
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 10 May 2017
Sec. Evolutionary and Population Genetics
Volume 8 - 2017 | https://doi.org/10.3389/fpls.2017.00751
 Nicoletta Ferradini1
Nicoletta Ferradini1 Hovirag Lancioni2
Hovirag Lancioni2 Renzo Torricelli1
Renzo Torricelli1 Luigi Russi1
Luigi Russi1 Isabella Dalla Ragione1
Isabella Dalla Ragione1 Irene Cardinali2
Irene Cardinali2 Gianpiero Marconi1Mauro Gramaccia3Luciano Concezzi3
Gianpiero Marconi1Mauro Gramaccia3Luciano Concezzi3 Alessandro Achilli4
Alessandro Achilli4 Fabio Veronesi1
Fabio Veronesi1 Emidio Albertini1*
Emidio Albertini1*Pear is one of the oldest fruit tree crops and the third most important temperate fruit species. Its domestication took place independently in the Far East (China) and in the Caucasus region. While the origin of Eastern Asian cultivars is clear, that of European cultivars is still in doubt. Italy has a wealth of local varieties and genetic resources safeguarded by several public and private collections to face the erosion caused by the introduction of improved varieties in specialized orchards. The objectives of the present study were: (i) to characterize the existing germplasm through nuclear (SSR) and (ii) to clarify the genetic divergence between local and cultivated populations through chloroplast DNA (cpDNA) markers in order to provide insights into phylogenetic relationships of Pyrus spp. For this reason, 95 entries from five different germplasm collections, including nine European, Mediterranean and Eastern Asian species, were analyzed, and the intergenic accD-psaI sequences were compared to the worldwide distributed dataset encompassing a total of 298 sequences from 26 different Pyrus species. The nine nuclear SSRs were able to identify a total of 179 alleles, with a loci polymorphism P = 0.89. Most of the variation (97%) was found within groups. Five accessions from different sources were confirmed to be the same. Eight out of 20 accessions of unknown origin were identified, and six synonyms were detected. Locus NH030a was found to be monomorphic in all the cultivated accessions and in reference species interfertile with P. communis, leading to hypothesize selection pressures for adaptation to cultivation. The cpDNA sequences of the 95 accessions were represented by 14 haplotypes, six of which (derived from P. communis, P. cossonii and P. ussuriensis) are recorded here for the first time and may suggest the ancient origin of some local varieties. The network analysis of the 298 cpDNA sequences allowed two different haplogroups, Eastern and Western Eurasia, to be defined, supporting recent views of a clear division between Occidental and Oriental species. By combining the results from nuclear and uniparental markers, it was possible to better define many unknown accessions.
Pear belongs to the Rosaceae family, the Maloideae subfamily, and is the third most important temperate fruit species after grape and apple. It is widespread throughout the world with China, the United States, Italy, Argentina, and Spain being the most important producers. Annual pear production in the world is about 25 million tons, 3% of which is produced in Italy, making it the first European producer (FAO, 2014).
The genus Pyrus is characterized by a high genetic variability with several species and thousands of cultivars that can be divided into two major groups, the Occidental (European) and the Oriental (Asian) pears. Being cultivated for more than 3,000 years, pear is also one of the oldest fruit crops in the world (Wu et al., 2013, 2014). The primary centers of origin and of domestication of the genus Pyrus are China and the region from Asia Minor to the Middle East, in the Caucasus Mountains, while a secondary center is located in Central Asia (Vavilov, 1951; Zukovskij, 1962; Silva et al., 2014). Even if the most cultivated species are P. ussuriensis, P. pyrifolia, P. bretschneideri, P. communis, and P. sinkiangensis, to date as many as 22 species have been well recognized (Wu et al., 2013). The basic chromosome number of the Maloideae subfamily is x = 17, a significantly higher number than that of other Rosaceae species (x = 7 or x = 9), thus suggesting a possible polyploidization event (Sax, 1931). According to Silva et al. (2014), the Pyrus genus could be the result of a hybridization between two primitive forms of Rosaceae: Prunoideae (x = 8) and Spiraeodeae (x = 9). The majority of cultivated pears are diploid (2n = 2x = 34), but a few cultivars of P. communis and Pyrus × bretschneideri are also known to be polyploids. It is also believed that cultivated European pears derive from two wild pears, P. pyraster and P. caucasica, which are interfertile with domesticated forms (Zohary and Hopf, 2000).
Despite its presence in Europe from prehistoric time and its economic importance, the exact origin of the European pear is still unknown; on the contrary, places and times of domestication for Asian pears are clearer (Silva et al., 2014).
From the historical point of view, Homer was the first to mention pear cultivation, followed by Marco Porcio Catone (253–150 BC), Varrone, Virgilio, Celso, and by Plinius the Elder, who described almost all of the 40 varieties present in the Roman Empire at their times (Hedrick, 1921; Layne and Quamme, 1975; Columella, 1977; Silva et al., 2014). In the Italian peninsula, after the fall of the Roman Empire, agriculture underwent intense changes, accompanied by modification of the landscape (Andreolli, 1990). In such a context, pears were basically cultivated only in vegetable gardens, mainly for family consumption, honey production and quality wood (Gaulin, 2007). In the Middle Ages the Benedictine monks played a major role in selection, conservation and cultivation of pear varieties, role maintained also in the following period (XVI century) when métayage contracts contributed to increasing the diversification of cultivation (Anselmi, 2000). Starting from the XVIII century many varieties were systematically introduced into Central Italy from other Italian areas, as well as from foreign countries, as documented by botanists and agronomists of that period, such as Felici (1565), Durante (1585), and Caporali (1599). The presence of French/Belgian (Decana del Comizio, Abate Fetel, Curato, Bergamotte Esperen, Passa Crassana) and English (William) varieties was historically well documented and widespread in many other areas, increasing pear diversity and most likely changing of their names. In more recent decades, the introduction of modern and more productive pear cultivars grown in specialized orchards has caused a drastic decrease in diversity and the genetic erosion of ancient cultivars.
Assessment of the existing genetic diversity is a preliminary step in order to plan for its safeguard and its use in breeding programs. Several studies based on molecular markers have estimated the diversity in Pyrus, including modern cultivars and germplasm accessions. RFLPs (Iketani et al., 1998), RAPDs (Oliveira et al., 1999; Teng et al., 2001, 2002), AFLPs (Monte-Corvo et al., 2000; Dolatowski et al., 2004; Bao et al., 2008) and SSRs (Kimura et al., 2002; Bao et al., 2007; Wünsch and Hormaza, 2007; Miranda et al., 2010; Gasi et al., 2013; Akçay et al., 2014; Zheng et al., 2014; Urrestarazu et al., 2015) have been widely used to estimate the genetic diversity and the relationships between pear cultivars, wild forms, and related species. Some ancient Italian pear accessions have also been characterized using morphological and SSR markers (Martinelli et al., 2009).
Recently, in order to reconstruct the phylogeny and the evolution of several genera, scientists have directed their attention to the variability of the chloroplast DNA (cpDNA) (Crosby and Smith, 2012; Vieira et al., 2014; Pervaiz et al., 2015). Due to its peculiarities, such as uniparental inheritance, absence of recombination and high level of genetic diversity, cpDNA analyses are very useful for assessing the phylogenetic structure of different populations (Nock et al., 2011; Scarcelli et al., 2011; Yang et al., 2013; Wheeler et al., 2014). Noncoding DNA regions of cpDNA have been successfully applied to phylogenetic studies of plants at lower taxonomic levels, as in the Rosaceae taxa (Corriveau and Coleman, 1988), and in Pyrus in particular (Kimura et al., 2003; Katayama et al., 2007, 2012).
Moreover, uniparentally inherited genomes are sensitive to historical bottlenecks (Morgante et al., 1997) and cpDNA markers might provide some genetic information about a species. Therefore, a combined analysis of nuclear SSR markers and cpDNA haplotypes could provide a more comprehensive view of population structure and demographic history (Burban and Petit, 2003; Petit et al., 2005).
The present research aims to provide further insight into the phylogenetic relationships of Pyrus, starting from native local accessions across the entire genus. This comparative analysis involved 95 pear accessions (prevalently classified as P. communis, but also P. pyraster, P. caucasica and some Asian species), and all intergenic segment accD-psaI recorded in GenBank, encompassing a total of 298 sequences of 26 different Pyrus species from both cultivated and wild populations. Integrating nuclear and cpDNA markers, it thus represents one of the first comprehensive phylogenetic and phylogeographic analyses of different pear populations distributed from East to West in the Eurasian region. It is expected to shed light on the geographical origins and ancestral populations of cultivated pear species, in order to clarify the genetic divergence between local and cultivated populations and to define appropriate management of the local genetic resources.
Ninety-five accessions from European and Asiatic Pyrus spp. were included in this study (Table 1 and Table S1). Many of the 95 accessions were well documented by historically reliable sources. For others, lacking some information, the original name, like that given by the donor, was maintained. In all other cases they were named Unknown. Therefore, based on the initial information, the 95 accessions used in this study were grouped into reference species (RS), commercial varieties (CV), local varieties (LV) and unknown accessions (UA), where CV and RS were used as controls. Moreover, 75 accessions were divided into three geographic groups, following the classification of Fideghelli (2007): Mediterranean (2), East-Asian (4), and European (69 in total: 24 CV, 2 RS, 43 LV). Because the remaining 20 accessions could not be assigned to any geographic group, they were grouped as Unknown (Table S1).
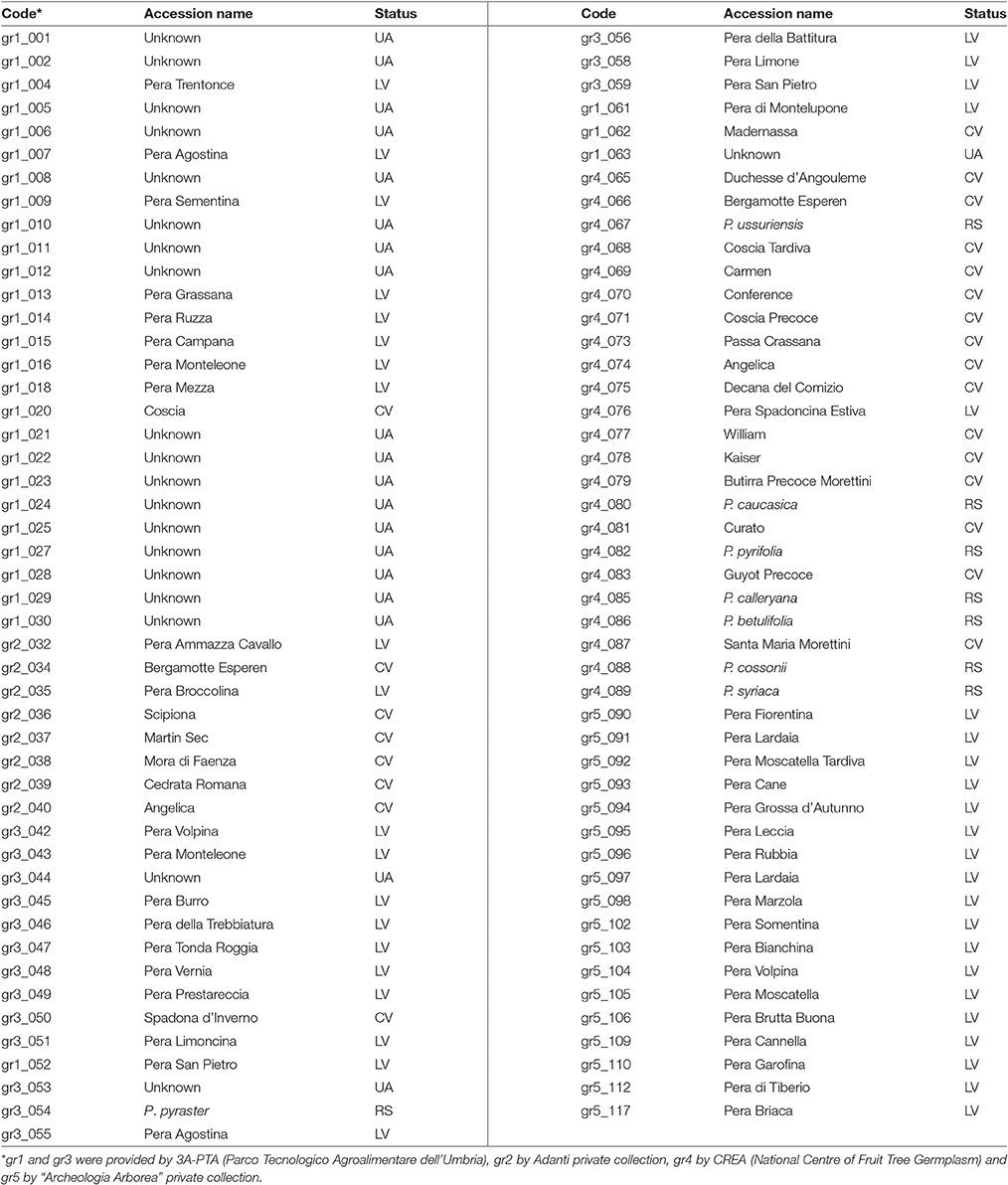
Table 1. Names, status and codes of the 95 pear accessions used in the present study.
Total genomic DNA was isolated from young leaves using the DNeasy Plant Kit (Qiagen) following the protocol provided by the manufacturer. Nine pear SSR primer combinations (Yamamoto et al., 2002a,b) were used (Table S2). PCRs were carried out with the Type-it Microsatellite PCR Kit (Qiagen) containing 1X Type-it master mix with 0.2 μM of each forward and reverse primer and 20 ng of DNA and H2O to a final volume of 20 μl. Amplification was performed as follow: an initial step at 95°C for 5 min followed by 30 cycles at 95°C for 30 s, 54–62°C for 30 s, and 72°C for 30 s, and a final extension at 72°C for 10 min.
PCR products were separated and analyzed on a 3130 XL DNA Analyzer (Applied Biosystems). The size of the amplified products was determined on internal standard DNA (GeneScan 500 Liz, Thermo Fischer Scientific) and the scorable peaks were assigned by GeneMapper software (Applied Biosystems).
The hypervariable accD-psaI intergenic spacer was amplified by PCR using accD F2 (Zheng et al., 2014) and psaI 75R (Small et al., 1998) primers. PCR reactions contained: 1X Phusion HF Buffer, 200 μM dNTPs each, 0.5 μM each primer, 0.2 U Phusion Taq DNA polymerase (Thermo Fischer Scientific), 30 ng genomic DNA and H2O to a final volume of 50 μl. PCR amplification was carried out by GeneAmp PCR system 9700 (Applied Biosystems) programmed as follow: 98°C for 30 s, followed by 30 cycles of 98°C for 10 s, 66°C for 15 s, 72°C for 30 s, and then 72°C for 10 min.
The purified PCR products of about 1,000 bp were sequenced at the Polo di Innovazione Genomica, Genetica e Biologia (Perugia, Italy). The new cpDNA sequences were recorded in GenBank with accession numbers from KY606436-KY606530.
The statistical analysis of the SSR data matrix included (i) the estimation of observed (Ho) and expected (He) heterozygosity (Nei, 1978), (ii) the F statistics (Fis and Fst) (Weir and Cockerham, 1984), and (iii) the analysis of molecular variance (AMOVA) by estimating the fraction of the genetic variation among and within populations (Excoffier et al., 1992; Michalakis and Excoffier, 1996). The software packages GENODIVE (Meirmans and Van Tienderen, 2004) and SPAGeDi1.2 (Hardy and Vekemans, 2002) were used for these purposes, being able to analyze data files containing diploid and triploid accessions.
SSR data were also converted to a binary data matrix by assigning “0” to the absence of a defined allele and “1” to its presence, and were used to estimate a similarity matrix using the coefficient of Dice (Dice, 1945), and the individuals were clustered by the unweighted pair group method with arithmetic mean (UPGMA) and validated by 1,000 bootstrap replicates using PAST software (Hammer et al., 2001).
The SSR profiles of the 95 accessions were used to investigate the population structure through the Bayesian model-based clustering procedure of STRUCTURE ver. 2.2.3 (Pritchard et al., 2000). The analyses were based on an admixture ancestral model with correlated allele frequencies, and the number of K clusters was determined by simulating a range of K-values starting from one to ten. A burn-in and a run length of the Monte Carlo Markov Chain (MCMC) of 200,000 and 500,000 iterations for data collection with 10 runs per K-value were used. The best K-value was determined through the ΔK method (Evanno et al., 2005) by using Structure Harvester ver. 0.6.193 application (Earl and vonHoldt, 2012). A second step analysis of STRUCTURE was then performed separately at these K-values with 600,000 burning period and 1,000,000 MCMC repeats after burning. The 95 individuals were assigned to the groups according to their highest membership coefficient, considering a strong affinity when the assigning probability (qI) was ≥0.80 (Breton et al., 2008; Pereira-Lorenzo et al., 2008; Miranda et al., 2010; Urrestarazu et al., 2012).
In order to analyze the cpDNA sequences, forward and reverse sequences from each sample were assembled and aligned using Sequencher™ 5.10 (Gene Codes Corporation). Accession gr4_077 P. communis cultivar William was selected as a reference sequence (GenBank accession number KY606501). It is one of the most common and suitable exemplars of European pear cultivars.
Different cpDNA sequence variation parameters were estimated by using DnaSP 5.1 software (Librado and Rozas, 2009). AMOVA was carried out as reported above for the SSR data. The 95 sequences were also compared to all accD-psaI data recorded in GenBank: 203 Pyrus sequences from 26 species and 4 putative inter-specific hybrids. The final dataset included 298 Pyrus accessions from three major geographic areas: Europe, the Mediterranean Area and Asia; the latter included Eastern and Middle Eastern subgroups (Tables S3, S4). A sequence from Malus domestica was included and used as an outgroup (Katayama et al., 2012). Indels of different lengths at the same position were separately treated and coded with the number of inserted or deleted nucleotides (Table S5). The final alignment was compared to the reference sequence, thus allowing sequence classification in different haplotypes. The evolutionary relationships among haplotypes were visualized through the construction of a Median-Joining network using the software Network 4.6 (http://www.fluxus-engineering.com/), each indel was considered as a single mutational event and partitioned by using different indel codes (Table S6).
As for principal component analysis (PCA), performed by XLSTAT (2011) statistical software, each species was considered as a discrete variable, the initial dataset was converted into principal components (PCs) and it was possible to graphically display the relationships among the intergenic regions accD-psaI of all sequences.
Scorable amplicons were produced for all nine nuclear SSRs, with a total of 179 alleles. The average number of alleles per locus was 20, ranging from five (NH030a) to 29 (NH023a), but the number of effective alleles per locus was significantly lower (NAe = 6.8). At locus NH030a, out of five alleles, allele 167 showed a frequency of 0.97; thus loci polymorphism was P = 0.89 (Cavalli-Sforza and Bodmer, 1971).
As many as 24 individuals out of 95 (23%) showed only one locus with a third allele. Ten genotypes showed 2 loci with a third allele (9.5%), while nine individuals showed a third allele at more than 2 loci (Table S7). In particular, except for locus NH030a, all other loci showed at least one genotype with three alleles. NH026a and NH023a identified 16 and 18 individuals with three alleles, respectively; NH029a had only two, while the other loci identified between seven and ten individuals with three alleles.
The AMOVA carried out with all loci showed that most of the existing variability was within groups (97%) rather than among groups (Table S8), which is in agreement with many outbreeding species and similar studies in pear (Jiang et al., 2009; Miranda et al., 2010; Wolko et al., 2015; Wuyun et al., 2015). Nonetheless, differences among the 4 groups (CV, LV, RS, and UA) were found in terms of the effective number of alleles and levels of heterozygosity.
The 95 individuals in the present study were not a panmictic population, as classically defined in population genetics. Therefore, the indices used here (number of effective alleles, observed and expected heterozygosity, FIS and FST) as estimates of existing genetic variability should be considered with caution. It is worth noting that the effective number of alleles (NAe) is a measure of the genetic variability (Zouros, 1979), also valid in the presence of small samples (Nielsen et al., 2003). In our data the highest average number of effective alleles was found in RS (NAe = 12.4), while the lowest was in CV (4.8) (Table 2); it is interesting to note that the values of LV and UA were intermediate and similar to one another (6.9).
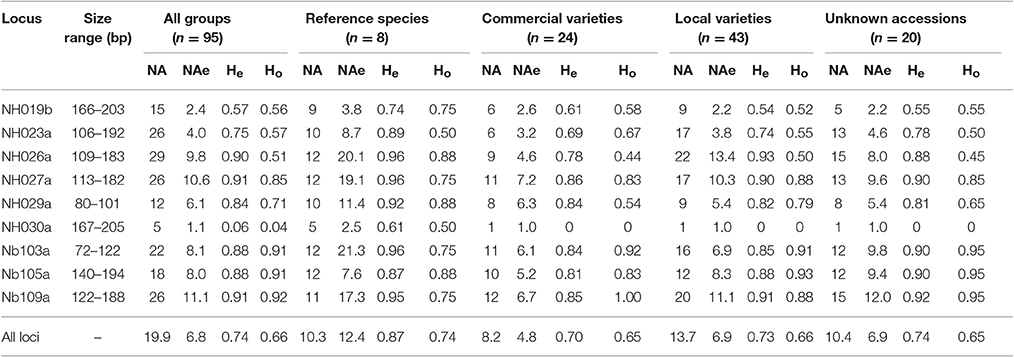
Table 2. Range of SSR allele size per locus, number of alleles (NA) and number of effective alleles per locus (NAe), expected (He) and observed (Ho) heterozygosity in all individuals and in each group based on available information, as estimated with SPAGeDi 1.5.
A similar trend was observed in terms of heterozygosity. The average expected heterozygosity, considering all of the pear accessions, was He = 0.74, ranging from 0.87 in RS to 0.70 in CV. The highest mean expected value of heterozygosity for all accessions was found at loci Nb109a and NH027a (He = 0.91), while the lowest was at locus NH030a (He = 0.063). Noticeably, at this locus, He in the reference species was 0.61, while in all other groups (LV, CV, and UA) He = 0, monomorphic and due to the fixation of allele 167. In addition, in RS, NH026a, NH027a, and Nb103a loci showed He-values as high as 0.96 (Table 2).
The mean FST-value equal to 0.014 (Table 3) indicates a moderate differentiation among the four groups (P < 0.0005), thus confirming that most of the variation is within groups; at locus NH030a the FST value of 0.278 indicates a significantly high genetic differentiation (P < 0.0002), suggesting that at this locus the selection for local adaptation could have been so strong as to restrict its variation to a single allele in all cultivated forms.
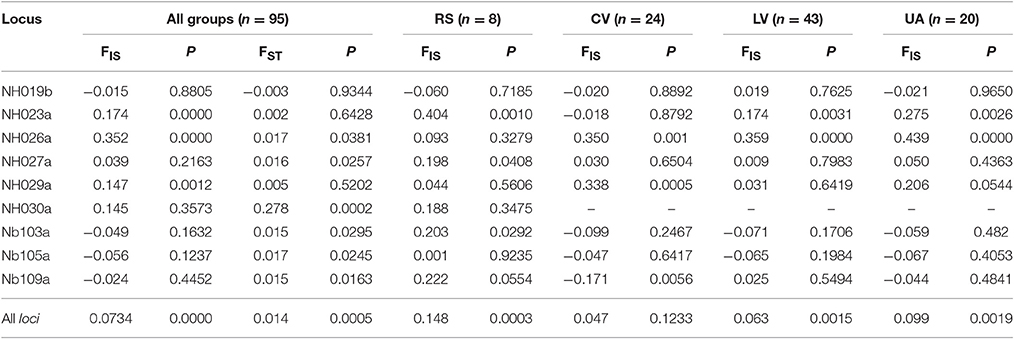
Table 3. F-Statistics as estimated by SPAGeDi 1.5 (Weir and Cockerham, 1984) of 95 entries of Pyrus spp. grouped by Reference species (RS), Commercial varieties (CV), Local varieties (LV), and Unknown accessions (UA), and based on the 9 simple sequence repeat loci (alone and all together).
The nine SSRs were also used to determine the genetic structure among the 95 accessions of Pyrus spp. The plot of the average log-likelihood values for Ks ranging from 1 to 10 and the distribution of ΔK-values (Evanno et al., 2005) according to K-values is shown in Figure S1. Two peaks were found, corresponding to K = 3 and K = 6, and the hierarchical genetic structure was investigated at K = 3. A threshold value qI ≥ 0.80 was used to assign individuals to the clusters (Figure 1). Structure at K = 3 was able to define 3 clusters (from 1A to 3A). Cluster 1A included mostly commercial cultivars and P. syriaca, one of the reference species able to intercross with P. communis. Cluster 2A included 27 accessions, almost all the rest of the reference species (six, except P. cossonii), most of LV provided by the “Archeologia Arborea” collection and only one commercial cultivar (Carmen). Cluster 3A included 22 accessions, mostly LV (12) and 7 CV, four of which were of French origin. Some accessions (11 LV, 6 CV, 5 UA, and P. cossonii) were clustered in the admixture group. Although this was not the object of the present study, as physiological and phenotypic information was scarce and incomplete, Cluster 1A included the majority of accessions characterized by summer ripening, Cluster 2A all accessions with an autumn-winter ripening period, while accessions in Cluster 3A were not clear cut in terms of ripening period.
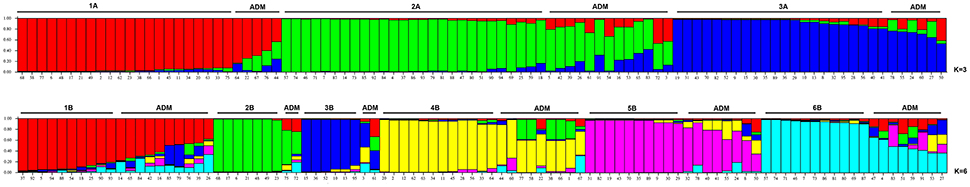
Figure 1. Structure classification at K = 3 and K = 6 of the 95 entries of Pyrus spp. The clusters are marked with different colors, while the accessions at P of (qI) ≤ 0.80 are grouped as Admixture (ADM).
The structure at K = 6 allowed six Clusters (from 1B to 6B, Figure 1) to be distinguished. Cluster 1B included some unknown accessions and some local varieties provided by “Archeologia Arborea” (gr5). Cluster 2B included two commercial cultivars. Pera Briaca, Pera di Montelupone, Pera Monteleone and Pera Ruzza were assigned to Cluster 3B, characterized by winter ripening and a round fruit shape (Table S1). Cluster 4B included only commercial cultivars. Cluster 5B was represented by two local varieties (Pera Broccolina and Pera Volpina) and some cultivars, all characterized by autumn-winter ripening. On the contrary, Cluster 3B included accessions characterized by winter ripening and a round fruit shape (Supplementary Table S1). Cluster 6B included four local varieties and five reference species (P. betulifolia, P. calleryana, P. caucasica, P. pyrifolia, and P. ussuriensis).
The accessions of Pyrus spp. were clustered by UPGMA following the similarity estimates of Dice's coefficient (Figure 2). Reference species were found in a different subcluster, quite distant from most accessions, with the exception of P. cossonii, P. pyraster, and P. syriaca. In particular, P. syriaca is clustered with Coscia, Coscia Tardiva, Decana del Comizio, Santa Maria Morettini and William, while P. cossonii and P. pyraster with some local varieties, such as Pera di Tiberio and Pera Rubbia.
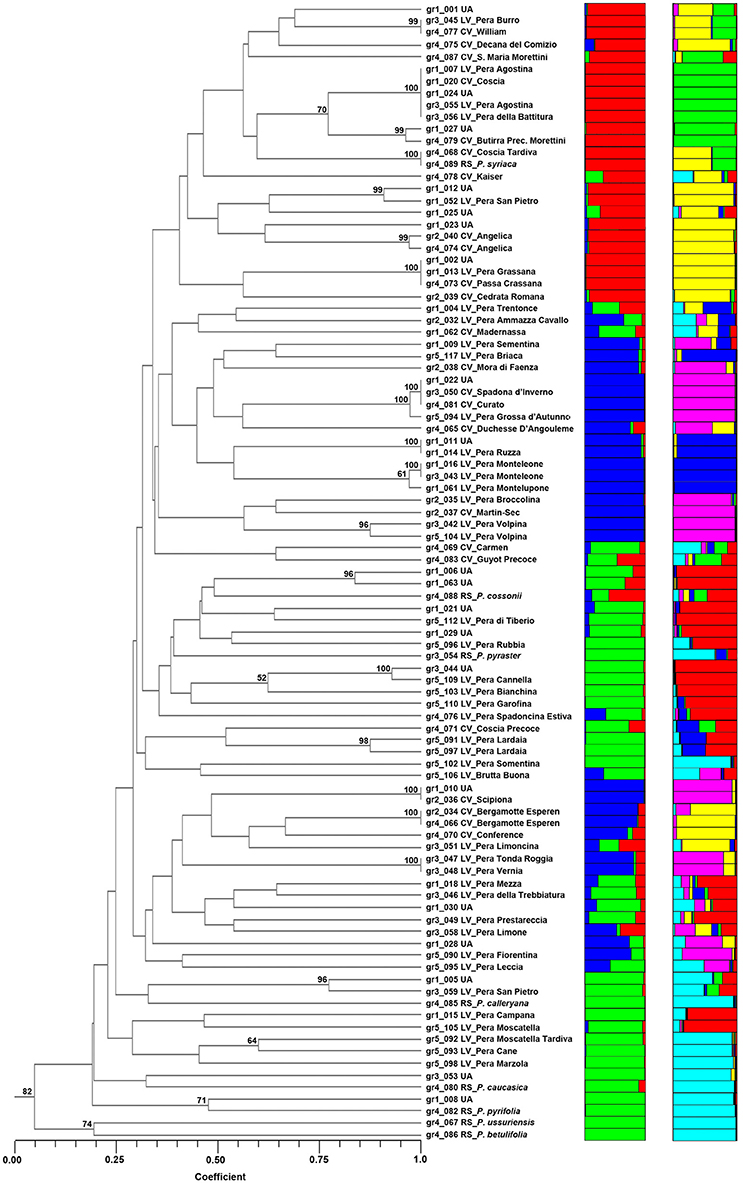
Figure 2. The dendrogram of the 95 accessions of Pyrus spp. as clustered by UPGMA (on the left) and by STRUCTURE at K = 3 and K = 6 (on the right). Numbers on the cluster node indicate its probability (%) obtained by bootstrap.
Several entries clustered at a similarity coefficient equal to 1. For example, the two accessions of Pera Agostina (gr1_007 and gr1_055) clustered with Coscia (gr1_020), Pera della Battitura (gr1_056) and one unknown accession (gr1_024), suggesting cases of synonymies with Coscia. As a matter of fact, “Agostina” in Italian means August, and “battitura” means “threshing,” most likely referring to the harvesting of cereals, normally occurring in summer, which is the ripening period of Coscia.
Similarly, Pera Grassana (gr1_013), Passa Crassana (gr4_073) and the unknown accession gr1_002 are all the same and likely to be Passa Crassana. Moreover, the present study confirmed that Spadona d'Inverno (gr3_050) and Curato (gr4_081) are synonyms and that gr1_022 (Unknown), clustering at a similarity of 1, is also likely to be the same accession. The same holds for Pera Tonda Roggia (gr3_047) and Pera Vernia (gr3_048), probably synonyms for the same genotype.
Furthermore, entry gr1_010 (Unknown) could be named Scipiona (gr2_036) and entry gr1_011 could be named Pera Ruzza (gr1_014). Entries gr1_016 and gr1_043, known as Pera Monteleone, are confirmed to have the same genotype and clustered together with Pera di Montelupone.
In addition to the clusters that joined at a similarity equal to 1, the analysis confirmed that the two accessions of Angelica clustered together, as well as the two accessions of Bergamotte Esperen and all the accessions of Pera Lardaia and Pera Volpina. The analysis summarized in Table S9 was also able to give insights for other accessions, such as Pera Grossa d'Autunno, similar to Curato and Spadona d'Inverno, and some unknown accessions that could be named as the closest commercial or local variety.
The 95 novel chloroplast DNA sequences ranged from 622 to 899 bps depending on the presence of ten indels (four deletions at nps 210, 322, 545, and 601, and six insertions at nps 305, 544, 600, 620, 636, and 665); gr4_088 was the only accession without indels and differed from the reference sequence William only by the transversion at np 567 (567T). After grouping the accessions into four categories (reference species, RS; commercial varieties, CV; local varieties, LV; and unknown accessions, UA), AMOVA was carried out on all haplotypes and the results obtained through the SSR were confirmed. In fact, most of the observed variance was attributable to differences among samples within groups (96.69%), rather than the variability among groups (3.31%).
The analyses was then extended to the entire dataset of 298 cpDNA sequences (95 from the present study and 203 from GenBank). A total of 75 haplotypes were identified and named from HT01 to HT75 (Table S3). The 95 accessions of the present study were represented by 14 haplotypes, the most frequent of which was haplotype HT06 (47%). Out of these 14 haplotypes, 8 were shared among different accessions of P. communis (HT02, HT06, HT07, HT08, HT09, HT11, HT12, HT13), P. caucasica (HT09), P. syriaca (HT06) and some unknown accessions (HT02, HT06, HT08, HT09, HT12, HT13), while 6 were novel (Table S10): one was found in P. ussuriensis (HT18), one in P. cossonii (HT19) and the other four (HT08, HT11, HT12, and HT13) were found in P. communis. As evident in Figure S2, the new haplotypes (circled in red), whose sequences have never been reported before, were present in about 28% of the whole P. communis species and this value rises up to 50% if the most common haplotype HT06 is excluded from the analysis. This means that half of the P. communis samples have sequences never reported before. It is worth noting that four novel haplotypes (HT08, HT11, HT12, and HT13) were well represented in many accessions from Central Italy, most of which were provided by “Archeologia Arborea” (13 out of the 18 accessions, Table S1).
The reconstructed network of the cpDNA intergenic region accD-psaI clearly defined the distribution of the 95 accessions in different branches. All sequences clustered into two main groups, hereafter called Western and Eastern haplogroups (W and E, respectively, Figure S3) and were discriminated by the deletion of 20 bp at np 601. The Eastern group included only the four RS from Eastern Asia (P. betulifolia, P. calleryana, P. pyrifolia, and P. ussuriensis). The Western group included prevalently European and Mediterranean species, and encompassed all the unknown accessions (N = 20). These latter samples showed the two most common Western haplotypes, namely HT06 and HT09, encompassing different Pyrus spp., and five other haplotypes, four of them shared with accessions of P. communis (Figure S3).
A clear-cut geographic subdivision was also observed when all available sequences from GenBank were included in the network (Figure 3). The dominant haplotypes for the Western and Eastern geographic groups were HT06 (including the reference sequence) and HT04, respectively. Out of a total of 298 samples (95 from this study and 203 from GenBank), 169 accessions belonged to the Western haplogroup (57%), mostly derived from European species (61%), Mediterranean species (16%) and only a small amount from Eastern Asia (1.2% including only P. pyrifolia) and from the Middle East (P. spinosa, P. salicifolia, and P. regelii; 8.3%). These two latter species were absent in the Eastern haplogroup which presented only P. pashia (15.5%) and one sequence from P. spinosa, as representative of Middle Eastern species (Figure 3). In addition, the Eastern haplogroup is prevalently characterized by species from Eastern Asia (68.2%), no Mediterranean species, and only three sequences from European species (P. caucasica, P. communis, and P. korshinskyi).
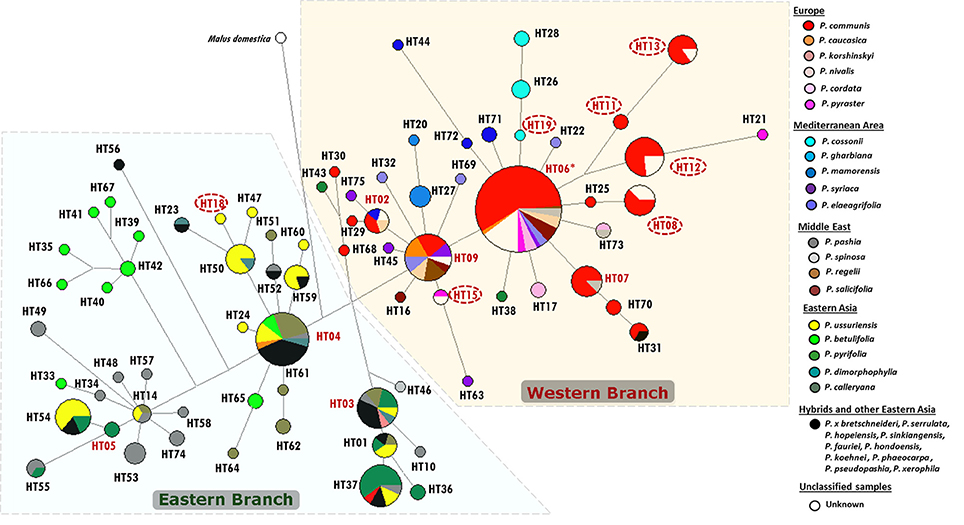
Figure 3. Median-joining network based on all available accD-psaI sequences in the Pyrus genus. Different species, each derived from specific geographic areas, are marked with different colors. Haplotype classification is detailed in Table S3 and Table S4. In particular, haplotypes derived from our samples are reported in red: circled when unique (see also Table S10), and not circled when shared with previously recorded sequences. The asterisk indicates the haplotype used as Reference Sequence.
The unknown accessions of the present study were included in the Western branch, and shared the same haplotypes of P. communis (HT08, HT12, and HT13), P. pyraster (HT15) and of other Pyrus spp. (HT02, HT06, and HT09).
The haplotype relationships were then summarized through principal component analysis (PCA) of the entire dataset. A preliminary PCA (not shown) was uninformative because of the distortion due to two evident outliers (P. betulifolia and P. pashia), both exclusively from the Eastern cluster in Figure 3, which moved far because of their unique haplotypes (HT33, HT35, HT39, HT40, HT41, HT42, HT65, HT66, HT67 for P. betulifolia, and HT10, HT34, HT48, HT49, HT53, HT57, HT58, HT74 for P. pashia). In order to better resolve the graph, a further analysis was performed without those two outliers (Figure 4). In the resulting PCA, some species split up into different quadrants: P. communis in the first, P. pyrifolia in the second, and P. calleryana and P. ussuriensis in the third one. The unknown accessions (grouped together here) were placed between P. communis and a mixed Western Eurasian group located in the center. Magnification of this group highlighted that the unknown accessions were nearby to a subgroup of four closely-related Middle Eastern (P. salicifolia and P. spinosa) and European (P. cordata and P. pyraster) species, which in turn were closer to P. caucasica and P. regelii, than to those from Eastern Asia and the Mediterranean area.
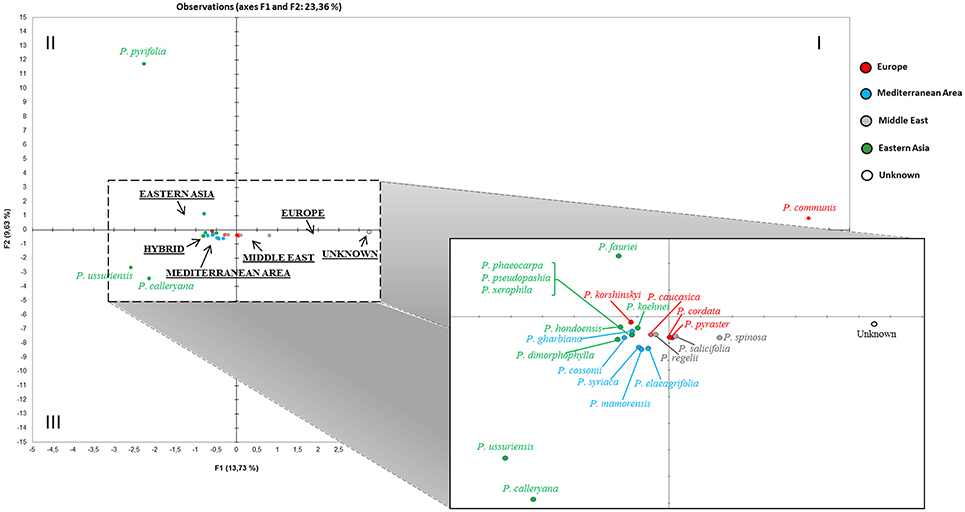
Figure 4. Two-dimensional region-based PCA plot obtained by including all available Pyrus cpDNA data (see also Table S3 and Table S4). The most divergent P. betulifolia and P. pashia, and all hybrids were excluded from the final PC analysis. The macrogeographic areas are underlined and represent the centroids of species derived from the area. A magnification of the central mixed group is also presented.
The possibility of identifying synonymous and homonymous accessions, sometimes even unknown genotypes, emphasized the importance of verifying germplasm collections with powerful tools such as molecular markers. This step is important in order to avoid redundancy in the collections, reduce their management costs and to be able to distribute true-to-type cultivars to nurseries. This was one of the goals of the present study, along with inference on the genetic structure and on the understanding of the geographic origin (phylogeny and evolutionary history) of pear accessions found in Central Italy. For both objectives reliable markers are essential for an accurate genetic identification, establishing genetic relationships among the accessions and improving the management and use of field-collected fruit tree germplasm.
The former task was partially achieved through nine SSR markers that displayed a high degree of polymorphism and discriminating power, a sufficient number to fingerprint germplasm collections (Urrestarazu et al., 2015). In summary, five accessions obtained from different sources were confirmed to be the same. Eight out of 20 accessions of unknown origin were identified. No homonymous accessions were present. Six synonyms were detected. In some instances, synonyms may be due to small changes in the names due to differences in local dialects (Pera Monteleone - Pera di Montelupone; Passa Crassana - Pera Grassana), and in other cases a variety may have been renamed to associate its ripening with some seasonal events (Coscia - Pera Agostina; Coscia - Pera della Battitura).
Understanding the genetic structure of pear accessions is more difficult to achieve than in most fruit tree species, as they are perennials and since ancient times their distribution has been generally human-mediated by clonal propagation (basically by grafting) in order to overcome the juvenile phase. The mode of reproduction, essentially outcrossing, and the human-mediated evolutionary processes (clonal propagation) played a critical role in the domestication and genetic variation found nowadays in most fruit tree species. For these species, domestication has been greatly affected by the propagation system, and studies in these fields, compared with annual species, are in progress. In evolutionary terms, by selecting and growing a high number of individuals with the same genotype (cultivar), a genetic bottleneck (Miller and Gross, 2011) has developed, which is expected to reduce the genetic variability of the species. Nevertheless, the selected genotype contains genetic variability in a potential state, conserved in the heterozygotic condition. Despite such observations, in perennial fruit tree crops the size of the genetic variation found within populations is generally significantly higher compared with that between populations, so that normally these populations are unstructured (Miller and Gross, 2011). High levels of heterozygosity and a high effective number of alleles per locus were found in the present study. By grouping the accessions into four categories (CV, LV, RS, and UA) the within group variation was as high as 97% compared with that among groups; even by grouping the genotypes by STRUCTURE the within variation was still as high as 87.5%, confirming a weak population structure (Loveless and Hamrick, 1984).
At locus NH030a, an FST-value of 0.278 was found, indicating a high differentiation between RS and all other cultivated forms. Looking at the alleles in these two groups it was found that allele 167 was fixed in all individuals of CV, LV, UA compared with RS. In addition, among the RS, P. caucasica, P. cossonii, P. pyraster, and P. syriaca showed allele 167 fixed. This was partially confirmed by the cluster analysis, particularly for P. cossonii and P. syriaca, grouped together with many cultivated accessions, of which they are interfertile. Interestingly, locus NH030a was described as being associated with two QTLs, Pfl-7-2 and Pfi-7-1, located on the 7th linkage group and involved in controlling fruit dimension (length) and fruit shape index, respectively (Zhang et al., 2013). This shed light on the SSR results and led to the hypothesis that allele fixation at this locus is most likely the result of human selection for fruit size. Moreover, this association is difficult to break if reproduction is carried out by clonal procedures.
Among the 95 accessions, 52 were diploids and 43 (45%) were putative polyploids, since at least 3 alleles were found in at least 1 of the 9 loci. Reference species were generally diploids, except for P. pyrifolia that is characterized by 5 loci with three alleles. But, as pointed out by Ferreira dos Santos et al. (2011), the amplification of three fragments in a single locus is not proof of the triploid status: mutational events (somatic mutations generating chimerical or mosaic states, or duplication events of a chromosome fragment) might give rise to non-real alleles. In our case, considering only individuals showing at least 2 or more loci with a third allele, the percentage of putative polyploids is 20%, a value similar to 18% reported by Ferreira dos Santos et al. (2011).
The uniparental genetic analysis confirmed the AMOVA on SSRs, with the size of the within group variability enormously greater than that among groups (96.7 vs. 3.3%, respectively).
Based on the available phylogenetic data, the large number of varieties of the cultivated European pears is most likely derived from one or two wild species (P. pyraster and/or P. caucasica), widely distributed throughout Europe (Vavilov, 1992). The analysis on the intergenic segment accD-psaI supported the opinion of a clear division between Occidental and Oriental species. The network analysis of 298 cpDNA sequences allowed two different haplogroups with a geographic prevalence in Eastern and Western Eurasia to be defined. The sequences of P. caucasica and P. pyraster presented mostly western haplotypes (HT06, HT09, HT15, and HT21); an eastern haplotype was identified only in P. caucasica (HT04). HT06 and HT09 were the most frequent and variable in the Western haplogroup and presented other different species belonging to all the geographic areas, except for Eastern Asia. Remarkably, one of our unknown accessions shared haplotype HT15 with an accession of P. pyraster, indicating a possible connection between them, confirmed also by their inclusion in the same STRUCTURE Cluster (Figures 1, 2A). Furthermore, haplotype HT21 (present only in P. pyraster and absent in all our 95 accessions) is closely related to HT12, one of the novel haplotypes identified here. HT12 includes ten accessions of P. communis (all LV, mostly from Archeologia Arborea) and three unknown accessions from Central Italy; the proximity of HT21 and HT12 suggests an ancient origin of them. In addition, haplotype HT12 and HT21 were closely clustered to HT11 and HT13, all represented by local varieties, again mostly from Archeologia Arborea, and further highlighting their possible ancient origin.
By combining the results from the SSRs and cpDNA, it was possible to better define many unknown accessions. For instance, HT07 included almost all local varieties (86%), two of them (Pera Tonda Roggia and Pera Vernia) were found as synonyms according to the SSR results. Haplotype HT08 included a total of eight accessions, four commercial varieties, one local variety and three unknown accessions. These unknown accessions could be classified as P. communis. Moreover, one of them (gr1_010) showed the same haplotype as Scipiona, which is a further validation of the similarity found by SSR, so that it might consider as the same genotype. Sharing the same haplotype (HT08) also confirmed that Curato and Spadona d'Inverno could be considered synonyms, while Pera Grossa d'Autunno, considered to be Spadona d'Inverno by the SSR profile, showed a different haplotype (HT11) and needs to be reconsidered.
HT12 included 13 accessions, ten local varieties and three unknown accessions. Therefore, the unknowns are expected to be P. communis; one of them (gr1_011) was confirmed to be Pera Ruzza, while gr1_44 was confirmed to be Pera Cannella. The same haplotype also included the two accessions of Pera Lardaia, the two accessions of Pera Monteleone and Pera di Montelupone, confirming once again the SSR results.
HT13 included only one unknown accession (gr1_025) and this could be ascribed to P. communis.
All together out of the 20 unknown accessions, seven could be classified at least at a species level, namely as P. communis (4 CV and 3 LV). These results confirm the usefulness of comparing the two molecular marker systems for evaluating the genetic variability of local and unknown accessions. The uniparental genetic system leads to reconstructing the phylogenetic relationships of different cultivars and species. The comparison between the haplotypes found in our accessions and that from GenBank highlighted that 99% of the accessions of P. communis, all Mediterranean accessions and all European accessions, P. cordata, P. nivalis, and P. pyraster, belonged to the Western clade, which also included some sequences with a Middle Eastern origin, but without any P. pashia representatives. This result also strengthens the hypothesis of the proximity of European pears to some species of Mediterranean and Middle Eastern origin.
EA and FV conceived the study; EA designed and coordinated the experiments; IR, MG, and LC chose and provided the germplasm; IR provided the historical and anthropological information; NF and GM performed the lab experiments; NF, RT and LR conducted the SSR data analysis; AA coordinated the analysis on the cpDNA; HL and IC performed the analysis on the cpDNA; NF, HL, RT, LR, IC, and EA wrote the manuscript; NF, HL, RT, LR, IC, GM, AA, and EA revised the manuscript.
The research was supported by “Convenzione con il Parco 3APTA 2014: servizio di conservazione e ampliamento delle banche regionali della biodiversità,” by the personal funding of Prof. Albertini (ALBPRESTAZ and ALBRICVAR) and by the Italian Ministry of Education, University and Research: Progetti Futuro in Ricerca 2012 (RBFR126B8I).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Authors would like to thank Adanti private collection (Bevagna, Perugia) and the National Center of Fruit Tree Germplasm (CREA, Rome) for providing germplasm material, and Dr. Mary F. Traynor for the English revision of the manuscript.
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.00751/full#supplementary-material
Akçay, M. E., Burak, M., Kazan, K., Yüksel, C., Mutaf, F., Bakir, M., et al. (2014). Genetic analysis of Anatolian pear germplasm by simple sequence repeats. Ann. Appl. Biol. 164, 441–452. doi: 10.1111/aab.12113
Andreolli, B. (1990). “Il Ruolo dell'Orticultura e della Frutticultura nelle Campagne Dell'alto Medioevo”, in L'ambiente Vegetale Nell'Alto Medioevo (Spoleto: CISAM - Centro Italiano Studi sull'Alto Medioevo), 175–219.
Anselmi, S. (2000). “Il farsi della piccola proprietà contadina”, in Chi ha Letame Non Avrà Mai Fame. Studi di Storia dell'Agricoltura, 1975–1999. Quaderni di “Proposte e ricerche.” (Ancona: Università Politecnica delle Marche), 71–175.
Bao, L., Chen, K., Zhang, D., Cao, Y., Yamamoto, T., and Teng, Y. (2007). Genetic diversity and similarity of pear (Pyrus L.) cultivars native to East Asia revealed by SSR (simple sequence repeat) markers. Genet. Resour. Crop. Evol. 54, 959–971. doi: 10.1007/s10722-006-9152-y
Bao, L., Chen, K., Zhang, D., Li, X., and Teng, Y. (2008). An assessment of genetic variability and relationships within Asian pears based on AFLP (amplified fragment length polymorphism) markers. Sci. Hortic. 116, 374–380. doi: 10.1016/j.scienta.2008.02.008
Breton, C., Pinatel, C., Médail, F., Bonhomme, F., and Bervillé, A. (2008). Comparison between classical and Bayesian methods to investigate the history of olive cultivars using SSR-polymorphisms. Plant Sci. 175, 524–532. doi: 10.1016/j.plantsci.2008.05.025
Burban, C., and Petit, R. J. (2003). Phylogeography of maritime pine inferred with organelle markers having contrasted inheritance. Mol. Ecol. 12, 1487–1495. doi: 10.1046/j.1365-294X.2003.01817.x
Cavalli-Sforza, L. L., and Bodmer, W. F. (1971). The Genetics of Human Populations. San Francisco, CA: Freeman W.H. Company.
Columella, L. (1977). L'arte dell'Agricoltura. Transl. by R. Calzecchi Onesti and ed. by C. Carena. Torino: Einaudi Editore.
Corriveau, J. L., and Coleman, A. W. (1988). Rapid screening method to detect potential biparental inheritance of plastid DNA and results for over 200 angiosperms. Am. J. Bot. 75, 1443–1458. doi: 10.2307/2444695
Crosby, K., and Smith, D. R. (2012). Does the mode of plastid inheritance influence plastid genome architecture? PLoS ONE 7:e46260. doi: 10.1371/journal.pone.0046260
Dice, L. R. (1945). Measures of the amount of ecologic association between species. Ecology 26, 297–302. doi: 10.2307/1932409
Dolatowski, J., Nowosielski, J., Podyma, W., Szymanka, M., and Zych, M. (2004). Molecular studies on the variability of Polish semi-wild pears (Pyrus) using AFLP. J. Fruit Ornam. Plant Res. 12, 331–337.
Earl, D. A., and vonHoldt, B. M. (2012). STRUCTURE HARVESTER: a website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 4, 359–361. doi: 10.1007/s12686-011-9548-7
Evanno, G., Regnaut, S., and Goudet, J. (2005). Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14, 2611–2620. doi: 10.1111/j.1365-294X.2005.02553.x
Excoffier, L., Smouse, P. E., and Quattro, J. M. (1992). Analysis of molecular variance inferred from metric distances among DNA haplotypes: application to human mitochondrial DNA restriction data. Genetics 131, 479–491.
FAO (Food and Agricultural Organization of the United Nation) (2014). Available online at: http://faostat.fao.org/beta/en/#data/QC/visualize.
Felici, C. (1565). “De l'insalata e piante che in qualunque modo vengono per cibo de l'homo,” in Scritti Naturalistici, ed G. Arbizzoni (Urbino: QuattroVenti), 92–93.
Ferreira dos Santos, A. R., Ramos-Cabrer, A. M., Díaz-Hernández, M. B., and Pereira-Lorenzo, S. (2011). Genetic variability and diversification process in local pear cultivars from northwestern Spain using microsatellites. Tree Genet. Genomes 7, 1041–1056. doi: 10.1007/s11295-011-0393-3
Fideghelli, C. (2007). “Origine ed Evoluzione”, in Il Pero, ed R. Angelini (Milano: Bayer CropScience), 2–17.
Gasi, F., Kurtovic, M., Kalamujic, B., Pojskic, N., Grahic, J., Kaiser, C., et al. (2013). Assessment of European pear (Pyrus communis L.) genetic resources in Bosnia and Herzegovina using microsatellite markers. Sci. Hortic. 157, 74–83. doi: 10.1016/j.scienta.2013.04.017
Gaulin, J.-L. (2007). “Trattati di Agronomia e Innovazione Agricola”, in Il Rinascimento Italiano e l'Europa: Vol. 3. Produzione e Tecniche, ed P. Braunstein and L. Molà (Vicenza: Angelo Colla Editore), 145–163.
Hammer, Ø., Harper, D. A. T., and Ryan, P. D. (2001). Past: paleontological statistics software package for education and data analysis. Palaeontol. Electr. 4:9. Available online at: http://palaeo-electronica.org/2001_1/past/issue1_01.htm
Hardy, O. J., and Vekemans, X. (2002). SPAGeDi: a versatile computer program to analyse spatial genetic structure at the individual or population levels. Mol. Ecol. Notes 2, 618–620. doi: 10.1046/j.1471-8286.2002.00305.x
Hedrick, U. P. (1921). “The pears of New York”, in Report of the New York Agricultural Experiment Station, (New York, NY: Department of Agriculture, Annual report 1921/2), 4–5.
Iketani, H., Manabe, T., Matsuta, N., Akihama, T., and Hayashi, T. (1998). Incongruence between RFLPs of chloroplast DNA and morphological classification in East Asia pear (Pyrus spp.). Genet. Resour. Crop Evol. 45, 533–539. doi: 10.1023/A:1008646016181
Jiang, Z., Tang, F., Huang, H., Hu, H., and Chen, Q. (2009). Assessment of genetic diversity of Chinese sand pear landraces (Pyrus pyrifolia Nakai) using simple sequence repeat markers. HortScience 44, 619–626.
Katayama, H., Adachi, S., Yamamoto, T., and Uematsu, C. (2007). A wide range of genetic diversity in pear (Pyrus ussuriensis var. aromatica) genetic resources from Iwate, Japan revealed by SSR and chloroplast DNA markers. Genet. Resour. Crop Evol. 54, 1573–1585. doi: 10.1007/s10722-006-9170-9
Katayama, H., Tachibana, M., Iketani, H., Zhang, S. L., and Uematsu, C. (2012). Phylogenetic utility of structural alterations found in the chloroplast genome of pear: hypervariable regions in a highly conserved genome. Tree Genet. Genomes 8, 313–326. doi: 10.1007/s11295-011-0442-y
Kimura, T., Iketani, H., Kotobuki, K., Matsuta, N., Ban, Y., Hayashi, T., et al. (2003). Genetic characterization of pear varieties revealed by chloroplast DNA sequences. J. Hortic. Sci. Biotechnol. 78, 241–247. doi: 10.1080/14620316.2003.11511612
Kimura, T., Shi, Y. Z., Shoda, M., Kotobuki, K., Matsuta, N., Hayashi, T., et al. (2002). Identification of Asian pear varieties by SSR analysis. Breed. Sci. 52, 115–121. doi: 10.1270/jsbbs.52.115
Layne, R. E. C., and Quamme, H. A. (1975). “Pears”, in Advances in Fruit Breeding, ed J. Janick and J. N. Moore (West Lafayette, IN: Purdue University Press), 38–70.
Librado, P., and Rozas, J. (2009). DnaSPv5: a software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452. doi: 10.1093/bioinformatics/btp187
Loveless, M. D., and Hamrick, J. L. (1984). Ecological determinants of genetic structure in plant populations. Annu. Rev. Ecol. Evol. Syst. 15, 65–95. doi: 10.1146/annurev.es.15.110184.000433
Martinelli, F., Busconi, M., Fogher, C., and Sebastiani, L. (2009). Development of an efficient regeneration protocol for pear rootstock Pyrodwarf and assessment of SSR variability in regenerating shoots. Caryologia 62, 62–68. doi: 10.1080/00087114.2004.10589667
Meirmans, P., and Van Tienderen, P. (2004). Genotype and GenoDive: two programs for the analysis of genetic diversity of asexual organisms. Mol. Ecol. Notes 4, 792–794. doi: 10.1111/j.1471-8286.2004.00770.x
Michalakis, Y., and Excoffier, L. (1996). A generic estimation of population subdivision using distances between alleles with special reference for microsatellite loci. Genetics 142, 1061–1064.
Miller, A. J., and Gross, B. L. (2011). From forest to field: perennial fruit crop domestication. Am. J. Bot. 98, 1389–1414. doi: 10.3732/ajb.1000522
Miranda, C., Urrestarazu, J., Santesteban, L. G., and Royo, J. B. (2010). Genetic diversity and structure in a collection of ancient Spanish pear cultivars assessed by microsatellite markers. J. Amer. Soc. Hortic. Sci. 135, 428–437.
Monte-Corvo, L., Cabrita, L., Oliveira, C., and Leitão, J. (2000). Assessment of genetic relationships among Pyrus species and cultivars using AFLP and RAPD markers. Genet. Resour. Crop Evol. 47, 257–265. doi: 10.1023/A:1008794809807
Morgante, M., Felice, N., and Vendramin, G. G. (1997). “Analysis of hyper-variable chloroplast microsatellites in Pinus halepensis reveals a dramatic genetic bottleneck,” in Molecular Tools for Screening Biodiversity: Plants and Animals, eds A. Karp, D. S. Ingram, and P. G. Isaac (London: Springer), 407–412.
Nei, M. (1978). Estimation of average heterozygosity and genetic distance from a small number of individuals. Genetics 89, 583–590.
Nielsen, R., Tarpy, D. R., and Reeve, H. K. (2003). Estimating effective paternity number in social insects and the effective number of alleles in a population. Mol. Ecol. 12, 3157–3164. doi: 10.1046/j.1365-294X.2003.01994.x
Nock, C. J., Waters, D. L., Edwards, M. A., Bowen, S. G., Rice, N., Cordeiro, M., et al. (2011). Chloroplast genome sequences from total DNA for plant identification. Plant Biotechnol. J. 9, 328–333. doi: 10.1111/j.1467-7652.2010.00558.x
Oliveira, C. M., Mota, M., Monte-Corvo, L., Goulão, L., and Silva, D. M. (1999). Molecular typing of Pyrus based on RAPD markers. Sci. Hortic. 79, 163–174. doi: 10.1016/S0304-4238(98)00205-2
Pereira-Lorenzo, S., Ramos-Cabrer, A. M., González-Díaz, A. J., and Díaz-Hernández, M. B. (2008). Genetic assessment of local apple cultivars from La Palma, Spain, using simple sequence repeats (SSRs). Sci. Hortic. 117, 160–166. doi: 10.1016/j.scienta.2008.03.033
Pervaiz, T., Sun, X., Zhang, Y., Tao, R., Zhang, J., and Fang, J. (2015). Association between chloroplast and mitochondrial DNA sequences in Chinese Prunus genotypes (Prunus persica, Prunus domestica, and Prunus avium). BMC Plant Biol. 15:4. doi: 10.1186/s12870-014-0402-4
Petit, R. J., Duminil, J., Fineschi, S., Hampe, A., Salvini, D., and Vendramin, G. G. (2005). Comparative organization of chloroplast, mitochondrial and nuclear diversity in plant populations. Mol. Ecol. 14, 689–701. doi: 10.1111/j.1365-294X.2004.02410.x
Pritchard, J. K., Stephens, M., and Donnelly, P. (2000). Inference of population structure using multilocus genotype data. Genetics 155, 945–959.
Scarcelli, N., Barnaud, A., Eiserhardt, W., Treier, U. A., Seveno, M., d'Anfray, A., et al. (2011). A set of 100 chloroplast DNA primer pairs to study population genetics and phylogeny in monocotyledons. PLoS ONE 6:e19954. doi: 10.1371/journal.pone.0019954
Silva, G. J., Souza, T. M., Barbieri, R. L., and Costa de Oliveira, A. (2014). Origin, domestication, and dispersing of pear (Pyrus spp.). Adv. Agric. 2014:8. doi: 10.1155/2014/541097
Small, R. L., Ryburn, J. A., Cronn, R. C., Seelanan, T., and Wendel, J. F. (1998). The tortoise and the hare: choosing between noncoding plastome and nuclear Adh sequences for phylogeny reconstruction in a recently diverged plant group. Am. J. Bot. 85, 1301–1315. doi: 10.2307/2446640
Teng, Y., Tanabe, K., Tamura, F., and Itai, A. (2001). Genetic relationships of pear cultivars in Xinjiang, China as measured by RAPD markers. J. Hortic. Sci. Biotechnol. 76, 771–779. doi: 10.1080/14620316.2001.11511444
Teng, Y., Tanabe, K., Tamura, F., and Itai, A. (2002). Genetic relationships of Pyrus species and cultivars native to East Asia revealed by randomly amplified polymorphic DNA markers. J. Am. Soc. Hortic. Sci. 127, 262–270.
Urrestarazu, J., Miranda, C., Santesteban, L. G., and Royo, B. (2012). Genetic diversity and structure of local apple cultivars from Northeastern Spain assessed by micro satellite markers. Tree Genet. Genomes 8, 1163–1180. doi: 10.1007/s11295-012-0502-y
Urrestarazu, J., Royo, J. B., Santesteban, L. G., and Miranda, C. (2015). Evaluating the influence of the microsatellite marker set on the genetic structure inferred in Pyrus communis L. PLoS ONE 10:e0138417. doi: 10.1371/journal.pone.0138417
Vavilov, N. I. (1951). The origin, variation, immunity and breeding of cultivated plants. Chron. Bot. 13, 1–364. doi: 10.1097/00010694-195112000-00018
Vavilov, N. I. (1992). Origin and Geography of Cultivated Plants. Transl. by Doris Love. Cambridge, UK: Cambridge University Press.
Vieira, L. N., Faoro, H., Fraga, H. P. F., Rogalski, M., Souza, E. M., Pedrosa, F. O., et al. (2014). An improved protocol for intact chloroplasts and cpDNA isolation in conifers. PLoS ONE 9:e84792. doi: 10.1371/journal.pone.0084792
Weir, B. S., and Cockerham, C. C. (1984). Estimating F-statistics for the analysis of population structure. Evolution 38, 1358–1370. doi: 10.2307/2408641
Wheeler, G. L., Dorman, H. E., Buchanan, A., Challagundla, L., and Wallace, L. E. (2014). A review of the prevalence, utility, and caveats of using chloroplast simple sequence repeats for studies of plant biology. Appl. Plant Sci. 2:1400059. doi: 10.3732/apps.1400059
Wolko, T., Bocianowski, J., Antkowiak, W., and Stomski, R. (2015). Genetic diversity and population structure of wild pear (Pyrus pyraster (L.) Burgsd.) in Poland. De Gruyter Open Life Sci. 10, 19–29. doi: 10.1515/biol-2015-0003
Wu, J., Li, L.-T., Li, M., Khan, M. A., Li, X.-G., Chen, H., et al. (2014). High-density genetic linkage map construction and identification of fruit-related QTLs in pear using SNP and SSR markers. J. Exp. Bot. 65, 5771–5781. doi: 10.1093/jxb/eru311
Wu, J., Wang, Z., Shi, Z., Zhang, S., Ming, R., Zhu, S., et al. (2013). The genome of the pear (Pyrus bretschneideri Rehd.). Genome Res. 23, 396–408. doi: 10.1101/gr.144311.112
Wünsch, A., and Hormaza, J. I. (2007). Characterization of variability and genetic similarity of European pear using microsatellite loci developed in apple. Sci. Hortic. 113, 37–43. doi: 10.1016/j.scienta.2007.02.002
Wuyun, T., Amo, H., Xu, J., Ma, T., Uematsu, C., and Katayama, H. (2015). Population structure of and conservation strategies for wild Pyrus ussuriensis Maxim. in China. PLoS ONE 10:e0133686. doi: 10.1371/journal.pone.0133686
XLSTAT (2011). Data Analysis and Statistics with Microsoft Excel. Addinsoft. Available online at: http://www.xlstat.com
Yamamoto, T., Kimura, T., Shoda, M., Ban, Y., Hayashi, T., and Matsuta, N. (2002a). Development of microsatellite markers in Japanese pear (Pyrus pyrifolia Nakai). Mol. Ecol. Notes 2, 14–16. doi: 10.1046/j.1471-8286.2002.00128.x
Yamamoto, T., Kimura, T., Shoda, M., Imai, T., Saito, T., Sawamura, Y., et al. (2002b). Genetic linkage maps constructed by using an interspecific cross between Japanese and European pears. Theor. Appl. Genet. 106, 9–18. doi: 10.1007/s00122-002-0966-5
Yang, J.-B., Tang, M., Li, H.-T., Zhang, Z.-R., and Li, D.-Z. (2013). Complete chloroplast genome of the genus Cymbidium: lights into the species identification, phylogenetic implications and population genetic analyses. BMC Evol. Biol. 13:84. doi: 10.1186/1471-2148-13-84
Zhang, R.-P., Wu, J., Li, X.-G., Khan, M. A., Chen, H., Korban, S. S., et al. (2013). An AFLP, SRAP and SSR Genetic Linkage Map and identification of QTLs for fruit traits in pear (Pyrus L.). Plant Mol. Biol. Rep. 31, 678–687. doi: 10.1007/s11105-012-0544-1
Zheng, X., Cai, D., Potter, D., Postman, J., Liu, J., and Teng, Y. (2014). Phylogeny and evolutionary histories of Pyrus L. revealed by phylogenetic trees and networks based on data from multiple DNA sequences. Mol. Phylogenet. Evol. 80, 54–65. doi: 10.1016/j.ympev.2014.07.009
Zohary, D., and Hopf, M. (2000). Domestication of Plants in the Old World. Oxford: University Press.
Zouros, E. (1979). Mutation rates, population sizes, and amounts of electrophoretic variation of enzyme loci in natural populations. Genetics 92, 623–646.
Keywords: Pyrus spp., SSR markers, chloroplast DNA, hypervariable intergenic region, local varieties, phylogeny, genetic resources
Citation: Ferradini N, Lancioni H, Torricelli R, Russi L, Dalla Ragione I, Cardinali I, Marconi G, Gramaccia M, Concezzi L, Achilli A, Veronesi F and Albertini E (2017) Characterization and Phylogenetic Analysis of Ancient Italian Landraces of Pear. Front. Plant Sci. 8:751. doi: 10.3389/fpls.2017.00751
Received: 13 February 2017; Accepted: 21 April 2017;
Published: 10 May 2017.
Edited by:
Alessio Mengoni, University of Florence, ItalyReviewed by:
Majid Talebi, Isfahan University of Technology, IranCopyright © 2017 Ferradini, Lancioni, Torricelli, Russi, Dalla Ragione, Cardinali, Marconi, Gramaccia, Concezzi, Achilli, Veronesi and Albertini. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Emidio Albertini, ZW1pZGlvLmFsYmVydGluaUB1bmlwZy5pdA==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.