 Abirami Ramalingam1
Abirami Ramalingam1 Himabindu Kudapa
Himabindu Kudapa Lekha T. Pazhamala
Lekha T. Pazhamala Wolfram Weckwerth
Wolfram Weckwerth Rajeev K. Varshney
Rajeev K. Varshney- 1International Crops Research Institute for the Semi-Arid Tropics (ICRISAT), Hyderabad, India
- 2Department of Ecogenomics and Systems Biology, University of Vienna, Vienna, Austria
- 3School of Plant Biology and Institute of Agriculture, The University of Western Australia, Crawley, WA, Australia
The crop legumes such as chickpea, common bean, cowpea, peanut, pigeonpea, soybean, etc. are important sources of nutrition and contribute to a significant amount of biological nitrogen fixation (>20 million tons of fixed nitrogen) in agriculture. However, the production of legumes is constrained due to abiotic and biotic stresses. It is therefore imperative to understand the molecular mechanisms of plant response to different stresses and identify key candidate genes regulating tolerance which can be deployed in breeding programs. The information obtained from transcriptomics has facilitated the identification of candidate genes for the given trait of interest and utilizing them in crop breeding programs to improve stress tolerance. However, the mechanisms of stress tolerance are complex due to the influence of multi-genes and post-transcriptional regulations. Furthermore, stress conditions greatly affect gene expression which in turn causes modifications in the composition of plant proteomes and metabolomes. Therefore, functional genomics involving various proteomics and metabolomics approaches have been obligatory for understanding plant stress tolerance. These approaches have also been found useful to unravel different pathways related to plant and seed development as well as symbiosis. Proteome and metabolome profiling using high-throughput based systems have been extensively applied in the model legume species, Medicago truncatula and Lotus japonicus, as well as in the model crop legume, soybean, to examine stress signaling pathways, cellular and developmental processes and nodule symbiosis. Moreover, the availability of protein reference maps as well as proteomics and metabolomics databases greatly support research and understanding of various biological processes in legumes. Protein-protein interaction techniques, particularly the yeast two-hybrid system have been advantageous for studying symbiosis and stress signaling in legumes. In this review, several studies on proteomics and metabolomics in model and crop legumes have been discussed. Additionally, applications of advanced proteomics and metabolomics approaches have also been included in this review for future applications in legume research. The integration of these “omics” approaches will greatly support the identification of accurate biomarkers in legume smart breeding programs.
Introduction
The legume crops such as chickpea (Cicer arietinum), common bean (Phaseolus vulgaris), cowpea (Vigna unguiculata), faba bean (Vicia faba), lentil (Lens culinaris), lupin (Lupinus luteus), mungbean (Vigna radiata), pea (Pisum sativum), peanut (Arachis hypogaea), pigeonpea (Cajanus cajan), and soybean (Glycine max) have greatly contributed in providing nutrition, food security, and environmental sustainability (Graham and Vance, 2003; Varshney et al., 2013a). However, a majority of them are grown in marginal environments, subjected to abiotic (e.g., drought, heat, cold, salinity, waterlogging, heavy metal toxicities etc.) and biotic stresses (e.g., anthracnose, bean rust, bacterial blight, Fusarium wilt etc.), thus limiting their productivity (Dita et al., 2006; Varshney and Tuberosa, 2013; Rodziewicz et al., 2014). Moreover, these environmental conditions severely affect rhizobia-legume symbiosis, which contributes to ~45% of nitrogen required for agriculture (Karmakar et al., 2015). Therefore, for the development of superior varieties with enhanced stress tolerance, it is extremely important to understand stress response mechanisms in legumes which include changes in gene expression, and the consequent variations in the transcriptome, proteome and metabolome.
The availability of high throughput and cost effective next generation sequencing (NGS) platforms as well as high throughput genotyping technologies, have facilitated the generation of massive genomic data for model as well as crop legumes. These platforms have been vital in producing the genome sequence assemblies for the following legumes: adzuki bean (Kang et al., 2015; Yang et al., 2015), chickpea (Varshney et al., 2013b), common bean (Schmutz et al., 2014), Lotus (Sato et al., 2008), Medicago (Young et al., 2011), mung bean (Kang et al., 2014), pigeonpea (Varshney et al., 2012), and soybean (Schmutz et al., 2010). Furthermore, whole genome-resequencing data are also becoming readily available for mining superior alleles (Varshney et al., 2009; Lam et al., 2010). Similarly, transcriptomics/gene expression studies, using a range of platforms, have been valuable for identifying candidate genes associated with tolerance/resistance to different stresses as well as several agronomic traits (Kudapa et al., 2013; Campbell et al., 2014; Brasileiro et al., 2015).
Although QTLs/candidate genes/alleles have been used in breeding programmes, it has been observed that structural/expression variation identified at the genetic level are not always translated into the “predicted” phenotype. Additionally, mechanisms involved in stress tolerance can be complicated, e.g., the involvement of metabolites, multigenes and post-translational modifications (PTM) which cannot be investigated by genomics or transcriptomics approaches (Mazzucotelli et al., 2008; Weckwerth, 2011a). In this context, proteomics and metabolomics are promising approaches to enhance our understanding of functional molecules on specific aspects of multigene families and PTMs, instead of analyzing the genetic code (DNA) or transcript (RNA) abundance which may not correlate with their corresponding proteins (Weckwerth, 2011b; Hossain et al., 2012).
Proteomics, defined as the high-throughput study of proteins, has taken the lead in plant biological research and stress responses, especially due to the increasing number of plant genomes being sequenced and released (Pandey and Mann, 2000; Weckwerth, 2011b; Jorrín-Novo et al., 2015). In addition, advancements in mass spectrometry (MS), quantitative methods and bioinformatics approaches have allowed comprehensive identification, quantitation, validation and characterization for a wide range of proteins from specific organ/tissue/cells (Glinski and Weckwerth, 2006). The information obtained through these advanced approaches are valuable for deciphering protein structure and complex mechanisms such as enzymatic and regulatory functions of proteins coded by specific genes (Bachi and Bonaldi, 2008; Wienkoop et al., 2010a; Nanjo et al., 2011; Abdallah et al., 2012). Furthermore, proteomics approaches provide valuable information such as protein levels associated with stress tolerance, the modifications in proteomes under stress that link transcriptomics and metabolomics analyses as well as the role of expressed genes in the functionally translated regions of the genome linked to traits of interest (Kosová et al., 2011). Many proteomics based publications, especially related to plant development and other biological phenomenon such as symbiosis in legumes are available in the model legumes and Arabidopsis thaliana, as well as in some crop plants such as rice (Oryza sativa), wheat (Triticum aestivum), maize (Zea mays), soybean, tomato (Solanum lycopersicum) and tobacco (Nicotiana tabacum) (Jorrín-Novo et al., 2015).
In addition to proteomics, metabolomics is another important approach of functional genomics in which the identification and quantitation of metabolomes (collection of metabolites or small molecules) within a cell, tissue or organism produced through cellular metabolism, connects the cellular biochemical activity with the phenotype (Weckwerth, 2003). Major plant metabolomics approaches include metabolic fingerprinting, metabolite profiling and targeted analysis (Fiehn et al., 2000; Halket et al., 2005; Shulaev, 2006). Depending on the objective of study, different metabolomics approaches or a combination of approaches are applied. Furthermore, the use of MS, bioinformatics tools and softwares, allows metabolites to be measured quickly, simultaneously, in large numbers from a small amount of sample, which are spatially localized within the biological material (Bhalla et al., 2005; Patti et al., 2012). Since metabolites are closer to the phenotype, they reflect gene expressions and different regulatory processes more accurately and metabolomics is a powerful tool to study plant molecular phenotypes in response to stresses (Scherling et al., 2010; Arbona et al., 2013; Doerfler et al., 2013). For example, under abiotic stress conditions, the plant metabolism is affected due to factors such as inhibition of metabolic enzymes, shortage of substrate, extreme demand for specific compounds and a combination of these factors. Thus, the plant undergoes metabolic reprogramming to adapt to the predominant stress conditions through the production of anti-stress components such as compatible solutes, antioxidants and stress-responsive proteins (Wienkoop et al., 2008a; Obata and Fernie, 2012; Doerfler et al., 2014). In crop breeding programs, there has been great interest in using metabolites as selection biomarkers, since metabolites integrate the complex interaction between genotype and the environment (Fernie and Schauer, 2009).
With proteomics and metabolomics emerging as cutting edge functional biology disciplines for understanding plant adaptation mechanisms to stresses at cellular and developmental stages in different plant systems, there has been great interest in applying the knowledge to understand responses in different crop plants. These approaches, integrated with information obtained from genomics data, allow accurate identification of candidate genes and pathways involved in important agronomic traits that can be applied in crop breeding programs (Langridge and Fleury, 2011).
In this article different proteomics and metabolomics tools available and the use of these approaches in legumes especially to understand stress response mechanism, cellular and developmental processes and symbiosis, have been discussed. The assemblage of information and resources available for these aspects of legume proteomics and metabolomics will facilitate our understanding and utilization of these resources for legume crop improvement programs.
Proteomics Approaches and Challenges for Legume Research
Proteomics approaches have been implemented according to the objective of the study and are based on certain criteria such as (1) descriptive proteomics for classification of proteins, (2) comparative proteomics for comparison of protein profiles (genotypes, cells, organs, developmental stages, stress response, etc., (3) PTMs that determine how proteins are modified, (4) Protein-protein interactions (PPI) for identifying protein complexes and interacting networks, (5) proteinomics for studying protein structure and functional groups, and (6) translational proteomics that involves transfer of methodology and knowledge for crop improvement (Jorrín-Novo et al., 2015). Data obtained through above mentioned approaches together with bioinformatics, provide significant information on biological processes and stress tolerance mechanisms that can be applied in crop breeding programs (Salekdeh and Komatsu, 2007; Nanjo et al., 2011; Hu et al., 2015).
Furthermore, the comprehensive analysis using differential proteomics in complex systems has demanded development of new technologies to study the cell proteome. Each approach has advantages and disadvantages from both the conceptual and the methodological perspectives. Several approaches are being used to study proteomics in plants and these include electrophoresis and/or chromatography combined with chemical or metabolic labeling and MS. In the following section, we have discussed the gel-based system which is significant for differential expression profiling for studying legume stress responses, and the advantages of the technologically more advanced gel-free, quantitative systems. The application of these approaches for protein profiling in model and crop legumes for understanding stress responses have been addressed with successful examples where proteomics approaches have been utilized for the advancement of crop legumes research. In the interest of channeling the usage of more advanced approaches for legume crop improvement research, the application of more advanced approaches applied in other plant systems, have also been addressed.
Gel-Based System for Protein Differential Expression Profiling
Gel-based systems have been widely applied for protein differential expression analysis which involves the use of two-dimensional gel electrophoresis (2D-GE) or two-dimensional difference gel electrophoresis (2D-DIGE) for protein separation coupled with MS applications for the identification and quantitation of proteins (Kosová et al., 2011; Subramanian and Smith, 2013). The 2D-GE method has an advantage of resolving and visualizing thousands of spots corresponding to different molecular forms of same proteins. However, it has low sensitivity and is not suited for protein quantitation (Marouga et al., 2005; Abdallah et al., 2012). It was found that the use of fluorescent dyes in place of Coomassie can considerably increase the sensitivity and compatibility for MS analyses (Vanderschuren et al., 2013). These factors have led to the usage of a more refined gel electrophoresis method, called the 2D-DIGE which is based on pre-labeling of protein mixtures with fluorescence. Thus, 2D-DIGE is a much more sensitive method, which allows protein detection at sub-picomolar levels for highly precise quantitative proteomic studies (Marouga et al., 2005). Few studies have been reported in legumes, which employed 2D-DIGE for monitoring early response to symbiosis and pathogenesis, such as in the root proteome in Medicago (Schenkluhn et al., 2010) and parasitic infections in pea (Castillejo et al., 2012). The advantages and disadvantages of 2D-GE and 2D-DIGE have been reviewed before (see Griffin and Aebersold, 2001; Marouga et al., 2005; Abdallah et al., 2012).
For identification of proteins separated by 2D-GE, MS based techniques were found to have high accuracy, resolution and sensitivity but the sample preparation seemed to be laborious (van Wijk, 2001; Subramanian and Smith, 2013). Earlier, the usage of techniques such as matrix-assisted laser desorption ionization time-of-flight (MALDI-TOF) MS and electrospray ionization (ESI) MS with tandem MS (MS/MS) overcame the slower and less sensitive methods (Griffin and Aebersold, 2001; Griffin et al., 2001; van Wijk, 2001). In particular, the application of tandem MS methods provided sequence information for more peptides accurately for the identification of proteins and PTMs (Pandey and Mann, 2000; Bachi and Bonaldi, 2008). The use of MALDI ionization with two TOF analysers, (MALDI TOF/TOF) and liquid chromatography tandem MS (LC-MS/MS) have been fairly new technologies for protein identification that enhanced the pipeline for de novo assembly. The availability of these tools, together with the genome sequences of more plants and accessibility of sequence databases, intensified the identification of protein spots in different plants (see Jorrín-Novo et al., 2015). The most commonly used pipeline for the identification of proteins involves comparing the MS/MS spectra to the reference databases (Romero-Rodríguez et al., 2014). Although SEQUEST, MASCOT and X!Tandem are the widely used database search programs for protein identification, MassMatrix is also suited for tandem MS data (Xu and Freitas, 2009; Senkler and Braun, 2012).
Gel-Free Proteomics Tools and Approaches for Quantitation
Shot-gun proteomics is a gel-free “bottom-up” strategy, in which complex peptide fractions produced after proteolytic digestion of proteins are analyzed by LC-MS/MS. The protein identification rates are enhanced using different peptide or protein fractionation strategies (Abdallah et al., 2012). This approach allows high throughput and comprehensive investigation, by providing an overview of the organelle or cell-type proteomes (Glinski and Weckwerth, 2006). For large-scale proteomics, a multidimensional chromatography technique called multidimensional protein identification technology (MudPIT) has been used to separate complex protein samples prior to MS analyses to increase the proteome coverage and dynamic range (Washburn et al., 2001; Bachi and Bonaldi, 2008).
MS based protein quantitation strategies may include untargeted quantitation, (database dependent or database independent protein identification) or targeted absolute quantitation, which are important for biomarker discovery and protein stoichiometries in protein complexes (Wienkoop et al., 2008b; Weckwerth, 2011b; Schmidt and Urlaub, 2012). Relative quantitation is an unbiased, large-scale screening strategy, which is useful to detect proteins involved in unknown regulatory processes, protein modifications and mechanisms in systems biology. MS-based methods for protein quantitation can be label (stable-isotope in vivo or in vitro label) or non-label based (see Bachi and Bonaldi, 2008; Abdallah et al., 2012). Label based proteomics for relative quantitation consists of chemical labeling and metabolic labeling. The iTRAQ (isobaric tags for relative and absolute quantitation), is a chemical labeling approach and has been applied to study salinity stress tolerance in the phloem sap proteome of Cucumis sativus (Fan et al., 2015), leaf proteome of Vitis vinifera to understand heat stress response (Liu et al., 2014) and the root proteome of rice to investigate aluminum stress response (Wang et al., 2014). The platforms available for relative quantitation include Mass Accuracy Precursor Alignment (MAPA), ProtMax, MASS WESTERN, and PROMEX (Hoehenwarter et al., 2008; Weckwerth et al., 2014). Recently, a rapid shot-gun LC-MS approach for relative quantitation called full-scan (FS) selective peptide extraction (Selpex) was successfully used for generating a reference library of targeted peptides from leaf tissue of Medicago (Castillejo et al., 2014).
Selective reaction monitoring (SRM) or multiple reaction monitoring (MRM), which involves the use of triple-quadrupole (QQQ) MS is a quantitative, targeted and label-free approach, that complements untargeted shotgun methods due to its reliable quantitation of low abundance proteins in complex mixtures (Lange et al., 2008; Wienkoop et al., 2010b; Schmidt and Urlaub, 2012). This approach has been useful to measure a predetermined set of proteins that could constitute cellular networks or candidate biomarkers across samples consistent and reproducibly (Wienkoop et al., 2010b; Picotti and Aebersold, 2012). In Medicago, the MRM approach was applied for the absolute quantitation of sucrose synthase isoforms and N-metabolism enzymes in symbiotic root nodules as well as in the analysis of nodule metabolism under drought stress (Wienkoop et al., 2008b; Larrainzar et al., 2009).
Protein Profiling in Legumes
Proteomic studies have been carried out predominantly in Medicago to investigate stress tolerance, seed physiology, plant growth, development and symbiosis which has immense significance in agricultural research (Colditz and Braun, 2010; Jorrín-Novo et al., 2015). A majority of studies have been carried out to investigate abiotic stress responses, e.g., drought tolerance in shoots, leaves and roots using gel-based as well as non-gel based approaches (Table 1). For instance, 26 differentially expressed proteins were identified using 2D-GE and ESI-LC-MS/MS approaches from leaf samples subjected to drought stress (Aranjuelo et al., 2011). The study revealed the regulation process involved in the synthesis of amino acids pertaining to osmoregulation. Using non-gel based LC-MS/MS approaches, root nodule proteome and symbiotic nitrogen fixation was studied under drought stress in Medicago (Larrainzar et al., 2007, 2009). The former study (Larrainzar et al., 2007) not only identified a large number of nodule proteins, but also grouped the drought responsive proteins from plant and bacteroid which was helpful in understanding the mechanism in which each symbiotic member responded to drought stress. The later study (Larrainzar et al., 2009) was important for the relative quantification of root nodule proteins and absolute quantification of a key enzyme in sucrose metabolism to understand the regulation of nitrogen fixation under drought stress. In order to understand the roles of diverse LEA proteins in developing seeds of Medicago, differential profiling was effectively used to identify proteins that may have a role in desiccation tolerance and seed longevity (Chatelain et al., 2012). The study of arbuscular mycorrhizal symbiosis in Medicago roots identified differential accumulation of 96 membrane proteins which could have a role during symbiosis when compared between the mycorrhizal and non-mycorrhizal roots. The study has also identified proteins that stimulated changes in membrane trafficking (Abdallah et al., 2014).
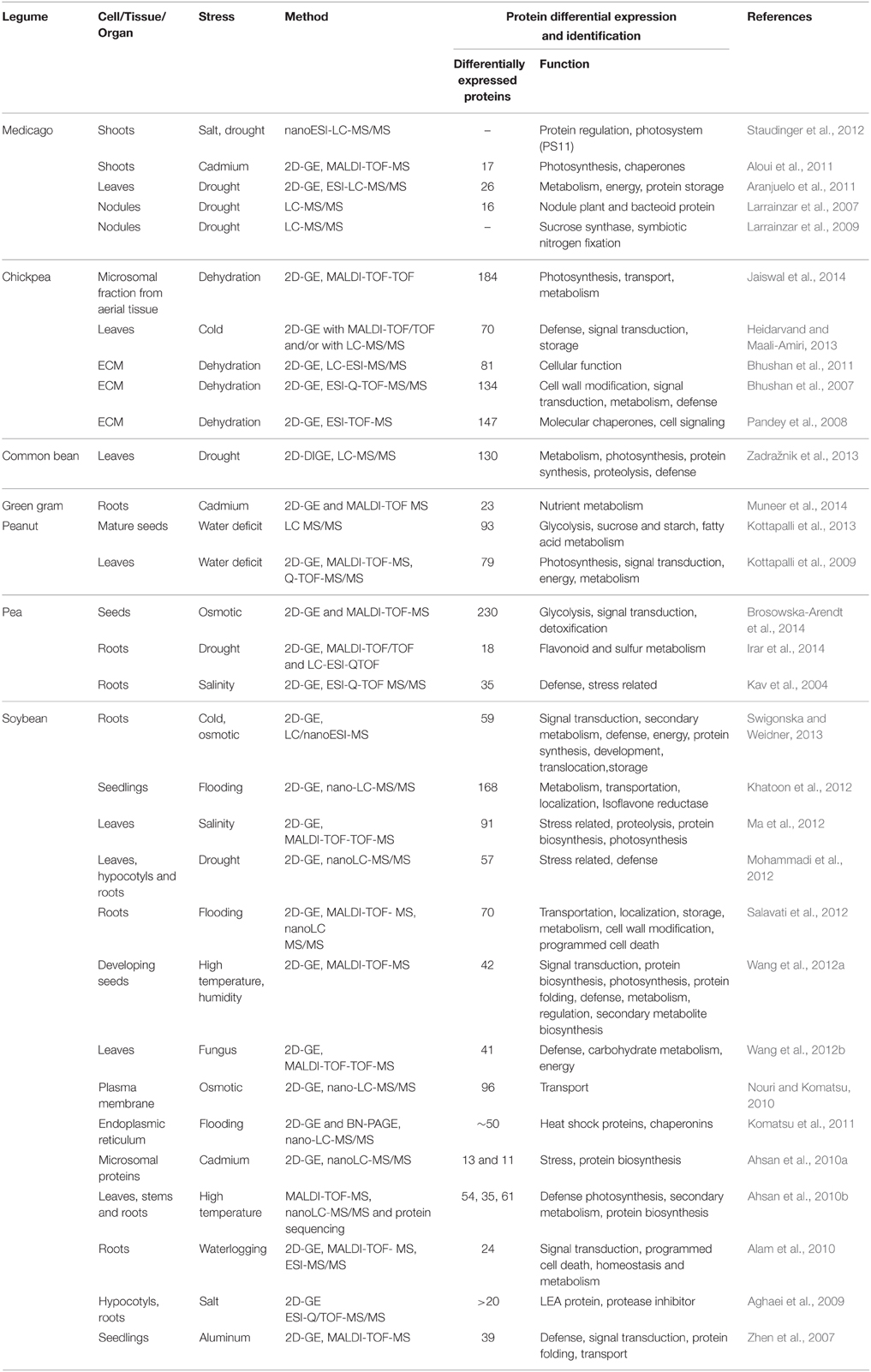
Table 1. Key studies on protein differential expression analysis in response to various stress in some model and crop legumes.
In addition, there is a significant contribution of proteomics for studying abiotic stress in soybean at subcellular, organ and whole plant levels. The methodologies applied and the major discoveries in these studies have been reviewed in a number of articles (see Hossain et al., 2013; Hossain and Komatsu, 2014a,b). A few important examples of proteomics approaches used for analyzing soybean abiotic stress are described here. For instance, osmotic stress in the plasma membrane of soybean seedling was studied using both gel-based and nanoLC MS/MS approaches (Nouri and Komatsu, 2010). While the former technique identified four up-regulated and eight down-regulated protein spots, the latter approach, identified 11 up-regulated and 75 down-regulated proteins, of which 7 were identified in both the studies. In dissecting the mechanism of Cadmium (Cd) uptake and distribution in soybean, 2D-GE and LC-MS/MS approaches were employed on contrasting soybean cultivars to identify 13 and 11 differentially expressed proteins, respectively. In this study, highly up-regulated proteins associated with lignin biosynthesis indicated that xylem lignification may be preventing the translocation of Cd (Ahsan et al., 2010a). For deciphering the mechanism of heat tolerance in soybean seedlings, 2D-GE, MALDI-TOF-MS, LC-MS/MS and protein sequencing were applied in which 54, 35 and 61 proteins were differentially expressed respectively in leaves, stems and roots, respectively (Ahsan et al., 2010b). The study showed that heat shock proteins (HSPs) and antioxidant defense related proteins were induced and identified different proteins involved in tissue specific and common defense mechanisms.
In other legume crops, there are reports of significant discoveries being made for understanding abiotic stress responses through protein profiling. In the case of chickpea, drought is one of the most important abiotic stresses that severely affect its productivity, for which efforts have been made to dissect the genetic basis of tolerance (Thudi et al., 2014; Varshney et al., 2014). Toward understanding proteomic response to dehydration stress in chickpea, a few protein profiling studies have been made available (Table 1). For instance, the investigation of a dehydration-responsive microsomal proteome with 2D-GE and MALDI-TOF/TOF identified 184 proteins that showed significant differential expression (Jaiswal et al., 2014). This study was significant in identifying a novel component involved in dehydration signaling called CaSUN1. In a different study, using 2D-GE with MALDI-TOF/TOF and LC-MS/MS, proteomic changes were identified at early stage of cold stress in chickpea leaves (Heidarvand and Maali-Amiri, 2013). The analysis indicated that energy resources and primary metabolites respond by recreating a new homeostasis in preparation for long-term cold stress adaptation. In an earlier study, 81 dehydration responsive proteins were identified from profiling analysis of the chickpea organellar proteome in two contrasting genotypes, with 2D-GE and LC-ESI-MS/MS (Bhushan et al., 2011). The study proposed that cell wall restructuring and the control of reactive oxygen species were mainly responsible for better adaptability to the stress.
A few studies have also been conducted in pea through protein profiling for understanding abiotic stress response and are being described here. For example, 2D-GE, MALDI-TOF/TOF, and LC-ESI-QTOF was employed to dissect the signaling pathway leading to inhibition of biological nitrogen fixation under drought stress (Irar et al., 2014). The study successfully identified 18 nodule proteins regulated by both pea and rhizobium genomes under drought stress and unraveling the SNF regulation machinery in nodules. In a different study, changes in protein accumulation in germinating seeds under different osmotic conditions were monitored with 2D-GE and MALDI-TOF-MS (Brosowska-Arendt et al., 2014). This work showed that under optimal conditions, proteins associated to glycolysis, fatty acids synthesis and detoxification under osmotic stress considerably decreased, while proteins involved in signal transduction and protection were accumulated. Similarly, investigation of salinity stress response in pea with 2D-GE and ESI-Q-TOF MS/MS approach revealed significant differential expression of 35 proteins, of which 10 pathogenesis-related (PR) proteins was reported for the first time to be involved in salinity stress response, possibly involving a new signal transduction pathway (Kav et al., 2004).
Protein Reference Maps in Legumes
Protein reference maps have been developed for model and crop legumes, which identify as many proteins as possible in a particular tissue or cell cultures at a given point of time. This provides useful insights into important plant processes, such as stress tolerance, nutrient uptake and symbiotic association with rhizobia (Brechenmacher et al., 2009). These resources will allow further studies for efficient proteomics applications for the crop legumes, especially through the classification and characterization of proteins related to development and stress tolerance important for unambiguous candidate gene identifications. Reference maps have been established in Medicago for cell suspension cultures, roots, leaves, stems, flowers and pods (Watson et al., 2003). A protein reference map generated for cell suspension culture of Medicago consisted of 1367 proteins with 907 unique accessions, which could identify a complete tricarboxylic acid cycle, a nearly complete glycolytic pathway, a partial ubiquitin pathway, enzymes involved in secondary metabolism through functional annotations (Lei et al., 2005). Other specialized proteome maps generated include Medicago mitochondria (Dubinin et al., 2011); cell wall (Gokulakannan and Niehaus, 2010); embryonic cell cultures generated from single protoplasts (Imin et al., 2004) and roots (Mathesius et al., 2011).
Lotus proteome reference maps for nodules and roots consist of 780 and 790 protein spots identified with 2D-GE, with 45% of the corresponding unique gene accessions common in both the tissues (Dam et al., 2014). The study showed that PTMs were more prominent in nodules rather than in roots. In addition to this, higher levels of proteins such as pathogen-related 10, heat shock, and redox processes related were found in the nodules prior to nitrogen fixation and nodulin related proteins were prevalent in mature nitrogen fixing nodules. In Lotus proteome reference maps were also generated for pod and seed development (Nautrup-Pedersen et al., 2010). Similar reference maps were also developed in crop legumes such as soybean and peanut. In soybean, a comprehensive proteome reference map was generated with 5702 proteins identified for a single root hair cell (Brechenmacher et al., 2012). A previous reference map in root hair cells identified 1492 proteins (Brechenmacher et al., 2009). Other proteome maps generated in soybean includes root apex and differentiated root zone (Mathesius et al., 2011); seed filling (Hajduch et al., 2005), leaf (Xu et al., 2006), and response to pathogen invasion (Mithöfer et al., 2002). Similarly, leaf proteome reference map was developed in peanut (Katam et al., 2010). In chickpea, reference maps were generated for understanding the complexity of plant nuclear proteins (Pandey et al., 2006) and membrane proteins (Jaiswal et al., 2012). In the case of pea, reference maps have been developed for vegetative tissues (Schiltz et al., 2004) and mature seeds (Bourgeois et al., 2009).
Proteome reference maps generated through gel-based approaches consist of gel images in which selected “spots” were linked with protein identity information with arrows and numbers (Senkler and Braun, 2012). However, web-based resources with interactive features of the reference maps have also been made available. Examples of these resources include the “Seed Proteome,” the “Rice proteome database,” and the “Arabidopsis seed proteome.” Recently, for constructing proteome reference map, a software tool called “GelMap” was developed (Rode et al., 2011). This tool was used for generating a 3D GelMap of Arabidopsis complex 1 in which its unique proteins constituents has been defined (Peters et al., 2013).
Legume Databases for Proteomics Analyses
Various databases have been developed that store a large resource of plant proteins from the proteome reference maps of legumes and other plants, such as the plant proteomics database PROMEX (Hummel et al., 2007; Wienkoop et al., 2012). A continuously growing database, PROMEX consists of 116,364 tryptic peptide product ion spectra entries of 48,218 different peptide sequence entries from Lotus, Medicago, common bean and soybean as well as other plants such as Arabidopsis, rice, etc. The database could be searched for whole experiments with an experimental ID, meta-information and single proteins and their corresponding peptide reference spectra. Furthermore, new LC-MS/MS analyses can be searched against this spectral library (http://promex.pph.univie.ac.at/promex/). LegProt (http://bioinfo.noble.org/manuscript-support/legumedb) is a legume specific protein database consisting of amino acid sequences translated from predicted gene models and 6-frame translations of tentative consensus sequences expressed sequence tags (ESTs) (Lei et al., 2011). The ProteomeXchange contains the dataset of seed phosphoproteins from Lotus (PXD000053, http://proteomecentral.proteomexchange.org/cgi/GetDataset?ID=PXD00053) and has been valuable for understanding the regulatory mechanisms of seed germination in legumes (Ino et al., 2014). This dataset contains a total of 721 phosphopeptides from 343 phosphoproteins in cotyledons and 931 phosphopeptides from 473 phosphoproteins in hypocotyls.
The Medicago PhosphoProtein Database (MPPD, http://phospho.Medicago.wisc.edu), contains 3457 unique phosphopeptides with 3404 non-redundant sites of phosphorylation on 829 proteins. This database represents the most comprehensive Medicago phosphorylation data, which allows browsing of identified proteins, searching proteins of interests, in addition to conducting BLAST searches of the database using peptide sequences and phosphorylation motifs as queries (Rose et al., 2012a). The Soybean Proteome Database (SPD, http://proteome.dc.affrc.go.jp/Soybean/) consists of a repository of functional analysis of abiotic stresses (flooding, drought, and salt). In total, it consists of 23 reference maps of soybean and proteins collected from several organs, tissues, and organelles of soybean (Ohyanagi et al., 2012). Recently, another database for storage, allergen, and anti-nutritional proteins from cultivated soybean called Soybean Protein Database (SoyProDB; http://bioinformatics.towson.edu/Soybean_Seed_Proteins_2D_Gel_DB/Home.aspx) has also been developed (Tavakolan et al., 2013). In the case of yellow lupin (Lupinus luteus L.), a seed-protein catalog has been developed. In this study, 736 proteins corresponding to 152 unique proteins have been deposited in the WORLD-2DPAGE repository (http://world-2dpage.expasy.org/repository/0066/; Ogura et al., 2014).
Post-Translational Modifications (PTMs) in Legumes
PTMs are required for the functionality of proteins that regulate processes and the subcellular localization that could be analyzed using proteomics approaches (Pandey and Mann, 2000; Seo and Lee, 2004; Downes and Vierstra, 2005). Although mostly identified through MS approaches, PTM analyses are not straightforward as protein identifications, the reason being peptide analyses do not show the expected molecular mass and therefore more protein samples are required (Pandey and Mann, 2000). However, advancement in phosphoproteomics procedures, e.g., the enrichment of phosphopeptides using immobilized metal affinity chromatography (IMAC) or proteins with aluminum hydroxide or titanium dioxide called metal oxide affinity chromatography (MOAC) (Wolschin et al., 2005) have improved the identification efficiency (Ndassa et al., 2006; Chen et al., 2010; Hoehenwarter et al., 2013; Beckers et al., 2014). Among the PTMs, phosphorylation is the major post-translational regulatory processes in all eukaryotes followed by ubiquitin and SUMO (Small Ubiquitin-like MOdifier) conjugations (Mazzucotelli et al., 2008).
The root proteome of the Medicago genotype, Jemalong A173457, revealed unique phosphopeptides that covers 3404 non-redundant sites of in vivo phosphorylation on 829 proteins (Grimsrud et al., 2010). The large scale phosphoproteomic study identified multiple sites of phosphorylation on a number of crucial proteins in rhizobial symbiosis initiation such as SICKLE, NUCLEOPORIN133 and INTERACTING PROTEIN OF DM13. Further, the rapid Nod-factors (NF) induced changes in the phosphorylation levels of 13,506 phosphosites in 7739 proteins was recently measured for rhizobia-legume symbiosis in Medicago which was found useful for quantifying phosphorylation, specifically associated with NF-signaling (Rose et al., 2012b).
In chickpea, a differential phosphoproteomic study in response to dehydration stress identified 91 phosphoproteins that are likely to be involved in cell defense, photosynthesis, photorespiration, molecular chaperones and ion transport. The study also identified multiple sites of phosphorylation in key regulatory and functional proteins (Subba et al., 2013). Recently, a nucleus-specific phosphoproteome map of 107 identified phosphoproteins was constructed in chickpea, which identified a collection of phosphoproteins involved in many cellular functions such as protein folding, signaling, gene regulation, DNA replication/repair/modification, metabolism, etc. (Kumar et al., 2014). In an earlier study, the PTMs of αAI (α-amylase inhibitor) were compared among transgenic pea and chickpea expressing αAI from common bean, with the processed form of the protein from several bean varieties. The αAI proteins displayed microheterogeneity due to differences in glycan addition frequency, variation in glycan processing and differences in C-terminal exopeptidase activity (Campbell et al., 2011). Thus, PTMs in common beans were also investigated on three seed defensive proteins αAI-1, αAI-2, and arcelin-5 (Young et al., 1999). The data showed that the proteolytic cleavage is required for the activation of the proteins, which resulted in the loss of the terminal Asn residue in αAI-1, and a minimum of seven residues from the C-termini of all three proteins. Additionally, a significant difference in the glycosylation patterns of αAI-1 and αAI-2 has been reported, although the proteins showed high sequence homology. Similarly in pea, the presence of multiple trypsin inhibitors (TI) isoforms were attributed to PTMs and particularly post-translational processing at the C-terminus during the desiccation stage of seed development resulted in the appearance of TI isoforms in pea (Domoney et al., 1995).
Histone modifications and histone variants are known to be vital for various biological processes. In this context, variants of soybean histone, H3 and H4 and their PTMs were reported, which revealed several distinct variants of soybean histone and their modifications that were different from Arabidopsis (Wu et al., 2009). The study thus, provides important biological information toward understanding histone modifications and their functional relevance in legumes.
This area of research possesses great promise for legume improvement as it is supported by advanced proteomics technologies, in particular, developments in the strategies for detection and selective isolation of proteins with known function. As discussed above functional properties of proteins are often regulated by PTMs of proteins and the numerous techniques developed can be applied to the global identification of PTMs and their processing sites in legumes.
PPI Approaches and Application in Legume Research
PPIs provide functional knowledge about proteins by analysing the interacting partners and interactomics, which is the large-scale study of PPI networks using high throughput methods. This provides valuable insights for understanding cellular function, metabolism and signaling mechanisms (Braun et al., 2013; Stasi et al., 2015). Interactomes (maps of PPI) are constructed on the basis of experimental data and computational prediction of interactions (Stasi et al., 2015). Approaches used in PPI analyses and mapping can be genetic, biochemical or proteomics-based. The strategies involve direct interactions with binary methods such as yeast two-hybrid (Y2H) or split ubiquitin, analyses of protein complexes with co-immunoprecipitation or affinity purification followed by MS (AP-MS), imaging and database analyses (Pandey and Mann, 2000; Fukao, 2012; Braun et al., 2013; Stasi et al., 2015). The identification of minor PPIs are also essential for studying transient interactions and proteins of low-abundance, which may be functionally relevant (Fukao, 2012). In recent years, many proteins have been identified with advances in MS technologies and large-scale proteomics, however, the consequence of this is the increased number of false positive protein identifications. Therefore, for the accurate appraisal of PPI identification, the inclusion of independent experimental validation is required (Fukao, 2012). Y2H system is sensitive in detecting transient and unstable interactions, and is suitable for PPI mapping and generating high-throughput data with a fine resolution to understand cellular process at the systemic level. However, it has limitations with incidences of false-positives and false-negative interactions. The PPIs are usually validated with techniques such as pull-down assays, co-immunoprecipitation, in-situ hybridization (von Mering et al., 2002; Parrish et al., 2006; Brückner et al., 2009).
Although limited PPI based studies have been undertaken in legumes, the studies involving stress signaling networks, nodule formation and symbiosis have provided valuable information in understanding these processes. The Y2H system has been mostly used in model legumes to study symbiosis and nodulation mechanisms and in the crop legumes mainly to study stress responses (Table 2). For example in pea, using the Y2H system and validation using in planta co-immunoprecipitation, the PPI observed among the heterotrimeric G proteins and GPCR protein were shown to be relevant in the salt and heat stress signaling pathways (Misra et al., 2007). In another study, the exposure to salt and cold simultaneously stimulated the expression of genes encoding a protein kinase (PsCIPK) and a calcineurin B-like protein (PsCBL) in pea (Mahajan et al., 2006). Although immunofluorescence and confocal microscopy showed that the PsCBL was localized in the cytosol and PsCIPK in the outer membrane, Y2H analysis indicated that both the protein products interacted and this was supported with Western blots. In chickpea, a few candidate interactors were identified for the 1R-MYB for drought tolerance with the Y2H system (Ramalingam et al., 2015).
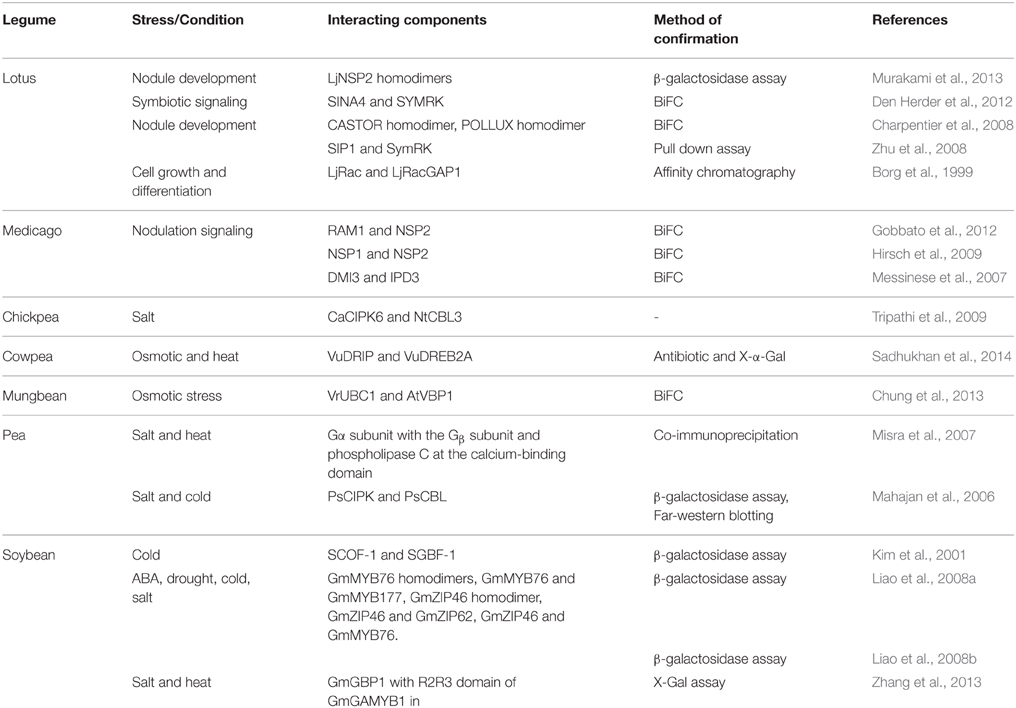
Table 2. PPI analyses in some model and crop legume using Y2H and validated with other approaches.
Bimolecular fluorescence complementation (BiFC) uses the yellow fluorescent protein (YFP), split into two-non-overlapping N-terminal (YN) and C-terminal (YC) fragments, where each fragment is cloned in-frame to the gene of interest, allowing the expression of fusion proteins. PPI detected in planta causes the detection of yellow fluorescence, which is not found for non-interacting pairs or non-fused YN/YC (Bracha-Drori et al., 2004). Although identification of PPI by screening a cDNA library is usually performed in yeast, BiFC technology was applied to screen an Arabidopsis cDNA library against a bait protein in planta since subcellular compartmentation and protein modifications differ between plant and yeast cells (Lee et al., 2012).
A challenging area in crops research would be the development of a proteome-wide PPI maps to understand the complex biological pathways and cellular networks. A high-throughput, Y2H system (HTP-YTH), suitable for mapping PPIs was described by Fang et al. (2002) to screen PPI in plants. This system involves a yeast gap-repair cloning and a selectivity that reduces false positives and negative clones with automation in laboratory procedures. This system has been used to study the defense signal transduction pathway in rice, where more than 100 genes were selected as “baits” for HTP-YTH screening in which many known and novel PPIs were identified. In Arabidopsis, the first binary PPI map for the interactome network of plants was developed (Arabidopsis Interactome Mapping Consortium, 2011). The construction of these maps in legumes should facilitate system biology approaches and will greatly benefit legume crop improvement programs.
Metabolomics Approaches in Legumes
Plants synthesize specialized metabolites that define the biochemical phenotype of a cell or tissue and can be viewed as the end products of gene expression (Sumner et al., 2003). Quantitative and qualitative measurements of cellular metabolites provide a broad view of the biochemical status of an organism that could be used to monitor and assess gene function (Fiehn et al., 2000). Furthermore, metabolomics contributes significantly to the study of stress biology by identifying different compounds such as by-products of stress metabolism, stress signal transduction molecules, molecules that are part of plant acclimation process etc. (Weckwerth, 2003; Larrainzar et al., 2009). The identified metabolic compounds could be further studied by direct measurement or correlating with the changes in transcriptome and proteome expression and confirmed by mutant analysis. In this section, recent applications of metabolomics approaches in the area of legume development, symbiosis and stress response are discussed.
Metabolomics is a rapidly developing technology and at present metabolic fingerprinting and metabolite profiling approaches are being used. For wide coverage of the vast range of metabolites present, several analytical techniques involving separation and detection are implemented (Scherling et al., 2010; Doerfler et al., 2013, 2014). The separation technique is selective for certain groups of metabolites which includes gas chromatography (GC) for volatile and primary metabolites such as sugars and amino acids (Weckwerth, 2011a), LC for mainly secondary metabolites (Scherling et al., 2010; Weckwerth, 2011a), capillary electrophoresis (CE) for ionic metabolites to be separated (Soga et al., 2003; Soga, 2007) and ultra-performance liquid chromatography (UPLC). UPLC is a powerful technique which has high resolution, sensitivity and throughput than conventional high performance liquid chromatography (HPLC). The MS analysers have been commonly used for metabolite profiling, particularly those that provide accurate mass measurements such as FTICR_MS, Orbitrap-MS or TOF-MS due to its fast scan times with improved deconvolution, run times and high mass accuracy (Scherling et al., 2010; Weckwerth, 2010, 2011a; Obata and Fernie, 2012; Arbona et al., 2013; Doerfler et al., 2013, 2014). GC-MS has been widely used in plant metabolomics research and electron impact (EI) supports strong interfacing of GC with MS which allows fragmentation patterns to be highly reproducible. Major metabolomics approaches and their applications in legumes are described below and are summarized in Table 3.
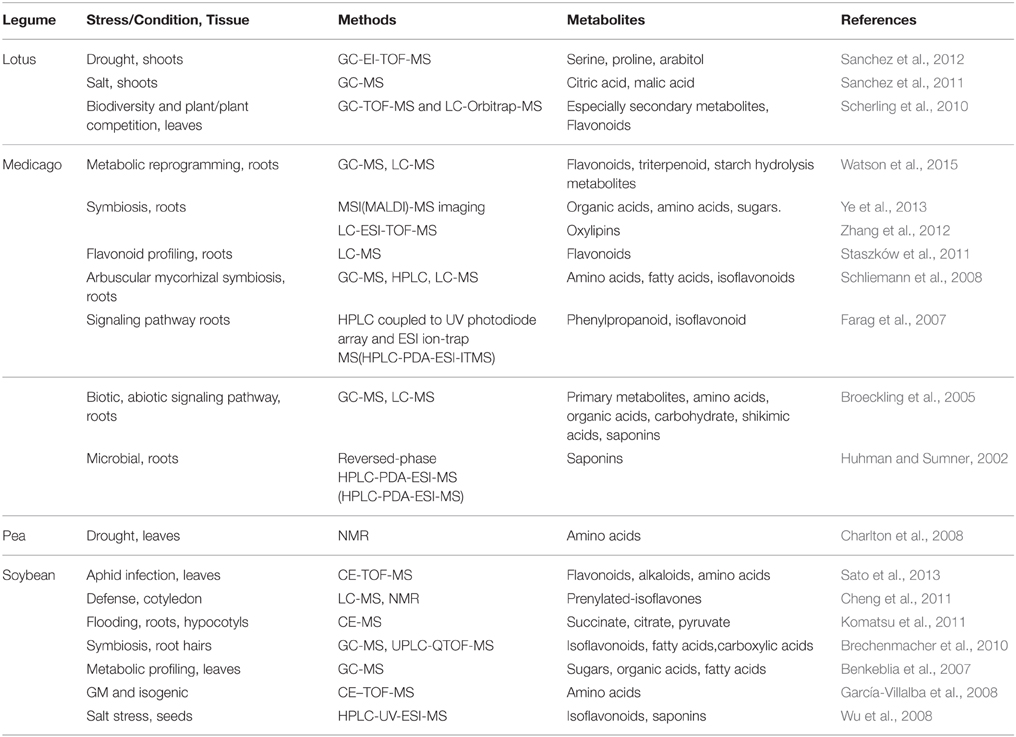
Table 3. Application of metabolomics approaches in stress, development and symbiotic processes in some legumes.
Metabolite Profiling
Metabolite profiling is the simultaneous measurement of all or a set of metabolites in a given sample. Several analytical techniques such as nuclear magnetic resonance (NMR), GC-MS, LC-MS, capillary electrophoresis–MS (CE-MS) and Fourier transform infrared (FT-IR) spectroscopy have been reported for analysing the data from metabolite profiling (Sumner et al., 2003; Weckwerth, 2003; Shulaev, 2006). The advantages and disadvantages of each technique for metabolite profiling have been discussed previously (Fiehn et al., 2000; Roessner et al., 2000; Sumner et al., 2003; Weckwerth, 2003; Weckwerth et al., 2004a,b; Shulaev, 2006). The use of metabolic profiling has been limited in crop legumes but this approach has been successfully demonstrated in model legumes. For example, in Medicago, untargeted quantitative MS approach was used to profile metabolites treated with rhizobial Nod factors to study the metabolic changes between the symbionts (Zhang et al., 2013). The study showed decrease in concentration of (9)-HODE class of oxylipins upon Nod factor treatment in planta and together with jasmonic acid inhibited Nod factor signaling. This suggests an important role for the oxylipin pathway in Nod factor signaling in symbiosis. In a different study, under early salt and drought stress conditions, the involvement of certain metabolites in nutritional priming through symbiotic interaction of nodulated plants and N-fertilized Medicago has been reported (Staudinger et al., 2012).
Similarly, to understand drought acclimation in model and forage legumes, a comprehensive and progressive reprogramming of metabolic pathways were suggested for increased water stress in Lotus (Sanchez et al., 2012). Using GC coupled to electron impact ionization (EI)-TOF-MS (GC-EI-TOF-MS), this study reported gradual increase in most of the soluble molecules profiled. In addition, comparative metabolomics between the Lotus species showed the presence of metabolites that were conserved and unique in response to drought stress. Metabolite profiling using a combination of ionomic and GC-MS was conducted for the shoots of extremophile Lotus species, adapted to highly saline coastal regions and was compared with that of cultivated glycophytic grassland forage Lotus species, to understand salt tolerance mechanisms. The extremophile Lotus species was identified to have higher salt levels with a differential rearrangement of shoot nutrient levels upon salt exposure (Sanchez et al., 2011). In a similar study, the accumulation of alanine under anoxic conditions was examined in Lotus, which is highly tolerant to water logging (Rocha et al., 2010). High accumulation of succinate, alanine and the direct co-substrates for alanine synthesis, glutamate and gamma aminobutyric acid (GABA) in the roots of Lotus during water logging was reported. Whereas, majority of amino acids that are derived from TCA cycle intermediate were found to be decreased, which support earlier findings that the metabolic equilibriums are expected to drive the metabolic flux from glycolysis, via alanine synthesis and oxoglutarate to succinate, which prevents the accumulation of pyruvate activating fermentation leading to ATP production by succinyl- CoA ligase. In another study in Lotus, GC-TOF-MS (primary metabolism) and LC-Fourier-Transformation-MS (secondary metabolism) was applied to study plant/plant competition responses in a large biodiversity experiment (Scherling et al., 2010). Significant effects in Lotus were not associated with primary metabolism (sugars, amino acids, organic acids) but with the secondary metabolism. A significant gradient of several putative flavonoids structures showed a high correlation to increasing biodiversity in the close environment of the individual plant (Scherling et al., 2010).
As described above, plants show a variety of metabolic responses against varied abiotic stresses. With the availability of genomics platforms, scientists are now able to opt for metabolomics for studying metabolites involving non-biased approaches. However, it would be important to study if there are any common metabolic responses associated with all the abiotic stresses or if the responses are specific to the stress. In this context, Komatsu et al. (2011) identified 81 metabolites related to the mitochondria under flooding stress in roots and hypocotyls of soybean using CE-MS which showed that the TCA-cycle-related metabolites, glycolysis related metabolites, GABA, pyruvate, NAD, NADH and amino acids increased, while ATP decreased. This kind of accumulation/increase in GABA and amino acids was also reported in Lotus unlike TCA cycle related metabolites were found to be low in Lotus and high in soybean. These studies support that integrative analysis is required on the metabolite accumulation within the context of understanding metabolic responses to stress.
Phosphorous is an essential component of energy metabolism, signaling molecules, and structural macromolecules. Therefore, studies have been conducted to understand the role of phosphorus in stress response metabolite profiling of common bean roots and nodules under P starvation (Hernández et al., 2007). Increase in the levels of most of the amino acids and several sugars was reported in P-stressed roots. It was suggested that the accumulation of sugars may be partitioned preferentially to P-stressed roots to support the expression of P stress-induced genes. On the other hand reduced amounts of organic acids are reported in P-starved roots that likely reflect exudation of these metabolites from the roots into the rhizosphere (Hernández et al., 2007). The metabolic response of P-starved nodules is in contrast to that of P-starved roots in common bean. It was reported that amino acids, N-containing metabolites and sugars were decreased, while organic acids were accumulated in P-deficient nodules (Hernández et al., 2009). Such contrasting response may be due to the suppression of N supply from fixed N2 under environmental limitations such as P-starvation in nodules. Recently, metabolite profiling study in chickpea revealed 49 primary metabolites in contrasting salt stress responsive cultivars (Dias et al., 2015).
One drawback with the metabolite profiling is that the data obtained is not sufficient to determine the regulation mechanisms of the pathways of interest. In order to overcome this issue, integrated analysis of metabolomics data together with that of proteomic and transcriptomic data need to be performed. For example in Medicago, an integrated metabolomics and transcriptomic approach was found useful to study metabolic reprogramming of the border cells in roots through cumulative and pathway specific datasets (Watson et al., 2015). This integrative approach showed that there were significant differences in the levels of phytohormone, supported by variation in lipoxygenases and auxin responsive transcripts in the border cells and root tips. Additionally, this approach identified metabolic resources for growth and development redirected to the border cells for the accumulation of specialized metabolites that were defense and symbiosis related. Similarly, Komatsu et al. (2011) used an integrative proteomic and metabolomics approach which was useful in identifying the expression and regulation of components linked to flooding stress in soybean seedlings. Larrainzar et al. (2009) integrated GC-TOF-MS metabolite profiling with untargeted and targeted proteomics to reveal nodule metabolic responses under drought stress and recovery by re-watering in Medicago. Metabolite profiling was able to show a highly pronounced reprogramming of metabolism during drought response and the ability of the nodules to recover completely after re-watering.
Targeted and Untargeted Metabolite Analysis
Metabolites are analyzed either using a targeted or untargeted method (Patti et al., 2012). The targeted method is used to analyse a specified set of metabolites which targets one or more pathways of interest and involves the setup of selected reaction monitoring methods of the standard compounds of the metabolite of interest, followed by the extraction of metabolites from the sample and analysis. The data obtained provides quantitation based on standardized methods for the metabolites of interest. This method has been widely used to follow the dynamics of a limited number of metabolites known to be involved in a particular stress and also for comparative metabolite profiling of a large number of known metabolites. For example, in a single chromatographic run, highly parallel targeted assays based on SRM can be used for sensitive simultaneous analysis of over 100 metabolites (see Bajad and Shulaev, 2007). Alternatively, quantitative profiling may provide in vivo enrichment of metabolites with stable isotopes like C-13 and N-15. This can be possible only by growing plants or plant cells in liquid media containing N-15-labeled inorganic nitrogen sources or C-13-labeled carbon dioxide or glucose (Hegeman et al., 2007; Huege et al., 2007). Metabolic labeling combined with MS has been successfully used for quantitative metabolic profiling in microorganisms (Mashego et al., 2004; Lafaye et al., 2005; Wu et al., 2005).
On the other hand, the untargeted metabolite profiling is often used for global and broader applications, e.g., understanding cellular metabolism. In this approach, metabolites are isolated from samples followed by the LC-MS analysis. The data obtained is processed using bioinformatics and values for peaks of interest are searched against metabolite databases for possible identity. These metabolite identities are confirmed using tandem MS (MS/MS) data and retention time data compared with standard components. Untargeted metabolite profiling in Lotus demonstrated a major and reproducible change of the metabolic phenotype in the course of salt acclimatization, which was most evident for amino acid, sugars and organic acid metabolism (Sanchez et al., 2008). Accumulation of amino acids and other nitrogen-containing compounds is a remarkable biochemical feature of almost all plant stress responses reported so far. However, the main disadvantage of untargeted profiling is that it is a semi-quantitative method and provides relative concentration data based on the same “surrogate” internal standard. These semi-quantitative data have to be further validated using targeted quantitative assays.
Metabolic Fingerprinting
Metabolic fingerprinting is mainly used to identify metabolic signatures, for example, finding patterns associated with a particular stress response without precise quantitation/identification of different metabolites in the given sample. Features specific to a fingerprint can be identified using a variety of pattern recognition and multivariate statistical analysis (e.g., principal component analysis (PCA), self-organizing maps (SOMs) and hierarchical clustering, discriminant function analysis (DFA), ANOVA etc.) on the data (Sumner et al., 2003). Different analytical techniques, including NMR spectroscopy (Krishnan et al., 2005), MS (Goodacre et al., 2003), and FT-IR (Johnson et al., 2003) may be used to perform metabolic finger printing. Of these, MS is advantages over NMR spectroscopy because of the low sensitivity of NMR, which makes it difficult to detect low abundance cellular metabolites. On contrary, MS has high resolving power compared to NMR, providing higher sensitivity and lower detection limit but generates more complex spectrum because of its results in the form of discriminant ions which remains as a challenge for data validation. Moreover, a larger subset of metabolites associated with the phenotype can be identified using MS.
There have been few studies that reported the application of these approaches in legumes. Metabolic finger printing has been utilized to study drought in seven different model and forage species of the Lotus genus. Analysis using PCA of the metabolite features regardless metabolite identification status yielded sample “fingerprints,” which classified primarily according to the genotype. Infrared (IR) spectroscopy has been used to obtain a snapshot of the sample metabolome (typically low-molecular-weight compounds) at a given time. This study identified metabolic compounds responsible for rapid fermentation for the efficient conservation of forage proteins (Johnson et al., 2004). In another study, NMR-based approach has been used for metabolic fingerprinting of 21 grass and legume cultivars. Applying PCA, variation between cultivars and the magnitude of changes in the metabolic fingerprint between the spring growth and the second regrowth was elucidated in the study. Furthermore, variation in metabolic compounds such as malic acid, choline, and glucose was reported due to seasonal change (Bertram et al., 2010).
Metabolomics Databases for Legumes
As with transcriptomics and proteomics data, metabolomics approaches also generate huge datasets that require specialized data mining and bioinformatics tools. It is imperative to integrate functional genomics data to comprehensively study biological components, using a systemic approach, e.g., through the mathematical modeling of biological systems. Metabolomics data handling, mining and analysis etc. have been improved tremendously, due to advances in bioinformatics tools. In this scenario, several databases have been developed for plant metabolomics data analysis. For example, a metabolic pathway reconstruction was used to generate a pathway database for Medicago called MedicCyc (http://www.noble.org/MedicCyc/) which features more than 250 pathways with related genes, enzymes and metabolites (Urbanczyk-Wochniak and Sumner, 2007). The database contains Medicago specific pathways including isoflavonoid, lignin and triterpene saponin biosynthesis which were added or modified based on literature and in-house expertise. MedicCyc is designed to visualize functional genomics datasets from Medicago within the biological context of metabolic pathways and has been believed that this is best achieved through the visualization of data within the biological context of metabolic pathways in legumes. The pathways were engineered to enable the correlated visualization of integrated functional genomics data. Another database, Soybean Knowledge Base (SoyKB) (http://soykb.org) has been reported to be a comprehensive resource for soybean translational genomics and contains integrative information on soybean genomics, transcriptomics, proteomics and metabolomics (Joshi et al., 2014). This is a web resource that would not only be useful for soybean translational genomics, but also for legume crop improvement programs. Also, a database for plant metabolomics, PlantMetabolomics.org (PM) (http://www.plantmetabolomics.org) was developed (Bais et al., 2010). This database represents metabolomics data generated from Arabidopsis through an integration of experiments compiled from different platforms with visualization tools. PlantMetabolomics has been widely used for exploring, visualizing and downloading plant metabolomics data and well-annotated metabolomics datasets which is useful for establishing metabolomics as a functional genomics tool in legumes.
Metabolomics for Crop Breeding
There is growing interest in using metabolites as selection markers in crop breeding programs, because metabolite biomarkers have been linked with strong environmentally-controlled traits (Steinfath et al., 2010). Mapping and metabolomics genome wide association studies (mGWAS) have been conducted to develop “metabotypes” using metabotype quantitative trait locus (mQTL) (association of genomic markers and metabolic markers) which enabled the associations between metabolic concentrations and genetic polymorphisms. Overall, plant metabolomics has benefited from a rich array of pre-existing methodological approaches and bioanalytical knowledge for the characterization of the chemically diverse classes of metabolites. However, speedy progress in the application of these approaches in legumes will be quite useful for legume improvement.
Summary and Future Outlook
The development in the area of proteomics and metabolomics has enhanced the power of “omics” with the possibility of studying at different levels of plant regulations, namely transcriptome, proteome and the metabolome (Shulaev et al., 2008). Plant growth, development and stress responses are not straightforward to be able to understand by just looking into one or two level(s), which has been done traditionally. For instance, to understand the molecular basis of stress physiology and biochemistry, the quantitative correlation of different protein groups and classes of metabolites with stress levels are required for the identification of candidate bio-markers (Weckwerth, 2011b; Rodziewicz et al., 2014). Moreover, with the recent developments in the technologies, availability of legume genomics and protein databases, it would be possible to have efficient and high throughput identification of stress related proteins in crop legumes (Hiremath et al., 2011; Kosová et al., 2011; Varshney et al., 2013a; Hossain and Komatsu, 2014a,b).
Legume crops are widely cultivated in the semi-arid tropics where various abiotic and biotic stresses pose severe threats to the productivity. The crops are subjected to not only one stress, but also a combination of stresses at a given point of time under natural field conditions. To sustain legume cultivation under these environments, crop improvement programs requires resourceful methods such as integrated “omics” approaches to understand stress responses at the molecular level such as cellular mechanisms, signaling pathways, biochemical processes. Such studies have largely been initiated in model plants such as Arabidopsis. For instance, transcriptomics and the metabolomics data revealed a different pattern of defense response in Arabidopsis subjected to a combination of drought and heat stress. It was found that proline (Pro) was accumulated in response to drought stress, but during the combination of stresses, Pro was replaced by sucrose as osmoprotectants. Similar studies needs to be undertaken in legume crops such as chickpea, pigeonpea and peanut, which are of agronomic value especially in the semi-arid tropics.
In this review, the available proteomics and metabolomics resources for legume research and their applications for further our understanding of the stress biology in model as well as crop legumes have been provided (Figure 1). A large number of protein reference maps for various tissues are already available for model legume crops that will greatly support comparative proteomics approaches. There is a great need to generate proteome maps from crop legumes that would greatly help in precise understanding of various cellular processes and signaling pathways. These resources would improve the functional annotations of the gene models in identifying novel ORFs as in the case of Arabidopsis (Castellana et al., 2008) and also validates the existing annotation (Agrawal et al., 2012, 2013; Dash et al., 2015; Walley and Briggs, 2015). Moreover, it is now possible to uncover the regulation of these processes by studying the post-translational modifications. Further, huge amount of information on metabolomics data related to stress responses in legumes have been generated which includes a large number of metabolic pathways under stress conditions. The integrative analyses of data through genomics, transcriptomics, proteomics and metabolomics will be important for a system biology approach and for efficient application in legume crop improvement. At this point, it would be worthwhile to caution that combining and integrating omics data together is challenging, because of the difficulty in correlating the data due to the differences in the time for quenching metabolism. In addition, some errors can be introduced due to the nature of the samples used and the lack of appropriate mathematical models that would allow identification of various biochemical and signaling pathways involved in stress response. Therefore, there is a need to advance at all the fronts, i.e., sampling strategy, data generation, data analysis, etc. in different disciplines so that precise system biology approach can be deployed to understand molecular basis of different traits related to plant biology as well as breeding applications.
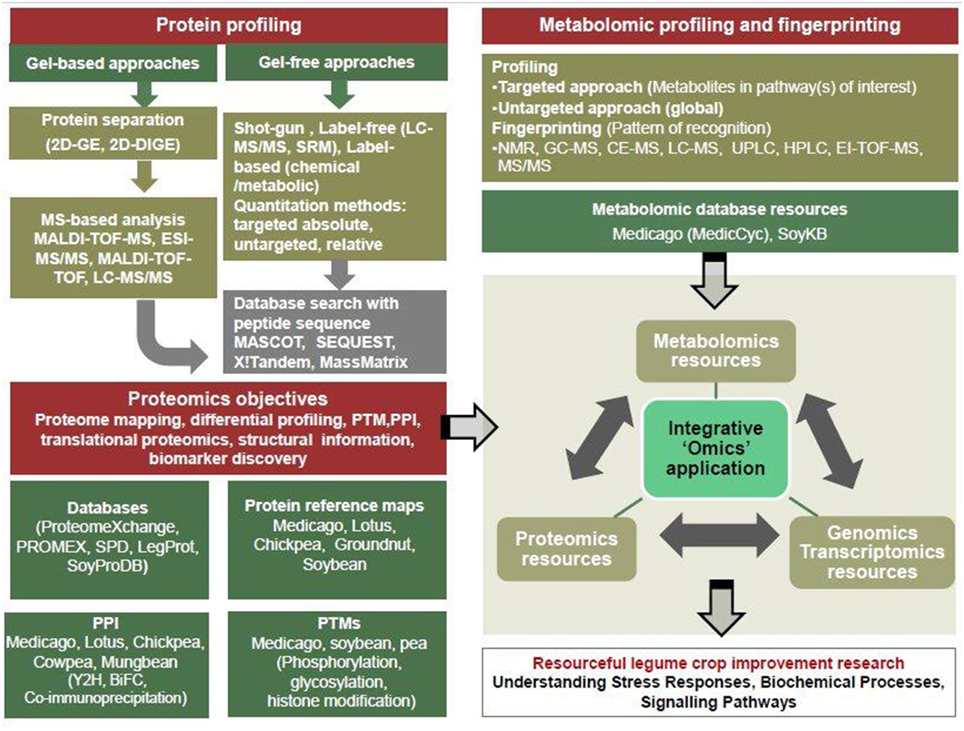
Figure 1. Significant advances in proteomic and metabolomics for integrative “omics” approaches in legume crop research.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
This work has been undertaken as part of the CGIAR Research Program on Grain Legumes. ICRISAT is a member of CGIAR Consortium.
References
Abdallah, C., Dumas-Gaudot, E., Renaut, J., and Sergeant, K. (2012). Gel-based and gel-free quantitative proteomics approaches at a glance. Int. J. Plant Genomics 2012:494572. doi: 10.1155/2012/494572
Abdallah, C., Valot, B., Guillier, C., Mounier, A., Balliau, T., Zivy, M., et al. (2014). The membrane proteome of Medicago truncatula roots displays qualitative and quantitative changes in response to arbuscular mycorrhizal symbiosis. J. Proteomics 108, 354–368. doi: 10.1016/j.jprot.2014.05.028
Aghaei, K., Ehsanpour, A. A., Shah, A. H., and Komatsu, S. (2009). Proteome analysis of soybean hypocotyl and root under salt stress. Amino Acids 36, 91–98. doi: 10.1007/s00726-008-0036-7
Agrawal, G. K., Pedreschi, R., Barkla, B. J., Bindschedler, L. V., Cramer, R., Sarkar, A., et al. (2012). Translational plant proteomics: A perspective. J. Proteomics 75, 4588–4601. doi: 10.1016/j.jprot.2012.03.055
Agrawal, G. K., Sarkar, A., Righetti, P. G., Pedreschi, R., Carpentier, S., Wang, T., et al. (2013). A decade of plant proteomics and mass spectrometry: Translation of technical advancements to food security and safety issues. Mass Spectrom. Rev. 32, 335–365. doi: 10.1002/mas.21365
Ahsan, N., Donnart, T., Nouri, M. Z., and Komatsu, S. (2010b). Tissue-specific defense and thermo-adaptive mechanisms of soybean seedlings under heat stress revealed by proteomic approach. J. Proteome Res. 9, 4189–4204. doi: 10.1021/pr100504j
Ahsan, N., Nanjo, Y., Sawada, H., Kohno, Y., and Komatsu, S. (2010a). Ozone stress-induced proteomic changes in leaf total soluble and chloroplast proteins of soybean reveal that carbon allocation is involved in adaptation in the early developmental stage. Proteomics 10, 2605–2619. doi: 10.1002/pmic.201000180
Alam, I., Lee, D. G., Kim, K. H., Park, C. H., Sharmin, S. A., Lee, H., et al. (2010). Proteome analysis of soybean roots under water logging stress at an early vegetative stage. J. Biosci. 35, 49–62. doi: 10.1007/s12038-010-0007-5
Aloui, A., Recorbet, G., Robert, F., Schoefs, B., Bertrand, M., Henry, C., et al. (2011). Arbuscular mycorrhizal symbiosis elicits shoot proteome changes that are modified during cadmium stress alleviation in Medicago truncatula. BMC Plant Biol. 11:75. doi: 10.1186/1471-2229-11-75
Arabidopsis Interactome Mapping Consortium (2011). Evidence for network evolution in an Arabidopsis interactome map. Science 333, 601–607. doi: 10.1126/science.1203877
Aranjuelo, I., Molero, G., Erice, G., Avice, J. C., and Nogués, S. (2011). Plant physiology and proteomics reveals the leaf response to drought in alfalfa (Medicago sativa L.). J. Exp. Bot. 62, 111–123. doi: 10.1093/jxb/erq249
Arbona, V., Manzi, M., de Ollas, C., and Gómez-Cadenas, A. (2013). Metabolomics as a tool to investigate abiotic stress tolerance in plants. Int. J. Mol. Sci. 14, 4885–4911. doi: 10.3390/ijms14034885
Bachi, A., and Bonaldi, T. (2008). Quantitative proteomics as a new piece of the systems biology puzzle. J. Proteomics 71, 357–367. doi: 10.1016/j.jprot.2008.07.001
Bais, P., Moon, S. M., He, K., Leitao, R., Dreher, K., Walk, T., et al. (2010). PlantMetabolomics. org: A web portal for plant metabolomics experiments. Plant Physiol. 152, 1807–1816. doi: 10.1104/pp.109.151027
Bajad, S., and Shulaev, V. (2007). Highly-parallel metabolomics approaches using LC-MS for pharmaceutical and environmental analysis. Trends Analyt. Chem. 26, 625–636. doi: 10.1016/j.trac.2007.02.009
Beckers, G. J., Hoehenwarter, W., Röhrig, H., Conrath, U., and Weckwerth, W. (2014). Tandem metal-oxide affinity chromatography for enhanced depth of phosphoproteome analysis. Methods Mol. Biol. 1072, 621–632. doi: 10.1007/978-1-62703-631-3_42
Benkeblia, N., Shinano, T., and Osaki, M. (2007). Metabolite profiling and assessment of metabolome compartmentation of soybean leaves using non-aqueous fractionation and GC-MS analysis. Metabolomics 3, 297–305. doi: 10.1007/s11306-007-0078-y
Bertram, H. C., Weisbjerg, M. R., Jensen, C. S., Pedersen, M. G., Didion, T., Petersen, B. O., et al. (2010). Seasonal changes in the metabolic fingerprint of 21 grass and legume cultivars studied by nuclear magnetic resonance-based metabolomics. J. Agric. Food Chem. 58, 4336–4341. doi: 10.1021/jf904321p
Bhalla, R., Narasimhan, K., and Swarup, S. (2005). Metabolomics and its role in understanding cellular responses in plants. Plant Cell Rep. 24, 562–571. doi: 10.1007/s00299-005-0054-9
Bhushan, D., Jaiswal, D. K., Ray, D., Basu, D., Datta, A., Chakraborty, S., et al. (2011). Dehydration-responsive reversible and irreversible changes in the extracellular matrix: Comparative proteomics of chickpea genotypes with contrasting tolerance. J. Proteome Res. 10, 2027–2046. doi: 10.1021/pr200010f
Bhushan, D., Pandey, A., Choudhary, M. K., Datta, A., Chakraborty, S., and Chakraborty, N. (2007). Comparative proteomics analysis of differentially expressed proteins in chickpea extracellular matrix during dehydration stress. Mol. Cell. Proteomics 6, 1868–1884. doi: 10.1074/mcp.M700015-MCP200
Borg, S., Pødenphant, L., Jensen, T. J., and Poulsen, C. (1999). Plant cell growth and differentiation may involve GAP regulation of Rac activity. FEBS Lett. 453, 341–345. doi: 10.1016/S0014-5793(99)00750-4
Bourgeois, M., Jacquin, F., Savois, V., Sommerer, N., Labas, V., Henry, C., et al. (2009). Dissecting the proteome of pea mature seeds reveals the phenotypic plasticity of seed protein composition. Proteomics 9, 254–271. doi: 10.1002/pmic.200700903
Bracha-Drori, K., Shichrur, K., Katz, A., Oliva, M., Angelovici, R., Yalovsky, S., et al. (2004). Detection of protein–protein interactions in plants using bimolecular fluorescence complementation. Plant J. 40, 419–427. doi: 10.1111/j.1365-313X.2004.02206.x
Brasileiro, A. C., Morgante, C. V., Araujo, A. C., Leal-Bertioli, S. C., Silva, A. K., Martins, A. C., et al. (2015). Transcriptome profiling of wild Arachis from water-limited environments uncovers drought tolerance candidate genes. Plant Mol. Biol. Rep. 1–17. doi: 10.1007/s11105-015-0882-x
Braun, P., Aubourg, S., Van Leene, J., De Jaeger, G., and Lurin, C. (2013). Plant protein interactomes. Annu. Rev. Plant Biol. 64, 161–187. doi: 10.1146/annurev-arplant-050312-120140
Brechenmacher, L., Lee, J., Sachdev, S., Song, Z., Nguyen, T. H. N., Joshi, T., et al. (2009). Establishment of a protein reference map for soybean root hair cells. Plant Physiol. 149, 670–682. doi: 10.1104/pp.108.131649
Brechenmacher, L., Lei, Z., Libault, M., Findley, S., Sugawara, M., Sadowsky, M. J., et al. (2010). Soybean metabolites regulated in root hairs in response to the symbiotic bacterium Bradyrhizobium japonicum. Plant Physiol. 153, 1808–1822. doi: 10.1104/pp.110.157800
Brechenmacher, L., Nguyen, T. H. N., Hixson, K., Libault, M., Aldrich, J., Pasa-Tolic, L., et al. (2012). Identification of soybean proteins from a single cell type: The root hair. Proteomics 12, 3365–3373. doi: 10.1002/pmic.201200160
Broeckling, C. D., Huhman, D. V., Farag, M. A., Smith, J. T., May, G. D., Mendes, P., et al. (2005). Metabolic profiling of Medicago truncatula cell cultures reveals the effects of biotic and abiotic elicitors on metabolism. J. Exp. Bot. 56, 323–336. doi: 10.1093/jxb/eri058
Brosowska-Arendt, W., Gallardo, K., Sommerer, N., and Weidner, S. (2014). Changes in the proteome of pea (Pisum sativum L.) seeds germinating under optimal and osmotic stress conditions and subjected to post-stress recovery. Acta Physiol. Plant 36, 795–807. doi: 10.1007/s11738-013-1458-8
Brückner, A., Polge, C., Lentze, N., Auerbach, D., and Schlattner, U. (2009). Yeast two-hybrid, a powerful tool for systems biology. Int. J. Mol. Sci. 18, 2763–2788. doi: 10.3390/ijms10062763
Campbell, P. M., Reiner, D., Moore, A. E., Lee, R. Y., Epstein, M. M., and Higgins, T. J. V. (2011). Comparison of the α-amylase inhibitor-1 from common bean (Phaseolus vulgaris) varieties and transgenic expression in other legumes post-translational modifications and immunogenicity. J. Agric. Food Chem. 59, 6047–6054. doi: 10.1021/jf200456j
Campbell, R., Pont, S. D., Morris, J. A., McKenzie, G., Sharma, S. K., Hedley, P. E., et al. (2014). Genome-wide QTL and bulked transcriptomic analysis reveals new candidate genes for the control of tuber carotenoid content in potato (Solanum tuberosum L.). Theor. Appl. Genet. 127, 1917–1933. doi: 10.1007/s00122-014-2349-0
Castellana, N. E., Payne, S. H., Shen, Z., Stanke, M., Bafna, V., and Briggs, S. P. (2008). Discovery and revision of Arabidopsis genes by proteogenomics. Proc. Natl. Acad. Sci. U.S.A. 105, 21034–21038. doi: 10.1073/pnas.0811066106
Castillejo, M. Á., Fernández-Aparicio, M., and Rubiales, D. (2012). Proteomic analysis by two-dimensional differential in gel electrophoresis (2D DIGE) of the early response of Pisum sativum to Orobanche crenata. J. Exp. Bot. 63, 107–119. doi: 10.1093/jxb/err246
Castillejo, M. A., Staudinger, C., Egelhofer, V., and Wienkoop, S. (2014). Medicago truncatula proteomics for systems biology: Novel rapid shotgun LC-MS approach for relative quantification based on full-scan selective peptide extraction (Selpex). Methods Mol. Biol. 1072, 303–313. doi: 10.1007/978-1-62703-631-3_22
Charlton, A. J., Donarski, J. A., Harrison, M., Jones, S. A., Godward, J., Oehlschlager, S., et al. (2008). Responses of the pea (Pisum sativum L.) leaf metabolome to drought stress assessed by nuclear magnetic resonance spectroscopy. Metabolomics 4, 312–327. doi: 10.1007/s11306-008-0128-0
Charpentier, M., Bredemeier, R., Wanner, G., Takeda, N., Schleiff, E., and Parniske, M. (2008). Lotus japonicus CASTOR and POLLUX are ion channels essential for perinuclear calcium spiking in legume root endosymbiosis. Plant Cell 20, 3467–3479. doi: 10.1105/tpc.108.063255
Chatelain, E., Hundertmark, M., Leprince, O., Gall, S. L., Satour, P., Deligny-Penninck, S., et al. (2012). Temporal profiling of the heat-stable proteome during late maturation of Medicago truncatula seeds identifies a restricted subset of late embryogenesis abundant proteins associated with longevity. Plant Cell Environ. 35, 1440–1455. doi: 10.1111/j.1365-3040.2012.02501.x
Chen, Y., Hoehenwarter, W., and Weckwerth, W. (2010). Comparative analysis of phytohormone−responsive phosphoproteins in Arabidopsis thaliana using TiO2−phosphopeptide enrichment and mass accuracy precursor alignment. Plant J. 63, 1–17. doi: 10.1111/j.1365-313x.2010.04218.x
Cheng, J., Yuan, C., and Graham, T. L. (2011). Potential defense-related prenylated isoflavones in lactofen-induced soybean. Phytochem. 72, 875–881. doi: 10.1016/j.phytochem.2011.03.010
Chung, E., Cho, C. W., So, H. A., Kang, J. S., Chung, Y. S., and Lee, J. H. (2013). Overexpression of VrUBC1, a mung bean E2 ubiquitin-conjugating enzyme, enhances osmotic stress tolerance in Arabidopsis. PLoS ONE 8:e66056. doi: 10.1371/journal.pone.0066056
Colditz, F., and Braun, H. P. (2010). Medicago truncatula proteomics. J. Proteomics 73, 1974–1985. doi: 10.1016/j.jprot.2010.07.004
Dam, S., Dyrlund, T. F., Ussatjuk, A., Jochimsen, B., Nielsen, K., Goffard, N., et al. (2014). Proteome reference maps of the Lotus japonicus nodule and root. Proteomics 14, 230–240. doi: 10.1002/pmic.201300353
Dash, S., Campbell, J. D., Cannon, E. K. S., Cleary, A. M., Huang, W., Kalberer, S. R., et al. (2015). Legume information system (LegumeInfo. org): A key component of a set of federated data resources for the legume family. Nucleic Acids Res. gkv1159. doi: 10.1093/nar/gkv1159
Den Herder, G., Yoshida, S., Antolín-Llovera, M., Ried, M. K., and Parniske, M. (2012). Lotus japonicus E3 ligase SEVEN IN ABSENTIA4 destabilizes the symbiosis receptor-like kinase SYMRK and negatively regulates rhizobial infection. Plant Cell 24, 1691–1707. doi: 10.1105/tpc.110.082248
Dias, D. A., Hill, C. B., Jayasinghe, N. S., Atieno, J., Sutton, T., and Roessner, U. (2015). Quantitative profiling of polar primary metabolites of two chickpea cultivars with contrasting responses to salinity. J. Chromatogr. B 1000, 1–13. doi: 10.1016/j.jchromb.2015.07.002
Dita, M. A., Rispail, N., Prats, E., Rubiales, D., and Singh, K. B. (2006). Biotechnology approaches to overcome biotic and abiotic stress constraints in legumes. Euphytica 147, 1–24. doi: 10.1007/s10681-006-6156-9
Doerfler, H., Lyon, D., Nägele, T., Sun, X., Fragner, L., Hadacek, F., et al. (2013). Granger causality in integrated GC–MS and LC–MS metabolomics data reveals the interface of primary and secondary metabolism. Metabolomics 9, 564–574. doi: 10.1007/s11306-012-0470-0
Doerfler, H., Sun, X., Wang, L., Engelmeier, D., Lyon, D., and Weckwerth, W. (2014). mzGroupAnalyzer-Predicting pathways and novel chemical structures from untargeted high-throughput metabolomics data. PLoS ONE 9:e96188. doi: 10.1371/journal.pone.0096188
Domoney, C., Welham, T., Sidebottom, C., and Firmin, J. L. (1995). Multiple isoforms of Pisum trypsin inhibitors result from modification of two primary gene products. FEBS Lett. 360, 15–20. doi: 10.1016/0014-5793(95)00070-P
Downes, B., and Vierstra, R. D. (2005). Post-translational regulation in plants employing a diverse set of polypeptide tags. Biochem. Soc. Trans. 33, 393–400. doi: 10.1042/BST0330393
Dubinin, J., Braun, H. P., Schmitz, U., and Colditz, F. (2011). The mitochondrial proteome of the model legume Medicago truncatula. Biochim. Biophys. Acta 1814, 1658–1668. doi: 10.1016/j.bbapap.2011.08.008
Fan, H., Xu, Y., Du, C., and Wu, X. (2015). Phloem sap proteome studied by iTRAQ provides integrated insight into salinity response mechanisms in cucumber plants. J. Proteomics 125, 54–67. doi: 10.1016/j.jprot.2015.05.001
Fang, Y., Macool, D. J., Xue, Z., Heppard, E. P., Hainey, C. F., Tingey, S. V., et al. (2002). Development of a high-throughput yeast two-hybrid screening system to study protein-protein interactions in plants. Mol. Genet. Genomics 267, 142–153. doi: 10.1007/s00438-002-0656-7
Farag, M. A., Huhman, D. V., Lei, Z., and Sumner, L. W. (2007). Metabolic profiling and systematic identification of flavonoids and isoflavonoids in roots and cell suspension cultures of Medicago truncatula using HPLC–UV–ESI–MS and GC–MS. Phytochemistry 68, 342–354. doi: 10.1016/j.phytochem.2006.10.023
Fernie, A. R., and Schauer, N. (2009). Metabolomics-assisted breeding: A viable option for crop improvement? Trends Genet. 25, 39–48. doi: 10.1016/j.tig.2008.10.010
Fiehn, O., Kopka, J., Dörmann, P., Altmann, T., Trethewey, R. N., and Willmitzer, L. (2000). Metabolite profiling for plant functional genomics. Nat. Biotechnol. 18, 1157–1161. doi: 10.1038/81137
Fukao, Y. (2012). Protein–protein interactions in plants. Plant Cell Physiol. 53, 617–625. doi: 10.1093/pcp/pcs026
García-Villalba, R., León, C., Dinelli, G., Segura-Carretero, A., Fernández-Gutiérrez, A., Garcia-Cañas, V., et al. (2008). Comparative metabolomic study of transgenic versus conventional soybean using capillary electrophoresis–time-of-flight mass spectrometry. J. Chromatogr. A 1195, 164–173. doi: 10.1016/j.chroma.2008.05.018
Glinski, M., and Weckwerth, W. (2006). The role of mass spectrometry in plant systems biology. Mass Spectrom. Rev. 25, 173–214. doi: 10.1002/mas.20063
Gobbato, E., Marsh, J. F., Vernié, T., Wang, E., Maillet, F., Kim, J., et al. (2012). A GRAS-type transcription factor with a specific function in mycorrhizal signaling. Curr. Biol. 22, 2236–2241. doi: 10.1016/j.cub.2012.09.044
Gokulakannan, G. G., and Niehaus, K. (2010). Characterization of the ‘Medicago truncatula’ cell wall proteome in cell suspension culture upon elicitation and suppression of plant defense. J. Plant Physiol. 167, 1533–1541. doi: 10.1016/j.jplph.2010.06.023
Goodacre, R., York, E. V., Heald, J. K., and Scott, I. M. (2003). Chemometric discrimination of unfractionated plant extracts analyzed by electrospray mass spectrometry. Phytochemistry 62, 859–863. doi: 10.1016/S0031-9422(02)00718-5
Graham, P. H., and Vance, C. P. (2003). Legumes: Importance and constraints to greater use. Plant Physiol. 131, 872–877. doi: 10.1104/pp.017004
Griffin, T. J., and Aebersold, R. (2001). Advances in proteome analysis by mass spectrometry. J. Biol. Chem. 276, 45497–45500. doi: 10.1074/jbc.R100014200
Griffin, T. J., Gygi, S. P., Rist, B., Aebersold, R., Loboda, A., Jilkine, A., et al. (2001). Quantitative proteomic analysis using a MALDI quadrupole time-of-flight mass spectrometer. Anal. Chem. 73, 978–986. doi: 10.1021/ac001169y
Grimsrud, P. A., den Os, D., Wenger, C. D., Swaney, D. L., Schwartz, D., Sussman, M. R., et al. (2010). Large-scale phosphoprotein analysis in Medicago truncatula roots provides insight into in vivo kinase activity in legumes. Plant Physiol. 152, 19–28. doi: 10.1104/pp.109.149625
Hajduch, M., Ganapathy, A., Stein, J. W., and Thelen, J. J. (2005). A systematic proteomic study of seed-filling in soybean: Establishment of high resolution two-dimensional reference maps, expression profiles, and an interactive proteome database. Plant Physiol. 137, 1397–1419. doi: 10.1104/pp.104.056614
Halket, J. M., Waterman, D., Przyborowska, A. M., Patel, R. K., Fraser, P. D., and Bramley, P. M. (2005). Chemical derivatization and mass spectral libraries in metabolic profiling by GC/MS and LC/MS/MS. J. Exp. Bot. 56, 219–243. doi: 10.1093/jxb/eri069
Hegeman, A. D., Schulte, C. F., Cui, Q., Lewis, I. A, Huttlin, E. L., Eghbalnia, H., et al. (2007). Stable isotope assisted assignment of elemental compositions for metabolomics. Anal. Chem. 79, 6912–6921. doi: 10.1021/ac070346t
Heidarvand, L., and Maali-Amiri, R. (2013). Physio-biochemical and proteome analysis of chickpea in early phases of cold stress. J. Plant Physiol. 170, 459–469. doi: 10.1016/j.jplph.2012.11.021
Hernández, G., Ramírez, M., Valdés-López, O., Goffard, N., Weiller, G., Aparicio-Fabre, R., et al. (2009). Global changes in the transcript and metabolic profiles during symbiotic nitrogen fixation in phosphorus-stressed common bean plants. Plant Physiol. 151, 1221–1238. doi: 10.1104/pp.109.143842
Hernández, G., Ramírez, M., Valdés-López, O., Tesfaye, M., Graham, M. A., Czechowski, T., et al. (2007). Phosphorus stress in common bean: Root transcript and metabolic responses. Plant Physiol. 144, 752–767. doi: 10.1104/pp.107.096958
Hiremath, P. J., Farmer, A., Cannon, S. B., Woodward, J., Kudapa, H., Tuteja, R., et al. (2011). Large−scale transcriptome analysis in chickpea (Cicer arietinum L.), an orphan legume crop of the semi−arid tropics of Asia and Africa. Plant Biotechnol. J. 9, 922–931. doi: 10.1111/j.1467-7652.2011.00625.x
Hirsch, S., Kim, J., Muñoz, A., Heckmann, A. B., Downie, J. A., and Oldroyd, G. E. (2009). GRAS proteins form a DNA binding complex to induce gene expression during nodulation signaling in Medicago truncatula. Plant Cell 21, 545–557. doi: 10.1105/tpc.108.064501
Hoehenwarter, W., Thomas, M., Nukarinen, E., Egelhofer, V., Röhrig, H., Weckwerth, W., et al. (2013). Identification of novel in vivo MAP kinase substrates in Arabidopsis thaliana through use of tandem metal oxide affinity chromatography. Mol. Cell Proteomics 12, 369–380. doi: 10.1074/mcp.M112.020560
Hoehenwarter, W., van Dongen, J. T., Wienkoop, S., Steinfath, M., Hummel, J., Erban, A., et al. (2008). A rapid approach for phenotype-screening and database independent detection of cSNP/protein polymorphism using mass accuracy precursor alignment. Proteomics 8, 4214–4225. doi: 10.1002/pmic.200701047
Hossain, Z., Khatoon, A., and Komatsu, S. (2013). Soybean proteomics for unravelling abiotic stress response mechanism. J. Proteome Res. 12, 4670–4684. doi: 10.1021/pr400604b
Hossain, Z., and Komatsu, S. (2014a). Potentiality of soybean proteomics in untying the mechanism of flood and drought stress tolerance. Proteomes 2, 107–127. doi: 10.3390/proteomes2010107
Hossain, Z., and Komatsu, S. (2014b). Soybean proteomics. Methods Mol. Biol. 1072, 315–331. doi: 10.1007/978-1-62703-631-3_23
Hossain, Z., Nouri, M. Z., and Komatsu, S. (2012). Plant cell organelle proteomics in response to abiotic stress. J. Proteome Res. 11, 37–48. doi: 10.1021/pr200863r
Hu, J., Rampitsch, C., and Bykova, N. V. (2015). Advances in plant proteomics toward improvement of crop productivity and stress resistance. Front. Plant Sci. 6:209. doi: 10.3389/fpls.2015.00209
Huege, J., Sulpice, R., Gibon, Y., Lisec, J., Koehl, K., and Kopka, J. (2007). GC-EI-TOF-MS analysis of in vivo carbon-partitioning into soluble metabolite pools of higher plants by monitoring isotope dilution after 13CO2 labelling. Phytochemistry 68, 2258–2272. doi: 10.1016/j.phytochem.2007.03.026
Huhman, D. V., and Sumner, L. W. (2002). Metabolic profiling of saponins in Medicago sativa and Medicago truncatula using HPLC coupled to an electrospray ion-trap mass spectrometer. Phytochemistry 59, 347–360. doi: 10.1016/S0031-9422(01)00432-0
Hummel, J., Niemann, M., Wienkoop, S., Schulze, W., Steinhauser, D., Selbig, J., et al. (2007). ProMEX: A mass spectral reference database for proteins and protein phosphorylation sites. BMC Bioinformatics 8:216. doi: 10.1186/1471-2105-8-216
Imin, N., De-Jong, F., Mathesius, U., Van Noorden, G., Saeed, N. A., Wang, X. D., et al. (2004). Proteome reference maps of Medicago truncatula embryogenic cell cultures generated from single protoplasts. Proteomics 4, 1883–1896. doi: 10.1002/pmic.200300803
Ino, Y., Ishikawa, A., Nomura, A., Kajiwara, H., Harada, K., and Hirano, H. (2014). Phosphoproteome analysis of Lotus japonicus seeds. Proteomics 14, 116–120. doi: 10.1002/pmic.201300237
Irar, S., González, E. M., Arrese-Igor, C., and Marino, D. (2014). A proteomic approach reveals new actors of nodule response to drought in split-root grown pea plants. Physiol. Plant 152, 634–645. doi: 10.1111/ppl.12214
Jaiswal, D. K., Mishra, P., Subba, P., Rathi, D., Chakraborty, S., and Chakraborty, N. (2014). Membrane-associated proteomics of chickpea identifies Sad1/UNC-84 protein (CaSUN1), a novel component of dehydration signaling. Sci. Rep. 4, 4177. doi: 10.1038/srep04177
Jaiswal, D. K., Ray, D., Subba, P., Mishra, P., Gayali, S., Datta, A., et al. (2012). Proteomic analysis reveals the diversity and complexity of membrane proteins in chickpea (Cicer arietinum L.). Proteome Sci. 10, 59–5956. doi: 10.1186/1477-5956-10-59
Johnson, H. E., Broadhurst, D., Goodacre, R., and Smith, A. R. (2003). Metabolic fingerprinting of salt-stressed tomatoes. Phytochemistry 62, 919–928. doi: 10.1016/S0031-9422(02)00722-7
Johnson, H. E., Broadhurst, D., Kell, D. B., Theodorou, M. K., Merry, R. J., and Griffith, G. W. (2004). High-throughput metabolic fingerprinting of legume silage fermentations via Fourier transform infrared spectroscopy and chemometrics. Appl. Environ. Microbiol. 70, 1583–1592. doi: 10.1128/AEM.70.3.1583-1592.2004
Jorrín-Novo, J. V., Pascual, J., Sánchez-Lucas, R., Romero-Rodríguez, M. C., Rodríguez-Ortega, M. J., Lenz, C., et al. (2015). Fourteen years of plant proteomics reflected in Proteomics: Moving from model species and 2DE−based approaches to orphan species and gel-free platforms. Proteomics 15, 1089–1112. doi: 10.1002/pmic.201400349
Joshi, T., Fitzpatrick, M. R., Chen, S., Liu, Y., Zhang, H., Endacott, R. Z., et al. (2014). Soybean knowledge base (SoyKB): A web resource for integration of soybean translational genomics and molecular breeding. Nucleic Acids Res. 42, D1245–D1252. doi: 10.1093/nar/gkt905
Kang, Y. J., Kim, S. K., Kim, M. Y., Lestari, P., Kim, K. H., Ha, B. K., et al. (2014). Genome sequence of mungbean and insights into evolution within Vigna species. Nat. Commun. 5, 5443. doi: 10.1038/ncomms6443
Kang, Y. J., Satyawan, D., Shim, S., Lee, T., Lee, J., Hwang, W. J., et al. (2015). Draft genome sequence of adzuki bean, Vigna angularis. Sci. Rep. 5:8069. doi: 10.1038/srep08069
Karmakar, K., Rana, A., Rajwar, A., Sahgal, M., and Johri, B. N. (2015). “Legume-rhizobia symbiosis under stress,” in Plant Microbes Symbiosis: Applied Facets, ed N. K. Arora (New Delhi; Heidelberg: Springer), 241–258.
Katam, R., Basha, S. M., Suravajhala, P., and Pechan, T. (2010). Analysis of peanut leaf proteome. J. Proteome Res. 9, 2236–2254. doi: 10.1021/pr901009n
Kav, N. N. V., Srivastava, S., Goonewardene, L., and Blade, S. F. (2004). Proteome−level changes in the roots of Pisum sativum in response to salinity. Ann. Appl. Biol. 145, 217–230. doi: 10.1111/j.1744-7348.2004.tb00378.x
Khatoon, A., Rehman, S., Hiraga, S., Makino, T., and Komatsu, S. (2012). Organ-specific proteomics analysis for identification of response mechanism in soybean seedlings under flooding stress. J. Proteomics 75, 5706–5723. doi: 10.1016/j.jprot.2012.07.031
Kim, J. C., Lee, S. H., Cheong, Y. H., Yoo, C. M., Lee, S. I., Chun, H. J., et al. (2001). A novel cold−inducible zinc finger protein from soybean, SCOF−1, enhances cold tolerance in transgenic plants. Plant J. 25, 247–259. doi: 10.1046/j.1365-313x.2001.00947.x
Komatsu, S., Yamamoto, A., Nakamura, T., Nouri, M. Z., Nanjo, Y., Nishizawa, K., et al. (2011). Comprehensive analysis of mitochondria in roots and hypocotyls of soybean under flooding stress using proteomics and metabolomics techniques. J. Proteome Res. 10, 3993–4004. doi: 10.1021/pr2001918
Kosová, K., Vítámvás, P., Prášil, I. T., and Renaut, J. (2011). Plant proteome changes under abiotic stress–Contribution of proteomics studies to understanding plant stress response. J. Proteomics 74, 1301–1322. doi: 10.1016/j.jprot.2011.02.006
Kottapalli, K. R., Rakwal, R., Shibato, J., Burow, G., Tissue, D., Burke, J., et al. (2009). Physiology and proteomics of the water-deficit stress response in three contrasting peanut genotypes. Plant Cell Environ. 32, 380–407. doi: 10.1111/j.1365-3040.2009.01933.x
Kottapalli, K. R., Zabet-Moghaddam, M., Rowland, D., Faircloth, W., Mirzaei, M., Haynes, P. A., et al. (2013). Shotgun label-free quantitative proteomics of water-deficit-stressed midmature peanut (Arachis hypogaea L.) seed. J. Proteome Res. 12, 5048–5057. doi: 10.1021/pr400936d
Krishnan, P., Kruger, N. J., and Ratcliffe, R. G. (2005). Metabolite fingerprinting and profiling in plants using NMR. J. Exp. Bot. 56, 255–265. doi: 10.1093/jxb/eri010
Kudapa, H., Ramalingam, A., Nayakoti, S., Chen, X., Zhuang, W. J., Liang, X., et al. (2013). Functional genomics to study stress responses in crop legumes: Progress and prospects. Funct. Plant Biol. 40, 1221–1233. doi: 10.1071/FP13191
Kumar, R., Kumar, A., Subba, P., Gayali, S., Barua, P., Chakraborty, S., et al. (2014). Nuclear phosphoproteome of developing chickpea seedlings (Cicer arietinum L.) and protein-kinase interaction network. J. Proteomics 105, 58–73. doi: 10.1016/j.jprot.2014.04.002
Lafaye, A., Labarre, J., Tabet, J. C., Ezan, E., and Junot, C. (2005). Liquid chromatography-mass spectrometry and 15N metabolic labeling for quantitative metabolic profiling. Anal. Chem. 77, 2026–2033. doi: 10.1021/ac048657g
Lam, H. M., Xu, X., Liu, X., Chen, W., Yang, G., Wong, F., et al. (2010). Resequencing of 31 wild and cultivated soybean genomes identifies patterns of genetic diversity and selection. Nat. Genet. 42, 1053–1059. doi: 10.1038/ng.715
Lange, V., Picotti, P., Domon, B., and Aebersold, R. (2008). Selected reaction monitoring for quantitative proteomics: A tutorial. Mol. Sys. Biol. 4, 222. doi: 10.1038/msb.2008.61
Langridge, P., and Fleury, D. (2011). Making the most of ‘omics’ for crop breeding. Trends Biotechnol. 29, 33–40. doi: 10.1016/j.tibtech.2010.09.006
Larrainzar, E., Wienkoop, S., Scherling, C., Kempa, S., Ladrera, R., Arrese-Igor, C., et al. (2009). Carbon metabolism and bacteroid functioning are involved in the regulation of nitrogen fixation in Medicago truncatula under drought and recovery. Mol. Plant Microbe Interact. 22, 1565–1576. doi: 10.1094/MPMI-22-12-1565
Larrainzar, E., Wienkoop, S., Weckwerth, W., Ladrera, R., Arrese-Igor, C., and González, E. M. (2007). Medicago truncatula root nodule proteome analysis reveals differential plant and bacteroid responses to drought stress. Plant Physiol. 144, 1495–1507. doi: 10.1104/pp.107.101618
Lee, L. Y., Wu, F. H., Hsu, C. T., Shen, S. C., Yeh, H. Y., Liao, D. C., et al. (2012). Screening a cDNA library for protein–protein interactions directly in planta. Plant Cell 24, 1746–1759. doi: 10.1105/tpc.112.097998
Lei, Z., Dai, X., Watson, B. S., Zhao, P. X., and Sumner, L. W. (2011). A legume specific protein database (LegProt) improves the number of identified peptides, confidence scores and overall protein identification success rates for legume proteomics. Phytochemistry 72, 1020–1027. doi: 10.1016/j.phytochem.2011.01.026
Lei, Z., Elmer, A. M., Watson, B. S., Dixon, R. A., Mendes, P. J., and Sumner, L. W. (2005). A two-dimensional electrophoresis proteomic reference map and systematic identification of 1367 proteins from a cell suspension culture of the model legume Medicago truncatula. Mol. Cell Proteomics 4, 1812–1825. doi: 10.1074/mcp.D500005-MCP200
Liao, Y., Zou, H. F., Wang, H. W., Zhang, W. K., Ma, B., Zhang, J. S., et al. (2008a). Soybean GmMYB76, GmMYB92, and GmMYB177 genes confer stress tolerance in transgenic Arabidopsis plants. Cell Res. 18, 1047–1060. doi: 10.1038/cr.2008.280
Liao, Y., Zou, H. F., Wei, W., Hao, Y. J., Tian, A. G., Huang, J., et al. (2008b). Soybean GmbZIP44, GmbZIP62 and GmbZIP78 genes function as negative regulator of ABA signaling and confer salt and freezing tolerance in transgenic Arabidopsis. Planta 228, 225–240. doi: 10.1007/s00425-008-0731-3
Liu, G. T., Ma, L., Duan, W., Wang, B. C., Li, J. H., Xu, H. G., et al. (2014). Differential proteomic analysis of grapevine leaves by iTRAQ reveals responses to heat stress and subsequent recovery. BMC Plant Biol. 14:110. doi: 10.1186/1471-2229-14-110
Ma, H., Song, L., Shu, Y., Wang, S., Niu, J., Wang, Z., et al. (2012). Comparative proteomic analysis of seedling leaves of different salt tolerant soybean genotypes. J. Proteomics 75, 1529–1546. doi: 10.1016/j.jprot.2011.11.026
Mahajan, S., Sopory, S. K., and Tuteja, N. (2006). Cloning and characterization of CBL−CIPK signalling components from a legume (Pisum sativum). FEBS J. 273, 907–925. doi: 10.1111/j.1742-4658.2006.05111.x
Marouga, R., David, S., and Hawkins, E. (2005). The development of the DIGE system: 2D fluorescence difference gel analysis technology Anal. Bioanal. Chem. 382, 669–678. doi: 10.1007/s00216-005-3126-3
Mashego, M. R., Wu, L., Van Dam, J. C., Ras, C., Vinke, J. L., Van Winden, W. A., et al. (2004). MIRACLE: Mass isotopomer ratio analysis of U-13C-labeled extracts. A new method for accurate quantification of changes in concentrations of intracellular metabolites. Biotechnol. Bioeng. 85, 620–628. doi: 10.1002/bit.10907
Mathesius, U., Djordjevic, M. A., Oakes, M., Goffard, N., Haerizadeh, F., Weiller, G. F., et al. (2011). Comparative proteomic profiles of the soybean (Glycine max) root apex and differentiated root zone. Proteomics 11, 1707–1719. doi: 10.1002/pmic.201000619
Mazzucotelli, E., Mastrangelo, A. M., Crosatti, C., Guerra, D., Stanca, A. M., and Cattivelli, L. (2008). Abiotic stress response in plants: When post-transcriptional and post-translational regulations control transcription. Plant Sci. 174, 420–431. doi: 10.1016/j.plantsci.2008.02.005
Messinese, E., Mun, J. H., Yeun, L. H., Jayaraman, D. K., Rougé, P., Barre, A., et al. (2007). A novel nuclear protein interacts with the symbiotic DMI3 calcium-and calmodulin-dependent protein kinase of Medicago truncatula. Mol. Plant Microbe Interact. 20, 912–921. doi: 10.1094/MPMI-20-8-0912
Misra, S., Wu, Y., Venkataraman, G., Sopory, S. K., and Tuteja, N. (2007). Heterotrimeric G-protein complex and G-protein-coupled receptor from a legume (Pisum sativum): Role in salinity and heat stress and cross-talk with phospholipase C. Plant J. 51, 656–669. doi: 10.1111/j.1365-313X.2007.03169.x
Mithöfer, A., Müller, B., Wanner, G., and Eichacker, L. A. (2002). Identification of defence-related cell wall proteins in Phytophthora sojae-infected soybean roots by ESI-MS/MS. Mol. Plant Pathol. 3, 163–166. doi: 10.1046/j.1364-3703.2002.00109.x
Mohammadi, P., Moieni, A., Hiraga, S., and Komatsu, S. (2012). Organ-specific proteomic analysis of drought-stressed soybean seedlings. J. Proteomics 75, 1906–1923. doi: 10.1016/j.jprot.2011.12.041
Muneer, S., Hakeem, K. R., Mohamed, R., and Lee, J. H. (2014). Cadmium toxicity induced alterations in the root proteome of green gram in contrasting response towards iron supplement. Int. J. Mol. Sci. 15, 6343–6355. doi: 10.3390/ijms15046343
Murakami, Y., Imaizumi-Anraku, H., Kouchi, H., Kawagushi, M., and Kawasaki, S. (2013). The transcription activation and homodimerization of Lotus japonicus Nod factor Signaling Pathway2 protein. Plant Signal Behav. 8:e26457. doi: 10.4161/psb.26457
Nanjo, Y., Skultety, L., Uváčková, L. U., Klubicová, K., Hajduch, M., and Komatsu, S. (2011). Mass spectrometry-based analysis of proteomic changes in the root tips of flooded soybean seedlings. J. Proteome Res. 11, 372–385. doi: 10.1021/pr200701y
Nautrup-Pedersen, G., Dam, S., Laursen, B. S., Siegumfeldt, A. L., Nielsen, K., Goffard, N., et al. (2010). Proteome analysis of pod and seed development in the model legume Lotus japonicus. J. Proteome Res. 9, 5715–5726. doi: 10.1021/pr100511u
Ndassa, Y. M., Orsi, C., Marto, J. A., Chen, S., and Ross, M. M. (2006). Improved immobilized metal affinity chromatography for large-scale phosphoproteomics applications. J. Proteome Res. 5, 2789–2799. doi: 10.1021/pr0602803
Nouri, M. Z., and Komatsu, S. (2010). Comparative analysis of soybean plasma membrane proteins under osmotic stress using gel−based and LC MS/MS−based proteomics approaches. Proteomics 10, 1930–1945. doi: 10.1002/pmic.200900632
Obata, T., and Fernie, A. R. (2012). The use of metabolomics to dissect plant responses to abiotic stresses. Cell. Mol. Life Sci. 69, 3225–3243. doi: 10.1007/s00018-012-1091-5
Ohyanagi, H., Sakata, K., and Komatsu, S. (2012). Soybean Proteome Database 2012: Update on the comprehensive data repository for soybean proteomics. Front. Plant Sci. 3:110. doi: 10.3389/fpls.2012.00110
Ogura, T., Ogihara, J., Sunairi, M., Takeishi, H., Aizawa, T., Olivos-Trujillo, M. R., et al. (2014). Proteomic characterization of seeds from yellow lupin (Lupinus luteus L.). Proteomics 14, 1543–1546. doi: 10.1002/pmic.201300511
Pandey, A., Chakraborty, S., Datta, A., and Chakraborty, N. (2008). Proteomics approach to identify dehydration responsive nuclear proteins from chickpea (Cicer arietinum L.). Mol. Cell. Proteomics 7, 88–107. doi: 10.1074/mcp.M700314-MCP200
Pandey, A., Choudhary, M. K., Bhushan, D., Chattopadhyay, A., Chakraborty, S., Datta, A., et al. (2006). The nuclear proteome of chickpea (Cicer arietinum L.) reveals predicted and unexpected proteins. J. Proteome Res. 5, 3301–3311. doi: 10.1021/pr060147a
Pandey, A., and Mann, M. (2000). Proteomics to study genes and genomes. Nature 405, 837–846. doi: 10.1038/35015709
Parrish, J. R., Gulyas, K. D., and Finley, R. L. (2006). Yeast two-hybrid contributions to interactome mapping. Curr. Opin. Plant Biol. 17, 387–393. doi: 10.1016/j.copbio.2006.06.006
Patti, G. J., Yanes, O., and Siuzdak, G. (2012). Innovation: Metabolomics: The apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol. 13, 263–269. doi: 10.1038/nrm3314
Peters, K., Belt, K., and Braun, H. P. (2013). 3D gel map of Arabidopsis complex I. Front. Plant Sci. 4:153. doi: 10.3389/fpls.2013.00153
Picotti, P., and Aebersold, R. (2012). Selected reaction monitoring-based proteomics: Workflows, potential, pitfalls and future directions. Nat. Methods 9, 555–566. doi: 10.1038/nmeth.2015
Ramalingam, A., Kudapa, H., Pazhamala, L. T., Garg, V., and Varshney, R. K. (2015). Gene expression and Yeast two-hybrid studies of a 1R-MYB transcription factor mediating drought stress response in root tissues of chickpea (Cicer arietinum L.). Front. Plant Sci. 6:1117. doi: 10.3389/fpls.2015.01117
Rocha, M., Licausi, F., Araújo, W. L., Nunes-Nesi, A., Sodek, L., Fernie, A. R., et al. (2010). Glycolysis and the tricarboxylic acid cycle are linked by alanine aminotransferase during hypoxia induced by waterlogging of Lotus japonicus. Plant Physiol. 152, 1501–1513. doi: 10.1104/pp.109.150045
Rode, C., Senkler, M., Klodmann, J., Winkelmann, T., and Braun, H. P. (2011). GelMap - a novel software tool for building and presenting proteome reference maps. J. Proteomics 74, 2214–2219. doi: 10.1016/j.jprot.2011.06.017
Rodziewicz, P., Swarcewicz, B., Chmielewska, K., Wojakowska, A., and Stobiecki, M. (2014). Influence of abiotic stresses on plant proteome and metabolome changes. Acta Physiol. Plant 36, 1–19. doi: 10.1007/s11738-013-1402-y
Roessner, U., Wagner, C., Kopka, J., Trethewey, R. N., and Willmitzer, L. (2000). Technical advance: Simultaneous analysis of metabolites in potato tuber by gas chromatography-mass spectrometry. Plant J. 23, 131–142. doi: 10.1046/j.1365-313x.2000.00774.x
Romero-Rodríguez, M. C., Pascual, J., Valledor, L., and Jorrín-Novo, J. (2014). Improving the quality of protein identification in non-model species. Characterization of Quercus ilex seed and Pinus radiata needle proteomes by using SEQUEST and custom databases. J. Proteomics 105, 85–91. doi: 10.1016/j.jprot.2014.01.027
Rose, C. M., Venkateshwaran, M., Grimsrud, P. A., Westphall, M. S., Sussman, M. R., Coon, J. J., et al. (2012a). Medicago PhosphoProtein Database: A repository for Medicago truncatula phosphoprotein data. Front. Plant Sci. 3:122. doi: 10.3389/fpls.2012.00122
Rose, C. M., Venkateshwaran, M., Volkening, J. D., Grimsrud, P. A., Maeda, J., Bailey, D. J., et al. (2012b). Rapid phosphoproteomic and transcriptomic changes in the rhizobia-legume symbiosis. Mol. Cell. Proteomics 11, 724–744. doi: 10.1074/mcp.M112.019208
Sadhukhan, A., Panda, S. K., and Sahoo, L. (2014). The cowpea RING ubiquitin ligase VuDRIP interacts with transcription factor VuDREB2A for regulating abiotic stress responses. Plant Physiol. Biochem. 83, 51–56. doi: 10.1016/j.plaphy.2014.07.007
Salavati, A., Khatoon, A., Nanjo, Y., and Komatsu, S. (2012). Analysis of proteomic changes in roots of soybean seedlings during recovery after flooding. J. Proteomics 75, 878–893. doi: 10.1016/j.jprot.2011.10.002
Salekdeh, G. H., and Komatsu, S. (2007). Crop proteomics: Aim at sustainable agriculture of tomorrow. Proteomics 7, 2976–2996. doi: 10.1002/pmic.200700181
Sanchez, D. H., Lippold, F., Redestig, H., Hannah, M. A., Erban, A., Krämer, U., et al. (2008). Integrative functional genomics of salt acclimatization in the model legume Lotus japonicus. Plant J. 53, 973–987. doi: 10.1111/j.1365-313X.2007.03381.x
Sanchez, D. H., Pieckenstain, F. L., Escaray, F., Erban, A., Kraemer, U. T. E., Udvardi, M. K., et al. (2011). Comparative ionomics and metabolomics in extremophile and glycophytic Lotus species under salt stress challenge the metabolic pre-adaptation hypothesis. Plant Cell Environ. 34, 605–617. doi: 10.1111/j.1365-3040.2010.02266.x
Sanchez, D. H., Schwabe, F., Erban, A., Udvardi, M. K., and Kopka, J. (2012). Comparative metabolomics of drought acclimation in model and forage legumes. Plant Cell Environ. 35, 136–149. doi: 10.1111/j.1365-3040.2011.02423.x
Sato, D., Akashi, H., Sugimoto, M., Tomita, M., and Soga, T. (2013). Metabolomic profiling of the response of susceptible and resistant soybean strains to foxglove aphid, Aulacorthum solani Kaltenbach. J. Chromatogr. B 925, 95–103. doi: 10.1016/j.jchromb.2013.02.036
Sato, S., Nakamura, Y., Kaneko, T., Asamizu, E., Kato, T., Nakao, M., et al. (2008). Genome structure of the legume, Lotus japonicus. DNA Res. 15, 227–239. doi: 10.1093/dnares/dsn008
Schenkluhn, L., Hohnjec, N., Niehaus, K., Schmitz, U., and Colditz, F. (2010). Differential gel electrophoresis (DIGE) to quantitatively monitor early symbiosis-and pathogenesis-induced changes of the Medicago truncatula root proteome. J. Proteomics 73, 753–768. doi: 10.1016/j.jprot.2009.10.009
Scherling, C., Roscher, C., Giavalisco, P., Schulze, E. D., and Weckwerth, W. (2010). Metabolomics unravel contrasting effects of biodiversity on the performance of individual plant species. PLoS ONE 5:e12569. doi: 10.1371/journal.pone.0012569
Schiltz, S., Gallardo, K., Huart, M., Negroni, L., Sommerer, N., and Burstin, J. (2004). Proteome reference maps of vegetative tissues in pea. An investigation of nitrogen mobilization from leaves during seed filling. Plant Physiol. 135, 2241–2260. doi: 10.1104/pp.104.041947
Schliemann, W., Ammer, C., and Strack, D. (2008). Metabolite profiling of mycorrhizal roots of Medicago truncatula. Phytochemistry 69, 112–146. doi: 10.1016/j.phytochem.2007.06.032
Schmidt, C., and Urlaub, H. (2012). Absolute quantification of proteins using standard peptides and multiple reaction monitoring. Methods Mol. Biol. 893, 249–265. doi: 10.1007/978-1-61779-885-6_17
Schmutz, J., Cannon, S. B., Schlueter, J., Ma, J., Mitros, T., Nelson, W., et al. (2010). Genome sequence of the palaeopolyploid soybean. Nature 463, 178–183. doi: 10.1038/nature08670
Schmutz, J., McClean, P. E., Mamidi, S., Wu, G. A., Cannon, S. B., Grimwood, J., et al. (2014). A reference genome for common bean and genome-wide analysis of dual domestications. Nat. Genet. 46, 707–713. doi: 10.1038/ng.3008
Senkler, M., and Braun, H. P. (2012). Functional annotation of 2D protein maps: The GelMap portal. Front. Plant Sci. 14:87. doi: 10.3389/fpls.2012.00087
Seo, J., and Lee, K. J. (2004). Post-translational modifications and their biological functions: Proteomic analysis and systematic approaches. J. Biochem. Mol. Biol. 37, 35–44. doi: 10.5483/BMBRep.2004.37.1.035
Shulaev, V. (2006). Metabolomics technology and bioinformatics. Brief Bioinform. 7, 128–139. doi: 10.1093/bib/bbl012
Shulaev, V., Cortes, D., Miller, G., and Mittler, R. (2008). Metabolomics for plant stress response. Physiol. Plant. 132, 199–208. doi: 10.1111/j.1399-3054.2007.01025.x
Soga, T. (2007). Capillary electrophoresis-mass spectrometry for metabolomics. Methods Mol. Biol. 358, 129–137. doi: 10.1007/978-1-59745-244-1_8
Soga, T., Ohashi, Y., Ueno, Y., Naraoka, H., Tomita, M., and Nishioka, T. (2003). Quantitative metabolome analysis using capillary electrophoresis mass spectrometry. J. Proteome Res. 2, 488–494. doi: 10.1021/pr034020m
Stasi, M., De Luca, M., and Bucci, C. (2015). Two-hybrid-based systems: Powerful tools for investigation of membrane traffic machineries. J. Biotechnol. 202, 105–117. doi: 10.1016/j.jbiotec.2014.12.007
Staszków, A., Swarcewicz, B., Banasiak, J., Muth, D., Jasiñski, M., and Stobiecki, M. (2011). LC/MS profiling of flavonoid glycoconjugates isolated from hairy roots, suspension root cell cultures and seedling roots of Medicago truncatula. Metabolomics 7, 604–613. doi: 10.1007/s11306-011-0287-2
Staudinger, C., Mehmeti, V., Turetschek, R., Lyon, D., Egelhofer, V., and Wienkoop, S. (2012). Possible role of nutritional priming for early salt and drought stress responses in Medicago truncatula. Front. Plant Sci. 3:285. doi: 10.3389/fpls.2012.00285
Steinfath, M., Strehmel, N., Peters, R., Schauer, N., Groth, D., Hummel, J., et al. (2010). Discovering plant metabolic biomarkers for phenotype prediction using an untargeted approach. Plant Biotechnol. J. 8, 900–911. doi: 10.1111/j.1467-7652.2010.00516.x
Subba, P., Barua, P., Kumar, R., Datta, A., Soni, K. K., Chakraborty, S., et al. (2013). Phosphoproteomic dynamics of chickpea (Cicer arietinum L.) reveals shared and distinct components of dehydration response. J. Proteome Res. 12, 5025–5047. doi: 10.1021/pr400628j
Subramanian, S., and Smith, D. L. (2013). “A proteomics approach to study soybean and its symbiont Bradyrhizobium japonicum- a review,” in A Comprehensive Survey of International Soybean Research—Genetics, Physiology, Agronomy and Nitrogen Relationships, ed J. Board (Rijeka: INTECH Open Access Publisher), 978–953.
Sumner, L.W., Mendes, P., and Dixon, R. A. (2003). Plant metabolomics: Large-scale phytochemistry in the functional genomics era. Phytochemistry 62, 817–836. doi: 10.1016/S0031-9422(02)00708-2
Swigonska, S., and Weidner, S. (2013). Proteomic analysis of response to long-term continuous stress in roots of germinating soybean seeds. J. Plant Physiol. 170, 470–479. doi: 10.1016/j.jplph.2012.11.020
Tavakolan, M., Alkharouf, N. W., Khan, F. H., and Natarajan, S. (2013). SoyProDB: A database for the identification of soybean seed proteins. Bioinformation 9, 165–167. doi: 10.6026/97320630009165
Thudi, M., Upadhyaya, H. D., Rathore, A., Gaur, P. M., Krishnamurthy, L., Roorkiwal, M., et al. (2014). Genetic dissection of drought and heat tolerance in chickpea through genome-wide and candidate gene-based association mapping approaches. PLoS ONE 9:e96758. doi: 10.1371/journal.pone.0096758
Tripathi, V., Parasuraman, B., Laxmi, A., and Chattopadhyay, D. (2009). CIPK6, a CBL-interacting protein kinase is required for development and salt tolerance in plants. Plant J. 58, 778–790. doi: 10.1111/j.1365-313X.2009.03812.x
Urbanczyk-Wochniak, E., and Sumner, L. W. (2007). MedicCyc: A biochemical pathway database for Medicago truncatula. Bioinformatics 23, 1418–1423. doi: 10.1093/bioinformatics/btm040
Vanderschuren, H., Lentz, E., Zainuddin, I., and Gruissem, W. (2013). Proteomics of model and crop plant species: Status, current limitations and strategic advances for crop improvement. J. Proteomics. 93, 5–19. doi: 10.1016/j.jprot.2013.05.036
van Wijk, K. J. (2001). Challenges and prospects of plant proteomics. Plant Physiol. 126, 501–508. doi: 10.1104/pp.126.2.501
Varshney, R. K., Chen, W., Li, Y., Bharti, A. K., Saxena, R. K., Schlueter, J. A., et al. (2012). Draft genome sequence of pigeonpea (Cajanus cajan), an orphan legume crop of resource-poor farmers. Nat. Biotechnol. 30, 83–89. doi: 10.1038/nbt.2022
Varshney, R. K., Close, T. J., Singh, N. K., Hoisington, D. A., and Cook, D. R. (2009). Orphan legume crops enter the genomics era! Curr. Opin. Plant Biol. 12, 202–210. doi: 10.1016/j.pbi.2008.12.004
Varshney, R. K., Mohan, S. M., Gaur, P. M., Gangarao, N. V., Pandey, M. K., Bohra, A., et al. (2013a). Achievements and prospects of genomics-assisted breeding in three legume crops of the semi-arid tropics. Biotechnol. Adv. 31, 1120–1134. doi: 10.1016/j.biotechadv.2013.01.001
Varshney, R. K., Song, C., Saxena, R. K., Azam, S., Yu, S., Sharpe, A. G., et al. (2013b). Draft genome sequence of chickpea (Cicer arietinum) provides a resource for trait improvement. Nat. Biotechnol. 31, 240–246. doi: 10.1038/nbt.2491
Varshney, R. K., Thudi, M., Nayak, S. N., Gaur, P. M., Kashiwagi, J., Krishnamurthy, L., et al. (2014). Genetic dissection of drought tolerance in chickpea (Cicer arietinum L.). Theor. Appl. Genet. 127, 445–462. doi: 10.1007/s00122-013-2230-6
Varshney, R. K., and Tuberosa, R. (2013). “Translational genomics in crop breeding for biotic stress resistance: An introduction” in Translational Genomics for Crop Breeding: Biotic Stress, Vol. 1, eds R. K. Varshney and R. Tuberosa (Chichester, UK: John Wiley & Sons), 1–9.
von Mering, C., Krause, R., Snel, B., Cornell, M., Oliver, S. G., Fields, S., et al. (2002). Comparative assessment of large-scale data sets of protein–protein interactions. Nature 417, 399–403. doi: 10.1038/nature750
Walley, J. W., and Briggs, S. P. (2015). Dual use of peptide mass spectra: Protein atlas and genome annotation. Curr. Plant Biol. 2, 21–24. doi: 10.1016/j.cpb.2015.02.001
Wang, L., Ma, H., Song, L., Shu, Y., and Gu, W. (2012a). Comparative proteomics analysis reveals the mechanism of pre-harvest seed deterioration of soybean under high temperature and humidity stress. J. Proteomics 75, 2109–2127. doi: 10.1016/j.jprot.2012.01.007
Wang, Y., Yuan, X., Hu, H., Liu, Y., Sun, W., Shan, Z., et al. (2012b). Proteomic analysis of differentially expressed proteins in resistant soybean leaves after Phakopsora pachyrhizi infection. J. Phytopathol. 160, 554–560. doi: 10.1111/j.1439-0434.2012.01949.x
Wang, Z. Q., Xu, X. Y., Gong, Q. Q., Xie, C., Fan, W., Yang, J. L., et al. (2014). Root proteome of rice studied by iTRAQ provides integrated insight into aluminum stress tolerance mechanisms in plants. J. Proteomics 98, 189–205. doi: 10.1016/j.jprot.2013.12.023
Washburn, M. P., Wolters, D., and Yates, J. R. (2001). Large-scale analysis of the yeast proteome by multidimensional protein identification technology. Nat. Biotechnol. 19, 242–247. doi: 10.1038/85686
Watson, B. S., Asirvatham, V. S., Wang, L., and Sumner, L. W. (2003). Mapping the proteome of barrel medic (Medicago truncatula). Plant Physiol. 131, 1104–1123. doi: 10.1104/pp.102.019034
Watson, B. S., Bedair, M. F., Urbanczyk-Wochniak, E., Huhman, D. V., Yang, D. S., Allen, S. N., et al. (2015). Integrated metabolomics and transcriptomics reveal enhanced specialized metabolism in Medicago truncatula root Border Cells. Plant Physiol. 167, 1699–1716. doi: 10.1104/pp.114.253054
Weckwerth, W. (2003). Metabolomics in systems biology. Annu Rev Plant Biol. 54, 669–689. doi: 10.1146/annurev.arplant.54.031902.135014
Weckwerth, W. (2010). Metabolomics: An integral technique in systems biology. Bioanalysis 2, 829–836. doi: 10.4155/bio.09.192
Weckwerth, W. (2011a). Unpredictability of metabolism- the key role of metabolomics science in combination with next-generation genome sequencing. Anal. Bioanal. Chem. 400, 1967–1978. doi: 10.1007/s00216-011-4948-9
Weckwerth, W. (2011b). Green systems biology- From single genomes, proteomes and metabolomes to ecosystems research and biotechnology. J. Proteomics 75, 284–305. doi: 10.1016/j.jprot.2011.07.010
Weckwerth, W., Loureiro, M. E., Wenzel, K., and Fiehn, O. (2004a). Differential metabolic networks unravel the effects of silent plant phenotypes. Proc. Natl. Acad. Sci. U.S.A. 101, 7809–7814. doi: 10.1073/pnas.0303415101
Weckwerth, W., Wenzel, K., and Fiehn, O. (2004b). Process for the integrated extraction, identification and quantification of metabolites, proteins and RNA to reveal their co−regulation in biochemical networks. Proteomics 4, 78–83. doi: 10.1002/pmic.200200500
Weckwerth, W., Wienkoop, S., Hoehenwarter, W., Egelhofer, V., and Sun, X. (2014). “From proteomics to systems biology: MAPA, MASS WESTERN, PROMEX, and COVAIN as a user-oriented platform,” in Plant Proteomics: Methods and Protocols, eds J. V. Jorrin-Novo, S. Komatsu, W. Weckwerth, and S. Wienkoop (New York, NY: Springer, LLC), 15–27.
Wienkoop, S., Baginsky, S., and Weckwerth, W. (2010a). Arabidopsis thaliana as a model organism for plant proteome research. J. Proteomics 73, 2239–2248. doi: 10.1016/j.jprot.2010.07.012
Wienkoop, S., Larrainzar, E., Glinski, M., González, E. M., Arrese-Igor, C., and Weckwerth, W. (2008b). Absolute quantification of Medicago truncatula sucrose synthase isoforms and N-metabolism enzymes in symbiotic root nodules and the detection of novel nodule phosphoproteins by mass spectrometry. J. Exp. Bot. 59, 3307–3315. doi: 10.1093/jxb/ern182
Wienkoop, S., Morgenthal, K., Wolschin, F., Scholz, M., Selbig, J., and Weckwerth, W. (2008a). Integration of metabolomic and proteomic phenotypes analysis of data covariance dissects starch and RFO metabolism from low and high temperature compensation response in Arabidopsis thaliana. Mol. Cell. Proteomics 7, 1725–1736. doi: 10.1074/mcp.M700273-MCP200
Wienkoop, S., Staudinger, C., Hoehenwarter, W., Weckwerth, W., and Egelhofer, V. (2012). ProMEX- a mass spectral reference database for plant proteomics. Front. Plant. Sci. 3:125. doi: 10.3389/fpls.2012.00125
Wienkoop, S., Weiß, J., May, P., Kempa, S., Irgang, S., Recuenco-Munoz, L., et al. (2010b). Targeted proteomics for Chlamydomonas reinhardtii combined with rapid subcellular protein fractionation, metabolomics and metabolic flux analyses. Mol Biosyst. 6, 1018–1031. doi: 10.1039/b920913a
Wolschin, F., Wienkoop, S., and Weckwerth, W. (2005). Enrichment of phosphorylated proteins and peptides from complex mixtures using metal oxide/hydroxide affinity chromatography (MOAC). Proteomics 5, 4389–4397. doi: 10.1002/pmic.200402049
Wu, L., Mashego, M. R., van Dam, J. C., Proell, A. M., Vinke, J. L., Ras, C., et al. (2005). Quantitative analysis of the microbial metabolome by isotope dilution mass spectrometry using uniformly 13C-labeled cell extracts as internal standards. Anal. Biochem. 336, 164–171. doi: 10.1016/j.ab.2004.09.001
Wu, T., Yuan, T., Tsai, S. N., Wang, C., Sun, S. M., Lam, H. M., et al. (2009). Mass spectrometry analysis of the variants of histone H3 and H4 of soybean and their post-translational modifications. BMC Plant Biol. 9:98. doi: 10.1186/1471-2229-9-98
Wu, W., Zhang, Q., Zhu, Y., Lam, H. M., Cai, Z., and Guo, D. (2008). Comparative metabolic profiling reveals secondary metabolites correlated with soybean salt tolerance. J. Agric. Food Chem. 56, 11132–11138. doi: 10.1021/jf8024024
Xu, C., Garrett, W. M., Sullivan, J., Caperna, T. J., and Natarajan, S. (2006). Separation and identification of soybean leaf proteins by two-dimensional gel electrophoresis and mass spectrometry. Phytochemistry 67, 2431–2440. doi: 10.1016/j.phytochem.2006.09.002
Xu, H., and Freitas, M. A. (2009). MassMatrix: A database search program for rapid characterization of proteins and peptides from tandem mass spectrometry data. Proteomics 9, 1548–1555. doi: 10.1002/pmic.200700322
Yang, K., Tian, Z., Chen, C., Luo, L., Zhao, B., Wang, Z., et al. (2015). Genome sequencing of adzuki bean (Vigna angularis) provides insight into high starch and low fat accumulation and domestication. Proc. Natl. Acad. Sci. U.S.A. 112, 13213–13218. doi: 10.1073/pnas.1420949112
Ye, H., Gemperline, E., Venkateshwaran, M., Chen, R., Delaux, P. M., Howes-Podoll, M., et al. (2013). MALDI mass spectrometry-assisted molecular imaging of metabolites during nitrogen fixation in the Medicago truncatula–Sinorhizobium meliloti symbiosis. Plant J. 75, 130–145. doi: 10.1111/tpj.12191
Young, N. D., Debellé, F., Oldroyd, G. E., Geurts, R., Cannon, S. B., Udvardi, M. K., et al. (2011). The Medicago genome provides insight into the evolution of rhizobial symbioses. Nature 480, 520–524. doi: 10.1038/nature10625
Young, N. M., Thibault, P., Watson, D. C., and Chrispeels, M. J. (1999). Post-translational processing of two K-amylase inhibitors and an arcelin from the common bean, Phaseolus vulgaris. FEBS Lett. 446, 203–206. doi: 10.1016/S0014-5793(99)00212-4
Zadražnik, T., Hollung, K., Egge-Jacobsen, W., Megliè, V., and Šuštar-Vozliè, J. (2013). Differential proteomic analysis of drought stress response in leaves of common bean (Phaseolus vulgaris L.). J. Proteomics 78, 254–272. doi: 10.1016/j.jprot.2012.09.021
Zhang, N., Venkateshwaran, M., Boersma, M., Harms, A., Howes-Podoll, M., den Os, D., et al. (2012). Metabolomic profiling reveals suppression of oxylipin biosynthesis during the early stages of legume–rhizobia symbiosis. FEBS Lett. 586, 3150–3158. doi: 10.1016/j.febslet.2012.06.046
Zhang, Y., Zhao, L., Li, H., Gao, Y., Li, Y., Wu, X., et al. (2013). GmGBP1, a homolog of human ski interacting protein in soybean, regulates flowering and stress tolerance in Arabidopsis. BMC Plant Biol. 13:21. doi: 10.1186/1471-2229-13-21
Zhen, Y., Qi, J. L., Wang, S. S., Su, J., Xu, G. H., Zhang, M. S., et al. (2007). Comparative proteome analysis of differentially expressed proteins induced by Al toxicity in soybean. Physiol. Plant 131, 542–554. doi: 10.1111/j.1399-3054.2007.00979.x
Keywords: abiotic and biotic stresses, developmental process, functional genomics, signaling pathways, stress tolerance
Citation: Ramalingam A, Kudapa H, Pazhamala LT, Weckwerth W and Varshney RK (2015) Proteomics and Metabolomics: Two Emerging Areas for Legume Improvement. Front. Plant Sci. 6:1116. doi: 10.3389/fpls.2015.01116
Received: 14 August 2015; Accepted: 25 November 2015;
Published: 24 December 2015.
Edited by:
Francesca Sparvoli, Institute of Agricultural Biology and Biotechnology, ItalyReviewed by:
Hao Peng, Washington State University, USALuca Espen, Università degli Studi di Milano, Italy
Copyright © 2015 Ramalingam, Kudapa, Pazhamala, Weckwerth and Varshney. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rajeev K. Varshney, ci5rLnZhcnNobmV5QGNnaWFyLm9yZw==