 Sandhya Verma
Sandhya Verma Rajesh K. Gazara
Rajesh K. Gazara Praveen K. Verma
Praveen K. Verma- Plant Immunity Laboratory, National Institute of Plant Genome Research, New Delhi, India
Transcription factors (TFs) are the key players in gene expression and their study is highly significant for shedding light on the molecular mechanisms and evolutionary history of organisms. During host–pathogen interaction, extensive reprogramming of gene expression facilitated by TFs is likely to occur in both host and pathogen. To date, the knowledge about TF repertoire in filamentous fungi is in infancy. The necrotrophic fungus Ascochyta rabiei, that causes destructive Ascochyta blight (AB) disease of chickpea (Cicer arietinum), demands more comprehensive study for better understanding of Ascochyta-legume pathosystem. In the present study, we performed the genome-wide identification and analysis of TFs in A. rabiei. Taking advantage of A. rabiei genome sequence, we used a bioinformatic approach to predict the TF repertoire of A. rabiei. For identification and classification of A. rabiei TFs, we designed a comprehensive pipeline using a combination of BLAST and InterProScan software. A total of 381 A. rabiei TFs were predicted and divided into 32 fungal specific families of TFs. The gene structure, domain organization and phylogenetic analysis of abundant families of A. rabiei TFs were also carried out. Comparative study of A. rabiei TFs with that of other necrotrophic, biotrophic, hemibiotrophic, symbiotic, and saprotrophic fungi was performed. It suggested presence of both conserved as well as unique features among them. Moreover, cis-acting elements on promoter sequences of earlier predicted A. rabiei secretome were also identified. With the help of published A. rabiei transcriptome data, the differential expression of TF and secretory protein coding genes was analyzed. Furthermore, comprehensive expression analysis of few selected A. rabiei TFs using quantitative real-time polymerase chain reaction revealed variety of expression patterns during host colonization. These genes were expressed in at least one of the time points tested post infection. Overall, this study illustrates the first genome-wide identification and analysis of TF repertoire of A. rabiei. This work would provide the basis for further studies to dissect role of TFs in the molecular mechanisms during A. rabiei–chickpea interactions.
Introduction
A very adaptable and economical source of protein available to mankind is legumes. Apart from having high nutritional value, legumes also serve as great natural soil fertilizers. They provide nitrogen to the crops by fixing atmospheric nitrogen, thereby, decreasing the use of artificial nitrogen fertilizers. This ultimately minimizes the side effects that artificial fertilizers impose on our environment. However, like all plants, legumes are also challenged by various biotic and abiotic stresses causing major yield loss worldwide. The defense responses of plants in combating these environmental stresses are crucial for completing their lifecycle successfully.
Chickpea is one of the important legume crops grown worldwide. Its global production is estimated to be 14 million metric tons (FAO, 2014). India is the largest producer with annual production of about 10 million tons that accounts for 70% of the total world production. However, chickpea faces Ascochyta blight (AB) as a major constraint to its production. It can result in 100% crop mortality and complete yield loss (Singh et al., 1984). Due to several epidemics of AB, substantial losses have been reported from India, Pakistan, Australia, Spain (Pande et al., 2005), Canada (Chandirasekaran et al., 2009), Latin America (Kaiser et al., 2000), and United States (Kaiser et al., 1994). AB is a foliar disease caused by the necrotrophic fungus Ascochyta rabiei (Pass.) Labrouse [teleomorph: Didymella rabiei (Kovachevski) v. Arx]. This fungus belongs to dothideomycetes class of filamentous fungi. It hibernates on crop residues and seeds between seasons and readily gets transmitted through infected seeds and airborne ascospores. The conidia present in the field sometimes leads to several cycles of infection in the same season when favorable conditions prevail. A. rabiei penetrates the host directly through the cuticle and sometimes through stomata as well, eventually resulting in necrosis (Jayakumar et al., 2005). Despite after several pathological and molecular studies, the pathogenicity mechanisms of A. rabiei are still poorly understood. However in recent years, an in-depth and path-breaking research has been done in A. rabiei that changed many of the conventional concepts. One such example is a report where it was revealed that the solanapyrones toxins of A. rabiei are not required for pathogenicity (Kim et al., 2015). This suggests that necrotrophic fungi exploit the cell death machinery of the host plant for pathogenesis, which abandons the earlier concept that necrotrophic fungi solely relies on lytic enzymes for causing necrosis. Recently, the transcriptome analysis of A. rabiei has been carried out and a number of transcripts and putatively secreted proteins coding transcripts up-regulated during infection were identified (Fondevilla et al., 2015).
In order to solve the mystery of plant–pathogen interaction, the intricate biological mechanisms of the pathogen needs to be well addressed. Various signaling components and downstream factors play crucial role in connecting the specific pathways into an interlaced regulatory network. Among such components, transcription factors (TFs) are vital for regulating numerous mechanisms and responses. TFs are proteins that have at least one DNA-binding domain (DBD) and an activation domain (AD). The DBD binds to the sequence-specific cis-acting elements present in promoters of the target genes. On the other hand, the AD triggers transcription from promoter by recruiting the transcriptional machinery. Besides these, other domains might occur to facilitate homo/hetero-dimerization or the binding with co-activators or co-repressors. The DBDs of TFs are usually highly conserved and are the basis of classification of TFs into superclasses and classes (Stegmaier et al., 2004). By contrast, the ADs are far less conserved.
Knowledge about the TFs repertoire in phytopathogenic fungi is still very limited. Most of the studies have been carried out in Saccharomyces cerevisiae and Schizosaccharomyces pombe (Spitz and Furlong, 2012). With the rise in ease and availability of whole genome and transcriptome data, genome-wide identification of TFs prove to be highly beneficial to get insights of the TF repertoire present in an organism. Genome-wide search and functional identification of TFs has been carried out in the mycorrhizal fungus Tuber melanosporum (Montanini et al., 2011). A bioinformatics approach in combination with functional analysis in yeast and transcriptome profiling was performed to identify T. melanosporum TFs. Montanini et al. (2011) found that T. melanosporum consists of 102 homologs of previously characterized TFs, 57 homologs of hypothetical TFs, and 42 putative TFs apparently specific to Tuber. About one-fifth of the in silico predicted TFs of T. melanosporum were validated by yeast screen. Moreover, 29 TFs were up-regulated in ectomycorrhiza or fruiting bodies. There are other numerous studies and databases related to genome-wide search of TFs in plants (Guo et al., 2008; Mochida et al., 2010), mouse (Kanamori et al., 2004; Zheng et al., 2008), Drosophila melanogaster (Adryan and Teichmann, 2006), human (Lee et al., 2007; Zheng et al., 2008; Fulton et al., 2009), and rat (Zheng et al., 2008). Similarly, AnimalTFDB is a comprehensive TF database for 50 animal species ranging from Caenorhabditis elegans to human (Zhang et al., 2012). TRANSFAC database and its module TRANSCompel are available to study transcriptional gene regulation in eukaryotes (Matys et al., 2006). The DBD database consists of predicted TF repertoires for 930 completely sequenced genomes of eukaryotes and prokaryotes (Wilson et al., 2008). Still there are limited resources available to study TFs in case of phytopathogenic fungi.
With the available genome of A. rabiei (Verma et al., 2016), we have the unique prospect to identify and study a global view of putative TFs repertoire present in A. rabiei, for the first time. In the present study, the aim was to obtain the largest possible catalog of DBD-containing proteins which are the bona fide transcriptional regulators encoded by A. rabiei genome. The A. rabiei putative TF repertoire has been compared with that of several other filamentous fungi. Gene structure and phylogenetic analyses have been performed to shed light on the basic information about putative TFs of A. rabiei. In order to gain clues regarding expression profiles of these putative TFs and their relevance in regulation of A. rabiei secretome during infection, we explored the published transcriptomic profiles in the mycelium and infected chickpea leaves (Fondevilla et al., 2015). The results obtained herein gives us insight into the putative TF repertoire of A. rabiei and provides a significant basis for future studies on functional characterization of TFs involved in various biological processes of the phytopathogenic fungi.
Materials and Methods
Data Collection and Identification of A. rabiei Transcription Factors
The list of TFs of selected 30 fungal species was obtained from the publicly available databases: DBD release 2.01 and FTFD v1.22. To acquire the protein sequences of TFs, the proteomes of these fungi were downloaded from JGI. Using the protein IDs, the corresponding protein sequences of TFs were retrieved for proteomes and were used as a database. A total of 10,596 protein sequences of A. rabiei (Supplementary Dataset S1) were used as queries to identify all possible TFs by performing BLASTP (e-value cutoff ≤ 1e-5) search against TFs of 30 fungal species as database. This resulted in 1,854 hits as subject sequences. InterProScan v5.21-60.03 was employed to identify TF DBDs belonging to 12 superfamilies and 37 PFAM families in fungi in the resultant 1,854 hits. A total of 392 protein sequences had DBDs and these sequences were used to perform BLASTP with FTFD at default parameters. Out of 392, 376 protein sequences returned significant hits, while 16 protein sequences did not show any hits. These 16 protein sequences were further used as queries to perform BLAST searches in NCBI and 5 of them showed significant similarity to fungal TFs in the database. Therefore, a total of 381 putative TFs were predicted.
Gene Structure Analysis, Domain Organization, and Phylogenetic Analysis
The exon and intron structures of individual TF coding genes belonging to different families were illustrated with the help of Gene Structure Display Server v2.04 (Hu et al., 2015) by aligning the cDNA sequences with the corresponding genomic DNA sequences. The functional motifs or domains of putative TF protein sequences were analyzed using CDvist5 (Adebali et al., 2015). For phylogenetic analysis, multiple sequence alignments of the full-length protein sequences were performed using MUSCLE v3.8.316 with maxiters set at 1000. The acquired alignment was used to carry out phylogenetic analyses using Bayesian Inference (BI) implemented in MrBayes v3.2.6 (Ronquist and Huelsenbeck, 2003). The protein sequence alignments were run over 3,000,000 generations under a mixed amino acid substitution model with two independent runs and each containing four Markov Chain Monte Carlo (MCMC) chains. To estimate the posterior probabilities for each node, a sampling frequency was set at 300 iterations with MCMC left at default settings. The consensus tree was finally generated with the help of Sumt function of MrBayes. By removing the burn-in generations for each run the posterior probabilities were estimated. All the phylogenetic trees were visualized using FigTree v1.47.
De Novo Motif Discovery
The RSAT suite for fungi (Regulatory Sequence Analysis Tools8) was used to identify motifs enriched in the 1 kb promoter sequences of genes encoding secretory proteins using pleosporales as the background model and 6-, 7-, or 8-bp-long seeds on both the strands. Once we had the cis-regulatory elements identified across the promoter regions, annotation of cis-regulatory elements were carried out by scanning UniPROBE database9 (Newburger and Bulyk, 2009).
Identification of Differentially Expressed Genes
RNA-seq reads available for A. rabiei grown “in medium” and “in planta” at time points 12, 36, and 96 hours post inoculation (hpi) were downloaded from NCBI (Fondevilla et al., 2015). Reads were mapped on the genome of A. rabiei using TopHat2.1.0 software (Kim et al., 2013). Transcriptional levels were estimated with Cufflinks v2.1.1 (Trapnell et al., 2013) and normalized by fragments per kilobase of transcript per million mapped reads (FPKM). The differential expression between in medium and in planta at different time points was determined by Cuffdiff v2.2.1 (Trapnell et al., 2013). The transcripts with difference of at least two-fold change along with P-value ≤ 0.05 were considered to be significantly differentially expressed. The P-value was generated by Benjamini–Hochberg correction for multiple tests running in the background of Cuffdiff v2.2.1.
Culture Conditions and Plant Infection
Vegetative mycelia of A. rabiei isolate ArD2 (Indian Type Culture Collection No. 4638) were grown on potato dextrose agar (PDA; Difco Laboratories, United states) for 20 days in dark. For harvesting fungal tissue, A. rabiei spores were grown in potato dextrose broth (PDB; Difco Laboratories, United states) for 5 days in dark at 22°C in an incubator shaker at 120 rpm. Mycelial samples were collected, immediately frozen in liquid nitrogen and stored at -80°C. For plant infection, 21 days old chickpea seedlings (Pusa-362) were spray inoculated with A. rabiei spore suspensions of concentration 2 × 105 spores/ml. Infected leaves and stems were harvested at 12, 24, 72, and 144 hpi with three biological replicates and stored as above.
RNA Isolation and Quantitative Real-Time PCR
Total RNA was isolated from A. rabiei infected chickpea samples using the TRIzol® reagent (Invitrogen, United states). The contaminating genomic DNA was removed by treating samples with RNase-free RQ1 DNase (Promega, United states) as per the manufacturer’s instruction. One microgram of total RNA primed with Oligo-dT was used for first-strand cDNA synthesis using High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, United states), according to given instructions in the manual. Primer pairs were designed from the untranslated regions (UTRs) of the target genes using Primer Express® (version 3.0) software with the default parameters. For the internal control, elongation factor1-alpha (ArEF1a) was used. Each primer combination gave specific amplification of single desired band. Moreover, only one melting temperature was observed for each primer pair while dissociation curve testing. Quantitative real-time PCR (qRT-PCR) was carried out using the PowerUpTM SYBR® Green Master Mix (ABI) in a 7900HT Real-Time PCR System. Each reaction mixture contained 10 μl of SYBR® Green PCR Master Mix, 2 μl of cDNA, and 18 pmol of each primer in a final volume of 20 μl. The thermal cycling parameters were 2 min at 50°C, 10 min at 95°C, and 40 cycles of 15 s at 95°C, and 1 min at 60°C. Each reaction was performed in triplicate. Transcripts of each gene and reference gene ArEF1a were amplified using the primers listed in Supplementary Table S1. The obtained values for each gene were then normalized according to the CT values of ArEF1a (Singh et al., 2012; Nizam et al., 2014b). Relative gene expression levels were calculated using the ΔΔCT method.
Results
Genome-Wide Identification and Classification of A. rabiei Transcription Factors
There are no bioinformatics tools available to predict the putative TFs in filamentous fungi. Therefore, a systematic workflow was employed in order to identify the putative TFs encoded by A. rabiei genome (Figure 1). For this, first of all, a comprehensive list of 30 different fungal species was prepared (Table 1). These fungal species were selected on the basis of their ecology and lifestyle. The list consisted of 26 fungal species belonging to Dothideomycetes, Eurotiomycetes, Leotiomycetes, Sordariomycetes, Saccharomycetes, and Schizosaccharomycetes classes of ascomycetes and also 4 fungal species from the phylum basidiomycetes. They represented different forms of lifestyle such as necrotrophy, hemibiotrophy, biotrophy, saprotrophy, symbiosis, and animal pathogens. These criteria were chosen to ensure maximum possible coverage while predicting the putative TFs of A. rabiei. The list of predicted TFs of these 30 fungal species was extracted from DBD: a TF prediction database (Wilson et al., 2008) and Fungal Transcription Factor Database (FTFD) (Park et al., 2008). Since these databases provided only the list of TFs and not their protein sequences, thus, the proteomes of the 30 fungal species were downloaded to obtain the corresponding protein sequences. Now these predicted TFs of the 30 fungal species was used as database to predict putative TFs of A. rabiei. BLASTP search was carried out using 10,596 protein sequences of A. rabiei as query against TFs of 30 species as database. A total of 1,854 hits were obtained. In fungi, 12 superfamilies and 37 PFAM families of TF DBDs are predicted to exist (Shelest, 2008). Considering this, SUPERFAMILY (Gough et al., 2001) and PFAM family (Finn et al., 2014) search were employed on 1,854 resultant hits using InterProScan (Jones et al., 2014). A total of 392 proteins were predicted to have TF DBDs from 12 superfamilies and 37 PFAM families. To minimize false predictions, each of the 392 putative TFs was employed for BLAST search in FTFD database. Sixteen putative TFs that failed to show any match in FTFD database were searched in NCBI database and were selected if they displayed significant homology with any known TF. From these analyses, a set of 381 proteins were obtained with significant hits (Supplementary Dataset S2) and were designated as the putative TFs of A. rabiei.
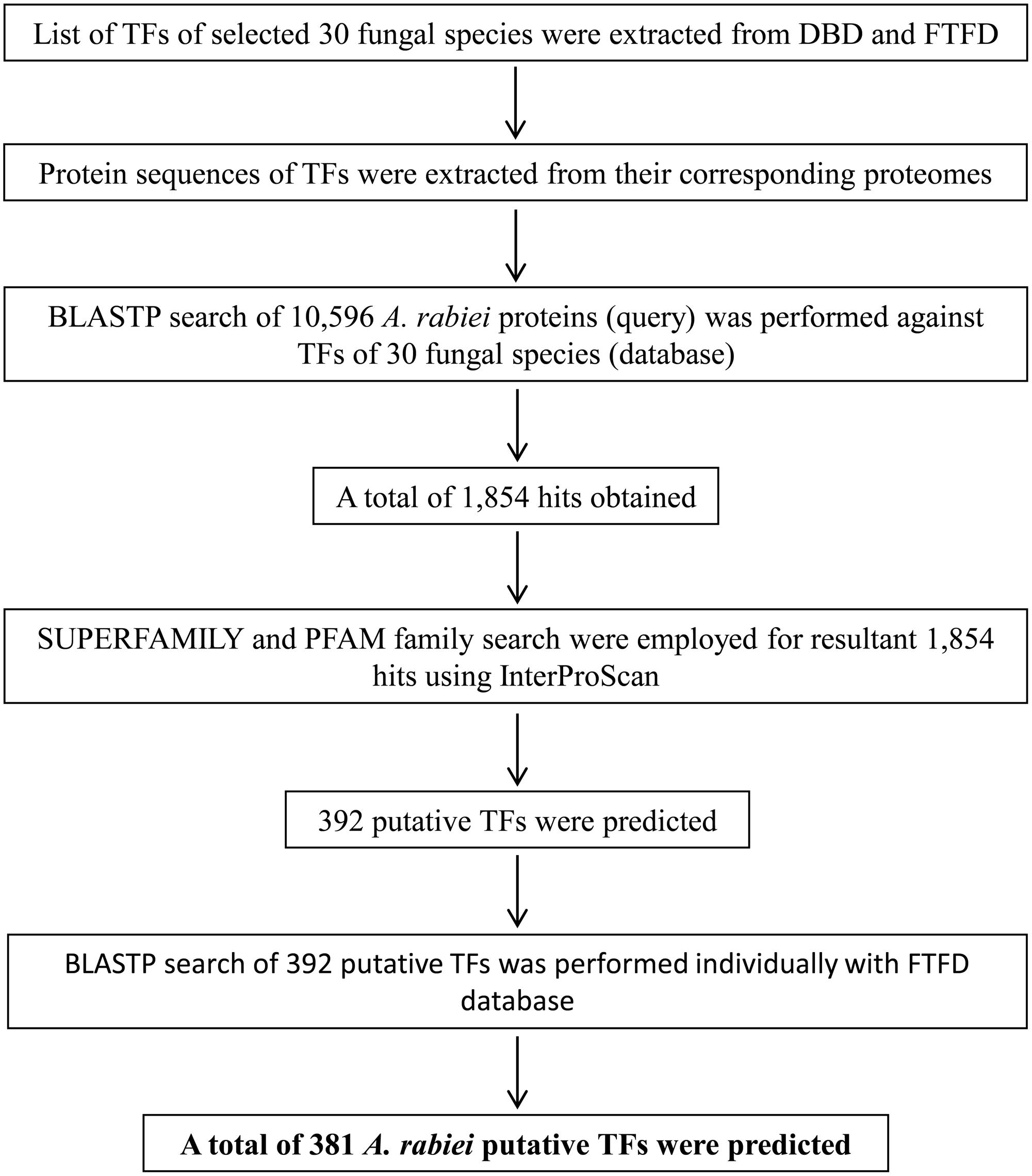
FIGURE 1. Overview of the computational pipeline used to identify the putative TFs of A. rabiei. Based on sequence similarity with characterized TFs from other fungi and the presence of conserved DNA-binding domain regions, proteins were assigned as TFs. A set of 381 putative TFs were predicted in the genome of A. rabiei.
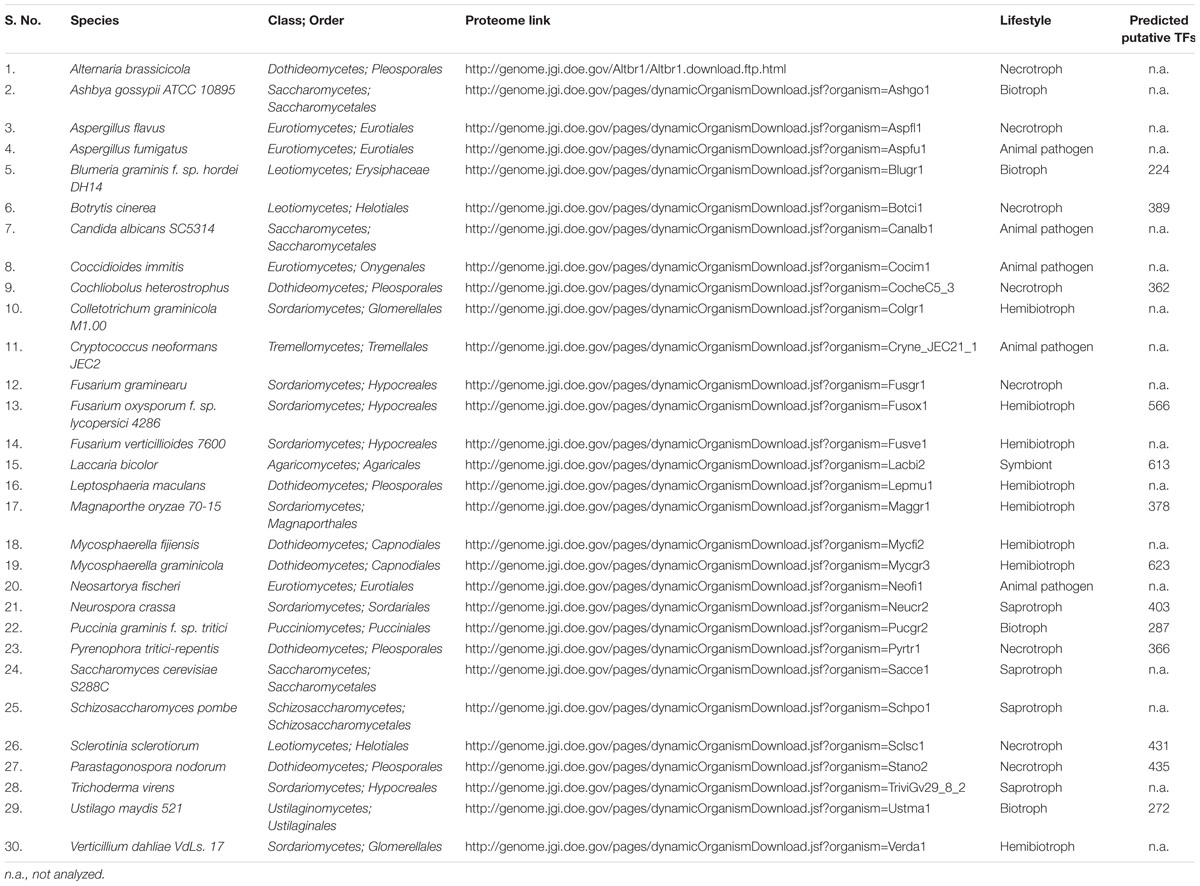
TABLE 1. List of fungal species used in this study.
Among the 381 putative TFs of A. rabiei, 142 and 76 proteins showed significant matches in SUPERFAMILY and PFAM family databases, respectively, whereas 163 proteins were common in both (Figure 2A). These 381 putative TFs were then classified and annotated on the basis of 83 InterPro terms present in the InterPro database (Finn et al., 2017), which is also the basis of fungal TF annotation in FTFD database. It grouped 381 A. rabiei putative TFs into 32 families (Supplementary Table S2). The maximum number of putative TFs (150) belonged to zinc-cluster superfamily [Zn(II)2Cys6], which is the largest class of fungal-specific domains. This was followed by C2H2 type zinc finger domains having 61 putative TFs. The third abundant family of putative TFs in A. rabiei was nucleic acid-binding-OB-fold with 45 proteins.
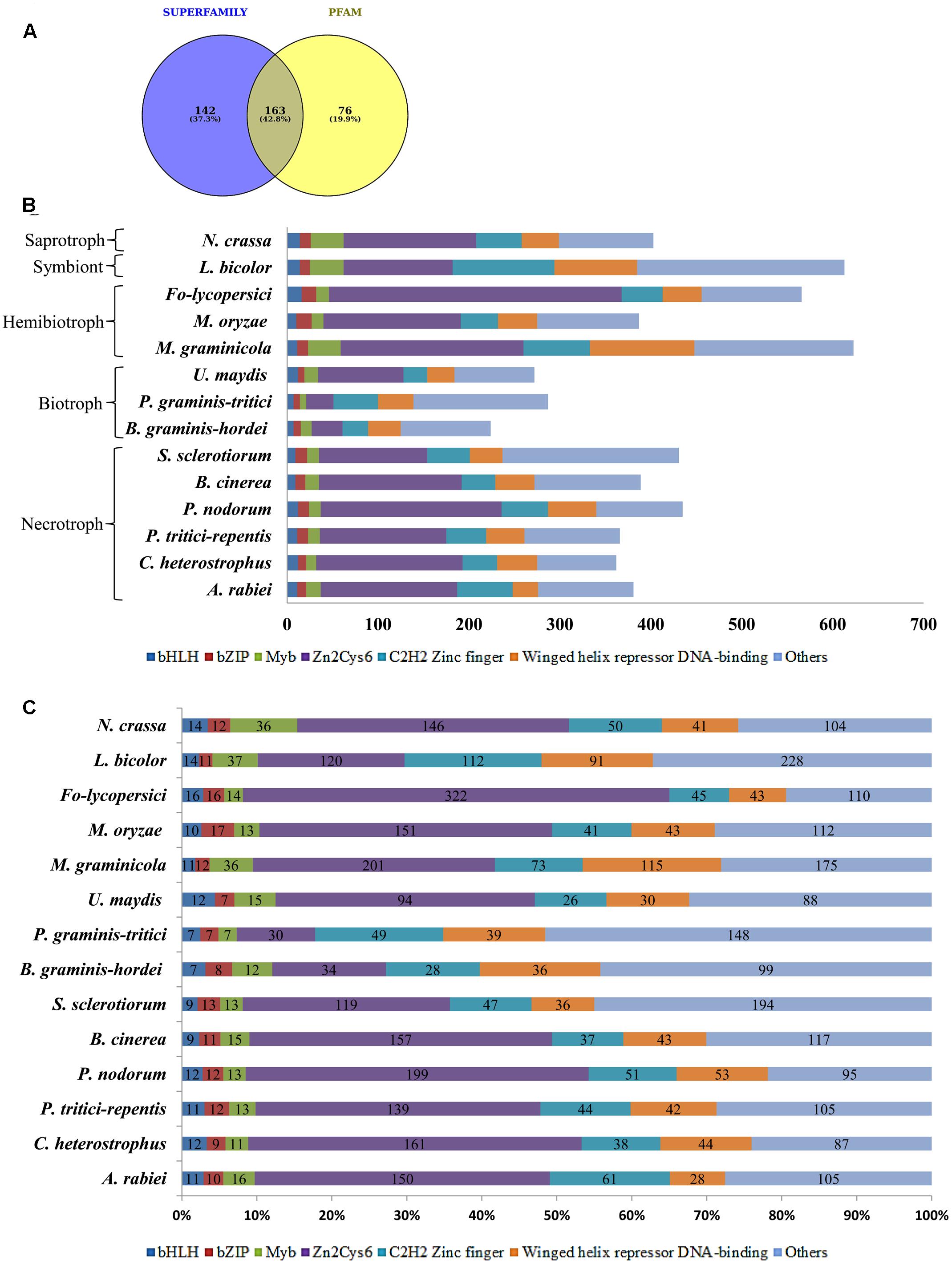
FIGURE 2. Distribution of A. rabiei putative TFs among different structural categories. (A) Out of 381 putative TFs predicted, 142 had significant matches with fungi specific 12 superfamilies and 76 had similarities with fungi specific 37 PFAM families (Shelest, 2008). Whereas 163 proteins showed matches in both SUPERFAMILY and PFAM family database. (B) The A. rabiei putative TFs were compared with TF repertoire of other necrotrophs, biotrophs, hemibiotrophs, symbiont, and saprotroph. The DNA binding domains considered for comparison were winged helix repressor DNA-binding, C2H2 zinc finger, [Zn(II)2Cys6], Myb, bZIP, and bHLH domains. The less represented DNA binding domains were categorized under others. (C) The relative abundance of each of the selected TF families across the fungal species is shown. The numbers inside the bars of graph are the absolute number of TFs in that family.
Comparison of A. rabiei Transcription Factors with Other Fungal Species
In order to compare A. rabiei putative TF repertoire with other fungal genomes, 13 fungal species were selected representing the major lifestyles. Following the same workflow as for A. rabiei, putative TFs were predicted for these 13 fungal species (Table 1). The number of predicted TFs in closely related necrotrophic fungi Cochliobolus heterostrophus and Pyrenophora tritici-repentis were 362 and 366, respectively, similar to the 381 putative TFs of A. rabiei. A set of 389 putative TFs were predicted in Botrytis cinerea. In Parastagonospora nodorum and Sclerotinia sclerotiorum, 435 and 431 putative TFs were predicted, respectively. Interestingly, the predicted putative TFs in selected biotrophic fungi, i.e., Blumeria graminis f. sp. hordei (224), Puccinia graminis f. sp. tritici (287), and Ustilago maydis (272) were comparatively fewer than the number of putative TFs predicted in necrotrophic fungi. Also, the hemibiotrophic fungi Mycosphaerella graminicola (623) and Fusarium oxysporum f. sp. lycopersici (566), and symbiont Laccaria bicolor (613) had significantly higher number of predicted TFs. However, the hemibiotrophic fungi Magnaporthe oryzae (378) and saprotroph Neurospora crassa (403) had putative TFs close to putative TFs predicted for A. rabiei.
The predicted TFs from 13 fungal species were classified and annotated on the basis of 83 InterPro terms (Supplementary Table S2). All the fungal species, except B. graminis f. sp. hordei and P. graminis f. sp. tritici, had the highest number of putative TFs belonging to zinc-cluster superfamily [Zn(II)2Cys6]. The HMG (High Mobility Group) and AraC type helix-turn-helix family TFs were abundant in A. rabiei similar to M. graminicola and L. bicolor. The Myb TFs were also more in A. rabiei as compared to other necrotrophs. Also, C2H2 zinc finger TFs were significantly higher in A. rabiei unlike most of the selected fungal species, particularly biotrophic fungi (Figures 2B,C). The winged helix repressor DNA-binding family was little underrepresented in A. rabiei compared to other fungi. This indicates that A. rabiei putative TFs are distributed among different classes where the total number of predicted TFs and their distribution into distinct classes differ marginally from those of the selected biotrophic, hemibiotrophic, and symbiotic fungi.
Gene Structure Analysis of Most Prevalent A. rabiei Transcription Factor Families
To explore the structural diversity of most abundant putative TFs of A. rabiei, we analyzed their exon–intron organization. In the most prevalent zinc-cluster superfamily [Zn(II)2Cys6] having 150 putative TFs, 10 genes were intronless while 28 had single intron (Supplementary Figure S1). The intron phases for all the genes were also analyzed. There are three different phase classes to which introns can be assigned: phase 0, 1, and 2. Phase 0 intron locates between two codons while phase 1 intron splits codons between the first and second nucleotides, and intron is said to be phase 2 when it splits codons between the second and third nucleotides. Half of the single intronic genes encoding zinc-cluster [Zn(II)2Cys6] TFs had intron phase 0. In C2H2 zinc finger TFs, 13 genes were equally found intronless and single intronic with phase 1 intron as the most prevalent one (Supplementary Figure S2). The third most abundant putative TF family, nucleic acid-binding-OB-fold, had 6 intronless genes and 13 single intron genes with most of them having intron phase 1 (Supplementary Figure S3). Out of 28 winged helix repressor DNA-binding TFs, 2 were intronless and 6 were single intron genes with phase 1 as the most common intron phase (Figure 3A). In Myb TF family, intronless genes were 6 and single intron genes were 5 and had intron phase 0 in most of them (Figure 3B). Altogether, this analysis reveals that single intron genes with intron phase either 0 or 1 were predominant in the abundantly found putative TFs of A. rabiei.
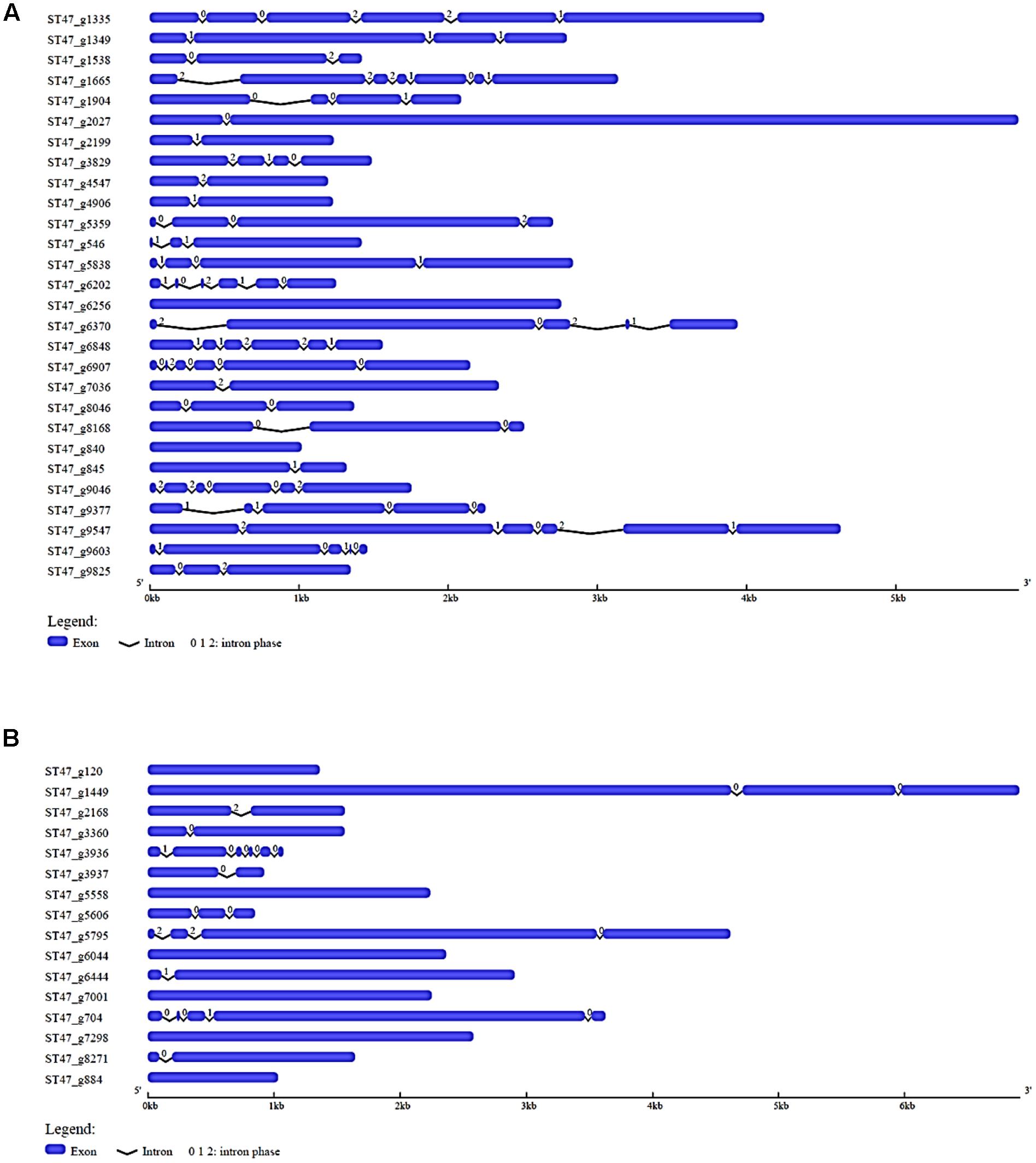
FIGURE 3. Gene structure analysis. The exon–intron organization is shown for (A) winged helix repressor DNA-binding, and (B) Myb family of A. rabiei putative TFs. Exons and introns are represented by blue rectangles and black lines, respectively. The numbers 0, 1, and 2 represent the intron phase.
Domain Organization and Phylogenetic Analysis of Myb, bHLH, and bZIP Transcription Factor Families
Owing to the significant biological functions of TFs, we studied their domain architecture and phylogenetic relationships to gain evolutionary insights. The three families of putative TFs, Myb, bHLH, and bZIP, showed presence of characteristic domains in their protein sequences (Figure 4). The Myb TFs had numerous low-complexity regions which are the regions containing little diversity in their amino acid composition (Figure 4A). Basic helix-loop-helix domain was present in each of the members of bHLH TFs; however, the size of domain was varying (Figure 4B). Similarly, all bZIP TFs of A. rabiei had a bZIP domain of uniform size with coiled coil regions overlapping with the bZIP domain (Figure 4C). Both these domains are important for the dimerization of the proteins.

FIGURE 4. Structural analysis of putative TFs. The protein domains of (A) Myb, (B) bHLH, and (C) bZIP family of A. rabiei putative TFs are shown. The domains are denoted by black outlined hollow rectangles, whereas coiled coil regions and low complexity regions are represented by green and pink rectangles, respectively.
Phylogenetic analysis of Myb TFs of A. rabiei with other closely related necrotrophic fungi, i.e., C. heterostrophus, P. tritici-repentis and P. nodorum was performed. It showed that two major clades were formed in C. heterostrophus, P. tritici-repentis, and P. nodorum (Figure 5). On the contrary, A. rabiei had Myb TFs grouped into single clade only. The bHLH TFs showed much conserved distribution in the selected fungal species. They were distributed into two major clades in all of the four fungi (Figure 6). Similarly, the bZIP TFs were also divided into two clades (Figure 7). The bHLH and bZIP TFs particularly showed very similar pattern of phylogenetic tree between A. rabiei and C. heterostrophus. In the same way, P. tritici-repentis and P. nodorum had identical phylogenetic relationship of these TFs. This suggests that these putative TFs have evolved in highly conserved manner in A. rabiei and other closely related necrotrophic fungi.
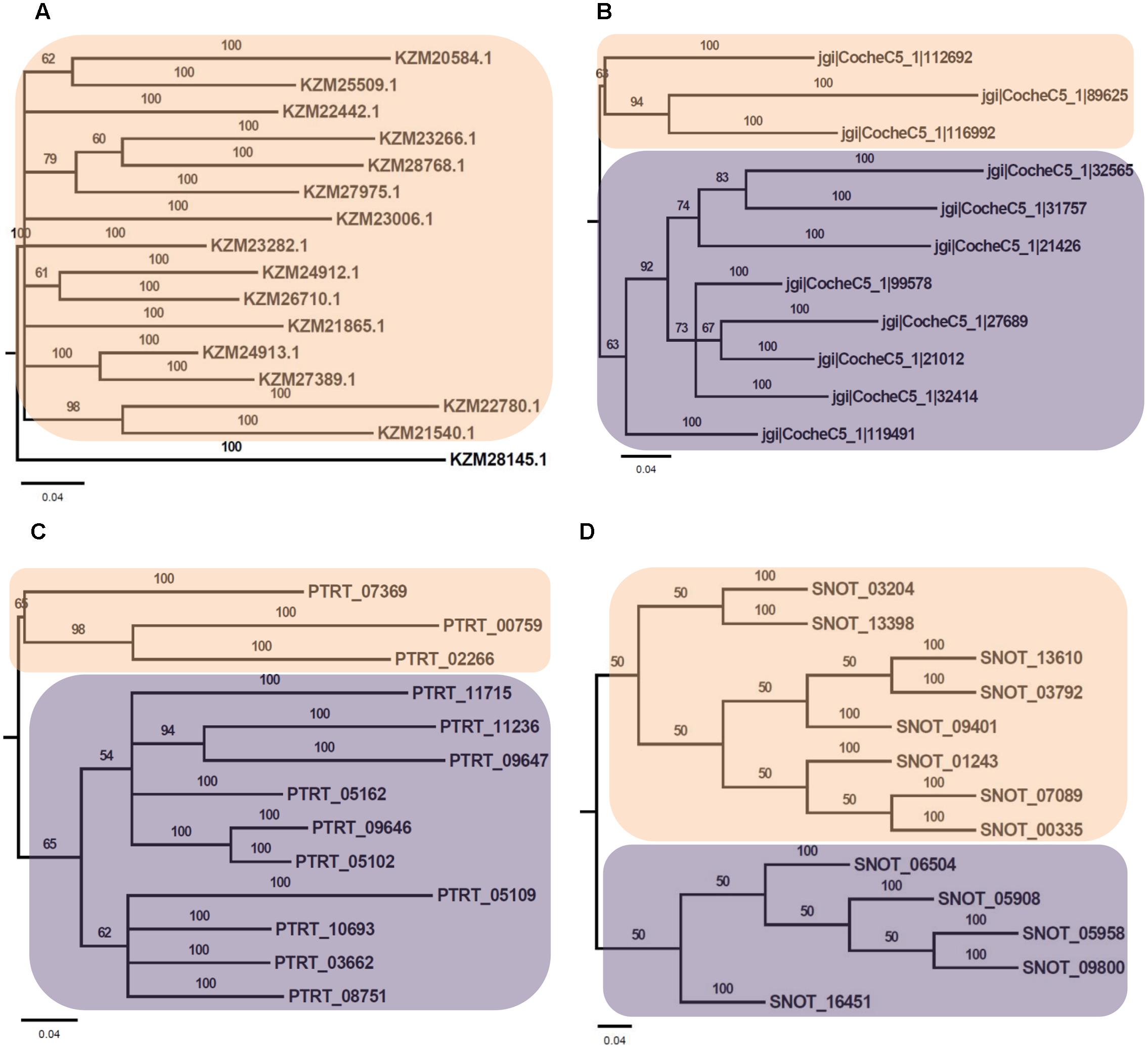
FIGURE 5. Phylogenetic analysis of Myb family of putative TFs. (A) The evolutionary relationship of Myb family of A. rabiei putative TFs was compared to that of (B) C. heterostrophus, (C) P. tritici-repentis, and (D) P. nodorum based on Bayesian inference analysis. Each clade is highlighted by colored rectangular block. The Bayesian posterior probabilities are indicated at the nodes.
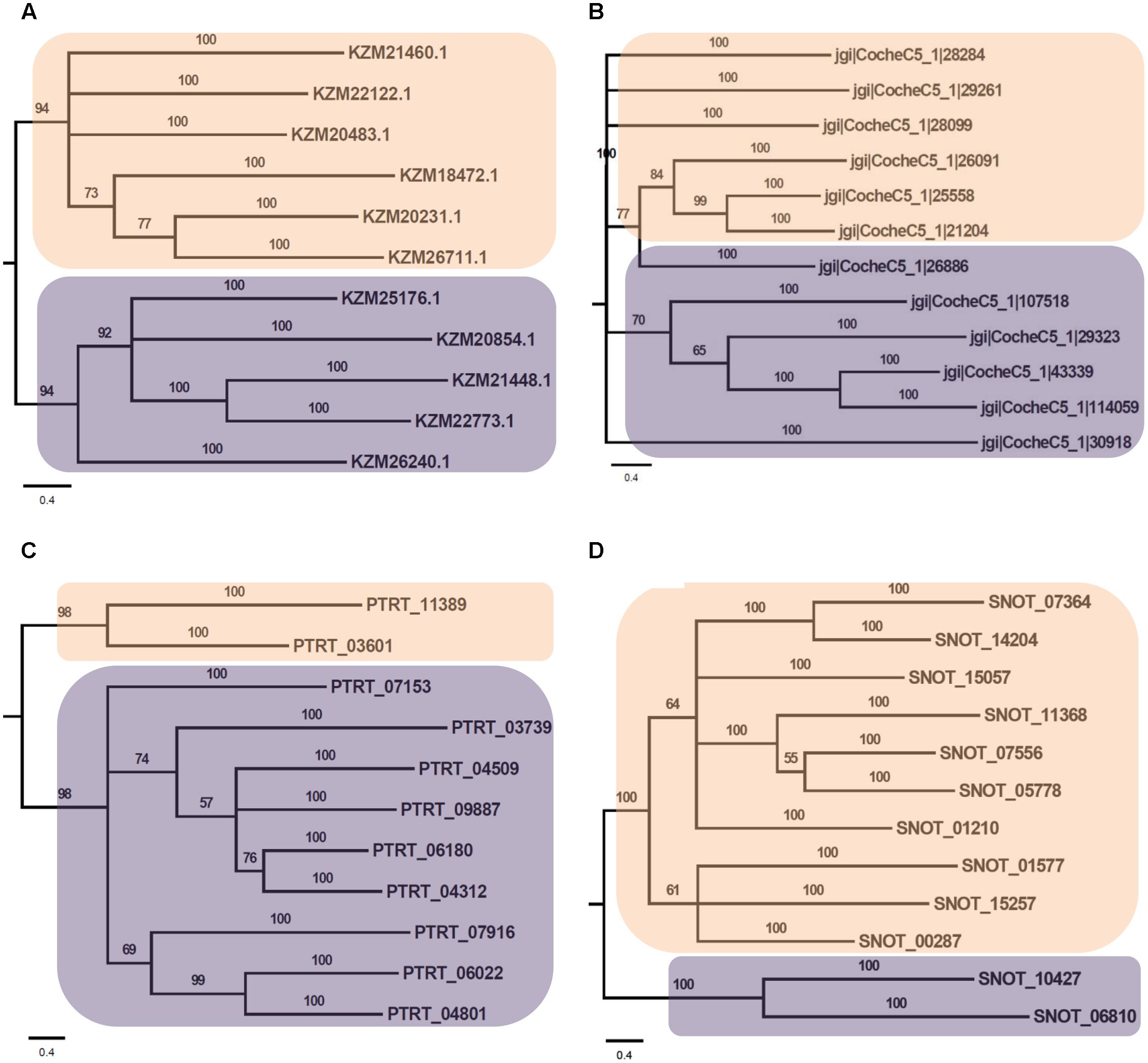
FIGURE 6. Phylogenetic analysis of bHLH family of putative TFs. (A) The evolutionary relationship of bHLH family of A. rabiei putative TFs was compared to that of (B) C. heterostrophus, (C) P. tritici-repentis, and (D) P. nodorum based on Bayesian inference analysis. Each clade is highlighted by colored rectangular block. The Bayesian posterior probabilities are indicated at the nodes.
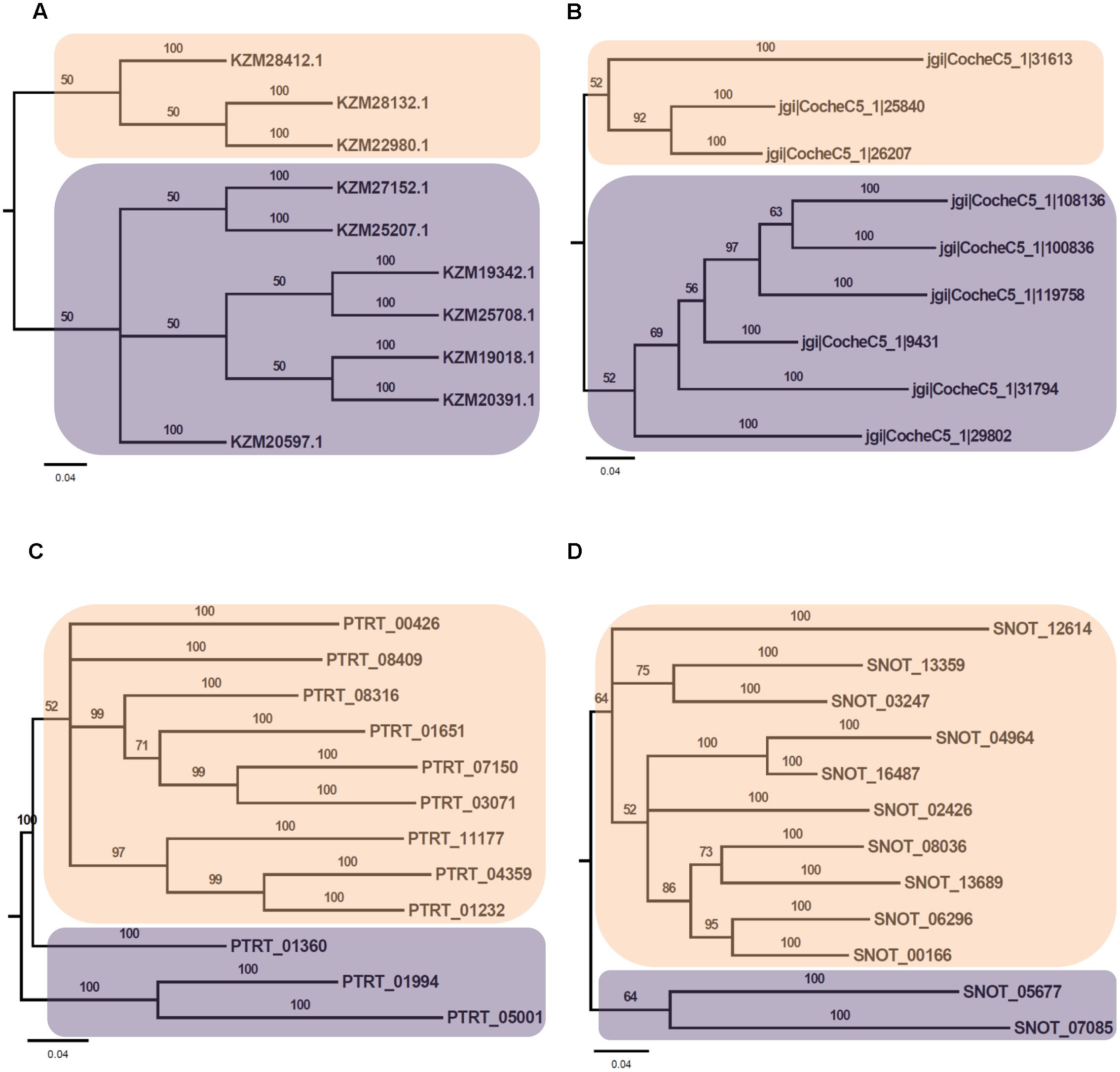
FIGURE 7. Phylogenetic analysis of bZIP family of putative TFs. (A) The evolutionary relationship of bZIP family of A. rabiei putative TFs was compared to that of (B) C. heterostrophus, (C) P. tritici-repentis, and (D) P. nodorum based on Bayesian inference analysis. Each clade is highlighted by colored rectangular block. The Bayesian posterior probabilities are indicated at the nodes.
Cis-Regulatory Elements in Promoter of A. rabiei Genes Encoding Secretory Proteins
We earlier reported the necrotrophic effector repertoire of A. rabiei (Verma et al., 2016). A set of 758 putative secretory proteins were predicted to constitute the secretome. Therefore, we investigated the cis-regulatory elements present in the promoter sequences of those A. rabiei genes that encode secretory proteins. This would aid in identifying the putative TFs that regulate the co-ordinated expression of A. rabiei secretome. For this, up to 1 kb 5′ flanking sequence of the genes encoding putative secretory proteins were selected and any other gene sequences occurring in these promoter regions were discarded. A scan in promoters of the 758 genes was performed using RSAT suite (Medina-Rivera et al., 2015) to obtain highly frequent DNA patterns known to be the binding sites of TFs. After scanning, the 10 most frequent motifs were identified (Figure 8 and Table 2). The motif CATCAACACCAC was the most recurrent motif that was predicted to bind to the promoter regions of 234 genes out of 758 genes encoding secretory proteins and had 316 numbers of occurrences. This was followed by CATCTCCACCAC motif that was identified in the promoters of 222 genes with 303 instances of occurrences. The third most abundant motif was TCCTTCCCC, which was present at 216 promoter sequences and had 273 matches.
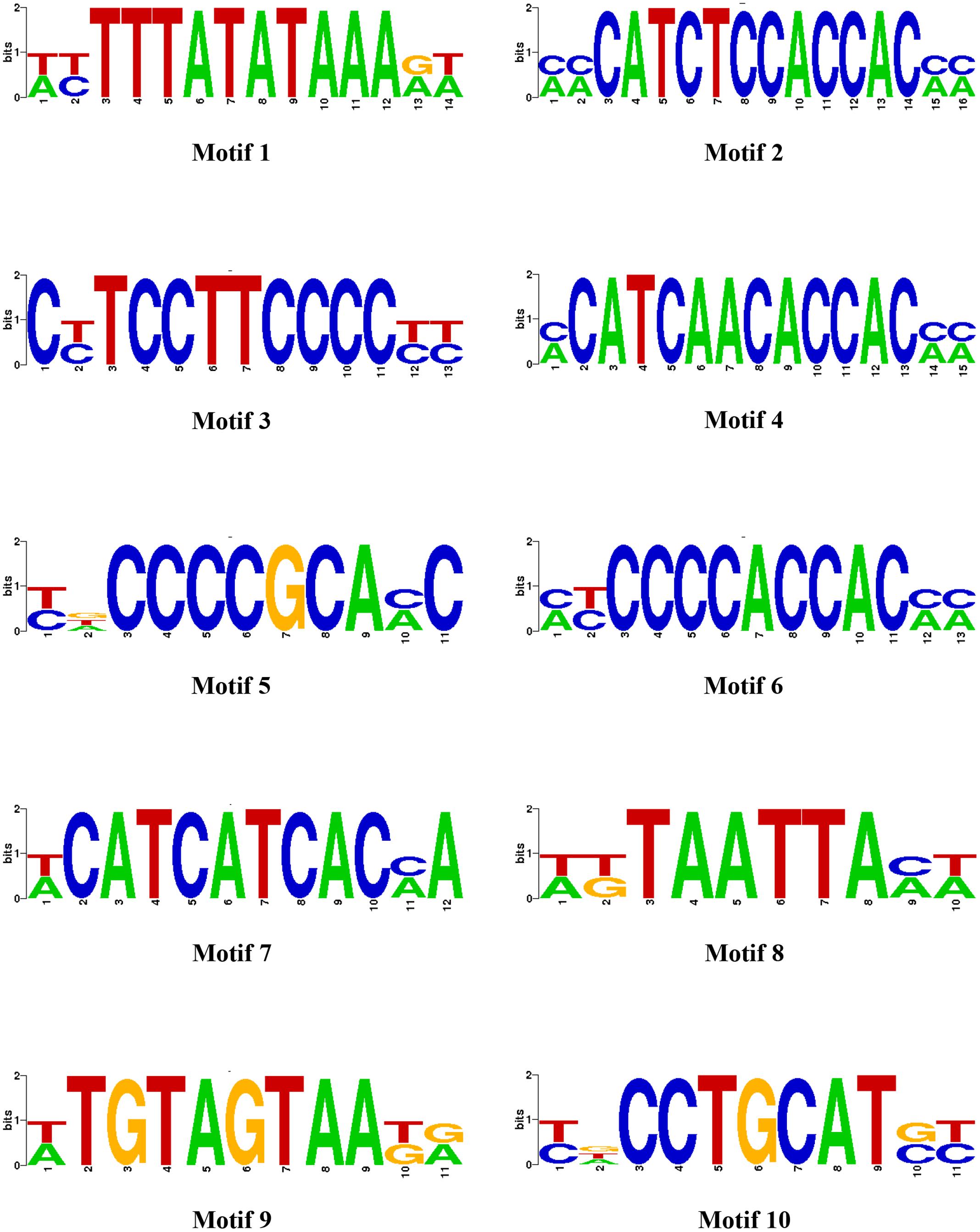
FIGURE 8. The most enriched cis-regulatory elements in the promoter sequences of secretory protein coding genes. Ten most abundant motifs in the promoter sequences of genes encoding A. rabiei secretory proteins are shown, as identified by RSAT suite for fungi (Regulatory Sequence Analysis).
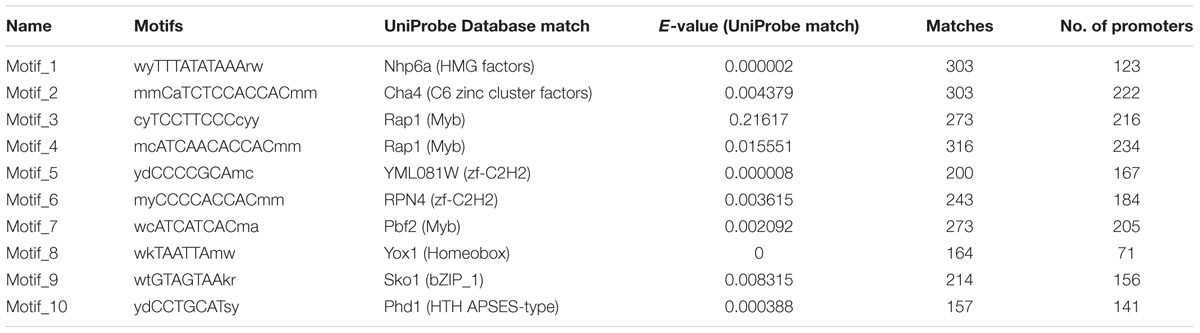
TABLE 2. Ten most enriched cis-regulatory elements identified in the promoter sequences of secretory protein coding genes.
Once we identified the most frequent cis-regulatory elements across the promoter regions of the genes encoding secretory proteins of A. rabiei, the TFs known to bind on these sites were identified. For this, the motifs were scanned for their corresponding TFs using UniPROBE database (Newburger and Bulyk, 2009). It revealed that Myb TFs were the corresponding TFs that bind to CATCAACACCAC motif (Table 2). For the second most abundant motif CATCTCCACCAC, [Zn(II)2Cys6] zinc cluster TFs were predicted to bind. The Myb TFs were also predicted to bind to the motif TCCTTCCCC. Among the 10 most frequent motifs identified, the corresponding TFs for 3 motifs were Myb TFs. In addition, two cis-regulatory elements were regulated by C2H2 zinc finger TFs. Likewise, the [Zn(II)2Cys6] zinc cluster, bZIP, APSES, HMG, and homeobox TFs were predicted to bind on 1 of the 10 motifs identified. This suggests that the secretome of A. rabiei is regulated by an array of TFs, mainly Myb TFs.
Differentially Expressed Transcription Factor Genes of A. rabiei during Host Infection
Recently, the transcriptome data of A. rabiei during plant invasion was reported (Fondevilla et al., 2015). We utilized this data in order to identify the differentially expressed genes (DEGs) encoding predicted TFs during host infection. The RNA-seq reads of “in medium” and “in planta” samples at time points 12, 36, and 96 hpi were mapped on A. rabiei genome and expression values were calculated in the terms of FPKM. A total of 13 putative TF encoding genes were found differentially expressed (Figure 9). Interestingly, seven genes were found to be expressed exclusively in medium and were not at all expressed during in planta conditions (Figure 9). Likewise six genes which were expressed during in planta conditions had no expression in medium. Four putative TF encoding genes were found expressed exclusively at 96 hpi whereas only one putative TF (bZIP) encoding gene was expressing at all the three time points during plant infection (Figure 9). The DEGs mainly belonged to [Zn(II)2Cys6] zinc cluster, C2H2 zinc finger and nucleic acid-binding-OB-fold family of TFs.
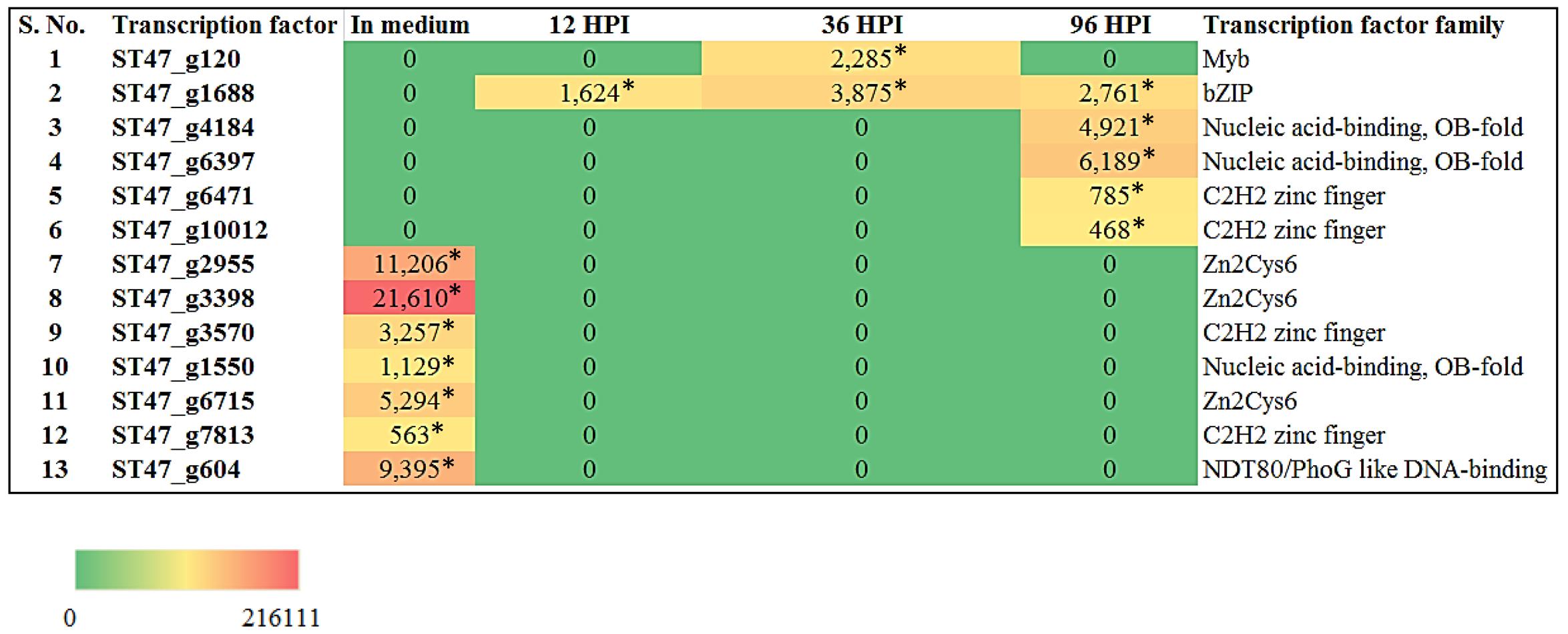
FIGURE 9. Heat map representation of A. rabiei putative TFs. The expression of A. rabiei putative TF genes is shown during vegetative growth (in medium) and host invasion [12, 36, and 96 hours post inoculation (hpi)]. For this, A. rabiei transcriptome data was utilized (Fondevilla et al., 2015). Expression of putative TFs genes are denoted in the terms of FPKM values. Values with asterisk represent P-value < 0.005 at which differentially expressed genes (DEGs) were found.
We also investigated the expression of genes encoding secretory proteins of A. rabiei that we have predicted earlier (Verma et al., 2016). A set of 34 putative secretory protein coding genes were found differentially expressed (Figure 10). During in planta conditions, 27 genes showed differential expression at different time points and these genes were not expressed when the fungus was grown in in vitro condition. At 96 hpi, most of the secretory protein coding genes (26) were up-regulated and out of these, 23 genes were exclusively expressed at this time point of infection. On the other hand, two secretory protein coding genes were differentially expressed at all the three time points of infection. Interestingly, there were seven genes which were expressed only during in medium condition. This suggests that A. rabiei exhibits a much orchestrated spatial and temporal distribution of secretory proteins during its lifecycle.
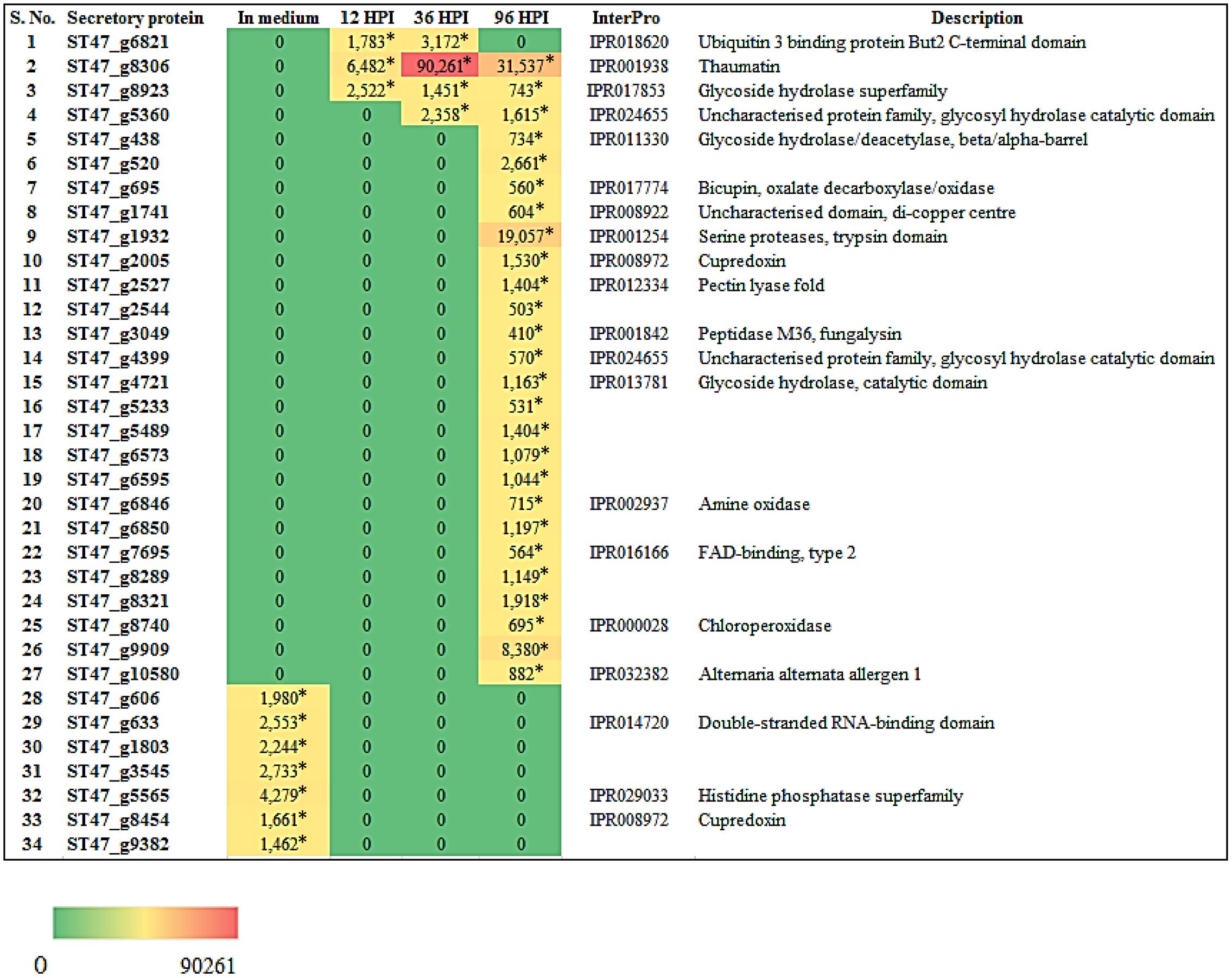
FIGURE 10. Heat map representation of A. rabiei putative secretory protein coding genes. The expression pattern of genes encoding A. rabiei putative secretory proteins is shown during vegetative growth (in medium) and host invasion (12, 36, and 96 hpi). For this, A. rabiei transcriptome data was utilized (Fondevilla et al., 2015). Expression of putative secretory protein coding genes are denoted in the terms of FPKM values. Values with asterisk represent P-value < 0.05 at which DEGs were found.
In order to get the insights of putative TFs that regulate the secretory proteins of A. rabiei, we searched for the presence of most frequent cis-regulatory elements in the promoters of secretory protein encoding genes which were found up-regulated during in medium and in planta conditions. Twelve secretory proteins had CATCAACACCAC motif in their promoters and this motif is the binding site for Myb TFs, particularly Rap1 (Supplementary Dataset S3). Similarly, CCTGCAT motif was found in promoters of 11 secretory proteins and is the binding site of APSES TF (Phd1). The bZIP and C2H2 zinc finger TFs appeared to regulate expression of 7 secretory proteins due to the presence of their binding sites on promoter sequences. It indicates that an array of putative TFs regulate secretory proteins during host invasion and in vitro condition.
Expression Analysis of A. rabiei Transcription Factors by qRT-PCR
For transcriptomic studies of A. rabiei–chickpea pathosystem (Fondevilla et al., 2015), A. rabiei isolate P4 (originated from Kaljebrin, Syria) was used to inoculate susceptible chickpea cultivar “Blanco Lechoso.” In our laboratory, we use ArD2 isolate of A. rabiei (obtained from Indian Agricultural Research Institute, New Delhi, India) representing pathotype 2 to infect highly susceptible chickpea cultivar Pusa-362. Therefore, we investigated whether the putative TFs exhibit similar expression pattern in A. rabiei–chickpea pathosystem when the pathotype of A. rabiei and the chickpea cultivar were different. For this, the expression profile of seven different putative TFs during host colonization was assayed by qRT-PCR at 12, 24, 72, and 144 hpi (Figure 11). All the selected genes were found differentially expressed during infection. In comparison with other putative TF genes, the abundance of ST47_g4184 transcripts (nucleic acid-binding-OB-fold) was highest at almost all the time points. Its expression was highest at 12 hpi that drastically reduced at 24 hpi and then again gradually increased at 72 and 144 hpi suggesting a bi-phasic induction. Similarly, ST47_g10012 (C2H2 zinc finger) also showed maximum expression at 12 and 144 hpi. In terms of expression pattern, the transcripts of ST47_g120 (Myb), ST47_g1688 (bZIP), ST47_g3570 (C2H2 zinc finger), ST47_g4184 and ST47_g6471 (C2H2 zinc finger) showed maximum expression at 12 hpi followed by 144 hpi suggesting that these genes were getting induced during initial phases of pathogenesis. While ST47_g3398 [Zn(II)2Cys6] and ST47_g10012 appeared to play role during later stages of pathogenesis as they showed maximum transcript abundance at 144 hpi. Overall, these results indicate that TFs may exhibit differential expression in a temporal manner specific to the pathotype of A. rabiei and the cultivar of chickpea.
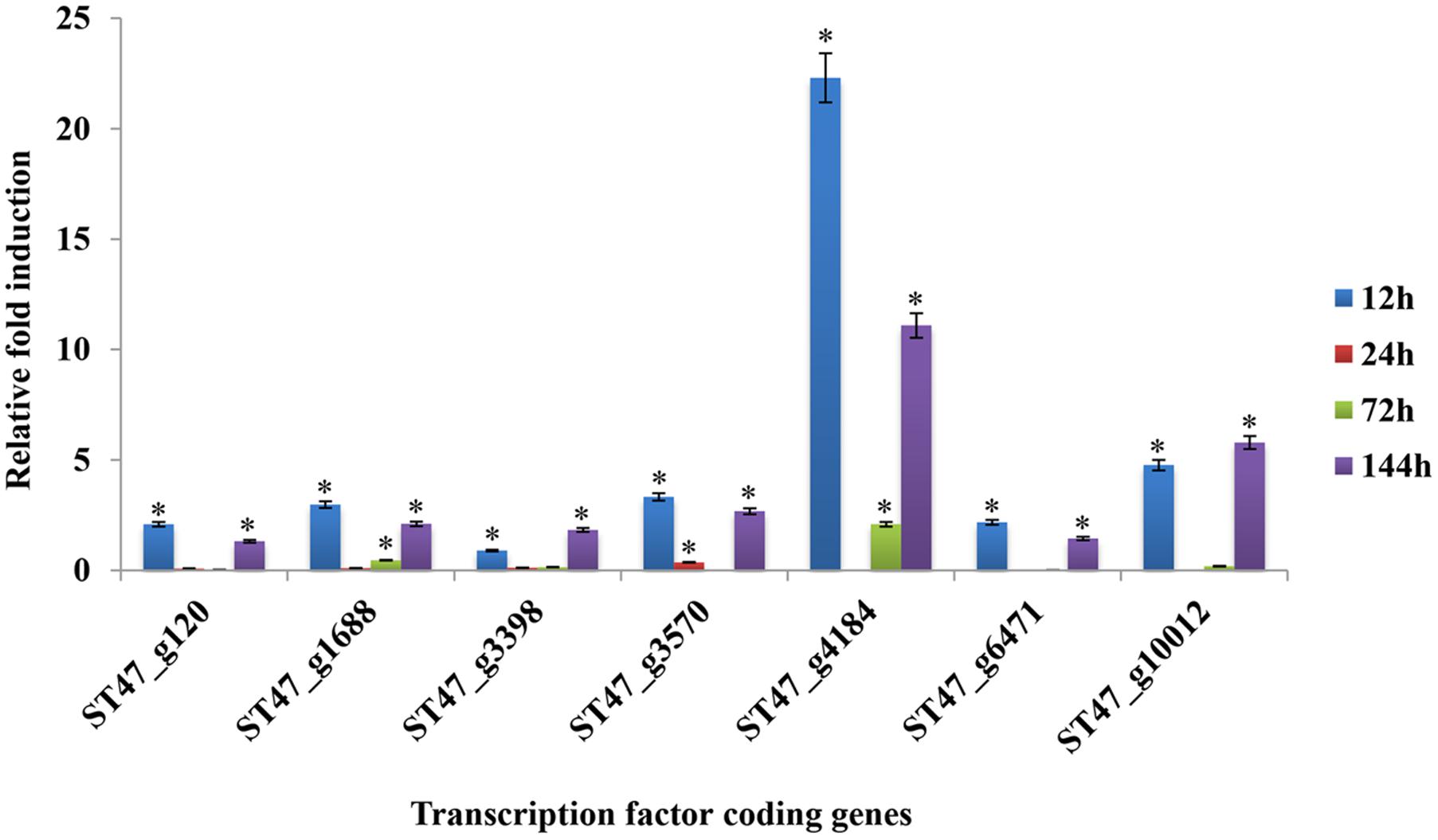
FIGURE 11. Expression profiles of putative TF genes assayed by qRT-PCR during host colonization. Bar diagrams representing the expression pattern of seven putative TF genes are shown as the fold-change compared to the control. Expression was analyzed in A. rabiei-infected chickpea samples at 12, 24, 72, and 144 hpi. ArEF1a gene was used as an internal reference gene. Asterisks denote significant difference compared with the expression level at 0 hpi (p < 0.05).
Discussion
Whenever a pathogen attacks host plant, extensive reprogramming of gene expression facilitated by TFs occur in both the host and pathogen (Verma et al., 2013). Several distinct reports suggest the crucial role played by TFs in the growth, development and virulence of filamentous fungi. Consequently, the attention has been recently focused to study TFs since they regulate an array of pathogenicity and stress responsive genes. In yeast and filamentous fungi, a basic leucine zipper (bZIP) TF Activating Protein 1 (AP-1) acts as a transcriptional activator in response to oxidative stress (Wu and Moye-Rowley, 1994; Toone and Jones, 1998; Reverberi et al., 2008) and carries out multiple functions as a redox regulator (Fernandes et al., 1997; Karin et al., 1997). The yeast AP-1 family of TFs consists of Yap1 in S. cerevisiae, Pap1 in S. pombe, Cap1 in Candida albicans, and Kap1 in Kluyveromyces lactis (Toone et al., 2001). Later, Yap1 orthologs were also identified in Aspergillus nidulans, Aspergillus fumigatus, and Aspergillus parasiticus where they act as key players in cellular defense against oxidative stress (Asano et al., 2007; Reverberi et al., 2008, 2012). However, in necrotrophic phytopathogenic fungus B. cinerea, a bZIP TF (BcAtf1) regulates expression of catalase B but does not contribute to osmotic and oxidative stress tolerance (Temme et al., 2012). In M. oryzae, MST12, a homolog of the yeast TF Ste12, plays crucial role in host penetration and colonization (Park et al., 2002). Few other specific TFs, for instance Cochliobolus carbonum ccSNF1, regulates in planta expression of cell wall degrading enzymes (Tonukari et al., 2000). In Fusarium graminearum, the pathogenicity and sexual development is largely affected by a Myb-like transcription factor MYT3 (Kim et al., 2014). Similarly, the AbPf2 TF of Alternaria brassicicola is an important regulator of pathogenesis and it does not affect other cellular processes (Cho et al., 2013). Therefore, TFs are vital for survival and completion of the lifecycle.
Ascochyta spp. are highly devastating pathogens causing severe losses in production of legumes worldwide. However, not much is known about the pathogenicity mechanisms of this necrotrophic fungus. There are only few reports available which are not quite sufficient to shed light on understanding of A. rabiei–chickpea pathosystem. Despite extensive pathological studies, the nature and extent of pathogenic variability in A. rabiei have not been clearly established. The mechanisms by which A. rabiei infects and colonizes chickpea plants remain poorly understood. Though, with the recent advances in genomics, transcriptomics, and molecular studies of A. rabiei, it is now possible to carry out in depth investigations of A. rabiei pathogenicity. The genetic manipulations/transformations of A. rabiei have made identification of gene functions possible (Nizam et al., 2010). Similarly, various comprehensive studies of A. rabiei gene families have been performed in order to gain the molecular and evolutionary insights (Nizam et al., 2014a,b; Kim et al., 2015).
With the present study, we have further expanded the understanding of A. rabiei as a pathogen. Since TFs have a major role in fungal development, pathogenesis and response to the environment, we have identified and classified the putative TF repertoire of A. rabiei. This detailed analysis was achievable mainly due to the availability of A. rabiei genome sequencing data. A comprehensive computational approach was implemented that resulted in identification of a set of 381 proteins as the putative TFs of A. rabiei. In fungi, usually the TFs comprise 0.5–8% of the gene content and can be classified on the basis of structure of their DBDs (Stegmaier et al., 2004; Wilson et al., 2008). In A. rabiei, TFs constitute 3.5% of the gene content. Among the A. rabiei putative TFs predicted, [Zn(II)2Cys6] family of TFs was overwhelmingly predominated. The [Zn(II)2Cys6] TFs are usually the most abundant TFs in fungi as revealed by few studies analyzing 37 known fungal transcription regulator-related PFAM domains in several ascomycete and basidiomycete genomes (Shelest, 2008; Todd et al., 2014). A variety of cellular and metabolic processes are regulated by this class of TFs. In model filamentous ascomycete fungus Podospora anserina, the two zinc cluster proteins RSE2 and RSE3 regulates expression of genes encoding alternative oxidase, phosphoenolpyruvate carboxykinase, fructose-1,6-biphosphatase, alternative NADH dehydrogenase, a [Zn(II)2Cys6] TF, a flavohemoglobin, and various hydrolases (Bovier et al., 2014). Necrotrophic fungus B. cinerea has a [Zn(II)2Cys6] TF, BcGaaR that induces D-galacturonic acid (GalA)-inducible genes and promotes growth of B. cinerea on GalA (Zhang et al., 2015). In F. graminearum, [Zn(II)2Cys6] TF EBR1 regulates virulence and apical dominance of the hyphal tip (Zhao et al., 2011). Furthermore, a high-throughput gene knockout of 104 [Zn(II)2Cys6] TF genes in M. oryzae was performed that suggested their significance in growth, asexual and infection-related development, pathogenesis and response to nine abiotic stresses (Lu et al., 2014). It also revealed that [Zn(II)2Cys6] TFs involved in pathogenicity frequently tend to function in multiple development stages. During hemibiotrophic Colletotrichum lindemuthianum-bean interaction, CLTA1 (a fungal zinc cluster TF) regulates biotrophic phase specific genes facilitating the biotrophy to necrotrophy switch (Dufresne et al., 2000). Therefore, the prevalence of [Zn(II)2Cys6] family of TFs in A. rabiei suggests that they might play important role in survival and virulence of A. rabiei.
In A. rabiei, the C2H2 zinc finger class was second most abundant class which represents a smaller but major regulator class in the ascomycetes. These are involved in development (Kwon et al., 2010) and calcium signaling in A. nidulans (Hagiwara et al., 2008). Another C2H2 TF, MtfA, is a regulator of secondary metabolism and morphogenesis in A. nidulans (Ramamoorthy et al., 2013). Moreover, a conserved C2H2 TF (RsrA) coordinates a NapA mediated oxidative response in Aspergillus fungi (Bok et al., 2014). In A. fumigatus, GipA induces production of immunosuppressive secondary metabolite gliotoxin (Schoberle et al., 2014). In A. parasiticus and Aspergillus flavus, deletion of MSNA resulted in enhanced production of conidia, ROS, aflatoxin, and kojic acid (Roze et al., 2004). In addition, the C2H2 TFs may use multiple recognition motifs to control gene expression (Han et al., 2016). This indicates that C2H2 zinc finger class of TFs in A. rabiei might be the major contributors in regulating the growth and developmental processes.
Comparison of A. rabiei putative TFs with that of other necrotrophic, biotrophic, hemibiotrophic, symbiotic, and saprotrophic fungi suggested a conserved as well as unique distribution of TFs among different classes in all the selected fungi. This indicates toward the evolutionary specificity of TFs depending on the lifestyle and host of the fungi. Similar indications were also provided when phylogenetic analysis of A. rabiei Myb, bHLH and bZIP TF families was compared with other closely related necrotrophic fungi, i.e., C. heterostrophus, P. tritici-repentis, and P. nodorum. It suggested formation of well conserved clades in these TF families among the closely related necrotrophic fungi. The domain organization of Myb, bHLH, and bZIP TF families of A. rabiei showed presence of characteristic domains as well as numerous low-complexity regions. Various studies have suggested that these regions show significant divergence across protein families. Proteins containing low-complexity regions have more binding partners across different protein–protein interaction networks (Coletta et al., 2010). The low-complexity regions positioned at center of protein sequence are related to transcription-related gene ontology terms, whereas terminally located low-complexity regions are associated with translation and stress response-related terms (Coletta et al., 2010).
The TFs of bZIP family of A. rabiei had bZIP domains superimposed with coiled coil regions. Several TFs of bHLH and Myb family also had coiled coil regions. The architecture of a particular coiled-coil domain governs its oligomerization state, rigidity and ability to function as a molecular recognition system. In fungi, Myb TFs play an important role in cell differentiation, development and pathogenicity (Arratia-Quijada et al., 2012; Kim et al., 2014). Similarly, basic helix-loop-helix (bHLH) TFs are also known to be involved in development (Murre et al., 1994). The highly functionally conserved bZIP TFs ensure proper growth, development, sulfur metabolism, and iron homeostasis in fungi (Guo et al., 2011; Kong et al., 2015; Sun et al., 2016). They are highly stress responsive and are vital for pathogenicity in a reactive oxygen species-dependent manner. The gene structure of [Zn(II)2Cys6], C2H2, nucleic acid-binding-OB-fold, winged helix repressor DNA-binding and Myb family of TFs suggested that the genes with single intron had intron phase either 0 or 1 predominantly. The three classes of intron phases are far from even distribution. Phase 0 introns are usually present in excess (Long et al., 1995) and the inferred evolution of intron phase distribution showed that the proportion of phase 0 introns increased over evolution (Nguyen et al., 2006). Therefore, the excess of phase 0 introns in A. rabiei putative TF genes indicates toward the evolutionary development of this pathogen over the years.
Earlier, we predicted a set of 758 proteins in the secretome of A. rabiei (Verma et al., 2016). Since the secretory proteins are largely involved in the pathogenesis and deriving nutrition from the host, the cis-regulatory elements present in promoter sequences of secretory proteins coding genes of A. rabiei were identified. The secretome coding genes of A. rabiei were predicted to be significantly regulated by Myb TFs followed by [Zn(II)2Cys6] zinc cluster TFs. Fungal genomes have at least 37 PFAM families of TF (Shelest, 2008). So far, 12 TFs from four families (Zinc finger, APSES, WOPR, and Fork head) have been found to regulate the gene expression of candidate effectors (Tan and Oliver, 2017). A central role for Zn-finger TF in effector expression was studied in A. brassicicola where a [Zn(II)2Cys6] zinc cluster TF (AbPf2) regulates expression of 106 genes, out of which 33 genes encode secreted proteins, including eight putative effector proteins (Cho et al., 2013). Plants challenged with Δabpf2 mutants had elevated expression levels of photosynthesis, pentose phosphate pathway and primary metabolism related genes but decreased levels of defense-related genes. In P. nodorum, AbPf2 ortholog PnPf2 positively regulates the necrotrophic effectors SnToxA and SnTox3 expression (Rybak et al., 2017). Likewise, PtrPf2 controls the expression of P. tritici-repentis ToxA, a near-identical copy of SnToxA (Rybak et al., 2017). This indicates that Pf2 TF exhibits a conserved role in regulating the effectors. Furthermore, SnStuA (an APSES bHLH TF) was found to regulate the expression of SnTox3 (IpCho et al., 2010). In Leptosphaeria maculans, the expression of AvrLm4-7, AvrLm1, and AvrLm6 is regulated by LmStuA (Soyer et al., 2015). The example of WOPR TFs was found in Verticillium dahlia and Cladosporium fulvum. The VdSge1 TF is required for full expression of the Cys-rich effector Ave1 (Santhanam and Thomma, 2013). Similarly, CfWor1 TF primarily regulates development of Cladosporium, but also indirectly controls expression of a subset of effector genes (Ökmen et al., 2014). In U. maydis, a fork head TF named Fox1 acts as a positive regulator of six candidate effector genes (Zahiri et al., 2010). Over all, these studies indicate the significance of TFs in regulating the effectors that ultimately govern the pathogenesis. The A. rabiei secretome was predicted to be mainly regulated by Myb TFs, therefore, it would be highly significant to identify the potential Myb TFs that controls the expression of A. rabiei effectors.
In order to understand significance of A. rabiei secretome and putative TFs during host colonization, the RNA-seq data of A. rabiei during in medium and in planta conditions were analyzed (Fondevilla et al., 2015). Thirty-four secretory protein coding genes and seven putative TF genes were differentially expressed. Most of these secretory protein coding genes had binding sites of Myb TFs (particularly Rap1) in their promoter sequences. This is consistent with our computational prediction suggesting Myb TFs as the major regulators of A. rabiei secretome. The qRT-PCR analysis of few A. rabiei putative TFs showed a lot of differences in expression profiling as compared to RNA-seq data. The putative TFs ST47_3398 and ST47_3570, which were not expressing during in planta conditions in RNA-seq data, showed expression in qRT-PCR analysis. Furthermore, RNA-seq data showed that majority of putative TFs were expressed during later stages of infection. By contrast, according to qRT-PCR analysis almost all selected putative TFs were expressed in early stages of infection as well. Biphasic kinetics of gene expression is commonly observed in both pathogen and host during infection (Schneider et al., 2011; Singh et al., 2012). This suggests that the expression profile of these putative TFs could be differential depending on the pathotype of A. rabiei and the cultivar of chickpea. Further investigation in this direction is a prerequisite to determine if TFs function discretely in different pathotypes of A. rabiei.
Since TFs as regulators play a crucial role in the life cycle of fungi, their presence or absence may offer opportunities or enforce limitations on the natural habitat of fungal species. The present study provides a platform to study A. rabiei TFs in detail. This will offer better insight into the evolution of regulatory mechanisms in A. rabiei. Comparative studies of A. rabiei TFs in other pathotypes will decipher their contribution in determining the pathotypes of this fungus. The extensive knowledge obtained will aid in designing successful strategies to control this devastating pathogen and prevent further crop losses.
Author Contributions
SV, RG, and PV designed the experiments; RG and SV performed bioinformatics analysis; SV performed the experiments; SV, RG, and PV analyzed data; and SV, RG, and PV wrote the manuscript.
Funding
This study was supported by research grant (BT/PR7164/PBD/16/1016/2012) of Department of Biotechnology, Government of India and core grant from National Institute of Plant Genome Research, New Delhi, India.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgment
SV acknowledges National Institute of Plant Genome Research, New Delhi, India for providing short-term research fellowship.
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.01037/full#supplementary-material
Footnotes
- ^ http://www.transcriptionfactor.org
- ^ http://ftfd.snu.ac.kr/
- ^ http://www.ebi.ac.uk/Tools/pfa/iprscan5/
- ^ http://gsds.cbi.pku.edu.cn
- ^ http://cdvist.utk.edu
- ^ http://www.ebi.ac.uk/Tools/msa/muscle/
- ^ http://tree.bio.ed.ac.uk/software/figtree/
- ^ http://www.rsat.eu/
- ^ http://thebrain.bwh.harvard.edu/uniprobe/
References
Adebali, O., Ortega, D. R., and Zhulin, I. B. (2015). CDvist: a webserver for identification and visualization of conserved domains in protein sequences. Bioinformatics 31, 1475–1477. doi: 10.1093/bioinformatics/btu836
Adryan, B., and Teichmann, S. A. (2006). FlyTF: a systematic review of site-specific transcription factors in the fruit fly Drosophila melanogaster. Bioinformatics 22, 1532–1533.
Arratia-Quijada, J., Sánchez, O., Scazzocchio, C., and Aguirre, J. (2012). FlbD, a Myb transcription factor of Aspergillus nidulans, is uniquely involved in both asexual and sexual differentiation. Eukaryot. Cell 11, 1132–1142. doi: 10.1128/EC.00101-12
Asano, Y., Hagiwara, D., Yamashino, T., and Mizuno, T. (2007). Characterization of the bZip-type transcription factor NapA with reference to oxidative stress response in Aspergillus nidulans. Biosci. Biotechnol. Biochem. 71, 1800–1803.
Bok, J. W., Wiemann, P., Garvey, G. S., Lim, F. Y., Haas, B., Wortman, J., et al. (2014). Illumina identification of RsrA, a conserved C2H2 transcription factor coordinating the NapA mediated oxidative stress signaling pathway in Aspergillus. BMC Genomics 15:1011. doi: 10.1186/1471-2164-15-1011
Bovier, E., Sellem, C. H., Humbert, A., and Sainsard-Chanet, A. (2014). Genetic and functional investigation of Zn(2)Cys(6) transcription factors RSE2 and RSE3 in Podospora anserina. Eukaryot. Cell 13, 53–65. doi: 10.1128/EC.00172-13
Chandirasekaran, R., Warkentin, T. D., Gan, Y., Shirtliffe, S., Gossen, B. D., Tar’an, B., et al. (2009). Improved sources of resistance to ascochyta blight in chickpea. Can. J. Plant Sci. 89, 107–118. doi: 10.4141/CJPS07210
Cho, Y., Ohm, R. A., Grigoriev, I. V., and Srivastava, A. (2013). Fungal specific transcription factor AbPf2 activates pathogenicity in Alternaria brassicicola. Plant J. 75, 498–514. doi: 10.1111/tpj.12217
Coletta, A., Pinney, J. W., Solís, D. Y. W., Marsh, J., Pettifer, S. R., and Attwood, T. K. (2010). Low-complexity regions within protein sequences have position-dependent roles. BMC Syst. Biol. 4:43. doi: 10.1186/1752-0509-4-43
Dufresne, M., Perfect, S., Pellier, A. L., Bailey, J. A., and Langin, T. (2000). A GAL4-like protein is involved in the switch between biotrophic and necrotrophic phases of the infection process of Colletotrichum lindemuthianum on common bean. Plant Cell 12, 1579–1589.
FAO (2014). FAO Statistical Databases. Available at: http://www.fao.org/faostat/en/#data/QC
Fernandes, L., Rodrigues-Pousada, C., and Struhl, K. (1997). Yap, a novel family of eight bZIP proteins in Saccharomyces cerevisiae with distinct biological functions. Mol. Cell. Biol. 17, 6982–6993.
Finn, R. D., Attwood, T. K., Babbitt, P. C., Bateman, A., Bork, P., Bridge, A. J., et al. (2017). InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 45, D190–D199. doi: 10.1093/nar/gkw1107
Finn, R. D., Bateman, A., Clements, J., Coggill, P., Eberhardt, R. Y., Eddy, S. R., et al. (2014). Pfam: the protein families database. Nucleic Acids Res. 42, 222–230. doi: 10.1093/nar/gkt1223
Fondevilla, S., Krezdorn, N., Rotter, B., Kahl, G., and Winter, P. (2015). In planta identification of putative pathogenicity factors from the chickpea pathogen Ascochyta rabiei by de novo transcriptome sequencing using RNA-Seq and massive analysis of cDNA ends. Front. Microbiol. 6:1329. doi: 10.3389/fmicb.2015.01329
Fulton, D. L., Sundararajan, S., Badis, G., Hughes, T. R., Wasserman, W. W., Roach, J. C., et al. (2009). TFCat: the curated catalog of mouse and human transcription factors. Genome Biol. 10:R29. doi: 10.1186/gb-2009-10-3-r29
Gough, J., Karplus, K., Hughey, R., and Chothia, C. (2001). Assignment of homology to genome sequences using a library of hidden markov models that represent all proteins of known structure. J. Mol. Biol. 313, 903–919.
Guo, A. Y., Chen, X., Gao, G., Zhang, H., Zhu, Q. H., Liu, X. C., et al. (2008). PlantTFDB: a comprehensive plant transcription factor database. Nucleic Acids Res. 36, D966–D969.
Guo, M., Chen, Y., Du, Y., Dong, Y., Guo, W., Zhai, S., et al. (2011). The bZIP transcription factor MoAP1 mediates the oxidative stress response and is critical for pathogenicity of the rice blast fungus Magnaporthe oryzae. PLoS Pathog. 7:e1001302. doi: 10.1371/journal.ppat.1001302
Hagiwara, D., Kondo, A., Fujioka, T., and Abe, K. (2008). Functional analysis of C2H2 zinc finger transcription factor CrzA involved in calcium signaling in Aspergillus nidulans. Curr. Genet. 54, 325–338. doi: 10.1007/s00294-008-0220-z
Han, B. Y., Foo, C. S., Wu, S., and Cyster, J. G. (2016). The C2H2-ZF transcription factor Zfp335 recognizes two consensus motifs using separate zinc finger arrays. Genes Dev. 30, 1509–1514. doi: 10.1101/gad.279406.116
Hu, B., Jin, J., Guo, A. Y., Zhang, H., Luo, J., and Gao, G. (2015). GSDS 2.0: an upgraded gene feature visualization server. Bioinformatics 31, 1296–1297. doi: 10.1093/bioinformatics/btu817
IpCho, S. V., Tan, K.-C., Koh, G., Gummer, J., Oliver, R. P., Trengove, R. D., et al. (2010). The transcription factor StuA regulates central carbon metabolism, mycotoxin production, and effector gene expression in the wheat pathogen Stagonospora nodorum. Eukaryot. Cell 9, 1100–1108. doi: 10.1128/EC.00064-10
Jayakumar, P., Gossen, B. D., Gan, Y. T., Warkentin, T., and Banniza, S. (2005). Ascochyta blight of chickpea: infection and host resistance mechanisms. Can. J. Plant Pathol. 27, 499–509. doi: 10.1080/07060660509507251
Jones, P., Binns, D., Chang, H. Y., Fraser, M., Li, W., McAnulla, C., et al. (2014). InterProScan 5: genome-scale protein function classification. Bioinformatics 30, 1236–1240. doi: 10.1093/bioinformatics/btu031
Kaiser, W. J., Coca, F. W., and Vega, S. O. (2000). First report of Ascochyta blight of chickpea in Latin America. Plant Dis. 84, 102–102. doi: 10.1094/pdis.2000.84.1.102c
Kaiser, W. J., Muehlbauer, F. J., and Hannan, R. M. (1994). “Experience with ascochyta blight of chickpea in the united-states,” in Expanding the Production and Use of Cool Season Food Legumes, Vol. 19, eds F. J. Muehlbauer and W. J. Kaiser (Dordrecht: Springer), 849–858. doi: 10.1007/978-94-011-0798-3_52
Kanamori, M., Konno, H., Osato, N., Kawai, J., Hayashizaki, Y., and Suzuki, H. (2004). A genome-wide and nonredundant mouse transcription factor database. Biochem. Biophys. Res. Commun. 322, 787–793.
Karin, M., Liu, Z., and Zandi, E. (1997). AP-1 function and regulation. Curr. Opin. Cell Biol. 9, 240–246. doi: 10.1016/S0955-0674(97)80068-3
Kim, D., Pertea, G., Trapnell, C., Pimentel, H., Kelley, R., and Salzberg, S. (2013). TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14:R36. doi: 10.1186/gb-2013-14-4-r36
Kim, W., Park, C. M., Park, J. J., Akamatsu, H. O., Peever, T. L., Xian, M., et al. (2015). Functional analyses of the diels-alderase gene sol5 of Ascochyta rabiei and Alternaria solani indicate that the solanapyrone phytotoxins are not required for pathogenicity. Mol. Plant Microbe Interact. 28, 482–496. doi: 10.1094/MPMI-08-14-0234-R
Kim, Y., Kim, H., Son, H., Choi, G. J., Kim, J. C., and Lee, Y. W. (2014). MYT3, a Myb-like transcription factor, affects fungal development and pathogenicity of Fusarium graminearum. PLoS ONE 9:e94359. doi: 10.1371/journal.pone.0094359
Kong, S., Park, S. Y., and Lee, Y. H. (2015). Systematic characterization of the bZIP transcription factor gene family in the rice blast fungus, Magnaporthe oryzae. Environ. Microbiol. 17, 1425–1443. doi: 10.1111/1462-2920.12633
Kwon, N. J., Garzia, A., Espeso, E. A., Ugalde, U., and Yu, J. H. (2010). FlbC is a putative nuclear C2H2 transcription factor regulating development in Aspergillus nidulans. Mol. Microbiol. 77, 1203–1219. doi: 10.1111/j.1365-2958.2010.07282.x
Lee, A. P., Yang, Y., Brenner, S., and Venkatesh, B. (2007). TFCONES: a database of vertebrate transcription factor-encoding genes and their associated conserved noncoding elements. BMC Genomics 8:441. doi: 10.1186/1471-2164-8-441
Long, M., Rosenberg, C., and Gilbert, W. (1995). Intron phase correlations and the evolution of the intron/exon structure of genes. Proc. Natl. Acad. Sci. U.S.A. 92, 12495–12499. doi: 10.1073/pnas.92.26.12495
Lu, J., Cao, H., Zhang, L., Huang, P., and Lin, F. (2014). Systematic analysis of Zn2Cys6 transcription factors required for development and pathogenicity by high-throughput gene knockout in the rice blast fungus. PLoS Pathog. 10:e1004432. doi: 10.1371/journal.ppat.1004432
Matys, V., Kel-Margoulis, O. V., Fricke, E., Liebich, I., Land, S., Barre-Dirrie, A., et al. (2006). TRANSFAC and its module TRANSCompel: transcriptional gene regulation in eukaryotes. Nucleic Acids Res. 34, D108–D110. doi: 10.1093/nar/gkj143
Medina-Rivera, A., Defrance, M., Sand, O., Herrmann, C., Castro-Mondragon, J. A., Delerce, J., et al. (2015). RSAT 2015: regulatory sequence analysis tools. Nucleic Acids Res. 43, W50–W56. doi: 10.1093/nar/gkv362
Mochida, K., Yoshida, T., Sakurai, T., Yamaguchi-Shinozaki, K., Shinozaki, K., and Tran, L. S. (2010). LegumeTFDB: an integrative database of Glycine max, Lotus japonicus and Medicago truncatula transcription factors. Bioinformatics 26, 290–291. doi: 10.1093/bioinformatics/btp645
Montanini, B., Levati, E., Bolchi, A., Kohler, A., Morin, E., Tisserant, E., et al. (2011). Genome-wide search and functional identification of transcription factors in the mycorrhizal fungus Tuber melanosporum. New Phytol. 189, 736–750. doi: 10.1111/j.1469-8137.2010.03525.x
Murre, C., Bain, G., van Dijk, M. A., Engel, I., Furnari, B. A., Massari, M. E., et al. (1994). Structure and function of helix-loop-helix proteins. Biochim. Biophys. Acta 1218, 129–135. doi: 10.1016/0167-4781(94)90001-9
Newburger, D. E., and Bulyk, M. L. (2009). UniPROBE: an online database of protein binding microarray data on protein-DNA interactions. Nucleic Acids Res. 37, D77–D82. doi: 10.1093/nar/gkn660
Nguyen, H. D., Yoshihama, M., and Kenmochi, N. (2006). Phase distribution of spliceosomal introns: implications for intron origin. BMC Evol. Biol. 6:69. doi: 10.1186/1471-2148-6-69
Nizam, S., Gazara, R. K., Verma, S., Singh, K., and Verma, P. K. (2014a). Comparative structural modeling of six old yellow enzymes (OYEs) from the necrotrophic fungus Ascochyta rabiei: insight into novel OYE classes with differences in cofactor binding, organization of active site residues and stereopreferences. PLoS ONE 9:e95989. doi: 10.1371/journal.pone.0095989
Nizam, S., Singh, K., and Verma, P. K. (2010). Expression of the fluorescent proteins DsRed and EGFP to visualize early events of colonization of the chickpea blight fungus Ascochyta rabiei. Curr. Genet. 56, 391–399. doi: 10.1007/s00294-010-0305-3
Nizam, S., Verma, S., Borah, N. N., Gazara, R. K., and Verma, P. K. (2014b). Comprehensive genome-wide analysis reveals different classes of enigmatic old yellow enzyme in fungi. Sci. Rep. 4:4013. doi: 10.1038/srep04013
Ökmen, B., Collemare, J., Griffiths, S., van der Burgt, A., Cox, R., and de Wit, P. J. G. M. (2014). Functional analysis of the conserved transcriptional regulator CfWor1 in Cladosporium fulvum reveals diverse roles in the virulence of plant-pathogenic fungi. Mol. Microbiol. 92, 10–27. doi: 10.1111/mmi.12535
Pande, S., Siddique, K. H. M., Kishore, G. K., Bayaa, B., Gaur, P. M., Gowda, C. L. L., et al. (2005). Ascochyta blight of chickpea (Cicer arietinum L.): a review of biology, pathogenicity, and disease management. Aust. J. Agric. Res. 56, 317–332. doi: 10.1071/ar04143
Park, G., Xue, G. Y., Zheng, L., Lam, S., and Xu, J. R. (2002). MST12 regulates infectious growth but not appressorium formation in the rice blast fungus Magnaporthe grisea. Mol. Plant Microbe Interact. 15, 183–192. doi: 10.1094/MPMI.2002.15.3.183
Park, J., Park, J., Jang, S., Kim, S., Kong, S., Choi, J., et al. (2008). FTFD: an informatics pipeline supporting phylogenomic analysis of fungal transcription factors. Bioinformatics 24, 1024–1025. doi: 10.1093/bioinformatics/btn058
Ramamoorthy, V., Dhingra, S., Kincaid, A., Shantappa, S., Feng, X., and Calvo, A. M. (2013). The putative C2H2 transcription factor MtfA is a novel regulator of secondary metabolism and morphogenesis in Aspergillus nidulans. PLoS ONE 8:e74122. doi: 10.1371/journal.pone.0074122
Reverberi, M., Gazzetti, K., Punelli, F., Scarpari, M., Zjalic, S., Ricelli, A., et al. (2012). Aoyap1 regulates OTA synthesis by controlling cell redox balance in Aspergillus ochraceus. Appl. Microbiol. Biotechnol. 95, 1293–1304. doi: 10.1007/s00253-012-3985-4
Reverberi, M., Zjalic, S., Ricelli, A., Punelli, F., Camera, E., Fabbri, C., et al. (2008). Modulation of antioxidant defense in Aspergillus parasiticus is involved in aflatoxin biosynthesis: a role for the ApyapA gene. Eukaryot. Cell 7, 988–1000. doi: 10.1128/EC.00228-07
Ronquist, F., and Huelsenbeck, J. P. (2003). Mrbayes 3: bayesian phylogenetic inference under mixed models. Bioinformatics 19, 1572–1574. doi: 10.1093/bioinformatics/btg180
Roze, L. V., Miller, M. J., Rarick, M., Mahanti, N., and Linz, J. E. (2004). A novel cAMP-response element, CRE1, modulates expression of nor-1 in Aspergillus parasiticus. J. Biol. Chem. 279, 27428–27439. doi: 10.1074/jbc.M400075200
Rybak, K., See, P. T., Phan, H. T. T., Syme, R. A., Moffat, C. S., Oliver, R. P., et al. (2017). A functionally conserved Zn2Cys6 binuclear cluster transcription factor class regulates necrotrophic effector gene expression and host specific virulence of two major Pleosporales fungal pathogens of wheat. Mol. Plant Pathol. 18, 420–434. doi: 10.1111/mpp.12511
Santhanam, P., and Thomma, B. P. (2013). Verticillium dahliae Sge1 differentially regulates expression of candidate effector genes. Mol. Plant Microbe Interact. 26, 249–256. doi: 10.1094/MPMI-08-12-0198-R
Schneider, K. T., van de Mortel, M., Bancroft, T. J., Braun, E., Nettleton, D., Nelson, R. T., et al. (2011). Biphasic gene expression changes elicited by Phakopsora pachyrhizi in soybean correlates with fungal penetration and haustoria formation. Plant Physiol. 157, 355–371. doi: 10.1104/pp.111.181149
Schoberle, T. J., Nguyen-Coleman, C. K., Herold, J., Yang, A., Weirauch, M., Hughes, T. R., et al. (2014). A novel C2H2 transcription factor that regulates gliA expression interdependently with GliZ in Aspergillus fumigatus. PLoS Genet. 10:e1004336. doi: 10.1371/journal.pgen.1004336
Shelest, E. (2008). Transcription factors in fungi. FEMS Microbiol. Lett. 286, 145–151. doi: 10.1111/j.1574-6968.2008.01293.x
Singh, K., Nizam, S., Sinha, M., and Verma, P. K. (2012). Comparative transcriptome analysis of the necrotrophic fungus Ascochyta rabiei during oxidative stress: insight for fungal survival in the host plant. PLoS ONE 7:e33128. doi: 10.1371/journal.pone.0033128
Singh, K. B., Reddy, M. V., and Nene, Y. L. (1984). International testing of chickpeas for resistance to Ascochyta blight. Plant Dis. 68, 782–784. doi: 10.1094/PD-69-782
Soyer, J. L., Hamiot, A., Ollivier, B., Balesdent, M. H., Rouxel, T., and Fudal, I. (2015). The APSES transcription factor LmStuA is required for sporulation, pathogenic development and effector gene expression in Leptosphaeria maculans. Mol. Plant Pathol. 16, 1000–1005. doi: 10.1111/mpp.12249
Spitz, F., and Furlong, E. E. (2012). Transcription factors: from enhancer binding to developmental control. Nat. Rev. Genet. 13, 613–626. doi: 10.1038/nrg3207
Stegmaier, P., Kel, A. E., and Wingender, E. (2004). Systematic DNA-binding domain classification of transcription factors. Genome Inform. 15, 276–286.
Sun, Y., Wang, Y., and Tian, C. (2016). bZIP transcription factor CgAP1 is essential for oxidative stress tolerance and full virulence of the poplar anthracnose fungus Colletotrichum gloeosporioides. Fungal Genet. Biol. 95, 58–66. doi: 10.1016/j.fgb.2016.08.006
Tan, K.-C., and Oliver, R. P. (2017). Regulation of proteinaceous effector expression in phytopathogenic fungi. PLoS Pathog. 13:e1006241. doi: 10.1371/journal.ppat.1006241
Temme, N., Oeser, B., Massaroli, M., Heller, J., Simon, A., Collado, I. G., et al. (2012). BcAtf1, a global regulator, controls various differentiation processes and phytotoxin production in Botrytis cinerea. Mol. Plant Pathol. 13, 704–718. doi: 10.1111/j.1364-3703.2011.00778.x
Todd, R. B., Zhou, M., Ohm, R. A., Leeggangers, H. A. C. F., Visser, L., and de Vries, R. P. (2014). Prevalence of transcription factors in ascomycete and basidiomycete fungi. BMC Genomics 15:214. doi: 10.1186/1471-2164-15-214
Tonukari, N. J., Scott-Craig, J. S., and Walton, J. D. (2000). The Cochliobolus carbonum SNF1 gene is required for cell wall-degrading enzyme expression and virulence in maize. Plant Cell 12, 237–247. doi: 10.1105/tpc.12.2.237
Toone, W. M., and Jones, N. (1998). Stress-activated signalling pathways in yeast. Genes Cells 3, 485–498. doi: 10.1046/j.1365-2443.1998.00211.x
Toone, W. M., Morgan, B. A., and Jones, N. (2001). Redox control of AP-1-like factors in yeast and beyond. Oncogene 20, 2336–2346. doi: 10.1038/sj.onc.1204384
Trapnell, C., Hendrickson, D. G., Sauvageau, M., Goff, L., Rinn, J. L., and Pachter, L. (2013). Differential analysis of gene regulation at transcript resolution with RNA-seq. Nat. Biotechnol. 31, 46–53. doi: 10.1038/nbt.2450
Verma, S., Gazara, R. K., Nizam, S., Parween, S., Chattopadhyay, D., and Verma, P. K. (2016). Draft genome sequencing and secretome analysis of fungal phytopathogen Ascochyta rabiei provides insight into the necrotrophic effector repertoire. Sci. Rep. 6:24638. doi: 10.1038/srep24638
Verma, S., Nizam, S., and Verma, P. K. (2013). “Biotic and abiotic stress signaling in plants,” in Stress Signaling In Plants: Genomics and Proteomics Perspective, Vol. 1, eds M. Sarwat, A. Ahmad, M. Z. Abdin (New York, NY: Springer Science), 25–49. doi: 10.1007/978-1-4614-6372-6_2
Wilson, D., Charoensawan, V., Kummerfeld, S. K., and Teichmann, S. A. (2008). DBD - taxonomically broad transcription factor predictions: new content and functionality. Nucleic Acids Res. 36, D88–D92. doi: 10.1093/nar/gkm964
Wu, A. L., and Moye-Rowley, W. S. (1994). GSH1, which encodes gamma-glutamylcysteine synthetase, is a target gene for yAP-1 transcriptional regulation. Mol. Cell. Biol. 14, 5832–5839. doi: 10.1128/MCB.14.9.5832
Zahiri, A., Heimel, K., Wahl, R., Rath, M., and Kamper, J. (2010). The Ustilago maydis forkhead transcription factor Fox1 is involved in the regulation of genes required for the attenuation of plant defenses during pathogenic development. Mol. Plant Microbe Interact. 23, 1118–1129. doi: 10.1094/MPMI-23-9-1118
Zhang, H. M., Chen, H., Liu, W., Liu, H., Gong, J., Wang, H., et al. (2012). AnimalTFDB: a comprehensive animal transcription factor database. Nucleic Acids Res. 40, D144–D149. doi: 10.1093/nar/gkr965
Zhang, L., Lubbers, R. J. M., Simon, A., Stassen, J. H. M., Ribera, P. R. V., Viaud, M., et al. (2015). A novel Zn2Cys6 transcription factor BcGaaR regulates D-galacturonic acid utilization in Botrytis cinerea. Mol. Microbiol. 100, 247–262. doi: 10.1111/mmi.13314
Zhao, C., Waalwijk, C., de Wit, P. J. G. M., van der Lee, T., and Tang, D. (2011). EBR1, a Novel Zn(2)Cys(6) transcription factor, affects virulence and apical dominance of the hyphal tip in Fusarium graminearum. Mol. Plant Microbe Interact. 24, 1407–1418. doi: 10.1094/MPMI-06-11-0158
Keywords: plant–pathogen interaction, Ascochyta rabiei, necrotrophic fungi, transcription factors, cis-acting elements, secretome
Citation: Verma S, Gazara RK and Verma PK (2017) Transcription Factor Repertoire of Necrotrophic Fungal Phytopathogen Ascochyta rabiei: Predominance of MYB Transcription Factors As Potential Regulators of Secretome. Front. Plant Sci. 8:1037. doi: 10.3389/fpls.2017.01037
Received: 31 March 2017; Accepted: 30 May 2017;
Published: 14 June 2017.
Edited by:
Weidong Chen, Agricultural Research Service (USDA), United StatesReviewed by:
Matthew Denton-Giles, Curtin University, AustraliaR. Varma Penmetsa, University of California, Davis, United States
Copyright © 2017 Verma, Gazara and Verma. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Praveen K. Verma, cGt2QG5pcGdyLmFjLmlu
†These authors have contributed equally to this work.