 Carlos M. Rodríguez López
Carlos M. Rodríguez López Mike J. Wilkinson
Mike J. Wilkinson
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Plant Sci. , 05 June 2015
Sec. Plant Biotechnology
Volume 6 - 2015 | https://doi.org/10.3389/fpls.2015.00397
This article is part of the Research Topic Recent Advances of Epigenetics in Crop Biotechnology View all 18 articles
Increasing crop production at a time of rapid climate change represents the greatest challenge facing contemporary agricultural research. Our understanding of the genetic control of yield derives from controlled field experiments designed to minimize environmental variance. In spite of these efforts there is substantial residual variability among plants attributable to Genotype × Environment interactions. Recent advances in the field of epigenetics have revealed a plethora of gene control mechanisms that could account for much of this unassigned variation. These systems act as a regulatory interface between the perception of the environment and associated alterations in gene expression. Direct intervention of epigenetic control systems hold the enticing promise of creating new sources of variability that could enhance crop performance. Equally, understanding the relationship between various epigenetic states and responses of the crop to specific aspects of the growing environment (epigenetic fingerprinting) could allow for a more tailored approach to plant agronomy. In this review, we explore the many ways in which epigenetic interventions and epigenetic fingerprinting can be deployed for the improvement of crop production and quality.
The sustained growth in food production over the 50 years since the start of the green revolution can be at least partly explained by the introduction of molecular approaches to crop breeding (Evenson and Gollin, 2000). Systematic marker-assisted introgression has now become a mainstay of genetic improvement programs (Collard and Mackill, 2008) and yet some of the most successful varieties of several crops have arisen spontaneously, and have been identified by simple phenotypic selection. These so-called ‘sports’ are far more common in crops that are propagated vegetatively, and can often form a substantial proportion of the varieties grown. The source of the observed phenotypic divergence in sports is often assumed to have a genetic rather than epigenetic origin (Schmitz et al., 2013). In either case, the genetic divergence between sports and their progenitor lines is inevitably minimal, and so are notoriously difficult to differentiate using conventional molecular markers (Breto et al., 2001). The reality is that for the vast majority of instances we do not fully understand how phenotypic variability can be explained at the molecular level (Ball, 2013). This uncertainty is often exacerbated by poor trait definition and a lack of genomic resolution (King et al., 2010) but may sometimes arise from a mistaken presumption of genetic rather than epigenetic causality (Breto et al., 2001; Rois et al., 2013). Ever since Waddington (1942) first proposed the term epigenotype to describe the interface between genotype and phenotype, the science of epigenetics has been progressively adding more layers of complexity to our knowledge of how information is stored and utilized within the living cell. Recent years has seen a dramatic increase in the depth of understanding of how epigenetic control mechanisms operate. There is now growing desire to better understand the stability and role of epigenetic regulatory systems in controlling development, shaping the phenotype, and determining the physiological resilience of higher organisms surviving in fluctuating environments (Geyer et al., 2011; Bräutigam et al., 2013).
Epigenetic processes can affect a phenotype without altering the genetic code (Bird, 2007) and can operate in a number of ways to alter the availability or efficacy of DNA sequences for transcription; determine transcript identity or amend the longevity of mRNA transcripts in the cell (for review, see Chahwan et al., 2011) or by changing the stability or activity of protein products. The many epigenetic mechanisms that mediate these effects include modifications of histone tags, ATP-dependent chromatin remodeling, polycomb/trithorax protein complexes, chemical modification on DNA bases and regulatory processes directing mRNA degradation and alterations to DNA chemistry driven by small RNA molecules, with circular RNA as the latest addition (Wilusz and Sharp, 2013) to the many small RNAs that fulfill this role (i.e., lncRNA, siRNA, microRNA). This array of processes is clearly interconnected and almost certainly acts in a complex, interactive and redundant fashion (Grant-Downton and Dickinson, 2005; Berger, 2007). Describing all the methods developed to study all the mentioned epigenetic layers is outside the scope of this review and we will instead focus on the potential role of the best-studied epigenetic mechanism, DNA methylation, as a route to elicit new advances in crop improvement.
Applied epigenetics is an area of science that is evolving rapidly and spawning new opportunities for the enhancement of crop production. DNA methylation involves the addition of a methyl group to carbon 5 of cytosine bases (forming 5-methylcytosine, 5mC). In plants, DNA methylation can occur in three contexts (i.e., CG, CHG, or CHH, H = a nucleotide other than G). DNA methylation occurring within promoters or coding regions typically act to repress gene transcription. RNA-directed DNA Methylation (RdDM) is an important mechanism by which plants can achieve targeted DNA methylation to reduce expression of a particular gene (Wassenegger et al., 1994). This form of gene silencing is directed by small interfering RNAs (siRNAs) and is often associated with the silencing of transposable elements (TEs). However, the system can also repress the expression of endogenous genes, especially those positioned close to TEs. RdDM relies on the activity of DICER-like 3 (DCL3), Argonaute 4 (AGO4) and the DNA-dependent RNA polymerases Pol IV, and Pol V and the RNA-dependent polymerase RDR2. Collectively, the products of these genes direct the DOMAIN REARRANGED METHYLTRANSFERASE 2 (DRM2) protein to add methyl groups to Cytosines within the targeted region and so repress expression (Naumann et al., 2011). In this way the expression of genes that regulate development or cell metabolism can be altered (Becker and Weigel, 2012). The first and most direct means of exploiting this relationship is through the deliberate perturbation of global methylation patterns via exogenous interventions. This can be achieved in several ways. Most simply, chemical inhibitors of DNA methyltransferases such as 5-azacytidine or decitabine can be used to cause partial, genome-wide DNA demethylation (Stresemann and Lyko, 2008) and so generate new ‘epigenetic’ variants that hopefully include epi-alleles that confer desirable changes to crop phenotype. Amoah et al. (2012) used this strategy when they applied 5-azacytidine to seedlings of rapeseed (Brassica napus) and generated novel lines that exhibited increased seed protein content. This blind tactic for the release of new variation is perhaps most analogous to mutation breeding and relies on the screening of similarly large numbers of individuals to yield positive results. It nevertheless offers the tangible benefit of not requiring a deep understanding of the mechanisms involved.
A more directed approach to epigenetic intervention is made possible by reference to the relationship between changes in the growing environment and associated changes in methylation-driven gene expression. One system by which plants can increase their resilience to challenge by biotic or abiotic threats is by intensifying the responsiveness of their immune system after recognition of specific signals from their environment. This so-called ‘priming’ provides potentially long-lasting protection and is based on eliciting a faster and/or stronger reaction upon subsequent challenge by the same or related stressor (Conrath, 2011). The primed response is made possible by increased sensitivity of previously exposed plants to signal molecules such as b-aminobutyric acid (BABA), volatile organic compounds associated with herbivore damage or to strain-specific pathogen effectors (Pastor et al., 2013). Several studies indicate that the primed response of plants to pathogen attack is mediated through early and strong activation of immune response systems such as the Salicylic Acid (SA) pathway (Kohler et al., 2002; Jung et al., 2009) and the Jasmonic Acid pathway (Turlings and Ton, 2006; Heil and Ton, 2008). It is now becoming clear that RdDM-associated DNA methylation is sometimes implicated in the improved responsiveness of primed plants. For instance, Agorio and Vera (2007) showed that AGO4 is required for full resistance in Arabidopsis against Pseudomonas syringae and by implication RdDM-mediated methylation. Yu et al. (2013) showed that some TEs become demethylated in Arabidopsis following exposure to P. syringae and that this change is associated with restricted multiplication and vascular propagation of the pathogen. The authors inferred that the widespread demethylation of the TEs may have caused prime transcriptional activation of some defense genes. Other studies have similarly shown that manipulation of the growing environment can also evoke DNA methylation-mediated changes to the expression of genes that can influence yield, such as stomatal development (Tricker et al., 2012) or aspects of product quality such as vitamin E levels (Quadrana et al., 2014). Whatever the mechanism of operation leading to these effects, the ability to enhance the defensive capability of crop plants through the prior exposure to signal molecules or to disabled or denign pathogens has innate appeal. This prospect is most immediately tangible for clonal crops, where the effect of the conditioning treatment on methylation-mediated changes to phenotype need not pass through a filial generation. For most seed crops, however, there is the need that the induced changes to methylation status remains stable across generations for methylation-based priming to have practical utility. There is now growing evidence to suggest that at least some environmentally induced methylation marks can remain stable between generations, implying that intergenerational plant priming may also be possible.
Molinier et al. (2006) provided the first compelling evidence that environmentally induced epigenetic change can be retained over subsequent generations that were naïve to the eliciting factor. In this case, exposure to UV and flagellin (an elicitor of plant defenses) was seen to cause Arabidopsis to respond by increasing homologous recombination as detected by restoration of transgene function. Whilst the authors were unable to assign the effect to a particular epigenetic mechanism, they were able to demonstrate that the effect did not require presence of the transgene, was dominant, could be inherited from either parent and persisted for at least four filial generations. Boyko et al. (2007) subsequently found that progeny of tobacco mosaic virus (TMV)-infected plants show reduced methylation levels of R-gene-like genes, and enhanced resistance to different pathogens (Kathiria et al., 2010). Likewise, Slaughter et al. (2012) demonstrated that Arabidopsis exposed to localized infection by an avirulent strain of P. syringae or priming-inducing treatments with BABA produce descendants that are more resistant to Hyaloperonospora arabidopsidis. These and many other examples of transgenerational priming of resistance (for review, see Pastor et al., 2013) imply that it may be possible to supply the grower communities with seed lots as well as clonal cuttings that are primed to enhance tolerance to biotic or abiotic stresses. Delivery of such a service will depend on stability of the effect, ability to assure that the expected change to DNA methylation has occurred, and most importantly, that there are no yield penalties associated with the priming event itself. Certainly, Luna et al. (2012) demonstrated that whilst the asymmetric DNA methyltransferase (drm1drm2cmt3) triple mutant of Arabidopsis (blocked for RdDM-dependent DNA methylation function) is more resistant to biotrophic pathogens such as H. arabidopsidis and P. syringae, it is also more susceptible to the necrotrophic fungus Alternaria brassicicola. Thus, it is entirely plausible that some beneficial changes that are induced by priming may come at the expense of some associated detrimental features. The nature of such interactions will no doubt emerge with time and effort.
There are also more direct ways in which transgenerational stability of epi-alleles could ultimately be integrated into crop breeding efforts. In a landmark paper, Hauben et al. (2009) demonstrated that it is possible to obtain stable epigenotypes exhibiting improved energy use efficiency (an important yield determinant) through recurrent phenotypic selection of isogenic B. napus lines. Furthermore, crosses between these genetically identical but epigenetically divergent lines generated hybrids with a 5% yield increase on top of heterosis. Tricker et al. (2013a) for showed that environmentally induced epi-alleles associated with drought and low relative humidity tolerance can become fixed and remain stable over several generations. These observations raise the scope of targeted management of the growing environment during breeding to deliberately elicit and fix epigenetic changes responsible for control of a particular trait or developmental process. The high likelihood that genotypes will vary in their capacity to become primed or to remain stably fixed in a desired state (Daymond et al., 2011) provides scope for simultaneous genetic and epigenetic selection for (or against) aspects of plant plasticity and resilience. To our knowledge, this type of profiling has yet to be formally incorporated into commercial breeding efforts. In the following sections we therefore explore a range of specific approaches that hold promise to enhance contemporary crop improvement efforts.
At a more fundamental level, Cortijo et al. (2014) have provided an elegant illustration of how the transgenerational stability of some induced methylation marks can be usefully exploited for forward genetics efforts when they were able to construct linkage maps to describe the epigenetic basis of complex traits, so-called epiQTL analysis (Long et al., 2011; Cortijo et al., 2014). This strategy has the significant potential advantage over conventional QTL analysis by circumventing the need for functional mutational differences between parental genotypes of mapping populations used for forward genetics.
DNA methylation-dependent gene regulation plays an important role in orchestrating cellular differentiation and development (Rogers and Rogers, 1995; Manning et al., 2006; Henderson and Jacobsen, 2007; Feng et al., 2010; Ito et al., 2010; Yaish et al., 2011) and also provides the basis for genome–environment interactions that confer agility and plasticity of gene expression, and mediates molecular response to fluctuations in the living environment (Amoah et al., 2012). The genomes of almost all phyla include at least one alternate form of chemically modified base (Hattman, 2005), including N6- methyladenine (m6A), N4-methylcytosine (m4C), 5-methylcytosine (5mC), and 5-hydroxymethylcytosine (5hmC) (Figure 1). Of these, 5mC is by far the best studied and was originally thought to be the only functional base modification found in higher organisms (Kriaucionis and Heintz, 2009). Environmentally induced changes in 5mC have also been shown to be at least partially stable between filial generations (Tricker et al., 2013a; Cortijo et al., 2014). We are just starting to understand the mechanisms that either prevent or permit the inheritance of such epigenetic changes (Iwasaki and Paszkowski, 2014). The value of a particular 5mC as a biomarker for a particular physiological or developmental state relies partly on the consistency its association with each particular state but also on its stability. There is considerable variation in the extent to which a locus shows both consistency and stability. For example, in tomato, a spontaneous epi-allele (cnr) is responsible for the inhibition of fruit ripening in some epi-mutant lines (Manning et al., 2006). The methylation status of sites within this locus are highly predictive of the observed phenotype and reversions (demethylation and associated phenotypic change) occur at a frequency of roughly one in 1000. In comparison, mutability of the epigenetic silencing of the DWARF1 gene in rice occurs in around 1 in 10 plants (Miura et al., 2009). Overall, it appears that DNA methylation patterns do not fluctuate randomly between generations or in response to the environment but neither are they completely stable (Becker and Weigel, 2012). It will therefore desirable to identify specific sites or loci that are both stable and predictable for a particular state to maximize the capacity to apply epifingerprinting techniques across a wide range of germplasm and also between laboratories.
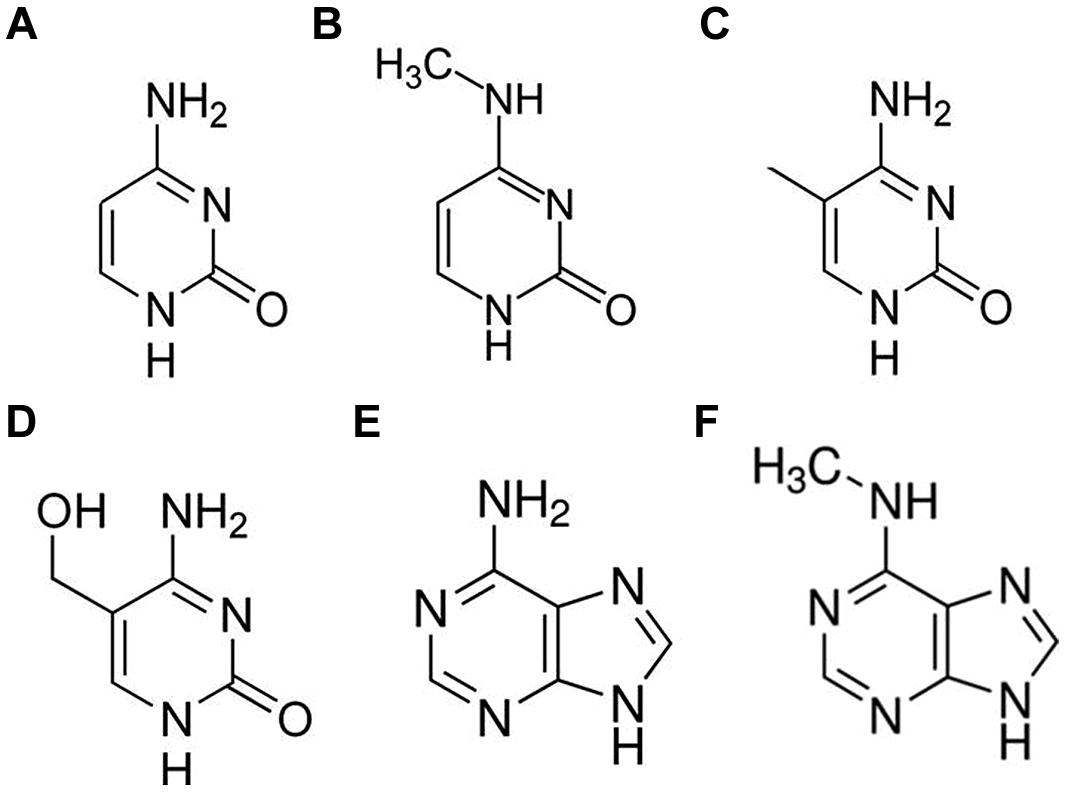
FIGURE 1. Molecular structures of DNA bases: cytosine (A), N4-methylcytosine (m4C; B), 5-methylcytosine (5mC; C), 5-hydroxymethylcytosine (5hmC; D), adenine (E), and N6-methyladenine (m6A; F).
It is now emerging that other modified bases are also present in at least some eukaryotic organisms. These most notably include m6A and 5hmC, although relatively little is currently known about the distribution or function of these bases in plants (Ashapkin et al., 2002). The methylated modification of adenine, m6A, was first discovered in Escherichia coli and has since been found in a wide range of prokaryotes and simple eukaryotes (e.g., prokaryotes Dunn and Smith, 1955; ciliates, Hattman, 2005). In prokaryotes, it appears that m6A induces DNA conformational changes that alter protein–DNA interactions (Sternberg, 1985). There is indirect evidence that m6A may also be present in mammals (Polaczek et al., 1998; Ashapkin et al., 2002) although this has yet to be demonstrated unequivocally. There is stronger evidence for the presence of m6A in plants (Ashapkin et al., 2002), including the identification of a putative adenine DNA methyltransferase gene in the genome of Arabidopsis thaliana (Sternberg, 1985). While it is unclear whether m6A is essential for the regulation of eukaryotic genes, the detection of m6A residues in the DNA methylation maintenance gene DRM2 (Ashapkin et al., 2002) implies that this possibility is at least plausible, and that the presence and location of this modified base could be used for diagnostic purposes.
The alternate modification of cytosine, 5hmC, is present both in the nuclear (Kriaucionis and Heintz, 2009) and mitochondrial (Shock et al., 2011) genomes of mammals. This form of the base is far less abundant than 5mC and is typically more highly tissue-specific (Muenzel et al., 2011), perhaps implying a role in tissue differentiation and development. In plants, 5hmC has only been reported in the genome of chloroplasts (Moricová et al., 2013) although more recent publications demonstrate that it is either absent or present at undetectably low levels in plants (Erdmann et al., 2015). This form of cytosine has been proposed as an intermediate in either the active or passive demethylation of 5mC (Huang et al., 2010). However, recent evidence leads some to suggest that it may have an important functional role in its own right, at least in animals (Robertson et al., 2011a; Wu et al., 2011). Moreover, under high resolution melting (HRM) conditions 5mC has been shown to elicit a stabilizing effect to the double stranded DNA structure (Rodríguez López et al., 2010a); a feature that accords with its reported effect on the fine structure of DNA (Heinemann and Hahn, 1992). In contrast, spectroscopic (Thalhammer et al., 2011), calorimetric (Wanunu et al., 2011), and HRM (Rodríguez López et al., 2012a) analyses have all suggested that presence of the alternate base modifications (5hmC and m6A) in the DNA could reverse the stabilizing effect of 5mC. Whether or not the changes to DNA thermostability induced by 5hmC have functional impact on gene expression is still a matter of conjecture. Certainly, some authors have reported that 5mC hydroxylation is associated with the activation of gene transcription (Ito et al., 2010; Thalhammer et al., 2011; Wanunu et al., 2011) while others argue that any contribution to transcriptional activation or repression is highly context-dependent (Wu et al., 2011). Whatever role (if any) that these alternate base modifications play in gene regulation, it is already clear that they are far less abundant, if present at all, in the plant genome than 5mC and so probably hold only limited value as diagnostic marks for epifingerprinting purposes. It is therefore the distribution of 5mC in the genome that has formed the focus of attempts to link epifingerprints to the physiological, developmental, or stress status of higher organisms, including crop plants.
An array of methods has been developed to describe the global pattern of 5mC across the genome (for extensive reviews on the subject, see Tost and Gut, 2009; Chaudhry, 2010; Plongthongkum et al., 2014). All methods carry their own limitations (Rodríguez López et al., 2010a) but can be broadly grouped into three functional types that: (1) indicate the methylation status of a specific sequence; (2) reveal the degree and patterning of DNA methylation across partly characterized genomes; (3) facilitate the discovery and sequencing of new epialleles (Fraga and Esteller, 2002; Dahl and Guldberg, 2003).
The ability to propagate elite or desirable clones is an essential part of the seed production industry. The advent of reliable in vitro systems for the replication and regeneration of plant materials has led to their widespread deployment for propagation (Bertrand et al., 2011; Etienne et al., 2012), germplasm conservation (Fang et al., 2009), and breeding purposes (Henry, 1998), as well as for more fundamental research on model species (Berdasco et al., 2008; De-la-Peña et al., 2012; Moricová et al., 2013). For micropropagation and genetic transformation systems to be efficient, it is necessary that the plants recovered from them are genetically and epigenetically faithful to the original stock material. Trueness-to-type is of particular importance when propagating elite genotypes of high value crops such as grapevine: especially in traditional vine areas where high clone quality is a prerequisite (Schellenbaum et al., 2008). In comparison to genetic somaclonal variation, divergence between DNA methylation patterns is generally wider among regenerated plants and can be directly associated with ‘plastic’ phenotypic variation (Miguel and Marum, 2011). The loss of epigenetic fidelity during micropropagation has been a major source of economic damage in several crops. For instance, in oil palm, mantled inflorescence syndrome was found to be associated with global changes to C-methylation status during micropropagation, and caused catastrophic reductions in yield among all affected plants and incurred huge costs to the industry (Matthes et al., 2001). Many studies have reported global changes to the distribution of cytosine methylation can be induced by in vitro culture spanning an impressive array of species in a wide taxonomic spread. Examples include: tobacco (Nicotiana tabacum, Schmitt et al., 1997); rice (Oryza sativa, Xiong et al., 1999); strawberry (Fragaria Xananassa, Hao et al., 2002); potato (Solanum tuberosum, Joyce and Cassells, 2002; A. thaliana, Bardini et al., 2003); oil palm (Elaeis guineensis, Jaligot et al., 2004); and cocoa (Theobroma cacao, Rodríguez López et al., 2010b). On the other hand, some forms of such ‘somaclonal variation’ may offer a source of valuable new variation that has potential applications in plant breeding (Henry, 1998). Furthermore, different studies have shown that epigenetic regulation plays an important role during plant development in vitro (Rodríguez López et al., 2010b; Nic-Can et al., 2013). Regardless of whether the change in methylation status evokes a desirable or unwanted outcome, there is clearly great value in the ability to detect these changes or at least to predict the scale of any phenotypic or physiological divergence. Advances in our understanding of the links between gene expression and phenotype mean that the ambition may now turn from simply viewing these plants as a new source of variation for breeding and toward a more targeted approach that deliberately manipulates the process for use in crop improvement efforts.
The majority of agricultural land is cultivated with commodity crops that are either highly inbred or clonal. These genetically invariant populations nevertheless exhibit measurable morphological or developmental plasticity, even when grown under controlled conditions, which may be at least partly explained by stable epigenomic states (Hauben et al., 2009). It has recently been argued that these epigenetic sources of variation may even be greater than those attributable to genetic causes (Hirsch et al., 2013; Schmitz et al., 2013). Several authors have linked genotype-specific changes to DNA methylation to yield components or to other agronomically desirable traits (e.g., Gourcilleau et al., 2010; Alonso et al., 2014; Table 1). The first classic example of a single epiallelic gene variant was attributed to hypermethylation of the CYCLOIDEA gene of Linaria vulgaris; a state which causes radial symmetry of previously bilaterally symmetric flowers (Cubas et al., 1999). Other epigenetic variants have subsequently emerged with features that have economic potential. For instance, the hypomethylation of the rice gene FIE1 induces its ectopic expression and results in a dwarf and flower-aberrant phenotype (Zhang et al., 2012). Goettel and Messing (2013) reported that cytosine methylation of a gene (P1-rr) encoding for a Myb-like transcription factor that mediates pigmentation in floral organs and grains, is negatively correlated with transcription and pigment levels. These mutations are thought to have arisen spontaneously by somatic epi-mutation and later became fixed after repeated passage through meiosis.

TABLE 1. Examples of plant genes involved in agronomic traits affected by DNA methylation.
Systematic selection for fixed epi-loci is not the only possible source of new varietal material with potential to improve crop production or quality. Environmentally induced epi-alleles also offer an important potential source of exploitable variation. For many inbreeding and clonal crops, environmentally induced epigenetic variation can sometimes outweigh genetic variation, with such changes being induced by exposure to various aspects of the living environment (Raj et al., 2011; Tricker et al., 2012; Hirsch et al., 2013). These properties can lead to an epigenetic convergence of populations when grown under similar conditions (Schulz et al., 2014) but can also lead to spontaneous divergence of fixed epigenetic states (Becker et al., 2011). Tricker et al. (2013b) proposed an approach in which the deliberate manipulation of the specific aspects of the growing environment could be used to induce desirable changes in tolerance to low humidity and periodic drought. Nevertheless, the disentanglement of this kind of epigenetic variation from the genetic background that underpins the capacity to produce new variability continues to pose major technical difficulties (Cortijo et al., 2014) and is probably still some way from commercial reality.
For vegetatively propagated perennial crops such as grapevine (Zufferey et al., 2000) or Pinus radiata (Fraga et al., 2001) the need to fix between generations is circumvented. For these crops there is a long association between productivity and quality characteristics and plant age. The possibility that this relationship has an epigenetic basis and so is amiable for manipulation is especially appealing. Certainly, it is known that DNA methylation changes progressively during maturation and aging, for both plants and animals species (Theiss and Follmann, 1980; Quemada et al., 1987; Fraga et al., 2005). There is also evidence that these changes are associated with altered expression of genes that are implicated in morphological changes in plants (Galaud et al., 1993) and animals (Zhang et al., 2002). More specifically, the extent of genomic DNA methylation in pine is a strong indicator of aging and can provide molecular evidence of reinvigoration (Fraga et al., 2001). Thus, there is scope to manipulate the methylation status of crop genomes either chemically using methyltransferase inhibitors, by exposure to signaling molecules or by manipulation of the growing environment. Individuals exhibiting stable, rejuvenated methylation profiles, and associated phenotypes could then be selected and used for commercial planting.
In addition to the generation of new variation there is also considerable scope for deploying epigenetic fingerprinting approaches to improve the efficacy of agronomic or prophylactic interventions. Plants are sessile organisms and so unable to avoid abiotic or biotic stresses. They must instead rely on rapid and effective stress response systems to withstand harmful changes to the living environment to enhance their chances of survival. Plants have amassed an array of mechanisms for detecting and then responding to stresses in ways that can include substantial amendments to key metabolic pathways (Madlung and Comai, 2004). Such responses can be activated in a number of ways including the adjustment of the transcriptional control of genes through differential cytosine methylation (Aceituno et al., 2008).
Several authors have noted that large numbers of biotic and abiotic stresses induce global changes to the methylation patterns of plants (Stokes et al., 2002; Boyko and Kovalchuk, 2008, 2011; Chinnusamy and Zhu, 2009). This feature means there is often a clear relationship between the detection of a particular stress by a plant and overall C-methylation profile. This property means that there is scope for the use of C-methylation fingerprinting approaches as a tool to diagnose the early onset or asymptomatic exposure of a crop to a range of stresses. Several workers have demonstrated that diagnostic changes in methylation fingerprints are associated with exposure to a wide range of abiotic stresses including drought (Raj et al., 2011; Tricker et al., 2013b), low relative humidity, (Tricker et al., 2012), low temperatures (Pan et al., 2011), salt and heavy metals (Choi and Sano, 2007; Verhoeven et al., 2010), and low nutrient levels (Verhoeven et al., 2010). The same is seemingly also true for exposure to biotic stresses, with changed DNA methylation profiles also being reported following plant–herbivore (Verhoeven et al., 2010, Herrera and Bazaga, 2013) and plant–pathogen interactions (Mason et al., 2008; Boyko and Kovalchuk, 2011). These observations have yet to be used as a basis to develop a robust set of methylation markers to routinely diagnose exposure of crops to these stresses but this aspiration appears both attractive and tractable within a relatively short time period.
There is also opportunity to use C-methylation profiling to gain better understanding of the relationship between the stress and the physiological response of the plant to that stress. Herrera and Bazaga (2013) reported that phenotypic changes adopted by the plant in response to stress (such as prickly leaves induced by herbivory) positively correlated to global changes in DNA methylation. Resistance to Rhizoctonia solani in maize is similarly linked to global shifts in DNA methylation (Li et al., 2011). Sequence characterisation of these differentially methylated loci may ultimately provide a useful route through which to discover candidate genes that are implicated in these responses. This approach has been adopted in other cases. For instance in rice, where resistance to bacterial blight is linked to plant age, it has been shown that acquired resistance is regulated by the hypo/hypermethylation of several loci. Such methylation changes correlate with the expression levels of several genes including a putative Gag-Pol polyprotein, a putative RNA helicase of the Ski2 subfamily and a putative receptor-like protein kinase (Sha et al., 2005). There has also been interest in tracking changes in DNA methylation associated with virus silencing in plants (English et al., 1996).
The apparent stability of some C-methylation sites following induction allows for stress detection long after initial exposure and means that carefully selected epimarkers potentially provide a more robust source of a posteriori stress diagnosis than more ephemeral changes within the cell such as the abundance of mRNA (transcriptomics), proteins (proteomics), or metabolites (metabolomics). Furthermore, this ‘memory of stress’ is not limited to cells and cell lineages but as described above can also persist through filial generations. Boyko and Kovalchuk (2011) showed that changes to the DNA methylation patterns of plants associated with continuous interactions with pathogens were successfully transmitted and fixed in their progeny seemingly also potentially allowing for the diagnosis of parental stress exposure.
Looking ahead, it seems inevitable that in the relatively near future there will be methylation markers developed for many crops able to track developmental progression and also the exposure and response of the plants to the stresses they are experiencing. The long-term possibility of using these markers as sentinels of health and developmental state leads to the enticing prospect that they may ultimately be integrated into models to predict yield. If applied onto a broader scale, it is even possible that epigenetic fingerprinting of airborne pollen samples for signatures of stress could eventually augment existing monitoring of the landscape for the effects of climate change or to track new epidemiological events, and so facilitate more timely and targeted interventions.
The high market value of ‘top end’ agricultural products used for nutritional or medicinal properties frequently attracts fraudulent labeling of lesser products with lower quality or commercial value (Mader et al., 2011). Certifying the authenticity and origin of such products is a legal requirement in many jurisdictions to avoid unfair competition and assure consumers protection against fraudulent practices (Reid et al., 2006). Although there is an increasing demand by consumers for high quality food products (Luykx and van Ruth, 2008), the majority of authentication techniques for food products have focussed on species or varietal identification or on the chemical composition of processed foods (Sentandreu and Sentandreu, 2011). However, quality traits of plant products are not only influenced by the choice of species or cultivars. In some agricultural products, quality can be primarily determined by the harvested components of the crop used to generate a product (Srancikova et al., 2013) or else by climate, location, crop age, management systems used to cultivate the crop (e.g., industrialized versus organic farming, manure versus N-fertilizer; Posner et al., 2008). Equally, soil conditions, as well as the interactions of different environmental conditions or “terroir” can be viewed as important quality determinants of products such as wine (van Leeuwen et al., 2004). These conditions affect plant composition variables such as dry matter content and furthermore starch, crude protein, amino acids, nitrate, sugars, and citric acid (Müller and Hippe, 1987). The measurement of such components has often necessitated development of a series of independent tests to detect fraudulent labeling. The use of methylation profiles as a diagnostic tool relating to several different aspects of crop quality is therefore appealing because it provides a ‘plant’s perspective of the growing environment.’ This area of methylation profiling is still untested but would be especially alluring if evidence can be provided to distinguish between agronomic practices (such as those used for organic farming) that are currently primarily verified only by certification.
New evidence is now emerging to suggest that this may be possible. For example, Boyko et al. (2010) showed that exposure of A. thaliana to a range of mild abiotic modifications (salt around the roots, UVC, cold, heat, and flood) could be detected by reproducible changes in DNA methylation patterns. Similarly, in clonally propagated poplar grown under different conditions of water availability, differences in genome-wide DNA methylation paralleled differences in transcriptome, suggesting an epigenomic basis for the clone history-dependent divergence (Raj et al., 2011) and illustrating the plausibility of epigenetic profiling to characterize watering regime. Indeed, cultivation conditions of a wide variety of plants have now been shown to induce differences at methylome level (i.e., Dandelions, Verhoeven et al., 2010; mangrove, Lira-Medeiros et al., 2010; alligator weed, Li et al., 2013). These findings open the door for deploying epigenetic profiling approaches to diagnose growth conditions and geographical region of origin of otherwise identical crops and theirs processed products.
It is therefore tempting to speculate that quality traits associated with crop management may also be detectable using the same C-methylation markers. There is equally scope also to differentiate between products generated from parts of the plant with different market value. Certainly, it is now well established that different cell types or tissues within an organism can have markedly different methylation profiles (Baron et al., 2006; Feng et al., 2010; Rodríguez López et al., 2010b, 2012b) and that the use of epigenetic markers has been proven to be an effective means of generating organ-specific epigenetic markers as a tool for identifying the tissue of origin in plant (Rodríguez López et al., 2010b) and animal (Rodríguez López et al., 2012b) products. This gives rise to the prospect of simplifying global methylation patterns to generate generate smaller numbers of highly diagnostic epimarkers for use in food quality assessment. Such markers could not only have potential value in identifying the cultivating system and product composition, but also to other factors affecting quality such as storage, transport and processing conditions.
Epigenetic control mechanisms provide the crop plant with an ability to respond to the many and varied challenges posed to them by an ever-changing growing environment during growth and development. Of all these mechanisms, histone tail modifications and DNA methylation are by far the better studied. Of the two, DNA methylation way of action is the better understood, the easier to analyze and the one with most associated epialleles in the literature.
We have shown that the deliberate manipulation of this relationship through direct (chemical) and indirect (environmental) interventions holds the potential to generate new and useful variability to the crop. In some cases the induced changes can alter the genome regulatory system of the crop in such a way as to allow it to better cope with particular, anticipated stress types. The capability to fix at least some of these states across generations offers the tantalizing possibility of a targeted system of epigenetic breeding to augment existing breeding efforts, and has particular appeal for long-lived clonal crops. We have also shown that gaining a better understanding of the relationship between the stress elicitor and the changed epigenetic state offers new opportunities for the identification of candidate genes that are important in conferring resilience against important stresses. Such stable epigenetic markers, especially if associated to commercially interesting traits, can be of interest to plant breeders. Apart from variations in the gene sequence, epigenetic variation may contribute to commercially interesting traits.
However, it is perhaps as a diagnostic tool of stress that there is the greatest source of unexplored opportunity for short-term step improvements to crop management and production. A plethora of works have shown that there is a clear and strong relationship between a vast array of stresses and the C-methylation status of crop plants. Conversion of these global differences into specific diagnostic epimarks of stress detection and stress-induced physiological response by the crop plants offers a range of opportunities for the improvement in varietal selection, crop management, for the control of pests and disease, and to control and regulate the quality of agricultural products. Moreover, the methylome epifingerprinting can be considered as a measure of the phenotype of the crop’s genome. Such an ‘epiphenotype’ not only provides a new diagnostic tool to study stress responses and developmental progression but also provides a useful bridge that allows direct functional relationships to be inferred between the growing environment and associated genome regulation. In the medium term we expect the collective impact of these developments to enable substantive advances in crop production and protection; an epigreen revolution (Figure 2).
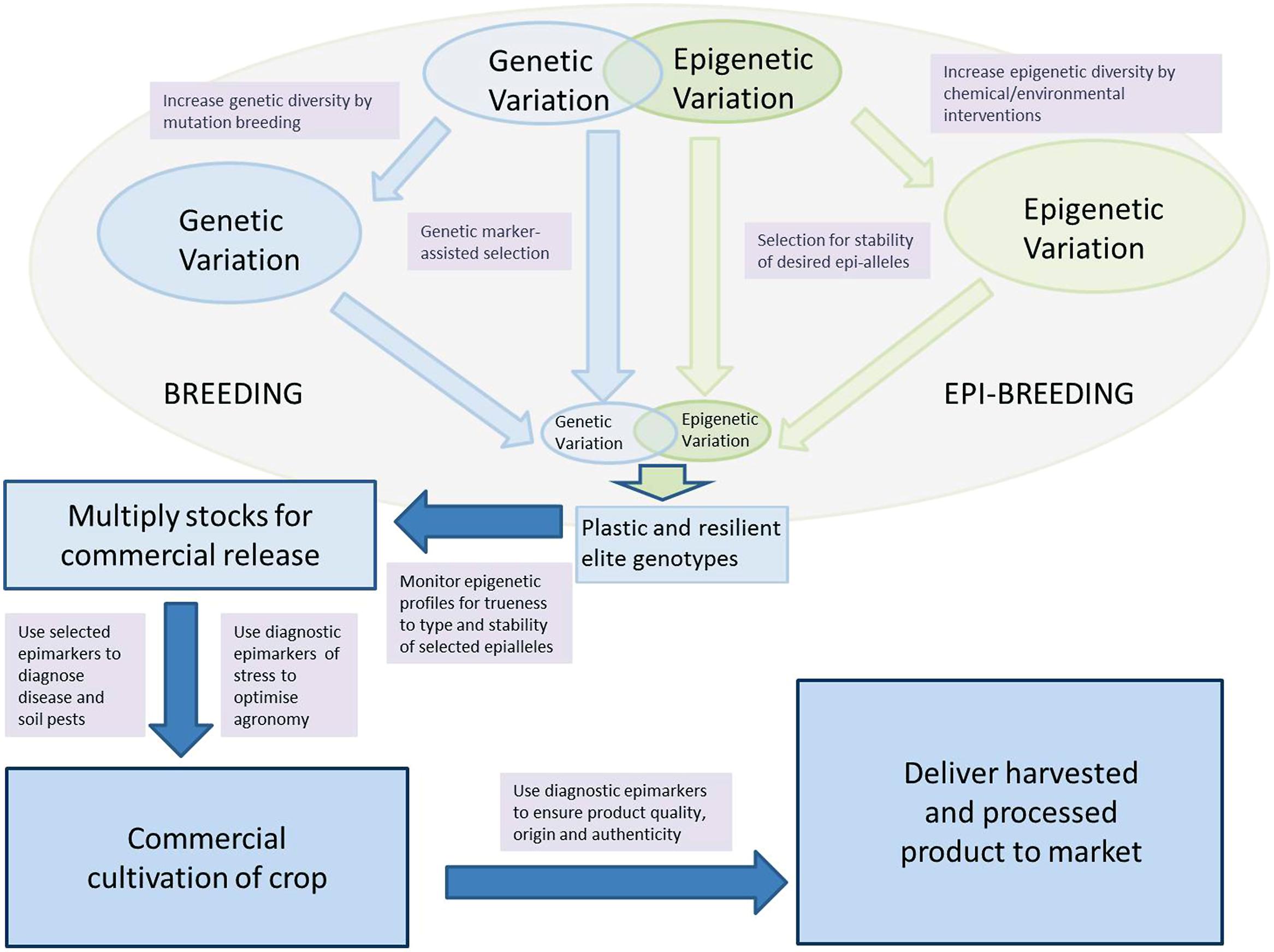
FIGURE 2. Epi-fingerprinting and epi-interventions for improved crop production and food quality: schematic illustration on how epi-fingerprinting and epigenetic interventions could potentially impact on various parts of the agricultural supply chain. The model starts with a combined breeding and epi-breeding approach to varietal production and is followed by seed/clone multiplication systems that uses epigenetic profiling techniques to minimize appearance of off-types. Cultivation of the crop is augmented by agronomic and pest/disease management strategies that utilize epi-fingerprinting to diagnose/optimize the health status of the crop. At delivery to market, epi-fingerprinting is used to authenticate products and to ensure quality.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Aceituno, F. F., Nick, M., Seung, Y. R., and Rodrigo, A. G. (2008). The rules of gene expression in plants: organ identity and gene body methylation are key factors for regulation of gene expression in Arabidopsis thaliana. BMC Genomics 9:438–451. doi: 10.1186/1471-2164-9-438
Agorio, A., and Vera, P. (2007). ARGONAUTE4 is required for resistance to Pseudomonas syringae in Arabidopsis. Plant Cell 198, 3778–3790. doi: 10.1105/tpc.107.054494
Alonso, C., Perez, R., Bazaga, P., Medrano, M., and Herrera, C. M. (2014). Individual variation in size and fecundity is correlated with differences in global DNA cytosine methylation in the perennial herb Helleborusfoetidus (Ranunculaceae). Am. J. Bot. 101, 1309–1313. doi: 10.3732/ajb.1400126
Amoah, S., Kurup, S., Rodríguez López, C. M., Welham, S. J., Powers, S. J., Hopkins, C. J., et al. (2012). A hypomethylated population of Brassica rapa for Forward and reverse Epi-Genetics. BMC Plant Biol. 12:193–210. doi: 10.1186/1471-2229-12-193
Ashapkin, V. V., Kutueva, L. I., and Vanyushin, B. F. (2002). The gene for domains rearranged methyltransferase (DRM2) in Arabidopsis thaliana plants is methylated at both cytosine and adenine residues. FEBS Lett. 532, 367–372. doi: 10.1016/S0014-5793(02)03711-0
Avivi, Y., Morad, V., Ben-Meir, H., Zhao, J., Kashkush, K., Tzfira, T., et al. (2004). Reorganization of specific chromosomal domains and activation of silent genes in plant cells acquiring pluripotentiality. Dev. Dyn. 230, 12–22. doi: 10.1002/dvdy.20006
Bardini, M., Labra, M., Winfield, M., and Sala, F. (2003). Antibiotic-induced DNA methylation changes in calluses of Arabidopsis thaliana. Plant Cell Tissue Organ Cult. 72, 157–162. doi: 10.1023/A:1022208302819
Baron, U., Türbachova, I., Hellwag, A., Eckhardt, F., Berlin, K., Hoffmuller, U., et al. (2006). DNA methylation analysis as a tool for cell typing. Epigenetics 1, 55–60. doi: 10.4161/epi.1.1.2643
Becker, C., Hagmann, J., Muller, J., Koenig, D., Stegle, O., Borgwardt, K., et al. (2011). Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature 480, 245–249. doi: 10.1038/nature10555
Becker, C., and Weigel, D. (2012). Epigenetic variation: origin and transgenerational inheritance. Curr. Opin. Plant Biol 15, 562–567. doi: 10.1016/j.pbi.2012.08.004
Berdasco, M., Alcazar, R., García-Ortiz, M. V., Ballestar, E., Fernández, A. F., Teresa Roldán-Arjona, T., et al. (2008). Promoter DNA hypermethylation and gene repression in undifferentiated Arabidopsis cells. PLoS ONE 3:e3306. doi: 10.1371/journal.pone0003306.g003
Berger, S. L. (2007). The complex language of chromatin regulation during transcription. Nature 447, 407–412. doi: 10.1038/nature05915
Bertrand, B., Alpizar, E., Llara, L., Santacreo, R., Hidalgo, M., Quijano, J. M., et al. (2011). Performance of Coffea arabica F1 hybrids in agroforestry and full-sun cropping systems in comparison with pure lines varieties. Euphytica 181, 147–158. doi: 10.1007/s10681-011-0372-7
Boyko, A., Blevins, T., Yao, Y., Golubov, A., Bilichak, A., Ilnytskyy, Y., et al. (2010). Transgenerational adaptation of Arabidopsis to stress requires DNA methylation and the function of Dicer-like proteins. PLoS ONE 5:e9514. doi: 10.1371/journal.pone.0009514
Boyko, A., Kathiria, P., Zemp, F. J., Yao, Y., Pogribny, I., and Kovalchuk, I. (2007). Transgenerational changes in the genome stability and methylation in pathogen-infected plants: (virus-induced plant genome instability). Nucleic Acids Res. 35, 1714–1725. doi: 10.1093/nar/gkm029
Boyko, A., and Kovalchuk, I. (2008). Epigenetic control of plant stress response. Environ. Mol. Mutagen. 49, 61–72. doi: 10.1002/em.20347
Boyko, A., and Kovalchuk, I. (2011). Genetic and epigenetic effects of plant–pathogen interactions: an evolutionary perspective. Mol. Plant 4, 1014–1023. doi: 10.1093/mp/ssr022
Bräutigam, K., Vining, K. J., Lafon-Placette, C., Fossdal, C. G., Mirouze, M., Gutiérrez Marcos, J., et al. (2013). Epigenetic regulation of adaptive responses of forest tree species to the environment. Ecol. Evol. 3, 399–415. doi: 10.1002/ece3.461
Breto, M. P., Ruiz, C., Pina, J. A., and Asíns, M. J. (2001). The diversification of citrus clementina hort. ex tan., a vegetatively propagated crop species. Mol. Phylogen. Evol. 21, 285–293. doi: 10.1006/mpev.2001.1008
Chahwan, R., Wontakal, S. N., and Roa, S. (2011). The multidimensional nature of epigenetic information and its role in disease. Discov. Med. 11, 233–243.
Chaudhry, M. A. (2010). Strategies for detecting genomic DNA methylation: a survey of US patents. Recent Pat. DNA Gene. Seq. 4, 79–85. doi: 10.2174/187221510793205719
Chinnusamy, V., and Zhu, J.-K. (2009). Epigenetic regulation of stress responses in plants. Curr. Opin. Plant Biol. 12, 133–139. doi: 10.1016/j.pbi.2008.12.006
Choi, C.-S., and Sano, H. (2007). Abiotic-stress induces demethylation and transcriptional activation of a gene encoding a glycerophosphodiesterase-like protein in tobacco plants. Mol. Genet. Genomics 277, 589–600. doi: 10.1007/s00438-007-0209-1
Collard, B. C. Y., and Mackill, D. J. (2008). Marker-assisted selection: an approach for precision plant breeding in the twenty-first century. Philos. Trans. R. Soc. Lond. B Biol. Sci. 363, 557–572. doi: 10.1098/rstb.2007.2170
Conrath, U. (2011). Molecular aspects of defence priming. Trends Plant Sci. 16, 524–531. doi: 10.1016/j.tplants.2011.06.004
Cortijo, S., Wardenaar, R., Colomé-Tatché, M., Gilly, A., Etcheverry, M., Labadie, K., et al. (2014). Mapping the epigenetic basis of complex traits. Science 343, 1145–1148. doi: 10.1126/science.1248127
Cubas, P., Vincent, C., and Coen, E. (1999). An epigenetic mutation responsible for natural variation in floral symmetry. Nature 401, 157–161. doi: 10.1038/43657
Da, K., Nowak, J., and Flinn, B. (2012). Potato cytosine methylation and gene expression changes induced by a beneficial bacterial endophyte, Burkholderia phytofirmans strain PsJN. Plant Physiol. Biochem. 50:24e34. doi: 10.1016/j.plaphy.2011.09.013
Dahl, C., and Guldberg, P. (2003). DNA methylation analysis techniques. Biogerontology 4, 233–250. doi: 10.1023/A:1025103319328
Das, O. P., and Messing, J. (1994). Variegated phenotype and developmental methylation changes of a maize allele originating from epimutation. Genetics 136, 1121–1141.
Daymond, A. J., Tricker, P. J., and Hadley, P. (2011). Genotypic variation in photosynthesis in cacao is correlated with stomatal conductance and leaf nitrogen. Biol. Plant. 55, 99–104. doi: 10.1007/s10535-011-0013-y
De-la-Peña, C., Nic-Can, G. I., Ojeda, G., Herrera-Herrera, J. L., López-Torres, A., Wrobel, K., et al. (2012). KNOX1 is expressed and epigenetically regulated during in vitro conditions in Agave spp. BMC Plant Biol. 12:203–214. doi: 10.1186/1471-2229-12-203
Dubin, M. J., Zhang, P., Meng, D., Remigereau, M.-S., Osborne, E. J., Casale, F. P., et al. (2014). DNA methylation variation in Arabidopsis has a genetic basis and shows evidence of local adaptation. Elife 4:e05255. doi: 10.7554/eLife.05255
Dunn, D. B., and Smith, J. D. (1955). Occurrence of a new base in the deoxyribonucleic acid of a strain of Bacterium Coli. Nature 175, 336–337. doi: 10.1038/175336a0
English, J. J., Mueller, E., and Baulcombe, D. C. (1996). Suppression of virus accumulation in transgenic plants exhibiting silencing of nuclear genes. Plant Cell 8, 179–188. doi: 10.1105/tpc.8.2.179
Erdmann, R. M., Souza, A. L., Clish, C. B., and Gehring, M. (2015). 5-Hydroxymethylcytosine is not present in appreciable quantities in Arabidopsis DNA. G3 (Bethesda) 5, 1–8. doi: 10.1534/g3.114.014670
Etienne, H., Bertrand, B., Montagnon, C., Bobadilla Landey, R., Dechamp, E., Jourdan, I., et al. (2012). Un exemple de transfert technologique réussi en micropropagation: la multiplication de Coffea arabica parembryogenèse somatique. Cah. Agric. 21, 115–124.
Evenson, R. E., and Gollin, D. (2000). Assessing the impact of the green revolution, 1960 to 2000. Science 300, 758–762. doi: 10.1126/science.1078710
Fang, J.-Y., Wetten, A., Adu-Gyamfi, R., Wilkinson, M., and Rodríguez-López, C. (2009). Use of secondary somatic embryos promotes genetic fidelity in cryopreservation of cocoa (Theobroma cacao L.). Agric. Food Sci. 18, 152–159. doi: 10.2137/145960609789267579
Feng, S., Jacobsen, S. E., and Reik, W. (2010). Epigenetic reprogramming in plant and animal development. Science 330, 622–627. doi: 10.1126/science.1190614
Fraga, M. F., Ballestar, E., Paz, M. F., Ropero, S., Setien, F., Ballestar, M. L., et al. (2005). Epigenetic differences arise during the lifetime of monozygotic twins. Proc. Natl. Acad. Sci. U.S.A. 102, 10604–10609. doi: 10.1073/pnas.0500398102
Fraga, M. F., and Esteller, M. (2002). DNA Methylation: a profile of methods and applications. BioTechniques 33, 632–649. doi: 10.3389/fgene.2011.00074
Fraga, M. F., Rodriguez, R., and Canal, M. J. (2001). Genomic DNA methylation–demethylation during aging and reinvigoration of Pinus radiata. Tree Physiol. 22, 813–816. doi: 10.1093/treephys/22.11.813
Galaud, J.-P., Gaspar, T., and Boyer, N. (1993). Effect of anti-DNA methylation drugs on growth, level of methylated DNA, peroxidase activity and ethylene production of Bryonia dioica internodes. Physiol. Plant. 87, 528–534. doi: 10.1111/j.1399-3054.1993.tb02503.x
Geyer, K. K., Rodríguez López, C. M., Heald, J., Wilkinson, M. J., and Hoffmann, K. F. (2011). Cytosine methylation regulates oviposition in the pathogenic blood fluke Schistosoma mansoni. Nat. Commun. 9, 424–434. doi: 10.1038/ncomms1433
Goettel, W., and Messing, J. (2013). Paramutagenicity of a p1 epiallele in maize. Theor Appl Genet. 126, 159–177. doi: 10.1007/s00122-012-1970-z
Gourcilleau, D., Bogeat-Triboulot, M. B., Le Thiec, D., Lafon-Placette, C., Delaunay, A., El-Soud, W. A., et al. (2010). DNA methylation and histone acetylation: genotypic variations in hybrid poplars, impact of water deficit and relationships with productivity. Ann. For. Sci. 67, 208–217. doi: 10.1051/forest/2009101
Grant-Downton, R. T., and Dickinson, H. G. (2005). Epigenetics and its implications for plant biology: 1. The epigenetic network in plants. Ann. Bot. 96, 1143–1164. doi: 10.1093/aob/mci273
Grossniklaus, U., Vielle-Calzada, J. P., Hoeppner, M. A., and Gagliano, W. B. (1998). Maternal control of embryogenesis by medea, a Polycomb group gene in Arabidopsis. Science 280, 446–450. doi: 10.1126/science.280.5362.446
Gutierrez-Marcos, J. F., Costa, L. M., Biderre-Petit, C., Khbaya, B., O’Sullivan, D. M., Wormald, M., et al. (2004). Maternally expressed gene 1 is a novel maize endosperm transfer cell-specific gene with a maternal parent-of-origin pattern of expression. Plant Cell 16, 1288–1301. doi: 10.1105/tpc.019778
Hao, Y. J., You, C. X., and Deng, X. X. (2002). Analysis of ploidy and the patterns of amplified length polymorphism and methylation sensitive amplification polymorphism in strawberry plants recovered from cryopreservation. Cryoletters 23, 37–46.
Hattman, S. (2005). DNA-[Adenine] methylation in lower eukaryotes. Biochemistry (Moscow) 70, 550–558. doi: 10.1007/s10541-005-0148-6
Hauben, M., Haesendonckx, B., Standaert, E., Van Der Kelen, K., Azmi, A., Akpo, H., et al. (2009). Energy use efficiency is characterized by an epigenetic component that can be directed through artificial selection to increase yield. Proc. Natl. Acad. Sci. U.S.A. 106, 20109–20114. doi: 10.1073/pnas.0908755106
Heil, M., and Ton, J. (2008). Long-distance signalling in plant defence. Trends Plant Sci. 13, 264–272. doi: 10.1016/j.tplants.2008.03.005
Heinemann, U., and Hahn, M. (1992). C-C-A-G-G-C-m5C-T-G-G. Helical fine structure, hydration, and comparison with C-C-A-G-G-C-C-T-G-G. J. Biol. Chem. 267, 7332–7341.
Henderson, I. R., and Jacobsen, S. E. (2007). Epigenetic inheritance in plants. Nature 447, 418–424. doi: 10.1038/nature05917
Henry, R. J. (1998). “Molecular and biochemical characterization of somaclonal variation,” in Somaclonal Variation and Induced Mutations in Crop Improvement, eds S. M. Jain, D. S. Brar, and B. S. Ahloowalia (Dordrecht: Kluwer Academic Publisher), 485–499. doi: 10.1007/978-94-015-9125-6_24
Herrera, C. M., Bazaga, P. (2013). Epigenetic correlates of plant phenotypic plasticity: DNA methylation differs between prickly and nonprickly leaves in heterophyllous Ilex aquifolium (Aquifoliaceae) trees. Bot. J. Linnean Soc. 171, 441–452. doi: 10.1111/boj.12007
Hirsch, S., Baumberger, R., and Grossniklaus, U. (2013). Epigenetic variation, inheritance, and selection in plant populations. Cold Spring. Harb. Symp. Quant. Biol. 77, 97–104. doi: 10.1101/sqb.2013.77.014605
Huang, Y., Pastor, W. A., Shen, Y., Tahiliani, M., Liu, D. R., and Rao, A. (2010). The behaviour of 5-hydroxymethylcytosine in bisulfite sequencing. PLoS ONE 5:e8888. doi: 10.1371/journal.pone.0008888
Ito, H. (2012). Small RNAs and transposon silencing in plants. Dev. Growth Diff. 54, 100–107. doi: 10.1111/j.1440-169X.2011.01309.x
Ito, S., D’Alessio, A. C., Taranova, O. V., Hong, K., Sowers, L. C., and Zhang, Y. (2010). Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification. Nature 466, 1129–1133. doi: 10.1038/nature09303
Iwasaki, M., and Paszkowski, J. (2014). Identification of genes preventing transgenerational transmission of stress-induced epigenetic states. Proc. Natl. Acad Sci. U.S.A. 111, 8547–8552. doi: 10.1073/pnas.1402275111
Jacobsen, S. E., and Meyerowitz, E. M. (1997). Hypermethylated SUPERMAN epigenetic alleles in Arabidopsis. Science 277, 1100–1103. doi: 10.1126/science.277.5329.1100
Jaligot, E., Beule, T., Baurens, F. C., Billote, N., and Rival, A. (2004). Search for methylation-sensitive amplification polymorphisms associated with the “mantled” variant phenotype in oil palm (Elaeis guineensis Jacq.). Genome 47, 224–228. doi: 10.1139/g03-085
Jeon, J., Choi, J., Lee, G.-W., Park, S.-Y., Huh, A., Dean, R. A., et al. (2015). Genome-wide profiling of DNA methylation provides insights into epigenetic regulation of fungal development in a plant pathogenic fungus, Magnaporthe oryzae. Sci. Rep. 5, 8567. doi: 10.1038/srep08567
Joyce, S. M., and Cassells, A. C. (2002). Variation in potato microplant morphology in vitro and DNA methylation. Plant Cell Tissue Organ Cult. 70, 125–137. doi: 10.1023/A:1016312303320
Jung, H. W., Tschaplinski, T. J., Wang, L., Glazebrook, J., and Greenberg, J. T. (2009). Priming in systemic plant immunity RID D-4021-2009. Science 324, 89–91. doi: 10.1126/science.1170025
Kathiria, P., Sidler, C., Golubov, A., Kalischuk, M., Kawchuk, L. M., and Kovalchuk, I. (2010). Tobacco mosaic virus infection results in an increase in recombination frequency and resistance to viral, bacterial, and fungal pathogens in the progeny of infected tobacco plants. Plant Physiol. 153, 1859–1870. doi: 10.1104/pp.110.157263
Kimatu, J. N., Diarso, M., Song, C., Agboola, R. S., Pang, J., Qi, X., et al. (2011). DNA cytosine methylation alterations associated with aluminium toxicity and low pH in Sorghum bicolor. Afr. J. Agric. Res. 6, 4579–4593. doi: 10.5897/AJAR11.954
King, G. J., Amoah, S., and Kurup, S. (2010). Exploring and exploiting epigenetic variation in crops. Genome 53, 856–868. doi: 10.1139/G10-059
Kinoshita, T., Miura, A., Choi, Y. H., Kinoshita, Y., Cao, X. F., Jacobsen, S. E., et al. (2004). One-way control of FWA imprinting in Arabidopsis endosperm by DNA methylation. Science 303, 521–523. doi: 10.1126/science.1089835
Kohler, A., Schwindling, S., and Conrath, U. (2002). Benzothiadiazole-induced primingfor potentiated responses to pathogen infection, wounding, and infiltration ofwater into leaves requires the NPR1/NIM1 gene in Arabidopsis. Plant Physiol. 128, 1046–1056. doi: 10.1104/pp.010744
Kohler, C., and Makarevich, G. (2006). Epigenetic mechanisms governing seed development in plants. EMBO Rep. 7, 1223–1227. doi: 10.1038/sj.embor.7400854
Kriaucionis, S., and Heintz, N. (2009). The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 324, 929–930. doi: 10.1126/science.1169786
Li, L., Ming, Z. Z., DaiBo, L., Jing, W., Jian, G., MaoJun, Z., et al. (2011). Analysis of infection process and methylation-sensitive amplified polymorphism in Zea mays genome stressed by Rhizoctoniasolani. J. Agric. Biotechnol. 19, 243–249.
Li, R.-Q., Huang, J.-Z., Zhao, H.-J., Fu, H.-W., Li, Y.-F., Liu, G.-Z., et al. (2014). A down-regulated epi-allele of the genomes uncoupled 4 gene generates a xantha marker trait in rice. Theor. Appl. Genet. 127, 2491–2501. doi: 10.1007/s00122-014-2393-9
Li, W., Chen, W., Qi, X., Wang, Q., and Chen, J. (2013). Variation of cytosine methylation in response to water availability in two contrasting growth types of an amphibious plant Alternantheraphiloxeroides. Biochem. Syst. Ecol. 50, 175–181. doi: 10.1016/j.bse.2013.03.053
Lira-Medeiros, C. F., Parisod, C., Fernandes, R. A., Mata, C. S., Cardoso, M. A., and Ferreira, P. C. (2010). Epigenetic variation in mangrove plants occurring in contrasting natural environment. PLoS ONE 5:e10326. doi: 10.1371/journal.pone.0010326
Long Y Xia, W., Li, R. Y., Wang, J., Shao, M. Q., Feng, J., King, G. J., et al. (2011). Epigentic QTL mapping in Brassica napus. Genetics 189, 1093–10102. doi: 10.1534/genetics.111.131615
Luna, E., Bruce, T. J. A., Roberts, M. R., Flors, V., and Ton, J. (2012). Next generation systemic acquired resistance. Plant Physiol. 158, 844–853. doi: 10.1104/pp.111.187468
Luo, M., Bilodeau, P., Koltunow, A., Dennis, E. S., Peacock, W. J., and Chaudhury, A. M. (1999). Genes controlling fertilization-independent seed development in Arabidopsis thaliana. Proc. Natl Acad. Sci. U.S.A. 96, 296–301. doi: 10.1073/pnas.96.1.296
Luykx, D. M. A. M., and van Ruth, S. M. (2008). An overview of analytical methods for determining the geographical origin of food products. Food Chem. 107, 897–911. doi: 10.1016/j.foodchem.2007.09.038
Mader, E., Ruzicka, J., Schmiderer, C., and Novak, J. (2011). Quantitative high-resolution melting analysis for detecting adulterations. Anal. Biochem. 409, 153–155. doi: 10.1016/j.ab.2010.10.009
Madlung, A., and Comai, L. (2004). The effect of stress on genome regulation and structure. Ann. Bot. 94, 481–495. doi: 10.1093/aob/mch172
Manning, K., Tör, M., Poole, M., Hong, Y., Thompson, A. J., King, G. J., et al. (2006). A naturally occurring epigenetic mutation in a gene encoding an SBP-box transcription factor inhibits tomato fruit ripening. Nat. Genet. 38, 948–952. doi: 10.1038/ng1841
Mason, G., Noris, E., Lanteri, S., Acquadro, A., Accotto, G. P., and Portis, E. (2008). Potentiality of methylation-sensitive amplification polymorphism (MSAP) in identifying genes involved in tomato response to tomato yellow leaf curl sardinia virus. Plant Mol. Biol. Rep. 26, 156–173. doi: 10.1007/s11105-008-0031-x
Matthes, M., Singh, R., Cheah, S. C., and Karp, A. (2001). Variation in oil palm (Elaeis guineensis Jacq.) tissue culture-derived regenerants revealed by AFLPs with methylation-sensitive enzymes. Theor. Appl. Genet. 102, 971–979. doi: 10.1007/s001220000491
Miguel, C., and Marum, L. (2011). An epigenetic view of plant cells cultured in vitro: somaclonal variation and beyond. J. Exp. Bot. 62, 3713–3725. doi: 10.1093/jxb/err155
Miura, K., Agetsuma, M., Kitano, H., Yoshimura, A., Matsuoka, M., Jacobsen, S. E., et al. (2009). A metastable DWARF1 epigenetic mutant affecting plant stature in rice. Proc. Natl. Acad. Sci. U.S.A. 106, 11218–11223. doi: 10.1073/pnas.0901942106
Molinier, J., Ries, G., Zipfel, C., and Hohn, B. (2006). Transgeneration memory of stress in plants. Nature 442, 1046–1049. doi: 10.1038/nature05022
Moricová, P., Ondøej, V., Navrátilová, B., and Luhová, L. (2013). Changes of DNA methylation and hydroxymethylation in plant protoplast cultures. Acta Biochim. Pol. 60, 33–36.
Muenzel, M., Globisch, D., and Carell, T. (2011). 5-Hydroxymethylcytosine, the sixth base of the genom. Angew. Chem. Int. Ed. 50, 6460–6468. doi: 10.1002/anie.201101547
Müller, K., and Hippe, J. (1987). “Influence of differences in nutrition on important quality characteristics of some agricultural crops. Plant and Soil Interfaces and Interactions,” in Developments in Plant and Soil Sciences, ed. A. Van Diest (Amsterdam: Springer), 28, 35–45.
Naumann, U., Daxinger, L., Kanno, T., Eun, C., Long, Q., Lorkovic, Z. J., et al. (2011). Genetic evidence that DNA methyltransferase DRM2 hasa direct catalytic role in RNA-directed DNA methylation in Arabidopsis thaliana. Genetics 187, 977–979. doi: 10.1534/genetics.110.125401
Nic-Can, G. I., López-Torres, A., Barredo-Pool, F., Wrobel, K., Loyola-Vargas, V. M., Rojas-Herrera, R., et al. (2013). New insights into somatic embryogenesis: LEAFY COTYLEDON1, BABY BOOM1 and WUSCHEL-RELATED HOMEOBOX4 are epigenetically regulated in Coffea canephora. PLoS ONE 8:e72160. doi: 10.1371/journal.pone.0072160
Ohad, N., Margossian, L., Hsu, Y. C., Williams, C., Repetti, P., and Fischer, R. L. (1996). A mutation that allows endosperm development without fertilization. Proc. Natl Acad. Sci. U.S.A. 93, 5319–5324. doi: 10.1073/pnas.93.11.5319
Pan, Y., Wang, W., Zhao, X., Zhu, L., Fu, B., Li, Z., et al. (2011). DNA methylation alterations of rice in response to cold stress. Plant Omics J. 4, 364–369.
Pastor, V., Luna, E., Mauch-Mani, B., Ton, J., and Flors, V. (2013). Primed plants do not forget. Environ. Exp. Bot. 94, 46–56 doi: 10.1016/j.envexpbot.2012.02.013
Plongthongkum, N., Diep, D. H., and Zhang, K. (2014). Advances in the profiling of DNA modifications: cytosine methylation and beyond. Nat. Rev. Genet. 15, 647–661. doi: 10.1038/nrg3772
Polaczek, P., Kwan, K., and Campbell, J. L. (1998). GATC motifs may alter the conformation of DNA depending on sequence context and N6-adenine methylation status: possible implications for DNA-protein recognition. Mol. Genet. Genomics 258, 488–493. doi: 10.1007/s004380050759
Posner, J. L., Baldock, J. O., and Hedtcke, J. L. (2008). Organic and conventional production systems in the wisconsin integrated cropping systems trials: I. Productivity 1990–2002. Agron. J. 100, 253–260. doi: 10.2134/agrojnl2007.0058
Quadrana, L., Almeida, J., Asís, R., Duffy, R., Guadalupe Dominguez, P., Bermúdez, L., et al. (2014). Natural occurring epialleles determine vitamin E accumulation in tomato fruits. Nat. Commun. 5:3027. doi: 10.1038/ncomms5027
Quemada, H., Roth, E. J., and Lark, K. G. (1987). Changes in methylation of tissue cultured soybean cells detected by digestion with the restriction enzymes HpaII and MspI. Plant Cell Rep. 6, 63–66. doi: 10.1007/BF00269741
Raj, S., Bräutigam, K., Hamanishi, E. T., Wilkins, O., Thomasd, B. R., Schroeder, W. R., Mansfield, S. D., et al. (2011). Clone history shapes Populus drought responses. Proc. Natl. Acad. Sci. U.S.A. 108, 12521–12526. doi: 10.1073/pnas.1103341108
Reid, L. M., O’Donell, C. P., and Downell, G. (2006). Recent technological advances for the determination of food authenticity. Trends Food Sci. Technol. 17, 344–353. doi: 10.1016/j.tifs.2006.01.006
Robertson, J., Robertson, A. B., and Klungland, A. (2011a). The presence of 5-hydroxymethylcytosine at the gene promoter and not in the gene body negatively regulates gene expression. Biochem. Biophys. Res. Commun. 411, 40–43. doi: 10.1016/j.bbrc.2011.06.077
Rodríguez López, C. M., Guzman Asenjo, B., Lloyd, A. J., and Wilkinson, M. (2010a). Direct detection and quantification of methylation in nucleic acid sequences using high-resolution melting analysis. Anal. Chem. 82, 9100–9108. doi: 10.1021/ac1024057
Rodríguez López, C. M., Wetten, A. C., and Wilkinson, M. J. (2010b). Progressive erosion of genetic and epigenetic variation in callus-derived cocoa (Theobroma cacao) plants. New Phytol. 186, 56–868. doi: 10.1111/j.1469-8137.2010.03242.x
Rodríguez López, C. M., Lloyd, A. J., Leonard, K., and Wilkinson, M. J. (2012a). Differential effect of three base modifications on DNA thermostability revealed by high resolution melting. Anal. Chem. 84, 7336–7342. doi: 10.1021/ac301459x
Rodríguez López, C. M., Morán, P., Lago, F., Espiñeira, M., Beckmann, M., and Consuegra, S. (2012b). Detection and quantification of tissue of origin in salmon and veal products using methylation sensitive AFLPs. Food Chem. 131, 1493–1498. doi: 10.1016/j.foodchem.2011.09.120
Rogers, J. C., and Rogers, S. W. (1995). Comparison of the effects of N6-methyldeoxyadenosine and N5-methyldeoxycytosine on transcription from nuclear gene promoters in barley. Plant J. 7, 221–233. doi: 10.1046/j.1365-313X.1995.7020221.x
Rois, A. S., Rodríguez López, C. M., Cortinhas, A., Erben, M., Espirito Santo, D., Wilkinson, M. J., et al. (2013). Epigenetic rather than genetic factors may explain phenotypic divergence between coastal populations of diploid and tetraploid (Limonium sp., Plumbaginaceae) in Portugal. BMC Plant Biol. 13:205–220. doi: 10.1186/1471-2229-13-205
Schellenbaum, P., Mohler, V., Wenzel, G., and Walter, B. (2008). Variation in DNA methylation patterns of grapevine somaclones (Vitisvinifera L.). BMC Plant Biology. 8:78–98. doi: 10.1186/1471-2229-8-78
Schmitt, F., Oakeley, E. J., and Jost, J. P. (1997). Antibiotics induce genome-wide hypermethylation in cultured NicotianaTabacum plants. J. Biol. Chem. 272, 1534–1540. doi: 10.1074/jbc.272.3.1534
Schmitz, R. J., He, Y., Valdés-López, O., Khan, S. M., Joshi, T., Urich, M. A., et al. (2013). Epigenome-wide inheritance of cytosine methylation variants in a recombinant inbred population. Genome Res. 23, 1663–1674. doi: 10.1101/gr.152538.112
Schulz, B., Eckstein, R. L., and Durka, W. (2014). Epigenetic variation reflects dynamic habitat conditions in a rare floodplain herb. Mol. Ecol. 23, 3523–3537. doi: 10.1111/mec.12835
Sentandreu, M. A., and Sentandreu, E. (2011). Peptide biomarkers as a way to determine meat authenticity. Meat Sci. 89, 280–285. doi: 10.1016/j.meatsci.2011.04.028
Sha, A. H., Lin, X. H., Huang, J. B., and Zhang, D. P. (2005). Analysis of DNA methylation related to rice adult plant resistance to bacterial blight based on methylation-sensitive AFLP (MSAP) analysis. Mol. Genet. Genomics 273, 484–490. doi: 10.1007/s00438-005-1148-3
Shan, X., Wang, X., Yang, G., Wu, Y., Su, S., Li, S., et al. (2013). Analysis of the DNA methylation of maize (Zea mays L.) in response to cold stress based on methylation-sensitive amplified polymorphisms. J. Plant Biol. 56, 32–38. doi: 10.1007/s12374-012-0251-3
Shiba, H., Kakizaki, T., Iwano, M., Tarutani, Y., Watanabe, M., Isogai, A., et al. (2006). Dominance relationships between self-incompatibility alleles controlled by DNA methylation. Nat. Genet. 38, 297–299. doi: 10.1038/ng1734
Shock, L. S., Thakkar, P. V., Peterson, E. J., Moran, R. G., and Taylor, S. M. (2011). DNA methytransferase 1, cytosine methylation, and cytosine hydroxymethlation in mammalian mitochondria. Proc. Natl. Acad. Sci. U.S.A. 108, 3630–3635. doi: 10.1073/pnas.1012311108
Slaughter, A., Daniel, X., Flors, V., Luna, E., Hohn, E., and Mauch-Mani, B. (2012). Descendants of primed Arabidopsis plants exhibit resistance to biotic stress. Plant Physiol. 158, 835–843. doi: 10.1104/pp.111.191593
Song, Y., Ma, K., Bo, W., Zhang, Z., and Zhang, D. (2012). Sex-specific DNA methylation and gene expression in andromonoecious poplar. Plant Cell Rep. 31, 1393–1405. doi: 10.1007/s00299-012-1255-7
Srancikova, A., Horvathova, E., and Kozics, K. (2013). Biological effects of four frequently used medicinal plants of Lamiaceae. Neoplasma 60, 585–597. doi: 10.4149/neo_2013_076
Sternberg, N. (1985). Evidence that adenine methylation influences DNA-protein interactions in Escherichia coli. J. Bacteriol. 164, 490–493.
Stokes, T. L., Kunkel, B. N., and Richards, E. J. (2002). Epigenetic variation in Arabidopsis disease resistance. Genes Dev. 16, 171–182. doi: 10.1101/gad.952102
Stresemann, C., and Lyko, F. (2008). Modes of action of the DNA methyltransferase inhibitors azacytidine and decitabine. Int. J. Cancer 123, 8–13. doi: 10.1002/ijc.23607
Thalhammer, A., Hansen, A. S., El-Sagheer, A. H., Brown, T., and Schofield, C. (2011). Hydroxylation of methylated CpGdinucleotides reverses stabilisation of DNA duplexes by cytosine 5-methylation. J. Chem. Commun. 47, 5325–5327. doi: 10.1039/c0cc05671e
Theiss, G., and Follmann, H. (1980). 5-Methylcytosine formation in wheat embryo DNA. Biochem. Biophys. Res. Commun. 94, 291–297. doi: 10.1016/S0006-291X(80)80219-1
Tiwari, S., Schulz, R., Ikeda, Y., Dytham, L., Bravo, J., Mathers, L., et al. (2008). MATERNALLY EXPRESSED PAB C-TERMINAL, a novel imprinted gene in Arabidopsis, encodes the conserved C-terminal domain of polyadenylate binding proteins. Plant Cell 20, 2387–2398. doi: 10.1105/tpc.108.061929
Tost, J., and Gut, I. G. (2009). “Molecular Techniques for DNA methylation studies,” in Molecular Diagnostics, 2nd Edn, eds G. Patrinos and W. Ansorge (Amsterdam: Academic Press), 199–228.
Tricker P., Gibbings, G., Rodríguez López, C. M., Hadley, P., and Wilkinson, M. J. (2012). Low relative humidity triggers RNA-directed de novo DNA methylation and suppression of genes controlling stomatal development. J. Exp. Bot. 63, 3799–3813. doi: 10.1093/jxb/ers076
Tricker, P., Rodríguez López, C. M., Gibbings, G., Hadley, P., and Wilkinson, M. J. (2013a). Transgenerational, dynamic methylation of stomata genes in response to low relative humidity. Int. J. Mol. Sci. 14, 6674–6689. doi: 10.3390/ijms14046674
Tricker, P., Rodríguez López, C. M., Hadley, P., Wagstaff, C., and Wilkinson, M. J. (2013b). Pre-conditioning the epigenetic response to high vapour pressure deficit increases the drought tolerance in Arabidopsis thaliana. Plant Signal. Behav. 8:e25974. doi: 10.4161/psb.25974
Turlings, T. C. J., and Ton, J. (2006). Exploiting scents of distress: the prospect of manipulat-ing herbivore-induced plant odours to enhance the control of agricultural pests. Curr. Opin. Plant Biol. 9, 421–427. doi: 10.1016/j.pbi.2006.05.010
van Leeuwen, C., Friant, F., Choné, X., Tregoat, O., Koundouras, S., and Dubourdieu, D. (2004). Influence of climate, aoil, and cultivar on terroir. Am. J. Enol. Viticul. 55, 207–217.
Verhoeven, K. J. F., Jansen, J. J., van Dijk, P. J., and Biere, A. (2010). Stress-induced DNA methylation changes and their heritability in asexual dandelions. New Phytol. 185, 1108–1118. doi: 10.1111/j.1469-8137.2009.03121.x
Wanunu, M., Cohen-Karni, D., Johnson, R. R., Fields, L., Benner, J., Peterman, N., et al. (2011). Discrimination of methylcytosine from hydroxymethylcytosine in DNA molecules. J. Am. Chem. Soc. 133, 486–492. doi: 10.1021/ja107836t
Wassenegger, M., Heimes, S., Riedel, L., and Sanger, H. (1994). RNA-directed de-novo methylation of genomic sequences in plants. Cell 76, 567–576. doi: 10.1016/0092-8674(94)90119-8
Wilusz, J. E., and Sharp, P. A. (2013). A Circuitous Route to Noncoding RNA. Science 2013, 340, 440–441. doi: 10.1126/science.1238522
Wu, H., D’Alessio, A. C., Ito, S., Wang, Z., Cui, K., Zhao, K., et al. (2011). Genome-wide analysis of 5-hydroxymethylcytosine distribution reveals its dual function in transcriptional regulation in mouse embryonic stem cells. Genes Dev. 25, 679–684. doi: 10.1101/gad.2036011
Xiong, L. Z., Xu, C. G., Maroof, M. A. S., and Zhang, Q. F. (1999). Patterns of cytosine methylation in an elite rice hybrid and its parental lines, detected by a methylation sensitive amplification polymorphism technique. Mol. Gen. Genet. 261, 439–446. doi: 10.1007/s004380050986
Yaish, M. W., Colasanti, J., and Rothstein, S. J. (2011). The role of epigenetic processes in controlling flowering time in plants exposed to stress. J. Exp. Bot. 62, 3727–3735. doi: 10.1093/jxb/err177
Yu, A., Lepere, G., Jay, F., Wang, J. Y., Bapaume, L., Wang, Y., et al. (2013). Dynamics and biological relevance of DNA demethylation in Arabidopsis antibacterial defense. Proc. Natl. Acad. Sci. U.S.A. 110, 2389–2394. doi: 10.1073/pnas.1211757110
Zhang, L., Cheng, Z., Qin, R., Qiu, Y., Wang, J.-L., Cui, X., et al. (2012). Identification and characterization of an Epi-Allele of FIE1 reveals a regulatory linkage between two epigenetic marks in rice. Plant Cell 24, 4407–4421. doi: 10.1105/tpc.112.102269
Zhang, L., Peng, Y., Wei, X., Dai, Y., Yuan, D., Lu, Y., et al. (2014). Small RNAs as important regulators for the hybrid vigour of super-hybrid rice. J. Exp. Bot. 65, 5989–6002. doi: 10.1093/jxb/eru337
Zhang, Z., Deng, C., Lu, Q., and Richardson, B. (2002). Age-dependent DNA methylation changes in the ITGAL (CD11a) promoter. Mech. Ageing Dev. 123, 1257–1268. doi: 10.1016/S0047-6374(02)00014-3
Keywords: Fingerprinting, epigenetics, crop biotechnology, crop plants, crop quality, crop protection, crop improvement, priming
Citation: Rodríguez López CM and Wilkinson MJ (2015) Epi-fingerprinting and epi-interventions for improved crop production and food quality. Front. Plant Sci. 6:397. doi: 10.3389/fpls.2015.00397
Received: 15 March 2015; Accepted: 18 May 2015;
Published online: 05 June 2015.
Edited by:
Raúl Alvarez-Venegas, Centro de Investigación y de Estudios Avanzados del Instituto Politécnico Nacional, MexicoReviewed by:
Paula Casati, Centro de Estudios Fotosinteticos y Bioquímicos, ArgentinaCopyright © 2015 Rodríguez López and Wilkinson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Carlos M. Rodríguez López, Plant Research Centre, School of Agriculture, Food and Wine, Faculty of Sciences, University of Adelaide, Waite Campus, PMB1, Glen Osmond, Adelaide, SA 5064, Australia,Y2FybG9zLnJvZHJpZ3VlemxvcGV6QGFkZWxhaWRlLmVkdS5hdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.