 Atsushi Fukushima
Atsushi Fukushima Shigehiko Kanaya
Shigehiko Kanaya Kozo Nishida
Kozo Nishida- 1RIKEN Center for Sustainable Resource Science, Tsurumi, Yokohama, Japan
- 2Japan Science and Technology Agency, National Bioscience Database Center, Tokyo, Japan
- 3Graduate School of Information Science, Nara Institute of Science and Technology, Nara, Japan
- 4Laboratory for Biochemical Simulation, RIKEN Quantitative Biology Center, Osaka, Japan
One of the ultimate goals in plant systems biology is to elucidate the genotype-phenotype relationship in plant cellular systems. Integrated network analysis that combines omics data with mathematical models has received particular attention. Here we focus on the latest cutting-edge computational advances that facilitate their combination. We highlight (1) network visualization tools, (2) pathway analyses, (3) genome-scale metabolic reconstruction, and (4) the integration of high-throughput experimental data and mathematical models. Multi-omics data that contain the genome, transcriptome, proteome, and metabolome and mathematical models are expected to integrate and expand our knowledge of complex plant metabolisms.
Introduction
Plants are a paramount source of food, energy, and valuable compounds. The developing field of plant systems biology has provided outstanding insights into how these products are synthesized; its ultimate goal is an understanding of the genotype-phenotype relationship in cellular systems (Kell, 2002; Benfey and Mitchell-Olds, 2008; Weckwerth, 2011). Recent technical advances in high-throughput sequencing and various analytical instruments have made it possible to comprehensively measure and analyze genes, transcripts, proteins, and metabolites (Fukushima et al., 2009; Lei et al., 2011; Lucas et al., 2011; Stitt, 2013). These omics technologies are not only platforms that monitor the cellular inventory, but they also provide the opportunity to evaluate cellular behaviors from a multi-level perspective and enhance our understanding of plant systems (Krouk et al., 2010; Saito and Matsuda, 2010; Dhondt et al., 2013).
Major effective and efficient approaches to analyze omics data are network- and pathway analysis (for example, see, Ramanan et al., 2012; Carter et al., 2013). The former is based on the network concept derived from mathematical graph theory and typically represents a biological component (e.g., a gene) as a node and physical-, genetic-, and/or functional interactions as a link in the network to visualize and interpret the omics data (“data-driven approach”). On the other hand, pathway analysis is a knowledge-based approach that involves the associated biochemical pathway. Enrichment analysis approaches can be combined with pathway analysis to evaluate whether a particular molecular group is significantly over-represented. Examples are gene set enrichment analysis (Hung et al., 2012), Metabolite Set Enrichment Analysis (MSEA) (Xia and Wishart, 2010), and other functional enrichment analyses using gene ontology (GO) and biochemical pathways (for comprehensive reviews see Chagoyen and Pazos, 2013 or Khatri et al., 2012).
For a holistic view of plant metabolisms, measuring the metabolic flux by experimental flux analysis, e.g., metabolic flux analysis (MFA) (Libourel and Shachar-Hill, 2008; Sweetlove et al., 2014) or in silico flux modeling, e.g., flux balance analysis (FBA) (Kruger and Ratcliffe, 2012; Junker, 2014) is also important. FBA is a constraint-based approach for predicting flux through reactions in a quantitative manner (Orth et al., 2010; Sweetlove and Ratcliffe, 2011); it complements experimental flux analysis. It does not use knowledge of kinetic parameters from metabolic reactions but relies solely on the stoichiometric balance assuming steady-state conditions. These models can be extended to a level that almost fully includes the metabolism. Indeed, the past few years have seen an increase in the use of genome-scale metabolic models in plants (Collakova et al., 2012; Seaver et al., 2012; De Oliveira Dal'molin and Nielsen, 2013). Integrated network analysis by combining omics data with mathematical models has become popular. In this review we focus on the latest cutting-edge computational advances for analyzing omics networks and performing pathway analysis. We highlight (1) network visualization tools, (2) pathway analyses, (3) genome-scale metabolic reconstruction, and (4) the integration of high-throughput experimental data with mathematical modeling. These topics correspond to interaction-based and constraint-based approaches to the mathematical modeling of cellular networks as classified by Stelling (2004), Lewis et al. (2012).
Network Visualization and Pathway Analysis Tools for Interaction-based Approaches
The relationship between the biological components of a biological network includes four types of interactions: physical interactions (e.g., drug targets Yildirim et al., 2007 and protein-protein interactions Brandao et al., 2009), genetic interactions (Costanzo et al., 2010), and functional interactions (e.g., biochemical/signaling pathways Caspi et al., 2012; Kanehisa et al., 2014). Interaction-based approaches such as topological analysis (e.g., shortest path search Yu et al., 2014, centrality analysis Carrera et al., 2009, and network module detection Altaf-Ul-Amin et al., 2006), correlation network analysis (Provart, 2012), or enrichment analysis (Hung et al., 2012) have been used to construct and analyze biological networks from omics data. For example, GeneMANIA (Montojo et al., 2010; Zuberi et al., 2013) is a web-based interaction network for the visualization of physical, genetic, and functional interactions. Network visualization tools (e.g., igraph, http://igraph.org/) can not only describe a biological network, but also calculate and perform computational analysis (for a comprehensive review see Gehlenborg et al., 2010). Furthermore, network visualization tools assist the database client and facilitate data integration (Table 1).
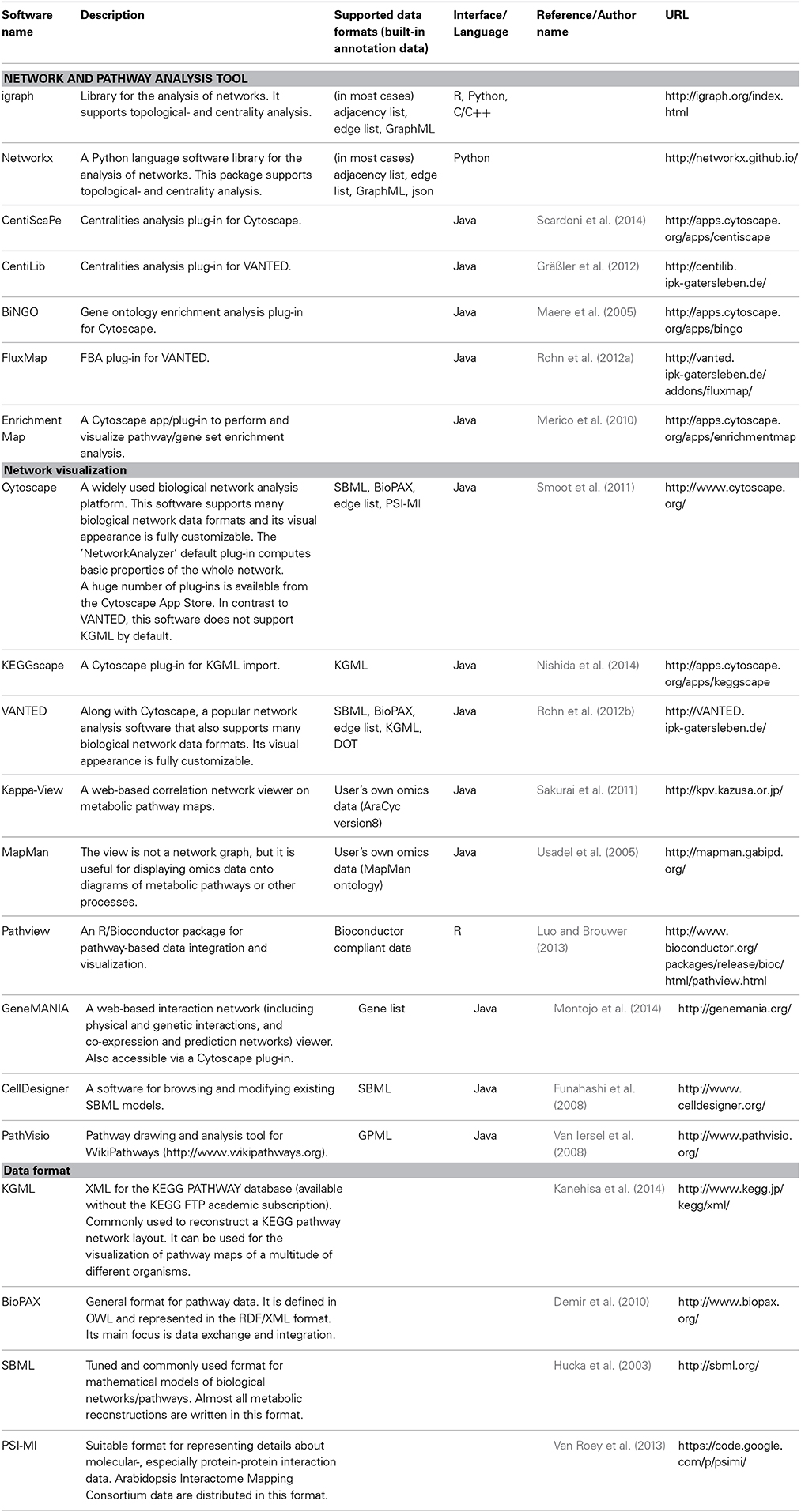
Table 1. List of software discussed in this review: Network tools for metabolic system biology analysis and related data formats.
Network Visualization and Pathway Analysis Tools
Data analysis of biological networks by graph representations includes topological analysis (for an example see, Toubiana et al., 2013). For functional networks, correlation and enrichment analyses can be used. Correlation analysis is based on associations between biological components (e.g., genes and metabolites). The Pearson correlation coefficient is a special case of association that evaluates linear relationships among molecular abundances (Kusano and Fukushima, 2013). Enrichment analysis uses a given molecular group such as gene ontology and biochemical pathways. Some network visualization tools implement these approaches while others involve independent, plug-in software modules (e.g., Cytoscape Smoot et al., 2011 and VANTED Rohn et al., 2012b). Cytoscape apps/plug-ins include BiNGO (Maere et al., 2005) for GO enrichment analysis and FluxMap for FBA (Rohn et al., 2012a). Network analysis platforms such as Enrichment map (Merico et al., 2010) feature system flexibility and expandability for omics data. Most network visualization tools manage and visualize network data that correspond to the type of interaction. For example, when performing a quality check of protein-protein interaction data generated from a high-throughput yeast two-hybrid screening system, these tools can visualize a giant network component from a large number of interactions (for example see Arabidopsis Interactome Mapping Consortium, 2011). For functional interactions, when mapping transcriptomics profiles onto metabolic pathways, a pathway-level representation of the gene expressions involved can be assessed (Usadel et al., 2005; Sakurai et al., 2011). The network visualization tool requires a sophisticated function, structured and controlled functional categories, and vocabularies, to inspect the profile data on a pathway. Two typical functional categories are the Kyoto Encyclopedia of Genes and Genomes (KEGG) (Kanehisa et al., 2014) and GO, which can be used to evaluate perturbed pathways in the omics data. For physical interaction networks, concerted efforts are being made to share data formats that visualize biological networks such as the PSI-MI format (Van Roey et al., 2013) in IntAct (Kerrien et al., 2012).
Network/Pathway Data Formats
There are many data formats for functional interaction networks, especially biochemical pathway databases. As KEGG XML (KGML) (Kanehisa et al., 2014), BioPAX (Demir et al., 2010), and SBML (Hucka et al., 2003) are available for pathway data exchanges, a network visualization tool that implements and supports these data formats as import and export functions is desirable. For example, AraCyc (Zhang et al., 2005) and Arabidopsis Reactome (Tsesmetzis et al., 2008) are also represented in the BioPAX format (Table 1). BioPAX is defined in Web Ontology Language (OWL); it contains the most comprehensive ontology for representing pathway knowledge. It can also serve as a Resource Description Framework (RDF) for describing information on the world-wide-web (Jupp et al., 2014) and it is expected to utilize semantic data integration. According to Strömbäck and Lambrix (2005), SBML (Hucka et al., 2003) is the most widely used and finely tuned format for mathematical models (e.g., the FBA model). KEGG pathways are manually drawn and the layout is created by domain experts. Because KGML includes all KEGG pathway layout information, it uses another SBML-based software/database to reconstruct a pathway map. Although it is a de facto standard network visualization tool and supports most data formats, Cytoscape (Smoot et al., 2011) cannot seamlessly integrate all data resources irrespective of the data schema and controlled ontology. There are considerable community-wide efforts in the sustainable development and integration of various database resources with RDF (for example see BioHackathon Katayama et al., 2014, the Rhea database Alcantara et al., 2012, and Path2Models Buchel et al., 2013). The number of Wiki-based databases (Arita, 2009) is also increasing; this community curation process includes WikiPathways (Hanumappa et al., 2013) with PathVisio (Van Iersel et al., 2008) and LipidBank (http://jcbl.jp/wiki/Category:LB). Currently, several pathway resources are often combined with SBML to use FBA (see Section Genome-scale metabolic reconstruction in plants and constraint-based approaches).
Visualization of Omics Data for Exploring Biological Networks
Optimal network visualization tools must allow the seamless integration of multiple data resources and their comparison, irrespective of differences in the data formats generated by primary data providers. However, with currently available network visualization tools, the integration of different data resources remains difficult although the visualization of omics data has been partially achieved with tools such as VANTED and Pathview (Luo and Brouwer, 2013). As an example, to visualize metabolomic data we used Cytoscape with its app/plug-in KEGGscape (http://apps.cytoscape.org/apps/keggscape) (Nishida et al., 2014) and VANTED. KEGGscape supports KEGG pathway files in KGML format and reproduces KEGG pathway diagrams as a standard network object in Cytoscape. Users can easily integrate their own datasets with biologist-friendly KEGG pathway diagrams. Figure 1 is a network representation of the time-series metabolome (Espinoza et al., 2010) in Arabidopsis thaliana using KEGGscape. We integrated the metabolite profiles and the tricarboxylic acid (TCA) cycle with the KEGG compound IDs as the keys. Although Cytoscape and VANTED are different in design, both tools can visualize the same figure (Supplemental Figure S1) and we posit that they will be widely used to visualize omics-profiles on pathway maps. Such network visualization allows users to consider pathway-related profile variations that cannot be inferred immediately from the profile.
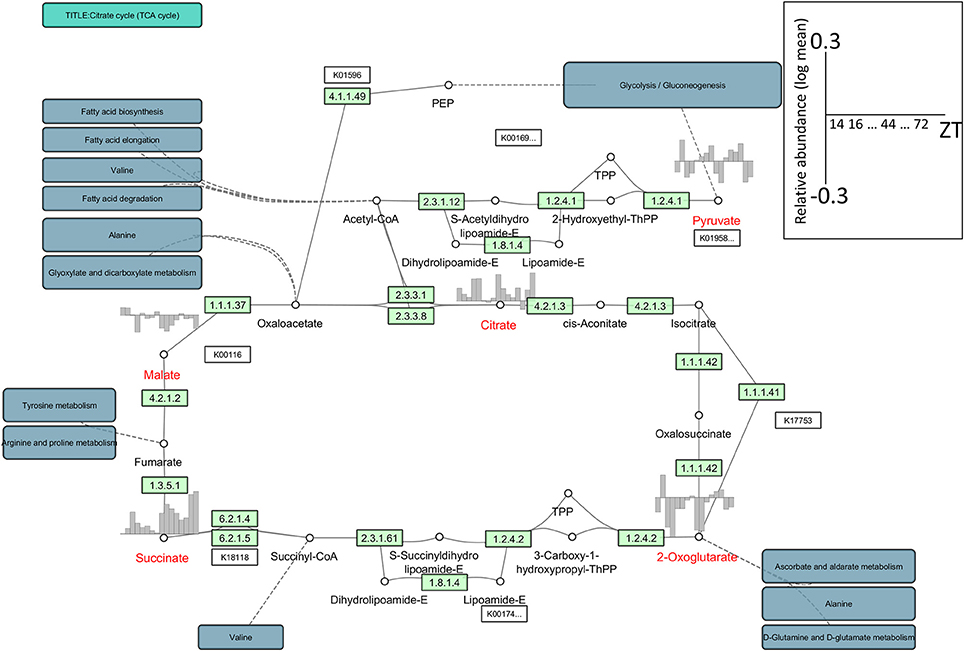
Figure 1. An example of the network representation of time-series metabolome data in Arabidopsis using KEGGscape (http://apps.cytoscape.org/apps/keggscape). The datasets were sampled with 4-h resolution under a 16 h/8 h light/dark cycle at 20°C (Espinoza et al., 2010). We used the KEGG pathway map (ath00020), the tricarboxylic acid (TCA) cycle, or the citrate cycle. We queried MetMask (Redestig et al., 2010) for a list of KEGG compound IDs associated with a list of predefined metabolite names and picked up the most pathway-mapped KEGG compound ID for each metabolite. Metabolite names shown in red represent detected compounds in the dataset. The diurnal changes were visualized on bar charts ranging from −0.3 to 0.3 in log-mean values. ZT, Zeitgeber time.
Genome-Scale Metabolic Reconstruction in Plants and Constraint-based Approaches
Both MFA and FBA use stoichiometric simulation to estimate and predict cellular metabolic flux. Although MFA with 13C labeling is the most promising approach to characterize metabolic phenotypes in a cell, technological issues prevent its application to complete metabolisms. In this section we focus on genome-scale metabolic reconstruction and FBA.
A Genome-Scale Metabolic Reconstruction
Due to the already extremely large and growing amount of genomic sequences yielded by high-throughput techniques, metabolism reconstruction from an organism's genome sequence has become possible (Thiele and Palsson, 2010). Although the metabolism has been reconstructed for only a few of the sequenced plant genomes, it has been modeled in some plants and crops (Seaver et al., 2012; De Oliveira Dal'molin and Nielsen, 2013). The first step in metabolic reconstruction from genome sequences involves the collection and integration of compounds, enzymes, genes, and curated published pathway databases. Subsequently, gene-protein-reaction (GPR) relationships in an organism (Fell et al., 2010) are identified and a stoichiometric matrix consisting of substances and reactions is generated. This draft metabolism requires further curation including metabolic gap filling and FBA. To explore flux states computationally, FBA uses the optimization of an objective function and predicts the growth rate of an organism or the production rate of industrially and medicinally important metabolites (Feist and Palsson, 2010). The collection of information on the biomass including proteins, amino acids, and lipid(s) as an objective function is required. Because these steps tend to be time-consuming, rapid algorithms for reconstructing genome-scale metabolisms have been developed (Chen et al., 2012; Kim et al., 2012). On SEED (Henry et al., 2010) and PlantSEED (Seaver et al., 2014) servers a significant number of genome-scale metabolisms in different organisms has already been reconstructed. In this review we do not present a comprehensive review of software tools/algorithms involved in reconstructing a genome-scale model and FBA.
Reconstructed Plant Metabolisms
The first genome-scale models in plants were designed and published for barley seeds and heterotrophic Arabidopsis cells. Grafahrend-Belau et al. (2009) constructed a compartmentalized barley seed metabolism model and performed mathematical simulations to investigate storage patterns that included responses to environmental and genetic perturbations (Grafahrend-Belau et al., 2009). Their comparison of published data for grain yields and growth rates with in silico data showed good reproducibility, indicating the usefulness of their model for predicting the seed storage metabolism. Poolman et al. (2009) generated an Arabidopsis genome-scale metabolic model using the AraCyc database (Poolman et al., 2009). They demonstrated that only 15% of the reactions in the reconstructed network (“minimal network”) were required to produce amino acids, nucleotides, and other biomass components. For the Arabidopsis metabolism, two other models are available, i.e., AraGEM (De Oliveira Dal'molin et al., 2010) and the model of Radrich (Radrich et al., 2010). In addition, 7 tissue-specific models for Arabidopsis have been presented (Mintz-Oron et al., 2012). The model of Poolman et al. (2009) was extended and updated to include more information on the subcellular localization of enzymes and transport reactions (Cheung et al., 2013) and to model the leaf metabolism over a day-night diel cycle (Cheung et al., 2014). The approach with MFA demonstrated a marked improvement in the quantitative match between predicted- and experimentally-estimated fluxes. To assess the central carbon partitioning and enzyme costs precisely, Arnold and Nikoloski (2014) newly reconstructed the Arabidopsis metabolism based on genomic and bibliomic data that included biochemical, genomic, and genetic information on compartmentalization and transport processes. Their model produced all amino acids and was able to estimate various cell performances (Arnold and Nikoloski, 2014). De Oliveira Dal'molin et al. (2010) constructed a genome-scale metabolic model for C4 plants (C4GEM) (Dal'molin et al., 2010), Saha et al. (2011) modeled the maize metabolism that contains maize-specific GPR (Saha et al., 2011), and Grafahrend-Belau et al. (2013) developed multi-scale metabolic modeling (MMM) for predicting the plant metabolism at the whole plant level; their barley model has provided significant insights into the metabolic capacity for yield stability and crop improvement (Grafahrend-Belau et al., 2013).
Integration of High-throughput Experimental Data with Mathematical Modeling
The integration of omics data and mathematical models is a promising approach to gain a better understanding of plant metabolisms (Bordbar et al., 2014; Saha et al., 2014). Integrated concepts involving FBA make it possible to predict genotype-phenotype relationships and to gain important insights into the metabolic network capacity of an organism (Blazier and Papin, 2012). For example, an integrated model in which gene expression was combined with a metabolic network (ME model) in Escherichia coli increased the accuracy for predicting feasible and computable phenotypes that respond to optimal growth conditions (Lerman et al., 2012). Karr et al. (2012) showed that a whole-cell model in Mycoplasma genitalium was useful for describing protein-DNA binding and correlations between DNA replication and its initiation. Their findings indicate that the integrated approach makes it possible to study previously unknown biological processes in a cell. These earlier studies demonstrated that high-throughput omics data are available as a constraint parameter for generating high-quality metabolic models. The model-building algorithm (MBA) developed by Jerby et al. (2010) is used to construct tissue-specific metabolisms from generic models and omics data (Jerby et al., 2010). Gene Inactivity Moderated by Metabolism and Expression (GIMME) (Becker and Palsson, 2008) is based on the premise that gene expression data correlate with metabolic fluxes and the user's pre-defined threshold of expression levels; GIMME removes reactions with expression levels lower than the threshold from the model and evaluates metabolic capacities. iMAT (Folger et al., 2011) is similar to GIMME; it is based on the discretization of input expression data and returns predictive optimal flux with confidence values over all network reactions. Metabolic Adjustment by Differential Expression (MADE) (Jensen and Papin, 2011) uses significant changes in transcript levels between two or more conditions classified into so-called “switch” approaches. This is then used to identify on/off reaction fluxes based on threshold expression levels in the constraint-based models (Hyduke et al., 2013; Saha et al., 2014).
Two other approaches exist, they are known as “valve” approaches and they allow the use of gene expression data to limit the maximum activity of an enzyme. The first, E-FLUX (Colijn et al., 2009), uses maximum flux constraints as a function of measured transcript levels without binalization of the expression data. The other approach is GIM3E, it does not apply arbitrary cutoffs for expression levels (Schmidt et al., 2013). Protein data can also be included. PROM (Chandrasekaran and Price, 2010) invokes a threshold to determine whether an enzyme is in its active or inactive state and uses information about regulatory interactions including transcription factor-target gene interactions. Integrative Omics-Metabolic Analysis (IOMA) (Yizhak et al., 2010) integrates proteomic and metabolomic data into a genome-scale metabolic model by evaluating kinetic rate equations subject to quantitative omics measurements. Machado and Herrgard (2014) who systematically evaluated different methods for the integration of transcriptome data into constraint-based models reported that no robust approaches worked well under all examinations (Machado and Herrgard, 2014). In plant science, Topfer et al. (2013) performed E-FLUX on the Arabidopsis genome-scale models created by Mintz-Oron et al. (2012) and used high-resolution time-series transcriptome data (Caldana et al., 2011) to investigate metabolic capacities in response to different environmental changes (Topfer et al., 2013). Their optimization-based approach was able to characterize many aspects of the metabolic behaviors and functions in response to a changing environment. In an attempt to integrate metabolome data with constraint-based mathematical models, Nagele and Weckwerth (2013) developed a complementary approach to obtain a comprehensive view of metabolic capacities in Arabidopsis leaves (Nagele and Weckwerth, 2013). Using experimentally accessible metabolites and the Mintz-Oron model (Mintz-Oron et al., 2012) they derived a metabolic model that yielded an overview of metabolic phenotypes perturbed by genetic and environmental differences.
Future Perspectives
Metabolic network models have contributed to the study of metabolic capacity in response to environmental and genetic perturbations and to the identification of feasible metabolic networks in an organism. They provided important clues about genotype-phenotype relationships. Reconstruction of the metabolism from genome sequences is a non-trivial task that requires not only effective computational tools but also integrated knowledge-based systems. For comprehensive reconstructions, improved technologies, including more sophisticated algorithms and tools, better software frameworks for multiple omics data analyses, improved visualization of biological networks, and more effective integration of data with mathematical models are needed. Multi-omics data that include the genome, transcriptome, proteome, and metabolome plus mathematical modeling can be expected to deepen our knowledge of complex plant metabolisms and to illuminate unexplored biological processes.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Acknowledgments
We apologize to authors whose work is not cited due to space limitations. The research activity of Atsushi Fukushima is partly supported by a Grant-in-Aid for Young Scientists (B; grant no. 26850024 to Atsushi Fukushima) from the Ministry of Education, Culture, Sports, Science and Technology, Japan. We thank Dr. Kansuporn Sriyudthsak for a critical reading of the manuscript and Ms. Ursula Petralia for editorial assistance.
Supplementary Material
The Supplementary Material for this article can be found online at: http://www.frontiersin.org/journal/10.3389/fpls.2014.00598/abstract
References
Alcantara, R., Axelsen, K. B., Morgat, A., Belda, E., Coudert, E., Bridge, A., et al. (2012). Rhea–a manually curated resource of biochemical reactions. Nucleic Acids Res. 40, D754–D760. doi: 10.1093/nar/gkr1126
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Altaf-Ul-Amin, M., Shinbo, Y., Mihara, K., Kurokawa, K., and Kanaya, S. (2006). Development and implementation of an algorithm for detection of protein complexes in large interaction networks. BMC Bioinformatics 7:207. doi: 10.1186/1471-2105-7-207
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Arabidopsis Interactome Mapping Consortium. (2011). Evidence for network evolution in an Arabidopsis interactome map. Science 333, 601–607. doi: 10.1126/science.1203877
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Arita, M. (2009). A pitfall of wiki solution for biological databases. Brief. Bioinform. 10, 295–296. doi: 10.1093/bib/bbn053
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Arnold, A., and Nikoloski, Z. (2014). Bottom-up metabolic reconstruction of Arabidopsis and its application to determining the metabolic costs of enzyme production. Plant Physiol. 165, 1380–1391. doi: 10.1104/pp.114.235358
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Becker, S. A., and Palsson, B. O. (2008). Context-specific metabolic networks are consistent with experiments. PLoS Comput. Biol. 4:e1000082. doi: 10.1371/journal.pcbi.1000082
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Benfey, P. N., and Mitchell-Olds, T. (2008). From genotype to phenotype: systems biology meets natural variation. Science 320, 495–497. doi: 10.1126/science.1153716
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Blazier, A. S., and Papin, J. A. (2012). Integration of expression data in genome-scale metabolic network reconstructions. Front. Physiol. 3:299. doi: 10.3389/fphys.2012.00299
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Bordbar, A., Monk, J. M., King, Z. A., and Palsson, B. O. (2014). Constraint-based models predict metabolic and associated cellular functions. Nat. Rev. Genet. 15, 107–120. doi: 10.1038/nrg3643
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Brandao, M. M., Dantas, L. L., and Silva-Filho, M. C. (2009). AtPIN: Arabidopsis thaliana protein interaction network. BMC Bioinformatics 10:454. doi: 10.1186/1471-2105-10-454
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Buchel, F., Rodriguez, N., Swainston, N., Wrzodek, C., Czauderna, T., Keller, R., et al. (2013). Path2Models: large-scale generation of computational models from biochemical pathway maps. BMC Syst. Biol. 7:116. doi: 10.1186/1752-0509-7-116
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Caldana, C., Degenkolbe, T., Cuadros-Inostroza, A., Klie, S., Sulpice, R., Leisse, A., et al. (2011). High-density kinetic analysis of the metabolomic and transcriptomic response of Arabidopsis to eight environmental conditions. Plant J. 67, 869–884. doi: 10.1111/j.1365-313X.2011.04640.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Carrera, J., Rodrigo, G., Jaramillo, A., and Elena, S. F. (2009). Reverse-engineering the Arabidopsis thaliana transcriptional network under changing environmental conditions. Genome Biol. 10:R96. doi: 10.1186/gb-2009-10-9-r96
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Carter, H., Hofree, M., and Ideker, T. (2013). Genotype to phenotype via network analysis. Curr. Opin. Genet. Dev. 23, 611–621. doi: 10.1016/j.gde.2013.10.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Caspi, R., Altman, T., Dreher, K., Fulcher, C. A., Subhraveti, P., Keseler, I. M., et al. (2012). The MetaCyc database of metabolic pathways and enzymes and the BioCyc collection of pathway/genome databases. Nucleic Acids Res. 40, D742–D753. doi: 10.1093/nar/gkr1014
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chagoyen, M., and Pazos, F. (2013). Tools for the functional interpretation of metabolomic experiments. Brief. Bioinform. 14, 737–744. doi: 10.1093/bib/bbs055
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chandrasekaran, S., and Price, N. D. (2010). Probabilistic integrative modeling of genome-scale metabolic and regulatory networks in Escherichia coli and Mycobacterium tuberculosis. Proc. Natl. Acad. Sci. U.S.A. 107, 17845–17850. doi: 10.1073/pnas.1005139107
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Chen, N., Del Val, I. J., Kyriakopoulos, S., Polizzi, K. M., and Kontoravdi, C. (2012). Metabolic network reconstruction: advances in in silico interpretation of analytical information. Curr. Opin. Biotechnol. 23, 77–82. doi: 10.1016/j.copbio.2011.10.015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cheung, C. Y., Poolman, M. G., Fell, D. A., Ratcliffe, R. G., and Sweetlove, L. J. (2014). A diel flux balance model captures interactions between light and dark metabolism during Day-Night cycles in C3 and crassulacean acid metabolism leaves. Plant Physiol. 165, 917–929. doi: 10.1104/pp.113.234468
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Cheung, C. Y., Williams, T. C., Poolman, M. G., Fell, D. A., Ratcliffe, R. G., and Sweetlove, L. J. (2013). A method for accounting for maintenance costs in flux balance analysis improves the prediction of plant cell metabolic phenotypes under stress conditions. Plant J. 75, 1050–1061. doi: 10.1111/tpj.12252
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Colijn, C., Brandes, A., Zucker, J., Lun, D. S., Weiner, B., Farhat, M. R., et al. (2009). Interpreting expression data with metabolic flux models: predicting Mycobacterium tuberculosis mycolic acid production. PLoS Comput. Biol. 5:e1000489. doi: 10.1371/journal.pcbi.1000489
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Collakova, E., Yen, J. Y., and Senger, R. S. (2012). Are we ready for genome-scale modeling in plants? Plant Sci. 191–192, 53–70. doi: 10.1016/j.plantsci.2012.04.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Costanzo, M., Baryshnikova, A., Bellay, J., Kim, Y., Spear, E. D., Sevier, C. S., et al. (2010). The genetic landscape of a cell. Science 327, 425–431. doi: 10.1126/science.1180823
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dal'molin, C. G., Quek, L. E., Palfreyman, R. W., Brumbley, S. M., and Nielsen, L. K. (2010). C4GEM, a genome-scale metabolic model to study C4 plant metabolism. Plant Physiol. 154, 1871–1885. doi: 10.1104/pp.110.166488
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Demir, E., Cary, M. P., Paley, S., Fukuda, K., Lemer, C., Vastrik, I., et al. (2010). The BioPAX community standard for pathway data sharing. Nat. Biotechnol. 28, 935–942. doi: 10.1038/nbt.1666
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
De Oliveira Dal'molin, C. G., and Nielsen, L. K. (2013). Plant genome-scale metabolic reconstruction and modelling. Curr. Opin. Biotechnol. 24, 271–277. doi: 10.1016/j.copbio.2012.08.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
De Oliveira Dal'molin, C. G., Quek, L. E., Palfreyman, R. W., Brumbley, S. M., and Nielsen, L. K. (2010). AraGEM, a genome-scale reconstruction of the primary metabolic network in Arabidopsis. Plant Physiol. 152, 579–589. doi: 10.1104/pp.109.148817
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Dhondt, S., Wuyts, N., and Inze, D. (2013). Cell to whole-plant phenotyping: the best is yet to come. Trends Plant Sci. 18, 428–439. doi: 10.1016/j.tplants.2013.04.008
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Espinoza, C., Degenkolbe, T., Caldana, C., Zuther, E., Leisse, A., Willmitzer, L., et al. (2010). Interaction with diurnal and circadian regulation results in dynamic metabolic and transcriptional changes during cold acclimation in Arabidopsis. PLoS ONE 5:e14101. doi: 10.1371/journal.pone.0014101
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Feist, A. M., and Palsson, B. O. (2010). The biomass objective function. Curr. Opin. Microbiol. 13, 344–349. doi: 10.1016/j.mib.2010.03.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fell, D. A., Poolman, M. G., and Gevorgyan, A. (2010). Building and analysing genome-scale metabolic models. Biochem. Soc. Trans. 38, 1197–1201. doi: 10.1042/BST0381197
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Folger, O., Jerby, L., Frezza, C., Gottlieb, E., Ruppin, E., and Shlomi, T. (2011). Predicting selective drug targets in cancer through metabolic networks. Mol. Syst. Biol. 7:501. doi: 10.1038/msb.2011.35
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Fukushima, A., Kusano, M., Redestig, H., Arita, M., and Saito, K. (2009). Integrated omics approaches in plant systems biology. Curr. Opin. Chem. Biol. 13, 532–538. doi: 10.1016/j.cbpa.2009.09.022
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Funahashi, A., Matsuoka, Y., Jouraku, A., Morohashi, M., Kikuchi, N., and Kitano, H. (2008). CellDesigner 3.5: a versatile modeling tool for biochemical networks. Proc. IEEE 96, 1254–1265. doi: 10.1109/JPROC.2008.925458
Gehlenborg, N., O'donoghue, S. I., Baliga, N. S., Goesmann, A., Hibbs, M. A., Kitano, H., et al. (2010). Visualization of omics data for systems biology. Nat. Methods 7, S56–S68. doi: 10.1038/nmeth.1436
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Grafahrend-Belau, E., Junker, A., Eschenroder, A., Muller, J., Schreiber, F., and Junker, B. H. (2013). Multiscale metabolic modeling: dynamic flux balance analysis on a whole-plant scale. Plant Physiol. 163, 637–647. doi: 10.1104/pp.113.224006
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Grafahrend-Belau, E., Schreiber, F., Koschutzki, D., and Junker, B. H. (2009). Flux balance analysis of barley seeds: a computational approach to study systemic properties of central metabolism. Plant Physiol. 149, 585–598. doi: 10.1104/pp.108.129635
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Gräßler, J., Koschützki, D., and Schreiber, F. (2012). CentiLib: comprehensive analysis and exploration of network centralities. Bioinformatics 28, 1178–1179. doi: 10.1093/bioinformatics/bts106
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hanumappa, M., Preece, J., Elser, J., Nemeth, D., Bono, G., Wu, K., et al. (2013). WikiPathways for plants: a community pathway curation portal and a case study in rice and arabidopsis seed development networks. Rice (N. Y). 6:14. doi: 10.1186/1939-8433-6-14
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Henry, C. S., Dejongh, M., Best, A. A., Frybarger, P. M., Linsay, B., and Stevens, R. L. (2010). High-throughput generation, optimization and analysis of genome-scale metabolic models. Nat. Biotechnol. 28, 977–982. doi: 10.1038/nbt.1672
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hucka, M., Finney, A., Sauro, H. M., Bolouri, H., Doyle, J. C., Kitano, H., et al. (2003). The systems biology markup language (SBML): a medium for representation and exchange of biochemical network models. Bioinformatics 19, 524–531. doi: 10.1093/bioinformatics/btg015
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hung, J. H., Yang, T. H., Hu, Z., Weng, Z., and Delisi, C. (2012). Gene set enrichment analysis: performance evaluation and usage guidelines. Brief. Bioinform. 13, 281–291. doi: 10.1093/bib/bbr049
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Hyduke, D. R., Lewis, N. E., and Palsson, B. O. (2013). Analysis of omics data with genome-scale models of metabolism. Mol. Biosyst. 9, 167–174. doi: 10.1039/c2mb25453k
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jensen, P. A., and Papin, J. A. (2011). Functional integration of a metabolic network model and expression data without arbitrary thresholding. Bioinformatics 27, 541–547. doi: 10.1093/bioinformatics/btq702
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jerby, L., Shlomi, T., and Ruppin, E. (2010). Computational reconstruction of tissue-specific metabolic models: application to human liver metabolism. Mol. Syst. Biol. 6:401. doi: 10.1038/msb.2010.56
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Junker, B. H. (2014). Flux analysis in plant metabolic networks: increasing throughput and coverage. Curr. Opin. Biotechnol. 26, 183–188. doi: 10.1016/j.copbio.2014.01.016
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Jupp, S., Malone, J., Bolleman, J., Brandizi, M., Davies, M., Garcia, L., et al. (2014). The EBI RDF platform: linked open data for the life sciences. Bioinformatics 30, 1338–1339. doi: 10.1093/bioinformatics/btt765
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kanehisa, M., Goto, S., Sato, Y., Kawashima, M., Furumichi, M., and Tanabe, M. (2014). Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res. 42, D199–D205. doi: 10.1093/nar/gkt1076
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Karr, J. R., Sanghvi, J. C., Macklin, D. N., Gutschow, M. V., Jacobs, J. M., Bolival, B. Jr., et al. (2012). A whole-cell computational model predicts phenotype from genotype. Cell 150, 389–401. doi: 10.1016/j.cell.2012.05.044
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Katayama, T., Wilkinson, M. D., Aoki-Kinoshita, K. F., Kawashima, S., Yamamoto, Y., Yamaguchi, A., et al. (2014). BioHackathon series in 2011 and 2012: penetration of ontology and linked data in life science domains. J. Biomed. Semantics 5:5. doi: 10.1186/2041-1480-5-5
Kell, D. B. (2002). Genotype-phenotype mapping: genes as computer programs. Trends Genet. 18, 555–559. doi: 10.1016/S0168-9525(02)02765-8
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kerrien, S., Aranda, B., Breuza, L., Bridge, A., Broackes-Carter, F., Chen, C., et al. (2012). The IntAct molecular interaction database in 2012. Nucleic Acids Res. 40, D841–D846. doi: 10.1093/nar/gkr1088
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Khatri, P., Sirota, M., and Butte, A. J. (2012). Ten years of pathway analysis: current approaches and outstanding challenges. PLoS Comput. Biol. 8:e1002375. doi: 10.1371/journal.pcbi.1002375
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kim, T. Y., Sohn, S. B., Kim, Y. B., Kim, W. J., and Lee, S. Y. (2012). Recent advances in reconstruction and applications of genome-scale metabolic models. Curr. Opin. Biotechnol. 23, 617–623. doi: 10.1016/j.copbio.2011.10.007
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Krouk, G., Crawford, N. M., Coruzzi, G. M., and Tsay, Y. F. (2010). Nitrate signaling: adaptation to fluctuating environments. Curr. Opin. Plant Biol. 13, 266–273. doi: 10.1016/j.pbi.2009.12.003
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kruger, N. J., and Ratcliffe, R. G. (2012). Pathways and fluxes: exploring the plant metabolic network. J. Exp. Bot. 63, 2243–2246. doi: 10.1093/jxb/ers073
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Kusano, M., and Fukushima, A. (2013). Current challenges and future potential of tomato breeding using omics approaches. Breed. Sci. 63, 31–41. doi: 10.1270/jsbbs.63.31
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lei, Z., Huhman, D. V., and Sumner, L. W. (2011). Mass spectrometry strategies in metabolomics. J. Biol. Chem. 286, 25435–25442. doi: 10.1074/jbc.R111.238691
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lerman, J. A., Hyduke, D. R., Latif, H., Portnoy, V. A., Lewis, N. E., Orth, J. D., et al. (2012). In silico method for modelling metabolism and gene product expression at genome scale. Nat. Commun. 3:929. doi: 10.1038/ncomms1928
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lewis, N. E., Nagarajan, H., and Palsson, B. O. (2012). Constraining the metabolic genotype-phenotype relationship using a phylogeny of in silico methods. Nat. Rev. Microbiol. 10, 291–305. doi: 10.1038/nrmicro2737
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Libourel, I. G., and Shachar-Hill, Y. (2008). Metabolic flux analysis in plants: from intelligent design to rational engineering. Annu. Rev. Plant Biol. 59, 625–650. doi: 10.1146/annurev.arplant.58.032806.103822
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Lucas, M., Laplaze, L., and Bennett, M. J. (2011). Plant systems biology: network matters. Plant Cell Environ. 34, 535–553. doi: 10.1111/j.1365-3040.2010.02273.x
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Luo, W., and Brouwer, C. (2013). Pathview: an R/Bioconductor package for pathway-based data integration and visualization. Bioinformatics 29, 1830–1831. doi: 10.1093/bioinformatics/btt285
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Machado, D., and Herrgard, M. (2014). Systematic evaluation of methods for integration of transcriptomic data into constraint-based models of metabolism. PLoS Comput. Biol. 10:e1003580. doi: 10.1371/journal.pcbi.1003580
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Maere, S., Heymans, K., and Kuiper, M. (2005). BiNGO: a Cytoscape plugin to assess overrepresentation of gene ontology categories in biological networks. Bioinformatics 21, 3448–3449. doi: 10.1093/bioinformatics/bti551
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Merico, D., Isserlin, R., Stueker, O., Emili, A., and Bader, G. D. (2010). Enrichment map: a network-based method for gene-set enrichment visualization and interpretation. PLoS ONE 5:e13984. doi: 10.1371/journal.pone.0013984
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Mintz-Oron, S., Meir, S., Malitsky, S., Ruppin, E., Aharoni, A., and Shlomi, T. (2012). Reconstruction of Arabidopsis metabolic network models accounting for subcellular compartmentalization and tissue-specificity. Proc. Natl. Acad. Sci. U.S.A. 109, 339–344. doi: 10.1073/pnas.1100358109
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Montojo, J., Zuberi, K., Rodriguez, H., Kazi, F., Wright, G., Donaldson, S. L., et al. (2010). GeneMANIA Cytoscape plugin: fast gene function predictions on the desktop. Bioinformatics 26, 2927–2928. doi: 10.1093/bioinformatics/btq562
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Montojo, J., Zuberi, K., Rodriguez, H., Bader, G. D., and Morris, Q. (2014). GeneMANIA: fast gene network construction and function prediction for Cytoscape. F1000Res 3:153. doi: 10.12688/f1000research.4572.1
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nagele, T., and Weckwerth, W. (2013). A workflow for mathematical modeling of subcellular metabolic pathways in leaf metabolism of Arabidopsis thaliana. Front. Plant Sci. 4:541. doi: 10.3389/fpls.2013.00541
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Nishida, K., Ono, K., Kanaya, S., and Takahashi, K. (2014). KEGGscape: a cytoscape app for pathway data integration [v1; ref status: approved with reservations 2, http://f1000r.es/3qe]. F1000 Research 3:144. doi: 10.12688/f1000research.4524.1
Orth, J. D., Thiele, I., and Palsson, B. O. (2010). What is flux balance analysis? Nat. Biotechnol. 28, 245–248. doi: 10.1038/nbt.1614
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Poolman, M. G., Miguet, L., Sweetlove, L. J., and Fell, D. A. (2009). A genome-scale metabolic model of Arabidopsis and some of its properties. Plant Physiol. 151, 1570–1581. doi: 10.1104/pp.109.141267
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Provart, N. (2012). Correlation networks visualization. Front. Plant Sci. 3:240. doi: 10.3389/fpls.2012.00240
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Radrich, K., Tsuruoka, Y., Dobson, P., Gevorgyan, A., Swainston, N., Baart, G., et al. (2010). Integration of metabolic databases for the reconstruction of genome-scale metabolic networks. BMC Syst. Biol. 4:114. doi: 10.1186/1752-0509-4-114
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Ramanan, V. K., Shen, L., Moore, J. H., and Saykin, A. J. (2012). Pathway analysis of genomic data: concepts, methods, and prospects for future development. Trends Genet. 28, 323–332. doi: 10.1016/j.tig.2012.03.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Redestig, H., Kusano, M., Fukushima, A., Matsuda, F., Saito, K., and Arita, M. (2010). Consolidating metabolite identifiers to enable contextual and multi-platform metabolomics data analysis. BMC Bioinformatics 11:214. doi: 10.1186/1471-2105-11-214
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rohn, H., Hartmann, A., Junker, A., Junker, B. H., and Schreiber, F. (2012a). FluxMap: a VANTED add-on for the visual exploration of flux distributions in biological networks. BMC Syst. Biol. 6:33. doi: 10.1186/1752-0509-6-33
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Rohn, H., Junker, A., Hartmann, A., Grafahrend-Belau, E., Treutler, H., Klapperstuck, M., et al. (2012b). VANTED v2: a framework for systems biology applications. BMC Syst. Biol. 6:139. doi: 10.1186/1752-0509-6-139
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Saha, R., Chowdhury, A., and Maranas, C. D. (2014). Recent advances in the reconstruction of metabolic models and integration of omics data. Curr. Opin. Biotechnol. 29C, 39–45. doi: 10.1016/j.copbio.2014.02.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Saha, R., Suthers, P. F., and Maranas, C. D. (2011). Zea mays iRS1563: a comprehensive genome-scale metabolic reconstruction of maize metabolism. PLoS ONE 6:e21784. doi: 10.1371/journal.pone.0021784
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Saito, K., and Matsuda, F. (2010). Metabolomics for functional genomics, systems biology, and biotechnology. Annu. Rev. Plant Biol. 61, 463–489. doi: 10.1146/annurev.arplant.043008.092035
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sakurai, N., Ara, T., Ogata, Y., Sano, R., Ohno, T., Sugiyama, K., et al. (2011). KaPPA-View4: a metabolic pathway database for representation and analysis of correlation networks of gene co-expression and metabolite co-accumulation and omics data. Nucleic Acids Res. 39, D677–D684. doi: 10.1093/nar/gkq989
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Scardoni, G., Tosadori, G., Faizan, M., Spoto, F., Fabbri, F., and Laudanna, C. (2014). Biological network analysis with CentiScaPe: centralities and experimental dataset integration [v1; ref status: approved with reservations 2, http://f1000r.es/3p0]. F1000Res 3:139. doi: 10.12688/f1000research.4477.1
Schmidt, B. J., Ebrahim, A., Metz, T. O., Adkins, J. N., Palsson, B. O., and Hyduke, D. R. (2013). GIM3E: condition-specific models of cellular metabolism developed from metabolomics and expression data. Bioinformatics 29, 2900–2908. doi: 10.1093/bioinformatics/btt493
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Seaver, S. M., Gerdes, S., Frelin, O., Lerma-Ortiz, C., Bradbury, L. M., Zallot, R., et al. (2014). High-throughput comparison, functional annotation, and metabolic modeling of plant genomes using the PlantSEED resource. Proc. Natl. Acad. Sci. U.S.A. 111, 9645–9650. doi: 10.1073/pnas.1401329111
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Seaver, S. M., Henry, C. S., and Hanson, A. D. (2012). Frontiers in metabolic reconstruction and modeling of plant genomes. J. Exp. Bot. 63, 2247–2258. doi: 10.1093/jxb/err371
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Smoot, M. E., Ono, K., Ruscheinski, J., Wang, P. L., and Ideker, T. (2011). Cytoscape 2.8: new features for data integration and network visualization. Bioinformatics 27, 431–432. doi: 10.1093/bioinformatics/btq675
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stelling, J. (2004). Mathematical models in microbial systems biology. Curr. Opin. Microbiol. 7, 513–518. doi: 10.1016/j.mib.2004.08.004
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Stitt, M. (2013). Systems-integration of plant metabolism: means, motive and opportunity. Curr. Opin. Plant Biol. 16, 381–388. doi: 10.1016/j.pbi.2013.02.012
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Strömbäck, L., and Lambrix, P. (2005). Representations of molecular pathways: an evaluation of SBML, PSI MI and BioPAX. Bioinformatics 21, 4401–4407. doi: 10.1093/bioinformatics/bti718
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sweetlove, L. J., Obata, T., and Fernie, A. R. (2014). Systems analysis of metabolic phenotypes: what have we learnt? Trends Plant Sci. 19, 222–230. doi: 10.1016/j.tplants.2013.09.005
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Sweetlove, L. J., and Ratcliffe, R. G. (2011). Flux-balance modeling of plant metabolism. Front. Plant Sci. 2:38. doi: 10.3389/fpls.2011.00038
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Thiele, I., and Palsson, B. O. (2010). A protocol for generating a high-quality genome-scale metabolic reconstruction. Nat. Protoc. 5, 93–121. doi: 10.1038/nprot.2009.203
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Topfer, N., Caldana, C., Grimbs, S., Willmitzer, L., Fernie, A. R., and Nikoloski, Z. (2013). Integration of genome-scale modeling and transcript profiling reveals metabolic pathways underlying light and temperature acclimation in Arabidopsis. Plant Cell 25, 1197–1211. doi: 10.1105/tpc.112.108852
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Toubiana, D., Fernie, A. R., Nikoloski, Z., and Fait, A. (2013). Network analysis: tackling complex data to study plant metabolism. Trends Biotechnol. 31, 29–36. doi: 10.1016/j.tibtech.2012.10.011
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Tsesmetzis, N., Couchman, M., Higgins, J., Smith, A., Doonan, J. H., Seifert, G. J., et al. (2008). Arabidopsis reactome: a foundation knowledgebase for plant systems biology. Plant Cell 20, 1426–1436. doi: 10.1105/tpc.108.057976
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Usadel, B., Nagel, A., Thimm, O., Redestig, H., Blaesing, O. E., Palacios-Rojas, N., et al. (2005). Extension of the visualization tool MapMan to allow statistical analysis of arrays, display of corresponding genes, and comparison with known responses. Plant Physiol. 138, 1195–1204. doi: 10.1104/pp.105.060459
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Van Iersel, M. P., Kelder, T., Pico, A. R., Hanspers, K., Coort, S., Conklin, B. R., et al. (2008). Presenting and exploring biological pathways with PathVisio. BMC Bioinformatics 9:399. doi: 10.1186/1471-2105-9-399
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Van Roey, K., Orchard, S., Kerrien, S., Dumousseau, M., Ricard-Blum, S., Hermjakob, H., et al. (2013). Capturing cooperative interactions with the PSI-MI format. Database (Oxford) 2013:bat066. doi: 10.1093/database/bat066
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Weckwerth, W. (2011). Green systems biology - From single genomes, proteomes and metabolomes to ecosystems research and biotechnology. J. Proteomics 75, 284–305. doi: 10.1016/j.jprot.2011.07.010
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Xia, J., and Wishart, D. S. (2010). MSEA: a web-based tool to identify biologically meaningful patterns in quantitative metabolomic data. Nucleic Acids Res. 38, W71–W77. doi: 10.1093/nar/gkq329
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yildirim, M. A., Goh, K. I., Cusick, M. E., Barabasi, A. L., and Vidal, M. (2007). Drug-target network. Nat. Biotechnol. 25, 1119–1126. doi: 10.1038/nbt1338
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yizhak, K., Benyamini, T., Liebermeister, W., Ruppin, E., and Shlomi, T. (2010). Integrating quantitative proteomics and metabolomics with a genome-scale metabolic network model. Bioinformatics 26, i255–i260. doi: 10.1093/bioinformatics/btq183
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Yu, X., Zheng, G., Shan, L., Meng, G., Vingron, M., Liu, Q., et al. (2014). Reconstruction of gene regulatory network related to photosynthesis in Arabidopsis thaliana. Front. Plant Sci. 5:273. doi: 10.3389/fpls.2014.00273
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zhang, P., Foerster, H., Tissier, C. P., Mueller, L., Paley, S., Karp, P. D., et al. (2005). MetaCyc and AraCyc. Metabolic pathway databases for plant research. Plant Physiol. 138, 27–37. doi: 10.1104/pp.105.060376
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Zuberi, K., Franz, M., Rodriguez, H., Montojo, J., Lopes, C. T., Bader, G. D., et al. (2013). GeneMANIA prediction server 2013 update. Nucleic Acids Res. 41, W115–W122. doi: 10.1093/nar/gkt533
Pubmed Abstract | Pubmed Full Text | CrossRef Full Text | Google Scholar
Keywords: network visualization, pathway analysis, flux balance analysis, genome-scale metabolic reconstruction, plant metabolism
Citation: Fukushima A, Kanaya S and Nishida K (2014) Integrated network analysis and effective tools in plant systems biology. Front. Plant Sci. 5:598. doi: 10.3389/fpls.2014.00598
Received: 01 August 2014; Accepted: 14 October 2014;
Published online: 04 November 2014.
Edited by:
Lee Sweetlove, University of Oxford, UKReviewed by:
Kris Morreel, University Ghent, BelgiumSabrina Kleessen, Max-Planck Institute of Molecular Plant Physiology, Germany
Copyright © 2014 Fukushima, Kanaya and Nishida. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) or licensor are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Atsushi Fukushima, RIKEN Center for Sustainable Resource Science, 1-7-22 Suehirocho, Tsurumi, Yokohama 230-0045, Japan e-mail:YXRzdXNoaS5mdWt1c2hpbWFAcmlrZW4uanA=