

- Australian Centre for Plant Functional Genomics, School of Botany, University of Melbourne, Parkville VIC, Australia
Abiotic stresses such as low water availability and high salinity are major causes of cereal crop yield losses and significantly impact on sustainability. Wheat and barley are two of the most important cereal crops (after maize and rice) and are grown in increasingly hostile environments with soil salinity and drought both expected to increase this century, reducing the availability of arable land. Barley and wheat are classified as glycophytes (salt-sensitive), yet they are more salt-tolerant than other cereal crops such as rice and so are good models for studying salt tolerance in cereals. The exploitation of genetic variation of phenotypic traits through plant breeding could significantly improve growth of cereals in salinity-affected regions, thus leading to improved crop yields. Genetic variation in phenotypic traits for abiotic stress tolerance have been identified in land races and wild germplasm but the molecular basis of these differences is often difficult to determine due to the complex genetic nature of these species. High-throughput functional genomics technologies, such as transcriptomics, metabolomics, proteomics, and ionomics are powerful tools for investigating the molecular responses of plants to abiotic stress. The advancement of these technologies has allowed for the identification and quantification of transcript/metabolites in specific cell types and/or tissues. Using these new technologies on plants will provide a powerful tool to uncovering genetic traits in more complex species such as wheat and barley and provide novel insights into the molecular mechanisms of salinity stress tolerance.
Introduction
Abiotic stresses such as drought, salinity, extreme temperatures and nutrient deficiency, and toxicity are major causes of plant stress and agriculture yield losses. With an increase in the loss of arable land due to drought and salinity it is imperative that we increase the crop yield per hectare. In terms of worldwide production maize, rice, wheat, and barley are the four most important crops (844, 672, 650, and 123 MT, respectively, 20101. Wheat and barley are grown in increasingly hostile environments with soil salinity and drought both expected to increase this century reducing the availability of arable land. With the global population expected to reach nine billion by 20502 an increase in global agriculture productivity will be needed to meet the increase in demand for global food supply. Around 80% of human food produced is comprised of crops, with this dominated by the cereals that comprise 50% of global food production (Langridge and Fleury, 2011). Since the 1960s global cereal production has undergone a linear increase of 30% per year (Tester and Langridge, 2010).However, in order to meet the required increase in food production, crop productivity will need to increase by 38% annually. To achieve this increase in food production it will be imperative to look for genetic improvement of crops, not only by using traditional plant breeding methods but also genetic modification technologies.
Complex traits such as an increased tolerance to abiotic stresses are multi-genic and thus are difficult to identify. The improvement of crops is largely dependent on exploiting genetic variation and this has been achieved in the past through traditional plant breeding methods (Langridge and Fleury, 2011). One approach to elucidate abiotic stress tolerance mechanisms is to identify naturally occurring genetic variation within a crop species by screening varieties, wild genotypes and landraces (Roy et al., 2011b). The use of forward genetics is a powerful approach to study traits contributing to abiotic stress tolerance, however, it is crucial that reliable and accurate quantitative phenotyping methods are employed to assist in the identification of these traits. In recent years, phenomics technologies have become much more advanced and high-throughput, non-destructive measurements can now be made to assist in accurate phenotyping (reviewed in Roy et al., 2011b). In conjunction, recent advances in high-throughput functional genomics technologies such as next generation sequencing (NGS), transcriptomics, metabolomics, and proteomics make it possible to use a systems biology approach to understand the response to environmental stress. In this review we will discuss the use of high-throughput functional genomics technologies to understand salinity stress tolerance mechanisms in plants focussing on cereal crops. Most functional genomics studies have used the model plant Arabidopsis and very few studies have been conducted on more genetically complex plant species such as wheat. However, with the rapid development of functional genomic tools we can now use this technology to identify abiotic stress tolerance mechanisms in cereal crops. We also highlight the need to use both spatial and temporal resolution to elucidate the molecular response to salinity (and other abiotic stresses). These studies will lead to a greater understanding of the plant response to salinity stress and thus the integration of this data will contribute to improving our ability to generate salt-tolerant crops.
Genetic Variation Insalt Stress Tolerance in Cereal Crops
Different plant species have a wide ranging capacity for salt tolerance from the very sensitive species (glycophytes) such as the model plant Arabidopsis to the very tolerant halophyticspecies such as Atriplex spp. (saltbush). Cereal crops are classified as glycophytes, however, different crop species can also have different capacities and mechanisms to tolerate salt stress, for example, rice is more sensitive than both barley and wheat(Munns and Tester, 2008). Within a species there can also be naturally occuring genetic variation in salt tolerance and this can be exploited for breeding of salt-tolerant crops (Roy et al., 2011b). It has been proposed that the temporal response of plantsto soil salinity occurs in two separate phases that have been termed as osmotic and ionic (Munns, 2002).The early phase (hours to days) is described as an osmotic stress, due to the low water potentialaround the roots, in response to saline soils and/or water deficit. Osmotic stress is described as a shoot ion independent stress and can result in cell dehydration and loss of cell turgor pressure, and can be characterized phenotypically by a reduction in root elongation, inhibition of photosynthesis and a reduction in shoot growth (Munns and Tester, 2008).In contrast, ionic stress occurs at a later stage (usually after weeks or months) and is a result of the accumulation of toxic concentrations of Na+ and Cl- in the cell cytoplasm resulting in decreased growth and yield. In response to salt stress, crop plants have evolved the following three tolerance mechanisms – (1) osmotic stress tolerance: ability to maintain water uptake and growth, (2) Na+ exclusion: exclusion of toxic ions from the shoot tissues, and (3) tissue tolerance: compartmentalisation of toxic ions into the vacuole or specific tissues (Munns and Tester, 2008).
One approach to identify adaptive traits to abiotic stresses is to screen for genetic diversity in populations (Figure 1). Genetic variation in salt tolerance has been shown inmany cereal crops including barley (Shavrukov et al., 2010) and wheat (James et al., 2008; Rahnama et al., 2011).The focus of most research for enhancing salt tolerance in plants has concentrated on the mechanisms that control Na+ exclusion (Munns and Tester, 2008). For example, in a study on durum wheat genotypes, a wide genetic variation was observed in their ability to exclude Na+ that was not present in modern cultivars (Munns et al., 2000). This led to the identification of two major genes for Na+ exclusion, Nax1 and Nax2 (Lindsay et al., 2004; James et al., 2006). The intergression of Nax2 from the parent line, Triticum monococcum, into durum wheat produced a salt-tolerant phenotype (James et al., 2012).
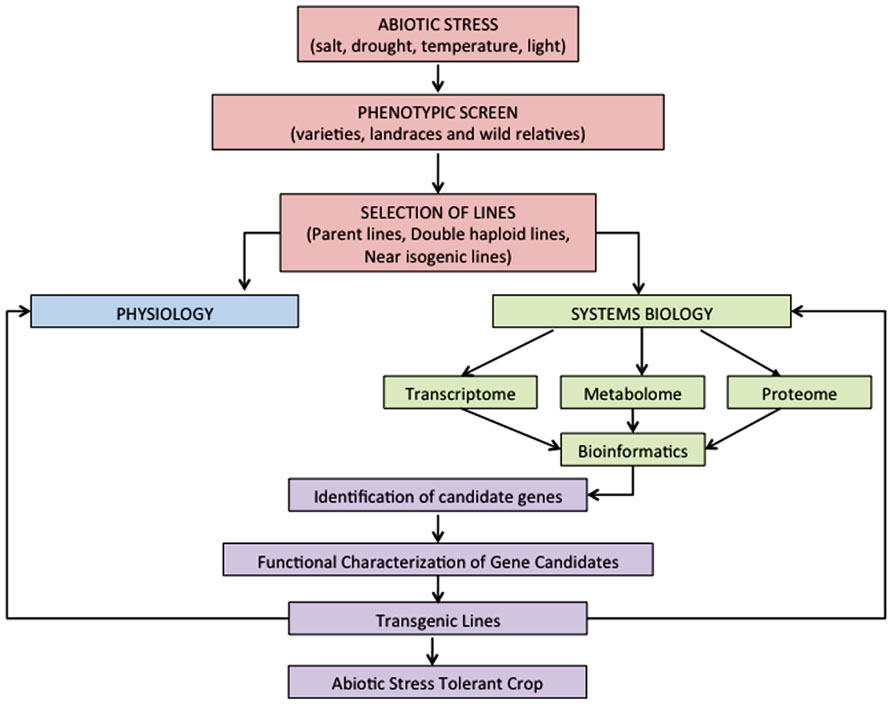
FIGURE 1. Proposed strategy for the integration of physiology and systems biology to gain insights into abiotic stress responses in cereals and the future development of abiotic stress tolerant crops.
It was originally thought that there was very little evidence of genetic variation in osmotic stress tolerance (Munns et al., 1995), so this area of research has largely been neglectedand there are limited studies reporting genetic variation in osmotic stress tolerance within a cereal species. A number of recent studies have identified genetic variation in osmotic stress tolerance by measuring shoot traits. In a study of 12 Triticum monococcum accessions, non-destructive assays were developed to distinguish the three mechanims of salinity tolerance; osmotic tolerance, Na+ exclusion and tissue tolerance (Rajendran et al., 2009). Different accessions appear to use different combinations of the three tolerance mechanisms to increase their salinity tolerance. This information can now be used for forward genetic screens to identify the molecular basis of these tolerance mechanims.
Stomatal conductance is known to be reduced immediately upon exposure of the roots to salinity, so the plant must be responding systemically to the osmotic impact.For example, in a screen of 50 durum varieties and landraces, two- to threefold differences in stomatal conductance were observed and higher stomatal conductance positively correlated with relative growth rate (James et al., 2008). Despite a number of recent phenotypic studies, the molecular mechanisms for osmotic stress tolerance remain unknown. Osmotic stress is initially detected by the roots upon exposure to a low water potential (either as a result of water deficit or salinity). There are few studies decribing genetic variation in the root growth response to osmotic stress, mainly due to the difficulties associated with visualizing and phenotyping roots (Gewin, 2010; Richards et al., 2010; Zhu et al., 2011). One example of genetic variation in root growth has been shown by the measurement ofroot elongation rate of eight barley genotypes (including barley cultivars, a landrace and wild barley) in response to the early component of salinity stress. In this study the wild barley, Hordeum vulgare ssp. spontaneum, was identified as the most tolerant (Shelden et al., 2013). The identification of genetic variation in root phenotypes will provide important information for future genetic characterisation and molecular studies to elucidate the molecular pathways involved in the early phase of salinity tolerance.
Utilizing Functional Genomics Approaches to Elucidate Plant Stress Responses
Functional genomics approaches such as genomics, transcriptomics, metabolomics, proteomics, and ionomics, have been extensively used to evaluate abiotic stress tolerance mechanisms in plants (Cramer et al., 2011). These platforms can be utilized to improve our ability to discover the genes and pathways that control specific traits in response to abiotic stress (Figure 1).
Plants are complex organisms with many different tissue and cell types and the interaction between these tissues and cells requires complex regulation. To gain a greater understanding of the complexities of cell-specific regulation, an increase in the sensitivity of functional genomic tools are required. To date, functional genomics approaches have mainly been utilized for studies of whole organs such as leaves and roots (Widodo et al., 2009), thus it is possible that important changes in both metabolites and gene expression go undetected because they are diluted out by the surrounding tissue. Combining a forward genetics approach with spatially resolved ‘omics approaches may greatly improve our chances of discovering gene candidates leading to the generation of abiotic stress tolerant crops.
Advancements in Functional Genomics Techniques for Spatial Resolution
Transcriptomics
Microarrays were the first available method for genome wide transcript expression profiling and have been used extensively in plant biology. Microarrays have been extensively used to generate transcriptional profiles in response to abiotic stresses in a range of plant species including, but not limited to, Arabidopsis (Kreps et al., 2002), Vitis vinifera L. (grapevine; Cramer et al., 2007) and Hordeum vulgare L. (barley; Walia et al., 2006). Studies of the transcriptional response to salt stress have been conducted in barley (Ueda et al., 2004, 2006; Walia et al., 2006, 2007; Gruber et al., 2009) and the model cereal Brachypodium (Kim et al., 2012) but due to the polyploidy genome, studies of wheat are more limited (Kawaura et al., 2006, 2008; Mott and Wang, 2007; Jamil et al., 2011; Garg et al., 2013).As the technology of microarrays and cell separation techniques has become more advanced it has become possible to do spatial profiling of plant tissues and cell-specific studies (Brady et al., 2007; Dinneny et al., 2008; Spollen et al., 2008). Cell-type specific transcript studies have been conducted in many plant species including Arabidopsis and the cereals maize, rice, barley, and soybean using microarrays (Pu and Brady, 2010; Long, 2011; Rogers et al., 2012). Despite a large number of microarray experiments, very few have led to the identification of stress tolerance pathways (Deyholos, 2010).
High-throughput, deep sequencing technology (RNA-Seq) provides a new strategy to detect and accurately quantitate changes in the transcriptome. NGS is still in its infancy in plant biology but is expected to supercede microarray based approaches as the technology becomes more developed. NGS technology is a powerful tool for gene expression profiling and has been used successfully in a number of organisms including mammalian systems (Mortazavi et al., 2008), yeast (Nagalakshmi et al., 2008) and plants (Lister et al., 2008) to show transcriptional activity. The use of NGS technology for expression studies is becoming more popular with studies reported in the model plant Arabidopsis (Weber et al., 2007) and several cereal species including Glycine max L. (soybean; Fan et al., 2013), Lolium perenne L. (perennial ryegrass; Studer et al., 2012), Triticum aestivum (wheat) endosperm (Gillies et al., 2012), Zea mays (maize; Li et al., 2010), Sorghum bicolor (sorghum; Dugas et al., 2011), Panicum virgatum L. (switchgrass; Wang et al., 2012) and the extremophile Thellungiella parvula (Dassanayake et al., 2011). Studies now show that RNA-Seq provides better quantitation and accuracy than microarrays (Jain, 2012). Theuse of RNA-Seq in plant biology has been comprehensively reviewed and will therefore not be reviewed here (Jain, 2011, 2012). There are only very few studies published utilizing NGS to study abiotic stress responses in plants (Deyholos, 2010). These include studies on salt stress (Molina et al., 2008; Fan et al., 2013), cold stress (Tamura and Yonemaru, 2010) and drought stress (Dugas et al., 2011; Dong et al., 2012; Vidal et al., 2012).
With the availability of a number of cereal genomes including Barley Sequencing Consortium (2012), sorghum (Paterson et al., 2009), maize (Schnable et al., 2009), rice (Project, 2005), and the cereal model Brachypodium (Vogel et al., 2010) it is now possible to use this information to conduct large scale NGS transcriptomic experiments in cereal crops. For example, in a study on Sorghum bicolor, RNA-Seq technology was utilized in combination with the sorghum genome sequence to analyze the root and shoot transcriptome in response to osmotic stress and abscisic acid (ABA; Dugas et al., 2011). The study identified over 28,000 unique genes that were transcriptionally regulated in response to osmotic stress and ABA. A collection of drought-regulated gene networks and transcription factors were identified that can provide a basis for further studies. The data in this study was also compared to other sequenced transcriptomes including maize, rice and Arabidopsis, identifying more than 50 differentially expressed drought responsive gene orthologs. The lack of fully sequenced genomes in plants with larger, more complex genomes, such as wheat (hexaploid), present many difficulties for these types of ‘omics studies. Utilizing these new technologies in combination with the completed genome sequences of cereals will providea powerful tool to uncovering genetic traits in more complex species such as wheat.
Proteomics
Proteomic studies of the response to salt stress have been conducted in many plant species including the cereals rice (Oryza sativa), wheat (Triticum durum and Triticum aestivum), barley (Hordeum vulgare), and maize (Zea mays; reviewed in Zhang et al., 2012). A number of comprehensive reviews describing proteomic responses to abiotic stress are available and therefore will not be reviewed here (Kosova et al., 2011; Roy et al., 2011a; Zhang et al., 2012). Single cell proteomic studies have been successful in mammalian systems where cells can be cultured to increase the starting material (Schirle et al., 2003; Diks and Peppelenbosch, 2004), but there are relatively few examples of single cell proteomic studies in plants, and especially in cereals, due to the difficulties in obtaining enough material. Single cell-type proteomic analyses have been reported for Arabidopsis guard cells (Zhao et al., 2008), trichomes (Wienkoop et al., 2004; Kryvych et al., 2011) and pollen (Holmes-Davis et al., 2005), soybean (Glycine max) root hairs (Wan et al., 2005) and tobacco (Nicotiana tabacum) trichomes (Amme et al., 2005). Tissue specific proteomic studies of the response to salinity include a comprehensive proteomics study of rice anthers (Sarhadi et al., 2012) wheat root seedlings (Guo et al., 2012) and rice plasma membranes (Cheng et al., 2009). Kamal et al. (2012) analyzed wheat chloroplast protein abundance only and correlated their salt-responsive behavior to numerous physiological parameters. However, in most of these studies only a small number of salt-responsive proteins were identified (Zhao et al., 2008).
Metabolomics
Metabolites are the building blocks for structural and enzymatic molecules and carry the energy required for growth and maintenance of living cells. Metabolites are a key link between genetic information and a phenotype and are a measure of the physiological state of an organism. In the past decade enormous efforts have been made to develop high-throughput and untargeted methodologies to analyze as many metabolites as possible. The resulting relatively new field of metabolomics uses a set of sophisticated analytical biochemistry tools for the identification and quantification of metabolites. The application of bioinformatics and statistical tools allows the extraction and analysis of multivariate data sets supporting biological interpretation. An inherent challenge of metabolomics derives from the fact that the metabolome is a simple descriptor for what is, in fact, huge chemical diversity. Hundreds and hundreds of compounds with different physical and chemical properties such as molecular weight, molecular size, polarity, stability, volatility, solubility, and many more, require the development of comprehensive and/or complementary extraction, separation, detection, and quantification techniques (Beckles and Roessner, 2012). A number of analytical technologies, including chromatographic separation techniques, such as liquid and gas chromatography, coupled to mass spectrometry and nuclear magnetic resonance (NMR) spectroscopy have been successfully used to analyze metabolites in many different organisms, tissues and biofluids (Roessner and Beckles, 2009).
Metabolomic analysis has been used extensively to study abiotic stress responses in plants (Urano et al., 2010; Obata and Fernie, 2012). Due to the importance of salinity and drought stress in agriculture, many metabolomic studies have been conducted on agriculturally important crops to gain insight into the effect of such stress on metabolic activity. Analyzing genotypes that have differing phenotypic responses to abiotic stress (sensitive vs. tolerant) will help elucidate the specific metabolic changes that are contributing to the increased tolerance of a genotype. For example, in a study conducted on two genotypes of barley that differ in salt tolerance, it was found that the landrace Sahara accumulates higher levels of metabolites involved in cellular protection in the leaves, corresponding with higher leaf Na+ concentrations compared to the less tolerant genotype, Clipper (Widodo et al., 2009).
To date, most metabolomics analyses have been carried out on bulked tissues (e.g., whole roots or leaves) due to sensitivity levels of the technologies employed. This means that the metabolic profile of different tissue and cell types, each likely to be characterized by a specific metabolite profile or a particular response to environmental or genetic stimuli, have to be measured together. Instrumentation with increased sensitivity will help substantially, but a major problem remains, that it is often difficult or even impossible to separate and isolate tissue types or single cell types from plant tissues. The first success of a single cell metabolomics approach has been reported in a study of Arabidopsis where cryo-sectioning was used to preserve cellular structures. Specific cell types were cut and collected using laser micro-dissection to obtain a sufficient number of cells to allow the detection of about 68 major metabolites by gas chromotography/mass spectrometry (GC-MS; Schad et al., 2005). In future, it may be possible to develop cell-type specific protoplasts that can be cultured to increase the amount of tissue for analysis (Keerberg et al., 2011). However, one issue in any such metabolomics analysis is that protoplasts are cell-wall free cells and their generation and culture may affect the physiological state of the cells and therefore the range and concentration of metabolites.
An important aim of the characterization of metabolites in plants is to understand the spatial distribution of single metabolites. Initial such analyses have includedindole-3-acetic acid (IAA) in Arabidopsis roots (Petersson et al., 2009) or ATP in Vicia faba embryos (Borisjuk et al., 2003), but these studies are now being extended to as many metabolites as possible. A very promising new approach to the spatial analysis of both proteins and metabolites is to raster across thin tissue slices and ionize any compound (peptides following proteolytic digestion or metabolites) using matrix-assisted laser desorption ionization (MALDI) coupled with analysis of every ionized molecule in a mass spectrometer (Kaspar et al., 2011). This technique, although not having cellular resolution, allows the determination of tissue-type metabolite distributions. This approach has been well developed for lipids (Vrkoslav et al., 2010; Horn et al., 2012) since they are easily ionized, while efforts are now being made to optimize this approach for proteins/peptides (Grassl et al., 2011) and small molecules (Jun et al., 2010; Lee et al., 2012; Peukert et al., 2012). However, there are currently no reports in the literature on applications of metabolomics to the spatial analysis of salinity responses in plants. Therefore this approach provides the potential to monitor the cell and tissue specific adaptation mechanisms in cereals and provide novel ideas for the development of more tolerant crop genotypes. Techniques such as laser micro-dissection (Yi et al., 2012), fluorescence-activated cell sorting (FACS; (Borges et al., 2012; Evrard et al., 2012) or MALDI based mass spectral imaging (Kaspar et al., 2011) may pave the way for such highly spatially resolved analyses.
Ionomics
The high-throughput analysis of elemental compositions is called ionomics and has been extensively used in the plant sciences for screening mutant collections, forward and reverse genetics approaches and investigating mechanisms of elemental or ion uptake, transport, compartmentalisation and exclusion (Baxter, 2009). In the area of abiotic stress research, elemental profiling provides a way to investigate a plant’s response to salinity, nutrient deficiencies and toxicities or hyper-accumulation. There are 13 minerals essential to all plants, including macronutrients, such as N and P, and micronutrients such as Na, K, B, Mn, Fe, and Ca. Several mineral deficiencies lead to dramatic growth retardation and possibly death, but excess amounts of some minerals can also be toxic. In both cases, plant metabolism is dramatically affected; plants are able to develop mechanisms in order to cope with either deficiency or toxicity. Analysis of the elemental composition is not only important when studying effects of high salt on a plant but also when identifying mechanisms of adaptation and tolerance to any osmotic stress such as drought. Some ions have hygroscopic properties and therefore are utilized to retain water in the cell in order to sustain turgor pressure upon osmotic stress such as water deficiency. There have been many reports on the effects of the expression of ion transporters in plants; studies aimed at determining the function of those transporters (Horie et al., 2009; Jiang et al., 2010; Xue et al., 2011) or increasing tolerance to either salinity or nutrient deficiencies or toxicities (Hauser and Horie, 2010; Plett and Moller, 2010; Horie et al., 2011; Mian et al., 2011).
The simultaneous analysis of the major macro-elements and many of the microelements can be achieved using inductively coupled plasma spectroscopy or optical emission spectroscopy coupled with mass spectrometry (ICP-MS and OES-MS, respectively). With appropriate sample preparation methods, these techniques allow the analysis of between 12 and 20 elements and ions simultaneously (Baxter, 2009). However, in most studies bulk tissue (e.g., leaves or roots) is extracted for elemental analysis, thus giving no information about spatial distribution of elemental composition. Emerging technologies that allow image distributions of elemental composition of biological tissues have now become available for plant tissue sections. Laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS) is a very sensitive and efficient technique that produces three-dimensional images of spatial element distributions in thin biological tissue sections (Becker, 2010). This well advanced technique for the analysis of elemental distributions in human or animal tissue sections allows the reconstruction of three-dimensional atlases of spatial concentrations of elements (Hare et al., 2012). In the plant sciences this technique has not been utilized extensively but it is easy to envisage using it in numerous productive applications.
Examples of Spatially Resolved ‘Omics for Identifying Abiotic Stress Tolerance Mechanisms in Cereals
There are very few examples of using spatially resolved functional genomics technologies to identify abiotic stress tolerance mechanisms in cereal crops. Utilizing emerging high resolution technologies to profile changes in a defined region of the plant could lead to novel insights into the molecular mechanisms of abiotic stress tolerance. The Arabidopsis root has been used as a model for understanding the mechanisms of cell-specific processes and has been extensively reviewed by Dinneny (2010). The development of green fluorescent protein (GFP) reporter lines enabled the characterization of 14 different cell types in the Arabidopsis root, revealing a complex pattern of spatial control of biological functions (Brady et al., 2007). The advancement of these technologies and our current understanding from model systems such as Arabidopsis can now be utilized to increase our understanding of more complex plant systems such as the cereal crops. This will allow development of crops with enhanced tolerance mechanisms to environmental stresses.
The importance of using spatial resolution for ‘omics studies of abiotic stress has been shown in a study on maize roots in response to water deficit (Yamaguchi and Sharp, 2010). The maize primary root has been extensively studied as a model crop system for understanding the root growth response to water deficit. Physiological studies demonstrated that the response to osmotic stress is not uniform in the apical region of the root; the root elongation rate varies spatially in the elongation zone in response to both water deficit (Sharp et al., 1988) and salinity (Bernstein and Kafkafi, 2002). Physiological studies were combined with transcriptomic and proteomic profiling for spatial and temporal profiling of the root zone (Yamaguchi and Sharp, 2010). Transcriptomic analysis was conducted on four regions of the root tip, designated as regions 1–4, defined by their differing spatial growth response to water deficit (Spollen et al., 2008). Only 7.5% of the differentially expressed transcripts were found to be the same between regions 1 and 2, indicating that the transcriptional response was dependent on the position along the root apex. A proteomic analysis of the cell wall proteins identified an increase in cell wall extension proteins and apoplastic reactive oxygen species in the most apical region of the root where root elongation rate is maintained (Zhu et al., 2007). This raises the possibility that these compounds regulate extension properties contributing to the maintenance of root elongation in response to water deficit. This work has led to important advances in understanding the regulation and adaptation of root growth to water deficit and highlights the importance of studying specific regions of the root. These studies clearly demonstrate the importance of combining physiological data with functional genomic data as well as emphasizing the importance of using spatial resolution.
Other studies integrating spatially resolved ‘omics in cereals are limited. Using laser-capture microdissection (LCM) spatially resolved transcriptomics was utilized to gain insight into the development of the maize leaf (Li et al., 2010).Along the developmental gradient, 64% of genes were differentially expressed, and 21% were differentially expressed between the bundle sheath and mesophyll cells. This study utilized the completed maize genome sequence and high-throughput Illumina sequencing for spatial profiling of the maize leave, providing novel information for understanding photosynthetic development in C4 plants. This information serves as a foundation for systems biology approaches and provides strong evidence for the importance of spatial profiling in plant studies.
Conclusions and Future Perspectives
To increase our understanding of a plant’s response to abiotic stress and identify the mechanisms involved in abiotic stress tolerance it is important to study both the physiology and molecular networks that are involved. The use of functional genomic tools in abiotic stress research has substantially extended our knowledge about the basic biology and physiology of abiotic stress responses, however, it has not yet allowed the identification of reproducible markers that could be used to determine if, and to what extent, the plant was under stress. By utilizing advances in both phenomics and functional genomics technologies it is becoming possible to identify cell and tissue specific changes in the molecular and biochemical networks in response to abiotic stress. This will lead to an increased understanding of the molecular mechanisms responsible for abiotic stress tolerance and the generation of abiotic stress tolerant crops.
Conflict of Interest Statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Footnotes
References
Amme, S., Rutten, T., Melzer, M., Sonsmann, G., Vissers, J. P. C., Schlesier, B., et al. (2005). A proteome approach defines protective functions of tobacco leaf trichomes. Proteomics 5, 2508–2518.
Barley Sequencing Consortium, T. I. B. G. S. (2012). A physical, genetic and functional sequence assembly of the barley genome. Nature 491, 711–716.
Baxter, I. (2009). Ionomics: studying the social network of mineral nutrients. Curr. Opin. Plant Biol. 12, 381–386.
Becker, J. S. (2010). “Imaging of metals, metalloids, and non-metals by laser ablation inductively coupled plasma mass spectrometry (LA-ICP-MS) in biological tissues,” in Mass Spectrometry Imaging: Principles and Protocols, eds S. S. Rubakhin and J. V. Sweedler (Totowa: Humana Press Inc), 51–82.
Beckles, D., and Roessner, U. (2012). “Plant metabolomics – applications and opportunities for agricultural biotechnology,” in Plant Biotechnology and Agriculture: Prospects for the 21st Century, eds A. Altmann and P. M. Hasegawa. (London: Elsevier), 67–78.
Bernstein, N., and Kafkafi, U. (2002). “Root growth under salinity stress,” in Plant Roots: The Hidden Half III edition, eds Y. Waisel, A. Eshel and U. Kafkafi (New York: Marcel Dekker, Inc.), 787–805.
Borges, F., Gardner, R., Lopes, T., Calarco, J. P., Boavida, L. C., Slotkin, R. K., et al. (2012). FACS-based purification of Arabidopsis microspores, sperm cells and vegetative nuclei. Plant Methods 8, 44.
Borisjuk, L., Rolletschek, H., Walenta, S., Panitz, R., Wobus, U., and Weber, H. (2003). Energy status and its control on embryogenesis of legumes: ATP distribution within Vicia faba embryos is developmentally regulated and correlated with photosynthetic capacity. Plant J. 36, 318–329.
Brady, S. M., Orlando, D. A., Lee, J.-Y., Wang, J. Y., Koch, J., Dinneny, J. R., et al. (2007). A high-resolution root spatiotemporal map reveals dominant expression patterns. Science 318, 801–806.
Cheng, Y., Qi, Y., Zhu, Q., Chen, X., Wang, N., Zhao, X., et al. (2009). New changes in the plasma-membrane-associated proteome of rice roots under salt stress. Proteomics 9, 3100–3114.
Cramer, G. R., Ergul, A., Grimplet, J., Tillett, R. L., Tattersall, E. A. R., Bohlman, M. C., et al. (2007). Water and salinity stress in grapevines: early and late changes in transcript and metabolite profiles. Funct. Integr. Genomics 7, 111–134.
Cramer, G. R., Urano, K., Delrot, S., Pezzotti, M., and Shinozaki, K. (2011). Effects of abiotic stress on plants: a systems biology perspective. BMC Plant Biol. 11:163. doi: 10.1186/1471-2229-11-163
Dassanayake, M., Oh, D. H., Haas, J. S., Hernandez, A., Hong, H., Ali, S., et al. (2011). The genome of the extremophile crucifer Thellungiella parvula. Nat. Genet. 43, 913–918.
Deyholos, M. K. (2010). Making the most of drought and salinity transcriptomics. Plant Cell Environ. 33, 648–654.
Diks, S. H., and Peppelenbosch, M. P. (2004). Single cell proteomics for personalised medicine. Trends Mol. Med. 10, 574–577.
Dinneny, J. R. (2010). Analysis of the salt-stress response at cell-type resolution. Plant Cell Environ. 33, 543–551.
Dinneny, J. R., Long, T. A., Wang, J. Y., Jung, J. W., Mace, D., Pointer, S., et al. (2008). Cell identity mediates the response of Arabidopsis roots to abiotic stress. Science 320, 942–945.
Dong, C. H., Li, C., Yan, X. H., Huang, S. M., Huang, J. Y., Wang, L. J., et al. (2012). Gene expression profiling of Sinapis alba leaves under drought stress and rewatering growth conditions with Illumina deep sequencing. Mol. Biol. Rep. 39, 5851–5857.
Dugas, D. V., Monaco, M. K., Olsen, A., Klein, R. R., Kumari, S., Ware, D., et al. (2011). Functional annotation of the transcriptome of Sorghum bicolor in response to osmotic stress and abscisic acid. BMC Genomics 12:514. doi: 10.1186/1471-2164-12-514
Evrard, A., Bargmann, B. O., Birnbaum, K. D., Tester, M., Baumann, U., and Johnson, A. A. (2012). Fluorescence-activated cell sorting for analysis of cell type-specific responses to salinity stress in Arabidopsis and rice. Methods Mol. Biol. 913, 265–276.
Fan, X. D., Wang, J. Q., Yang, N., Dong, Y. Y., Liu, L., Wang, F. W., et al. (2013). Gene expression profiling of soybean leaves and roots under salt, saline-alkali and drought stress by high-throughput Illumina sequencing. Gene 512, 392–402.
Garg, B., Puranik, S., Misra, S., Nath Tripathi, B., and Prasad, M. (2013). Transcript profiling identifies novel transcripts with unknown functions as primary response components to osmotic stress in wheat (Triticum aestivum L.). Plant Cell Tiss. Organ Cult. 113, 91–101.
Gillies, S. A., Futardo, A., and Henry, R. J. (2012). Gene expression in the developing aleurone and starchy endosperm of wheat. Plant Biotechnol. J. 10, 668–679.
Grassl, J., Taylor, N. L., and Millar, A. H. (2011). Matrix-assisted laser desorption/ionisation mass spectrometry imaging and its development for plant protein imaging. Plant Methods 7, 21.
Gruber, V., Blanchet, S., Diet, A., Zahaf, O., Boualem, A., Kakar, K., et al. (2009). Identification of transcription factors involved in root apex responses to salt stress in Medicago truncatula. Mol. Genet. Genomics 281, 55–66.
Guo, G. F., Ge, P., Ma, C. Y., Li, X. H., Lv, D. W., Wang, S. L., et al. (2012). Comparative proteomic analysis of salt response proteins in seedling roots of two wheat varieties. J. Proteomics 75, 1867–1885.
Hare, D. J., Lee, J. K., Beavis, A. D., Van Gramberg, A., George, J., Adlard, P. A., et al. (2012). Three-dimensional atlas of iron, copper, and zinc in the mouse cerebrum and brainstem. Anal. Chem. 84, 3990–3997.
Hauser, F., and Horie, T. (2010). A conserved primary salt tolerance mechanism mediated by HKT transporters: a mechanism for sodium exclusion and maintenance of high K+/Na+ ratio in leaves during salinity stress. Plant Cell Environ. 33, 552–565.
Holmes-Davis, R., Tanaka, C. K., Vensel, W. H., Hurkman, W. J., and Mccormick, S. (2005). Proteome mapping of mature pollen of Arabidopsis thaliana. Proteomics 5, 4864–4884.
Horie, T., Hauser, F., and Schroeder, J. I. (2009). HKT transporter-mediated salinity resistance mechanisms in Arabidopsis and monocot crop plants. Trends Plant Sci. 14, 660–668.
Horie, T., Sugawara, M., Okada, T., Taira, K., Kaothien-Nakayama, P., Katsuhara, M., et al. (2011). Rice sodium-insensitive potassium transporter, OsHAK5, confers increased salt tolerance in tobacco BY2 cells. J. Biosci. Bioeng. 111, 346–356.
Horn, P. J., Korte, A. R., Neogi, P. B., Love, E., Fuchs, J., Strupat, K., et al. (2012). Spatial mapping of lipids at cellular resolution in embryos of cotton. Plant Cell 24, 622–636.
Jain, M. (2011). A next-generation approach to the characterization of a non-model plant transcriptome. Curr. Sci. 101, 1435–1439.
Jain, M. (2012). Next-generation sequencing technologies for gene expression profiling in plants. Brief. Funct. Genomics 11, 63–70.
James, R. A., Blake, C., Zwart, A. B., Hare, R. A., Rathjen, A. J., and Munns, R. (2012). Impact of ancestral wheat sodium exclusion genes Nax1 and Nax2 on grain yield of durum wheat on saline soils. Funct. Plant Biol. 39, 609–618.
James, R. A., Davenport, R. J., and Munns, R. (2006). Physiological characterization of two genes for Na+ exclusion in durum wheat, Nax1 and Nax2. Plant Physiol. 142, 1537–1547.
James, R. A., Von Caemmerer, S., Condon, A. G. T., Zwart, A. B., and Munns, R. (2008). Genetic variation in tolerance to the osmotic stress component of salinity stress in durum wheat. Funct. Plant Biol. 35, 111–123.
Jamil, A., Riaz, S., Ashraf, M., and Foolad, M. R. (2011). Gene expression profiling of plants under salt stress. Crit. Rev. Plant Sci. 30, 435–458.
Jiang, X., Leidi, E. O., and Pardo, J. M. (2010). How do vacuolar NHX exchangers function in plant salt tolerance? Plant Signal. Behav. 5, 792–795.
Jun, J. H., Song, Z. H., Liu, Z. J., Nikolau, B. J., Yeung, E. S., and Lee, Y. J. (2010). High-spatial and high-mass resolution imaging of surface metabolites of Arabidopsis thaliana by laser desorption-ionization mass spectrometry using colloidal silver. Anal. Chem. 82, 3255–3265.
Kamal, A. H. M., Cho, K., Kim, D. E., Uozumi, N., Chung, K. Y., Lee, S. Y., et al. (2012). Changes in physiology and protein abundance in salt-stressed wheat chloroplasts. Mol. Biol. Rep. 39, 9059–9074.
Kaspar, S., Peukert, M., Svatos, A., Matros, A., and Mock, H. P. (2011). MALDI-imaging mass spectrometry – an emerging technique in plant biology. Proteomics 11, 1840–1850.
Kawaura, K., Mochida, K., and Ogihara, Y. (2006). Microarray analysis of salt-responsive genes in common wheat. Genes Genet. Syst. 81, 437–437.
Kawaura, K., Mochida, K., and Ogihara, Y. (2008). Genome-wide analysis for identification of salt-responsive genes in common wheat. Funct. Integr. Genomics 8, 277–286.
Keerberg, O., Ivanova, H., Keerberg, H., Parnik, T., Talts, P., and Gardestrom, P. (2011). Quantitative analysis of photosynthetic carbon metabolism in protoplasts and intact leaves of barley. Determination of carbon fluxes and pool sizes of metabolites in different cellular compartments. Biosystems 103, 291–301.
Kim, D. Y., Hong, M. J., Jang, J. H., and Seo, Y. W. (2012). cDNA-AFLP analysis reveals differential gene expression in response to salt stress in Brachypodium distachyon. Genes Genomics 34, 475–484.
Kosova, K., Vitamvas, P., Prasil, I. T., and Renaut, J. (2011). Plant proteome changes under abiotic stress – contribution of proteomics studies to understanding plant stress response. J. Proteomics 74, 1301–1322.
Kreps, J. A., Wu, Y., Chang, H.-S., Zhu, T., Wang, X., and Harper, J. F. (2002). Transcriptome changes for Arabidopsis in response to salt, osmotic, and cold stress. Plant Physiol. 130, 2129–2141.
Kryvych, S., Kleessen, S., Ebert, B., Kersten, B., and Fisahn, J. (2011). Proteomics – the key to understanding systems biology of Arabidopsis trichomes. Phytochemistry 72, 1061–1070.
Langridge, P., and Fleury, D. (2011). Making the most of ‘omics’ for crop breeding. Trends Biotechnol. 29, 33–40.
Lee, Y. J., Perdian, D. C., Song, Z. H., Yeung, E. S., and Nikolau, B. J. (2012). Use of mass spectrometry for imaging metabolites in plants. Plant J. 70, 81–95.
Li, P., Ponnala, L., Gandotra, N., Wang, L., Si, Y., Tausta, S. L., et al. (2010). The developmental dynamics of the maize leaf transcriptome. Nat. Genet. 42, 1060–1067.
Lindsay, M. P., Lagudah, E. S., Hare, R. A., and Munns, R. (2004). A locus for sodium exclusion (Nax1), a trait for salt tolerance, mapped in durum wheat. Funct. Plant Biol. 31, 1105–1114.
Lister, R., O’Malley, R. C., Tonti-Filippini, J., Gregory, B. D., Berry, C. C., Millar, A. H., et al. (2008). Highly integrated single-base resolution maps of the epigenome in Arabidopsis. Cell 133, 523–536.
Long, T. A. (2011). Many needles in a haystack: cell-type specific abiotic stress responses. Curr. Opin. Plant Biol. 14, 325–331.
Mian, A., Oomen, R., Isayenkov, S., Sentenac, H., Maathuis, F. J. M., and Very, A. A. (2011). Over-expression of an Na(+)- and K(+)-permeable HKT transporter in barley improves salt tolerance. Plant J. 68, 468–479.
Molina, C., Rotter, B., Horres, R., Udupa, S. M., Besser, B., Bellarmino, L., et al. (2008). SuperSAGE: the drought stress-responsive transcriptome of chickpea roots. BMC Genomics 9:553. doi: 10.1186/1471-2164-9-553
Mortazavi, A., Williams, B. A., Mccue, K., Schaeffer, L., and Wold, B. (2008). Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 5, 621–628.
Mott, I. W., and Wang, R. R. C. (2007). Comparative transcriptome analysis of salt-tolerant wheat germplasm lines using wheat genome arrays. Plant Sci. 173, 327–339.
Munns, R., Hare, R. A., James, R. A., and Rebetzke, G. J. (2000). Genetic variation for improving the salt tolerance of durum wheat. Austr. J. Agric. Res. 51, 69–74.
Munns, R., Schachtman, D. P., and Condon, A. G. (1995). The significance of a 2-phase growth-response to salinity in wheat and barley. Aust. J. Plant Physiol. 22, 561–569.
Munns, R., and Tester, M. (2008). Mechanisms of salinity tolerance. Annu. Rev. Plant Biol. 59, 651–681.
Nagalakshmi, U., Wang, Z., Waern, K., Shou, C., Raha, D., Gerstein, M., et al. (2008). The transcriptional landscape of the yeast genome defined by RNA sequencing. Science 320, 1344 –1349.
Obata, T., and Fernie, A. R. (2012). The use of metabolomics to dissect plant responses to abiotic stresses. Cell. Mol. Life Sci. 69, 3225–3243.
Paterson, A. H., Bowers, J. E., Bruggmann, R., Dubchak, I., Grimwood, J., Gundlach, H., et al. (2009). The Sorghum bicolor genome and the diversification of grasses. Nature 457, 551–556.
Petersson, S. V., Johansson, A. I., Kowalczyk, M., Makoveychuk, A., Wang, J. Y., Moritz, T., et al. (2009). An auxin gradient and maximum in the Arabidopsis root apex shown by high-resolution cell-specific analysis of IAA distribution and synthesis. Plant Cell 21, 1659–1668.
Peukert, M., Matros, A., Lattanzio, G., Kaspar, S., Abadia, J., and Mock, H. P. (2012). Spatially resolved analysis of small molecules by matrix-assisted laser desorption/ionization mass spectrometric imaging (MALDI-MSI). New Phytol. 193, 806–815.
Plett, D. C., and Moller, I. S. (2010). Na plus transport in glycophytic plants: what we know and would like to know. Plant Cell Environ. 33, 612–626.
Pu, L., and Brady, S. (2010). Systems biology update: cell type-specific transcriptional regulatory networks. Plant Physiol. 152, 411–419.
Rahnama, A., Munns, R., Poustini, K., and Watt, M. (2011). A screening method to identify genetic variation in root growth response to a salinity gradient. J. Exp. Bot. 62, 69–77.
Rajendran, K., Tester, M., and Roy, S. J. (2009). Quantifying the three main components of salinity tolerance in cereals. Plant Cell Environ. 32, 237–249.
Richards, R. A., Rebetzke, G. J., Watt, M., Condon, A. G., Spielmeyer, W., and Dolferus, R. (2010). Breeding for improved water productivity in temperate cereals: phenotyping, quantitative trait loci, markers and the selection environment. Funct. Plant Biol. 37, 85–97.
Roessner, U., and Beckles, D. M. (2009). “Metabolite measurements,” in Plant Metabolic Networks, ed. J. Schwender (New York: Springer), 39.
Rogers, E. D., Jackson, T., Moussaieff, A., Aharoni, A., and Benfey, P. N. (2012). Cell type-specific transcriptional profiling: implications for metabolite profiling. Plant J. 70, 5–17.
Roy, A., Rushton, P. J., and Rohila, J. S. (2011a). The potential of proteomics technologies for crop improvement under drought conditions. Crit. Rev. Plant Sci. 30, 471–490.
Roy, S. J., Tucker, E. J., and Tester, M. (2011b). Genetic analysis of abiotic stress tolerance in crops. Curr. Opin. Plant Biol. 14, 232–239.
Sarhadi, E., Bazargani, M. M., Sajise, A. G., Abdolahi, S., Vispo, N. A., Arceta, M., et al. (2012). Proteomic analysis of rice anthers under salt stress. Plant Physiol. Biochem. 58, 280–287.
Schad, M., Mungur, R., Fiehn, O., and Kehr, J. (2005). Metabolic profiling of laser microdissected vascular bundles of Arabidopsis thaliana. Plant Methods 1, 2.
Schirle, M., Heurtier, M. A., and Kuster, B. (2003). Profiling core proteomes of human cell lines by one-dimensional PAGE and liquid chromatography-tandem mass spectrometry. Mol. Cell. Proteomics 2, 1297–1305.
Schnable, P. S., Ware, D., Fulton, R. S., Stein, J. C., Wei, F., Pasternak, S., et al. (2009). The B73 maize genome: complexity, diversity, and dynamics. Science 326, 1112–1115.
Sharp, R. E., Silk, W. K., and Hsiao, T. C. (1988). Growth of the maize primary root at low water potentials.1. Spatial-distribution of expansive growth. Plant Physiol. 87, 50–57.
Shavrukov, Y., Gupta, N. K., Miyazaki, J., Baho, M. N., Chalmers, K. J., Tester, M., et al. (2010). HvNax3-a locus controlling shoot sodium exclusion derived from wild barley (Hordeum vulgare ssp spontaneum). Funct. Integr. Genomics 10, 277–291.
Shelden, M. C., Roessner, U., Sharp, R. E., Tester, M., and Bacic, A. (2013). Genetic variation in the root growth response of barley genotypes to salinity stress. Funct. Plant Biol. doi: 10.1071/FP12290 [Epub ahead of print].
Spollen, W. G., Tao, W., Valliyodan, B., Chen, K., Hejlek, L. G., Kim, J. J., et al. (2008). Spatial distribution of transcript changes in the maize primary root elongation zone at low water potential. BMC Plant Biol. 8:32. doi: 10.1186/1471-2229-8-32
Studer, B., Byrne, S., Nielsen, R. O., Panitz, F., Bendixen, C., Islam, M. S., et al. (2012). A transcriptome map of perennial ryegrass (Lolium perenne L.). BMC Genomics 13:140 doi: 10.1186/1471-2164-13-140
Tamura, K., and Yonemaru, J. (2010). Next-generation sequencing for comparative transcriptomics of perennial ryegrass (Lolium perenne L.) and meadow fescue (Festuca pratensis Huds.) during cold acclimation. Grassland Sci. 56, 230–239.
Tester, M., and Langridge, P. (2010). Breeding technologies to increase crop production in a changing world. Science 327, 818–822.
Ueda, A., Kathiresan, A., Bennett, J., and Takabe, T. (2006). Comparative transcriptome analyses of barley and rice under salt stress. Theor. Appl. Genet. 112, 1286–1294.
Ueda, A., Kathiresan, A., Inada, M., Narita, Y., Nakamura, T., Shi, W. M., et al. (2004). Osmotic stress in barley regulates expression of a different set of genes than salt stress does. J. Exp. Bot. 55, 2213–2218.
Urano, K., Kurihara, Y., Seki, M., and Shinozaki, K. (2010). ‘Omics’ analyses of regulatory networks in plant abiotic stress responses. Curr. Opin. Plant Biol. 13, 132–138.
Vidal, R. O., Do Nascimento, L. C., Mondego, J. M. C., Pereira, G. A. G., and Carazzolle, M. F. (2012). Identification of SNPs in RNA-seq data of two cultivars of Glycine max (soybean) differing in drought resistance. Genet. Mol. Biol. 35, 331–334.
Vogel, J. P., Garvin, D. F., Mockler, T. C., Schmutz, J., Rokhsar, D., Bevan, M. W., et al. (2010). Genome sequencing and analysis of the model grass Brachypodium distachyon. Nature 463, 763–768.
Vrkoslav, V., Muck, A., Cvacka, J., and Svatos, A. (2010). MALDI imaging of neutral cuticular lipids in insects and plants. J. Am. Soc. Mass Spectrom. 21, 220–231.
Walia, H., Wilson, C., Condamine, P., Ismail, A. M., Xu, J., Cui, X. P., et al. (2007). Array-based genotyping and expression analysis of barley cv. Maythorpe and Golden Promise. BMC Genomics 8:87. doi: 10.1186/1471-2164-8-87
Walia, H., Wilson, C., Wada, H., Condamine, P., Cui, X. P., and Close, T. J. (2006). Expression analysis of barley (Hordeum vulgare L.) during salinity stress. Funct. Integr. Genomics 6, 143–156.
Wan, J. R., Torres, M., Ganapathy, A., Thelen, J., Dague, B. B., Mooney, B., et al. (2005). Proteomic analysis of soybean root hairs after infection by Bradyrhizobium japonicum. Mol. Plant Microbe Interact. 18, 458–467.
Wang, Y., Zeng, X., Iyer, N. J., Bryant, D. W., Mockler, T. C., and Mahalingam, R. (2012). Exploring the switchgrass transcriptome using second-generation sequencing technology. PLoS ONE 7:e34225. doi: 10.1371/journal.pone.0034225
Weber, A. P., Weber, K. L., Carr, K., Wilkerson, C., and Ohlrogge, J. B. (2007). Sampling the Arabidopsis transcriptome with massively parallel pyrosequencing. Plant Physiol. 144, 32 –42.
Widodo, Patterson, J. H., Newbigin, E., Tester, M., Bacic, A., and Roessner, U. (2009). Metabolic responses to salt stress of barley (Hordeum vulgare L.) cultivars, Sahara and Clipper, which differ in salinity tolerance. J. Exp. Bot. 60, 4089–4103.
Wienkoop, S., Zoeller, D., Ebert, B., Simon-Rosin, U., Fisahn, J., Glinski, M., et al. (2004). Cell-specific protein profiling in Arabidopsis thaliana trichomes: identification of trichome-located proteins involved in sulfur metabolism and detoxification. Phytochemistry 65, 1641–1649.
Xue, S. W., Yao, X., Luo, W., Jha, D., Tester, M., Horie, T., et al. (2011). AtHKT1;1 mediates nernstian sodium channel transport properties in Arabidopsis root stelar cells. PLoS ONE 6:e24725. doi: 10.1371/journal.pone.0024725
Yamaguchi, M., and Sharp, R. E. (2010). Complexity and coordination of root growth at low water potentials: recent advances from transcriptomic and proteomic analyses. Plant Cell Environ. 33, 590–603.
Yi, L., Liang, Z. T., Peng, Y., Yao, X., Chen, H. B., and Zhao, Z. Z. (2012). Tissue-specific metabolite profiling of alkaloids in Sinomenii Caulis using laser microdissection and liquid chromatography-quadrupole/time of flight-mass spectrometry. J. Chromatogr. A 1248, 93–103.
Zhang, H., Han, B., Wang, T., Chen, S. X., Li, H. Y., Zhang, Y. H., et al. (2012). Mechanisms of plant salt response: insights from proteomics. J. Proteome Res. 11, 49–67.
Zhao, Z. X., Zhang, W., Stanley, B. A., and Assmann, S. M. (2008). Functional proteomics of Arabidopsis thaliana guard cells uncovers new stomatal signaling pathways. Plant Cell 20, 3210–3226.
Zhu, J., Ingram, P. A., Benfey, P. N., and Elich, T. (2011). From lab to field, new approaches to phenotyping root system architecture. Curr. Opin. Plant Biol. 14, 310–317.
Keywords: abiotic stress tolerance, functional genomics, spatial resolution, metabolomics, expression profiling
Citation: Shelden MC and Roessner U (2013) Advances in functional genomics for investigating salinity stress tolerance mechanisms in cereals. Front. Plant Sci. 4:123. doi: 10.3389/fpls.2013.00123
Received: 28 August 2012; Accepted: 16 April 2013;
Published online: 10 May 2013.
Edited by:
Kazuki Saito, RIKEN Plant Science Center; Chiba University, JapanReviewed by:
Katja Baerenfaller, Swiss Federal Institute of Technology Zurich, SwitzerlandKyonoshin Maruyama, Japan International Research Center for Agricultural Sciences, Japan
Copyright: © 2013 Shelden and Roessner. This is an open-access article distributed under the terms of the Creative Commons Attribution License, which permits use, distribution and reproduction in other forums, provided the original authors and source are credited and subject to any copyright notices concerning any third-party graphics etc.
*Correspondence: Ute Roessner, Australian Centre for Plant Functional Genomics, School of Botany, University of Melbourne, Parkville, VIC 3010, Australia. e-mail: u.roessner@unimelb.edu.au