
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 07 April 2025
Sec. Redox Physiology
Volume 16 - 2025 | https://doi.org/10.3389/fphys.2025.1562626
This article is part of the Research TopicRising Stars in Redox PhysiologyView all articles
 D. C. Eaton1
D. C. Eaton1 M. J. Romero2,3
M. J. Romero2,3 M. A. Matthay4
M. A. Matthay4 J. Hamacher5,6,7
J. Hamacher5,6,7 A. Advani8A. Wolf8
A. Advani8A. Wolf8 M. Abu Mraheil9T. Chakraborty9
M. Abu Mraheil9T. Chakraborty9 D. W. Stepp2
D. W. Stepp2 E. J. Belin de Chantemèle2
E. J. Belin de Chantemèle2 A. Kutlar8
A. Kutlar8 F. Kraft10
F. Kraft10 M. Zeitlinger11P. Kranke12S. Frank13Y. Su3,8,14
M. Zeitlinger11P. Kranke12S. Frank13Y. Su3,8,14 A. D. Verin2,8
A. D. Verin2,8 D. J. R. Fulton2,3
D. J. R. Fulton2,3 M. Ushio-Fukai2,3
M. Ushio-Fukai2,3 T. Fukai2,3
T. Fukai2,3 R. Lucas2,3,8*
R. Lucas2,3,8*The endothelium represents a crucial regulator of vascular homeostasis. Since endothelial cells mainly rely on glycolysis rather than on oxidative phosphorylation for their ATP generation, this allows capillaries to transport the maximum amount of oxygen to oxygen-starved tissues, where it can be used for energy generation. However, the occasionally high levels of oxygen and of reactive oxygen species (ROS) in the blood vessels requires a balancing act between pro- and anti-oxidative mechanisms in the endothelium. When this balance is disturbed by excessive oxidative stress, as can occur in bacterial and viral pneumonia, endothelial barrier function can be compromised. This review will discuss some of the recently discovered barrier-protective mechanisms during bacterial and viral pneumonia, mediated through the reduction of oxidative stress in lung capillaries by the epithelial sodium channel (ENaC).
The increased levels of oxygen over time in the earth’s atmosphere provided the crucial fuel for the evolution of complex, multicellular organisms with high energy demands. Energy production through aerobic respiration comes with the risk of the generation of harmful incompletely reduced reactive oxygen species (ROS). The three primary species include the superoxide anion (O2•−), hydrogen peroxide (H2O2) and the hydroxyl radical (HO•). All of these are all oxygen-containing compounds with reactive properties. ROS represent very potent oxidants that, when present at high concentrations, can cause physiologic dysfunction within the cells (Hsia et al., 2013). In order to minimize ROS toxicity, humans have evolved the circulatory system with hemoglobin in erythrocytes to deliver oxygen efficiently to tissues, thereby preventing the accumulation of excessive oxygen in other areas of the body (Ortiz-Prado et al., 2019). Pulmonary venous and capillary endothelial cells are particularly challenged by oxidative stress, since they are at times exposed to O2-rich environments. To maximize O2 delivery to tissues and perivascular cells, these cells use the glycolytic pathway (Xiao et al., 2021), rather than mitochondrial respiration -classically used by most other eukaryotic cells-as the main ATP-generating pathway.
Optimal gas exchange between the alveoli and the lung capillaries requires tight barriers. The endothelial barrier, made up of adherens junctions and tight junctions, is essential for controlling the leakage from blood vessels into tissues and moreover represents the first line of defense against inflammatory or infectious insults in the vasculature (Dejana, 2004). Dysregulated endothelial permeability significantly contributes to morbidity and mortality. This is particularly relevant in microvascular endothelial beds, like in lung capillary endothelial cells in the acute respiratory distress syndrome (ARDS) (Price and Garcia, 2024; Wick et al., 2024; Tzotzos et al., 2020; Gonzales et al., 2015) (the definition of which was recently updated (Matthay et al., 2024) and in microvascular endothelial cells in the blood brain barrier as was shown in mouse models of Alzheimer disease (Hossain et al., 2024).
The endothelial cells associated with different parts of the vasculature appear to have somewhat different properties from one vascular bed to another. Lung microvascular endothelial cells (MVEC) are not characterized as well as the endothelial cells of larger vessels, but they are adapted to be highly permeable to promote gas exchange and movement of low molecular weight organics and ions without normally being permeable to serum proteins. One membrane protein, the epithelial sodium channel (ENaC), has been described in several endothelial beds, but until recently its presence was not demonstrated in MVEC. Using commercially available primary cultures of human lung microvascular endothelial cells (HL-MVEC; Lonza Biosciences), our group recently described functional endothelial ENaC using electrophysiological and pharmacological methods (Czikora et al., 2017; Romero et al., 2024). Studies with endothelial ENaC-α knockout mice indicated an important role of the endothelial ENaC channel for capillary barrier function in models of bacterial pneumonia (Romero et al., 2024). Although mechanisms to counteract oxygen toxicity, including ROS-neutralizing antioxidant enzymes like superoxide dismutase (SOD), glutathione peroxidase and catalase have been well studied, this short review will discuss recent discoveries regarding the interaction between the epithelial sodium channel (ENaC) and pro-oxidative mechanisms in capillary endothelial cells and the relevance thereof for barrier function during bacterial and viral pneumonia.
As pointed out above, efficient gas exchange in the lungs requires a functional alveolar fluid clearance (AFC) capacity by type 1 and 2 alveolar epithelial cells (AT1/2), as well as the preservation of a tight alveolar-capillary barrier. This barrier not only facilitates diffusion of O2 and CO2 between the alveoli and the blood capillaries, but also controls movement of fluid, proteins and cells from the vascular compartment and interstitial space into the alveoli (Su et al., 2024). Adjacent endothelial cells express both adherens and tight junctions and these transmembrane adhesive proteins promote homophilic interactions that provide a pericellular zipper-like structure along the cell border. Cell adhesion at adherens junctions requires vascular endothelial cadherin (VE-cadherin), which is in turn linked to intracellular proteins like β-catenin, plakoglobin and p120. At tight junctions, adhesion is mediated by claudins, occludin and members of the junctional adhesion molecule (JAM) family (Dejana, 2004).
A common characteristic feature in autopsies of patients that succumbed to ARDS is the severely altered permeability of the alveolar-capillary barrier (Price and Garcia, 2024). The increase in permeability is a result of dynamic changes in cytoskeletal structure and adherens junction disorganization, such as the detachment of VE-cadherin from the actin cytoskeleton (Su et al., 2024; Gonzales et al., 2014). These cytoskeletal changes are due to myosin light chain phosphorylation and/or microtubule rearrangement. Either of these can be induced by pro-inflammatory factors, such as thrombin and TNF or by bacterial and viral compounds, such as LPS (Gonzales et al., 2014; Song et al., 2023), pneumolysin (Lucas et al., 2012a; Lucas et al., 2012b; Chen et al., 2014; Batori et al., 2022) or the S1 subunit of the SARS-CoV2 Spike protein, which contains the receptor binding domain for human ACE2 (Romero et al., 2023).
To improve gas exchange in patients with severe ARDS, high ventilator pressures are often required, but these can further aggravate barrier dysfunction and inflammation and as such induce ventilator-induced lung injury (VILI) (Johannes et al., 2014; Vieillard-Baron et al., 2016). Currently, barrier-strengthening Tie2-based strategies using the Tie2 agonist vasculotide are being evaluated in preclinical animal models combining bacterially-induced pneumonia with high ventilation pressures (Lask et al., 2022). Nonetheless, despite recent advances in the treatment of ARDS, i.e., low tidal volume ventilation (Amato et al., 1998; Brower et al., 2000), ventilation in a prone position (Gattinoni et al., 2001) and neuromuscular blockade (Huang et al., 2017), mortality remains high at about 40% (Wick et al., 2024). There is currently a complete lack of pharmacologic treatments for severe ARDS which is refractory to conventional therapy. Therefore the identification of novel therapeutic targets in ARDS associated with severe pneumonia remains an area of high priority.
A major complication in the development of pharmaceuticals to treat ARDS is the existence of hyper- and hypo-inflammatory patient phenotypes, each with distinct transcriptional and metagenomic features (Sinha et al., 2023; Neyton et al., 2024). A cohort of critically ill sepsis patients with hyperinflammatory ARDS had significantly increased levels of glycolytic metabolites such as lactate and pyruvate compared with patients with hypo-inflammatory ARDS, suggesting the involvement of excessive glycolysis in the pathology of hyper-inflammatory ARDS (Alipanah-Lechner et al., 2023). Moreover, following a transition from the hyper-to the hypo-inflammatory state survival of ARDS patients improved (van Amstel et al., 2024). A major challenge in developing novel pharmacological agents to treat ARDS is the recent observation of the divergent reactions to corticosteroid treatment in hyper-versus hypo-inflammatory phenotypes, reducing mortality in the former, but increasing it in the latter patient group (Sinha et al., 2021).
Major comorbidities that occur with ARDS are bacterial and viral pneumonia (Ha et al., 2024; Vaughn et al., 2024). One of the main etiological agents of mortality in children under 5 years of age worldwide and of community-acquired pneumonia (CAP) in the elderly is the facultative anaerobe, Gram-positive bacterium, Streptococcus pneumoniae. Mortality rates in individuals with CAP vary according to the treatment setting, with less than 1% in outpatient care, up to 18% in hospital wards and even reaching 47% in the intensive care unit (ICU) (Jain et al., 2015; Cavallazzi et al., 2020).
Pathological specimens from ARDS patients reveal diffuse alveolar damage. Moreover, animal studies of bacterial pneumonia-associated ARDS have demonstrated both alveolar epithelial and lung endothelial injury with accumulation of protein-rich fluid in the alveolar space (Zhang et al., 2024). The ability of pneumococci to promote lung disease in the human host depends not only on microbial virulence factors, such as the pore-forming toxin pneumolysin (Witzenrath et al., 2006) and H2O2 (Mraheil et al., 2021), but also on the age and on genetic and environmental factors. All of these affect the concerted ability of the immune system to clear bacteria on the one hand and the susceptibility to develop subsequent tissue damage on the other hand (Ochoa-Gondar et al., 2023).
Upon stimulation by inflammatory mediators such as TNF, otherwise quiescent endothelial cells become activated to produce significantly increased levels of NADPH oxidase-generated ROS (Gertzberg et al., 2004) with increased glycolysis mediated by 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase (PFKFB3) (Cao et al., 2019). ROS can in turn combine with nitric oxide (NO) to generate barrier-disruptive peroxynitrite (Neumann et al., 2006). Reduction of endothelial nitric oxide synthase (eNOS)-mediated NO generation was shown to promote pulmonary microvessel leakage in eNOS−/− mice (Predescu et al., 2005), suggesting a barrier-protective effect of eNOS-derived NO in the pulmonary microvasculature (Di Lorenzo et al., 2013). Increased ROS generation was shown to impair the expression and activity of eNOS (Neumann et al., 2004) and to increase the activity of arginase 1, an enzyme competing with eNOS for the common substrate arginine (Romero et al., 2008; Chandra et al., 2012). TNF appears to be critical for arginase 1 induction, since TNF−/− mice had significantly reduced endothelial arginase activity following ischemia and reperfusion (Gao et al., 2007). All of these events involve increased endothelial oxidative stress and can disrupt the vascular integrity by reducing adherens and tight junction protein expression to eventually induce endothelial cell death. Moreover, ROS can increase expression of adhesion molecules in endothelium and as such foster transendothelial migration of circulating leukocytes (Wang and Doerschuk, 2000). Severely restricted blood flow, as can occur in TNF-induced multiple organ failure or in the lungs following lung transplantation, activates the mechano-sensing machinery in the endothelium that generates NADPH oxidase-mediated ROS to subsequently drive neutrophil influx (Noel et al., 2013).
Endothelial cells express several ROS-generating enzymes, including the NADPH oxidases 1, 2, 4 and 5 (NOX-1, -2, -4 and -5). Of these, NOX-2, in particular, has been shown to contribute to endothelial ROS generation in inflammatory conditions associated with infection and ischemia (Drummond and Sobey, 2014; Violi et al., 2015). Notably, the presence of soluble NOX2-derived peptide (sNOX2-dp), a marker of NOX2 activation, is increased in the circulation of patients with pneumococcal pneumonia (Violi et al., 2015; Violi et al., 2020) and in those with peripheral artery disease (Loffredo et al., 2013). However, NOX2-derived ROS generation can also confer beneficial activities in endothelium, since it stimulates angiogenesis in a mouse model of myocardial infarction (Youn et al., 2019).
Pneumococcal infection can cause extensive ROS generation in pulmonary microvascular endothelium (Li et al., 2018). The pore-forming toxin pneumolysin is the most important virulence factor produced by S. pneumoniae (Anderson and Feldman, 2017). It has been proposed as the main inducer of ROS generation in mammalian cells (Martner et al., 2008). Upon pore formation following the binding to cholesterol-containing membrane surfaces, pneumolysin induces a rapid influx of Ca2+, which can activate protein kinase C-α (Lucas et al., 2012a) and this in turn potently activates NOX2 (Schröder et al., 2017; Shafique et al., 2017). NOX2 activation requires the formation of a complex consisting of two surface membrane proteins: p22phox and gp91phox with the four cytosolic proteins p47phox, p67phox, p40phox and the small GTPase Rac. The cytosolic subunits need to be phosphorylated by kinases like PKC in order to be recruited to the surface membrane (Schröder et al., 2017) (Figure 1). The role of NOX2 in pneumolysin-induced barrier dysfunction is also supported by the observation that a specific peptide inhibitor of NOX2 -gp91dstat-significantly blunts hyperpermeability induced by the toxin in human lung microvascular endothelial cell (HL-MVEC) monolayers, as measured by electrical cell substrate impedance sensing (Romero et al., 2024).
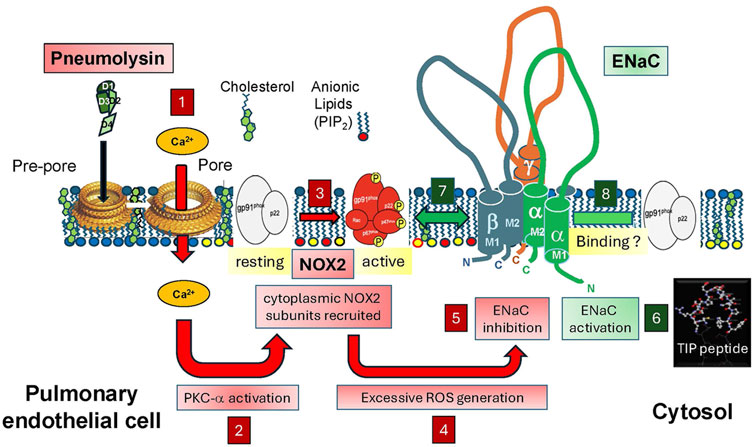
Figure 1. Upon autolysis, antibiotics treatment or upon induction by H2O2, S. pneumoniae (Sp.) releases the cholesterol-binding toxin pneumolysin, which, following pore formation, induces Ca2+ influx in infected cells (box 1). Increased intracellular Ca2+ activates PKC-α (box 2), which in turn phosphorylates NADPH oxidase 2 (NOX2) cytoplasmic subunits (box 3), thereby promoting their recruitment to the surface membrane, required for full activation of the enzyme. Increased NOX2 activity (box 4) promotes excessive generation of ROS in lung endothelial cells, which, together with the PKC-α activation, impairs ENaC activity (box 5). ENaC, activated by TIP peptide (box 6) can act as a repressor of NOX2 expression potentially upon binding to the gp91phox subunit (box 7), thus inhibiting assembly of the NOX2 complex or it can inhibit PKC-α-mediated phosphorylation of the subunits and thus NOX2 activation (box 8). Based on our findings, we hypothesize that during pneumococcal pneumonia, direct activation of ENaC by the TIP peptide reduces pneumococci-induced endothelial NOX2 activation and as such improves capillary barrier function, without affecting bactericidal activity of neutrophils, which do not express ENaC.
Another source of increased endothelial ROS generation are defective mitochondria. Apart from activating NOX2, pneumolysin also induces mitochondrial dysfunction which increases mitochondrial ROS generation in human lung MVEC (Romero et al., 2024; Shafique et al., 2017). It should be noted that a cross talk between mitochondrial ROS and NOX-derived ROS was recently suggested and that this intracellular communication could represent a ROS amplification mechanism in distinct subcellular compartments, relevant for activation of redox signaling (Shafique et al., 2017; Fukai and Ushio-Fukai, 2020).
Apart from superoxide generated by host cells upon pneumococcal-induced NOX2 activation, S. pneumoniae, which lacks catalase and common regulators of peroxide stress resistance, can by itself generate and release millimolar levels of H2O2 as a virulence factor (Mraheil et al., 2021). These H2O2 levels are high enough to kill or inhibit the growth of other common inhabitants of the respiratory tract, such as Haemophilus influenzae and Staphylococcus aureus, presumably as an evolutionary strategy to promote S. pneumoniae colonization (Regev-Yochay et al., 2006).
The main source of H2O2 generation in pneumococci is the enzyme pyruvate oxidase, which catalyzes the conversion of pyruvate to the phosphoryl donor, acetyl phosphate (Ac-P), while releasing CO2 and H2O2 as by-products (Mraheil et al., 2021). Another enzyme, lactate oxidase (LctO), positively impacts pyruvate flux through pyruvate oxidase, since it converts lactate to pyruvate. Importantly, actions of H2O2 overlap and complement those of pneumolysin in modulating the host immune responses and promoting organ injury (Pericone et al., 2000). As such, the release of pneumolysin, which normally occurs in the lungs by autolysis or after antibiotic-mediated lysis is defective in pneumococcal mutants that lack the pyruvate oxidase gene, whereas it is restored upon complementation of the enzyme. Since catalase supplementation, but not exogenous H2O2, prevents the release of pneumolysin in some pneumococcal strains, indicating the involvement of intracellular, rather than secreted H2O2 in this process (Bazant et al., 2023). Infection of H441 cells with wild type pneumococci was demonstrated to alter the kinome of the cells. This occurs at least partially through H2O2-mediated downregulation of Protein kinase B (Akt1) and activation of lymphocyte-specific tyrosine protein kinase (Lck) via H2O2-mediated phosphorylation (Bazant et al., 2024).
While there has been a focus on the pathogens causing pneumonia in recent years, there is an urgent need for research from the perspective of the host, especially in order to develop strategies to protect lung barrier function and alveolar fluid clearance. The problem is that ROS, such as H2O2 and superoxide play an important but highly complex role in pneumococcal pneumonia-associated ARDS. On the one hand, ROS generation is a conserved strategy of host phagocytic cells -primarily neutrophils, monocytes and macrophages-to facilitate clearance of bacteria at the infection site. Bacteria can be engulfed and enclosed in phagosomes, into which superoxide is released by activated NOX2. The resulting superoxide O2− then dismutates to H2O2 through the action of superoxide dismutase (SOD) in macrophages. H2O2 can then further be converted by myeloperoxidase (MPO) in neutrophils to hypochlorous acid, a highly microbicidal species (Winterbourn et al., 2016).
On the other hand, ROS generated in alveolar epithelial and capillary endothelial cells can impair alveolar liquid clearance mechanisms and contribute to dysfunction of the capillary endothelial barrier, respectively. In view of the dual role of NOX2 in pneumonia, strategies that globally reduce NOX2 activity bear the risk of increasing susceptibility to infections (Diebold et al., 2015). Novel therapies that can reduce deleterious oxidative stress in the alveolar epithelium and capillary endothelium, without significantly affecting anti-bacterial ROS generation in phagocytes seem therapeutically promising in pneumonia and ARDS.
Of all organs in the human body, the lungs are exposed to the highest levels of free O2, mainly originating from inspired air, with concomitant increased ROS production. Shortly after birth, the newborn lung must adapt from an embryonic fluid-secreting tissue to a fluid-absorbing organ, in order to allow for breathing normal ambient air. In particular, AT1/2 are exposed to the relatively high levels of free O2 in inspired air, which can lead to the generation of additional ROS. As such, physiological concentrations of H2O2 in the alveolar space in man are in the 1–10 μM range (Corradi et al., 2008).
Most ion transporters evolved from simpler channel-forming peptides following intragenic duplication events into more complex multi-domain transporters with diverse functions. The epithelial sodium channel (ENaC), consisting in its native configuration of three subunits: α, β and γ, is an example of such a multifunctional ion channel, since, apart from controlling blood pressure, it also modulates alveolar fluid clearance (AFC), by regulating vectorial Na+ transport in AT1/2 cells to control alveolar fluid balance and thereby assure normal gas exchange in the lungs (Matalon and O'Brodovich, 1999; Hummler et al., 1997).
Perinatal exposure of resident AT1/2 cells to O2-derived ROS was proposed to deliver a signal for initiating the uptake of Na+ by ENaC. This idea is supported by the observation that an increase in oxygen tension of up to 100 mmHg can augment Na+ transport within 6 h in fetal distal lung epithelial (FDLE) cells (Baines et al., 2001).
The overall Na+ transport capacity of ENaC is determined by the product of the channel’s surface density (N) and the mean open probability of each individual channel (Po); thus, by the product NPo. Na+ is then secreted in the interstitial space by the basolateral Na+-K+-ATPase. This vectorial Na+ transport is necessary to promote fluid clearance (AFC) from the newborn and the adult lung. The crucial role of ENaC in this process is underscored by the observation that mice lacking the pore-forming α subunit of ENaC die of alveolar flooding at birth (Hummler et al., 1996). Impairment of AFC capacity correlates with morbidity and mortality in patients with the acute respiratory distress syndrome (ARDS) (Ware and Matthay, 2001).
The increase in sodium transport postnatally is at least partially due to both an increase in ENaC open probability and in surface protein expression stimulated by ambient oxygen production of low levels of ROS. H2O2 activates PI3-kinases, which in turn generate the anionic phospholipids, such as phosphatidylinositol-3,4,5-trisphosphate (PIP3), which increases ENaC open probability and AFC (Kooijman et al., 2011; Pochynyuk et al., 2006; Yue et al., 2002). Long-term exposure to elevated O2 tension or moderate levels of ROS can increase activity of ENaC subunit gene promoters, since these contain redox-sensitive NF-κB and AP-1 response elements and can moreover promote total ENaC protein expression (Yue et al., 2002; Rafii et al., 1998; Thome et al., 2003). Apart from a direct sensitivity of ENaC in AT1/2 cells to O2, ROS generated by increased O2 tension, such as superoxide anion (O2−) also has the capacity to regulate ENaC as shown by the observation that in cellular models of alveolar fluid uptake addition of the cell-permeable O2− scavenger TEMPOL significantly decreases ENaC activity (Bremner et al., 2002).
Like AT1/2 cells, also human lung microvascular endothelial cells (HL-MVEC) express all three ENaC subunits (albeit at a significantly lower protein expression levels) (Yu et al., 2007). HL-MVEC generate electrophysiologically-identified cation channels with a conductance of 5 picoSiemens, and a current voltage relationship characteristic of functional ENaC channels (Romero et al., 2024). However, the role of ENaC and even the composition of sodium transporting channels in endothelium is not completely understood.
As discussed above, a low basal level of NOX2-mediated ROS generation is necessary for proper ENaC activity in AT1/2 cells (Takemura et al., 2010). HL-MVEC treated with specific ENaC-α siRNA or mouse lung ECs isolated from tamoxifen-inducible endothelial ENaC-α KO mice (produced by cross-breeding conditional Scnnalox/lox mice (Hummler et al., 2002) with tamoxifen-inducible VE-cadherin–CRE/ert2 driver mice (Wang et al., 2010) have a significantly higher protein expression level of the gp91phox NOX2 subunit than human lung MVEC transfected with scrambled siRNA or mouse lung ECs from control CRE driver mice or. Mouse lung endothelial cells from endothelial ENaC-α KO mice generated much higher levels of superoxide than cells from control animals upon stimulation with PMA (Romero et al., 2024). These findings indicate that ENaC-α inhibits NOX2 expression in lung endothelial cells from both mice and humans. Although further studies are required, recent findings from co-immunoprecipitation experiments in mouse lungs substantiate earlier findings of a direct binding between gp91phox and ENaC-α (Romero et al., 2024). Such an interaction might also occur in lung endothelial cells and could potentially interfere with NOX subunit assembly and successful NOX2 complex formation. An alternative mechanism by which activated endothelial ENaC could blunt NOX2 activity is through the inhibition of enzymes phosphorylating cytoplasmic subunits, necessary for their recruitment to the surface membrane and for complex assembly of the functional enzyme (Schröder et al., 2017). One such enzyme which is able to activate NOX2 is protein kinase C-α (PKC-α), the activity of which is increased by pneumolysin (Lucas et al., 2012b).
While these observations suggest that ENaC-α is necessary for normal endothelial cell function and serves to keep NOX2 activity at bay, the results do not necessarily show that hetero-multimeric ENaC is required. Classical ENaC in epithelial tissues consist of α, β, and γ subunits, but in renal endothelial cells there is some evidence that ENaC-α may form alternative combinations without one or more of the regulatory β or γ subunits (Mutchler et al., 2021; Trac et al., 2017). These non-classical channels can therefore also potentially alter endothelial function. To complicate the issue further, in human endothelial cells there is a fourth ENaC subunit, δ, which can substitute for ENaC-α to form sodium permeable ion channels (Ji et al., 2012). Whether any of these alternative constructs can alter endothelial function as native ENaC does, is however unclear to date. However, this does mean that results implicating ENaC in endothelial cells must be interpreted carefully especially when pharmacological agents are used to observe cellular responses to specific molecules.
In order to further investigate the effect of specific ENaC activation on endothelial NOX2 activity, we have used the TNF-derived TIP peptide (a.k.a. AP301, Solnatide), generated by our group (Lucas et al., 1994). This 17 residue cyclic synthetic peptide with the sequence CGQRETPEGAEAKPWYC, mimics the lectin-like domain of human TNF (Lucas et al., 1994; Elia et al., 2003) and increases ENaC activity upon binding to a helical structure in the C-terminal domain of the α subunit (Czikora et al., 2014; Lucas et al., 2016; Martin-Malpartida et al., 2022). Another direct peptide ENaC activator is compound S3969, which interacts with a specific binding pocket in the channel’s β-subunit (Lu et al., 2008; Sure et al., 2024). TIP peptide can increase the open probability as well as the surface expression of ENaC in human lung MVEC and in alveolar epithelial cells, even following treatment of the cells with pneumolysin (Romero et al., 2024; Lucas et al., 2016). Pneumolysin rapidly reduces ENaC open probability and moreover reduces surface expression of the crucial α subunit, possibly by decreasing lysine residue acetylation, which in turn increases its ubiquitination and degradation (Romero et al., 2024; Butler et al., 2015). Moreover, pneumolysin-induced mitochondrial ROS generation -which exerts cross-talk with NOX2-derived ROS- is blunted by TIP peptide in HL-MVEC (Romero et al., 2024).
It should be stressed that the TIP peptide is a specific and direct activator of ENaC, unlike other used indirect activators, such as aldosterone, the latter of which can also exert ENaC-independent effects, which may chronically affect vascular function through the paracrine release of histamine (Schjerning et al., 2013). The barrier-disruptive actions of pneumolysin on HL-MVEC barrier function involve NOX2 activation, since we showed that the specific inhibitor gp91dstat was protective (Romero et al., 2024). We found a similar protective activity towards pneumolysin-induced hyperpermeability in HL-MVEC monolayers with the TIP peptide, as such supporting our published hypothesis that activation of endothelial ENaC represses NOX2 expression and activity and at least partially as such strengthens capillary barriers in pneumococcal pneumonia (Czikora et al., 2017; Romero et al., 2024).
Although a barrier strengthening effect of ENaC-α in LPS-treated capillary endothelium was also reported in other studies (Sternak et al., 2018), other groups, often using aldosterone, have reported deleterious vascular stiffening actions of ENaC in studies ln large vessel endothelial cells (Jia et al., 2023; Zhang et al., 2022). TIP peptide inhibits PMA-induced phosphorylation of the cytoplasmic NOX2 subunit p47phox and it partially blunts PMA-induced ROS generation in COSp22phox cells, which express ENaC-α and NOX2, but not other NOX enzymes. By contrast, TIP peptide does not blunt PMA-induced ROS generation in primary bone marrow-derived mouse PMNs, which do not express ENaC-α (Romero et al., 2024). Taken together, these data demonstrate that ENaC, and especially its α subunit represents a novel repressor of PMA-induced NOX2 activity in microvascular endothelium, but not in PMNs, especially when activated by the TIP peptide (Figure 1).
In a mouse model of pneumococcal pneumonia-induced lung injury, a moderate dose of pneumolysin (1.5 μg/kg) or a low inoculum of D39 pneumococci (2 × 106 CFU) induces significantly higher capillary leak (measured as Evans Blue permeability from the vasculature into the alveoli) in tamoxifen-inducible endothelial ENaC-α KO mice as compared to control CRE driver mice. However, co-instillation of 2.5 mg/kg of TIP peptide significantly blunts capillary leak induced by a high inoculum of S. pneumoniae (107 CFU), without significantly affecting bacterial load in lung homogenates (Romero et al., 2024). These results demonstrate an important barrier-protective function for endothelial ENaC-α in murine pneumococcal pneumonia (Figure 1).
TIP peptide also blunts ROS generation and barrier dysfunction in HL-MVEC induced by the S1 subunit of the SARS-CoV2 spike protein (Romero et al., 2023), as such indicating that endothelial ENaC can also suppress endothelial oxidative stress in COVID-19 and possibly in long COVID, where the spike protein, which can be detected months later even after the virus is no longer present, was proposed to be involved in the pathology (Iba et al., 2024).
In a phase 1 clinical trial in healthy volunteers, inhalation of the ENaC-activator TIP peptide (a.k.a AP301, Solnatide) caused no noticeable side effects (Schwameis et al., 2014). A phase 2a double-blind trial was done in lung transplantation patients, the design of which was based on a preclinical study in rats with lung iso-transplantation showing potent anti-oxidative actions and lung function-promoting of the TIP peptide (Hamacher et al., 2010). Ischemia-reperfusion following lung transplantation was suggested to induce endothelial oxidative stress involving NOX2 activation following mechano-signaling (Chatterjee et al., 2015). The trial demonstrated a significant reduction in days on the ventilator in patients inhaling the peptide twice daily over 7 days, as compared to placebo (Aigner et al., 2017; Ware, 2017). A similar treatment pattern with the test compound in patients with ARDS revealed a significant reduction in extravascular lung water in ARDS patients with a sequential organ failure assessment (SOFA) score >11 (Krenn et al., 2017). Currently, a multi-center double-blind dose ascending phase 2b clinical trial is ongoing in a group of non-COVID and COVID ARDS patients receiving the TIP peptide (a.k.a. Solnatide) during invasive ventilation, organized by the Vienna-based Biotech company Apeptico. Results from this trial should provide further information regarding the peptide’s therapeutic potential (Schmid et al., 2021; Schmid et al., 2022).
Our data from mouse studies and cell studies with human lung MVEC have demonstrated that the epithelial sodium channel, and especially its α subunit, apart from playing a crucial role in vectorial Na+ transport and fluid clearance in the alveolar space, also represents a novel repressor of oxidative stress and a guardian of lung capillary barrier function during experimental pneumococcal and SARS-CoV2-induced pneumonia. These lung-protective mechanisms can be activated by a direct ENaC activator, the TNF-derived TIP peptide, in models of pneumococcal and SARS-CoV2-induced pneumonia. As a limitation of our studies, we should note that although we demonstrated ENaC expression and activity in human lung microvascular endothelial cells, expression of the channel in mouse lung capillaries has not been visualized so far. This may be complicated by low ENaC expression levels as well as by the existence of different capillary sub-phenotypes. Since capillary endothelial cells make up the majority of lung endothelial cells and since mice lacking endothelial ENaC alpha display significantly increased capillary leak as compared to control animals in pneumococcal pneumonia, this points to an important role of the channel in lung capillaries, although the involvement of other microvascular beds cannot be excluded. The blood-perfused isolated human lung (Ross et al., 2019) may represent a more translational model system for further evaluation of the observed effects of endothelial ENaC in pneumonia and ARDS in man.
Although other groups have demonstrated a protective effect of ENaC-α on capillary barrier function in the presence of LPS and on vasodilation capacity of large vessels (Sternak et al., 2018; Tarjus et al., 2017), more research is required to explain the reported deleterious role of endothelial ENaC in vascular stiffening (Jia et al., 2023; Zhang et al., 2022; Kusche-Vihrog et al., 2014). Whether these divergent observations regarding the role of ENaC in endothelial function can be explained by differences between capillary and large vessel endothelial cells, the use of direct (e.g., TIP peptide, S3969) versus indirect (e.g., aldosterone) activators of ENaC, by the existence of alternative ENaC-like channels in endothelium or by differences in the disease models used remains to be investigated.
Although increases in the Renin-Angiotensin System (RAS) can lead to increased absorption of Na+ through ENaC in the distal nephron, the TIP peptide -a direct ENaC activator-did not increase blood pressure in a mouse model of glomerulonephritis (Madaio et al., 2019). One possible explanation for this is that the TIP peptide, which mimics the lectin-like domain of TNF, can bind to uromodulin in the loops of Henle (Hession et al., 1987) before reaching the distal nephron (Madaio et al., 2019).
Phase 2 clinical trials with inhaled TIP peptide in lung transplantation (Hamacher et al., 2010) and ARDS patients (Krenn et al., 2017) have shown encouraging outcomes on lung function, indicating that activation of ENaC may be a promising approach in these pathologies. Negative effects of the treatment on vascular function or blood pressure have not been observed in these trials, although this may be linked to the rather modest number of subjects tested so far or to the limited leakage of the test compound into the blood circulation.
DE: Conceptualization, Investigation, Writing – original draft, Writing – review and editing, Data curation, Formal Analysis, Funding acquisition, Methodology, Project administration, Resources, Supervision, Validation. MR: Conceptualization, Investigation, Writing – original draft, Writing – review and editing, Data curation, Formal Analysis. MM: Conceptualization, Data curation, Investigation, Methodology, Resources, Validation, Writing – review and editing, Formal Analysis, Funding acquisition. JH: Conceptualization, Investigation, Writing – review and editing. AA: Investigation, Writing – original draft. AW: Investigation, Writing – original draft. MA: Investigation, Conceptualization, Writing – review and editing, Data curation. TC: Conceptualization, Investigation, Writing – review and editing. DS: Writing – review and editing, Conceptualization. EB: Writing – review and editing, Investigation. AK: Writing – review and editing, Conceptualization, Resources, Validation. FK: Investigation, Writing – review and editing, Formal Analysis, Methodology, Data curation. MZ: Conceptualization, Formal Analysis, Investigation, Methodology, Supervision, Validation, Writing – review and editing, Data curation. PK: Conceptualization, Writing – review and editing, Formal Analysis, Methodology, Supervision, Validation, Data curation. SF: Conceptualization, Writing – review and editing, Formal Analysis, Methodology, Supervision, Validation, Data curation. YS: Investigation, Writing – review and editing, Conceptualization, Formal Analysis, Methodology, Resources, Validation. AV: Conceptualization, Methodology, Writing – review and editing, Formal Analysis, Investigation, Resources, Validation. DF: Conceptualization, Methodology, Resources, Writing – review and editing, Formal Analysis, Investigation, Supervision, Writing – original draft. MU-F: Investigation, Methodology, Writing – review and editing, Validation, Resources, Writing – original draft. TF: Methodology, Validation, Writing – review and editing, Investigation, Resources, Writing – original draft. RL: Conceptualization, Resources, Validation, Writing – review and editing, Data curation, Formal Analysis, Funding acquisition, Investigation, Methodology, Project administration, Supervision, Visualization, Writing – original draft.
The author(s) declare that financial support was received for the research and/or publication of this article. Supported by National Heart, Lung, and Blood Institute grants R01HL157440 (A.D.V.), R01HL156646 and R01HL125926 (D.J.F.), R01HL158909 (A.D.V. and Y.S.), R01HL160014 (to MU.-F.), R01HL1740414, R01HL147550, R01HL147550-S1 (to T.F., M.U-F), R01HL138410 (RL.) and P01HL160557 (TF, M.U-F., DS., EBDC., DF., and RL); National Institute of Diabetes and Digestive and Kidney Diseases grant DK110409 (DCE.); American Heart Association award 22TPA971863 (to TF.) and 23TPA1072536 (RL.) and by the US Department of Veterans Affairs BX005350 (YS.) and Veterans Administration (VA) Merit Review Award 2I01BX001232 (to T.F). NHLBI funding for MAM: R35HL140026.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aigner C., Slama A., Barta M., Mitterbauer A., Lang G., Taghavi S., et al. (2017). Treatment of primary graft dysfunction after lung transplantation with orally inhaled AP301: a prospective, randomized pilot study. J. Heart Lung Transpl. 37, 225–231. doi:10.1016/j.healun.2017.09.021
Alipanah-Lechner N., Neyton L., Mick E., Willmore A., Leligdowicz A., Contrepois K., et al. (2023). Plasma metabolic profiling implicates dysregulated lipid metabolism and glycolytic shift in hyperinflammatory ARDS. Am. J. Physiol. Lung Cell Mol. Physiol. 324 (3), L297–L306. doi:10.1152/ajplung.00278.2022
Amato M. B., Barbas C. S., Medeiros D. M., Magaldi R. B., Schettino G. P., Lorenzi-Filho G., et al. (1998). Effect of a protective-ventilation strategy on mortality in the acute respiratory distress syndrome. N. Engl. J. Med. 338, 347–354. doi:10.1056/NEJM199802053380602
Anderson R., Feldman C. (2017). Pneumolysin as a potential therapeutic target in severe pneumococcal disease. J. Infect. 74, 527–544. doi:10.1016/j.jinf.2017.03.005
Baines D. L., Ramminger S. J., Collett A., Haddad J. J., Best O. G., Land S. C., et al. (2001). Oxygen-evoked Na+ transport in rat fetal distal lung epithelial cells. J. Physiol. 532 (Pt 1), 105–113. doi:10.1111/j.1469-7793.2001.0105g.x
Batori R. K., Chen F., Bordan Z., Haigh S., Su Y., Verin A. D., et al. (2022). Protective role of Cav-1 in pneumolysin-induced endothelial barrier dysfunction. Front. Immunol. 13, 945656. doi:10.3389/fimmu.2022.945656
Bazant J., Ott B., Hudel M., Hain T., Lucas R., Mraheil M. A. (2023). Impact of endogenous pneumococcal hydrogen peroxide on the activity and release of pneumolysin. Toxins (Basel) 15 (10), 593. doi:10.3390/toxins15100593
Bazant J., Weiss A., Baldauf J., Schermuly R. T., Hain T., Lucas R., et al. (2024). Pneumococcal hydrogen peroxide regulates host cell kinase activity. Front. Immunol. 15 (Jun 5), 1414195. doi:10.3389/fimmu.2024.1414195
Bremner H. R., Freywald T., O'Brodovich H. M., Otulakowski G. (2002). Promoter analysis of the gene encoding the beta-subunit of the rat amiloride-sensitive epithelial sodium channel. Am. J. Physiol. Lung Cell Mol. Physiol. 282 (1), L124–L134. doi:10.1152/ajplung.2002.282.1.L124
Brower R. G., Matthay M. A., Morris A., Schoenfeld D., Thompson B. T., Wheeler A., et al. (2000). Ventilation with lower tidal volumes as compared with traditional tidal volumes for acute lung injury and the acute respiratory distress syndrome. N. Engl. J. Med. 342, 1301–1308. doi:10.1056/NEJM200005043421801
Butler P. L., Staruschenko A., Snyder P. M. (2015). Acetylation stimulates the epithelial sodium channel by reducing its ubiquitination and degradation. J. Biol. Chem. 290 (20), 12497–12503. doi:10.1074/jbc.M114.635540
Cao Y., Zhang X., Wang L., Yang Q., Ma Q., Xu J., et al. (2019). PFKFB3-mediated endothelial glycolysis promotes pulmonary hypertension. Proc. Natl. Acad. Sci. U. S. A. 116 (27), 13394–13403. doi:10.1073/pnas.1821401116
Cavallazzi R., Furmanek S., Arnold F. W., Beavin L. A., Wunderink R. G., Niederman M. S., et al. (2020). The burden of community-acquired pneumonia requiring admission to ICU in the United States. Chest 158, 1008–1016. doi:10.1016/j.chest.2020.03.051
Chandra S., Romero M. J., Shatanawi A., Alkilany A. M., Caldwell R. B., Caldwell R. W. (2012). Oxidative species increase arginase activity in endothelial cells through the RhoA/Rho kinase pathway. Br. J. Pharmacol. 165 (2), 506–519. doi:10.1111/j.1476-5381.2011.01584.x
Chatterjee S., Fujiwara K., Pérez N. G., Ushio-Fukai M., Fisher A. B. (2015). Mechanosignaling in the vasculature: emerging concepts in sensing, transduction and physiological responses. Am. J. Physiol. Heart Circ. Physiol. 308 (12), H1451–H1462. doi:10.1152/ajpheart.00105.2015
Chen F., Kumar S., Yu Y., Aggarwal S., Gross C., Wang Y., et al. (2014). PKC-dependent phosphorylation of eNOS at T495 regulates eNOS coupling and endothelial barrier function in response to G+ -toxins. PLoS One 9 (7), e99823. doi:10.1371/journal.pone.0099823
Corradi M., Pignatti P., Brunetti G., Goldoni M., Caglieri A., Nava S., et al. (2008). Comparison between exhaled and bronchoalveolar lavage levels of hydrogen peroxide in patients with diffuse interstitial lung diseases. Acta Biomed. 79 (Suppl. 1), 73–78.
Czikora I., Alli A., Bao H. F., Kaftan D., Sridhar S., Apell H. J., et al. (2014). A novel tumor necrosis factor-mediated mechanism of direct epithelial sodium channel activation. Am. J. Respir. Crit. Care Med. 190 (5), 522–532. doi:10.1164/rccm.201405-0833OC
Czikora I., Alli A. A., Sridhar S., Matthay M. A., Pillich H., Hudel M., et al. (2017). Epithelial sodium channel-α mediates the protective effect of the TNF-derived TIP peptide in pneumolysin-induced endothelial barrier dysfunction. Front. Immunol. 8, 842. doi:10.3389/fimmu.2017.00842
Dejana E. (2004). Endothelial cell–cell junctions: happy together. Nat. Rev. Mol. Cell Biol. 5, 261–270. doi:10.1038/nrm1357
Diebold B. A., Smith S. M., Li Y., Lambeth J. D. (2015). NOX2 as a target for drug development: indications, possible complications, and progress. Antioxid. Redox Signal 23 (5), 375–405. doi:10.1089/ars.2014.5862
Di Lorenzo A., Lin M. I., Murata T., Landskroner-Eiger S., Schleicher M., Kothiya M., et al. (2013). eNOS-derived nitric oxide regulates endothelial barrier function through VE-cadherin and Rho GTPases. J. Cell Sci. 126 (Pt 24), 5541–5552. doi:10.1242/jcs.115972
Drummond G. R., Sobey C. G. (2014). Endothelial NADPH oxidases: which NOX to target in vascular disease? Trends Endocrinol. Metab. 25 (9), 452–463. doi:10.1016/j.tem.2014.06.012
Elia N., Tapponnier M., Matthay M. A., Hamacher J., Pache J. C., Brundler M. A., et al. (2003). Functional identification of the alveolar edema reabsorption activity of murine tumor necrosis factor-alpha. Am. J. Respir. Crit. Care Med. 168 (9), 1043–1050. doi:10.1164/rccm.200206-618OC
Fukai T., Ushio-Fukai M. (2020). Cross-talk between NADPH oxidase and mitochondria: role in ROS signaling and angiogenesis. Cells 9 (8), 1849. doi:10.3390/cells9081849
Gao X., Xu X., Belmadani S., Park Y., Tang Z., Feldman A. M., et al. (2007). TNF-alpha contributes to endothelial dysfunction by upregulating arginase in ischemia/reperfusion injury. Arterioscler. Thromb. Vasc. Biol. 27 (6), 1269–1275. doi:10.1161/ATVBAHA.107.142521
Gattinoni L., Tognoni G., Pesenti A., Taccone P., Mascheroni D., Labarta V., et al. (2001). Effect of prone positioning on the survival of patients with acute respiratory failure. N. Engl. J. Med. 345 (8), 568–573. doi:10.1056/NEJMoa010043
Gertzberg N., Neumann P., Rizzo V., Johnson A. (2004). NAD(P)H oxidase mediates the endothelial barrier dysfunction induced by TNF-alpha. Am. J. Physiol. Lung Cell Mol. Physiol. 286 (1), L37–L48. doi:10.1152/ajplung.00116.2003
Gonzales J. N., Gorshkov B., Varn M. N., Zemskova M. A., Zemskov E. A., Sridhar S., et al. (2014). Protective effect of adenosine receptors against lipopolysaccharide-induced acute lung injury. Am. J. Physiol. Lung Cell Mol. Physiol. 306 (6), L497–L507. doi:10.1152/ajplung.00086.2013
Gonzales J. N., Lucas R., Verin A. D. (2015). The acute respiratory distress syndrome: mechanisms and perspective therapeutic approaches. Austin J. Vasc. Med. 2 (1), 1009.
Ha A. W., Letsiou E., Dudek S. M. (2024). Can ENaC “TIP” the scales to reduce endothelial ROS and vascular leak during pneumococcal lung injury? Am. J. Respir. Cell Mol. Biol. doi:10.1165/rcmb.2024-0486ED
Hamacher J., Stammberger U., Roux J., Kumar S., Yang G., Xiong C., et al. (2010). The lectin-like domain of tumor necrosis factor improves lung function after rat lung transplantation--potential role for a reduction in reactive oxygen species generation. Crit. Care Med. 38 (3), 871–878. doi:10.1097/CCM.0b013e3181cdf725
Hession C., Decker J. M., Sherblom A. P., Kumar S., Yue C. C., Mattaliano R. J., et al. (1987). Uromodulin (Tamm-Horsfall glycoprotein): a renal ligand for lymphokines. Science 237 (4821), 1479–1484. doi:10.1126/science.3498215
Hossain M. S., Das A., Rafiq A. M., Deák F., Bagi Z., Outlaw R., et al. (2024). Altered copper transport in oxidative stress-dependent brain endothelial barrier dysfunction associated with Alzheimer's disease. Vasc. Pharmacol. 157, 107433. doi:10.1016/j.vph.2024.107433
Hsia C. C., Schmitz A., Lambertz M., Perry S. F., Maina J. N. (2013). Evolution of air breathing: oxygen homeostasis and the transitions from water to land and sky. Compr. Physiol. 3 (2), 849–915. doi:10.1002/cphy.c120003
Huang D. T., Angus D. C., Moss M., Thompson B. T., Ferguson N. D., Ginde A., et al. (2017). Design and rationale of the reevaluation of systemic early neuromuscular blockade trial for acute respiratory distress syndrome. Ann. Am. Thorac. Soc. 14 (1), 124–133. doi:10.1513/AnnalsATS.201608-629OT
Hummler E., Barker P., Gatzy J., Beermann F., Verdumo C., Schmidt A., et al. (1996). Early death due to defective neonatal lung liquid clearance in alpha-ENaC-deficient mice. Nat. Genet. 12 (3), 325–328. doi:10.1038/ng0396-325
Hummler E., Barker P., Talbot C., Wang Q., Verdumo C., Grubb B., et al. (1997). A mouse model for the renal salt-wasting syndrome pseudohypoaldosteronism. Proc. Natl. Acad. Sci. U. S. A. 94 (21), 11710–11715. doi:10.1073/pnas.94.21.11710
Hummler E., Merillat A. M., Rubera I., Rossier B. C., Beermann F. (2002). Conditional gene targeting of the Scnn1a (alphaENaC) gene locus. Genesis 32, 169–172. doi:10.1002/gene.10041
Iba T., Connors J. M., Levy J. H. (2024). What role does microthrombosis play in long COVID? Semin. Thromb. Hemost. 50 (4), 527–536. doi:10.1055/s-0043-1774795
Jain S., Self W. H., Wunderink R. G., Fakhran S., Balk R., Bramley A. M., et al. (2015). Community-acquired pneumonia requiring hospitalization among U.S. Adults. N. Engl. J. Med. 373, 415–427. doi:10.1056/NEJMoa1500245
Ji H. L., Zhao R. Z., Chen Z. X., Shetty S., Idell S., Matalon S. (2012). δ ENaC: a novel divergent amiloride-inhibitable sodium channel. Am. J. Physiol. Lung Cell Mol. Physiol. 303 (12), L1013–L1026. doi:10.1152/ajplung.00206.2012
Jia G., Hill M. A., Sowers J. R. (2023). Vascular endothelial mineralocorticoid receptors and epithelial sodium channels in metabolic syndrome and related cardiovascular disease. J. Mol. Endocrinol. 71 (3), e230066. doi:10.1530/JME-23-0066
Johannes A., Kredel M., Zollhoefer B., Schlegel N., Von Kirschbaum C., Brederlau J., et al. (2014). Influence of apneic oxygenation and minimal tidal volumes on ventilator-associated lung injury. Minerva Anestesiol. 80 (5), 526–536.
Kooijman E. E., Kuzenko S. R., Gong D., Best M. D., Folkesson H. G. (2011). Phosphatidylinositol 4,5-bisphosphate stimulates alveolar epithelial fluid clearance in male and female adult rats. Am. J. Physiol. Lung Cell Mol. Physiol. 301 (5), L804–L811. doi:10.1152/ajplung.00445.2010
Krenn K., Lucas R., Croizé A., Boehme S., Klein K. U., Hermann R., et al. (2017). Inhaled AP301 for treatment of pulmonary edema in mechanically ventilated patients with acute respiratory distress syndrome: a phase IIa randomized placebo-controlled trial. Crit. Care 21 (1), 194. doi:10.1186/s13054-017-1795-x
Kusche-Vihrog K., Jeggle P., Oberleithner H. (2014). The role of ENaC in vascular endothelium. Pflugers Arch. 466 (5), 851–859. doi:10.1007/s00424-013-1356-3
Lask A., Gutbier B., Kershaw O., Nouailles G., Gruber A. D., Müller-Redetzky H. C., et al. (2022). Adjunctive therapy with the Tie2 agonist Vasculotide reduces pulmonary permeability in Streptococcus pneumoniae infected and mechanically ventilated mice. Sci. Rep. 12, 15531. doi:10.1038/s41598-022-19560-3
Li X., Yu Y., Gorshkov B., Haigh S., Bordan Z., Weintraub D., et al. (2018). Hsp70 suppresses mitochondrial reactive oxygen species and preserves pulmonary microvascular barrier integrity following exposure to bacterial toxins. Front. Immunol. 9, 1309. doi:10.3389/fimmu.2018.01309
Loffredo L., Carnevale R., Cangemi R., Angelico F., Augelletti T., Di Santo S., et al. (2013). NOX2 up-regulation is associated with artery dysfunction in patients with peripheral artery disease. Int. J. Cardiol. 165 (1), 184–192. doi:10.1016/j.ijcard.2012.01.069
Lu M., Echeverri F., Kalabat D., Laita B., Dahan D. S., Smith R. D., et al. (2008). Small molecule activator of the human epithelial sodium channel. J. Biol. Chem. 283 (18), 11981–11994. doi:10.1074/jbc.M708001200
Lucas R., Magez S., De Leys R., Fransen L., Scheerlinck J. P., Rampelberg M., et al. (1994). Mapping the lectin-like activity of tumor necrosis factor. Science 263 (5148), 814–817. doi:10.1126/science.8303299
Lucas R., Sridhar S., Rick F. G., Gorshkov B., Umapathy N. S., Yang G., et al. (2012b). Agonist of growth hormone-releasing hormone reduces pneumolysin-induced pulmonary permeability edema. Proc. Natl. Acad. Sci. U. S. A. 109 (6), 2084–2089. doi:10.1073/pnas.1121075109
Lucas R., Yang G., Gorshkov B. A., Zemskov E. A., Sridhar S., Umapathy N. S., et al. (2012a). Protein kinase C-α and arginase I mediate pneumolysin-induced pulmonary endothelial hyperpermeability. Am. J. Respir. Cell Mol. Biol. 47 (4), 445–453. doi:10.1165/rcmb.2011-0332OC
Lucas R., Yue Q., Alli A., Duke B. J., Al-Khalili O., Thai T. L., et al. (2016). The lectin-like domain of TNF increases ENaC open probability through a novel site at the interface between the second transmembrane and C-terminal domains of the α-subunit. J. Biol. Chem. 291 (45), 23440–23451. doi:10.1074/jbc.M116.718163
Madaio M. P., Czikora I., Kvirkvelia N., McMenamin M., Yue Q., Liu T., et al. (2019). The TNF-derived TIP peptide activates the epithelial sodium channel and ameliorates experimental nephrotoxic serum nephritis. Kidney Int. 95 (6), 1359–1372. doi:10.1016/j.kint.2018.12.022
Martin-Malpartida P., Arrastia-Casado S., Farrera-Sinfreu J., Lucas R., Fischer H., Fischer B., et al. (2022). Conformational ensemble of the TNF-derived peptide solnatide in solution. Comput. Struct. Biotechnol. J. 20 (Apr 27), 2082–2090. doi:10.1016/j.csbj.2022.04.031
Martner A., Dahlgren C., Paton J. C., Wold A. E. (2008). Pneumolysin released during Streptococcus pneumoniae autolysis is a potent activator of intracellular oxygen radical production in neutrophils. Infect. Immun. 76 (9), 4079–4087. doi:10.1128/IAI.01747-07
Matalon S., O'Brodovich H. (1999). Sodium channels in alveolar epithelial cells: molecular characterization, biophysical properties, and physiological significance. Annu. Rev. Physiol. 61, 627–661. doi:10.1146/annurev.physiol.61.1.627
Matthay M. A., Arabi Y., Arroliga A. C., Bernard G., Bersten A. D., Brochard L. J., et al. (2024). A new global definition of acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 209 (1), 37–47. doi:10.1164/rccm.202303-0558WS
Mraheil M. A., Toque H. A., La Pietra L., Hamacher J., Phanthok T., Verin A., et al. (2021). Dual role of hydrogen peroxide as an oxidant in pneumococcal pneumonia. Antioxid. Redox Signal 34 (12), 962–978. doi:10.1089/ars.2019.7964
Mutchler S. M., Hasan M., Kohan D. E., Kleyman T. R., Tan R. J. (2021). Deletion of the gamma subunit of ENaC in endothelial cells does not protect against renal ischemia reperfusion injury. Int. J. Mol. Sci. 22 (20), 10914. doi:10.3390/ijms222010914
Neumann P., Gertzberg N., Johnson A. (2004). TNF-alpha induces a decrease in eNOS promoter activity. Am. J. Physiol. Lung Cell Mol. Physiol. 286 (2), L452–L459. doi:10.1152/ajplung.00378.2002
Neumann P., Gertzberg N., Vaughan E., Weisbrot J., Woodburn R., Lambert W., et al. (2006). Peroxynitrite mediates TNF-alpha-induced endothelial barrier dysfunction and nitration of actin. Am. J. Physiol. Lung Cell Mol. Physiol. 290 (4), L674-L684–L684. doi:10.1152/ajplung.00391.2005
Neyton L. P. A., Sinha P., Sarma A., Mick E., Kalantar K., Chen S., et al. (2024). Host and microbe blood metagenomics reveals key pathways characterizing critical illness phenotypes. Am. J. Respir. Crit. Care Med. 209 (7), 805–815. doi:10.1164/rccm.202308-1328OC
Noel J., Wang H., Hong N., Tao J. Q., Yu K., Sorokina E. M., et al. (2013). PECAM-1 and caveolae form the mechanosensing complex necessary for NOX2 activation and angiogenic signaling with stopped flow in pulmonary endothelium. Am. J. Physiol. Lung Cell Mol. Physiol. 305 (11), L805–L818. doi:10.1152/ajplung.00123.2013
Ochoa-Gondar O., Torras-Vives V., de Diego-Cabanes C., Satué-Gracia E. M., Vila-Rovira A., Forcadell-Perisa M. J., et al. (2023). Incidence and risk factors of pneumococcal pneumonia in adults: a population-based study. BMC Pulm. Med. 23, 200. doi:10.1186/s12890-023-02497-2
Ortiz-Prado E., Dunn J. F., Vasconez J., Castillo D., Viscor G. (2019). Partial pressure of oxygen in the human body: a general review. Am. J. Blood Res. 9 (1), 1–14.
Pericone C. D., Overweg K., Hermans P. W., Weiser J. N. (2000). Inhibitory and bactericidal effects of hydrogen peroxide production by Streptococcus pneumoniae on other inhabitants of the upper respiratory tract. Infect. Immun. 68, 3990–3997. doi:10.1128/iai.68.7.3990-3997.2000
Pochynyuk O., Tong Q., Staruschenko A., Ma H. P., Stockand J. D. (2006). Regulation of the epithelial Na+ channel (ENaC) by phosphatidylinositides. Am. J. Physiol. Ren. Physiol. 290 (5), F949–F957. doi:10.1152/ajprenal.00386.2005
Predescu D., Predescu S., Shimizu J., Miyawaki-Shimizu K., Malik A. B. (2005). Constitutive eNOS-derived nitric oxide is a determinant of endothelial junctional integrity. Am. J. Physiol. Lung Cell Mol. Physiol. 289 (3), L371–L381. doi:10.1152/ajplung.00175.2004
Price D. R., Garcia J. G. N. (2024). A razor's edge: vascular responses to acute inflammatory lung injury/acute respiratory distress syndrome. Annu. Rev. Physiol. 86, 505–529. doi:10.1146/annurev-physiol-042222-030731
Rafii B., Tanswell A. K., Otulakowski G., Pitkänen O., Belcastro-Taylor R., O'Brodovich H. (1998). O2-induced ENaC expression is associated with NF-kappaB activation and blocked by superoxide scavenger. Am. J. Physiol. 275 (4), L764–L770. doi:10.1152/ajplung.1998.275.4.L764
Regev-Yochay G., Trzcinski K., Thompson C. M., Malley R., Lipsitch M. (2006). Interference between Streptococcus pneumoniae and Staphylococcus aureus: in vitro hydrogen peroxide-mediated killing by Streptococcus pneumoniae. J. Bacteriol. 188, 4996–5001. doi:10.1128/JB.00317-06
Romero M. J., Platt D. H., Tawfik H. E., Labazi M., El-Remessy A. B., Bartoli M., et al. (2008). Diabetes-induced coronary vascular dysfunction involves increased arginase activity. Circ. Res. 102 (1), 95–102. doi:10.1161/CIRCRESAHA.107.155028
Romero M. J., Yue Q., Ahn W. M., Hamacher J., Zaidi Y., Haigh S., et al. (2024). Endothelial ENaC-α restrains oxidative stress in lung capillaries in murine pneumococcal pneumonia-associated acute lung injury. Am. J. Respir. Cell Mol. Biol. doi:10.1165/rcmb.2023-0440OC
Romero M. J., Yue Q., Singla B., Hamacher J., Sridhar S., Moseley A. S., et al. (2023). Direct endothelial ENaC activation mitigates vasculopathy induced by SARS-CoV2 spike protein. Front. Immunol. 14 (Aug 10), 1241448. doi:10.3389/fimmu.2023.1241448
Ross J. T., Nesseler N., Lee J. W., Ware L. B., Matthay M. A. (2019). The ex vivo human lung: research value for translational science. JCI Insight 4 (11), e128833. doi:10.1172/jci.insight.128833
Schjerning J., Uhrenholt T. R., Svenningsen P., Vanhoutte P. M., Skøtt O., Jensen B. L., et al. (2013). Histamine-dependent prolongation by aldosterone of vasoconstriction in isolated small mesenteric arteries of the mouse. Am. J. Physiol. Heart Circ. Physiol. 304 (8), H1094–H1102. doi:10.1152/ajpheart.00524.2012
Schmid B., Kranke P., Lucas R., Meybohm P., Zwissler B., Frank S. (2022). Safety and preliminary efficacy of sequential multiple ascending doses of solnatide to treat pulmonary permeability edema in patients with moderate to severe ARDS in a randomized, placebo-controlled, double-blind trial: preliminary evaluation of safety and feasibility in light of the COVID-19 pandemic. Trials 23 (1), 252. doi:10.1186/s13063-022-06182-3
Schmid B., Kredel M., Ullrich R., Krenn K., Lucas R., Markstaller K., et al. (2021). Safety and preliminary efficacy of sequential multiple ascending doses of solnatide to treat pulmonary permeability edema in patients with moderate-to-severe ARDS-a randomized, placebo-controlled, double-blind trial. Trials 22 (1), 643. doi:10.1186/s13063-021-05588-9
Schröder K., Weissmann N., Brandes R. P. (2017). Organizers and activators: cytosolic Nox proteins impacting on vascular function. Free Radic. Biol. Med. 109, 22–32. doi:10.1016/j.freeradbiomed.2017.03.017
Schwameis R., Eder S., Pietschmann H., Fischer B., Mascher H., Tzotzos S., et al. (2014). A FIM study to assess safety and exposure of inhaled single doses of AP301-A specific ENaC channel activator for the treatment of acute lung injury. J. Clin. Pharmacol. 54 (3), 341–350. doi:10.1002/jcph.203
Shafique E., Torina A., Reichert K., Colantuono B., Nur N., Zeeshan K., et al. (2017). Mitochondrial redox plays a critical role in the paradoxical effects of NAPDH oxidase-derived ROS on coronary endothelium. Cardiovasc Res. 113, 234–246. doi:10.1093/cvr/cvw249
Sinha P., Furfaro D., Cummings M. J., Abrams D., Delucchi K., Maddali M. V., et al. (2021). Latent class analysis reveals COVID-19-related acute respiratory distress syndrome subgroups with differential responses to corticosteroids. Am. J. Respir. Crit. Care Med. 204 (11), 1274–1285. doi:10.1164/rccm.202105-1302OC
Sinha P., Meyer N. J., Calfee C. S. (2023). Biological phenotyping in sepsis and acute respiratory distress syndrome. Annu. Rev. Med. 74, 457–471. doi:10.1146/annurev-med-043021-014005
Song L., Shi X., Kovacs L., Han W., John J., Barman S. A., et al. (2023). Calpain promotes LPS-induced lung endothelial barrier dysfunction via cleavage of talin. Am. J. Respir. Cell Mol. Biol. 69 (6), 678–688. doi:10.1165/rcmb.2023-0009OC
Sternak M., Bar A., Adamski M. G., Mohaissen T., Marczyk B., Kieronska A., et al. (2018). The deletion of endothelial sodium channel α (αENaC) impairs endothelium-dependent vasodilation and endothelial barrier integrity in endotoxemia in vivo. Front. Pharmacol. 9 (Apr 10), 178. doi:10.3389/fphar.2018.00178
Su Y., Lucas R., Fulton D. J. R., Verin A. D. (2024). Mechanisms of pulmonary endothelial barrier dysfunction in acute lung injury and acute respiratory distress syndrome. Chin. Med. J. Pulm. Crit. Care Med. 2 (2), 80–87. doi:10.1016/j.pccm.2024.04.002
Sure F., Einsiedel J., Gmeiner P., Duchstein P., Zahn D., Korbmacher C., et al. (2024). The small molecule activator S3969 stimulates the epithelial sodium channel by interacting with a specific binding pocket in the channel's β-subunit. J. Biol. Chem. 300 (4), 105785. doi:10.1016/j.jbc.2024.105785
Takemura Y., Goodson P., Bao H. F., Jain L., Helms M. N. (2010). Rac1-mediated NADPH oxidase release of O2- regulates epithelial sodium channel activity in the alveolar epithelium. Am. J. Physiol. Lung Cell Mol. Physiol. 298, L509–L520. doi:10.1152/ajplung.00230.2009
Tarjus A., Maase M., Jeggle P., Martinez-Martinez E., Fassot C., Loufrani L., et al. (2017). The endothelial αENaC contributes to vascular endothelial function in vivo. PLoS One 12 (9), e0185319. doi:10.1371/journal.pone.0185319
Thome U. H., Davis I. C., Nguyen S. V., Shelton B. J., Matalon S. (2003). Modulation of sodium transport in fetal alveolar epithelial cells by oxygen and corticosterone. Am. J. Physiol. Lung Cell Mol. Physiol. 284 (2), L376–L385. doi:10.1152/ajplung.00218.2002
Trac P. T., Thai T. L., Linck V., Zou L., Greenlee M., Yue Q., et al. (2017). Alveolar nonselective channels are ASIC1a/α-ENaC channels and contribute to AFC. Am. J. Physiol. Lung Cell Mol. Physiol. 312 (6), L797-L811–L811. doi:10.1152/ajplung.00379.2016
Tzotzos S. J., Fischer B., Fischer H., Zeitlinger M. (2020). Incidence of ARDS and outcomes in hospitalized patients with COVID-19: a global literature survey. Crit. Care 24 (1), 516. doi:10.1186/s13054-020-03240-7
van Amstel R. B. E., Bartek B., Vlaar A. P. J., Gay E., van Vught L. A., Cremer O. L., et al. (2024). Temporal transitions of the hyperinflammatory and hypoinflammatory phenotypes in critical illness. Am. J. Respir. Crit. Care Med. 211, 347–356. doi:10.1164/rccm.202406-1241OC
Vaughn V. M., Dickson R. P., Horowitz J. K., Flanders S. A. (2024). Community-acquired pneumonia: a review. JAMA 332 (15), 1282–1295. doi:10.1001/jama.2024.14796
Vieillard-Baron A., Matthay M., Teboul J. L., Bein T., Schultz M., Magder S., et al. (2016). Experts' opinion on management of hemodynamics in ARDS patients: focus on the effects of mechanical ventilation. Intensive Care Med. 42 (5), 739–749. doi:10.1007/s00134-016-4326-3
Violi F., Carnevale R., Calvieri C., Nocella C., Falcone M., Farcomeni A., et al. (2015). Nox2 up-regulation is associated with an enhanced risk of atrial fibrillation in patients with pneumonia. Thorax 70 (10), 961–966. doi:10.1136/thoraxjnl-2015-207178
Violi F., Oliva A., Cangemi R., Ceccarelli G., Pignatelli P., Carnevale R., et al. (2020). Nox2 activation in covid-19. Redox Biol. 36, 101655. doi:10.1016/j.redox.2020.101655
Wang Q., Doerschuk C. M. (2000). Neutrophil-induced changes in the biomechanical properties of endothelial cells: roles of ICAM-1 and reactive oxygen species. J. Immunol. 164 (12), 6487–6494. doi:10.4049/jimmunol.164.12.6487
Wang Y., Nakayama M., Pitulescu M. E., Schmidt T. S., Bochenek M. L., Sakakibara A., et al. (2010). Ephrin-B2 controls VEGF-induced angiogenesis and lymphangiogenesis. Nature 465 (7297), 483–486. doi:10.1038/nature09002
Ware L. B. (2017). Targeting resolution of pulmonary edema in primary graft dysfunction after lung transplantation: is inhaled AP301 the answer? J. Heart Lung Transpl. S1053-2498 (17), 189–191. doi:10.1016/j.healun.2017.11.013
Ware L. B., Matthay M. A. (2001). Alveolar fluid clearance is impaired in the majority of patients with acute lung injury and the acute respiratory distress syndrome. Am. J. Respir. Crit. Care Med. 163 (6), 1376–1383. doi:10.1164/ajrccm.163.6.2004035
Wick K. D., Ware L. B., Matthay M. A. (2024). Acute respiratory distress syndrome. BMJ 387, e076612. doi:10.1136/bmj-2023-076612
Winterbourn C. C., Kettle A. J., Hampton M. B. (2016). Reactive oxygen species and neutrophil function. Annu. Rev. Biochem. 85, 765–792. doi:10.1146/annurev-biochem-060815-014442
Witzenrath M., Gutbier B., Hocke A. C., Schmeck B., Hippenstiel S., Berger K., et al. (2006). Role of pneumolysin for the development of acute lung injury in pneumococcal pneumonia. Crit. Care Med. 34 (7), 1947–1954. doi:10.1097/01.CCM.0000220496.48295.A9
Xiao W., Oldham W. M., Priolo C., Pandey A. K., Loscalzo J. (2021). Immunometabolic endothelial phenotypes: integrating inflammation and glucose metabolism. Circ. Res. 129 (1), 9–29. doi:10.1161/CIRCRESAHA.120.318805
Youn S. W., Li Y., Kim Y. M., Sudhahar V., Abdelsaid K., Kim H. W., et al. (2019). Modification of cardiac progenitor cell-derived exosomes by miR-322 provides protection against myocardial infarction through nox2-dependent angiogenesis. Antioxidants (Basel) 8 (1), 18. doi:10.3390/antiox8010018
Yu L., Bao H. F., Self J. L., Eaton D. C., Helms M. N. (2007). Aldosterone-induced increases in superoxide production counters nitric oxide inhibition of epithelial Na channel activity in A6 distal nephron cells. Am. J. Physiol. Ren. Physiol. 293 (5), F1666–F1677. doi:10.1152/ajprenal.00444.2006
Yue G., Malik B., Yue G., Eaton D. C. (2002). Phosphatidylinositol 4,5-bisphosphate (PIP2) stimulates epithelial sodium channel activity in A6 cells. J. Biol. Chem. 277 (14), 11965–11969. doi:10.1074/jbc.M108951200
Zhang J., Yuan H. K., Chen S., Zhang Z. R. (2022). Detrimental or beneficial: role of endothelial ENaC in vascular function. J. Cell Physiol. 237, 29–48. doi:10.1002/jcp.30505
Keywords: NADPH oxidase, epithelial sodium channel, capillary endothelium, alveolar-capillary barrier function, pneumonia, TNF, TIP peptide
Citation: Eaton DC, Romero MJ, Matthay MA, Hamacher J, Advani A, Wolf A, Abu Mraheil M, Chakraborty T, Stepp DW, Belin de Chantemèle EJ, Kutlar A, Kraft F, Zeitlinger M, Kranke P, Frank S, Su Y, Verin AD, Fulton DJR, Ushio-Fukai M, Fukai T and Lucas R (2025) Endothelial ENaC as a repressor of oxidative stress and a guardian of lung capillary barrier function in bacterial and viral pneumonia. Front. Physiol. 16:1562626. doi: 10.3389/fphys.2025.1562626
Received: 17 January 2025; Accepted: 26 March 2025;
Published: 07 April 2025.
Edited by:
Miriam M. Cortese-Krott, Heinrich Heine University of Düsseldorf, GermanyReviewed by:
Dan Predescu, Rush University, United StatesCopyright © 2025 Eaton, Romero, Matthay, Hamacher, Advani, Wolf, Abu Mraheil, Chakraborty, Stepp, Belin de Chantemèle, Kutlar, Kraft, Zeitlinger, Kranke, Frank, Su, Verin, Fulton, Ushio-Fukai, Fukai and Lucas. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: R. Lucas, cmx1Y2FzQGF1Z3VzdGEuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.