 Pawel Mikulski1,2
Pawel Mikulski1,2 Olga Komarynets
Olga Komarynets Fabio Fachinelli
Fabio Fachinelli Andreas P.M. Weber
Andreas P.M. Weber Daniel Schubert
Daniel Schubert
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Plant Sci., 26 April 2017
Sec. Plant Genetics and Genomics
Volume 8 - 2017 | https://doi.org/10.3389/fpls.2017.00607
Polycomb Group (PcG) proteins mediate chromatin repression in plants and animals by catalyzing H3K27 methylation and H2AK118/119 mono-ubiquitination through the activity of the Polycomb repressive complex 2 (PRC2) and PRC1, respectively. PcG proteins were extensively studied in higher plants, but their function and target genes in unicellular branches of the green lineage remain largely unknown. To shed light on PcG function and modus operandi in a broad evolutionary context, we demonstrate phylogenetic relationship of core PRC1 and PRC2 proteins and H3K27me3 biochemical presence in several unicellular algae of different phylogenetic subclades. We focus then on one of the species, the model red alga Cyanidioschizon merolae, and show that H3K27me3 occupies both, genes and repetitive elements, and mediates the strength of repression depending on the differential occupancy over gene bodies. Furthermore, we report that H3K27me3 in C. merolae is enriched in telomeric and subtelomeric regions of the chromosomes and has unique preferential binding toward intein-containing genes involved in protein splicing. Thus, our study gives important insight for Polycomb-mediated repression in lower eukaryotes, uncovering a previously unknown link between H3K27me3 targets and protein splicing.
In the eukaryotic cells, transcription state is dependent on the underlying chromatin state. The fundamental structure of chromatin is based on a nucleosome, a complex of 147bp-long fragments of DNA wrapped around the core histone proteins (H2A, H2B, H3, H4). Chromatin state can be influenced by post-translational modifications deposited on the histones, either by direct structural changes in the nucleosomes or recruitment/displacement of secondary proteins involved in chromatin remodeling or transcription. The presence of particular histone modifications often determines the type of the chromatin and correlates with transcriptional activity of the target DNA. Among those, tri-methylation of lysine 27 on histone H3 (H3K27me3) and mono-ubiquitination of histone H2AK118/H2AK119 (H2AK118ub/H2AK119ub) are commonly associated with transcriptionally silent facultative heterochromatin.
Deposition of H3K27me3 and H2AK118ub/H2AK119ub is mediated by Polycomb group (PcG) proteins, whose function was initially shown to control developmentally regulated processes and maintain cell identity (Papp and Müller, 2006; Schuettengruber et al., 2007) in both, animals [reviewed in Schwartz and Pirrotta (2008)] and plants [reviewed in Köhler and Villar (2008)]. PcG proteins form distinct complexes, like Polycomb Repressive Complex 2 (PRC2), involved in H3K27 trimethylation [in metazoans Pcl-PRC2 complex (Nekrasov et al., 2007; Müller and Verrijzer, 2009)], and Polycomb Repressive Complex 1 (PRC1), responsible for H2A mono-ubiquitination. PRC2 in Drosophila melanogaster, where it was initially discovered, consists of four core subunits: Enhancer of zeste [E(z)], a catalytic component needed for methylation of H3K27; Extra Sex Combs (ESC), a WD40 motif-containing protein that scaffolds interactions within the complex; Suppressor of zeste (Su(z)12), a Zinc Finger subunit essential for binding of PRC2 to nucleosomes and p55, a nucleosome remodeling factor (Schwartz and Pirrotta, 2013). In turn, PRC1 is formed by: dRING/Sex Combs Extra (Sce); Posterior Sex Combs (Psc), both responsible for mono-ubiquitination activity; Polyhomeotic (Ph), essential for maintaining protein-protein interactions; and Polycomb (Pc), involved in recruitment of the complex to chromatin; and Sex comb on midleg (Scm), important for spreading of PcG silencing (Schwartz and Pirrotta, 2013). Both complexes are functionally connected with each other. In the canonical hierarchical model, initially introduced H3K27me3 mark is recognized by PRC1 and followed by H2A mono-ubiquitination to maintain the repression. However, it was shown that the mechanism of PcG-mediated repression can happen as well in the opposite order, with PRC1 introducing its modification prior to PRC2 activity (Yang et al., 2013; Del Prete et al., 2015).
Polycomb-group complexes were identified also in the plant kingdom. Core PRC1 complex in model flowering plant Arabidopsis thaliana consist of AtRING1a/b (equivalent to dRING/Sce), AtBMI1a/b/c and EMF1 (equivalent to Psc), and LHP1 (equivalent to Pc). Although a wide range of PRC1-associated proteins exists (Chen et al., 2016), it is unclear how the combinations of different subunits depend on specific temporal and spatial conditions. In contrast to PRC1, composition of the Arabidopsis PRC2 complex is better characterized. Arabidopsis PRC2 subunits underwent gene duplication, resulting in the presence of different, partially redundant or independently acting PRC2 complexes (Derkacheva and Hennig, 2014). In the Arabidopsis genome the following PRC2 components can be identified: three E(z)-homologs – CURLY LEAF (CLF), SWINGER (SWN), MEDEA (MEA); three Su(z)12-homologs – EMBRYONIC FLOWER 2 (EMF2), VERNALIZATION 2 (VRN2), FERTILIZATION INDEPENDENT SEED 2 (FIS2); as well as single gene subunits: ESC-homolog – FERTILIZATION INDEPENDENT ENDOSPERM (FIE) and p55-homologs – MULTICOPY SUPPRESSOR OF IRA 1-5 (MSI1-5).
One of the aspects of PRC2 biology concerns its origins and the abundance in lower eukaryotes. Due to the absence of PRC2 in the model unicellular species Saccharomyces cerevisiae and S. pombe (Lachner et al., 2004; Veerappan et al., 2008), the PRC2 appearance was previously thought to co-occur with the emergence of multicellularity. However, high conservation of PRC2 subunits in both, animal and plant lineages implies that PcG genes appeared already in the last common unicellular ancestor (Bowman et al., 2007), before those two kingdoms diverged. In an elegant study, Shaver et al (Shaver et al., 2010) identified the novel computational PRC2-homologs in several unicellular species and showed that E(z)-homolog in unicellular green algae Chlamydomonas reinhardtii is responsible for mono- and di-methylation of lysine 27 on histone H3. As the tri-methylation activity of Chlamydomonas E(z) homolog was not found, this study highlights important differences in the functional conservation of PRC2 components. For instance, although H3K27 tri-methylation is considered to be a prominent histone mark in PRC2-mediated repression, there is a range of H3K27 methylation levels catalyzed by PRC2 that varies between plant and Drosophila or mammals. In Arabidopsis, PRC2 controls H3K27 tri-methylation and H3K27 di-methylation in the euchromatin (Bastow et al., 2004; Lindroth et al., 2004; Sung et al., 2006), with mono-methylation being catalyzed by ATXR5 and ATXR6 (Jacob and Michaels, 2009; Jacob et al., 2009). However, in metazoans homologs of ATXR5/6 were not found and their PRC2 complexes mediate H3K27 methylation in all contexts (Müller et al., 2002; Ebert et al., 2004; Ferrari et al., 2014). Another difference between species is the type of DNA elements targeted by PRC2. Drosophila PRC2 targets both, genes and transposable elements/repetitive sequences (Yin et al., 2011), whereas in Arabidopsis the H3K27me3 mark is excluded from the majority of transposable elements/repetitive sequences (Lafos et al., 2011; Deleris et al., 2012; Park et al., 2012).
Even though PRC2-mediated repression has been extensively studied in higher plants and animals, its characterization in lower eukaryotes gathered much less attention. The existence of H3K27me3 in lower eukaryotes is largely unknown (Shaver et al., 2010) and distribution of the mark in the genome and its function were reported only for a handful of species, such as Neurospora crassa, Phaeodactylum tricornutum, or Tetrahymena thermophila. In N. crassa H3K27me3 is arranged into broad domains covering 774 genes connected with a full spectrum of functions (Jamieson et al., 2013). In T. thermophila, the mark associates with the developmentally regulated genome rearrangements as it occupies sequences eliminated during differentiation of the macronucleus (Liu et al., 2007). In P. tricornutum, H3K27me3 covers transposable elements and genes, and its targets are involved in, i.e., signal transduction, development and cell cycle control (Veluchamy et al., 2015).
Given the distinct degrees of phylogenetic conservation of PRC2 and PRC1, and poor characterization of H3K27me3 in lower eukaryotes, we sought to investigate the presence of Polycomb-group homologs and H3K27me3 abundance in several representative unicellular, photosynthetically active eukaryotes. Our scope included the representatives of red algae, green algae and Glaucophyta that vary in genome size, genome architecture, ecological niche and metabolism (Matsuzaki et al., 2004; Palenik et al., 2007; Worden et al., 2009; Merchant et al., 2010; Price et al., 2012). After an initial small-scale screen on H3K27me3 presence, we focused on one of the species, Cyanidioschyzon merolae, to study genome-wide occupation of H3K27me3 and characterize its targets. C. merolae is a unicellular red alga, living in highly acidic environment with high temperatures. It contains a small (16 Mb) genome which was fully sequenced as the first algal genome (Matsuzaki et al., 2004) and assembled as first 100% complete eukaryotic genome (Nozaki et al., 2007). Interestingly, the genome shows extremely simplified structure that contains almost exclusively intron-lacking genes (only 26 genes contain an intron), very low percentage (0,7%) of transposable elements and a novel class of a repetitive element, corresponding to the truncated ORF from White spot syndrome virus (WSV repeat) (Nozaki et al., 2007). Given its unicellularity, evolutionary ancestry, primitive architecture of the genome and availability of sequencing data, we argue that C. merolae is a suitable model for studying chromatin repression in an evolutionary context.
Our results confirmed the high conservation of PRC2 core members in lower eukaryotes and the widespread presence of H3K27me3 modification. We report that in C. merolae H3K27me3 mark targets both, genes and repetitive elements and is anti-correlated with transcriptional activity. We show that H3K27me3 distribution over its targets is not uniform, but can be grouped into several different clusters that correlate with different levels of repression. Furthermore, we demonstrate that H3K27me3 has a preferential chromosomal localization toward telomeric and subtelomeric regions. For the first time, we also reveal that H3K27me3 target genes are enriched in the functional class of intein-mediated protein splicing. Moreover, by deposition of RNA- and ChIP-sequencing data, we provide a resource for studies on the chromatin and transcriptome in the model red alga C. merolae.
Sequences of PRC1 and PRC2 components from A. thaliana and D. melanogaster were selected from TAIR10 and NCBI databases, respectively, and used as the queries. Homology searches were done using BLAST against organism-specific databases at: https://phytozome.jgi.doe.gov/pz/portal.html (C. reinhardtii v5.5 and Micromonas pusilla CCMP1545 v3.0), http://merolae.biol.s.u-tokyo.ac.jp/ (C. merolae), http://genome.jgi.doe.gov/Ostta4/Ostta4.home.html (Ostreococcus tauri v2.0), http://cyanophora.rutgers.edu/cyanophora/home.php (Cyanophora paradoxa).
The highest scoring candidate proteins were used for BLAST searches against the TAIR10 genome annotation to confirm a reciprocal match to the protein used as an initial query. The sequences were aligned using ClustalXv2.0 (Larkin et al., 2007) and conserved blocks were selected by gBlocks v0.91b (Talavera and Castresana, 2007). For substitution model estimation, ProtTestv3.4 was employed (Abascal et al., 2005). Phylogenetic trees were created using a Bayesian approach in Mr Bayesv3.2 (Ronquist et al., 2012), self-compiled developer version r1067 with implemented LG model. The runs were done with 20 mln generations. Sequences from D. melanogaster were selected as an out group. Consensus trees and alignments were visualized with Figtree v1.4.2 (Rambaut, 2009) and Jalviewv2 (Waterhouse et al., 2009), respectively.
Cyanidioschyzon merolae cells were grown at 42°C with shaking at 150 rpm. The cultures were kept under continuous light, with light intensity of 70 μmol.
Around 50.000 algae cells were diluted in Laemmli’s sample buffer, and 3–7 μl of total protein/lane was subjected to sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) using 12% SDS-polyacrylamide gel. The resolved proteins were transferred onto polyvinylidenedifluoride (PVDF) membranes (Immobilon-P membrane, Milipore) using a Vertical Electrophoresis Cell (Bio-Rad). Following transfer, membranes were washed with phosphate buffered saline containing 0.05% Tween-20 (PBST) and then blocked in 5% milk diluted in PBST for 1 h at room temperature. The membranes were then incubated overnight with anti_H3K27me3 (N 07-449, Millipore or C15410195, Diagenode) and anti H3 (ab1791-100, Abcam) rabbit polyclonal antibodies 1:2000 in PBST containing 5% milk at 4°C. After washing three times for 10 min in PBST, membranes were incubated with peroxidase-labeled secondary antibody for 1 h at room temperature. The membranes were washed three times for 10 min in PBST, incubated with SuperSignal West Femto Chemiluminescence Substrate (Thermo Scientific) for 1 min, and exposed for 1–2 min. Commercial histone extract from calf thymus (Sigma, #9064-47-5) was used as a positive control for H3K27me3 detection.
Pre-incubation of anti-H3K27me3 antibody (C15410195, Diagenode) and H3K27me3 peptide (Intavis) was done for 2 h in RT in PBST, with occasional mixing. The peptide-to-antibody molar ratio was 50:1. A control solution – PBST with just antibody at 2X final concentration – was used at the same time. At the end of the pre-incubation, 4% BSA blocking solution was added to the peptide/antibody mixture to a final concentration of 3% BSA, mixed briefly and added to the membranes. After washing three times for 10 min in PBST, membranes were incubated with peroxidase-labeled secondary antibody for 1 h at room temperature. The membranes were washed three times for 10 min in PBST, incubated with SuperSignal West Femto Chemiluminescence Substrate (Thermo Scientific) for 1 min, and exposed for 1–2 min.
Cyanidioschyzon merolae cells were grown until late log-phase. 40 ml samples were fixed [Formaldehyde, 1% (v/v)] for 10 min, until addition of glycine (to final concentration of 125 mM, 5 min incubation). Superfluous formaldehyde was removed by three washes with ice cold PBS buffer and the remaining cell pellet was resuspended in 4 ml of Extraction Buffer (50 mM Tris-HCl pH 8, 10 mM EDTA, 1% SDS) with Protease Inhibitor Cocktail (Roche). The samples were sonicated for 5 min with 30 s ON/30 s OFF cycle using Bioruptor Plus device (Diagenode) and cleared by two rounds of centrifugation (13000 rpm, 4°C, 10 min). Subsequent steps were performed as in the Plant ChIP-seq kit protocol (Diagenode) with higher volume of sample taken aside as an input (1:5 of chromatin for IP). Immunoprecipitation was done using anti-H3K27me3 Polyclonal Premium antibody (C15410195, Diagenode) and, as a negative control, IgG fraction from rabbit (C15410206, Diagenode). Quality and fragment size of immunoprecipitated DNA and input samples were measured using agarose gel electrophoresis and Bioanalyzer 2100 (Agilent Technologies).
DNA samples obtained from ChIP were used for H3K27me3 enrichment analysis for several target genes by real-time quantitative PCR. Reactions were prepared using KAPA SYBR FAST qPCR Mastermix (KapaBiosystems), according to the manufacturer’s protocol, and run on an iQ5 detection system (Biorad) using a 2-step program. Differences in H3K27me3 enrichment on target genes were scored by comparison of % recovery of input and standard error values. 5′- > 3′ sequences of oligonucleotides used for amplification were as follows: CmMADS- forward: GGATGAGAAAGCGAGAAATACGA and reverse: TCACAATGCCGATCTCACAG; CmEIF-4A – forward: TGTACGATATGATCCAGAGAAGAG and reverse: TGTAGATTTGCTCCTTGAAACC; Cm60S – forward: AAGTTTCGCTGTACGCTTGG and reverse: TAACCAGGACCATATCGCCG.
DNA library was prepared with MicroPlex Library Preparation Kit (Diagenode) and sequenced on HiSeq 2000 sequencer (Illumina Inc.). Quality of paired-end raw reads was assessed using FastQC v0.11.4. The reads were trimmed according to the quality and mapped to the reference in Bowtie v2.2.6, with standard options. Peaks were called using MACS v2.1 with exclusion of clonal reads. The new annotation from the C. merolae Genome Project v31 was employed as a reference. The correlation between ChIP-seq replicates was scored using Pearson correlation and shown in Supplementary Figure S3A. The data was deposited in the Gene Expression Omnibus under the series GSE93913.
Cyanidioschyzon merolae cells were grown until late log-phase. RNA was isolated using RNeasy Plant Mini Kit (Qiagen), following a standard protocol. cDNA synthesis and DNase treatment were performed using RevertAid First Strand cDNA Synthesis Kit (Thermo Scientific) with oligo(dT) primers, according to a standard manual. The quality and concentration of samples were measured spectrometrically using Nanodrop 1000 (Thermo Scientific) and electrophoretically using Bioanalyzer 2100 (Agilent Technologies). Libraries were constructed using TruSeq RNA Library Prep kit (Illumina) with gel-free library purification based on Agencourt AMPure XP beads (Beckman Coulter, Inc.). The samples were sequenced on HiSeq 2000 device (Illumina Inc.). Paired-end raw sequencing reads were analyzed using Galaxy implementations (usegalaxy.org) of FastQC program and Tuxedo protocol (Trapnell et al., 2012). The new annotation from C. merolae Genome Project v3 was employed as a reference. The correlation between replicates in RNA-seq experiment was scored using Pearson correlation and shown in Supplementary Figure S4. The data was deposited in the Gene Expression Omnibus, under the series GSE93912.
Identified H3K27me3 peaks were annotated by intersection with the reference using Bedtools v2.17 (Quinlan and Hall, 2010). Sequences from unannotated peaks were translated in silico to ORF in HMMER2GO v0.17 software and the longest ORF was used for homology searches by BLASTP/Pfam against respective protein databases. Alternatively, unannotated peak sequences were used for homology searches by BLASTN against NCBI nucleotide databases: nr/nt and ref_seq. Homology searches were performed in BLAST+ standalone package v2.2.28+ (Camacho et al., 2009). Alignment threshold was set as follows: >50% alignment length and <0.5 E-value. Complete genome records in nucleotide databases were used to form negative GI list and excluded from the BLASTN searches. Only the top alignment hit was included in the further analyses. Distance between peak and annotated genomic feature was obtained using ‘closest’ command from Bedtoolsv2.17. Mapping of the reads was visualized in IGV v2.3 (Thorvaldsdóttir et al., 2013). Clustering of H3K27me3 enrichment was done on deepTools2 (Ramirez et al., 2016). Gene ontology was inferred by using Singular Enrichment Analysis on the AgriGO server (Du et al., 2010) against complete GO list. Protein annotations were extracted from UniProtKB database and used as reference. To select significantly enriched GO terms, Fisher test with Yekutieli adjustment was used a statistical method. P-value was set to 0.05 and minimal number of entries kept at 5.
In order to assess evolutionary conservation of Polycomb complexes, we performed phylogenetic analysis in the species from various groups of lower plants. We focused on the representatives from subclades: Chlorophyta (green algae), Rhodophyta (red algae), Glaucophyta and Embryophyta. For comparison, a representative from Metazoa, D. melanogaster, was included as well. Using reciprocal BLAST searches with full length proteins from A. thaliana or D. melanogaster, we identified homologs of PRC1 and PRC2 members in several species (Table 1).
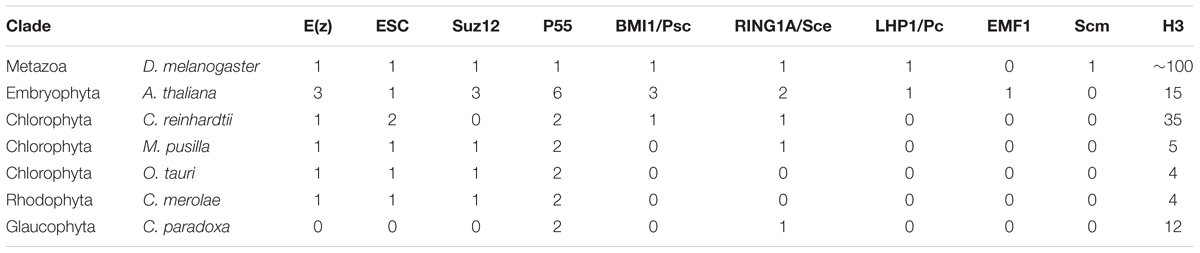
TABLE 1. Number of identified homologs of PRC2 and PRC1 components in selected species.
We found a general absence of PRC1 components: Psc/BMI1, Pc/LHP1, EMF1 and Scm in the algal genomes analyzed, with an exception of Psc/BMI1 homolog in C. reinhardtii. In contrast, the RING1 subunit is widely conserved, suggesting a mono-ubiquitylation activity unrelated to PRC1 in algae and protein moonlighting (Jeffery, 2003). In general, an abundance of RING1 homologs in chlorophytes, but the absence of other PRC1 components in lower branches of Archaeplastida used here is consistent with the previous studies (Hennig and Derkacheva, 2009; Berke and Snel, 2015; Chen et al., 2016) and agrees with the notion of lower PRC1 conservation and potential loss of the complex in several phylogenetic branches.
Homology searches on PRC2 revealed broad distribution of complex components: E(z), ESC and p55, consistently with results published elsewhere (Shaver et al., 2010; Butenko and Ohad, 2011; Kim et al., 2013). Importantly, p55 in higher eukaryotes has various functions and participates also in other complexes than PRC2 (i.e., chromatin assembly factor 1 [CAF1)]. Therefore it still remains to be proven whether a role of p55 homologs is Polycomb-related in lower plants. Moreover, we observed an absence of Suz12 from C. reinhardtii and C. paradoxa. Similarly to the other studies (Shaver et al., 2010), the lower abundance of this subunit shows that Suz12 was lost in several species, despite the presence of the other PRC2 components. Interestingly, we did not detect any core PRC2 members in the Cyanophora representative. Given that E(z) and ESC homologs were present in the representatives of Rhodophyta and Chlorophyta, it suggests the existence of PRC2 in the common algal ancestor and subsequent loss of the complex from Cyanophora. However, we could not exclude technical issues coming from incomplete annotation and incorrectly predicated protein models in the Cyanophora genome. In addition, we identified mostly one homolog per complex component in all screened species, apart from Arabidopsis, which adds another piece of evidence to the frequent gene duplication occurring in flowering plants.
Overall, our results agree with the ancient presence of core PRC2 components and the frequent losses of the Suz12 subunit and PRC1 complex members.
Phylogenetic analyses on the conserved domains or full sequences of core PRC2 homologs in lower and higher plants with the creation of Neighbor-Joining (NJ) trees revealed grouping into several distinct classes (Shaver et al., 2010; Kim et al., 2013; Huang et al., 2016). In a different approach, we extracted amino acid sequences from the most abundant PRC2 members, aligned them and automatically selected only the conserved blocks in the multiple alignment (see Material and Methods). With such prepared sequences we created Bayesian trees for E(z)z, ESC and Su(z)12 homologs. Consistently with the published data, the homologs formed defined clades. Bayesian trees for E(z) and ESC sequences generally resolved the evolutionary distance between groups (Figures 1A,B). In those cases, we found separation of representatives of Rhodophyta and Viridiplantae, with species from Chlorophyta and Embryophyta forming separate subclades within the latter. The exceptions included one of the ESC homologs from C. reinhardtii (CrESC.1), which clustered together with the C. merolae homolog, rather than the other green algae. In turn, our analysis on Su(z)12 detected distinct clades for the organisms from Embryophyta, Chlorophyta and Rhodophyta, without grouping of Arabidopsis and green algae sequences (Figure 1C). Moreover, we noted a phylogenetic distance between C. merolae and any other species for all the three proteins.
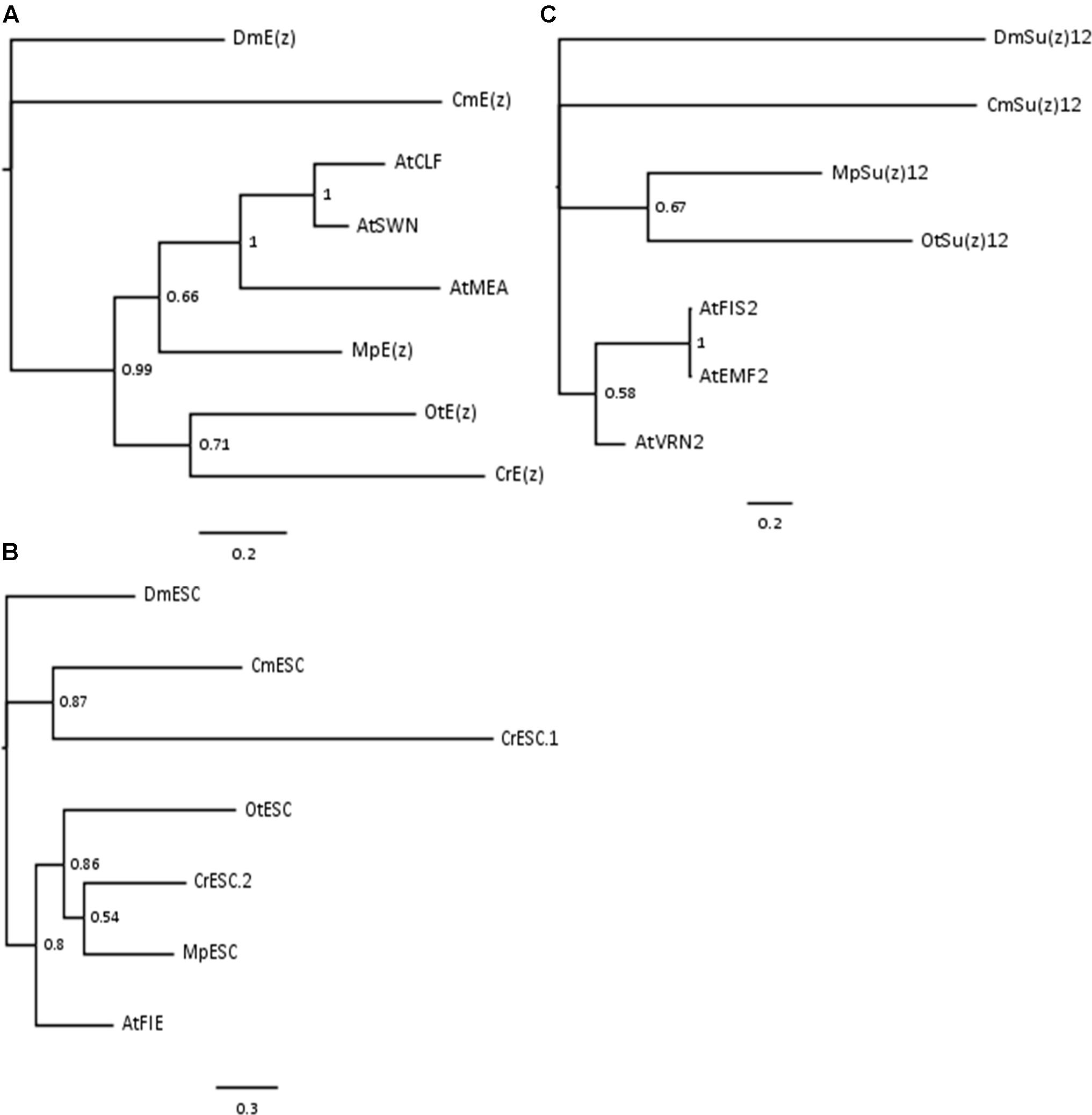
FIGURE 1. Bayesian-inferred phylogenetic trees on conserved blocks from homologs of core PRC2 components. Consensus trees were generated by MrBayes v2 and visualized in FigTree v1.4. Clade credibility values are depicted at the nodes. Branch length corresponds to the rate of expected mutations per site in the protein sequence, according to relevant scale bar below each tree. Sequences from D. melanogaster were selected as outgroups. Two-letter abbreviations represent species’ names: Dm – D. melanogaster; Cm – C. merolae; At – A. thaliana; Mp – M. pusilla; Ot – O. tauri; Cr – C. reinhardtii. (A) Phylogenetic tree on E(z) homologs. Conserved blocks selected from multiple alignment on following homologs (UniProtKB ID or protein/transcript name from resource databases indicated): DmE(z) – P42124; CmE(z) – CMQ156C; AtCLF – AT2G23380; AtSWN – AT4G02020; AtMEA – AT1G02580; MpE(z) – 59369; OtE(z) – 6642; CrE(z) – Cre17.g746247.t1.1. (B) Phylogenetic tree on ESC homologs. Homologs included in the analysis: DmESC – Q24338; CmESC – CMK173C; AtFIE – AT3G20740; MpESC – 49065; OtESC – 22117; CrESC.1 – Cre16.g693750.t1.1; CrESC.2 – Cre03.g180050.t1.1. (C) Phylogenetic tree on Su(z)12 homologs. Homologs included in the analysis – DmSu(z)12 – Q9NJG9; CmSu(z)12 – CML082C; AtFIS2 – AT2G35670; AtEMF2 – AT5G51230; AtVRN2 – AT4G16845; MpSu(z)12 – 9357; OtSu(z) – 13623.
In summary, our results on Bayesian phylogenetic trees on conserved sequence blocks are in agreement with the published NJ trees using the full length (Kim et al., 2013) or domain-only (Shaver et al., 2010) sequences. Given high conservation of domain architecture in core PRC2 components between the subclades (Shaver et al., 2010; Kim et al., 2013), we conclude that PRC2 is widely distributed and already evolved in a common unicellular ancestor.
As PRC2 has the canonical function to methylate lysine 27 in histone H3, we decided to analyze protein sequence conservation of histone H3. Amino acid sequences of algal histone H3 were retrieved from the databases of respective genome sequencing projects (see materials and methods) or NCBI, using the canonical histone H3.1 from Arabidopsis as a query (Okada et al., 2005). The results revealed a presence of multiple H3 genes in all of the species studied (Table 1). Noteworthy, we detected an equal amount of histone H3 gene copies as in the published reports for M. pusilla and C. reinhardtii (Cui et al., 2015). The number of H3 gene copies in D. was taken from the published results (McKay et al., 2015).
Next, the closest H3 homologs between the species were aligned using ClustalX and visualized with Jalview. The alignment revealed overall high amino acid conservation and a presence of lysine-27 in all studied species (Figure 2). Moreover, we noted that the sequences around lysine-27, including the underlying motif ARKS, does not show any sequence divergence with an exception of C. reinhardtii, which contains threonine-28 instead of serine-28. The results suggested that there is the potential for introduction of the H3K27me3 modification in most of species analyzed. However, in C. reinhardtii the presence of threonine-28 next to lysine-27 might not permit detection using commercial antibodies against the methylated H3K27. Moreover, a mass spectrometry analysis on Chlamydomonas histone H3 did not reveal the presence of the H3K27me3 modification (Shaver et al., 2010).

FIGURE 2. H3 sequence conservation. AtH3.1 was used a query for BLAST searches against databases of respective species. The closest homologs were selected and used for multiple alignment in ClustalX, visualized afterward in Jalview v2.9. Color-shading marks non-conserved residues. Red frame correspond to lysine-27, a residue typically modified by PRC2.
Given the phylogenetic conservation of PRC2 core components and high similarity of amino acid composition of histone H3 in the vast majority of the species, we sought to examine H3K27me3 presence in total protein extracts from selected organisms. Isolated proteins from crude extract were separated on a SDS-PAGE gel and detected using two independently raised anti-H3K27me3 antibodies. Our results revealed the presence of the H3K27me3 modification in M. pusilla, O. tauri, C. paradoxa, and C. merolae (Figures 3A,B). In order to decipher H3K27me3 relative abundance, we performed western blotting for histone H3 (Figure 3D) and calculated H3K27me3/H3 ratios based on band intensity quantification. We detected different relative amounts of the modification in the studied species (Figure 3E), with the lowest H3K27me3/H3 ratios for M. pusilla and O. tauri, intermediate for C. merolae and the highest for C. paradoxa.
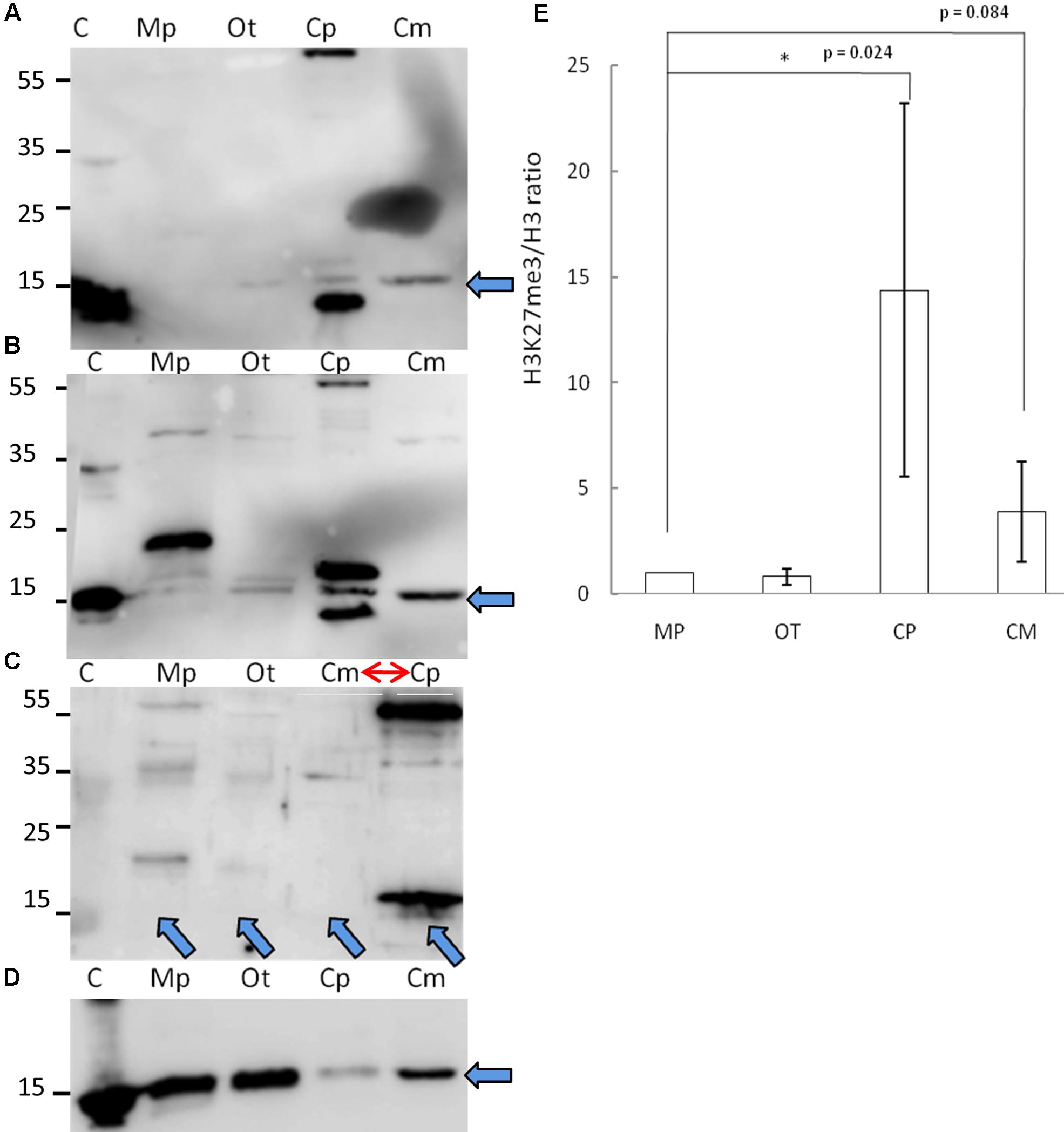
FIGURE 3. Biochemical detection of H3K27me3 histone mark. (A,B,D) Western blot analysis of H3K27me3 in different algae (C – histone powder control, Mp – M. pusilla, Ot – O. tauri, Cp – C. paradoxa, Cm – C. merolae). Membranes were incubated with antibodies against H3K27me3 [Millipore, N07-449 (A) and Diagenode, C15410206 (B)] and H3 [Abcam, ab1791-100 (D)]. The arrows indicate the band of interest. (C) Competition assay done after Western blotting with primary antibodies against H3K27me3 from (B). Note the order change of the samples (red arrow). (E) Quantification of Western blotting. Intensity of the bands was obtained using ImageJ. H3K27me3/H3 protein level ratio of three independent experiments is shown. P-values were calculated using Student t-test. A single asterisk indicates a significance level of p < 0.05. Error bars represent standard deviation.
Due to the fact that one of the antibodies detected proteins in the sizes not corresponding to histones, we performed protein competition assay for the confirmation of specific band reactivity. After preincubation with H3K27me3 peptide, we did not detect any band corresponding to this modification for a commercial histone extract from calf thymus (positive control), as well as for M. pusilla, O. tauri, and C. merolae (Figure 3C), suggesting a specificity of the antibody. Intensity decrease of the band corresponding to H3K27me3 for C. paradoxa was accompanied by the overall signal loss, including the bands of the other molecular weight proteins. Therefore, the presence of H3K27me3 in C. paradoxa remains inconclusive.
In summary, we were able to biochemically identify and confirm a presence of H3K27me3 in M. pusilla, O. tauri and C. merolae. This result is consistent with our phylogenetic analyses and suggests that the PRC2 complex is active in these species. For C. paradoxa, the biochemical results are unclear: potential absence of H3K27me3 mark would correspond to the lack of PRC2 homologs identified in its genome. Differences in relative H3K27me3 amounts between species imply, e.g., a distinct abundance of genomic targets of H3K27me3 or different H3K27me3 enrichment levels per genomic region. Further work should decipher an impact of such differences in the evolutionary context.
Based on the broad distribution of PRC2 genes, high sequence conservation of histone H3 and specific signal detected with the anti-H3K27me3 antibody on a protein blot, we selected one of the species, C. merolae, to investigate the H3K27me3 abundance further. Firstly, we asked whether the modification has similar targets as in the other organisms. Reciprocal BLAST searches between protein databases of C. merolae and A. thaliana were used to detect homologous genes of known A. thaliana PRC2 targets (Supplementary Figure S1). We selected a MADS box-containing gene CMA095C (CmMADS) as nearly all Arabidopsis MADS box genes carry H3K27me3. For negative controls, estimated not to be targeted by H3K27me3, homologous genes for EUKARYOTIC ELONGATION FACTOR EIF4A (CMK028C, CmEIF4A) and 60S RIBOSOMAL PROTEIN L23 (CMS262C, Cm60S) were taken.
Next, we performed Chromatin-immunoprecipitation (ChIP) using anti-H3K27me3 antibody and analyzed the expression of candidate genes by RT-PCR. As a result, we were able to show significant enrichment of H3K27me3 on CmMADS and low abundance of the mark on negative targets: CmEIF4A and Cm60S (Figure 4). Overall, we were able to identify positive target of H3K27me3 and show high enrichment of the mark comparing to negative loci.
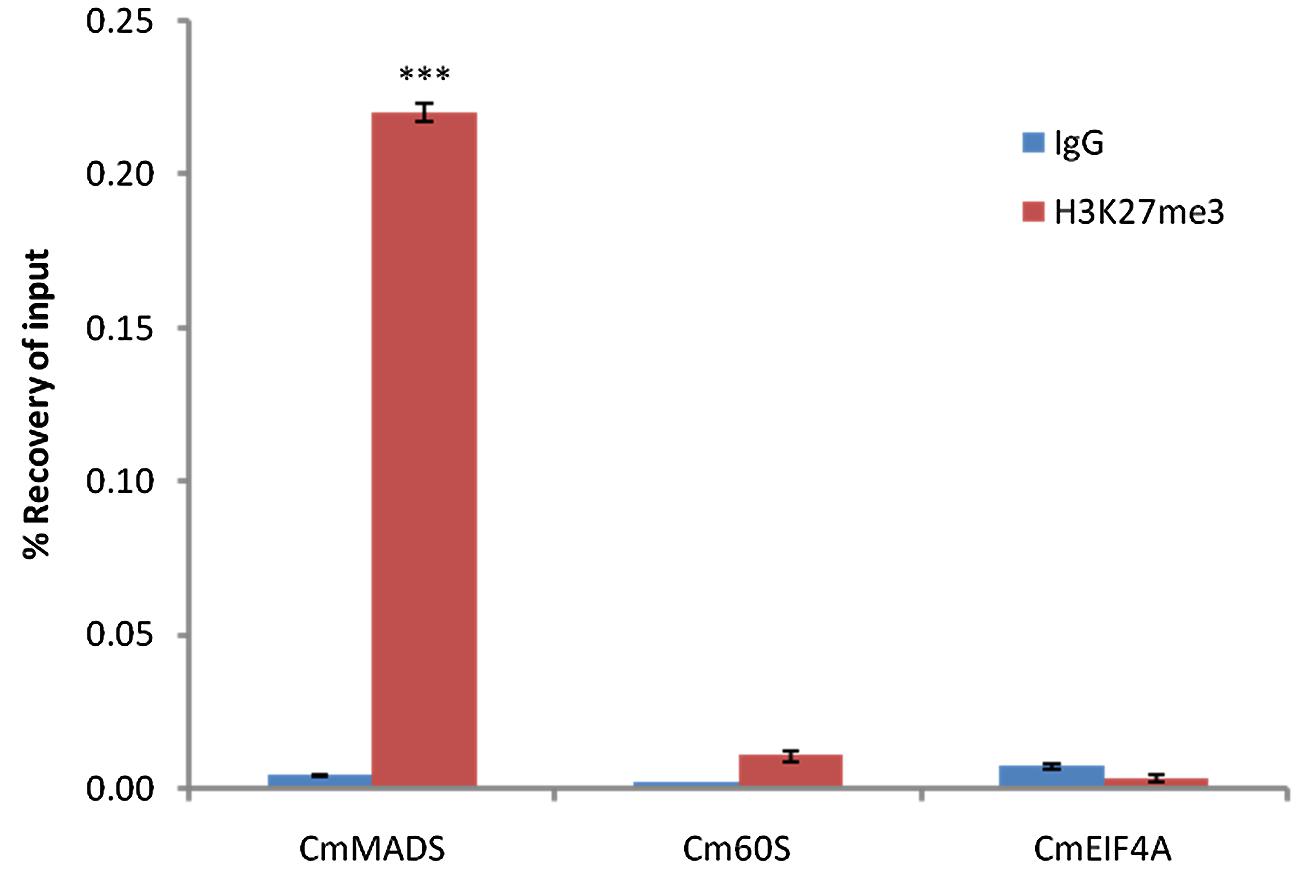
FIGURE 4. H3K27me3 abundance on selected genes from C. merolae. Chromatin immunoprecipitation was performed using H3K27me3 antibody (#C15410195, Diagenode) and IgG (#C15410206, Diagenode) for negative control, followed by RT-PCR analysis on a putative H3K27me3 target (CmMADS) and housekeeping genes as the putative non-H3K27me3 targets (Cm60S, CmEIF4A). Selected genes relate to following locus IDs from C. merolae genome project: CmMADS – CMA095C, Cm60S – CMS262C, CmEIF4A – CMK028C. Error bars correspond to the standard deviation from three biological replicates. Significance was calculated using Student’s t-test for the comparison between CmMADS and either, Cm60S, or CmEIF4A. A triple asterisk indicates the significance level of p < 0.001.
In order to characterize the targets of H3K27me3 and the distribution of H3K27me3 peaks in a genome-wide scale, we created a sequencing library and performed chromatin-immunoprecipitation coupled with sequencing (ChIP-seq) for the samples: H3K27me3-bound DNA, H3-bound DNA (both in triplicate) and input.
Cleaning raw sequencing reads, mapping to the reference genome and peak calling with input or H3 sample for normalization, let us identify more than 1300 H3K27me3 peaks per replicate (Figure 5A). The peaks cover 14% of total nuclear genome length, with an average peak size of 1792 bp. Good quality of the data was confirmed by a high number of H3K27me3 peaks in the overlaps between the biological replicates, as well as the high correlation values of the reads’ occupancy (Supplementary Figure S3)
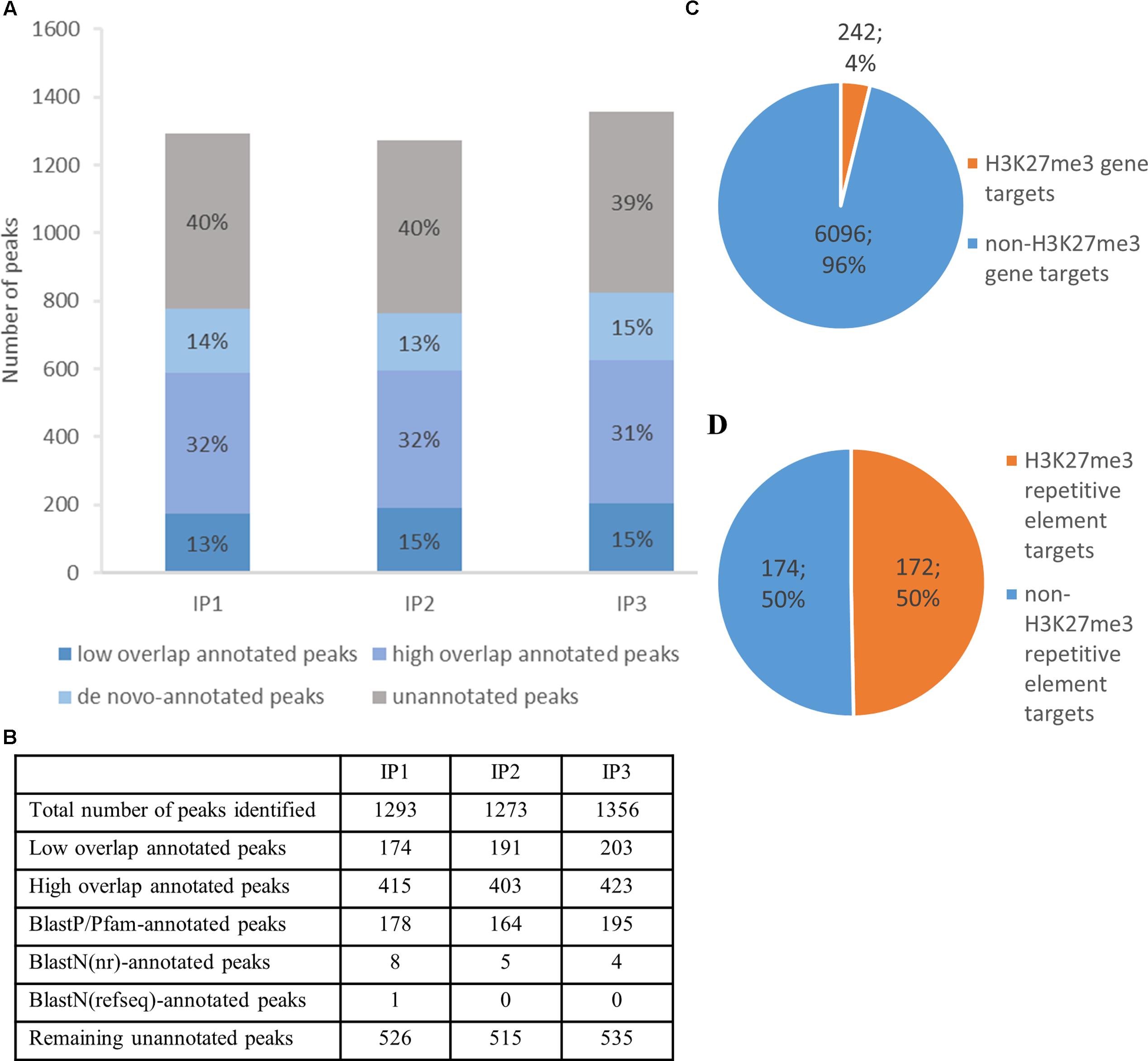
FIGURE 5. H3K27me3 genome-wide distribution and peak annotation. (A,B) – Number of peaks identified in three different biological replicates from chromatin immunoprecipitation experiment (IP1-3) in ‘high overlap annotated peaks,’ ‘low overlap annotated peaks’ category, ‘de novo-annotated’ and ‘unannotated’ categories (see main text). (C,D) – Characterization of H3K27me3 peaks according to genomic element type. High overlap H3K27me3 peaks cover 242 genes, which amounts to 4% of total gene number (C) or 172 repetitive elements, 50% of total repetitive element number (D). Both, loci of hypothetical proteins and transcripts, were included in total gene number.
Next, we compared H3K27me3-peak coordinates with the loci in C. merolae reference genome. We applied a threshold of 50% overlap of the locus length and successfully annotated ca. Six hundred peaks (‘high overlap annotated peaks’) (Figure 5A). The remaining peaks corresponded to the reference loci covered by H3K27me3 in less than 50% of their length (‘low overlap annotated peaks’) and to the unannotated peaks without any overlap with reference loci. We concluded that the unannotated peaks are the intergenic regions and/or uncharacterized genomic features missing in the current annotation. Considering the second possibility, we used several strategies to find de novo annotation of unknown peaks.
Firstly, we in silico translated sequences from the unannotated peaks and searched for open reading frames (ORFs) within them. We found ORFs for all of the unannotated peaks and used them for BLASTP/Pfam searches against the NCBI non-redundant protein sequence database (nr database) or Pfam protein database. Using a specific alignment threshold (alignment length > 50%; E-value < 0.5), we could annotate further 178, 164, and 195 peaks for biological replicate 1, 2, and 3, respectively (Figure 5B). In the second approach, we extracted DNA sequences of the unknown peaks and used BLASTN searches against the NCBI nucleotide collection (nr/nt database) and NCBI transcript reference sequences (refseq_rna database). The hits surpassing the alignment threshold (alignment length > 50%, E-value < 0.5) were compared to BLASTP/Pfam output, which allowed to annotate next 4 to 8 peaks, depending on the biological replicate (Figure 5B). In general, our sequence homology searches helped to detect de novo annotation of 13–15% peaks from their total number, depending on the biological replicate (Figure 5A).
We reasoned that the remaining unknown peaks after de novo annotation corresponded to the regions containing promoters and the other regulatory elements of neighboring annotated elements. Given the high density of genes along the chromosomes in C. merolae (Matsuzaki et al., 2004), such regions would reside close to 5′ or 3′ gene ends, but would not be present in the annotation. In order to validate this idea, we calculated average distances between gene locations and unannotated peaks. We compared distributions between peaks successfully aligned to BLASTP database (annotated peaks) and the ones showing no significant alignment (unannotated peaks), assuming that BLASTP-aligned peaks truly correspond to novel genes missing in our reference, rather than uncharacterized cis elements. However, we did not observe major differences between distances calculated for annotated and unannotated peaks (Supplementary Table S1), suggesting that unannotated peaks do not exclusively correspond to cis regulatory elements of known genomic elements. Further development of C. merolae genome annotation should deepen our understanding about these remaining uncharacterized H3K27me3 peaks.
Next, we sought to investigate the genomic feature profile underlying H3K27me3 peaks. We picked a stringent group of high overlap annotated targets, removed duplicates (two peaks spanning one genomic element) for each replicate and selected a consensus set of features that appear in any two out of three replicates. As a result, we found that H3K27me3 covers 242 genes, which amounts to 4% of C. merolae total gene number (Figure 5C). On the other hand, we also detected 172 H3K27me3-bound repetitive elements. Given very low repetitive element occupancy in the C. merolae genome (0,7% transposable elements + 5% WSV repeats) (Nozaki et al., 2007), H3K27me3 seems to be predominantly present on repetitive elements, covering as much as 50% of their total number (Figure 5D). We noted that the enrichment on both, genes and repetitive elements, is in concordance with the studies in Drosophila (Yin et al., 2011). Interestingly, the predominant enrichment of H3K27me3 on repetitive elements was found also in diatom P. tricornutum (Veluchamy et al., 2015), suggesting an ancestral role of Polycomb-mediated gene regulation in guarding the genome.
Next, we wanted to characterize the correlation of H3K27me3 enrichment with the expression of its targets. In order to decipher expression level on a genome-wide scale, we performed a RNA-seq experiment on reverse-transcribed RNA extracted from C. merolae cultures. The sequencing reads were mapped to the reference genome and expression level was assessed based on the FPKM values. We intersected the FPKM data with H3K27me3-binding profile and distinguished genes and repetitive elements for the analyses.
We found that both, H3K27me3-covered genes and H3K27me3-covered repetitive elements are on average significantly less expressed than non-H3K27me3 targets (Figure 6), confirming that the H3K27me3 modification in C. merolae is highly correlated with gene repression. Moreover, we noted that the general level of repetitive element expression is lower than gene expression, irrespectively of the H3K27me3 status. These results suggest the existence of additional mechanisms involved in repression of the repetitive elements.
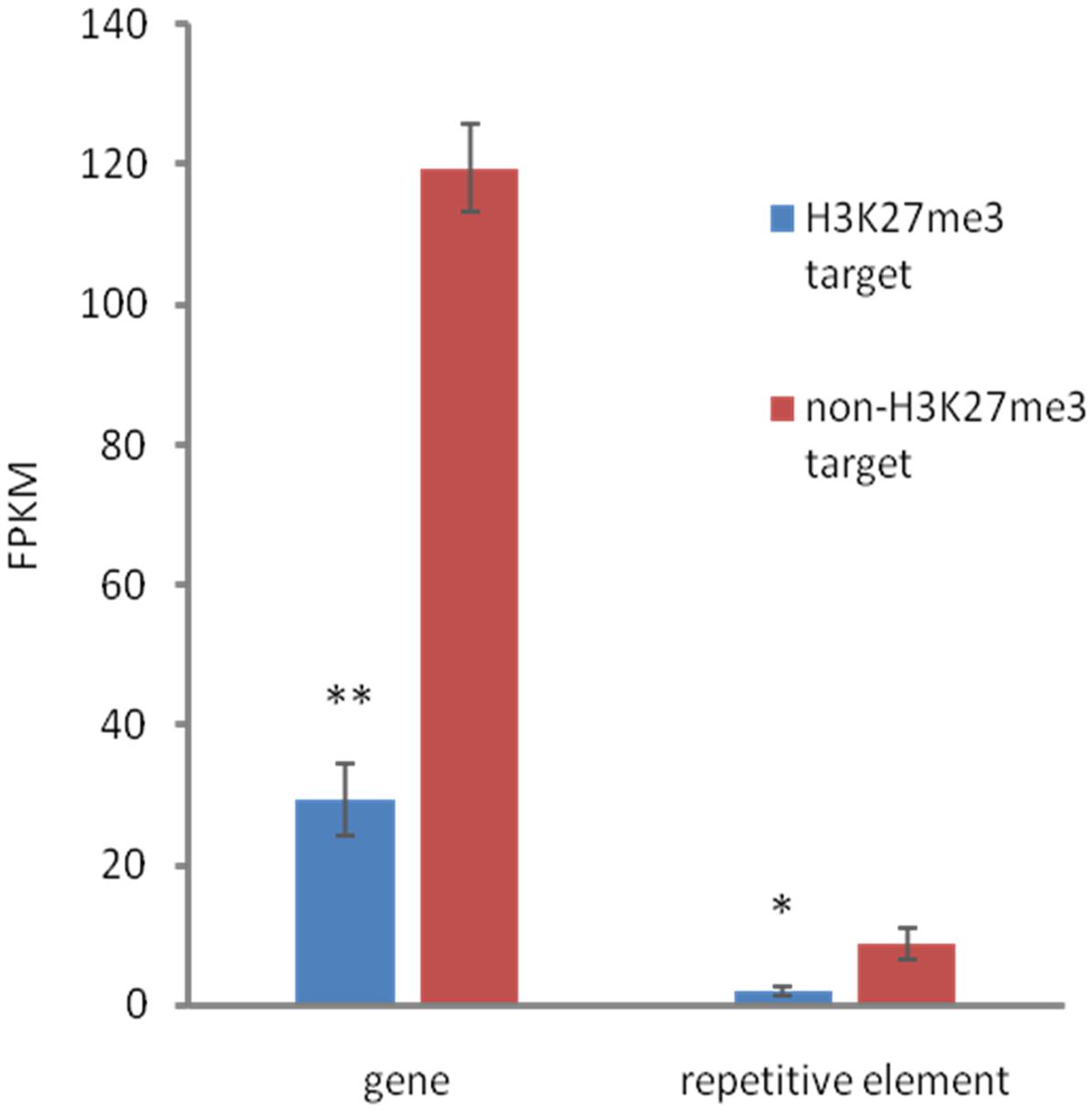
FIGURE 6. Relationship between expression and H3K27me3 presence. H3K27me3 occupancy is anti-correlated with expression level in both, genes and TEs. Expression was estimated based on FPKM values obtained from RNA-seq experiment. Student t-test was used to calculate significance. ∗∗p-value < 0.01, ∗p-value < 0.05. Error bars represent standard error (SE).
H3K27me3 enrichment has a broad distribution over gene bodies in plants (Zhang et al., 2007; Yang et al., 2014) or gene bodies and flanking regions in animals (Pauler et al., 2009; Cerase et al., 2014) and diatoms (Veluchamy et al., 2015). To inspect the distribution of the mark in C. merolae, we calculated average H3K27me3 enrichment over genic coordinates and observed that H3K27me3 covers gene bodies, 0.5 kb upstream and 0.5 kb downstream regions (Supplementary Figure S2). This general approach obscured whether all targets show similar, broad distribution of H3K27me3 or whether the differential enrichment subgroups are present among the targets. We therefore performed k-means clustering on the results. Our analysis identified four clusters with the preferential presence of H3K27me3 at gene bodies (cluster 1), the 0.5 kb downstream region (cluster 2), and the 0.5 kb upstream region (cluster 3) (Figure 7A). Cluster 4 contains non-H3K27me3 targets.
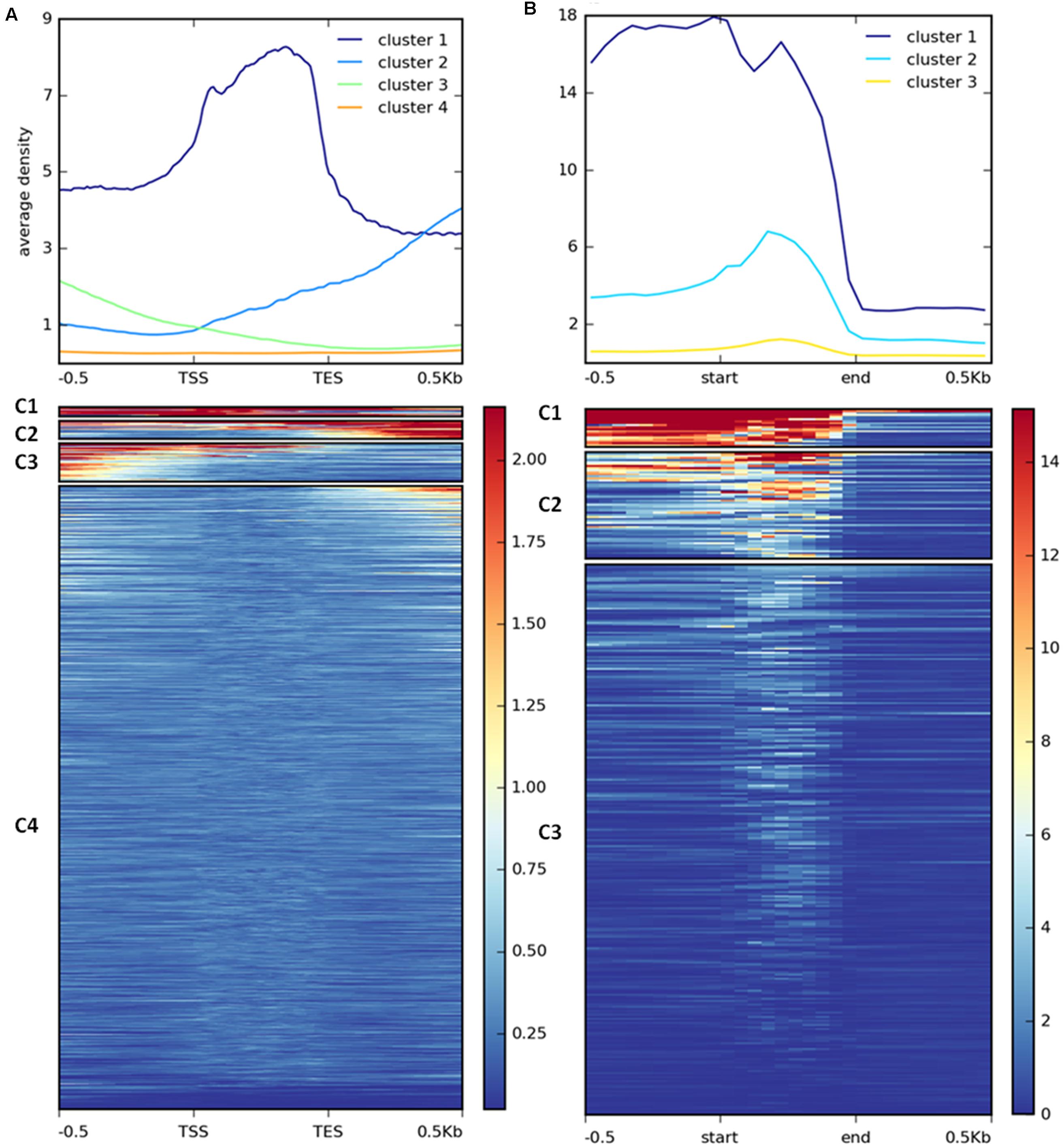
FIGURE 7. Clustering of H3K27me3 gene body enrichment. Coverage of ChIP-seq H3K27me3 reads normalized to input was calculated and used to generate matrix with gene (A) or repetitive element (B) locations as reference. The reads were scaled to 500 bp windows and flanking regions set to 500 bp. Matrix files served as an input for heatmap generation and K-means clustering in deepTools2.
As distinct H3K27me3 enrichment clusters impact target gene activity differently in some other species (Young et al., 2011; Veluchamy et al., 2015), we asked whether gene expression level varies between the H3K27me3 clusters in C. merolae. H3K27me3 differential enrichment compared with RNA-seq data revealed that clusters 1–3 correlate with the gene repression, albeit to an uneven extent. Cluster 1 correlated with the strongest repression level, whereas clusters 2 and 3 correlated with a similar, milder repression (Figure 8A). Thus, the strongest silencing effect was seen for H3K27me3 distribution at the gene bodies, similarly to the diatom P. tricornutum (Veluchamy et al., 2015), and the H3K27me3 enrichment at the flanking regions is correlated with the milder repression.
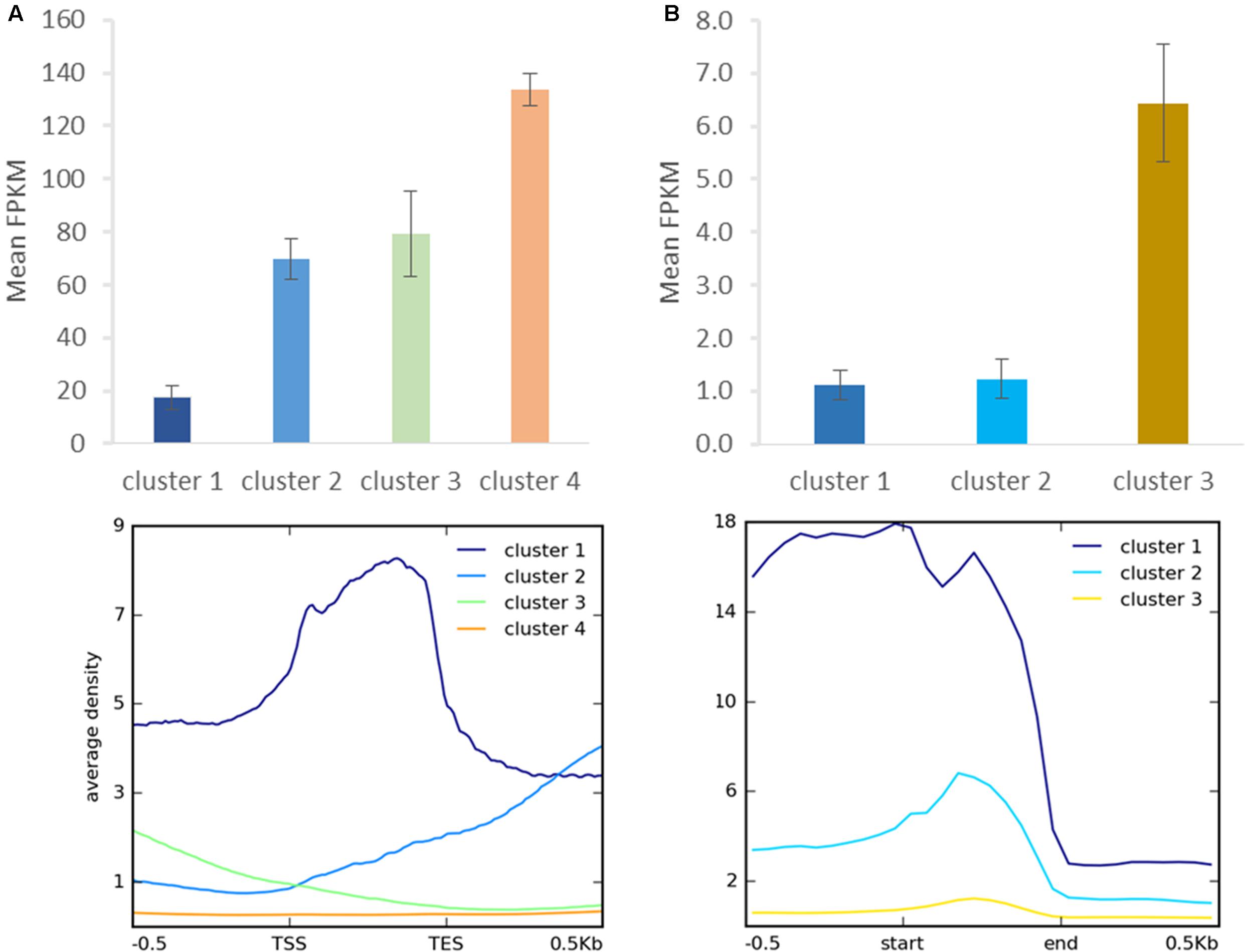
FIGURE 8. Relationship between H3K27me3 occupancy and expression after clustering analysis. Identified clusters (Figure 7) were correlated with expression level measured as FPKM in RNA-seq experiment. Expression level was determined for H3K27me3 occupancy clusters from genes (A) and repetitive elements (B). Error bars represent standard error.
We performed similar clustering analysis to characterize H3K27me3 enrichment also on the repetitive elements. We could distinguish two clusters with histone mark presence on 0.5 kb upstream region and gene bodies (cluster 1) or gene bodies only (cluster 2) (Figure 7B). Cluster 3 corresponds to non-H3K27me3 targets. In contrast to the genes, we found that cluster 1 and 2 correlate with reduced gene expression to the same extent (Figure 8B), suggesting that gene body H3K27me3 distribution has a predominant effect on gene repression. On the other hand, clusters present at repetitive elements are not fully analogous to those at genes as for the former we did not identify a cluster with enrichment exclusively at the flanking regions.
Looking at the whole-chromosome level of H3K27me3 distribution, we found a preferential enrichment of the mark on the chromosome ends (Figure 9). Such observation concerns 36 out 40 ends from 20 chromosomes. The exceptions, in which the closest H3K27me3 domain was detected only 4–5 kb away from the chromosome end, include: 5′ end of chromosome 3 and 3′ end of chromosomes 7, 14, 16 (Supplementary Table S2 distance). Telomeric repeats in plants span regions from 0.5 kb in Chlorella vulgaris to 150 kb in Nicotiana tabacum (Fajkus et al., 1995; Higashiyama et al., 1995). C. merolae telomere length is relatively short and varies from 400bp to 700bp (Nozaki et al., 2007). Hence, the H3K27me3-domains at the chromosome ends detected in C. merolae cover the regions of telomeres and subtelomeres, similarly to what has been shown for the fungi: N. crassa (Jamieson et al., 2013) and Cryptococcus neoformans (Dumesic et al., 2014).
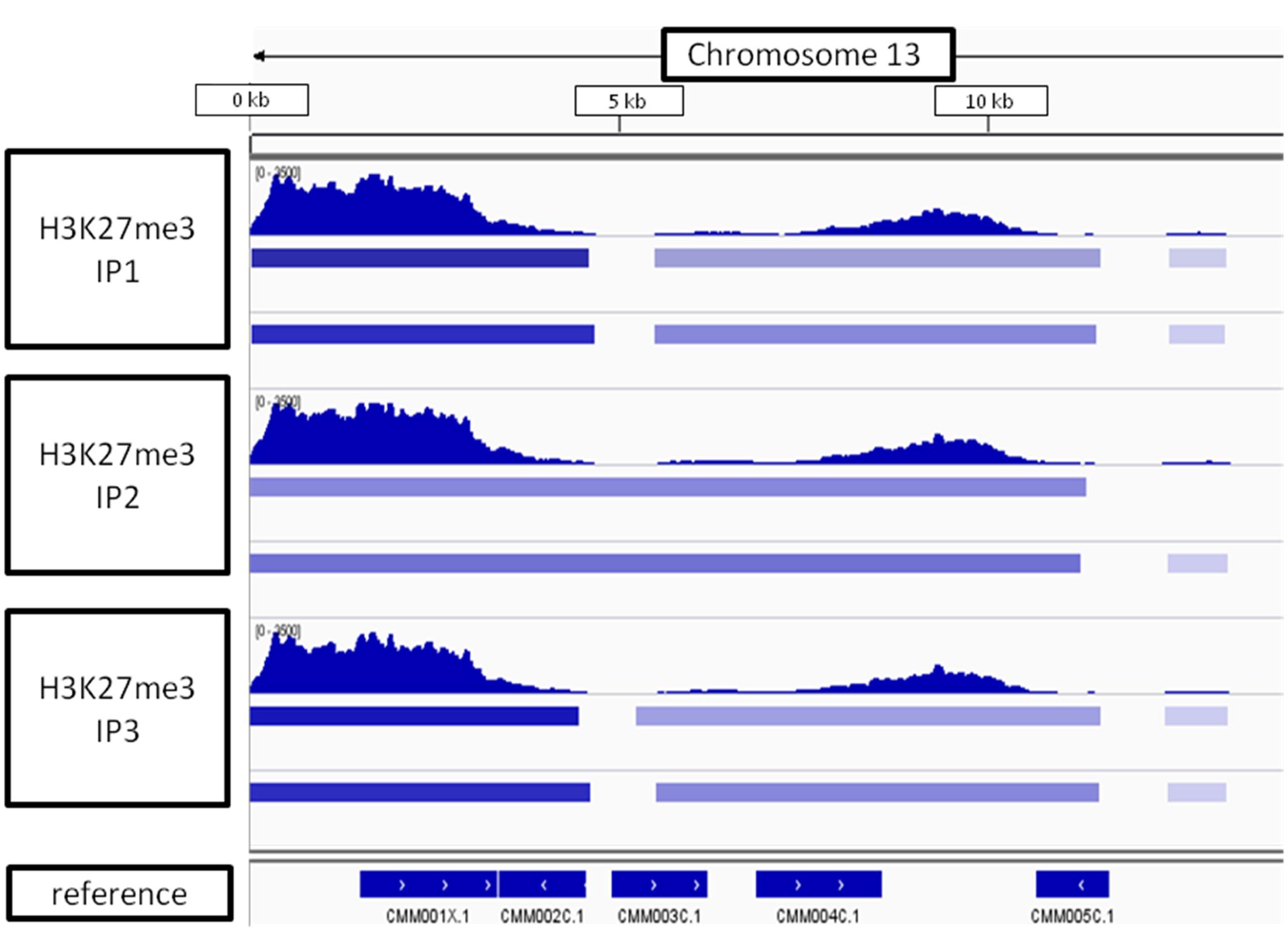
FIGURE 9. H3K27me3 domain enrichment on chromosome ends. 5′ end of chromosome 13 is used as an example. Results from three biological replicates (IP1-3) taken for sequencing are shown. Bars below H3K27me3 tracks correspond to peaks scored by MACS2 with input (top bar) or H3 (bottom bar) used for normalization. The lowest track correspond to C. merolae genome annotation (merolae.biol.s.u-tokyo.ac.jp). The data was visualized in IGV v2.3.
In higher eukaryotes, H3K27me3 is known to target, i.e., developmental, tissue-specific or stress-responsive genes (Boyer et al., 2006; Bracken et al., 2006; Zhang et al., 2007; Lafos et al., 2011; Kleinmanns and Schubert, 2014). We sought to investigate whether similar functional association can be found in C. merolae, a unicellular organism with primitive developmental program.
In order to decipher functional classification of H3K27me3 targets, we performed gene ontology analysis with Singular Enrichment Analysis from the AgriGO toolkit (Du et al., 2010). GO classes of H3K27me3-targets and full lists of C. merolae genes used as a reference were extracted from the UniProtKB-database. We found a GO annotation for 3085 genes (50,5% from total number of 6108, including hypothetical proteins and transcripts) in the reference with 144 GO-annotated genes among H3K27me3-targets (62,6% from total number of 230). Statistical significance was determined based on the adjusted p-value with Yekutieli test for FDR. For statistical tests, we selected only those GO classes that were represented by at least five entries (see material and methods). All three different GO sub-ontologies were taken into account: molecular function (GO:0003674), biological process (GO::0008150), and cellular component (GO::0008372).
We did not reveal any significantly (adjusted p-value < 0.05) enriched GO terms in cellular component sub-ontology. In contrast, we detected three GO term branches in biological process and one in molecular function sub-ontologies (Figure 10). Consistently with the known H3K27me3 function in the repression of developmental programs, we detected significantly enriched GO terms related to organismal process and development. Surprisingly, among enriched terms we found also those corresponding to protein maturation and intein-mediated protein splicing.
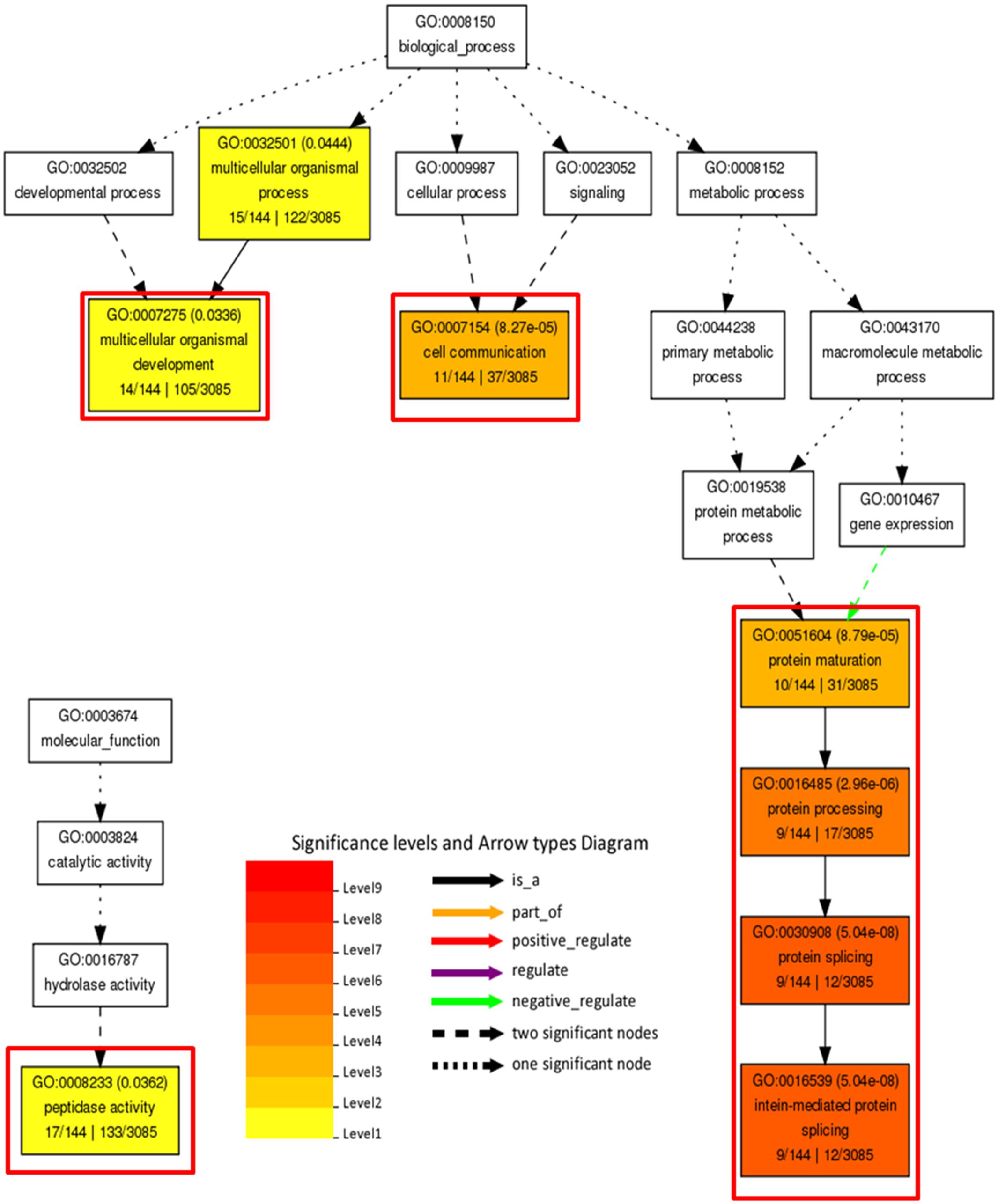
FIGURE 10. Gene ontology analysis on H3K27me3 target genes. Significantly enriched terms from molecular function and biological process subontologies (no significant hits found for cellular component subontology). Color coding represents significance level (1–9) below adjusted p-value threshold (p < 0.05). Adjusted p-values are shown next to GO IDs for each GO term. Relationships between GO terms are reflected in the arrow types. Red frames highlight GO terms connected with intein-mediated protein splicing. Numbers below subcategory name correspond to: GO subcategory gene number in query/total gene number in query | GO subcategory gene number in reference (TAIR10)/total gene number in reference.
Protein splicing is a protein maturation mechanism based on the excision of a protein fragment (intein) from a precursor and ligation of the flanking polypeptides (exteins) (Topilina and Mills, 2014). Comparative analyses of amino acid sequences revealed a homology of protein splice-junction motif from inteins to the C-terminal Hog domains in Hedgehog proteins, the secretory proteins controlling developmental processes in Metazoa (Jiang and Hui, 2008).
In concordance to the protein splicing mechanism and the function of intein-containing genes, we observed that the majority of protein entries present in significantly enriched GO terms (organismal process and development, cell communication and peptidase function) overlap with the protein splicing group (Supplementary Table S3). Moreover, we found that all of the H3K27me3 targets present in the protein splicing term indeed possess a Hog domain as the Hedgehog proteins.
Interestingly, the genes encoding Hedgehog proteins in C. merolae reside in the telomeric and subtelomeric regions of the chromosomes (Nozaki et al., 2007), which is in agreement to our results on the preferential H3K27me3 enrichment at the chromosome ends (Figure 9). Moreover, comparing with clustering data, we observed that Hedgehog loci were enriched in cluster 3 (Supplementary Table S4), bound by H3K27me3 preferentially to the upstream regions and moderately repressed.
Polycomb-group mediated regulation of gene expression in multicellular organisms is intensively studied in plants and has important roles in stress responses and developmental phase transitions (Köhler and Villar, 2008; Kleinmanns and Schubert, 2014). However, a model for Pc-G function in unicellular, photosynthetic organisms is currently lacking. We therefore performed homology searches and Bayesian phylogenetic tree analyses on the conserved alignment blocks. Our results confirmed a frequent absence of the PRC1 complex, ancient origin of PRC2 complex and the widespread distribution of core components of PRC2 in the green lineage [see also: (Shaver et al., 2010; Kim et al., 2013)]. We detected the PcG mark H3K27me3 in prasinophytes and red algae, arguing for the functionality of PRC2 in these organisms. Subsequent analyses of H3K27me3 occupancy in the model red alga, C. merolae, and correlative analyses with transcriptomic data revealed several important observations in this unicellular organism.
We observed that H3K27me3 in C. merolae is present on both, genes and repetitive elements, covering 4% of total gene number and 50% of total repetitive element number. The occupancy on both genomic types is in the agreement with the results from Drosophila and Phaeodactylum, suggesting an ancestral function of PcG in repetitive element silencing. As Arabidopsis H3K27me3 occupies preferentially genes, our results suggest also a partial divergence from PcG-mediated repetitive element repression in higher plants.
In Arabidopsis endosperm, the vicinity of repetitive elements was shown to impact H3K27me3-mediated gene imprinting (Weinhofer et al., 2010; Wolff et al., 2011). In contrast, the H3K27me3-covered genes in C. merolae are not in the vicinity of repetitive elements. However, we cannot exclude the possibility that the genome annotation is incomplete, with unknown types of repetitive elements being not reported.
We showed that H3K27me3 occupancy anti-correlates with the expression of its targets, consistently with its function in the multicellular organisms. As the other lysine-27 methylation marks, H3K27me1 and H3K27me2, have a repressive function in higher plants (Bastow et al., 2004; Jacob et al., 2009), similarly to the H3K27me3, the future steps should include profiling of those modifications in C. merolae genome. We also observed that the degree of repression is tightly correlated with the H3K27me3 profile at the genic level. The lowest expression was found for the genes covered by H3K27me3 over the gene body, whereas the targets with H3K27me3 occupancy on 5′ and 3′ flanking regions associate with intermediate repression.
Furthermore, H3K27me3 shows an enrichment on chromosome ends and covers a broad region including telomeric repeats and subtelomeric genes in C. merolae. Interestingly, several other species were reported to show an enrichment of H3K27me3 at the subtelomeric and telomeric regions (Smith et al., 2008; Vaquero-sedas et al., 2012). Moreover, Polycomb proteins were proved to be associated with telomere-binding factors (Zhou et al., 2013) and telobox motifs demonstrated to be enriched at PcG-peaks (Deng et al., 2013), arguing for a functional connection between PcG-mediated repression and chromosomal ends.
Our gene ontology analysis showed that H3K27me3 covers genes associated with intein-mediated protein splicing. Inteins are considered as ancient mobile elements present in all three domains of life (eukaryotes, archaea, bacteria) and viruses (Pietrokovski, 2001; Novikova et al., 2014). Hence, our results suggest that the H3K27me3-mediated repression might function in guarding the C. merolae genome. Such notion would be consistent with the predominant enrichment of H3K27me3 on the repetitive elements (Figure 5D). On the other hand, inteins may also develop to become post-translational regulatory elements in the course of evolution, as seen for the conditional protein splicing of Hog domains in animals (Shah and Muir, 2014). Therefore, it is of special importance to decipher the function of intein-containing Hedgehog genes and the conditions for their de-repression in C. merolae.
Interestingly, we also observed that the intein-containing genes reside in the subtelomeric regions and are moderately repressed by H3K27me3 that occupies upstream regions from their TSS. It is currently unclear how protein splicing, moderate H3K27me3-mediated repression and subtelomeric localization are inter-connected. It is feasible that the integration of intein-containing genes appeared relatively recently and that these ‘young’ H3K27me3 targets display H3K27me3 enrichment at regions upstream of their TSS, in agreement with their moderate repression. Comparative genomics between C. merolae and related species should be used to validate this idea. In turn, subtelomeres are dynamic regions and the objects of ectopic recombination events, which facilitate gene diversification, phenotypic diversity and adaptation to different environments, exemplified by an olfactory receptor gene family in humans (Linardopoulou et al., 2001), contingency systems in various pathogens (Barry et al., 2003) and carbon-source metabolism genes in S. cerevisiae (Carlson and Botstein, 1983; Louis et al., 1994; Brown et al., 2010). Hence, it is possible that the intein-containing subtelomeric H3K27me3-covered genes are the targets of rapid adaptive evolution. Characterization of their duplication frequency and diversification within their gene families should validate such notion.
Last but not the least, our results on H3K27me3 presence in various algae and genome-wide characterization of expression status and H3K27me3 occupancy in the model red alga, C. merolae, provide a resource for chromatin and transcriptomic studies. An existence of genetic tools, optimized growth conditions and the sequenced genome in C. merolae offer a possibility to decipher genome regulation in a broad evolutionary context, including the organisms with super-reduced genomes.
PM, OK, and DS designed the research. FF and AW designed C. merolae growth scheme. FF handled C. merolae cell cultures. PM performed bioinformatic analyses, chromatin immunoprecipitations and sequencing library preparations. OK performed competition assay and western blot experiments. PM and DS performed homology searches for H3K27me3 targets used in ChIP-RT-qPCR. The manuscript was written by PM and revised by the other authors.
PM, OK, and DS were supported by the DFG-funded SFB973, the Marie Curie International Training Network “EpiTRAITS” and by Freie Universität Berlin within the Excellence Initiative of the German Research Foundation.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
We thank Prof. Motomichi Matsuzaki for providing C. merolae genome annotation and Dr. Michael Grünstäudl for crucial insights in phylogenetic tree creation. We thank Julia Kleinmanns and Dorota Komar for critical revision of the manuscript.
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2017.00607/full#supplementary-material
Abascal, F., Zardoya, R., Posada, D., Miller, C. J., Lou, H. J., Johnson, A. D., et al. (2005). ProtTest: selection of best-fit models of protein evolution. Bioinformatics 21, 2104–2105. doi: 10.1093/bioinformatics/bti263
Barry, J. D., Ginger, M. L., Burton, P., and McCulloch, R. (2003). Why are parasite contingency genes often associated with telomeres? Int. J. Parasitol. 33, 29–45. doi: 10.1016/S0020-7519(02)00247-3
Bastow, R., Mylne, J. S., Lister, C., Lippman, Z., Martienssen, R. A., and Dean, C. (2004). Vernalization requires epigenetic silencing of FLC by histone methylation. Nature 427, 164–167. doi: 10.1038/nature02269
Berke, L., and Snel, B. (2015). The plant Polycomb repressive complex 1 (PRC1) existed in the ancestor of seed plants and has a complex duplication history. BMC Evol. Biol. 15:44. doi: 10.1186/s12862-015-0319-z
Bowman, J. L., Floyd, S. K., and Sakakibara, K. (2007). Green genes-comparative genomics of the green branch of life. Cell 129, 229–234. doi: 10.1016/j.cell.2007.04.004
Boyer, L. A., Plath, K., Zeitlinger, J., Brambrink, T., Medeiros, L. A., Lee, T. I., et al. (2006). Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 441, 349–353. doi: 10.1038/nature04733
Bracken, A. P., Dietrich, N., Pasini, D., Hansen, K. H., and Helin, K. (2006). Genome-wide mapping of Polycomb target genes unravels their roles in cell fate transitions. Genes Dev. 20, 1123–1136. doi: 10.1101/gad.381706.present
Brown, C. A., Murray, A. W., and Verstrepen, K. J. (2010). Rapid expansion and functional divergence of subtelomeric gene families in yeasts. Curr. Biol. 20, 895–903. doi: 10.1016/j.cub.2010.04.027
Butenko, Y., and Ohad, N. (2011). Polycomb-group mediated epigenetic mechanisms through plant evolution. Biochim. Biophys. Acta 1809, 395–406. doi: 10.1016/j.bbagrm.2011.05.013
Camacho, C., Coulouris, G., Avagyan, V., Ma, N., Papadopoulos, J., Bealer, K., et al. (2009). BLAST+: architecture and applications. BMC Bioinformatics 10:421. doi: 10.1186/1471-2105-10-421
Carlson, M., and Botstein, D. (1983). Organization of the SUC gene family in Saccharomyces. Mol. Cell. Biol. 3, 351–359. doi: 10.1128/MCB.3.3.351
Cerase, A., Smeets, D., Tang, Y. A., Gdula, M., Kraus, F., Spivakov, M., et al. (2014). Spatial separation of Xist RNA and polycomb proteins revealed by superresolution microscopy. Proc. Natl. Acad. Sci. U.S.A. 111, 2235–2240. doi: 10.1073/pnas.1312951111
Chen, D., Huang, Y., Ruan, Y., and Shen, W. H. (2016). The evolutionary landscape of PRC1 core components in green lineage. Planta 243, 825–846. doi: 10.1007/s00425-015-2451-9
Cui, J., Zhang, Z., Shao, Y., Zhang, K., Leng, P., and Liang, Z. (2015). Genome-wide identification, evolutionary, and expression analyses of histone H3 variants in plants. Biomed. Res. Int. 2015:341598. doi: 10.1155/2015/341598
Del Prete, S., Mikulski, P., Schubert, D., and Gaudin, V. (2015). One, two, three: Polycomb proteins hit all dimensions of gene regulation. Genes 6, 520–542. doi: 10.3390/genes6030520
Deleris, A., Stroud, H., Bernatavichute, Y., Johnson, E., Klein, G., Schubert, D., et al. (2012). Loss of the DNA methyltransferase MET1 Induces H3K9 hypermethylation at PcG target genes and redistribution of H3K27 trimethylation to transposons in Arabidopsis thaliana. PLoS Genet. 8:e1003062. doi: 10.1371/journal.pgen.1003062
Deng, W., Buzas, D. M., Ying, H., Robertson, M., Taylor, J., Peacock, W. J., et al. (2013). Arabidopsis Polycomb Repressive Complex 2 binding sites contain putative GAGA factor binding motifs within coding regions of genes. BMC Genomics 14:593. doi: 10.1186/1471-2164-14-593
Derkacheva, M., and Hennig, L. (2014). Variations on a theme: Polycomb group proteins in plants. J. Exp. Bot. 65, 2769–2784. doi: 10.1093/jxb/ert410
Du, Z., Zhou, X., Ling, Y., Zhang, Z., and Su, Z. (2010). agriGO: a GO analysis toolkit for the agricultural community. Nucleic Acids Res. 38, W64–W70. doi: 10.1093/nar/gkq310
Dumesic, P. A., Homer, C. M., Moresco, J. J., Pack, L. R., Shanle, E. K., Coyle, S. M., et al. (2014). Product binding enforces the genomic specificity of a yeast polycomb repressive complex. Cell 160, 204–218. doi: 10.1016/j.cell.2014.11.039
Ebert, A., Schotta, G., Lein, S., Kubicek, S., Krauss, V., Jenuwein, T., et al. (2004). Su(var) genes regulate the balance between euchromatin and heterochromatin in Drosophila. Genes Dev. 18, 2973–2983. doi: 10.1101/gad.323004
Fajkus, J., Kovarík, A., Královics, R., and Bezděk, M. (1995). Organization of telomeric and subtelomeric chromatin in the higher plant Nicotiana tabacum. Mol. Gen. Genet. 247, 633–638. doi: 10.1007/BF00290355
Ferrari, K. J., Scelfo, A., Jammula, S., Cuomo, A., Barozzi, I., Stützer, A., et al. (2014). Polycomb-dependent H3K27me1 and H3K27me2 regulate active transcription and enhancer fidelity. Mol. Cell 53, 49–62. doi: 10.1016/j.molcel.2013.10.030
Hennig, L., and Derkacheva, M. (2009). Diversity of Polycomb group complexes in plants: same rules, different players? Trends Genet. 25, 414–423. doi: 10.1016/j.tig.2009.07.002
Higashiyama, T., Maki, S., and Yamada, T. (1995). Molecular organization of Chlorella vulgaris chromosome I: presence of telomeric repeats that are conserved in higher plants. Mol. Gen. Genet. 246, 29–36. doi: 10.1007/BF00290130
Huang, Y., Chen, D.-H., Liu, B.-Y., Shen, W.-H., and Ruan, Y. (2016). Conservation and diversification of polycomb repressive complex 2 (PRC2) proteins in the green lineage. Brief. Funct. Genomics 16, 106–119. doi: 10.1093/bfgp/elw007
Jacob, Y., Feng, S., LeBlanc, C. A., Bernatavichute, Y. V., Stroud, H., Cokus, S., et al. (2009). ATXR5 and ATXR6 are H3K27 monomethyltransferases required for chromatin structure and gene silencing. Nat. Struct. Mol. Biol. 16, 763–768. doi: 10.1038/nsmb.1611
Jacob, Y., and Michaels, S. D. (2009). H3K27me1 is E(z) in animals, but not in plants. Epigenetics 4, 366–369. doi: 10.4161/epi.4.6.9713
Jamieson, K., Rountree, M. R., Lewis, Z. A., Stajich, J. E., and Selker, E. U. (2013). Regional control of histone H3 lysine 27 methylation in Neurospora. Proc. Natl. Acad. Sci. U.S.A. 110, 6027–6032. doi: 10.1073/pnas.1303750110
Jeffery, C. J. (2003). Moonlighting proteins: old proteins learning new tricks. Trends Genet. 19, 415–417. doi: 10.1016/S0168-9525(03)00167-7
Jiang, J., and Hui, C.-C. (2008). Hedgehog signaling in development and cancer. Dev. Cell 15, 801–812. doi: 10.1016/j.devcel.2008.11.010
Kim, E., Ma, X., and Cerutti, H. (2013). Gene silencing in microalgae?: mechanisms and biological roles. Bioresour. Technol. 184, 23–32. doi: 10.1016/j.biortech.2014.10.119
Kleinmanns, J. A., and Schubert, D. (2014). Polycomb and Trithorax group protein-mediated control of stress responses in plants. Biol. Chem. 395, 1291–1300. doi: 10.1515/hsz-2014-0197
Köhler, C., and Villar, C. B. R. (2008). Programming of gene expression by Polycomb group proteins. Trends Cell Biol. 18, 236–243. doi: 10.1016/j.tcb.2008.02.005
Lachner, M., Sengupta, R., Schotta, G., and Jenuwein, T. (2004). Trilogies of histone lysine methylation as epigenetic landmarks of the eukaryotic genome. Cold Spring Harb. Symp. Quant. Biol. 69, 209–218. doi: 10.1101/sqb.2004.69.209
Lafos, M., Kroll, P., Hohenstatt, M. L., Thorpe, F. L., Clarenz, O., and Schubert, D. (2011). Dynamic regulation of H3K27 trimethylation during Arabidopsis differentiation. PLoS Genet. 7:e1002040. doi: 10.1371/journal.pgen.1002040
Larkin, M. A., Blackshields, G., Brown, N. P., Chenna, R., Mcgettigan, P. A., McWilliam, H., et al. (2007). Clustal W and Clustal X version 2.0. Bioinformatics 23, 2947–2948. doi: 10.1093/bioinformatics/btm404
Linardopoulou, E., Mefford, H. C., Nguyen, O., Friedman, C., van den Engh, G., Farwell, D. G., et al. (2001). Transcriptional activity of multiple copies of a subtelomerically located olfactory receptor gene that is polymorphic in number and location. Hum. Mol. Genet. 10, 2373–2383. doi: 10.1093/hmg/10.21.2373
Lindroth, A. M., Shultis, D., Jasencakova, Z., Fuchs, J., Johnson, L., Schubert, D., et al. (2004). Dual histone H3 methylation marks at lysines 9 and 27 required for interaction with CHROMOMETHYLASE3. EMBO J. 23, 4286–4296. doi: 10.1038/sj.emboj.7600430
Liu, Y., Taverna, S. D., Muratore, T. L., Shabanowitz, J., Hunt, D. F., and Allis, C. D. (2007). RNAi-dependent H3K27 methylation is required for heterochromatin formation and DNA elimination in Tetrahymena. Genes Dev. 21, 1530–1545. doi: 10.1101/gad.1544207
Louis, E. J., Naumova, E. S., Lee, A., Naumov, G., and Haber, J. E. (1994). The chromosome end in yeast: its mosaic nature and influence on recombinational dynamics. Genetics 136, 789–802.
Matsuzaki, M., Misumi, O., Shin-I, T., Maruyama, S., Takahara, M., Miyagishima, S.-Y., et al. (2004). Genome sequence of the ultrasmall unicellular red alga Cyanidioschyzon merolae 10D. Nature 428, 653–657. doi: 10.1038/nature02398
McKay, D. J., Klusza, S., Penke, T. J. R., Meers, M. P., Curry, K. P., McDaniel, S. L., et al. (2015). Interrogating the function of metazoan histones using engineered gene clusters. Dev. Cell 32, 373–386. doi: 10.1016/j.devcel.2014.12.025
Merchant, S. S., Prochnik, S. E., Vallon, O., Harris, E. H., Karpowicz, J., Witman, G. B., et al. (2010). The Chlamydomonas genome reveals the evolution of key animal and plant functions. Science 318, 245–250. doi: 10.1126/science.1143609.The
Müller, J., Hart, C. M., Francis, N. J., Vargas, M. L., Sengupta, A., Wild, B., et al. (2002). Histone methyltransferase activity of a Drosophila Polycomb group repressor complex. Cell 111, 197–208. doi: 10.1016/S0092-8674(02)00976-5
Müller, J., and Verrijzer, P. (2009). Biochemical mechanisms of gene regulation by polycomb group protein complexes. Curr. Opin. Genet. Dev. 19, 150–158. doi: 10.1016/j.gde.2009.03.001
Nekrasov, M., Klymenko, T., Fraterman, S., Papp, B., Oktaba, K., Köcher, T., et al. (2007). Pcl-PRC2 is needed to generate high levels of H3-K27 trimethylation at Polycomb target genes. EMBO J. 26, 4078–4088. doi: 10.1038/sj.emboj.7601837
Novikova, O., Topilina, N., and Belfort, M. (2014). Enigmatic distribution, evolution, and function of inteins. J. Biol. Chem. 289, 14490–14497. doi: 10.1074/jbc.R114.548255
Nozaki, H., Takano, H., Misumi, O., Terasawa, K., Matsuzaki, M., Maruyama, S., et al. (2007). A 100%-complete sequence reveals unusually simple genomic features in the hot-spring red alga Cyanidioschyzon merolae. BMC Biol. 5:28. doi: 10.1186/1741-7007-5-28
Okada, T., Endo, M., Singh, M. B., and Bhalla, P. L. (2005). Analysis of the histone H3 gene family in Arabidopsis and identification of the male-gamete-specific variant AtMGH3. Plant J. 44, 557–568. doi: 10.1111/j.1365-313X.2005.02554.x
Palenik, B., Grimwood, J., Aerts, A., Rouzé, P., Salamov, A., Putnam, N., et al. (2007). The tiny eukaryote Ostreococcus provides genomic insights into the paradox of plankton speciation. Proc. Natl. Acad. Sci. U.S.A. 104, 7705–7710. doi: 10.1073/pnas.0611046104
Papp, B., and Müller, J. (2006). Histone trimethylation and the maintenance of transcriptional ON and OFF states by trxG and PcG proteins. Genes Dev. 20, 2041–2054. doi: 10.1101/gad.388706
Park, S., Oh, S., and van Nocker, S. (2012). Genomic and gene-level distribution of histone H3 dimethyl lysine-27 (H3K27me2) in Arabidopsis. PLoS ONE 7:e52855. doi: 10.1371/journal.pone.0052855
Pauler, F. M., Sloane, M. A., Huang, R., Regha, K., Koerner, M. V., Tamir, I., et al. (2009). H3K27me3 forms BLOCs over silent genes and intergenic regions and specifies a histone banding pattern on a mouse autosomal chromosome. Genome Res. 19, 221–233. doi: 10.1101/gr.080861.108.4
Pietrokovski, S. (2001). Intein spread and extinction in evolution. Trends Genet. 17, 465–472. doi: 10.1016/S0168-9525(01)02365-4
Price, D. C., Chan, C. X., Yoon, H. S., Yang, E. C., Qiu, H., Weber, A. P., et al. (2012). Cyanophora paradoxa genome elucidates origin of photosynthesis in algae and plants. Science 335, 843–847. doi: 10.1126/science.1213561
Quinlan, A. R., and Hall, I. M. (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. doi: 10.1093/bioinformatics/btq033
Rambaut, A. (2009). FigTree, a Graphical Viewer of Phylogenetic Trees. Available at: http://tree.bio.ed.ac.uk/software/figtree/
Ramirez, F., Ryan, D. P., Gruning, B., Bhardwaj, V., Kilpert, F., Richter, A. S., et al. (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, 160–165. doi: 10.1093/nar/gkw257
Ronquist, F., Teslenko, M., Van Der Mark, P., Ayres, D. L., Darling, A., Höhna, S., et al. (2012). Mrbayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 61, 539–542. doi: 10.1093/sysbio/sys029
Schuettengruber, B., Chourrout, D., Vervoort, M., Leblanc, B., and Cavalli, G. (2007). Genome regulation by Polycomb and Trithorax proteins. Cell 128, 735–745. doi: 10.1016/j.cell.2007.02.009
Schwartz, Y. B., and Pirrotta, V. (2008). Polycomb complexes and epigenetic states. Curr. Opin. Cell Biol. 20, 266–273. doi: 10.1016/j.ceb.2008.03.002
Schwartz, Y. B., and Pirrotta, V. (2013). A new world of Polycombs: unexpected partnerships and emerging functions. Nat. Rev. Genet. 14, 853–864. doi: 10.1038/nrg3603
Shah, N. H., and Muir, T. W. (2014). Inteins: nature’s gift to protein chemists. Chem. Sci. 5, 446–461. doi: 10.1039/C3sc52951g
Shaver, S., Casas-Mollano, J. A., Cerny, R. L., and Cerutti, H. (2010). Origin of the polycomb repressive complex 2 and gene silencing by an E(z) homolog in the unicellular alga Chlamydomonas. Epigenetics 5, 301–312. doi: 10.4161/epi.5.4.11608
Smith, K. M., Kothe, G. O., Matsen, C. B., Khlafallah, T. K., Adhvaryu, K. K., Hemphill, M., et al. (2008). The fungus Neurospora crassa displays telomeric silencing mediated by multiple sirtuins and by methylation of histone H3 lysine 9. Epigenetics Chromatin 1:5. doi: 10.1186/1756-8935-1-5
Sung, S., Schmitz, R. J., and Amasino, R. M. (2006). A PHD finger protein involved in both the vernalization and photoperiod pathways in Arabidopsis. Genes Dev. 20, 3244–3248. doi: 10.1101/gad.1493306
Talavera, G., and Castresana, J. (2007). Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 56, 564–577. doi: 10.1080/10635150701472164
Thorvaldsdóttir, H., Robinson, J. T., and Mesirov, J. P. (2013). Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief. Bioinform. 14, 178–192. doi: 10.1093/bib/bbs017
Topilina, N. I., and Mills, K. V. (2014). Recent advances in in vivo applications of intein-mediated protein splicing. Mob. DNA 5:5. doi: 10.1186/1759-8753-5-5
Trapnell, C., Roberts, A., Goff, L., Pertea, G., Kim, D., Kelley, D. R., et al. (2012). Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 7, 562–578. doi: 10.1038/nprot.2012.016
Vaquero-sedas, I., Luo, C., and Vega-palas, M. A. (2012). Analysis of the epigenetic status of telomeres by using ChIP-seq data. Nucleic Acids Res. 40:e163. doi: 10.1093/nar/gks730
Veerappan, C. S., Avramova, Z., and Moriyama, E. N. (2008). Evolution of SET-domain protein families in the unicellular and multicellular Ascomycota fungi. BMC Evol. Biol. 8:190. doi: 10.1186/1471-2148-8-190
Veluchamy, A., Rastogi, A., Lin, X., Lombard, B., Murik, O., Thomas, Y., et al. (2015). An integrative analysis of post-translational histone modifications in the marine diatom Phaeodactylum tricornutum. Genome Biol. 16:102. doi: 10.1186/s13059-015-0671-8
Waterhouse, A. M., Procter, J. B., Martin, D. M. A., Clamp, M., and Barton, G. J. (2009). Jalview Version 2–A multiple sequence alignment editor and analysis workbench. Bioinformatics 25, 1189–1191. doi: 10.1093/bioinformatics/btp033
Weinhofer, I., Hehenberger, E., Roszak, P., Hennig, L., and Köhler, C. (2010). H3K27me3 profiling of the endosperm implies exclusion of polycomb group protein targeting by DNA methylation. PLoS Genet. 6:e1001152. doi: 10.1371/journal.pgen.1001152
Wolff, P., Weinhofer, I., Seguin, J., Roszak, P., Beisel, C., Donoghue, M. T. A., et al. (2011). High-resolution analysis of parent-of-origin allelic expression in the Arabidopsis endosperm. PLoS Genet. 7:e1002126. doi: 10.1371/journal.pgen.1002126
Worden, A. Z., Lee, J.-H., Mock, T., Rouzé, P., Simmons, M. P., Aerts, A. L., et al. (2009). Green evolution and dynamic adaptations revealed by genomes of the marine picoeukaryotes Micromonas. Science 324, 268–272. doi: 10.1126/science.1167222
Yang, C., Bratzel, F., Hohmann, N., Koch, M., Turck, F., and Calonje, M. (2013). VAL-and AtBMI1-mediated H2Aub initiate the switch from embryonic to postgerminative growth in Arabidopsis. Curr. Biol. 23, 1324–1329. doi: 10.1016/j.cub.2013.05.050
Yang, H., Howard, M., and Dean, C. (2014). Antagonistic roles for H3K36me3 and H3K27me3 in the cold-induced epigenetic switch at Arabidopsis FLC. Curr. Biol. 24, 1793–1797. doi: 10.1016/j.cub.2014.06.047
Yin, H., Sweeney, S., Raha, D., Snyder, M., and Lin, H. (2011). A high-resolution whole-genome map of key chromatin modifications in the adult Drosophila melanogaster. PLoS Genet. 7:e1002380. doi: 10.1371/journal.pgen.1002380
Young, M. D., Willson, T. A., Wakefield, M. J., Trounson, E., Hilton, D. J., Blewitt, M. E., et al. (2011). ChIP-seq analysis reveals distinct H3K27me3 profiles that correlate with transcriptional activity. Nucleic Acids Res. 39, 7415–7427. doi: 10.1093/nar/gkr416
Zhang, X., Clarenz, O., Cokus, S., Bernatavichute, Y. V., Pellegrini, M., Goodrich, J., et al. (2007). Whole-genome analysis of histone H3 lysine 27 trimethylation in Arabidopsis. PLoS Biol. 5:e129. doi: 10.1371/journal.pbio.0050129
Keywords: Polycomb, PcG, H3K27me3, Cyanidioschyzon merolae, algae, intein, protein splicing, telomere
Citation: Mikulski P, Komarynets O, Fachinelli F, Weber APM and Schubert D (2017) Characterization of the Polycomb-Group Mark H3K27me3 in Unicellular Algae. Front. Plant Sci. 8:607. doi: 10.3389/fpls.2017.00607
Received: 18 February 2017; Accepted: 04 April 2017;
Published: 26 April 2017.
Edited by:
Dao-Xiu Zhou, Université Paris-Sud, FranceReviewed by:
Yuhai Cui, Agriculture and Agri-Food Canada, CanadaCopyright © 2017 Mikulski, Komarynets, Fachinelli, Weber and Schubert. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Daniel Schubert, ZGFuLnNjaHViZXJ0QGZ1LWJlcmxpbi5kZQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.