analyzed using the GenAlEx 6.5 (Peakall and Smouse, 2012). An UPGMA dendrogram of the 96 accessions, sections and species of Arachis was constructed using DARwin 6 (Perrier et al., 2003). Dissimilarity matrix was generated from the binary data using Jaccard's coefficient at 1,000 bootstraps and the dendrogram was generated using UPGMA method. Dendroscope (Huson et al., 2007) was used to depict the relationship among them. Based on Nei's (1972) genetic distance, PIC-value was also calculated using PowerMarker 3.25 (Liu and Muse, 2005).
Results
Discovery of Genome-Wide InDels in Two Diploid Ancestor Genomes
A total of 269,973 insertions and 245,250 deletions were identified by comparing the genome assemblies of both the progenitor species i.e., A. duranensis (A subgenome) and A. ipaënsis (B subgenome). The lowest number of insertions (14,054) were detected between homeologous pseudomolecules A08 and B08, while the highest number of insertions (36,732) were detected between homeologous pseudomolecules A03 and B03. Similarly, the lowest number of deletions (12,849) were detected between homeologous pseudomolecules A08 and B08 while the highest number of deletions (33,537) were detected between homeologous pseudomolecules A03 and B03 (Table 1, Figures 1, 2A,B). The number of deletions were lower than the number of insertions across corresponding pseudomolecules (Figures 1, 2A,B). The average size of insertion was 4.69 bp with the range of 1–105 bp, while, the average size of deletion was 4.50 bp and varied from 1 to 105 bp. It was important to observe that the number of InDels decreased gradually with increase in size of InDels. For instance, ~43.7% InDels were of 1 bp size and 39.9% were of 2–10 bp while remaining 16.4% were of 10–50 bp.
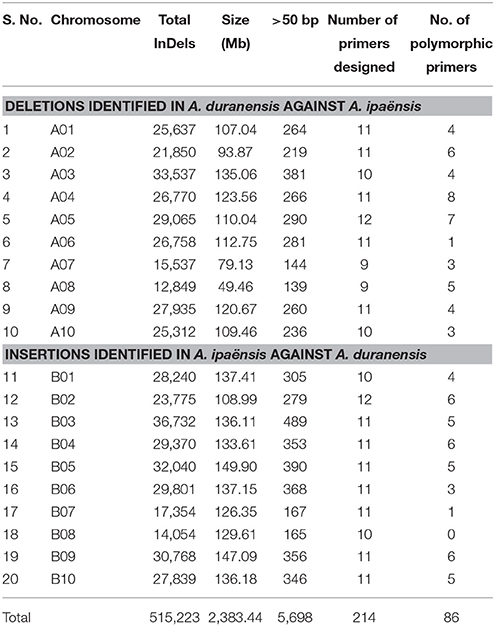
Table 1. Summary statistics of InDels discovery, primer design, and polymorphisms.
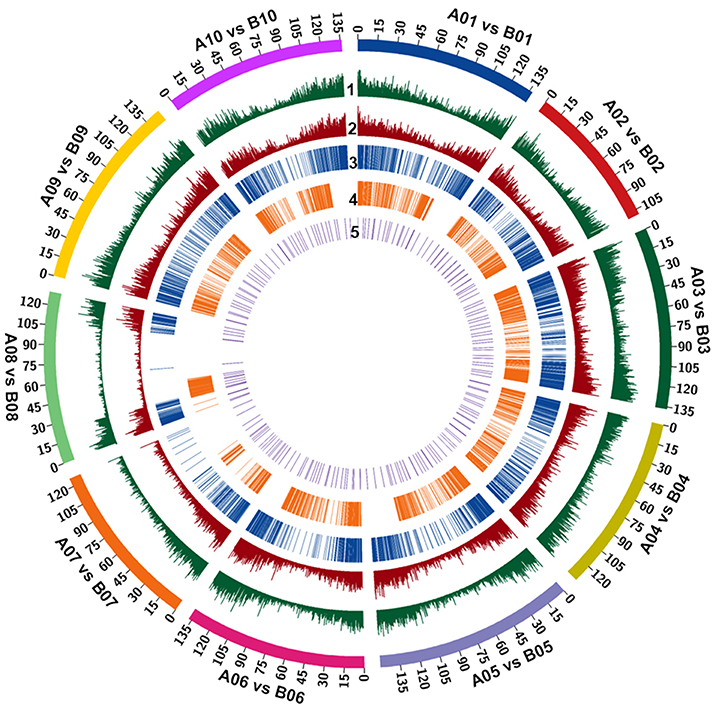
Figure 1. The frequency and relative distribution of 515,223 InDels physically mapped on A and B sub-genomes. The outermost circle denotes the different physical size (Mb) of 10 chromosomes of both sub-genomes coded with multiple colors as per the pseudomolecule size documented in groundnut genome. (1) All insertions density. (2) All deletions density. (3) insertions > 50. (4) deletions > 50. (5) Selected markers for genotyping.
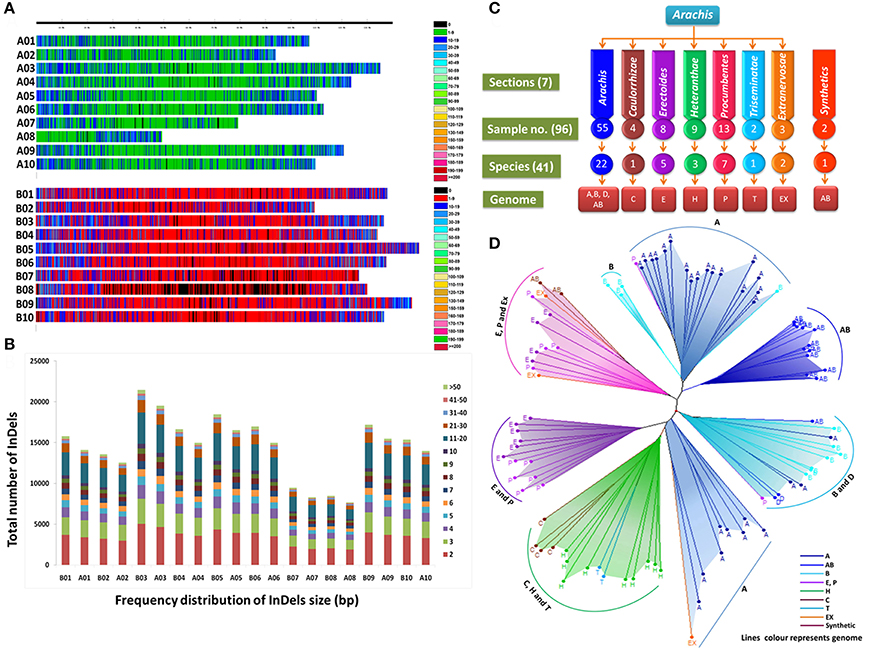
Figure 2. (A) InDels distribution pattern on A- and B- sub genomes. (B) Frequency distribution of InDels (bp) in A. duranensis and A. ipaënsis. (C) Number of samples taken for study from Arachis sections representatives of different species and genomes and (D) Grouping pattern of the accessions/genotypes based on polymorphic InDels.
InDel Marker Development and Their Validation
In order to develop user-friendly markers, InDels >50 bp were selected for primer designing and marker development. A total of such 5,698 InDels (3,218 insertions and 2,480 deletions) with >50 bp size were selected for further analysis and primer designing. Out of 5,698 selected InDels, primers could be designed for 5,519 InDels i.e., 3,111 insertions and 2,408 deletions (Supplementary Tables 3, 4). A total of 214 evenly distributed InDel markers with >50 bp size were selected for amplification and polymorphism study in a set of diverse germplasm. For these selected InDels, the physical distance between two InDel markers ranged from 0.06 to 42.18 Mb with an average of 6.54 Mb. Out of these 214 InDel markers, 86 (40.19%) markers found polymorphic in these tested genotypes (Table 1). These polymorphic InDels with the precise amplification, clear amplicons and length polymorphism were further used to study the phylogeny of 96 diverse accessions. The polymorphic information content (PIC) values of InDel markers ranged from 0.006 (Ai.B01_137090268) to 0.9951 (Ad.A01_10274031) with an average of 0.490.
InDel Markers Based Genetic Diversity among Seven Sections of Arachis Genus
The 96 Arachis accessions, belonging to 41 species from seven sections and two synthetics, were assessed for allelic diversity using genotyping data of 86 polymorphic InDel markers (Table 1; Figure 2C). In total, 174 alleles were identified (2.02 alleles per marker). Among the seven sections of genus Arachis, the Shannon–Weaver diversity index was 0.26. The section Arachis exhibited the highest diversity, with a Shannon–Weaver diversity index of 0.46 (Table 2). Subsequently, Erectoides and Procumbentes exhibited the high diversity with an average Shannon–Weaver diversity index of 0.34. The Triseminatae and Synthetic accessions exhibited relatively low diversity, with only 0.48 and 0.49 different alleles, respectively with the 1.09 effective alleles in accessions of both sections and Shannon–Weaver diversity index of 0.08 (Table 2). Within the seven sections, accessions from Procumbentes exhibited the highest diversity with a Shannon–Weaver diversity index of 0.18, followed by the accession of Triseminatae, Erectoides, and Heteranthae that exhibited high diversity with an average Shannon–Weaver diversity index of 0.16, 0.14, and 0.13, respectively. Among the accessions of Arachis section, low diversity was very much evident with an average Shannon–Weaver diversity index of 0.09 (Table 3). Shannon–Weaver diversity index was zero in 13 species as these species had only single representation.
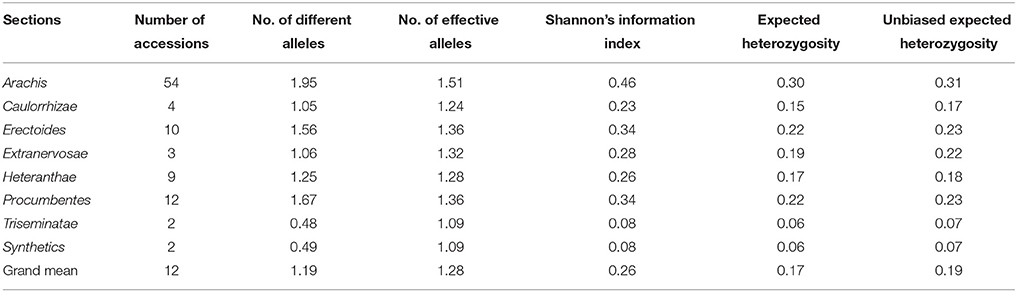
Table 2. Summary of genetic diversity and effective alleles among different Arachis sections.
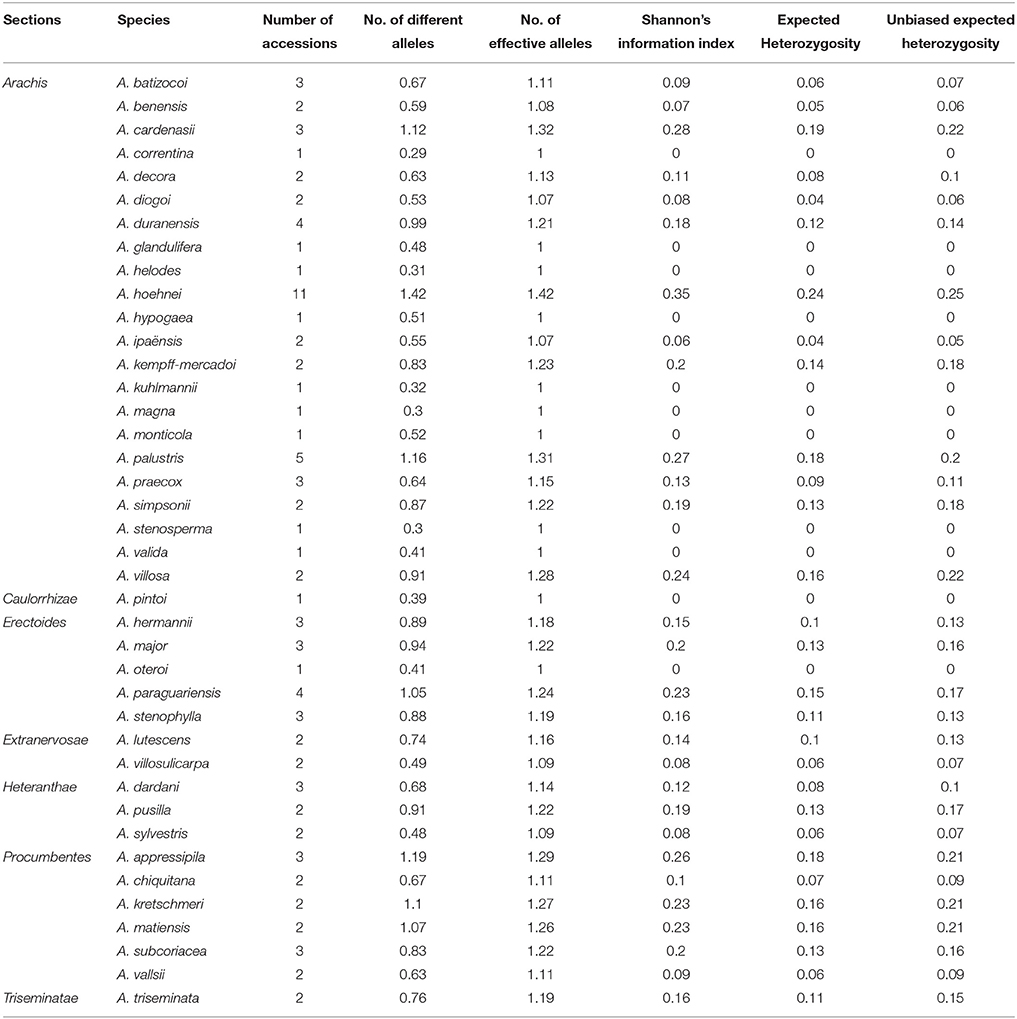
Table 3. Summary of genetic diversity and effective alleles among different Arachis species.
InDel Markers Based Estimation of Genetic Relatedness among Different Arachis Species
The analysis of molecular variance (AMOVA) was performed to assess genetic differentiation among sections and species (Table 4). It has been observed that ~15% (P = 0.001) molecular variation was attributed to genetic differentiation between the sections while remaining 85% among species within sections. These results indicated the presence of huge genetic variability in genus Arachis and between/within sections and species. Of the 86 InDel markers, 32 markers were amplified with the clear cut 50 bp difference between A. ipaënsis and A. duranensis. Out of these 32 subgenome discriminatory InDels, 16 markers had insertions while remaining 16 had deletions. These markers amplified well in tetraploid and diploid accessions of different sections (Supplementary Table 5). Four InDel markers viz. Ad.A10_44595144 (450 bp), Ai.B06_28217388 (265 bp), Ad.A04_62273881 (200 bp), and Ad.A08_24769399 (260 bp) specifically amplified in only Arachis section belonging to A-, B-, and AB- genome (Supplementary Table 6). Similarly, five markers namely Ad.A09_49136207 (275 bp), Ad.A09_49136207 (250 bp), Ai.B10_98116737 (250 bp), Ai.B09_119776002 (250 bp), and Ad.A06_365540 (310 bp) were able to amplify alleles in A, B, and AB genomes of section Arachis and P genome of section Procumbentes. Interestingly, from the above five markers, the marker Ad.A09_49136207 amplified 850 bp specific allele in some accessions of sections Caulorrhizae, Extranervosae, Heteranthae, and Triseminatae, which was not detected in remaining accessions of section Arachis. Subsequently, three markers namely Ai.B05_59490193 (280 bp), Ad.A01_10274031 (400 bp), and Ad.A10_77068321 (210 bp) could amplify genotypes of sections Arachis (A, B, and AB) and Heteranthae (H genome), while marker Ad.A10_77068321 (210 bp) could also amplify genotypes of D genome of Arachis section (Supplementary Table 6).
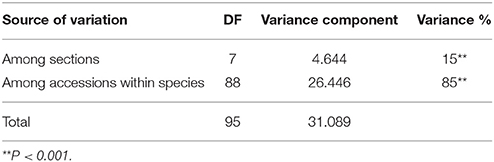
Table 4. Analysis of molecular variance among section and species.
Alleles Specific InDel Markers for Arachis Species and Genotypes
For the assessment of genetic diversity among species, the unique alleles provide a good index to discriminate different species in addition to the number of unique alleles in a population which is an elementary estimate of genetic distinctiveness and differentiating degree of speciation of the species/accessions (Chen et al., 2008). Allele bands specific to 41 different species were scored and analyzed. For instance marker, Ai.B04_11232665 amplified a unique 300 bp allele in species A. batizocoi of section Arachis (B-genome) which did not amplify in any other species (Supplementary Table 7). In addition to this, some markers specifically amplified in some genotypes of different species within and among sections. For instance, marker Ai.B02_41858043 (150 bp) amplified in A. duranensis (ICG 8138, A genome), and A. batizocoi (ICG 8211, B genome) of Arachis section, and A. stenophylla (ICG 8215, E genome) of Erectoides section. Among these 86 InDel markers, 13 markers amplified multiple alleles across all the 96 accessions. Different amplification events in different species with the same InDel markers manifested the distinct mutational histories of multiple alleles.
The Alleles of Cultivated Groundnut and Its Wild Relatives
Ascertaining the differences in genetic constitution between A. hypogaea and its diploid and tetraploid wild relatives is requisite to empathize the evolution of the cultivated groundnut. In addition to diploid wild progenitor species (A. duranensis and A. ipaënsis), the study also included both the tetraploid species i.e., A. monticola (AABB) and A. hypogaea (AABB) (Supplementary Table 8). In the exploration of the alleles for the cultivated and wild, the alleles Ai.B04_70511351 (200 bp), Ai.B04_11232665 (300), Ai.B03_14363004 (200), Ad.A01_90234020 (450), and Ad.A01_10274031 (250) were found specific to selected wild species A. stenosperma (ICG 8137), A. batizocoi (ICG 8209 and ICG 13160), A. duranensis (ICG 8138), A. helodes (ICG 8952) and A. monticola (ICG 8135), respectively. Similarly, two common alleles were also reported between wild species viz. Ad.A01_10274031 (400) in A. kuhlmannii (ICG 15144) and A. ipaënsis (ICG 8206), Ad.A09_119960897 (250) in A. helodes (ICG 8952) and A. palustris (ICG 15143), Ad.A09_49136207 (750) in A. helodes (ICG 8952) and A. simpsonii (ICG 15438), and Ai.B02_41858043 (150) in A. duranensis (ICG 8138) and A. batizocoi (ICG 8211).
Population Structure to Unravel Genetic Architecture
The population structure of the 96 accessions indicated three clusters i.e., Cluster I (13 accessions), Cluster II (48 accessions) and Cluster III (35 accessions). Cluster I had 13 accessions including 11 accessions from Arachis section and 1 accession each from Procumbentes and Erectoides (Figure 3). Cluster II consisted 48 accessions including 35 accessions from the Arachis section, 4 accessions from Procumbentes, 3 accessions from Erectoides, 2 each from Extranervosae, Heteranthae, and synthetics (ISATGR278-18 and ISATGR184). Cluster III consisted of 35 accessions which included 8 accessions from the Arachis section, 7 accessions from Procumbentes, 7 accessions each from Heteranthae and Erectoides, 4 accessions from Caulorrhizae, 2 from Triseminatae, and 1 from Extranervosae (Figure 3).
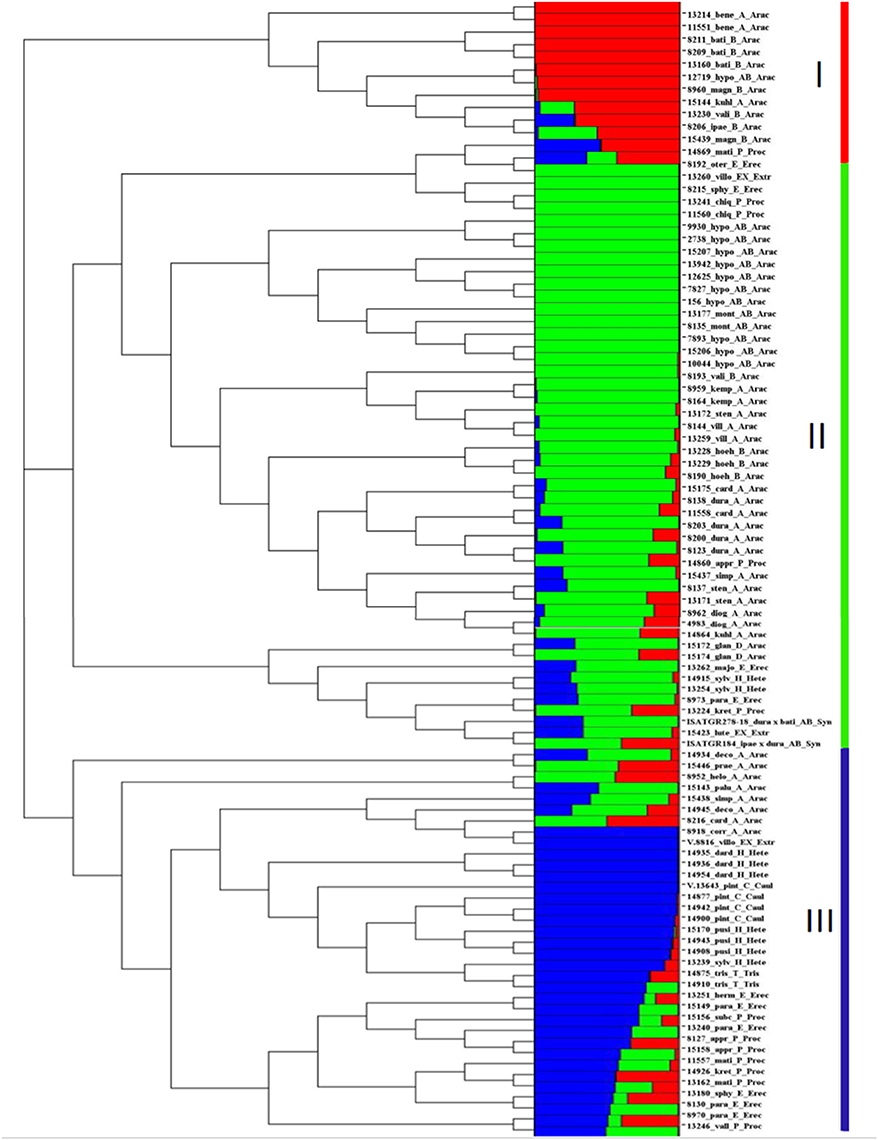
Figure 3. Dendrogram and population structure of 96 cultivated and wild Arachis accessions. The 96 Arachis accessions were classified into three clusters by structure analysis, I, II, and III, basically accompanying to the phylogenic dendrogram. Red, green, and blue represents the cluster I, II, and III, respectively. The proportion of each color of the horizontal bar represents the assignment possibilities to the specific cluster. The names of accessions and taxonomical information are given next to the horizontal bars, starting with the accession number followed by an abbreviated form of species name followed by respective genomes and sections. bati, A. batizocoi; bene, A. benensis; card, A. cardenasii; corr, A. correntina; deco, A. decora; chaco, A. chacoense; diog, A. diogoi; dura, A. duranensis; glan, A. glandulifera; helo, A. helodes; hoeh, A. hoehnei; hypo, A. hypogaea; ipaë, A. ipaënsis; kemp, A. kempff-mercadoi; kuhl, A. kuhlmannii; magn, A. magna; mont, A. monticola; palu, A. palustris; prae, A. praecox; simp, A. simpsonii; sten, A. stenosperma; vali, A. valida; vill, A. villosa; pint, A. pintoi; herm, A. hermannii; majo, A. major; oter, A. oteroi; para, A. paraguariensis; sten, A. stenophylla; lute, A. lutescens; villo, A. villosulicarpa; dard, A. dardani; pusi, A. pusilla; sylv, A. sylvestris; appr, A. appressipila; chiq, A. chiquitana; kret, A. kretschmeri; mati, A. matiensis; subc, A. subcoriacea; vall, A. vallsii; trise, A. triseminata; Arac, Arachis; Caul, Caulorrhizae; Erec, Erectoides; Extr, Extranervosae; Hete, Heteranthae; Proc, Procumbentes; Tris, Triseminatae; Synt: Synthetic.
Phylogenetic Analyses to Establish Genetic Relatedness
The phylogenetic analysis grouped the 96 Arachis accessions into 3 clusters (I, II, and III) corresponding to the structure analysis (Supplementary Table 9; Figure 3). The analysis also showed grouping of the accessions belonging to different genomic sections according to the affinity of their genomes. As expected, all the tetraploid genotypes were grouped together whereas the genotypes belonging to two diploid progenitor genomes (A and B subgenome) grouped separately (Figure 2D). Among other genomes, “P” and “E” genomes clustered together while “C,” “H,” and “T” genomes grouped together showing their genomic similarity with each other. A small separate group was also formed with representative accessions from “Ex,” “P,” and “E” genomes and also synthetics. Also, some accessions from “A” genome grouped with the accessions of “D” and “P” genomes. It was observed that the “D” genome accessions were grouped together with “B” genome indicating higher similarity with each other in comparison to “A” genome. In contrast, surprisingly three B genome accessions belonging to A. hoehnei species grouped with cluster dominated by accessions from A genome (Figure 2D).
For the analysis of pair-wise relationships between different sections of Arachis, dendrogram based on Nei's distance was constructed. Erectoides and Procumbentes were clustered together with a genetic distance of 0.011. Synthetics grouped exclusively indicated high distance from other sections (Triseminatae: 0.240, Caulorrhizae: 0.204, Heteranthae: 0.164, Arachis: 0.144, Procumbentes: 0.117, Erectoides: 0.109). Apart from synthetic genotypes, section Triseminatae also showed high distance from the other remaining sections with the genetic distances viz. Arachis: 0.133, Caulorrhizae: 0.127, Procumbentes: 0.122, Extranervosae: 0.120) (Supplementary Table 10; Figure 4A).
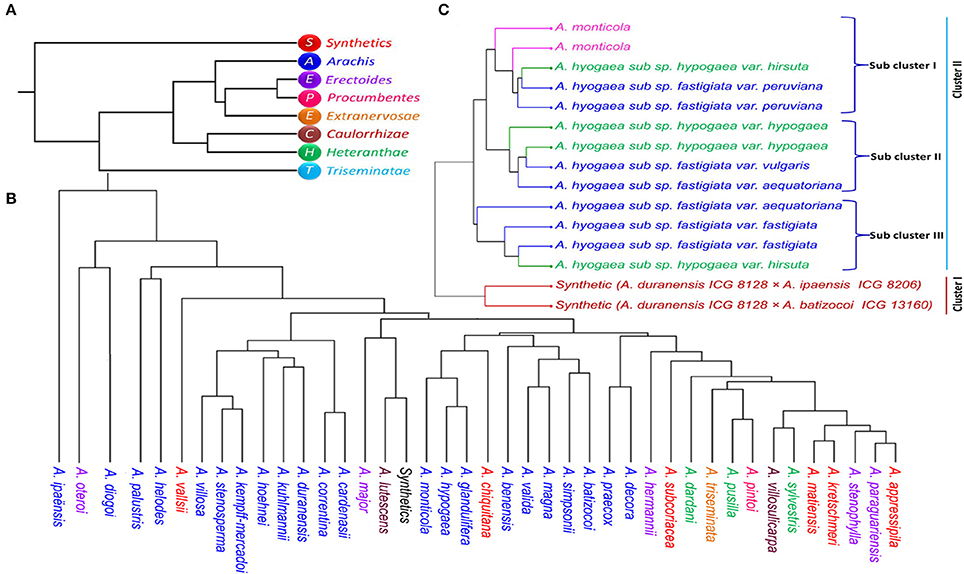
Figure 4. The dendrogram of (A) seven sections and synthetics (B) 22 species of Arachis (C) tetraploid genotypes including two synthetics generated from Nei's genetic distance matrix by UPGMA in PHYLIP.
Among the 41 species of Arachis genus studied for phylogeny separately, A. ipaënsis was highly distant from other species and had greater distance (A. duranensis: 0.742, A. oteroi 0.704, A. kempff-mercadoi: 0.656, A. major: 0.605, A. lutescens: 0.595, A. stenosperma: 0.590, A. villosa: 0.548, A. subcoriacea: 0.544, A. palustris: 0.515). On the other hand, A. oteroi also showed greater distance from many species (A. palustris: 0.659, A. hermannii: 0.574, A. subcoriacea: 0.564, A. batizocoi: 0.522) (Supplementary Table 11; Figure 4C). A. paraguariensis was found closer to A. appressipila, A. matiensis, A. stenophylla, A. sylvestris, and A. kretschmeri with the shortest distance of 0.039, 0.044, 0.045, 0.051, and 0.053, respectively. Interestingly both the tetraploid species, A. hypogaea and A. monticola clustered together with mere distance of 0.148 while their diploid progenitors (A. duranensis, AA, and A. ipaënsis, BB) showed high distance 0.742.
All the tetraploid accessions were evaluated for the differences and relatedness between cultivated, their wild relatives and synthetic genotypes. Two separate groups were formed for the synthetics and cultivated tetraploid genotypes. The second group containing cultivated genotypes clustered according to sub species i.e., sub sp. fastigiata and sub sp. hypogaea. For greater understanding, hierarchical analysis was performed which identified two major clusters. The first major cluster consisted synthetics and had maximum distance of 0.643 from the second major cluster (Figure 4B, Supplementary Table 12). The second cluster was separated into three sub clusters i.e., sub cluster I consisted A. monticola, two A. hypogaea sub sp. fastigiata var. peruviana and strangely one A. hypogaea sub sp. hypogaea var. hirsute. Sub cluster II consisted two A. hypogaea sub sp. fastigiata var. fastigiata with one A. hypogaea sub sp. fastigiata var. aequatoriana and one A. hypogaea sub sp. hypogaea var. hirsute. Sub cluster III consisted of two genotypes from A. hypogaea sub sp. hypogaea var. hypogaea with one A. hypogaea sub sp. fastigiata var. vulgaris and one A. hypogaea sub sp. fastigiata var. aequatoriana.
Discussion
Over the last decade, next-generation sequencing (NGS) technologies have revolutionized the availability of large-scale genetic markers and their deployment in trait discovery and breeding (Varshney, 2015; Pandey et al., 2016). InDel markers have been deployed in forensic and genetic studies in humans as well as in several plants/crops like wheat, rice, barley, mustard, citrus, tomato, and Arabidopsis (Yang et al., 2014; Lu et al., 2015; Zhou et al., 2015). The availability of draft genome assemblies of groundnut progenitors (Bertioli et al., 2016; Chen et al., 2016) have provided an excellent opportunity for initiating several genomic and genetic studies such as SNP discovery, gene prediction, gene expression, comparative genomics, genetic diversity, genetic mapping, and molecular breeding (Pandey et al., 2016). In this context, this study developed large-scale genome-wide InDel markers and demonstrated their utility in phylogenetic relationship among different species of Arachis genus.
Large Scale Genome-Wide InDels, an Important Resource for Genetic Studies and Breeding Applications in Groundnut
This study discovered large-scale InDels by comparing A subgenome (A. duranensis, accession V14167) and B subgenome (A. ipaënsis, accession K30076), and detected 5,698 InDels of more than 50 bp sizes indicating an abundance of InDels for genetic and breeding studies in groundnut. Accuracy in InDels identification basically depends on the quality of sequencing data, scheme, and parameters used for data extraction. One and two base pairs InDels were not included between A and B subgenomes in order to avoid over reckoning of small InDels due to sequencing errors as earlier experienced in tomato crop by Yang et al. (2014). In groundnut, identification of InDels have been reported from expressed sequence tags (ESTs) data and was used for studying genetic diversity in cultivated groundnuts (Liu et al., 2015). This is the first report on developing large scale genome-wide InDels using the sequence information of the diploid progenitors of groundnut. Groundnut is usually considered as a less diverse crop, and to explore more studies related to diversity analysis, several marker systems were used from time to time but their numbers were less or not optimum. In this study, we found 40.19% (86 InDels) polymorphism with these InDel markers which were higher than other earlier reported markers viz. start codon targeted polymorphism (SCoT) marker (38.2%) (Xiong et al., 2011); InDels developed from ESTs (33.3%) (Liu et al., 2015); AFLP markers (3.6%) (He and Prakash, 1997); RAPD markers (6.6%) (Subramanian et al., 2000); EST-SSR markers (10.4%) by Liang et al. (2009) and SSR markers (14.5%) Zhao et al. (2012). These InDel markers are a good genomic resource for groundnut research community and can be used in majority of the genetic and breeding applications in groundnut.
InDel Markers Provided Insights on Allele Diversity and Genetic Differentiation
For the better exploitation of wild species genes in groundnut improvement program, knowledge of genetic diversity in the Arachis germplasm is essential as indicated in several such studies (Barkley et al., 2007; Angelici et al., 2008). Our findings indicated the presence of lots of diversity within the Arachis section and its 23 species. Among the Arachis species of section's most diverse accessions was the A. hoehnei that belong to B subgenome, which was followed by accessions from the A subgenome (A. cardenasii, A. palustris, and A. villosa) with the highest Shannon–Weaver diversity index values. The A. hoehnei carrying B subgenome while A. villosa and A. cardenasii (both resistant to rust, LLS, and groundnut rosette) having A genome are considered in secondary gene pool as they are cross-compatible, having chromosome pairing, and hybrid fertility, due to this reason earlier they were considered as probable subgenomes contributors of A. hypogaea (Mallikarjuna et al., 2006). Although A. palustris is also highly diverse due to aneuploidy species (differences in basic chromosome number) but it would not be cross compatible with A. hypogaea, a barrier for the introduction of desirable diverse characters (Lavia, 1998). Differences in the genetic diversity within the Arachis section and between species of other sections could be due to the following three reasons or their combination: (1) polyploidization and its events create hurdle in gene mobilization from concerned diploid to cultivated species (Young et al., 1996), (2) combination of self- pollination and polyploidization in immediate past from one or a elite individual(s) of each diploid parental species (Halward et al., 1991), and (3) narrow genetic base induced by consistent use of elite cultivars and less use of exotic germplasm in breeding curricula (Knauft and Gorbet, 1989). This study showed high diversity and researchers need to found better ways for broadening the genetic base by introgressing desired genomic segments from wilds to cultivated groundnut.
Allele Specific Indels to Different Arachis Subgenomes Supporting Existence of K Genome
The species specific alleles confer particular species a unique identity in the population. Out of 86 InDel markers examined, 9 markers amplified 9 alleles, which were specific to genotype/accession and dissimilar for Arachis sections. The unique alleles were observed in the species/accessions indicating the various degrees of evolution and diversity of these species/accessions (Chen et al., 2008). We identified few markers that were specific to a particular genome like four markers amplified only Arachis section (A-, B-, AB- genome) which were not amplified in any other sections (representative of other than A-, B-, AB- genome). This indicated occurrence of new recombination events that might have shuffled the genomic sequences and created insertions and deletions sites due to domestication. Likewise, some markers amplified in Caulorrhizae, Extranervosae, Heteranthae, and Triseminatae sections, but not amplified in any accessions from Arachis section, these wild crop relatives of Arachis are endemic to South America, occurring in Bolivia, Argentina, Brazil, Paraguay, and Uruguay and are a rich source of specific alleles (Valls et al., 1985). This suggested that some genes/alleles specific to them were lost after natural selection or domestication events. In contrast to the markers amplified in Arachis section and as well as in section Procumbentes and Heteranthae. This result revealed sharing of some large genomic sequences between Arachis and Procumbentes sections, Arachis and Heteranthae sections which was not fully recombined during domestication and remained conserved. Likewise, two genotypes of A. batizocoi had a specific allele that was not found in any other genotypes or species. Our study is in quite an accordance with the earlier study (Leal-Bertioli et al., 2014) which claimed this species might have another genome “K” with more similarity to “B” genome. For getting insights into genetic constitution among A. hypogaea and its diploid and tetraploid wild congenators, we also considered specific alleles between them. We found five genotypes belonging to wild diploid species with one exception i.e., A. monticola with specific alleles within Arachis section. This indicated that these alleles were highly conserved in wild relatives and emerged during the speciation and evolution of groundnut but were restricted to wild or lost in domestication events. These wild resources can be a good source of mobilizing specific alleles from wild to diversify the genetic base of cultivated gene pools and to enrich economically significant traits.
InDels Established Genomic Affinities among Diverse Germplasm
The InDel-based phylogenetic study grouped all the seven taxonomic sections based on their genomic affinities with the exception of synthetic genotypes which grouped distinctly from all the sections. Arachis species is considered to be the most diverse, holding both annual and perennial species and distinct chromosome numbers, karyotype structures and ploidy levels as it was a group apart but very close to Erectoides and Procumbentes Krapovickas and Gregory, 1994). On the other hand, Heteranthae and Caulorrhizae grouped together indicating that some species of the sections Heteranthae and Caulorrhizae may be capable of producing hybrids with Arachis section while a substantial genetic isolation persists with the other sections (Bertioli et al., 2016). According to Krapovickas and Gregory (1994) sections, Extranervosae and Triseminatae are the most detached sections, however, their evolutionary place is yet to be decided (see Stalker, 2017). This study confirms the above assumption for Triseminatae as the most isolated section from the remaining sections. On the other hand, this study contradicts in case of Extranervosae which was found close to the Arachis, Procumbentes, and Erectoides. Nevertheless, the recent phylogenetic studies based on ribosomal DNA (rDNA) suggested sections Heteranthae, Extranervosae, and Triseminatae to be most primitive and section Arachis to be most recent while sections Procumbentes, Caulorrhizae, Erectoides, Rhizomatosae, and Trierectoides found in between these sections (Bechara et al., 2010; Wang et al., 2010). This study with the InDel markers indicates sections Heteranthae and Triseminatae to be the most primitive including Caulorrhizae; sections Erectoides and Procumbentes as intermediate and section Arachis as the most recent in origin.
The topology of the UPGMA dendrogram for all 44 species used in this study generated from InDel markers revealed that during the whole evolutionary courses of Arachis to date, there have been new recombinants occurring due to regular and frequent deletion process. Due to above possible reasons, all the species show higher affinity to A genome rather than B genome representative A. ipaënsis. This also indicated that the other B genome species, grouped close to A genome representative A. duarnensis, are distinct from A. ipaënsis due to greater difference in accumulation of deletions in the genome. The analysis of InDel markers showed grouping of species from D, K, and F genomes together with A genome species. The position of A. benensis distant from the B genome biological group also gives support to the validity of the F genome assignment. The position of the D, K, and F genomes closer to the A than to the B genome is worthy of further investigation.
A. monticola: A True Wild Species Escaped away from Cultivation or Ancestor?
Looking insights into the relationship of tetraploid genotypes/accessions based on the InDel markers, both the synthetic genotypes grouped separately as they were newly created and indicated their diverse genetic makeup. In addition, the dendrogram clearly showed that A. monticola and cultivated A. hypogaea (all botanical type) were grouped together with less or null genetic distance due to their close affinity. The complete homology observed here in the insertion and deletion sequences that manifested A. monticola and A. hypogaea tetraploid taxa very near and similar genome composition. The current finding also justifies the high rate of crossability and achieving fertile progenies reported by Krapovickas and Gregory (1994). Albeit, the dendrogram of tetraploid species, showed that A. monticola grouped separately from the other botanical types, which affirms the belief that it is a separate species from A. hypogaea. In a study, A. monticola was believed as a true wild species that had got away from cultivation which was not considered as a form of A. hypogaea (Krapovickas and Gregory, 1994; Bechara et al., 2010). This finding is also supported by the studies done on the basis of fruit structure which narrowly separate each seed, A. monticola was considered a discrete species from A. hypogaea (Krapovickas and Gregory, 1994). This attribute was not found in any cultivated groundnut and is conceived as a naive feature in the genus. These observations affirm the possibility that A. monticola is the immediate wild ancestor or an introgressive derivative between the A. hypogaea and wild species as reported in earlier studies (Gregory and Gregory, 1976; Moretzsohn et al., 2004; Koppolu et al., 2010).
Conclusion
The InDels are the second most abundant structural variations across the genome after SNPs and can serve as genetic markers for conducting genetic studies in labs with small to medium scale genotyping facility, especially with higher length polymorphism. This study successfully identified 515,223 InDels distributed across groundnut genome and designed primers for 5,698 InDels with >50 bp size. Further, randomly selected InDel markers were validated for their functionality and usefulness in studying the genetic relationship in a very diverse germplasm sets. The information on InDel markers is a very useful genomic resource for the groundnut research community for using them in an array of genetic and breeding applications.
Author Contributions
MKV and MKP: performed most of the experiments; SMK: performed InDel marker discovery and primer designing; MS, TN, and YS: generated genotyping data on diverse germplasm panel; MKV, MS, and VG: analyzed genotyping data and conducted genetic analysis; MKV and MKP: interpreted the results; MKV: drafted the MS; MKP and RKV: improved the manuscript; MKP and RKV: conceived, designed, and supervised the study and finalized the manuscript.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The authors are thankful to The Peanut Foundation, Bill, and Melinda Gates Foundation (Tropical Legumes III) and MARS Chocolate Inc., USA, and World Bank-assisted Karnataka Watershed Development Project-II (KWDP-II) funded by Government of Karnataka (GoK), India for financial assistance. Authors are thankful to Dr. Hari D. Upadhyaya for providing seeds of genotypes included in the study. The work reported in this article was undertaken as a part of the CGIAR Research Program on Grain Legumes. ICRISAT is a member of the CGIAR.
Supplementary Material
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fpls.2017.02064/full#supplementary-material
References
Altschul, S. F., Gish, W., Miller, W., Myers, E. W., and Lipman, D. J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. doi: 10.1016/S0022-2836(05)80360-2
Angelici, C. M. L. C. D., Hoshino, A. A., Nóbile, P. M., Palmieri, D. A., Valls, J. F. M., Gimenes, M. A., et al. (2008). Genetic diversity in section Rhizomatosae of the genus Arachis (Fabaceae) based on microsatellite markers. Genet. Mol. Biol. 31, 79–88. doi: 10.1590/S1415-47572008000100016
Angiuoli, S. V., and Salzberg, S. L. (2011). Mugsy: fast multiple alignments of closely related whole genomes, Bioinformatics 27, 334–342. doi: 10.1093/bioinformatics/btq665
Barkley, N. A., Dean, R. E., Pittman, R. N., Wang, M. L., Holbrook, C. C., and Pederson, G. A. (2007). Genetic diversity of cultivated and wild-type peanuts evaluated with M13-tailed SSR markers and sequencing. Genet. Res. 89, 93–106. doi: 10.1017/S0016672307008695
Bechara, M. D., Moretzsohn, M. C., Palmieri, D. A., Monteiro, J. P., Bacci, M. Jr., Martins, J. Jr., et al. (2010). Phylogenetic relationships in genus Arachis based on ITS and 5.8S rDNA sequences. BMC Plant Biol. 10:255. doi: 10.1186/1471-2229-10-255
Bertioli, D. J., Cannon, S. B., Froenicke, L., Huang, G., Farmer, A. D., Cannon, E. K., et al. (2016). The genome sequences of Arachis duranensis and Arachis ipaënsis, the diploid ancestors of cultivated peanut. Nat. Genet. 48, 438–446. doi: 10.1038/ng.3517
Chen, B.-y., Jiang, H.-f., Ren, X.-p., Liao, B.-s., and Huang, J.-q. (2008). Identific and molecular traits of Arachis species with resistance to bacterial wilt. Acta Agric. Boreali Sin. 23, 170–175. doi: 10.7668/hbnxb.2008.03.041
Chen, X., Li, H., Pandey, M. K., Yang, Q., Wang, X., Garg, V., et al. (2016). Draft genome of the peanut A-genome progenitor (Arachis duranensis) provides insights into geocarpy, oil biosynthesis, and allergens. Proc. Natl. Acad. Sci. U.S.A. 113, 6785–6790. doi: 10.1073/pnas.1600899113
Doyle, J. J., and Doyle, J. L. (1987). A rapid DNA isolation procedure for small quantities of fresh leaf tissue. Phytochem. Bull. 19, 11–15.
Ellegren, H. (2004). Microsatellites Simple sequences with complex evolution. Nat. Rev. Genet. 5, 435–445. doi: 10.1038/nrg1348
Gregory, W. C., and Gregory, M. P. (1976). “Groundnut,” in Evolution of Crop Plants, ed N. W. Simmonds (London: Longman Group Ltd.), 151–154.
Gupta, P. K., and Varshney, R. K. (2000). The development and use of microsatellite markers for genetics and plant breeding with emphasis on bread wheat. Euphytica 113, 163–185.
Halward, T. M., Stalker, H. T., LaRue, E. A., and Kochert, G. (1991). Genetic variation detectable with molecular markers among unadapted germ-plasm resources of cultivated peanut and related wild species. Genome 34, 1013–1020. doi: 10.1139/g91-156
He, G., and Prakash, C. S. (1997). Identification of polymorphic DNA markers in cultivated peanut (Arachis hypogaea L.). Euphytica 97, 143. doi: 10.1023/A:1002949813052
Huson, D. H., Richter, D. C., Rausch, C., Dezulian, T., Franz, M., and Rupp, R. (2007). Dendroscope: an interactive viewer for large phylogenetic trees. BMC Bioinformatics 8:460. doi: 10.1186/1471-2105-8-460
Knauft, D. A., and Gorbet, D. W. (1989). Genetic diversity among peanut cultivars. Crop Sci. 29, 1417–1422. doi: 10.2135/cropsci1989.0011183X002900060017x
Koppolu, R., Upadhyaya, H. D., Dwivedi, S. L., Hoisington, D. A., and Varshney, R. K. (2010). Genetic relationships among seven sections of genus Arachis studied by using SSR markers. BMC Plant Biol. 10:15. doi: 10.1186/1471-2229-10-15
Krapovickas, A., and Gregory, W. C. (1994). Taxonomia del genero Arachis (Leguminosae). Bonplandia 8, 1–186.
Krapovickas, A., and Gregory, W. C. (2007). Taxonomy of the genus Arachis (Leguminosae) translat by DE Williams, CE Simpson. Bonplandia 16, 1–205.
Krzywinski, M., Schein, J., Birol, I., Connors, J., Gascoyne, R., Horsman, D., et al. (2009). Circos: an information aesthetic for comparative genomics. Genome Res. 19, 1639–1645. doi: 10.1101/gr.092759.109
Lavia, G. I. (1998). Karyotypes of Arachis palustris and A. praecox (Section Arachis), two species with basic chromosome number x = 9. Cytologia 63, 177–181.
Leal-Bertioli, S. C. M., Santos, S. P., Dantas, K. M., Inglis, P. W., Nielen, S., Araujo, A. C., et al. (2014). Arachis batizocoi: a study of its relationship to cultivated peanut (A. hypogaea) and its potential for introgression of wild genes into the peanut crop using induced allotetraploids. Ann. Bot. 115, 237–249. doi: 10.1093/aob/mcu237
Liang, X., Chen, X., Hong, Y., Liu, H., Zhou, G., Li, S., et al. (2009). Utility of EST-derived SSR in cultivated peanut (Arachis hypogaea L.) and Arachis wild species. BMC Plant Biol. 9:35. doi: 10.1186/1471-2229-9-35
Liu, K., and Muse, S. V. (2005). PowerMarker: an integrated analysis environment for genetic marker analysis. Bioinformatics 21, 2128–2129. doi: 10.1093/bioinformatics/bti282
Liu, L., Dang, P. M., and Chen, C. Y. (2015). Development and utilization of indel markers to identify peanut (Arachis hypogaea) disease resistance. Front. Plant Sci. 6:988. doi: 10.3389/fpls.2015.00988
Lu, Y., Cui, X., Li, R., Huang, P., Zong, J., Yao, D., et al. (2015). Development of genome-wide insertion/deletion markers in rice based on graphic pipeline platform. J. Integr. Plant Biol. 57, 980–991. doi: 10.1111/jipb.12354
Mace, E. S., Buhariwalla, H. K., and Crouch, J. H. (2003). A high-throughput DNA extraction protocol for tropical molecular breeding programs. Plant Mol. Biol. Rep. 21, 459–460. doi: 10.1007/BF02772596
Mallikarjuna, N., Tandra, S. K., and Jadhav, D. R. (2006). Arachis hoehnei the probable B genome donor of Arachis hypogaea based on crossability, cytogenetical and molecular studies. J. SAT. Agri. Res. 2, 1–2.
Moghaddam, S. M., Song, Q., Mamidi, S., Schmutz, J., Lee, R., Cregan, P., et al. (2014). Developing market class specific InDel markers from next generation sequence data in Phaseolus vulgaris L. Front. Plant Sci. 5:185. doi: 10.3389/fpls.2014.00185
Moretzsohn, Mde. C., Hopkins, M. S., Mitchell, S. E., Kresovich, S., Valls, J. F., and Ferreira, M. E. (2004). Genetic diversity of peanut (Arachis hypogaea L.) and its wild relatives based on the analysis of hypervariable regions of the genome. BMC Plant Biol. 4:11. doi: 10.1186/1471-2229-4-11
Pandey, M. K., Gautami, B., Jayakumar, T., Sriswathi, M., Upadhyaya, H. D., Chennabyre Gowda, C. V., et al. (2012a). Highly informative genic and genomic SSR markers to facilitate molecular breeding in cultivated groundnut (Arachis hypogaea L.). Plant Breed. 131, 139–147. doi: 10.1111/j.1439-0523.2011.01911.x
Pandey, M. K., Monyo, E., Ozias-Akins, P., Liang, X., Guimarães, P., Nigam, S. N., et al. (2012b). Advances in Arachis genomics for peanut improvement. Biotechnol. Adv. 30, 639–651. doi: 10.1016/j.biotechadv.2011.11.001
Pandey, M. K., Roorkiwal, M., Singh, V. K., Ramalingam, A., Kudapa, H., Thudi, M., et al. (2016). Emerging genomic tools for legume breeding: current status and future prospects. Front. Plant Sci. 7:455. doi: 10.3389/fpls.2016.00455
Peakall, R., and Smouse, P. E. (2012). GenAlEx 6.5: genetic analysis in Excel. Population genetic software for teaching and research-an update. Bioinformatics 28, 2537–2539. doi: 10.1093/bioinformatics/bts460
Pearson, C. E., Edamura, N. K., and Cleary, J. D. (2005). Repeat instability: mechanisms of dynamic mutations. Nat. Rev. Genet. 6, 729–742. doi: 10.1038/nrg1689
Perrier, X., Flori, A., and Bonnot, F. (2003). “Data analysis methods,” in Genetic Diversity of Cultivated Tropical Plants, eds P. Hamon, M. Seguin, X. Perrier, and J. C. Glaszmann (Montpellier: Enfield Science Publishers), 43–76.
Pompanon, F., Bonin, A., Bellemain, E., and Taberlet, P. (2005). Genotyping errors: causes, consequences and solutions. Nat. Rev. Genet. 6, 847–859. doi: 10.1038/nrg1707
Pritchard, J. K., Stephens, M., and Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959.
Remington, D. L., Thornsberry, J. M., Matsuoka, Y., Wilson, L. M., Whitt, S. R., Doebley, J., et al. (2001). Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc. Natl. Acad. Sci. U.S.A. 98, 11479–11484. doi: 10.1073/pnas.201394398
Seijo, J. G., Lavia, G. I., Ferna'ndez, A., Krapovickas, A., Ducasse, D., and Moscone, E. A. (2004). Physical mapping of the 5S and 18S–25S rRNA genes by FISH as evidence that A. duranensis and A. ipaënsis are the wild diploid progenitors of A. hypogaea (Leguminosae). Am. J. Bot. 91, 1294–1303. doi: 10.3732/ajb.91.9.1294
Stalker, H. T. (2017). Utilizing wild species for peanut improvement. Crop Sci. 57, 1102–1120. doi: 10.2135/cropsci2016.09.0824
Subramanian, V., Gurtu, S., Nageswara Rao, R. C., and Nigam, S. N. (2000). Identification of DNA polymorphism in cultivated groundnut using random amplified polymorphic DNA (RAPD) assay. Genome 43, 656–660. doi: 10.1139/g00-034
Syvänen, A. C. (2001). Accessing genetic variation: genotyping single nucleotide polymorphisms. Nat. Rev. Genet. 2, 930–942. doi: 10.1038/35103535
Untergasser, A., Cutcutache, I., Koressaar, T., Ye, J., Faircloth, B. C., Remm, M., et al. (2012). Primer3 - new capabilities and interfaces. Nucleic Acids Res. 40:e115. doi: 10.1093/nar/gks596
Valls, J. F. M., and Simpson, C. E. (2005). New species of Arachis (Leguminosae) from Brazil, Paraguay and Bolivia. Bonplandia 14, 35–63. doi: 10.2307/41941266
Valls, J., Rao, V. R., Simpson, C., and Krapovickas, A. (1985). “Current status of collection and conservation of South American groundnut germplasm with emphasis on wild species of Arachis,” in Proceedings of an International Workshop on Cytogenetics of Arachis, ed J. P. Moss (Patancheru: ICRISAT Centre), 15–35.
Varshney, R. K. (2015). Exciting journey of 10 years from genomes to fields and markets: some 17 success stories of genomics-assisted breeding in chickpea, pigeonpea and groundnut. Plant Sci. 242, 98–107. doi: 10.1016/j.plantsci.2015.09.009
Varshney, R. K., Mohan, S. M., Gaur, P. M., Gangarao, N. V., Pandey, M. K., Bohra, A., et al. (2013). Achievements and prospects of genomics-assisted breeding in three legume crops of the semi-arid tropics. Biotechnol. Adv. 31, 1120–1134. doi: 10.1016/j.biotechadv.2013.01.001
Varshney, R. K., Nayak, S. N., May, G. D., and Jackson, S. A. (2009). Next-generation sequencing technologies and their implications for crop genetics and breeding. Trends Biotechnol. 27, 522–530.
Varshney, R. K., Pandey, M. K., Janila, P., Nigam, S. N., Sudini, H., Gowda, M. V. C., et al. (2014). Marker-assisted introgression of a QTL region to improve rust resistance in three elite and popular varieties of peanut (Arachis hypogaea L.). Theor. Appl. Genet. 127, 1771–1781. doi: 10.1007/s00122-014-2338-3
Wang, C. T., Wang, X. Z., Tang, Y. Y., Chen, D. Y., Cui, F. G., Zhang, J. C., et al. (2010). Phylogeny of Arachis based on internal transcribed spacer sequences. Genet. Resour. Crop Evol. 58, 311–319. doi: 10.1007/s10722-010-9576-2
Xiong, F., Zhong, R., Han, Z., Jiang, J., He, L., Zhuang, W., et al. (2011). Start codon targeted polymorphism for evaluation of functional genetic variation and relationships in cultivated peanut (Arachis hypogaea L.) genotypes. Mol. Biol. Rep. 38, 3487–3494. doi: 10.1007/s11033-010-0459-6
Yang, J., Wang, Y., Shen, H., and Yang, W. (2014). In silico identification and experimental validation of insertion-deletion polymorphisms in tomato genome. DNA Res. 21, 429–38. doi: 10.1093/dnares/dsu008
Young, N. D., Weeden, N. F., and Kochert, G. (1996). “Genome mapping in legumes (Family Fabaceae),” in Genome Mapping in Plants, ed N. H. Paterson (Austin, TX: Landes Biomedical Press), 211–277.
Zhao, Y. L., Prakash, C. S., and He, G. (2012). Characterization and compilation of polymorphic simple sequence repeat (SSR) markers of peanut from public database. BMC Res. Notes 5, 362–369. doi: 10.1186/1756-0500-5-362
Keywords: Arachis, InDel, genomes, phylogeny, sections
Citation: Vishwakarma MK, Kale SM, Sriswathi M, Naresh T, Shasidhar Y, Garg V, Pandey MK and Varshney RK (2017) Genome-Wide Discovery and Deployment of Insertions and Deletions Markers Provided Greater Insights on Species, Genomes, and Sections Relationships in the Genus Arachis. Front. Plant Sci. 8:2064. doi: 10.3389/fpls.2017.02064
Received: 27 June 2017; Accepted: 17 November 2017;
Published: 19 December 2017.
Edited by:
Eduard Akhunov, Kansas State University, United StatesReviewed by:
Robert Henry, The University of Queensland, AustraliaMartin Mascher, Leibniz-Institut für Pflanzengenetik und Kulturpflanzenforschung (IPK), Germany
Hengyou Zhang, University of North Carolina at Charlotte, United States
Copyright © 2017 Vishwakarma, Kale, Sriswathi, Naresh, Shasidhar, Garg, Pandey and Varshney. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manish K. Pandey, bS5wYW5kZXlAY2dpYXIub3Jn
Rajeev K. Varshney, ci5rLnZhcnNobmV5QGNnaWFyLm9yZw==
†Present Address: Manish K. Vishwakarma, Borlaug Institute for South Asia (BISA), Jabalpur, India