 Padma Nimmakayala1
Padma Nimmakayala1 Venkata L. Abburi1Thangasamy Saminathan1Aldo Almeida1Brittany Davenport1Joshua Davidson1
Venkata L. Abburi1Thangasamy Saminathan1Aldo Almeida1Brittany Davenport1Joshua Davidson1 C. V. Chandra Mohan Reddy1Gerald Hankins1Andreas Ebert2
C. V. Chandra Mohan Reddy1Gerald Hankins1Andreas Ebert2 Doil Choi3John Stommel4
Doil Choi3John Stommel4 Umesh K. Reddy1*†
Umesh K. Reddy1*†- 1Department of Biology, Gus R. Douglass Institute, West Virginia State University, Institute, WV, USA
- 2Genetic Resources and Seed Unit, Asian Vegetable Research and Development Center-The World Vegetable Center, Tainan, Taiwan
- 3Department of Plant Science, Plant Genomics and Breeding Institute, College of Agriculture and Life Sciences, Seoul National University, Seoul, South Korea
- 4Genetic Improvement of Fruits and Vegetables Laboratory (United States Department of Agriculture, Agricultural Research Service), Beltsville, MD, USA
Principal component analysis (PCA) with 36,621 polymorphic genome-anchored single nucleotide polymorphisms (SNPs) identified collectively for Capsicum annuum and Capsicum baccatum was used to characterize population structure and species domestication of these two important incompatible cultivated pepper species. Estimated mean nucleotide diversity (π) and Tajima's D across various chromosomes revealed biased distribution toward negative values on all chromosomes (except for chromosome 4) in cultivated C. baccatum, indicating a population bottleneck during domestication of C. baccatum. In contrast, C. annuum chromosomes showed positive π and Tajima's D on all chromosomes except chromosome 8, which may be because of domestication at multiple sites contributing to wider genetic diversity. For C. baccatum, 13,129 SNPs were available, with minor allele frequency (MAF) ≥0.05; PCA of the SNPs revealed 283 C. baccatum accessions grouped into 3 distinct clusters, for strong population structure. The fixation index (FST) between domesticated C. annuum and C. baccatum was 0.78, which indicates genome-wide divergence. We conducted extensive linkage disequilibrium (LD) analysis of C. baccatum var. pendulum cultivars on all adjacent SNP pairs within a chromosome to identify regions of high and low LD interspersed with a genome-wide average LD block size of 99.1 kb. We characterized 1742 haplotypes containing 4420 SNPs (range 9–2 SNPs per haplotype). Genome-wide association study (GWAS) of peduncle length, a trait that differentiates wild and domesticated C. baccatum types, revealed 36 significantly associated genome-wide SNPs. Population structure, identity by state (IBS) and LD patterns across the genome will be of potential use for future GWAS of economically important traits in C. baccatum peppers.
Introduction
Chile peppers (Capsicum spp.) are represented by at least 32 species, of which Capsicum annuum, Capsicum baccatum L. var. pendulum (Willd.) Eshbaugh, Capsicum chinense Jacq., Capsicum frutescens L., and Capsicum pubescens Ruiz & Pavon represent domesticated taxa (Heiser and Smith, 1953; Eshbaugh, 1980; Pickersgill, 1991; Bosland and Votava, 1999; Chiou and Hastorf, 2014). The eastern slopes of highland Bolivia are considered the origin of the Capsicum genus, which spread through the pre-Holocene Americas via dispersal by birds or through river flows. C. baccatum, with yellow spotted white flowers, is thought to have domesticated in lowland Bolivia or coastal Peru, whereas entirely white-flowered C. annuum was domesticated in Mexico (Eshbaugh, 1980; Andrews, 1984; Pickersgill, 1997; Aguilar-Meléndez et al., 2009b; Chiou and Hastorf, 2014). Within the C. baccatum complex, C. baccatum var. baccatum and C. baccatum var. pendulum represent the wild and domesticated forms of the species, respectively. C. baccatum var. pendulum extends northwards to Ecuador and southern Colombia and eastwards to south-eastern Brazil (Pickersgill, 1971).
Pepper germplasm is a valuable resource for investigating the still-unresolved question of whether similar domestication related changes occurred independently to result in parallel or convergent evolution in the domestication syndrome (Pickersgill, 2007). Because C. annuum and C. baccatum are sexually incompatible, the question cannot be resolved by crossing these genetically isolated domesticated peppers. However, genomic tools offer a plethora of opportunities to compare domestication footprints and determine whether complementary or different loci are involved (Pickersgill, 2007). C. baccatum var. pendulum is known for great variability in fruit quality traits, yield, pathogen resistance, and bioactive compounds (Yoon et al., 2006; Rodríguez-Burruezo et al., 2009; Do Rêgo et al., 2009; Eggink et al., 2014). Conventional plant breeding programs require costly investments in time, labor and land to develop improved cultivars; the application of genomic tools combined with next-generation sequencing could accelerate the genetic improvement of peppers. The use of C. baccatum and C. annuum species in interspecific breeding programs has been limited because of post-fertilization barriers.
Several studies mainly explored genetic distances and phylogenetic analysis in C. annuum (Lefebvre et al., 1993; Prince et al., 1995; Paran et al., 1998; Livingstone et al., 1999; Rodriguez et al., 1999; Patricia Toquica et al., 2003; Kim and Kim, 2005; Lefebvre, 2005; Portis et al., 2007; Aguilar-Meléndez et al., 2009a; Mimura et al., 2012; Hill et al., 2013; Nicolaï et al., 2013; González-Pérez et al., 2014). We have only a few reports of the genetic diversity and population structure of C. baccatum var. pendulum (Albrecht et al., 2011, 2012; Ibiza et al., 2012).
Genotyping by sequencing (GBS) is a reduced representation method, which utilizes next-generation sequencing to develop genome-wide single nucleotide polymorphisms (SNPs). SNPs generated by GBS have been successfully deployed for genetic diversity analysis and Genome-wide association studies (GWAS) in several crops (Poland and Rife, 2012; Narum et al., 2013; Liu et al., 2014; Nimmakayala et al., 2014, 2016; Guajardo et al., 2015; Otto et al., 2016). Increased marker density across the chromosomes facilitates to estimate genome-wide non-random association of allelic states across the chromosomes, which is known as Linkage disequilibrium (LD; Mackay and Powell, 2007; Reddy et al., 2014; Baird, 2015; Wang et al., 2015; Zanke et al., 2015). GWAS models are to scan genome-wide LD blocks to identify causal locus for trait of the interest, while involving population structure and identity by state (IBS) matrices as the cofactors to reduce spurious associations due to confounding effects of population stratification and polygenic background (Rafalski, 2010; Stich and Melchinger, 2010; Newell et al., 2011). The availability of genome-wide (SNPs) affords new opportunities in the current study to better resolve C. baccatum population structure, LD and diversity and dissect the population demographic history across the genome by comparison with another domesticated species, C. annuum. In addition, we utilized population structure analyses for a genome-wide association study (GWAS) of peduncle length, an important domestication trait.
Materials and Methods
Germplasm
A representative sample of 377 pepper accessions (283 C. baccatum and 94 diverse C. annuum accessions) collected from 32 countries across the world were obtained from the USDA-ARS, Germplasm Resource Information Network, Plant Genetic Resources Conservation Unit, Griffin, GA and World Vegetable Center (AVRDC, Shanhua, Taiwan) (Table S1). The C. annuum collection was comprised of 90 domesticated cultivars and 4 wild accessions. The C. baccatum collection had 218 lines of C. baccatum var. pendulum and 17 wild accessions (C. baccatum var. baccatum). Peduncle length (cm) was measured for 5 plants each of 217 accessions belonging to C. baccatum var. pendulum grown in a greenhouse in three replications.
Genotyping by Sequencing (GBS)
Genomic DNA was isolated from the seedlings using the DNeasy plant mini kit (QIAGEN, Germany), and GBS was as described (Elshire et al., 2011). DNA was treated with the restriction enzyme ApeKI, a type II restriction endonuclease, barcoded by accession, and sequenced on an Illumina HiSeq 2500 as described (Elshire et al., 2011). SNPs were identified using the TASSEL-GBS Discovery/Production pipeline (https://bitbucket.org/tasseladmin/tassel-5-source/wiki/Tassel5GBSv2Pipeline). Chromosomal assignment and position on the physical map of various SNPs were deduced from the C. annuum whole genome sequence (WGS) draft at http://peppergenome.snu.ac.kr. SNPs were designated by chromosome number and position (e.g., S10_172735351, which indicates an SNP located at position 172735351 on chromosome 10).
Genome-wide Divergence and Population Structure Analysis
Genetic diversity values were calculated by a neighbor-joining algorithm using TASSEL 5. In a second approach, we utilized IBS and principle component analysis (PCA) with the SNP & Variation Suite (SVS v8.1.5) (Golden Helix, Inc., Bozeman, MT, USA; www.goldenhelix.com). Observed nucleotide diversity (π) and Tajima's D were estimated by using TASSEL v5.0 with a sliding-window approach as described (Korneliussen et al., 2013). The fixation index (FST) was estimated on the basis of the Wright F statistic (Weir and Cockerham, 1984) with use of SVS v8.1.5.
Characterization of Linkage Disequilibrium (LD)
For GBS data, we considered only SNPs successfully mapped to the C. annuum WGS draft, because knowing the chromosome location of SNPs helps prevent spurious LD and thereby unreliable association mapping. Mapped SNPs were further filtered by call rate >90%. Before studying LD decay, haplotype blocks were calculated for all markers by using the default settings in SVS v8.1.5. Adjacent and pairwise measurements of LD for GBS data were calculated separately for SNPs in each chromosome. For computing LD, we used the expectation-maximization (EM) algorithm (Dempster et al., 1977) as an iterative technique for obtaining maximum likelihood estimates of sample haplotype frequencies.
GWAS Mapping
The PC matrix was constructed with the program “EIGENSTRAT” (http://genetics.med.harvard.edu/reich/Reich_Lab/) and the PCA correction technique; the method of stratification was as described (Price et al., 2006). IBS was calculated as described (Purcell et al., 2007). GWAS involved a single-locus mixed linear model (SLMM), a method that uses a forward and backward stepwise approach to select markers as fixed-effects covariates in the model (Segura et al., 2012), and implemented in SVS v8.1.5. We used a PC matrix to correct for population stratification and an IBS matrix to correct for a polygenic background. Manhattan plots for associated SNPs were visualized by using GenomeBrowse v1.0 (Golden Helix, Inc.). The SNP P-values from GWAS underwent false discovery rate (FDR) analysis (Storey, 2002).
Results
SNP Development
A total of 77,407 SNPs were isolated from the nucleotide sequence obtained for the 283 C. baccatum and 94 C. annuum accessions studied; a total of 8661, 8086, 9843, 6197, 5688, 7410, 5588, 5086, 4472, 5336, 5079, and 5961 SNPs were mapped to the WGS draft and located on chromosomes 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, and 12, respectively. We noted the presence of one SNP at every 35.6 kb across the genome, with average gap size of 31.7 kb and one SNP at every 104.4 kb in the coding regions. A total of 36,621 SNPs had minor allele frequency [MAF] ≥0.05, identified collectively for C. annuum and C. baccatum, and were used for various analyses in the current study. For C. baccatum, 13,129 SNPs had MAF ≥0.05; their chromosome distribution is listed in Table 1. In addition, we identified 26,697 SNPs located in various exons. SNP counts in exons of various genes were 2985, 3308, 3630, 2032, 1837, 2474, 1897, 1758, 1406, 1799, 1550, and 2021 on chromosomes 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 11, and 12, respectively.

Table 1. Chromosome-wise distribution 13,129 SNPs with MAF of ≥0.05 for C. baccatum collections.
Population Stratification
We used PCA of the 36,621 SNPs identified from C. baccatum and C. annuum with MAF ≥0.05 to characterize domesticated and wild C. annuum and C. baccatum peppers. PCA with first and second eigen vectors that explained 80% of the total variation produced two clusters of C. baccatum and C. annuum accessions (Figure 1). Tepin and Tepin Guatemala, two wild peppers belonging to C. annuum var. glabriusculum that are native to southern North America and northern South America, were close to CB-77, a wild C. baccatum pepper. Similarly, three other wild C. baccatum peppers, CB-93, CB-92, and CB-40, were intermediate between the major C. annuum and C. baccatum clusters. A third cluster comprised the remaining wild, semi-domesticated and crown shaped fruit type C. baccatum accessions that grouped with the domesticated large-fruited C. baccatum peppers. A separate PCA with 13,129 SNPs that were polymorphic for C. baccatum accessions resolved the population structure comprised by this group of C. baccatum accessions. This PCA identified 283 C. baccatum accessions in 3 distinct clusters (Figure 2). The middle cluster (cluster II) was parallel to the C. annuum cluster, and the wild species Tepin, Tepin Guatemala, CB-77, CB-93, CB-92, and CB-40 were found in the middle, which indicates intercrossing between wild C. annuum and C. baccatum peppers while or before domestication. PCA placement of various accessions are noted in Tables S2, S3.
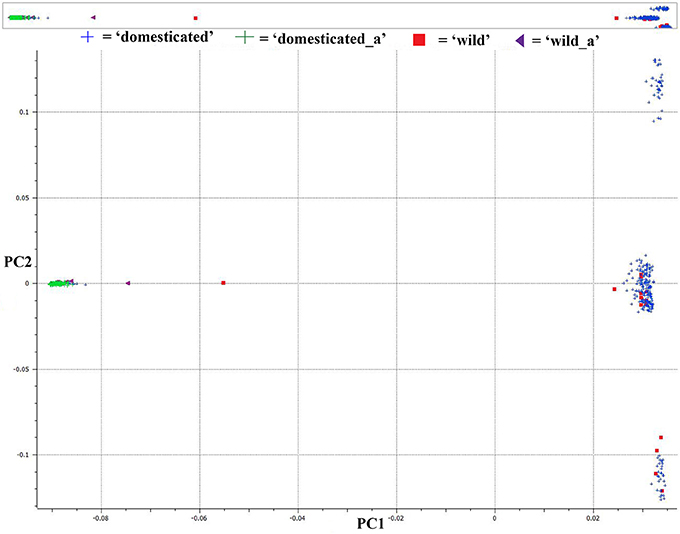
Figure 1. First and second principal component analysis (PCA) components for 36,621 single nucleotide polymorphisms (SNPs) in a set of 377 diverse pepper accessions (283 Capsicum baccatum and 94 C. annuum accessions). See Table S2 for a list of accessions and eigen values for respective positions of individual accessions in the figure.
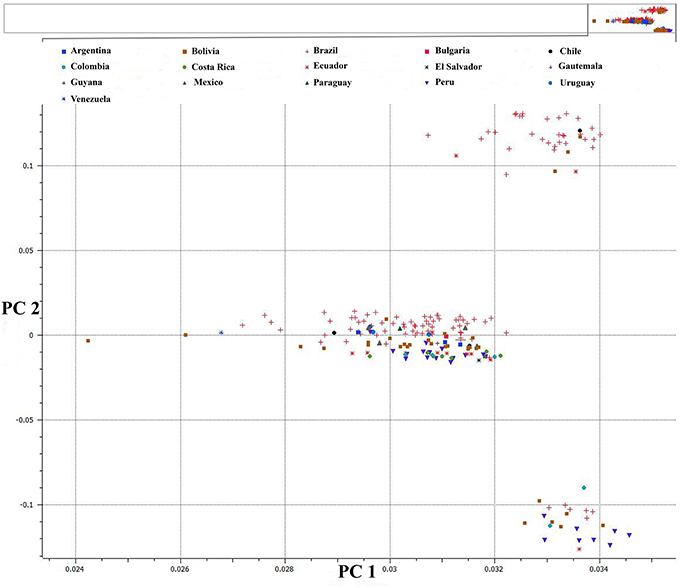
Figure 2. First and second PCA components for 13,129 SNPs within 283 C. baccatum accessions. See Table S3 for a list of accessions and eigen values for respective positions of individual accessions in the figure.
Fixation Index (FST) Distribution to Locate Positive Selection Footprints
FST was estimated with 95% confidence intervals between wild and domesticated C. annuum and C. baccatum. The FST between wild (C. annuum + C. baccatum) and domesticated (C. annuum + C. baccatum) accessions was 0.09 and 0.05, respectively. The FSTbetween domesticated C. annuum and C. baccatum was 0.78, which indicates genome-wide divergence. The FST between wild C. baccatum and wild C. annuum was 0.66. Crown-shaped fruited C. baccatum types are unique for this species group, and pairwise FST values with wild, semi-domesticated and domesticated were 0.10, 0.06, and 0.03, respectively, which indicates their closeness to domesticated types. FST-values for semi-domesticated with wild and domesticated C. baccatum types were 0.03 and 0.01, respectively. We present an overall FST distribution in a Manhattan plot for all chromosomes showing important chromosomal regions with the highest FST as peaks (Figure 3, Table S4). Based on FST values, peaks on chromosomes 1, 2, 3, 4, 5, 6, and 9 in the Manhattan plot might be the regions of positive selection and important for improvement.
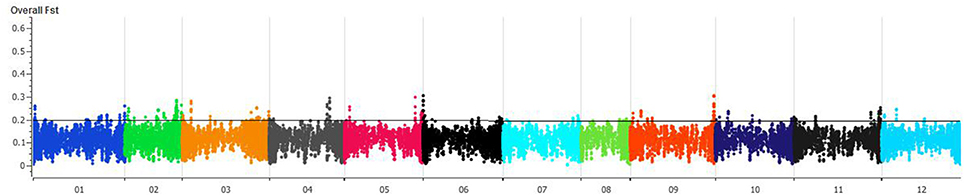
Figure 3. Manhattan plot of chromosome-wise overall fixation index (FST) values for 283 C. baccatum accessions. Individual FST-values are in Table S4.
Because of the strong population structure, we assessed patterns of variation separately for each group of domesticated accessions from the respective species when making inferences about the evolutionary dynamics of domestication. Crop domestication is often associated with “population bottlenecks” because of the limited number of founding individuals experiencing domestication events. These bottlenecks may be evident in pepper when comparing diversity between cultivated forms of C. annuum and C. baccatum. We estimated nucleotide diversity (π) and Tajima's D across various chromosomes to understand genome-wide bottleneck effects. The frequency of segregating SNPs as reflected by various chromosomal measures of mean π and Tajima's D is presented in Figure 4. For cultivated C. baccatum, chromosome 4 was positive for π and Tajima's D which indicates accumulation of rapid mutations on this chromosome. The remaining chromosomes were negative or nearly negative for Tajima's D, which indicates bottlenecks in domestication. In contrast, C. annuum chromosomes were positive for Tajima's D on all chromosomes except chromosome 8, which indicates differential evolution after the domestication or the influence of diverse breeding.
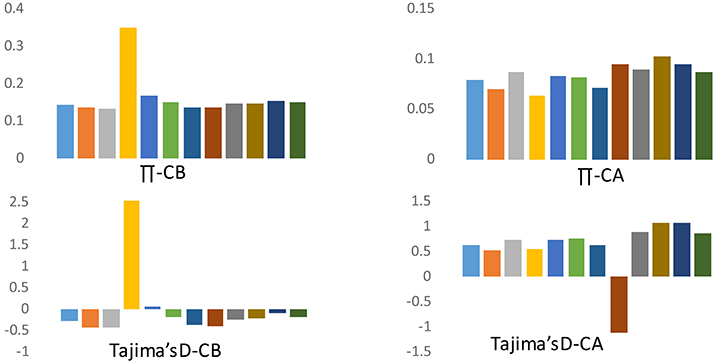
Figure 4. Frequency spectrum for chromosomal means for nucleotide diversity (π) and Tajima's D for C. annuum (CA) and C. baccatum (CB) domesticated accessions.
LD Analysis for C. baccatum
We conducted an extensive LD analysis on the entire dataset of C. baccatum accessions on all adjacent marker pairs within a chromosome or within a haplotype block. Haplotype distribution is important to understand patterns of genetic variation of C. baccatum gene pools and has a wide range of applications. The 2 major processes that shape haplotype structure are the domestication process and breeding history. We used “minimize historical recombination,” a block-defining algorithm developed by Gabriel et al. (2002). The upper confidence boundary was set to 0.98 and the lower boundary to 0.70. SNPs with MAF <0.05 were omitted. Maximum block length was set to 160 kb. The expectation maximization (EM) algorithm was used for haplotype estimation, with convergence tolerance 0.0001, and frequency threshold 0.01. Maximum EM iterations were set to 50. We identified 1742 haplotypes containing 4420 SNPs, with a range of 9–2 SNPs per haplotype (Table S5). The results provided values for both the EM algorithm (Dempster et al., 1977) and composite haplotype method (CHM; Weir and Cockerham, 1996). Squared-allele frequency correlations (r2) and LD estimate (D′) for the EM and CHM methods are in Table S6. We created LD plots by using marker-pair associations of adjacent SNPs within a chromosome, within a haplotype block, and within genes (Figure 5). The length of individual LD blocks varied among chromosomes, with regions of high and low LD interspersed (Table 2). The genome-wide average LD block was 99.1 kb. The largest LD block, of 13,021 kb, was on chromosome 11. Pairwise LD was estimated by r2 and we compared the pattern of decay at different levels. With pair-wise analysis considering adjacent SNPs across chromosomes, most SNP associations were within 50 kb (Figure 5). The second analysis based on adjacent SNPs within haplotypes revealed most associations within 20 kb (Figure S1). The third analysis of SNPs located in genes revealed most associations within 5 kb (Figure S2).
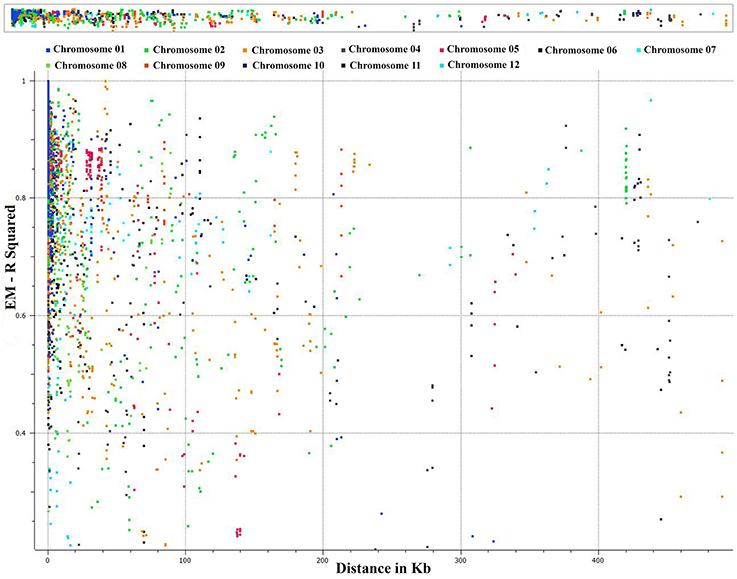
Figure 5. Genome-wide distribution of marker associations (r2) based on expectation-maximization (EM) analysis for adjacent SNPs across chromosomes showing most SNP associations (LD) decay within 50 kb.
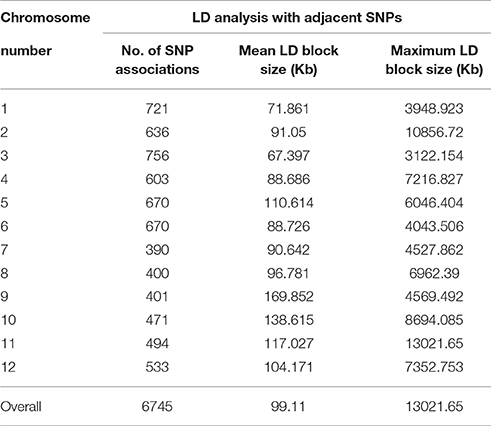
Table 2. Chromosome-wise distribution of LD blocks for C. baccatum var. pendulum.
GWAS for Peduncle Length
Peduncle length is the prime differentiating trait between wild and domesticated forms of C. baccatum. Mean peduncle lengths for respective accessions are listed in Table S7. The cultivated form of C. baccatum, var. pendulum, is named based on the epithet related to pendant fruits. In our GWAS, 36 SNPs located on chromosomes 1, 2, 3, 4, 6, 7, 8, 9, 10, and 11 were identified as significantly associated with peduncle length and cumulatively explained 21% of the total variation (Figure 6). Four SNPs located in the intergenic space between the oxidoreductase family protein/arogenate dehydrogenase on chromosome 7 explained 10.6% of the total variation. Chromosome number, map position, P-value, regression beta, FDR correction, variance explained, call rate, and minor/major allele frequencies for all significantly associated SNPs are in Table S8.
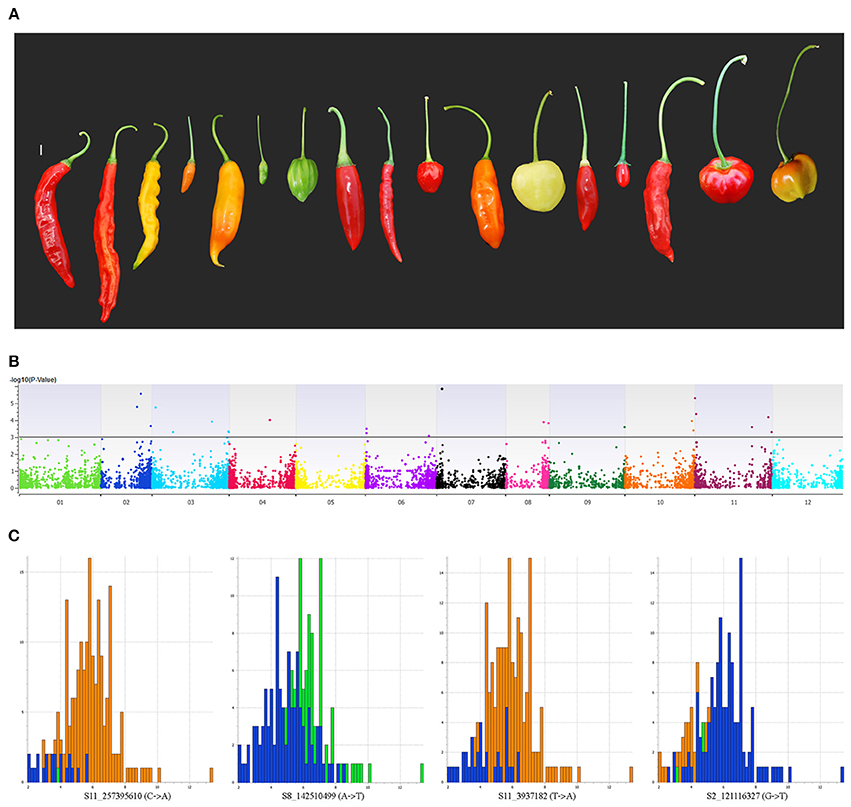
Figure 6. Manhattan plot of the genome-wide association study for peduncle length in C. baccatum var. pendulum. (A) Range of observed peduncle length. (B) Chromosome coordinates are on the X-axis, with the negative log-10 of the association P-value for each SNP on the Y-axis. High negative log-10 indicates strong association with the trait. Histograms show effects of significantly associated SNPs for peduncle length. (C) Four SNPs located in the intergenic space between the oxidoreductase family protein/arogenate dehydrogenase on chromosome 7 that explained 10.6% of the total variation for peduncle length.
Candidate Gene Selection
The predicted gene set from the annotated C. annuum cv. CM334 reference genome (Kim et al., 2014) was used to characterize the genes containing SNPs or nearby SNPs. Eleven candidate genes containing SNPs in exons or promoters were significantly associated with peduncle length, and 12 more SNPs in introns or intergenic regions of candidate genes were proposed. GWAS details and strengths of association of SNPs are in Table S8. Details of annotation for various associated SNPs, their location in various genes and type of mutation (synonymous or non-synonymous) are in Table 3.
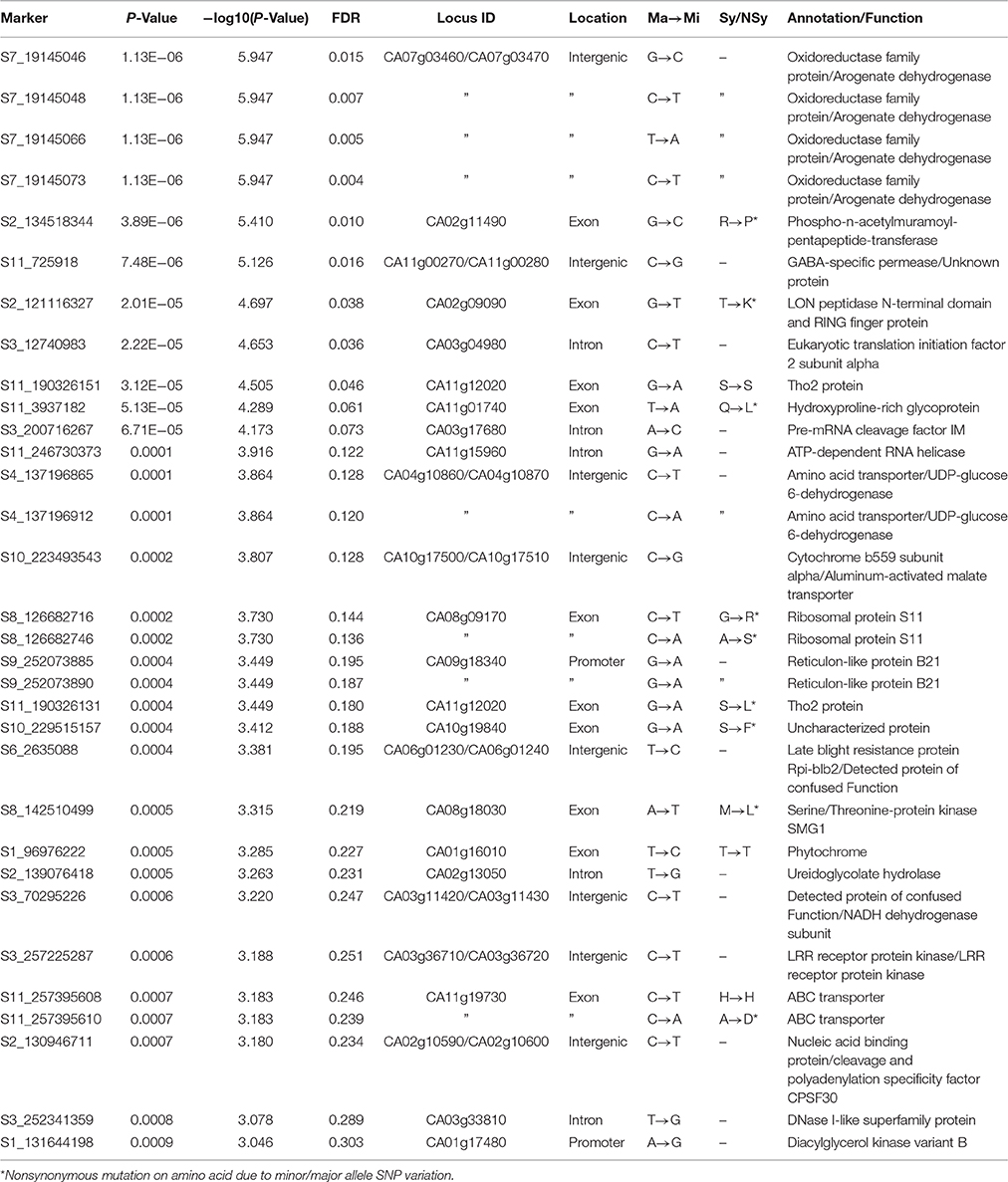
Table 3. Annotation of significantly associated SNPs for peduncle length in C. baccatum var. pendulum.
Discussion
The cultivated pepper species, C. baccatum, known as aji or Peruvian hot pepper, is a valuable source of novel genes that has not yet been analyzed for genome-wide diversity and population structure (Albrecht et al., 2012). Our genome-wide diversity analysis showed that many domesticated C. baccatum var. pendulum from western Bolivia/Peru and eastern Brazil/Paraguay cluster with most wild-type C. baccatum var. baccatum, suggesting that they may be the ancestral cluster. The flow of the river Rio Mizque from the south to join the Amazon is through lowland tropical Bolivia and the Amazon Basin and thus includes both the range of the C. baccatum group and a portion of the range of the C. annuum group (Eshbaugh, 1980). McLeod et al. (1982) suggested that the white-flowered ancestor migrated to dry areas of southern Bolivia, to produce the C. baccatum group, and the wild form in the wetter Amazon basin developed into the wild progenitor for C. annuum.
Our comparative divergence analysis across the chromosomes for C. annuum and C. baccatum revealed that chromosome 4 of C. baccatum had a unique divergence history, and for C. annuum, chromosome 8 showed a differential evolution when comparing mean π and Tajima's D for various chromosomes. In addition, biased distribution of Tajima's D toward negative values on all chromosomes (except chromosome 4) in cultivated C. baccatum indicates a population bottleneck during domestication or through the breeding histories, or the speciation of C. baccatum might have occurred with relatively narrow genetic diversity. In contrast, C. annuum chromosomes showed positive Tajima's D on all chromosomes except chromosome 8, which indicates that speciation or domestication of C. annuum might have occurred at multiple sites, contributing to wider genetic diversity as discussed by Kraft et al. (2014). Subsequent spread of C. annuum cultivars across the world and exposure to diverse breeding programs or selection in conjunction with diverse ecological adaptation might explain such rapid population size expansion and recovery from the bottleneck effects. The genome size of C. annuum types was estimated to be 3691 Mbp and C. baccatum was 4048 Mbp, which indicates wide divergence between these 2 cultivated pepper genomes (Belletti et al., 1998). Tang et al. (2006) concluded that unusually divergent genomic regions between closely related rice species are informative about species incompatibility or reproductive barriers resulting in partial fertility. Similar to the current findings, several reports implicated newly recruited polymorphisms as causing highly divergent genomic regions that may control traits associated with reproductive incompatibility or ecological adaptation (Wu, 2001; Wu and Ting, 2004).
Current advances in genome sequencing for identifying genome-wide SNPs and mapping them to WGS drafts allowed for scanning of LD decay across the genome. LD, the non-random association of alleles at different loci and germplasm panels that represent genome-wide cultivar diversity (power of association panel), plays an integral role in GWAS and determines the density of SNPs required for GWAS (Flint-Garcia et al., 2003; Nicolas et al., 2016). Low to moderate LD (decay within 100 kb) such as that observed for the C. baccatum panel in our study must utilize high SNP density (Kovi et al., 2015). In this study, we noted the highest LD for chromosome 11. One explanation for such variable LD is the “Bulmer effect,” whereby high LD regions are generally associated with selective sweeps harboring important genes underlying domestication (Bulmer, 1971; Kovi et al., 2015). The stochastic process that generates LD during selective sweeps is because of a spontaneous mutation leading to an advantageous effect or LD decays with recombination with a diverse haplotype and further segregation (Baird, 2015).
GWAS for Peduncle Length
Wild C. baccatum has a relatively restricted distribution confined to southern Peru, Bolivia, and southern Brazil (Eshbaugh, 1970). C. baccatum var. pendulum is a widely distributed cultivated plant found throughout western South America and now spreading worldwide (Eshbaugh, 1970). Wild C. baccatum has red, erect, and non-persistent fruits, and C. baccatum var. pendulum has red, orange, yellow, green, or brown fruits that are pendant and persistent. Because the peduncle is the most differentiating trait between domesticated and wild C. baccatum species, we performed GWAS for peduncle length. We associated 36 SNPs with the trait peduncle. Four of these SNPs clustered with candidate genes on chromosome 7. Annotation for some of these associated SNP-containing sequences revealed their location in various genes, so these genes might play a role in peduncle length, peduncle architecture and C. baccatum domestication.
Length of peduncle is determined by the cell number or cell size, although it is indirectly regulated by hormones and multiple pathways. Kinases play important roles in plant growth and development. Peduncle associated SNPs in the current study were located in leucine-rich repeat receptor like kinases (LRR-RLKs), serine/threonine protein kinase, ABC transporter gene and RING finger protein, which may play important roles in growth and development as well as cell wall integrity and elongation as has been shown in other plants (Lally et al., 2001; Arunyawat et al., 2007; Guo et al., 2009; Gish and Clark, 2011; Ghosh et al., 2013). Plant cell walls contain a glycoprotein component rich in the otherwise rare amino acid hydroxyproline and accumulation of this amino acid was positively correlated with cell elongation in pea epicotyls (Flint-Garcia et al., 2003). In the current study, we also associated a marker S11_725918 on GABA (γ-aminobutyric acid), a ubiquitous non-protein amino acid. An Arabidopsis GABA gene mutant pop2 exhibited defects in hypocotyl cell elongation and pollen-tube elongation via influence on cell-wall–related genes (Bulmer, 1971).
Our study describes the utility of SNPs generated by GBS for genome-wide divergence and LD patterns between C. annuum and C. baccatum. Mapping all the SNPs to the C. annuum reference genome helped to identify homologous SNPs between the two incompatible cultivated pepper genomes, which was further useful to reduce ascertainment bias, so this SNP set was useful in estimating genome-wide population differentiation and allele sharing between the two genomes. Furthermore, the SNPs anchored to the C. annuum genome may not be in the same order in the C. baccatum genome because some genomic regions may not be co-linear to the C. annuum genome because of genome rearrangements. In a comparison of C. baccatum and C. annuum linkage maps, Lee et al. (2016) identified two major reciprocal translocations between chromosomes 3 and 5 and between chromosomes 3 and 9, as well as translocations between chromosomes 1 and 8.
Such uncertain positions of SNPs can be corrected only when the whole genomesequence is available for C. baccatum genome. This SNP panel and the results pertaining to population structure, IBS and LD decay analyses will facilitate routine use of GWAS for identification of genes associated with various economically important traits in Peruvian peppers. Our identification of SNPs associated with fruit peduncle length demonstrates opportunities for utilization of GWAS in crop improvement.
Author Contributions
UR, PN, JS, GH, and AE designed the study and drafted the manuscript. PN, VA, JD, and BD conducted peduncle phenotyping. PN, VA, AA, JD, and BD extracted DNA and assisted to generate genome-wide SNPs. DC provided whole genome sequence draft and mapped SNPs to the genome. UR, PN, CR, TS, AA, and VA performed population structure and GWAS analysis.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
The study received funding from USDA-NIFA (2010-02419 and 2012-02617), NIH Grant P20RR016477 to the West Virginia IDeA Network for Biomedical Research Funding, Raman postdoctoral fellowship to CR by University Grants Commission, Government of India and the Gus R. Douglass Institute (graduate research assistantship to AV and BD). DC was supported by the Agricultural Genome Center of Next-Generation Biogreen 21 Program (PJ011275).
Supplementary Material
The Supplementary Material for this article can be found online at: http://journal.frontiersin.org/article/10.3389/fpls.2016.01646/full#supplementary-material
Table S1. List of Capsicum annuum and C. baccatum pepper accessions used in the current study.
Table S2. Eigen values for the first two components from principle component analysis (PCA) estimated for various accessions belonging to C. annuum and C. baccatum.
Table S3. Eigen values for the first two PCA components estimated for various accessions belonging to C. baccatum.
Table S4. Fixation index (FST) values for individual SNPs across chromosomes of C. baccatum genome.
Table S5. Haplotype blocks across the cultivated C. baccatum genome.
Table S6. Linkage disequilibrium (LD) analysis of adjacent SNP pairs across the C. baccatum genome.
Table S7. Phenotypic data for mean peduncle length (cm) for 217 C. baccatum var. pendulum accessions.
Table S8. Details of significantly associated SNPs as revealed by genome-wide association study.
Figure S1. LD analysis (r2) based on adjacent SNPs within haplotypes showing most associations within 20 kb.
Figure S2. LD analysis (r2) of SNPs located in genes showing most associations within 5 kb.
References
Aguilar-Meléndez, A., Morrell, P. L., Roose, M. L., and Kim, S. C. (2009b). Genetic diversity and structure in semiwild and domesticated chiles (Capsicum annuum; Solanaceae) from Mexico. Am. J. Bot. 96, 1190–1202. doi: 10.3732/ajb.0800155
Aguilar-Meléndez, A., Morrell, P., Roose, M., and Kim, S. (2009a). Genetic diversity and structure in semiwild and domesticated chiles (Capsicum annuum; Solanaceae) from Mexico. Am. J. Bot. 96, 1190–1202. doi: 10.3732/ajb.0800155
Albrecht, E., Zhang, D., Mays, A., Saftner, R., and Stommel, J. (2012). Genetic diversity in Capsicum baccatum is significantly influenced by its ecogeographical distribution. BMC Genet. 13:68. doi: 10.1186/1471-2156-13-68
Albrecht, E., Zhang, D., Saftner, R., and Stommel, J. (2011). Genetic diversity and population structure of Capsicum baccatum genetic resources. Genet. Resour. Crop Evol. 59, 517–538. doi: 10.1007/s10722-011-9700-y
Arunyawat, U., Stephan, W., and Städler, T. (2007). Using multilocus sequence data to assess population structure, natural selection, and linkage disequilibrium in wild tomatoes. Mol. Biol. Evol. 24, 2310–2322. doi: 10.1093/molbev/msm162
Baird, S. J. (2015). Exploring linkage disequilibrium. Mol. Ecol. Resour. 15, 1017–1019. doi: 10.1111/1755-0998.12424
Belletti, P., Marzachì, C., and Lanteri, S. (1998). Flow cytometric measurement of nuclear DNA content in Capsicum (Solanaceae). Plant Syst. Evol. 209, 85–91. doi: 10.1007/BF00991526
Bosland, P., and Votava, E. (1999). Peppers: Vegetable and Spice Capsicums. Oxford, UK: CABI Publishing.
Bulmer, M. (1971). The effect of selection on genetic variability. Am. Nat. 105, 201–211. doi: 10.1086/282718
Chiou, K., and Hastorf, C. (2014). A systematic approach to species–level identification of chile pepper (Capsicum spp.) seeds: establishing the groundwork for tracking the domestication and movement of chile peppers through the Americas and beyond. Econ. Bot. 68, 316–336. doi: 10.1007/s12231-014-9279-2
Dempster, A. P., Laird, N. M., and Rubin, D. B. (1977). Maximum likelihood from incomplete data via the EM algorithm. J. R. Stat. Soc. Ser. B 39, 1–38.
Do Rêgo, E., Do Rêgo, M., Finger, F., Cruz, C., and Casali, V. (2009). A diallel study of yield components and fruit quality in chilli pepper (Capsicum baccatum). Euphytica 168, 275–287. doi: 10.1007/s10681-009-9947-y
Eggink, P. M., Tikunov, Y., Maliepaard, C., Haanstra, J. P. W., De Rooij, H., Vogelaar, A., et al. (2014). Capturing flavors from Capsicum baccatum by introgression in sweet pepper. Theor. Appl. Genet. 127, 373–390. doi: 10.1007/s00122-013-2225-3
Elshire, R. J., Glaubitz, J. C., Sun, Q., Poland, J. A., Kawamoto, K., Buckler, E. S., et al. (2011). A robust, simple genotyping-by-sequencing (GBS) approach for high diversity species. PLoS ONE 6:e19379. doi: 10.1371/journal.pone.0019379
Eshbaugh, W. (1970). A biosystematic and evolutionary study of Capsicum baccatum (Solanaceae). Brittonia 22, 31–43. doi: 10.2307/2805720
Eshbaugh, W. (1980). The taxonomy of the genus Capsicum (Solanaceae). Phytologia 47, 153–166. doi: 10.5962/bhl.part.4455
Flint-Garcia, S. A., Thornsberry, J. M., and Iv, B. (2003). Structure of linkage disequilibrium in plants*. Annu. Rev. Plant Biol. 54, 357–374. doi: 10.1146/annurev.arplant.54.031902.134907
Gabriel, S. B., Schaffner, S. F., Nguyen, H., Moore, J. M., Roy, J., Blumenstiel, B., et al. (2002). The structure of haplotype blocks in the human genome. Science 296, 2225–2229. doi: 10.1126/science.1069424
Ghosh, J. S., Chaudhuri, S., Dey, N., and Pal, A. (2013). Functional characterization of a serine-threonine protein kinase from Bambusa balcooa that implicates in cellulose overproduction and superior quality fiber formation. BMC Plant Biol. 13:128. doi: 10.1186/1471-2229-13-128
Gish, L. A., and Clark, S. E. (2011). The RLK/Pelle family of kinases. Plant J. 66, 117–127. doi: 10.1111/j.1365-313X.2011.04518.x
González-Pérez, S., Garcés-Claver, A., Mallor, C., Sáenz De Miera, L. E., Fayos, O., Pomar, F., et al. (2014). New insights into Capsicum spp relatedness and the diversification process of Capsicum annuum in Spain. PLoS ONE 9:e116276. doi: 10.1371/journal.pone.0116276
Guajardo, V., Solís, S., Sagredo, B., Gainza, F., Muñoz, C., Gasic, K., et al. (2015). Construction of high density sweet cherry (Prunus avium L.) linkage maps using microsatellite markers and SNPs detected by genotyping-by-sequencing (GBS). PLoS ONE 10:e0127750. doi: 10.1371/journal.pone.0127750
Guo, H., Li, L., Ye, H., Yu, X., Algreen, A., and Yin, Y. (2009). Three related receptor-like kinases are required for optimal cell elongation in Arabidopsis thaliana. Proc. Natl. Acad. Sci. U.S.A. 106, 7648–7653. doi: 10.1073/pnas.0812346106
Heiser, C. B., and Smith, P. G. (1953). The cultivated Capsicum peppers. Econ. Bot. 7, 214–227. doi: 10.1007/BF02984948
Hill, T. A., Ashrafi, H., Reyes-Chin-Wo, S., Yao, J., Stoffel, K., Truco, M. J., et al. (2013). Characterization of Capsicum annuum genetic diversity and population structure based on parallel polymorphism discovery with a 30K unigene Pepper GeneChip. PLoS ONE 8 e56200. doi: 10.1371/journal.pone.0056200
Ibiza, V., Blanca, J., Cañizares, J., and Nuez, F. (2012). Taxonomy and genetic diversity of domesticated Capsicum species in the Andean region. Genet. Resour. Crop Evol. 59, 1077–1088. doi: 10.1007/s10722-011-9744-z
Kim, D. H., and Kim, B.-D. (2005). Development of SCAR markers for early identification of cytoplasmic male sterility genotype in chili pepper (Capsicum annuum L.). Mol. Cells 20, 416–422.
Kim, S., Park, M., Yeom, S.-I., Kim, Y.-M., Lee, J. M., Lee, H.-A., et al. (2014). Genome sequence of the hot pepper provides insights into the evolution of pungency in Capsicum species. Nat. Genet. 46, 270–278. doi: 10.1038/ng.2877
Korneliussen, T. S., Moltke, I., Albrechtsen, A., and Nielsen, R. (2013). Calculation of Tajima's D and other neutrality test statistics from low depth next-generation sequencing data. BMC Bioinform. 14:289. doi: 10.1186/1471-2105-14-289
Kovi, M. R., Fjellheim, S., Sandve, S. R., Larsen, A., Rudi, H., Asp, T., et al. (2015). Population structure, genetic variation, and linkage disequilibrium in perennial ryegrass populations divergently selected for freezing tolerance. Front. Plant Sci. 6:929. doi: 10.3389/fpls.2015.00929
Kraft, K. H., Brown, C. H., Nabhan, G. P., Luedeling, E., Luna Ruiz, J. D. J., Coppens D'eeckenbrugge, G., et al. (2014). Multiple lines of evidence for the origin of domesticated chili pepper, Capsicum annuum, in Mexico. Proc. Natl. Acad. Sci. U.S.A. 111, 6165–6170. doi: 10.1073/pnas.1308933111
Lally, D., Ingmire, P., Tong, H.-Y., and He, Z.-H. (2001). Antisense expression of a cell wall–associated protein kinase, WAK4, inhibits cell elongation and alters morphology. Plant Cell 13, 1317–1332. doi: 10.1105/tpc.13.6.1317
Lee, Y. R., Yoon, J. B., and Lee, J. (2016). A SNP-based genetic linkage map of Capsicum baccatum and its comparison to the Capsicum annuum reference physical map. Mol. Breed. 36, 1–11. doi: 10.1007/s11032-016-0485-8
Lefebvre, V. (2005). “Molecular markers for genetics and breeding: development and use in pepper (Capsicum spp.),” in Molecular Marker Systems in Plant Breeding and Crop Improvement, eds H. Lörz and G. Wenzel (Berlin; Heidelberg: Springer-Verlag), 189–214.
Lefebvre, V., Palloix, A., and Rives, M. (1993). Nuclear RFLP between pepper cultivars (Capsicum annuum L.). Euphytica 71, 189–199. doi: 10.1007/BF00040408
Liu, H., Bayer, M., Druka, A., Russell, J. R., Hackett, C. A., Poland, J., et al. (2014). An evaluation of genotyping by sequencing (GBS) to map the Breviaristatum-e (ari-e) locus in cultivated barley. BMC Genomics 15:1. doi: 10.1186/1471-2164-15-104
Livingstone, K. D., Lackney, V. K., Blauth, J. R., Van Wijk, R., and Jahn, M. K. (1999). Genome mapping in Capsicum and the evolution of genome structure in the Solanaceae. Genetics 152, 1183–1202.
Mackay, I., and Powell, W. (2007). Methods for linkage disequilibrium mapping in crops. Trends Plant Sci. 12, 57–63. doi: 10.1016/j.tplants.2006.12.001
McLeod, M., Guttman, S., and Eshbaugh, W. (1982). Early evolution of chili peppers (Capsicum). Econ. Bot. 36, 361–368. doi: 10.1007/BF02862689
Mimura, Y., Inoue, T., Minamiyama, Y., and Kubo, N. (2012). An SSR-based genetic map of pepper (Capsicum annuum L.) serves as an anchor for the alignment of major pepper maps. Breed. Sci. 62, 93–98. doi: 10.1270/jsbbs.62.93
Narum, S. R., Buerkle, C. A., Davey, J. W., Miller, M. R., and Hohenlohe, P. A. (2013). Genotyping by sequencing in ecological and conservation genomics. Mol. Ecol. 22, 2841–2847. doi: 10.1111/mec.12350
Newell, M., Cook, D., Tinker, N., and Jannink, J.-L. (2011). Population structure and linkage disequilibrium in oat (Avena sativa L.): implications for genome-wide association studies. Theor. Appl. Genet. 122, 623–632. doi: 10.1007/s00122-010-1474-7
Nicolaï, M., Cantet, M., Lefebvre, V., Sage-Palloix, A.-M., and Palloix, A. (2013). Genotyping a large collection of pepper (Capsicum spp.) with SSR loci brings new evidence for the wild origin of cultivated C. annuum and the structuring of genetic diversity by human selection of cultivar types. Genet. Resour. Crop Evol. 60, 2375–2390. doi: 10.1007/s10722-013-0006-0
Nicolas, S. D., Péros, J.-P., Lacombe, T., Launay, A., Le Paslier, M.-C., Bérard, A., et al. (2016). Genetic diversity, linkage disequilibrium and power of a large grapevine (Vitis vinifera L) diversity panel newly designed for association studies. BMC Plant Biol. 16:74. doi: 10.1186/s12870-016-0754-z
Nimmakayala, P., Levi, A., Abburi, L., Abburi, V. L., Tomason, Y. R., Saminathan, T., et al. (2014). Single nucleotide polymorphisms generated by genotyping by sequencing to characterize genome-wide diversity, linkage disequilibrium, and selective sweeps in cultivated watermelon. BMC Genomics 15:767. doi: 10.1186/1471-2164-15-767
Nimmakayala, P., Tomason, Y. R., Abburi, V. L., Alvarado, A., Saminathan, T., Vajja, V. G., et al. (2016). Genome-wide differentiation of various melon horticultural groups for use in GWAS for fruit firmness and construction of a high resolution genetic map. Front. Plant Sci. 7:437. doi: 10.3389/fpls.2016.01437
Otto, L.-G., Brassac, J., Mondal, P., Preiss, S., Degenhardt, J., and Sharbel, T. F. (2016). Use of genotyping by sequencing (GBS) in chamomile (Matricaria recutita L.) to enhance breeding. Julius Kühn Arch. 17, 453. doi: 10.5073/jka.2016.453.004
Paran, I., Aftergoot, E., and Shifriss, C. (1998). Variation in Capsicum annuum revealed by RAPD and AFLP markers. Euphytica 99, 167–173. doi: 10.1023/A:1018301215945
Patricia Toquica, S., Rodríguez, F., Martínez, E., Cristina Duque, M., and Tohme, J. (2003). Molecular characterization by AFLPs of capsicum germplasm from the Amazon Department in Colombia. Genet. Resour. Crop Evol. 50, 639–647. doi: 10.1023/A:1024429320771
Pickersgill, B. (1971). Relationships between weedy and cultivated forms in some species of chili peppers (genus Capsicum). Evolution 25, 683–691. doi: 10.2307/2406949
Pickersgill, B. (1991). Cytogenetics and Evolution of Capsicum, L. Chromosome Engineering in Plants: Genetics, Breeding, Evolution. Part, B. Amsterdam: Elsevier.
Pickersgill, B. (1997). Genetic resources and breeding of Capsicum spp. Euphytica 96, 129–133. doi: 10.1023/A:1002913228101
Pickersgill, B. (2007). Domestication of plants in the americas: insights from mendelian and molecular genetics. Ann. Bot. 100, 925–940. doi: 10.1093/aob/mcm193
Poland, J. A., and Rife, T. W. (2012). Genotyping-by-sequencing for plant breeding and genetics. Plant Genome 5, 92–102. doi: 10.3835/plantgenome2012.05.0005
Portis, E., Nagy, I., Sasvári, Z., Stágel, A., Barchi, L., and Lanteri, S. (2007). The design of Capsicum spp. SSR assays via analysis of in silico DNA sequence, and their potential utility for genetic mapping. Plant Sci. 172, 640–648. doi: 10.1016/j.plantsci.2006.11.016
Price, A. L., Patterson, N. J., Plenge, R. M., Weinblatt, M. E., Shadick, N. A., and Reich, D. (2006). Principal components analysis corrects for stratification in genome-wide association studies. Nat. Genet. 38, 904–909. doi: 10.1038/ng1847
Prince, J. P., Lackney, V. K., Angeles, C., Blauth, J. R., and Kyle, M. M. (1995). A survey of DNA polymorphism within the genus Capsicum and the fingerprinting of pepper cultivars. Genome 38, 224–231. doi: 10.1139/g95-027
Purcell, S., Neale, B., Todd-Brown, K., Thomas, L., Manuel, A. R., Bender, D., Maller, J., et al. (2007). PLINK: a tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 81, 559–575. doi: 10.1086/519795
Rafalski, J. A. (2010). Association genetics in crop improvement. Curr. Opin. Plant Biol. 13, 174–180. doi: 10.1016/j.pbi.2009.12.004
Reddy, U. K., Nimmakayala, P., Levi, A., Abburi, V. L., Saminathan, T., Tomason, Y. R., et al. (2014). High-resolution genetic map for understanding the effect of genome-wide recombination rate on nucleotide diversity in watermelon. G3 4, 2219–2230. doi: 10.1534/g3.114.012815
Rodríguez-Burruezo, A., Prohens, J., Raigón, M. D., and Nuez, F. (2009). Variation for bioactive compounds in ají (Capsicum baccatum L.) and rocoto (C. pubescens R. & P.) and implications for breeding. Euphytica 170, 169–181. doi: 10.1007/s10681-009-9916-5
Rodriguez, J., Berke, T., Engle, L., and Nienhuis, J. (1999). Variation among and within Capsicum species revealed by RAPD markers. Theor. Appl. Genet. 99, 147–156. doi: 10.1007/s001220051219
Segura, V., Vilhjálmsson, B. J., Platt, A., Korte, A., Seren, U., Long, Q., et al. (2012). An efficient multi-locus mixed-model approach for genome-wide association studies in structured populations. Nat. Genet. 44, 825–830. doi: 10.1038/ng.2314
Stich, B., and Melchinger, A. E. (2010). An introduction to association mapping in plants. CAB Rev. 5, 1–9. doi: 10.1079/PAVSNNR20105039
Storey, J. D. (2002). A direct approach to false discovery rates. J. R. Stat. Soc. Ser. B 64, 479–498. doi: 10.1111/1467-9868.00346
Tang, T., Lu, J., Huang, J., He, J., McCouch, S. R., Shen, Y., et al. (2006). Genomic variation in rice: genesis of highly polymorphic linkage blocks during domestication. PLoS Genet. 2:e199. doi: 10.1371/journal.pgen.0020199
Wang, Y., Shahid, M. Q., Huang, H., and Wang, Y. (2015). Nucleotide diversity patterns of three divergent soybean populations: evidences for population-dependent linkage disequilibrium and taxonomic status of Glycine gracilis. Ecol. Evol. 5, 3969–3978. doi: 10.1002/ece3.1550
Weir, B. S., and Cockerham, C. (1996). Genetic Data Analysis II: Methods for Discrete Population Genetic Data. Sunderland, MA: Sinauer Assoc. Inc.
Weir, B. S., and Cockerham, C. C. (1984). Estimating F-statistics for the analysis of population structure. Evolution 38, 1358–1370. doi: 10.2307/2408641
Wu, C. I. (2001). The genic view of the process of speciation. J. Evol. Biol. 14, 851–865. doi: 10.1046/j.1420-9101.2001.00335.x
Wu, C.-I., and Ting, C.-T. (2004). Genes and speciation. Nat. Rev. Genet. 5, 114–122. doi: 10.1038/nrg1269
Yoon, J. B., Yang, D. C., Do, J. W., and Park, H. G. (2006). Overcoming two post-fertilization genetic barriers in interspecific hybridization between Capsicum annuum and C. baccatum for introgression of anthracnose resistance. Breed. Sci. 56, 31–38. doi: 10.1270/jsbbs.56.31
Keywords: population structure, linkage disequilibrium, haplotyping, genotyping by sequencing, genome-wide association mapping, peduncle length
Citation: Nimmakayala P, Abburi VL, Saminathan T, Almeida A, Davenport B, Davidson J, Reddy CVCM, Hankins G, Ebert A, Choi D, Stommel J and Reddy UK (2016) Genome-Wide Divergence and Linkage Disequilibrium Analyses for Capsicum baccatum Revealed by Genome-Anchored Single Nucleotide Polymorphisms. Front. Plant Sci. 7:1646. doi: 10.3389/fpls.2016.01646
Received: 14 August 2016; Accepted: 18 October 2016;
Published: 03 November 2016.
Edited by:
Thomas Debener, Leibniz University of Hanover, GermanyReviewed by:
Clint W. Magill, Texas A&M University, USAJundae Lee, Chonbuk National University, South Korea
Copyright © 2016 Nimmakayala, Abburi, Saminathan, Almeida, Davenport, Davidson, Reddy, Hankins, Ebert, Choi, Stommel and Reddy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Umesh K. Reddy, dXJlZGR5QHd2c3RhdGV1LmVkdQ==
†This author has contributed equally to this work.