 Marcos Gonzalez
Marcos Gonzalez Sarah Clayton
Sarah Clayton Eric Wauson
Eric Wauson Daniel Christian
Daniel Christian Quang-Kim Tran
Quang-Kim Tran
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 23 January 2025
Sec. Vascular Physiology
Volume 16 - 2025 | https://doi.org/10.3389/fphys.2025.1545044
This article is part of the Research Topic Insights in Vascular Physiology: 2024 View all 9 articles
The discovery of nitric oxide (NO) and the role of endothelial cells (ECs) in its production has revolutionized medicine. NO can be produced by isoforms of NO synthases (NOS), including the neuronal (nNOS), inducible (iNOS), and endothelial isoforms (eNOS), and via the non-classical nitrate-nitrite-NO pathway. In particular, endothelium-derived NO, produced by eNOS, is essential for cardiovascular health. Endothelium-derived NO activates soluble guanylate cyclase (sGC) in vascular smooth muscle cells (VSMCs), elevating cyclic GMP (cGMP), causing vasodilation. Over the past four decades, the importance of this pathway in cardiovascular health has fueled the search for strategies to enhance NO bioavailability and/or preserve the outcomes of NO’s actions. Currently approved approaches operate in three directions: 1) providing exogenous NO, 2) promoting sGC activity, and 3) preventing degradation of cGMP by inhibiting phosphodiesterase 5 activity. Despite clear benefits, these approaches face challenges such as the development of nitrate tolerance and endothelial dysfunction. This highlights the need for sustainable options that promote endogenous NO production. This review will focus on strategies to promote endogenous NO production. A detailed review of the mechanisms regulating eNOS activity will be first provided, followed by a review of strategies to promote endogenous NO production based on the levels of available preclinical and clinical evidence, and perspectives on future possibilities.
NO-containing compounds have been used in medicine for over 160 years; however, it was not until the early 1980s that NO was discovered as the ingredient that exerts the therapeutic effects and ECs as the key source of vascular NO (Katsuki et al., 1977; Arnold et al., 1977; Furchgott and Zawadzki, 1980; Ignarro et al., 1987; Palmer et al., 1987). We now know NO can be produced by three NO synthases and via nitrate-nitrite-NO conversion (Bredt et al., 1991; Janssens et al., 1992; Geller et al., 1993; Benjamin et al., 1994; Lundberg et al., 1994; Stuehr, 1997). The NO synthases are classified a neuronal NOS (encoded by NOS1), inducible NOS (encoded by NOS2), and eNOS (encoded by NOS3). Among these sources, endothelium-derived NO plays a vital role in regulating vascular tone, inhibiting inflammation, and preventing thrombosis (Alheid et al., 1987; Forstermann et al., 1989; Bredt and Snyder, 1990; Mitchell et al., 1991; Forstermann et al., 1991). Endothelium-derived NO diffuses into the circulation and the underlying VSMCs, where it activates sGC, which enhances cGMP, causing vasodilation (Arnold et al., 1977; Ignarro et al., 1986a; Ignarro et al., 1986b; Forstermann et al., 1994). The critical role of eNOS in controlling vascular tone was documented by findings that pharmacological inhibition of NOS causes hypertension (Rees et al., 1989) and deletion of NOS3 results in high blood pressure (Huang et al., 1995). Beyond activating sGC and enhancing cGMP, NO exerts numerous other effects. For example, endothelium-derived NO directly suppresses the electrical excitability of VSMCs by inhibiting the action of T-type and L-type voltage-gated Ca2+ channels in small arteries, and reduced NO availability can trigger transient depolarization in normally quiescent VSMCs leading to vasospasm (Smith et al., 2020). Actions of NO leading to smooth muscle relaxation are important for cardiovascular, respiratory, renal and digestive functions. NO is also critical in brain function as a neurotransmitter and immune responses.
Dysfunction and uncoupling of eNOS are associated with cardiovascular diseases (CVD) (Janaszak-Jasiecka et al., 2023; Heitzer et al., 2000a; Munzel et al., 2005), and increased eNOS expression and reversal of eNOS uncoupling in experimental models improves vascular function (Li et al., 2006). Approaches to increase NO bioavailability have been intensively researched. Some studies suggest that diets that are high in antioxidants or antioxidant supplementation can help preserve vascular health and prevent CVD by reducing oxidative stress and improving endothelial function. However, a blanket recommendation has not been made in clinical guidelines as there are needs for stronger evidence and determinations of effective doses and specific patient populations that might benefit (Varadharaj et al., 2017). Dietary nitrates and nitrites, found in foods such as beetroot and leafy greens, can be converted to NO and have shown promising results in improving vascular function and lowering blood pressure. However, these are not yet recommended in clinical guidelines for prevention or management of CVD (Blekkenhorst et al., 2018). Current options in clinical practice guidelines focus mainly on downstream components of NO signaling, such as NO inhalation, use of NO donors, sGC stimulators/activators, or inhibition of phosphodiesterase (PDE) 5 (Chen et al., 2002; Rapoport et al., 1987; Bonderman et al., 2014; Ignarro et al., 1982; Kraehling and Sessa, 2017; Lundberg et al., 2015). However, challenges such as short-lived effects and the development of nitrate tolerance and endothelial dysfunction have limited their efficacy (Rapoport et al., 1987; Munzel et al., 1995a; Munzel et al., 1995b; Erdmann et al., 2013; Knorr et al., 2011; Munzel et al., 2014; Munzel et al., 2013; Oelze et al., 2013). There is thus a strong need to develop new and improve existing approaches to promote the endogenous production of NO.
eNOS is a ∼133 kDa homo-dimeric oxidoreductase enzyme with an N-terminal oxygenase domain that binds L-arginine and tetrahydrobiopterin (BH4) and a C-terminal reductase domain that transfers electrons from nicotinamide adenine dinucleotide phosphate (NADPH) via flavin adenine dinucleotide (FAD) and flavin mononucleotide (FMN) (Figure 1) (Stuehr, 1997; Palmer et al., 1988; Stuehr et al., 2005; Marsden et al., 1992). Dimerization is essential for eNOS function, stabilizing the enzyme and ensuring efficient electron transfer (List et al., 1997). Ca2+-bound calmodulin (CaM) binds to the CaM-binding domain and initiates electron flow from the reductase domain to the oxygenase domain, where NO is synthesized from L-arginine (Busse and Mulsch, 1990; Venema et al., 1996). eNOS-mediated production of NO follows a two-step process: 1) hydroxylation of L-arginine to Nω-hydroxy-L-arginine, and 2) further oxidation to generate L-citrulline and NO. Tetrahydrobiopterin (BH4) plays a vital role in this reaction by stabilizing the eNOS dimer and preventing the formation of superoxide, a potentially harmful byproduct, ensuring NO synthesis proceeds efficiently (Xia et al., 1998a; Xia et al., 1998b). eNOS regulation is a highly complex process, integrating multiple layers of control to fine-tune NO production according to physiological needs. These layers include transcriptional regulation, post-translational modifications, and protein-protein interactions.
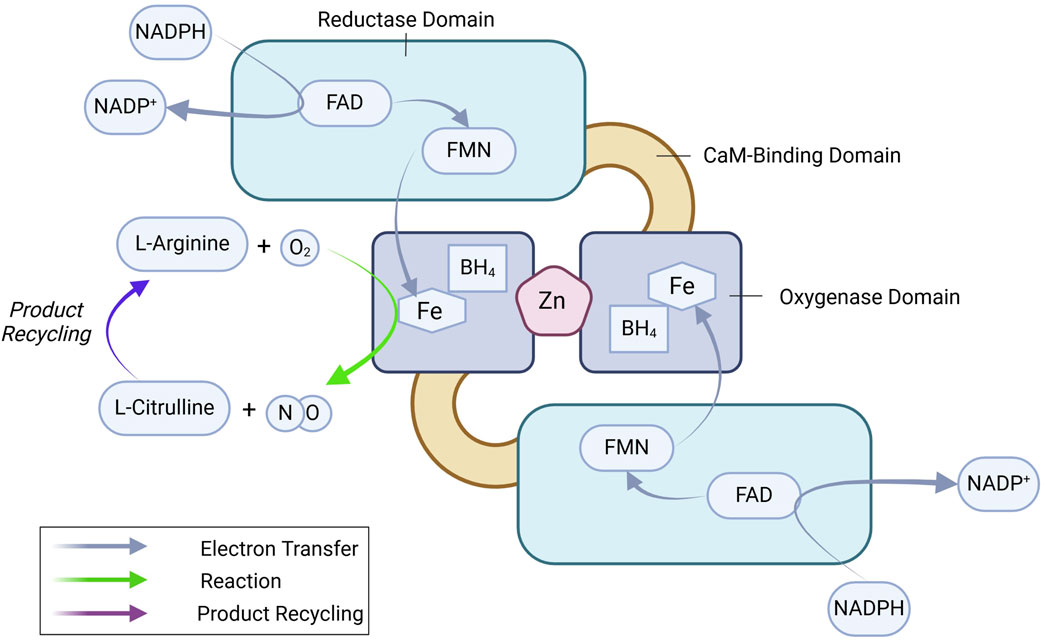
Figure 1. eNOS dimer and key enzymatic reactions leading to NO production. See text for details. BH4, tetrahydrobiopterin; FAD, flavin adenine dinucleotide; FMN, flavin mononucleotide; NADPH, nicotinamide adenine dinucleotide phosphate.
The NOS3 gene is located on chromosome 7 (7q35-36) and is regulated by a promoter region containing binding sites for multiple transcription factors, including Krüppel-like Factor 2 (KLF2), Specificity protein 1 (Sp1), Specificity protein 3 (Sp3), Ets1, mothers against decapentaplegic homolog-2 (Smad2), and Nuclear factor erythroid 2-related factor (Nrf2), among others (Karantzoulis-Fegaras et al., 1999; Laumonnier et al., 2000; Balligand et al., 2009; Fish and Marsden, 2006). These factors dynamically regulate eNOS expression in response to physiological signals. Initially thought to be constitutively expressed, NOS3 is now recognized to be highly responsive to various regulatory stimuli, which adjust its transcription to match the vascular environment’s needs (Balligand et al., 2009; Black et al., 1998; Mattsson et al., 1997; Nadaud et al., 1996; Nishida et al., 1992; Ou et al., 2005; Searles, 2006). Transcriptional regulation of eNOS can be categorized into upregulating, downregulating, and dual-regulating factors (Figure 2).
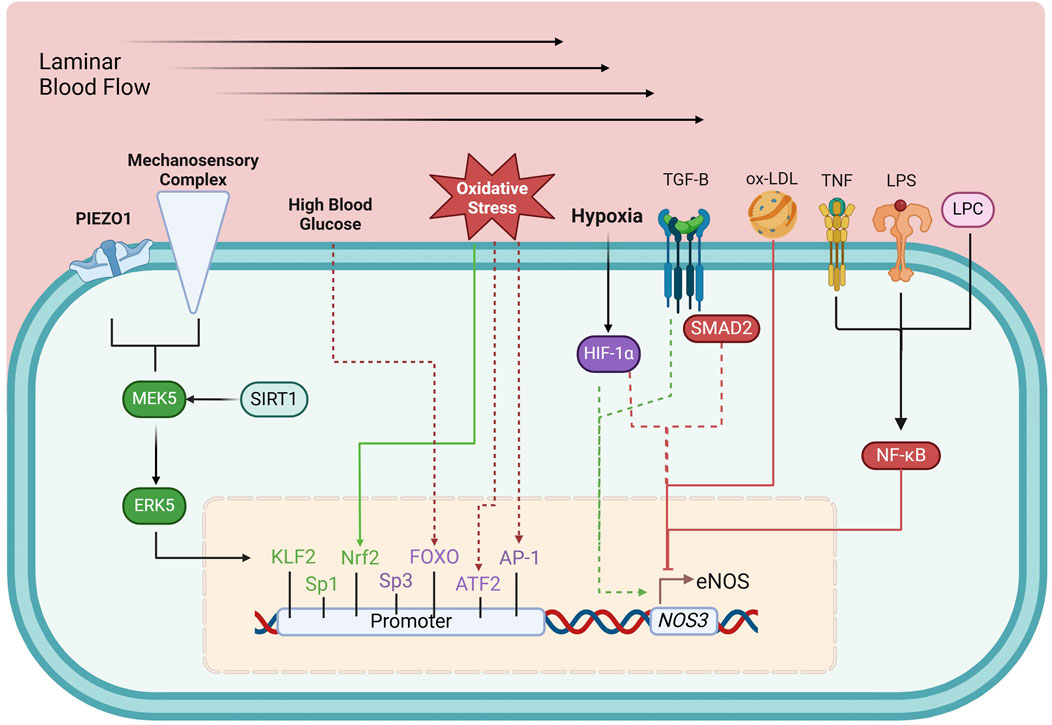
Figure 2. Transcriptional regulation of eNOS. Green boxes, upregulating factors; purple boxes, dual-regulating factors; red boxes, down-regulating factors; solid arrows, connection between environmental and metabolic elements to their respective regulating factors; dashed arrows, circumstantial elements that determine the effects of dual-regulating factors; AP-1, activator protein 1; ATF-2, activating transcription factor 2; ERK5, extracellular-regulated kinase 5; FOXO, forkhead box O; HIF-1α, hypoxia-inducible factor 1-α; KLF2, Krüppel-like factor 2; LPC, lysophosphatidylcholine; LPS, lipopolysaccharide; MEK5, Mitogen-activated protein kinase kinase 5; Nrf2, Nuclear factor erythroid 2-related factor; NF-κB, Nuclear Factor kappa-light-chain-enhancer of activated B cells; ox-LDL, oxidized low-density lipoprotein; SIRT1, sirtuin 1; SMAD2, mothers against decapentaplegic homolog-2; Sp1, specificity protein 1; Sp3, specificity protein 3; TGF-β, tumor growth factor β; TNF, tumor necrosis factor. See text for details.
Shear stress, the mechanical stimulus exerted on the endothelium by laminar blood flow, upregulates eNOS expression (Nadaud et al., 1996) by triggering PIEZO1 Ca2+ channels (Wang et al., 2016) and a mechanosensory complex leading to the activation of KLF2, which binds directly to the NOS3 promoter (Wang et al., 2010; Lin et al., 2005). Sp1 regulates basal NOS3 expression and responds to stimuli like growth factors and hypoxia, enhancing NO production to maintain vascular tone (Tang et al., 1995; Wariishi et al., 1995). Additionally, Nrf2, activated by oxidative stress, enhances NOS3 transcription by upregulating antioxidant response elements in the NOS3 promoter (Wu et al., 2019).
NF-κB (Nuclear Factor kappa-light-chain-enhancer of activated B cells) represses eNOS expression, particularly in inflammation. NF-κB is activated by pro-inflammatory stimuli such as tumor necrosis factor-alpha (TNF-α), lipopolysaccharide (LPS), and oxidized low-density lipoprotein (ox-LDL). These molecules trigger signaling events that lead to NF-κB translocation into the nucleus, where it inhibits eNOS transcription (Nishida et al., 1992; Neumann et al., 2004). LPS and ox-LDL, both associated with oxidative stress and vascular inflammation, promote NF-κB activation, further repressing eNOS expression (Liao et al., 1995; Lu et al., 1996).
Certain factors can both up- or downregulate eNOS expression depending on cellular conditions. Tumor growth factor β (TGF-β)/Smad2 can enhance eNOS transcription in a healthy endothelium but suppress it in chronic inflammation or vascular injury (Saura et al., 2002). Hypoxia-inducible factor-1α (HIF-1α) also exhibits dual regulation: during acute hypoxia, it stimulates eNOS expression to ensure adequate NO production but may suppress eNOS in chronic hypoxia, causing maladaptive vascular changes (McQuillan et al., 1994; Fish et al., 2010).
Post-transcriptional mechanisms offer additional precision by modulating the stability and translation of eNOS mRNA. Elements such as miRNAs (e.g., miR-92a) and long non-coding RNAs (lncRNAs) can either enhance or suppress NO production in response to physiological conditions (Lin et al., 2005; Miao et al., 2018; Suarez et al., 2007; Man et al., 2018; Bonauer et al., 2009).
eNOS activity is also intricately regulated by various PTMs, including phosphorylation, acetylation, S-nitrosylation, and palmitoylation. These modifications play critical roles in modulating eNOS enzymatic activity, localization, and interactions with other cellular components (Figure 3).
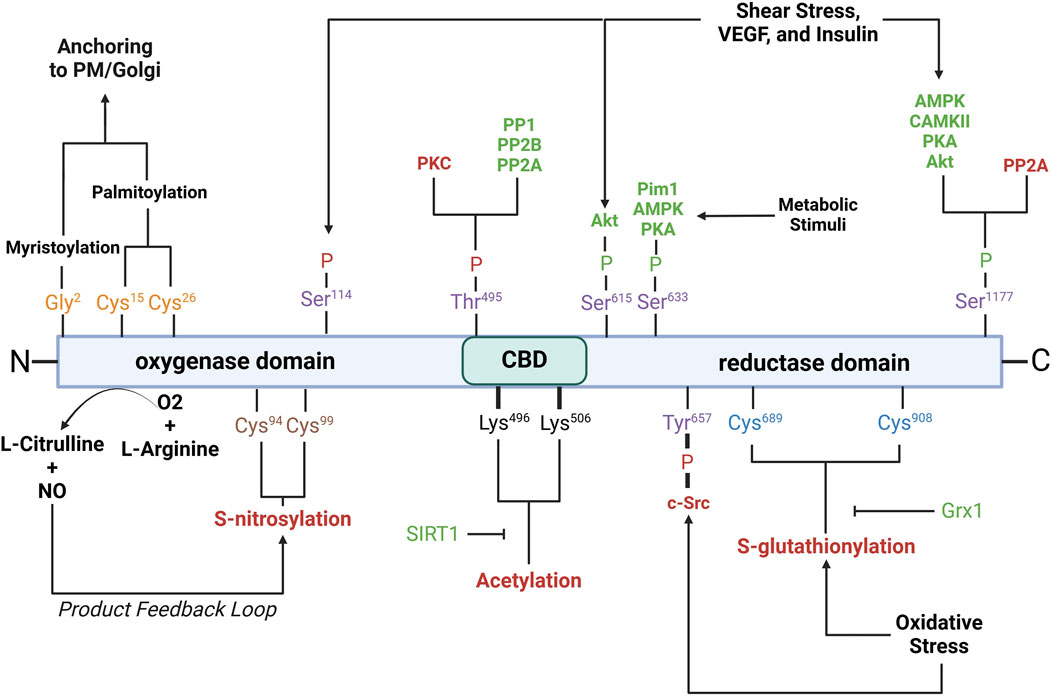
Figure 3. Schematic of eNOS domains with key residues involved in post-translational modifications. Purple residues, phosphorylation sites; yellow residues, myristoylation/palmitoylation sites; brown residues, S-nitrosylation sites; black residues, acetylation sites; blue residues, S-glutathionylation sites. Red- and green-letter enzymes, inhibitory and stimulatory effects on eNOS, respectively. Akt, protein kinase B; AMPK, AMP-activated protein kinase; CaMKII, calcium/calmodulin-dependent protein kinase II; c-Src, cellular sarcoma; Grx1, glutaredoxin; Pim1, proviral integration site for Moloney murine leukemia virus 1; PKA, protein kinase A; PKC, protein kinase C; PP1, protein phosphatase 1; PP2A, protein phosphatase 2a; PP2B, protein phosphatase 2b; PM, plasma membrane; SIRT1, sirtuin 1; VEGF, vascular endothelial growth factor. See text for further details.
Phosphorylation is a critical modification that modulates eNOS activity and is tightly regulated by various kinases and phosphatases in response to physiological cues such as shear stress, hypoxia, and growth factors. Serine 1177 is located on the C-terminal reductase domain and when phosphorylated, enhances eNOS activity by facilitating electron flow from NADPH to the heme domain, contributing to NO production (Fulton et al., 1999; Scotland et al., 2002; Kashiwagi et al., 2013; Li Q. et al., 2013; Park et al., 2016). Ser1177 phosphorylation is key positive regulator of eNOS function (Dimmeler et al., 1999; Tomada et al., 2014). Stimuli such as shear stress, vascular endothelial growth factor (VEGF), and insulin activate kinases including protein kinase B (Akt), AMP-activated protein kinase (AMPK), calcium/calmodulin-dependent protein kinase II (CaMKII), protein kinase A (PKA), and protein kinase G (PKG), which phosphorylate Ser1177 (Fulton et al., 1999; Dimmeler et al., 1999; Michell et al., 1999; Chen et al., 1999; Fleming et al., 2001; Atochin et al., 2007). Phosphorylation by Akt, in particular, is essential for eNOS activation in endothelial cells in response to VEGF and shear stress (Dimmeler et al., 1999; Di Lorenzo et al., 2013). Ser1177 phosphorylation also increases the Ca2+ sensitivity of the synthase, permitting CaM binding and enzyme activation at lower intracellular Ca2+ levels (Tran et al., 2009; Mount et al., 2007; McCabe et al., 2000). Serine 633 is phosphorylated in response to shear stress, exercise, and metabolic stimuli (Mount et al., 2007). Ser633 phosphorylation by PKA and AMPK during physical activity improves NO bioavailability and supports vascular homeostasis (Mount et al., 2007; Michell et al., 2002). Serine 615 phosphorylation enhances eNOS activity, working cooperatively with Ser1177 to enhance the binding affinity of the Ca2⁺-CaM complex to eNOS, a critical step for eNOS activation, which ensures a robust response to Ca2⁺ signals (Tran et al., 2009; Bauer et al., 2003). Threonine 495 phosphorylation, in contrast, inhibits eNOS by suppressing CaM binding (Fleming et al., 2001). Kinases like AMPK and PKC mediate this modification, especially during oxidative stress (Chen et al., 1999). Agonists such as bradykinin promote NO release by inducing Thr495 dephosphorylation, allowing CaM to activate eNOS (Harris et al., 2001). This dephosphorylation is mediated by calcineurin and inhibited by cyclosporine A (Harris et al., 2001). The balance between Thr495 phosphorylation and dephosphorylation is crucial for regulating eNOS activity and NO production. Serine 114 is phosphorylated in response to shear stress (Mount et al., 2007; Gallis et al., 1999). Though its role remains debatable, phosphorylation at Ser114 is considered a negative regulator of eNOS activity (Mount et al., 2007), supported by the observations that its dephosphorylation by VEGF treatment enhances eNOS function (Bauer et al., 2003) and that a phospho-null mutation here inhibits eNOS activity (Kou et al., 2002; Li et al., 2007).
Acetylation regulates eNOS interactions with other proteins, its plasma membrane localization, and overall enzymatic efficiency. Acetylation at lysine 609 affects eNOS interaction with heat shock protein 90 (Hsp90) and CaM, both essential for eNOS activation (Taubert et al., 2004). Lysine 609 acetylation is mediated by histone deacetylase 3 and inhibits eNOS activity by preventing proper electron transfer (Jung et al., 2010). In contrast, its deacetylation by sirtuin 1 (SIRT1) restores eNOS activity, enhancing eNOS-CaM interaction (Donato et al., 2011; Arunachalam et al., 2010).
S-nitrosylation is a reversible modification that constrains NO synthesis via a product feedback mechanism (Lipton et al., 1993; Erwin et al., 2005; Lima et al., 2010). S-nitrosylation involves the covalent attachment of a NO group to cysteine thiols, specifically Cys94 and Cys99 of eNOS, forming S-nitrosothiols (SNOs) (Erwin et al., 2006). Cys94 and Cys99 are located within the zinc tetrathiolate cluster (Erwin et al., 2005) that is important for the eNOS dimer interface; nevertheless, mutation of these sites does not disrupt dimer formation (Erwin et al., 2005). Paradoxically, agonist stimulation, which increases NO production, also promotes rapid denitrosylation of eNOS, in a similar timeframe as phosphorylation at Ser1177 (Erwin et al., 2005). S-nitrosylated eNOS exhibits reduced catalytic activity, which can be reversed with the release of NO. The subcellular localization of eNOS influences the degree of S-nitrosylation, with membrane-bound eNOS being more heavily nitrosylated than its cytosolic counterpart due to higher NO production at the membrane (Erwin et al., 2006).
Glutathionylation is a reversible post-translational modification in which the tripeptide glutathione attaches to cysteine residues in eNOS, notably Cys689 and Cys908, in the reductase domain (Chen et al., 2010). Glutathionylation is promoted by oxidative stress and results in decreased NO production and increased superoxide generation due to disrupted flavin-dependent electron transport (Chen et al., 2010; Crabtree et al., 2013). Fortunately, this modification is reversible through the action of glutaredoxin (Grx1), which interacts closely with eNOS (Chen et al., 2013). Loss of Grx1, either by oxidative stress or genetic silencing, increases eNOS glutathionylation and further NO synthesis (Chen et al., 2013).
Palmitoylation and myristoylation are lipid modifications that regulate the localization and activity of eNOS. Myristoylation, the irreversible attachment of myristic acid to Gly2, anchors eNOS to membranes such as the plasma membrane and Golgi apparatus (Liu et al., 1995; Sessa et al., 1995). Myristoylation is a prerequisite for palmitoylation, a reversible process where palmitic acid binds to Cys15 and Cys26, anchoring eNOS within plasmalemmal caveolae (Fernandez-Hernando et al., 2006). The reversible cycle of palmitoylation and depalmitoylation allows eNOS to dynamically shift between membrane locations in response to physiological signals (Yeh et al., 1999).
eNOS activity is intricately regulated through its interactions with various binding partners. These interactions play essential roles in modulating its localization, dimerization, and activation, ensuring that eNOS responds appropriately to cellular and environmental signals (Figure 4).
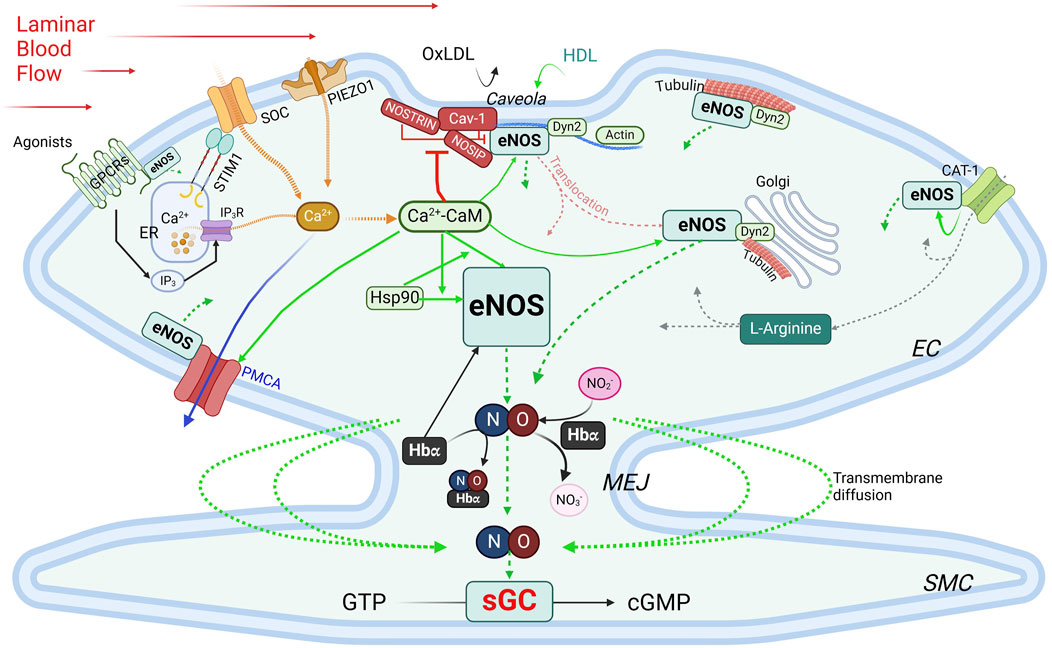
Figure 4. Regulation of eNOS by PPIs. Green boxes, stimulatory interacting partners; red boxes, inhibitory interacting partners. Green arrows indicate stimulatory interactions; dotted green arrows, NO production/diffusion; yellow dotted arrows, increase of intracellular Ca2+; solid blue arrow, Ca2+ extrusion; grey dotted arrows, L-Arginine transportation. See text for details. CAT-1, cationic anion transporter 1; Cav-1, caveolin-1; Ca2+-CaM, calcium-bound calmodulin; Dyn2, dynamin-2; EC, endothelial cells; ER, endoplasmic reticulum; GPCR, G protein-coupled receptor; Hbα, α subunit of hemoglobin; HDL, high-density lipoprotein; Hsp90, heat shock protein 90; IP3, inositol trisphosphate; IP3R, inositol trisphosphate receptor; MEJ, myoendothelial junctions; NOSIP, eNOS-interacting protein; NOSTRIN, eNOS trafficking inducer; ox-LDL, oxidized low-density lipoprotein; PMCA, plasma membrane Ca2+-ATPase; SMC, smooth muscle cell; SOC, store-operated Ca2+ channel; STIM1, stromal interaction protein 1.
Caveolin-1 (Cav-1) is the main caveolin of caveolae in endothelial cells (Feron et al., 1996; Garcia-Cardena et al., 1996; Ju et al., 1997). Cav-1 binds to eNOS at the caveolin-scaffolding domain (CSD, a.a. 81–101), preventing eNOS interaction with activators, thus inhibiting NO production under basal conditions (Garcia-Cardena et al., 1997; Michel et al., 1997). Physiological stimuli such as shear stress or G protein-coupled receptor (GPCR) agonists like bradykinin promote Ca2+ entry, leading to dissociation of the Cav-1-eNOS complex and allowing eNOS to be activated via phosphorylation by kinases such as Akt (Feron et al., 1998). Cav-1 deletion enhances endothelium-dependent relaxation and lowers blood pressure (Murata et al., 2007; Razani et al., 2001). Interaction with Cav-1, however, avoids excessive or aberrant eNOS activity, and Cav-1 deficiency can cause pulmonary hypertension and cardiomyopathy (Drab et al., 2001; Zhao et al., 2002). While plasma membrane localization is ideal for eNOS activation, this also exposes eNOS to external factors like ox-LDL and high-density lipoprotein (HDL). Ox-LDL can disrupt the cholesterol-rich environment in caveolae, thereby reducing NO production specifically from plasma membrane-bound eNOS, whereas Golgi-localized eNOS is more resistant (Zhang et al., 2006; Blair et al., 1999). HDL, on the other hand, provides cholesterol esters to maintain caveolae’s cholesterol, and via palmitoylation, retain eNOS at the plasma membrane (Uittenbogaard et al., 2000).
Ca2+-calmodulin eNOS activity is regulated by a complex and tightly regulated network of functionally and physically interacting proteins involved in Ca2+ signaling, including Ca2+ entry, the Ca2+ sensor CaM, and Ca2+ efflux. CaM, in its Ca2⁺-bound form (Ca2+-CaM), is required for activating eNOS by facilitating electron flow from the reductase domain to the oxygenase domain and promoting dimerization of the latter (Forstermann et al., 1991; Busse and Mulsch, 1990; Hellermann and Solomonson, 1997). The Ca2+ entry mechanisms in ECs play important roles in this process, as evidenced by the observations that removal of extracellular Ca2+ or inhibition of CaM suppresses agonist-induced NO production (Forstermann et al., 1991; Busse and Mulsch, 1990; Singer and Peach, 1982). Two major Ca2+ entry pathways are important for NO production: store-operated Ca2+ entry (SOCE), the main agonist-induced Ca2+ entry mechanism in ECs (Abdullaev et al., 2008; Tran, 2020), and mechanosensitive Ca2+ entry, stimulated by blood shear stress. For activation of SOCE, the stromal interaction molecule 1 (STIM1) is required (Roos et al., 2005; Soboloff et al., 2012). Vascular STIM1 plays opposing roles in the regulation of vascular tone; smooth muscle cell STIM1 is important for VSMC contractility, proliferation and the development of hypertension (Kassan et al., 2016). On the other hand, endothelial STIM1 plays a critical role in the activation of eNOS to produce NO, such that EC-specific deletion of the STIM1 gene impairs endothelium-dependent vasorelaxation and increases blood pressure (Nishimoto et al., 2018). Shear stress, a potent physiological stimulus of NO production, stimulates PIEZO1 mechanosensitive channel for Ca2+ entry (Wang et al., 2016; Ranade et al., 2014; Li J. et al., 2014). Once bound to Ca2+, CaM regulates eNOS activity via two important mechanisms. First, the Ca2+-CaM complex displaces eNOS from the inhibitory interaction with caveolin (Michel et al., 1997). Second, CaM binds eNOS at a canonical CaM-binding site encompassing amino acids 493–512 (Venema et al., 1996) with a Kd value of ∼0.2 nM (Tran et al., 2005). This interaction is inhibited at low Ca2+ concentration by the autoinhibitory domain (residues 595–639) (Chen and Wu, 2000). Upon increases in intracellular Ca2+, Ca2+-bound CaM binds eNOS, displacing the autoinhibitory loop and facilitating electron transfer between the two domains (Pollock et al., 1991; Nishida and Ortiz de Montellano, 1999). Phosphorylation at Ser615 within this loop reduces the concentration of Ca2+ required for eNOS-CaM interaction, alleviating the autoinhibitory effect (Tran et al., 2008). CaM binding also enhances eNOS phosphorylation at Ser1177, which, in combination with Ser615 phosphorylation, further increases the Ca2+ sensitivity of eNOS-CaM interaction and synthase activation (Fleming et al., 2001; Tran et al., 2009; Tsukahara et al., 1994; Fleming et al., 1997). These phosphorylation events facilitate significant eNOS-CaM interaction and synthase activity at basal level of intracellular Ca2+ and explain the effects of factors that promote NO production without triggering significant increases in global cytoplasmic Ca2+. Interaction with Ca2+ efflux channel – The plasma membrane Ca2+-ATPase (PMCA) is a key Ca2+ extrusion mechanism in ECs (Wang et al., 2002; Tran et al., 2003). eNOS directly interacts via residues 735 – 934 in its reductase domain with residues 428–651 in the catalytic domain of PMCA (Holton et al., 2010). This interaction enhances phosphorylation of Thr495 in the CaM-binding domain of eNOS (Holton et al., 2010), which reduces eNOS-CaM interaction (Fleming et al., 2001). The Ca2+/CaM-dependent phosphatase calcineurin is associated with the PMCA-eNOS complex, suggesting a potential role in the effect of PMCA expression on Thr495 phosphorylation status (Holton et al., 2010). Interestingly, PMCA activity is controlled by CaM interaction (Di Leva et al., 2008) and Ca2+ extrusion via PMCA moderates eNOS-CaM binding and synthase activation (Tran et al., 2016). Thus, CaM binding fine-tunes NO synthesis in response to subtle changes in the Ca2+ concentration surrounding eNOS and by control the activities of many of its interacting partners. With sub-nanomolar affinity for its interaction with CaM and limited abundance of CaM in ECs, eNOS expression level and activation in turn significantly influences the CaM-binding proteome in ECs (Tran et al., 2005; Tran et al., 2003). Treatment with 17β-estradiol or an agonist of the G protein-coupled estrogen receptor enhances CaM expression level substantially in ECs and promotes eNOS activity by moderating Ca2+ entry and efflux, enhancing eNOS-CaM interaction and associated eNOS phosphorylation (Tran, 2020; Tran et al., 2016; Fredette et al., 2017; Tran et al., 2015; Terry et al., 2017).
Interactions with components of the cytoskeleton and membrane-targeted proteins Actin interacts with an eight-amino acid motif (Jo et al., 2011; Higashi et al., 2002; Maier et al., 2000; Setoguchi et al., 2001; Fukuda et al., 2002; Holowatz and Kenney, 2011; Nystrom et al., 2004; Worthley et al., 2007) in the oxygenase domain of eNOS (Kondrikov et al., 2010). In in vitro assays, G-actin promotes eNOS activity more than does F actin (Su et al., 2003); however, a low G/F actin ratio appears to correlate with higher eNOS expression level (Searles et al., 2004). Dynamin-2 (Dyn2) is a GTP-binding protein in the caveolae and the Golgi (McClure and Robinson, 1996). Dynamin-2 interacts directly with eNOS in these compartments in ECs; this interaction is enhanced by Ca2+ and increases eNOS activity (Cao et al., 2001). NOSTRIN (eNOS trafficking inducer), a 506-a.a. protein enriched in vascular tissues, interacts with eNOS via an SH3 domain, promotes eNOS redistribution from the membrane, and inhibits synthase activity (Zimmermann et al., 2002). It also interacts with a region spanning a.a. 1- 61 of cav-1, N-terminally from the cav-1 scaffolding domain, thus forming a ternary complex with eNOS and cav-1 (Schilling et al., 2006). NOSTRIN interacts with dynamin-2 and is required for recruitment of eNOS to dynamin-containing structures (Icking et al., 2005). NOSIP (eNOS-interacting protein) is another protein residing in caveolae that interacts with eNOS and promotes its translocation from the plasma membrane and inhibit NO production (Dedio et al., 2001). In addition, eNOS associates with tubulin (Dedio et al., 2001), which plays an important role in its trafficking to the Golgi. Acetylation of α tubulin is involved in stabilizing microtubules where eNOS is associated in the Golgi and is phosphorylated for basal activity (Giustiniani et al., 2009). Interaction with GPCRs – Evidence from in vitro studies indicates that eNOS can interact with the juxtamembranous regions of AT1R, ETA, ETB and bradykinin B2 receptor (Marrero et al., 1999). While these interactions are likely important because activation of these receptors increases intracellular Ca2+, which is predicted to activate eNOS, they require further verification in vivo. Activity of eNOS is also regulated by its direct association with the cationic amino acid transporter (CAT)-1, the key transporter of L-arginine (Li et al., 2005). The promotion of eNOS activity by its interaction with CAT-1 is independent of L-arginine transport and is associated with enhanced phosphorylation at Ser1177 and Ser633 and reduced interaction with cav-1 (Li et al., 2005).
Heat shock protein 90 (Hsp90) is a molecular chaperone that stabilizes eNOS, thereby promoting its activation. The substrate-binding region of Hsp90 binds to the oxygenase domain of eNOS between a.a. 310–323 (Fontana et al., 2002; Xu H. et al., 2007). This interaction is increased by stimuli such as VEGF, histamine, estrogen, and shear stress (Garcia-Cardena et al., 1998; Russell et al., 2000; Venema et al., 1997). Hsp90 binding enhances phosphorylation at Ser1177 by recruiting kinases such as Akt, which further boosts NO production (Fontana et al., 2002). Hsp90 is critical for eNOS activity, and is involved in a reciprocal interactive relationship with eNOS and CaM: Hsp90 enhances both the magnitude and sensitivity of eNOS activation in response to Ca2+-CaM; in turn, the effect of Hsp90 to promote eNOS activity is enhanced by Ca2+-CaM complex (Takahashi and Mendelsohn, 2003). Hsp90 also exerts a CaM-independent effect to promote eNOS activity (Takahashi and Mendelsohn, 2003).
Hemoglobin alpha (Hbα) The α, but not β, subunit of hemoglobin is expressed in ECs in resistance blood vessels, where it is concentrated in the myoendothelial junctions (MEJs) between ECs and VSMCs (Straub et al., 2012). Hbα regulates NO availability via three mechanisms. First, it can bind and release NO and thus can act as both a reservoir and scavenger of NO, thereby regulating NO-mediated processes (Straub et al., 2014a; Keller et al., 2022; Straub et al., 2014b). In its ferrous (Fe2+) form, Hbα binds NO with high affinity, yielding nitrate and ferric (Fe3+) Hbα, which binds NO with much lower affinity (Straub et al., 2012). The reduction of Fe3+ Hbα to Fe2+ Hbα, catalyzed by cytochrome b5 reductase 3 (CytB5R3), thus enhances the scavenging action of NO by Hbα (Straub et al., 2012). Second, Hbα associates at its residues 34–43 with the oxygenase domain of eNOS in a macromolecular complex (Straub et al., 2014a), an interaction dictated by amino acids 36SFPT39 in the Hbα sequence (Keller et al., 2022). However, excessive NO scavenging in conditions of elevated Hb concentrations in the MEJs can reduce NO bioavailability and induce endothelial dysfunction (Denton et al., 2021), increasing vascular resistance (Straub et al., 2014a). Third, endothelial Hbα can function as a nitrite reductase, which produces NO through reduction of nitrite in hypoxic condition (Keller et al., 2022). These dynamic interactions help ensure that NO is available when needed, particularly in response to conditions like hypoxia and exercise, where vasodilation and increased tissue oxygen delivery are critical (Kim-Shapiro et al., 2005; Umbrello et al., 2013). In this context, it is worth noting that myoglobin is expressed in VSMCs and contributes to nitrite-dependent generation of NO in response to hypoxia independently of eNOS or iNOS (Totzeck et al., 2012).
The availability of substrates, co-factors, and the efficiency of product recycling are critical factors that dictate the outcome of eNOS activation. Disruptions in substrate availability, co-factor balance, or recycling processes—due to oxidative stress, metabolic disorders, or nutrient deficiencies—can impair eNOS activity and reduce NO bioavailability (Figure 5).
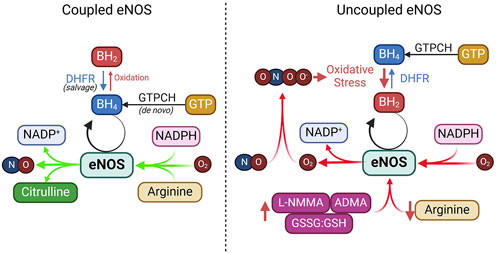
Figure 5. Coupled vs. uncoupled eNOS. See text for details. ADMA, asymmetric dimethylarginine; DHFR, dihydrofolate reductase; GTPCH, GTP cyclohydrolase I; GSSG, glutathione disulfide; GSH, glutathione; L-NMMA, NG-Monomethyl-L-arginine; NADPH, nicotinamide adenine dinucleotide phosphate.
L-arginine is the primary substrate for eNOS and is converted into NO and L-citrulline during the enzymatic reaction. Given its high affinity for eNOS (Km ∼2–3 μM) and an intracellular concentration above 100 μM, L-arginine is considered abundant in endothelial cells (Bode-Boger et al., 2007; Hardy and May, 2002). Nevertheless, extracellular L-arginine at millimolar concentrations can stimulate NO production (McDonald et al., 1997a; McDonald et al., 1997b). This has been considered the “arginine paradox.” Several mechanisms have been proposed to explain this phenomenon. One hypothesis involves the close association of eNOS with arginine transporters like CAT-1 and L-arginine recycling enzymes such as argininosuccinate lyase (ASL) (Erez et al., 2011). Other factors like competition from other amino acids, reduced uptake by cationic amino acid transporters (CATs), and elevated arginase activity—common in conditions such as hypertension and diabetes—can limit L-arginine availability to eNOS, diverting it away from NO production (McDonald et al., 1997a; McDonald et al., 1997b). Additionally, asymmetric dimethylarginine (ADMA), an endogenous inhibitor of eNOS, competes with L-arginine for binding, reducing NO synthesis (Boger et al., 1998a; Boger et al., 1998b). In addition to reducing NO synthesis, ADMA removes the suppression by NO of T-type voltage-dependent Ca2+ channels in VSMCs (Smith et al., 2020), triggering depolarizing Ca2+ spikes in VSMCs leading to vasoconstriction (Ng et al., 2024).
Tetrahydrobiopterin (BH4) is an essential cofactor for eNOS required for efficient electron transfer during the synthase’s catalytic cycle (Alp and Channon, 2004; Crabtree et al., 2009a; Bendall et al., 2014). BH4 keeps eNOS in a “coupled” state, in which it synthesizes NO rather than harmful superoxide (Alp and Channon, 2004; Crabtree et al., 2009b). BH4 can be produced through two pathways: de novo synthesis, regulated by the rate-limiting enzyme guanosine triphosphate cyclohydrolase I (GTPCH), or through the salvage pathway, where dihydrobiopterin (BH2) is recycled back to BH4 by dihydrofolate reductase (DHFR) (Crabtree et al., 2009b; Crabtree and Channon, 2011). Under oxidative stress, BH4 is rapidly oxidized into BH2 by superoxide anions or peroxynitrite, which is particularly strong during NO scavenging (Milstien and Katusic, 1999). This oxidative depletion of BH4 causes uncoupling of eNOS, where it produces superoxide instead of NO.
NADPH is a critical electron donor in eNOS and facilitates electron transfer through FAD and FMN for effective NO production. It also maintains the redox state of BH4, ensuring efficient NO synthesis (Reyes et al., 2015). Depletion of NADPH, as seen in glucose-6-phosphate dehydrogenase deficiency or due to excessive consumption by NADPH oxidases, leads to eNOS uncoupling and oxidative stress (Noreng et al., 2022). Additionally, activation of CD38 in post-ischemic heart injury can severely deplete NADPH, impairing eNOS function and disrupting NADPH-dependent BH4 salvage and synthesis (Reyes et al., 2015).
FAD and FMN, both derived from riboflavin (vitamin B2), are essential co-factors for eNOS’s electron transfer process. These flavins facilitate the flow of electrons from NADPH to the oxygenase domain, ensuring efficient NO production (Figure 1). Deficiency in FAD and FMN due to poor dietary intake or metabolic disorders can impair eNOS activity, resulting in reduced NO synthesis and increased oxidative stress.
Under certain conditions, eNOS can become “uncoupled,” shifting from producing NO to generating superoxide (O2⋅⁻), which not only reduces NO availability but also increases oxidative stress, reducing endothelial dysfunction. This section briefly explores the main causes of eNOS uncoupling and their therapeutic implications (Figure 5).
Suboptimal levels of BH4, or a decreased BH4/BH2 ratio, are significant contributors to eNOS uncoupling observed in hypertension, diabetes, and atherosclerosis (Vasquez-Vivar et al., 1998; Wever et al., 1997; Soltis and Cassis, 1991; Hong et al., 2001; Landmesser et al., 2003; Lee et al., 2009). Studies in animal models, such as hypertensive mice and rats and apolipoprotein E-deficient mice, have demonstrated that oxidative depletion of BH4 leads to increased eNOS-derived superoxide, which impairs vasorelaxation (Hong et al., 2001; Landmesser et al., 2003; Laursen et al., 2001). In humans, decreased BH4 levels and eNOS uncoupling have been linked to coronary artery disease, diabetes, and hypertension (Antoniades et al., 2007; Ismaeel et al., 2020; Heitzer et al., 2000b; Stroes et al., 1997). This imbalance is further exacerbated by oxidative stress, which directly oxidizes BH4 to BH2, reducing BH4 availability for NO synthesis (Vasquez-Vivar et al., 1998; Laursen et al., 2001; Guzik et al., 2002; Alp et al., 2003; Bendall et al., 2005). Additionally, reduced expression of GTP-cyclohydrolase 1, the rate-limiting enzyme in BH4 synthesis, and dihydrofolate reductase, the enzyme that recycles BH2 to BH4, further contributes to eNOS uncoupling in diabetes and hypercholesterolemia (Alp and Channon, 2004; Xu et al., 2009; Xu J. et al., 2007; Whitsett et al., 2007; Munzel and Daiber, 2017; Chuaiphichai et al., 2017).
L-arginine depletion is a significant contributor to eNOS uncoupling. Although intracellular L-arginine concentration typically far exceeds its Km for eNOS, obesity, diabetes, and cardiovascular diseases can cause L-arginine deficiency. In such cases, reduced availability of L-arginine limits its ability to act as a substrate for eNOS, favoring the production of ROS over that of NO (Bode-Boger et al., 2007; Yang and Ming, 2013). A key mechanism of L-arginine depletion is upregulation of arginase, an enzyme that competes with eNOS for L-arginine and converts it into urea and L-ornithine. Arginase induction is particularly prevalent in obesity, diabetes, and atherosclerosis (Lass et al., 2002). For example, oxidized LDL significantly increases ARG2 expression levels in ECs (∼20%) but increases its activity by ∼80% (Ryoo et al., 2006). Many other factors can upregulate the expression level and activity of arginase, such as lipopolysaccharide, TNF-α, glucose, thrombin, hypoxia and angiotensin II [reviewed in Pernow and Jung (2013)]. Inhibiting arginase or supplementing L-arginine can restore NO production and improve endothelial function in these conditions (Chicoine et al., 2004). Furthermore, imbalance between L-arginine and ADMA, an endogenous inhibitor of eNOS, leading to reduced L-arginine/ADMA ratio, contributes to eNOS uncoupling (Closs et al., 2012; Watson et al., 2016; Peyton et al., 2018).
Oxidative stress plays a pivotal role in disrupting endothelial function by promoting eNOS uncoupling. Excessive ROS overwhelms the antioxidant defense system and causes oxidative stress that damages lipids, proteins, and DNA, contributing to the development of CVD (Thannickal and Fanburg, 2000; Costa et al., 2021; Sharifi-Rad et al., 2020; Li H. et al., 2013). Risk factors such as dyslipidemia, diabetes, hypertension, obesity, and smoking increase ROS levels, increasing endothelial dysfunction and CVD progression (Cai and Harrison, 2000; Xiang et al., 2021; Jurcau and Ardelean, 2022). ROS also play a key role in ischemia-reperfusion injury (Xiang et al., 2021; Jurcau and Ardelean, 2022). In the vascular wall, enzymes such as NADPH oxidase, xanthine oxidase, and uncoupled eNOS generate ROS, particularly superoxide. Superoxide reacts with NO to form peroxynitrite (ONOO−), a toxic compound that depletes NO and worsens endothelial dysfunction (Li H. et al., 2014; Thomson et al., 1995; Victor et al., 2009). NADPH oxidases, particularly NOX2, are major sources of superoxide in diabetes, hypertension, smoking, and aging (Konior et al., 2014; Gray et al., 2013; Manea et al., 2018; Fukui et al., 1997; Marchi et al., 2016; Kim et al., 2014; Jiang et al., 2011). The oxidative depletion of BH4 by ROS, especially via NADPH oxidases, further exacerbates eNOS uncoupling (Konior et al., 2014; Zhang et al., 2020).
S-glutathionylation at Cys689 and Cys908 in the reductase domain results in eNOS uncoupling (Figure 3) (Chen et al., 2010; Chen et al., 2013; Suvorava et al., 2015; Zweier et al., 2011). This modification typically occurs under oxidative stress, when the ratio of reduced glutathione (GSH) to oxidized glutathione (GSSG) decreases (Suvorava et al., 2017; Benson et al., 2013; Daiber et al., 2002). S-glutathionylation disrupts the alignment of FAD and FMN, essential cofactors for eNOS’s electron transport, resulting in superoxide production instead of NO (Chen et al., 2010; Zweier et al., 2011). This mechanism of eNOS uncoupling is unique in that it occurs in the reductase domain, unlike other uncoupling mechanisms that occur in the oxygenase domain, and can be inhibited by L-NAME (Chen et al., 2010). In vivo studies have confirmed the association of eNOS S-glutathionylation with endothelial dysfunction in hypertension, aging, and cardiovascular diseases (Chen et al., 2010; Suvorava et al., 2015). In ex vivo rat aortic segments and spontaneously hypertensive rats, high levels of eNOS S-glutathionylation correspond with impaired vasodilation. S-glutathionylation can be reversed by Grx-1 (Chen et al., 2013; Shang et al., 2017).
The use of exogenous NO donors is limited by the short-lived nature of their effects and the development of nitrate tolerance. Enhancing endogenous NO production should provide physiological and more sustained effects. Current strategies involve promoting eNOS activity by providing essential substrates or cofactors or by pharmacologically upregulating eNOS expression and activity; this could also be done by preventing/reversing eNOS uncoupling. We will divide our review below of these approaches based on the level of available evidence.
L-arginine supplementation to promote NO production is based on the premise that in conditions of endothelial dysfunction, intracellular L-arginine levels may become suboptimal. It is also based on the “arginine paradox,” discussed above, that despite an intracellular L-arginine concentration much higher than its Km for eNOS activity, high extracellular concentration of L-arginine can still promote NO production. Additionally, L-arginine also competes with ADMA, an endogenous eNOS inhibitor (Boger et al., 1998c), thereby restoring the L-arginine/ADMA ratio that is critical for eNOS activity (Dong et al., 2011) (Figure 6).
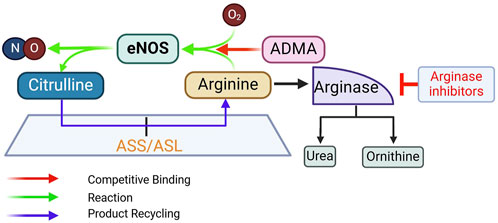
Figure 6. Rationale for supplementing L-arginine, inhibiting arginase, and supplementing L-citrulline to promote eNOS activity. ADMA, asymmetric dimethylarginine; ASL, argininosuccinate lyase; ASS, argininosuccinate synthase. See text for more details.
Early preclinical studies provided evidence that oral L-arginine supplementation improves acetylcholine (ACh)-induced vasorelaxation and NO production (Girerd et al., 1990; Cooke et al., 1991; Rossitch et al., 1991; Boger et al., 1995). Clinical studies began with intravascular injection of L-arginine in small groups of subjects. In hypercholesterolemic patients (n = 8, mean age 51.5) with slight luminal irregularities of the left anterior descending coronary artery (LAD), ACh-induced reduction in coronary blood flow, an indication of endothelial dysfunction, is improved by intracoronary injection of L-arginine (Drexler et al., 1991). In patients with diffuse atherosclerotic LADs (n = 13, age 56 ± 7.5), direct intracoronary L-arginine injection also ameliorates the ACh-induced vasoconstriction and reduction in blood flow (Dubois-Rande et al., 1992). In patients with critical peripheral limb ischemia (n = 10, age 68.3 ± 3.1), a single intravenous dose of L-arginine significantly increases blood flow, accompanied by urinary cGMP excretion (Bode-Boger et al., 1996). Overall, intravascular L-arginine delivery in humans offers acute improvement of endothelial function. In young healthy subjects (n = 80, age 25.4 ± 0.2), this effect is achieved with higher doses and is correlated with L-arginine plasma concentrations (Bode-Boger et al., 1998).
From clinical trials testing the effects of oral administration of L-arginine, a picture has emerged that short-term L-arginine administration (≤3 months) provides benefits, whereas longer supplementation (≥6 months) gives mixed results. For example, 4-week L-arginine supplementation improves reactive hyperemia in patients with hypertension and hyperhomocysteinemia (2.4 g/d, n = 25, age 40–65) (Reule et al., 2017); and flow-mediated dilation is improved by 3-month L-arginine supplementation in hypertensive subjects (2.4 g/d, n = 40, age 40–65) (Menzel et al., 2018). In patients with a history of coronary bypass surgery, 6-month supplementation with L-arginine (6.4 g/d, n = 32, age 65 ± 10) reduces ADMA levels, increases plasma cGMP, and improves reactive hyperemia compared to placebo (n = 32, age 64 ± 11) (Lucotti et al., 2009). However, 6-month supplementation with L-arginine in patients with peripheral arterial disease (3 g/d, n = 66, age 73 ± 9) does not increase NO synthesis or improve vascular reactivity vs. placebo (3 g/d, n = 67, age 72 ± 7) (Wilson et al., 2007) or alter vascular stiffness in post-myocardial infarction (MI) patients (3 × 3 g/day, n = 75, age 60.4 ± 12.9) (Schulman et al., 2006). This has led to the “arginine tolerance” hypothesis (Wilson et al., 2007), akin to the common nitrate tolerance phenomenon.
Given the state of current evidence, major cardiovascular guidelines do not include L-arginine supplementation for the prevention of CVD. Among explanations for the absence of effect of long-term L-arginine administration in some clinical trials, oxidative depletion of BH4 can result in eNOS uncoupling even with sufficient L-arginine supply (Xiong et al., 2014; Scalera et al., 2009). L-arginine also increases arginase expression, which in turn reduces L-arginine availability (Caldwell et al., 2018; Wang et al., 2006), a mechanism that may partly explain the arginine tolerance hypothesis. Nevertheless, interpretation of results of clinical trials for oral L-arginine should include whether the treatment regimens include other drugs that also promote eNOS function and whether there is baseline L-arginine deficiency. For example, in post-MI patients (n = 78, age 60.2 ± 14.2), addition of L-arginine (3 × 3 g/d) to the post-MI treatment regimen for 6 months does not improve vascular stiffness or left ventricular function compared to patients receiving placebo (n = 75, age 60.4 ± 12.9) (Schulman et al., 2006). However, in addition to the normal baseline L-arginine levels in both groups, this treatment regimen includes aspirin, which acetylates eNOS and promotes NO production (Taubert et al., 2004); clopidogrel, which stimulates eNOS phosphorylation (Schafer et al., 2011); an ACE inhibitor, which inhibits degradation of bradykinin, thereby triggering endothelial Ca2+ signals and activating eNOS (Bachetti et al., 2001); and a statin, which stabilizes eNOS mRNA and activates the PI3K/Akt pathway to activate the synthase (Wang et al., 2005). Similarly, in patients with hypertension and hyperhomocysteinemia (18 males and 7 females, age 40–65), reactive hyperemia is improved by 4 weeks oral L-arginine (Reule et al., 2017), administered in a mixture with pycnogenol, which itself activates eNOS (Fitzpatrick et al., 1998); α lipoic acid, which promotes eNOS recoupling (Sena et al., 2008); vitamin B2, which is a cofactor for eNOS; and folic acid, which activates eNOS and prevents its uncoupling (Stroes et al., 2000). The effects of co-treatment agents on eNOS may obscure or reduce the observed effect of L-arginine on endothelial function.
Arginase was identified in 1904 as the enzyme that hydrolyzes L-arginine into ornithine and urea (Kossel and Dakin, 1904). Two isoforms exist – Arg1 and Arg2. Arg1 is expressed abundantly in hepatic tissue, where it plays a major role in the urea cycle, whereas Arg2 is more predominant in extrahepatic tissues. Both isoforms are expressed in ECs (Buga et al., 1996); and many risk factors of CVD can increase the expression levels and activity of endothelial arginase (Pernow and Jung, 2013; Ryoo et al., 2008). Thus, arginase inhibition in ECs is predicted to increase L-arginine availability for NO production by eNOS (Figure 6).
Considering a high Km value for L-arginine as substrate for arginase (1–3 mM), it has been reasoned that the main function of arginase is to limit the intracellular accumulation of L-arginine; whereas given a low Km value for L-arginine as substrate of NOS (10–20 µM), arginase may not affect basal NO production, unless L-arginine level falls below 10–20 µM (Buga et al., 1996). Nevertheless, the Vmax for L-arginine of arginase is ∼1,000-fold higher than that of the NO synthases (Wu and Morris, 1998). So theoretically, arginase can compete with eNOS for L-arginine and affect NO synthesis. Indeed, treatment of ECs with L-arginine increases production of both urea and NO, while treatment with L-valine, which inhibits arginase [IC50 ∼ 6.2 ± 0.4 mM (Paik et al., 1978)], increases NO production only; consistently, combined treatment with L-arginine and L-valine shifts the balance towards NO synthesis over urea formation (Chicoine et al., 2004). Additionally, overexpression of both arginase isoforms reduces L-arginine availability (Li et al., 2001). Many arginase inhibitors with high affinity have been developed over the past several decades, such as N-hydroxy-L-arginine [NOHA, Ki = 3.6 µM for human ARG1 (Di Costanzo et al., 2010), 1.6 µM for human ARG2 (Colleluori and Ash, 2001)], N-hydroxy-nor-L-arginine [nor-NOHA, Kd = 0.47 µM for human ARG1 (Di Costanzo et al., 2010), 51 nM for human ARG2 (Colleluori and Ash, 2001)] and boronic acid derivatives like 2(S)-amino-6-hexanoic acid (ABH) [IC50 values of derivatives range 17–1,470 nM for human ARG1, 30–2,150 nM for human ARG2 (Collet et al., 2000; Ilies et al., 2011)] and S-(2-boronoethyl)-l-cysteine (BEC) [Ki = 0.4–0.6 µM (Busnel et al., 2005)]. A comprehensive review of arginase inhibitors can be found in Pudlo et al. (2017). In preclinical models, arginase inhibition prevents eNOS uncoupling and atherogenesis. In hypercholesterolemic mice, deletion of Arg2 or inhibition of arginase with BEC improves endothelial function and reduces atherosclerosis (Ryoo et al., 2008). Similarly, in diet-induced obesity models, arginase inhibition with ABH or deletion of Arg1 or Arg2 reverses vascular dysfunction (Bhatta et al., 2017; Atawia et al., 2019). In diabetic mice, the use of ABH effectively restores endothelial function (Pernow et al., 2015). Clinical studies in small groups of patients have shown effect of intravascular injection of nor-NOHA in several vascular beds in various conditions of EC dysfunction. Intra-arterial infusion of nor-NOHA (0.1 mg/min) improves endothelium-dependent vasorelaxation in radial artery following ischemia-reperfusion injury in male patients (average age 65) with coronary artery disease (CAD, n = 12) or CAD and type 2 diabetes (T2D, n = 12) (Kovamees et al., 2014). The same dose of nor-NOHA administration also improves endothelium-dependent vasorelaxation in forearm vessels in patients with heterozygous familial hypercholesterolemia (n = 12, age 32) and healthy controls (n = 12, age 30) (Kovamees et al., 2016a). Similarly, 0.1 mg/min infusion of nor-NOHA improves endothelium-dependent vasodilation in the forearm microvasculature in T2D patients (n = 12, age 66 ± 4), but not in age-matched healthy subjects (n = 12) (Kovamees et al., 2016b). Intracoronary infusion of nor-NOHA (0.1 mg/min) also enhances endothelium-dependent vasorelaxation in coronary arteries in patients (age 69 ± 3) with CAD (n = 16), or CAD and T2D (n = 16), but not in healthy subjects (n = 16) (Shemyakin et al., 2012). These effects appear to be more pronounced in aged than in younger subjects (n = 21, age 48–75) (Mahdi et al., 2019), and does not depend on efficacy of blood glucose-lowering therapy in T2D patients (n = 16, average age 64) (Mahdi et al., 2018). Nor-NOHA improves EC dysfunction in T2D patients (n = 26), but not in type 1 diabetes (T1D) patients (n = 13) with equal blood glucose levels (Tengbom et al., 2024).
Clinical trials testing the effect of oral arginase inhibitors on endothelial function are currently not available, due perhaps to the fact that most oral formulations of arginase inhibitors have poor oral bioavailability (Pudlo et al., 2017). Some clinical trials have begun to test new oral arginase inhibitors in cancer therapy, based on the high expression level of ARG1 in immunosuppressive cells. Examples include OATD-02, a recently developed ARG1/2 dual inhibitor with ∼15 nM affinity (Borek et al., 2023) and CB-1158, which has IC50 values of 86 nM and 296 nM for ARG1 and ARG2, respectively (Naing et al., 2024; Steggerda et al., 2017). However, using oral arginase inhibitors long-term to promote NO production may be challenged by the lack of isoform specificity, making it difficult to balance the roles of arginase isoforms in the urea cycle and NO production and potential disruption of nitrogen metabolism (Pudlo et al., 2017). While nor-NOHA is specific for ARG2, it is not available in oral formulations due to issues with stability and absorption with oral administration. Overall, due to the lack of clinical trials, arginase inhibitors are not currently recommended for the prevention or treatment of cardiovascular disease in clinical practice guidelines.
Oral L-arginine undergoes first-pass metabolism [∼38% in humans (Castillo et al., 1993)], whereas L-citrulline is converted into L-arginine via the argininosuccinate pathway, in which argininosuccinate synthase (ASS) catalyzes the formation of argininosuccinate from citrulline and aspartate; argininosuccinate is subsequently cleaved to form arginine and fumarate by ASL. Citrulline conversion to arginine takes place in multiple cell types, including ECs (Hecker et al., 1990; Wu and Meininger, 1993) (Figure 7). Oral L-citrulline has high intestinal absorption but minimal first-pass metabolism and renal reabsorption (Bahadoran et al., 2021), yielding more stable increases in plasma L-arginine levels. L-citrulline is also an inhibitor of arginase activity, as measured by the production of urea (Shearer et al., 1997). L-citrulline supplementation is thus a promising way to promote NO production (Yu et al., 1997; Agarwal et al., 2017; Osowska et al., 2004).
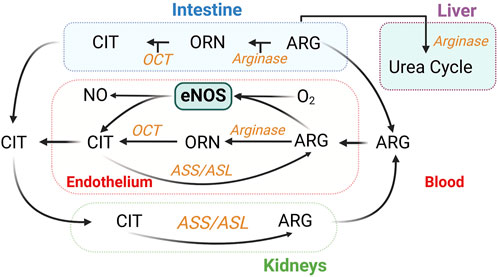
Figure 7. Inter-organ metabolism of L-citrulline (CIT) and L-arginine (ARG). Endothelium-derived NO synthesis takes place in all organs. Urea is also generated in the kidneys (Levillain, 2012). ASL, argininosuccinate lyase; ASS, argininosuccinate synthase; OCT, ornithine transcarbamylase; ORN, ornithine.
L-citrulline administration as a means to promote NO production has received significant attention recently. In cell models such as ECs exposed to hypoxia or high glucose, L-citrulline supplementation restores NO bioavailability and reduces ARG2 expression level and activity (Douglass et al., 2023; Tsuboi et al., 2018). In animal studies, L-citrulline increases NO production and improves vascular function, with positive effects on glucose tolerance, exercise capacity, and placental angiogenesis (Li et al., 2023; Eshreif et al., 2020). Several clinical studies in the past decade have examined the effects of L-citrulline administration in small cohorts with endothelial dysfunction. In patients with vasospastic angina (n = 22, age 41–64), L-citrulline supplementation (0.8 g/d, 4–8 weeks) increases brachial artery flow-mediated dilation, plasma NOx levels, and L-arginine/ADMA ratio (Morita et al., 2013). In T2D patients (n = 25, age 25–65), consumption of 1 g L-citrulline twice a day for 4 weeks reduces the T2D-induced increase in arginase activity and increases plasma nitrites, effects corroborated by improvement of ACh-induced relaxation in ex vivo mouse aortic rings incubated (24-h) in low or high glucose-containing media and increases in NO production in ECs treated with high glucose (Shatanawi et al., 2020). In hypertensive postmenopausal women (n = 14, age 61 ± 6), high-dose L-citrulline (10 g/d, 4 weeks) improves flow-mediated dilatation and aortic stiffness and reduces blood pressure (Maharaj et al., 2022). Most recently, in hypertensive postmenopausal women (n = 14, age 60 ± 1), L-citrulline (10 g/d, 4 weeks) reduces the increases in systolic blood pressure and pulse pressure induced by isometric handgrip exercise and the pressures of forward and backward aortic waves during postexercise muscle ischemia (Dillon et al., 2024).
Positive effects have been demonstrated of drastically different daily doses of L-citrulline in recent studies, from 0.8 g/d (Morita et al., 2013) to 10 g/d (Maharaj et al., 2022; Dillon et al., 2024). While side effects have not been reported, L-citrulline exerts a dose-dependent effect to activate arginase activity in a cell-based study (Douglass et al., 2023). This may limit its own effects via reduction in L-arginine availability. In addition, high doses of L-citrulline might also affect nitrogen metabolism, based on its complicated interorgan metabolism (Breuillard et al., 2015) (Figure 7). While there are promising results, more large-scale, long-term clinical trials are needed to establish the role of L-citrulline in the prevention or treatment of CVD.
Folic acid cannot be synthesized by the body and can only come from diet or supplementation. In addition to important role in the folate cycle that is involved in the reduction of BH2 to BH4, it is an important component in homocysteine metabolism via the methionine cycle (Blom and Smulders, 2011) (Figure 8), and is critical for neural tube development. Increases in plasma homocysteine concentration is strongly associated with cardiovascular disease, and supplementation of folic acid can lower plasma homocysteine levels (Kaye et al., 2020). Folic acid supplementation appears to have a variety of beneficial cardiovascular effects. These include reducing oxidative stress and inflammation, improving plasma lipid profile, and control of blood pressure and blood glucose levels (Asbaghi et al., 2021a; Asbaghi et al., 2021b; Asbaghi et al., 2022; Asbaghi et al., 2021c; Asbaghi et al., 2023).
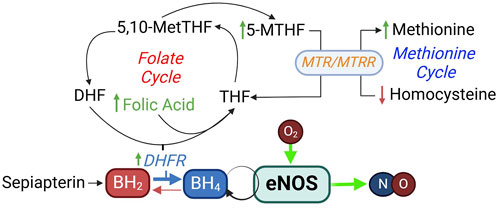
Figure 8. Folic acid in BH4 synthesis and homocysteine metabolism. DHF, dihydrofolate; DHFR, dihydrofolate reductase; MTHF, methylhydrofolate; MTR, methionine synthase; MTRR, methionine synthase reductase.
Studies in cells and animals have supported a direct role of folic acid to enhance NO availability via effects on eNOS and DHFR expression and activity (Stroes et al., 2000; Gao et al., 2009). In regard to eNOS function, 5-methyltetrahydrofolate (MTHF), the active form of folic acid, does not have an effect on pterin-free eNOS, but directly interferes with BH4-repleted, partially uncoupled eNOS, shifting it from superoxide production toward NO production (Stroes et al., 2000). Folic acid enhances DHFR expression and activity, thereby increasing BH4 and NO availability and reducing oxidant production in ECs and in mice (Gao et al., 2009). Some small-scale clinical trials (sample sizes ∼16–50 participants) have indicated improvement of endothelial function with folic acid supplementation. A recent meta-analysis of 21 randomized controlled trials on the effect of folic acid supplementation on endothelial function suggests that this intervention significantly improves flow-mediated dilation percentage (FMD%) and flow-mediated dilation, but not end-diastolic diameter (EDD) or intracellular adhesion molecule 1 (ICAM-1) expression (Zamani et al., 2023).
Due to its well-established role in neural tube development, folic acid supplementation is recommended in clinical guidelines (US Preventive Services Task Force, American College of Obstetricians and Gynecologists, American Academy of Family Physicians, American Academy of Pediatrics, and the Center for Disease Control and Prevention) for those who are planning to or could become pregnant (Force et al., 2023). However, given the lack of large-scale trials, folic acid supplementation is currently not recommended for cardiovascular disease prevention or treatment, although it may be considered in specific populations, such as those with elevated homocysteine levels (Kaye et al., 2020) or in regions with low dietary folate intake.
As reviewed above, BH4 deficiency is a common cause of eNOS uncoupling in many cardiovascular diseases (Figure 5) (Vasquez-Vivar et al., 1998; Wever et al., 1997; Soltis and Cassis, 1991; Hong et al., 2001; Landmesser et al., 2003; Lee et al., 2009). Sapropterin is a synthetic formulation of BH4. Sepiapterin is a stable precursor in the salvage pathway of BH4 biosynthesis, which converts sepiapterin to BH4 via the activities of sepiapterin reductase and DHFR (Figure 8). It has higher cell permeability than BH4 (Sawabe et al., 2008).
Many preclinical studies have examined effects of BH4 or sepiapterin administration on NO-dependent functions in the cardiovascular system. For example, BH4 administration following transverse aortic constriction (TAC) for 5 weeks in mice recouples eNOS and reduces oxidative stress, reverses cardiac hypertrophy and fibrosis, and improves cardiac function (Moens et al., 2008). Similarly, oral administration of sepiapterin for 8 weeks after TAC increases NO availability, inhibits oxidative stress, and reduces cardiomyocyte hypertrophy (Yoshioka et al., 2015). Of note, in the model of myocardial infarction, iNOS expression is upregulated, and oral sepiapterin administration inhibits nitrotyrosine formation and increases in nitrite and nitrate in wildtype, eNOS−/−, and nNOS−/− mice, but not iNOS−/− mice; these effects are associated with prevention of cardiac remodeling and dysfunction (Shimazu et al., 2011). Similar findings are observed in an MI model with streptozotocin-induced T2D (Jo et al., 2011). Multiple small-scale clinical studies (5–30 participants each) have shown BH4 supplementation can improve endothelial function in various cardiovascular conditions. Most early studies tested the effects of intravascular delivery of BH4. These studies showed that BH4 injection improves forearm blood flow in patients with hypercholesterolemia (Stroes et al., 1997), hypertension (Higashi et al., 2002) and T2D (Heitzer et al., 2000b). BH4 injection also increases coronary endothelial function in patients with CAD (Maier et al., 2000; Setoguchi et al., 2001; Fukuda et al., 2002), and increases skin blood flow responses to local heating, an indication of microvasculature EC function, in hypercholesterolemic patients (Stroes et al., 1997; Holowatz and Kenney, 2011). Nevertheless, in some studies, intraarterial infusion of BH4 fails to improve endothelial function (Nystrom et al., 2004; Worthley et al., 2007). Chronic oral supplementation of BH4 has only been examined in limited number of trials. In patients with poorly controlled hypertension (n = 16, age 57 ± 9), 400 mg/d oral BH4 for 4 weeks significantly improves brachial flow-mediated vasodilation and reduces BP (Porkert et al., 2008). In hypercholesterolemic patients (n = 22, age 60.8 ± 9.2), oral BH4 (400 mg bid) for 4 weeks normalizes ACh-induced vasodilation, vascular oxidative stress, and NO and superoxide production (Cosentino et al., 2008).
Despite evidence that BH4 and its analogs can improve endothelial function, the lack of large-scale trials and mixed outcomes of the published small-scale clinical studies have prevented their translation into current practice guidelines for CVD prevention and management (Heidenreich et al., 2022). Additionally, issues such as the oxidation of BH4 to inactive forms like BH2 limit its therapeutic potential (Cunnington et al., 2012). Of note, BH4 is also a cofactor of phenylalanine hydroxylase, which converts phenylalanine to tyrosine, a precursor to catecholamines. Large-scale trials are only available for BH4, sepiapterin or sapropterin in phenylketonuria (Muntau et al., 2024), an autosomal recessive disorder caused by deficiency of phenylalanine hydroxylase, leading to hyperphenylalaninemia, developmental delay, behavioral problems, and reduced quality of life (Blau et al., 2010).
Several compounds have been identified to enhance eNOS transcription and promote NO production. AVE3085 is a novel synthetic agent that acts as an eNOS transcription enhancer. Oral administration of AVE3085 for 7 days increases eNOS expression and activity, reverse eNOS uncoupling, reduces oxidative stress and improve endothelial function in diabetic mice (Cheang et al., 2011). Four-week administration of AVE3085 upregulates eNOS mRNA and protein levels, enhances eNOS phosphorylation and decreases nitrotyrosine formation, leading to improved endothelium-dependent vasorelaxation in hypertensive animals (Yang et al., 2011). These effects are absent in eNOS−/− mice (Cheang et al., 2011; Yang et al., 2011). AVE9488 is another eNOS transcription enhancer that has been shown to increase eNOS expression and activity, reverse eNOS uncoupling, protect against ischemia-reperfusion cardiac injury, and improve post-infarct function (Sasaki et al., 2006; Wohlfart et al., 2008; Frantz et al., 2009; Fraccarollo et al., 2008). More recently, LEENE, a long non-coding RNA induced downstream of KLF2/KLF4 has been shown to form proximity association with the eNOS promoter and enhances eNOS transcription through chromatin association (Miao et al., 2018). Atorvastatin increases LEENE expression, thereby upregulating eNOS expression, while inhibition of LEENE strongly enhances ECs-monocyte adhesion induced by pulsatile shear stress (Miao et al., 2018). No clinical trials have examined the effects of these transcription enhancers on endothelial function in humans.
Disrupting the inhibitory interaction between Cav-1 and eNOS (Figure 4) should promote eNOS activation and NO production. Cavnoxin, a 20-amino-acid peptide designed based on the caveolin scaffolding domain with alanine substitutions at key inhibitory residues (T90, T91, F92) (Bernatchez et al., 2005), releases eNOS from inhibition. In wild-type mice, cavnoxin administration significantly increases NO levels, reduces mean blood pressure, and improves vascular function, effects that are lost in eNOS−/− and Cav-1−/− mice (Bernatchez et al., 2011). Additionally, chronic cavnoxin treatment reduced atherosclerosis in ApoE-knockout mice and diabetic atherosclerotic models.
Disrupting Hbα-eNOS interaction can reduce the NO-scavenging action of Hbα (Figure 4). In addition, inhibiting CytB5R3 can increase NO availability by reducing Hb from its Fe2+ to Fe3+ form, thereby releasing NO from its high-affinity interaction. HbαX, a peptide based of the eNOS-interacting sequence of Hbα (a.a. 34–43), tagged to an HIV TAT cell-penetration sequence, has been developed to disrupt the interaction between Hbα and eNOS, which increases NO availability, acutely reducing blood pressure (Straub et al., 2014a). HbαX lowers blood pressure in mice with angiotensin II-induced hypertension and blunts vasoconstrictive response to phenylephrine in isolated human vessels (Keller et al., 2016). Pharmacological inhibition or genetic deletion of CytB5R3 increases NO availability in blood vessels (Straub et al., 2012).
DHFR plays a critical role in regenerating BH4 from its oxidized form BH2 (Figure 5). Enhancing DHFR activity will help maintain or increase BH4 levels, thus preventing eNOS uncoupling and promoting NO production. Genetic knockdown of DHFR decreases BH4 levels and increases BH2 levels, uncoupling eNOS and reducing NO production (Crabtree et al., 2009b; Crabtree et al., 2011). Oxidative stress, such as that induced by angiotensin II, downregulates DHFR, exacerbating eNOS uncoupling and reducing NO availability (Chalupsky and Cai, 2005). In addition, DHFR S-nitrosylation by eNOS-derived NO stabilizes DHFR, thereby preventing its degradation and ensuring its activity in recycling BH4 (Cai et al., 2015). This highlights the interdependence between eNOS-derived NO and DHFR stability. Challenges–Due to the lack of specific DHFR activators, there are no clinical studies that examined the possibility of stimulating its activity to increase NO availability. Antimetabolic drugs such as methotrexate or fluorodeoxyuridine and hydroxyurea stimulate DHFR promoter (Eastman et al., 1991); however, their actions are non-specific.
Nearly five decades have passed since NO was identified. Since eNOS was cloned (Marsden et al., 1992), our understanding of how NO availability is regulated has increased exponentially. Nevertheless, only general measures to improve eNOS function such as healthy lifestyle and physical exercise and approaches to amplify certain downstream components of NO signaling have made it to clinical practice guidelines. Among the salient indications, inhaled NO is used for acute pulmonary vasoreactivity testing and severe hypoxia in pulmonary arterial hypertension (PAH), nitrates are used extensively in ischemic heart disease and heart failure, sGC stimulators and activators are used for PAH, and PDE5 inhibitors are used for erectile dysfunction, PAH and benign prostatic hyperplasia.
This article has reviewed the basic regulatory mechanisms of eNOS activities and relevant approaches to improve endogenous NO production that have been tested to date but not yet approved clinically. We have highlighted the rationale, current evidence, and challenges that have prevented these approaches from entering the clinical armamentarium to improve endothelial function. Among the main challenges are the lack of specificity, lack of consistent efficacy of long-term treatment, and lack of large-scale clinical data.
Given this backdrop, what can be done? Obviously, additional studies and large-scale trials are needed in many cases. The lack of consistent outcomes of L-arginine supplementation could be due to multiple mechanisms, as discussed. Could intermittent L-arginine delivery, a strategy widely applied clinically to prevent nitrate tolerance to some degree, be considered and tested to reduce a potential “arginine tolerance”? In addition, because L-arginine supplementation increases arginase activity, which limits the efficacy, now that arginase inhibitors are available with better oral availability, L-arginine supplementation could be considered in combination with arginase inhibition. In regard to arginase inhibition, development of ARG2-specific inhibitors would help limit issues associated with disruption of the urea cycle in the liver, where ARG1 is more abundant. L-citrulline as a single supplementation appears to have yielded superior outcomes than L-arginine supplementation; however, large-scale trials are needed, and identifying the minimum effective dose range for L-citrulline is necessary, considering a potential effect of high doses on nitrogen balance. For peptide-based interventions, factors such as peptide stability and efficient delivery need to be addressed before clinical application can be considered.
Biochemically, it is worth noting that for NO-induced vasodilation, there does not need to be a high concentration of NO because of its picomolar affinity for sGC (Wu et al., 2022). Therefore, promoting NO production to a low level may preferentially promote vasodilation via the eNOS-NO-sGC pathway and prevent the formation of reactive nitrogen species. In addition, targeting interventions specifically to the endothelium is an ideal solution that is difficult to achieve but would reduce the required effective doses and limit many off-target effects. This would be particularly helpful for approaches aimed to target eNOS’s essential interacting partners and numerous transcriptional factors that are abundant also in other tissues.
MG: Writing–original draft, Writing–review and editing. SC: Writing–review and editing. EW: Writing–review and editing. DC: Writing–review and editing. Q-KT: Conceptualization, Funding acquisition, Writing–original draft, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by National Heart, Lung, and Blood Institute grant HL173818 (Q-KT), National Institute on Alcohol Abuse and Alcoholism grant AA031063 (DC), and Iowa Osteopathic and Educational Research grant IOER122204 (Q-KT). MG is supported by a scholarship from the PhD Program in Biomedical Sciences at Des Moines University.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdullaev I. F., Bisaillon J. M., Potier M., Gonzalez J. C., Motiani R. K., Trebak M. (2008). Stim1 and Orai1 mediate CRAC currents and store-operated calcium entry important for endothelial cell proliferation. Circ. Res. 103 (11), 1289–1299. doi:10.1161/01.RES.0000338496.95579.56
Agarwal U., Didelija I. C., Yuan Y., Wang X., Marini J. C. (2017). Supplemental citrulline is more efficient than arginine in increasing systemic arginine availability in mice. J. Nutr. 147 (4), 596–602. doi:10.3945/jn.116.240382
Alheid U., Frolich J. C., Forstermann U. (1987). Endothelium-derived relaxing factor from cultured human endothelial cells inhibits aggregation of human platelets. Thromb. Res. 47 (5), 561–571. doi:10.1016/0049-3848(87)90361-6
Alp N. J., Channon K. M. (2004). Regulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular disease. Arterioscler. Thromb. Vasc. Biol. 24 (3), 413–420. doi:10.1161/01.ATV.0000110785.96039.f6
Alp N. J., Mussa S., Khoo J., Cai S., Guzik T., Jefferson A., et al. (2003). Tetrahydrobiopterin-dependent preservation of nitric oxide-mediated endothelial function in diabetes by targeted transgenic GTP-cyclohydrolase I overexpression. J. Clin. Invest. 112 (5), 725–735. doi:10.1172/JCI17786
Antoniades C., Shirodaria C., Crabtree M., Rinze R., Alp N., Cunnington C., et al. (2007). Altered plasma versus vascular biopterins in human atherosclerosis reveal relationships between endothelial nitric oxide synthase coupling, endothelial function, and inflammation. Circulation 116 (24), 2851–2859. doi:10.1161/CIRCULATIONAHA.107.704155
Arnold W. P., Mittal C. K., Katsuki S., Murad F. (1977). Nitric oxide activates guanylate cyclase and increases guanosine 3':5'-cyclic monophosphate levels in various tissue preparations. Proc. Natl. Acad. Sci. U. S. A. 74 (8), 3203–3207. doi:10.1073/pnas.74.8.3203
Arunachalam G., Yao H., Sundar I. K., Caito S., Rahman I. (2010). SIRT1 regulates oxidant- and cigarette smoke-induced eNOS acetylation in endothelial cells: role of resveratrol. Biochem. Biophys. Res. Commun. 393 (1), 66–72. doi:10.1016/j.bbrc.2010.01.080
Asbaghi O., Ashtary-Larky D., Bagheri R., Moosavian S. P., Nazarian B., Afrisham R., et al. (2021a). Effects of folic acid supplementation on inflammatory markers: a grade-assessed systematic review and dose-response meta-analysis of randomized controlled trials. Nutrients 13 (7), 2327. doi:10.3390/nu13072327
Asbaghi O., Ashtary-Larky D., Bagheri R., Moosavian S. P., Olyaei H. P., Nazarian B., et al. (2021b). Folic acid supplementation improves glycemic control for diabetes prevention and management: a systematic review and dose-response meta-analysis of randomized controlled trials. Nutrients 13 (7), 2355. doi:10.3390/nu13072355
Asbaghi O., Ashtary-Larky D., Bagheri R., Nazarian B., Pourmirzaei Olyaei H., Rezaei Kelishadi M., et al. (2022). Beneficial effects of folic acid supplementation on lipid markers in adults: a GRADE-assessed systematic review and dose-response meta-analysis of data from 21,787 participants in 34 randomized controlled trials. Crit. Rev. Food Sci. Nutr. 62 (30), 8435–8453. doi:10.1080/10408398.2021.1928598
Asbaghi O., Ghanavati M., Ashtary-Larky D., Bagheri R., Rezaei Kelishadi M., Nazarian B., et al. (2021c). Effects of folic acid supplementation on oxidative stress markers: a systematic review and meta-analysis of randomized controlled trials. Antioxidants (Basel) 10 (6), 871. doi:10.3390/antiox10060871
Asbaghi O., Salehpour S., Rezaei Kelishadi M., Bagheri R., Ashtary-Larky D., Nazarian B., et al. (2023). Folic acid supplementation and blood pressure: a GRADE-assessed systematic review and dose-response meta-analysis of 41,633 participants. Crit. Rev. Food Sci. Nutr. 63 (13), 1846–1861. doi:10.1080/10408398.2021.1968787
Atawia R. T., Toque H. A., Meghil M. M., Benson T. W., Yiew N. K. H., Cutler C. W., et al. (2019). Role of arginase 2 in systemic metabolic activity and adipose tissue fatty acid metabolism in diet-induced obese mice. Int. J. Mol. Sci. 20 (6), 1462. doi:10.3390/ijms20061462
Atochin D. N., Wang A., Liu V. W., Critchlow J. D., Dantas A. P., Looft-Wilson R., et al. (2007). The phosphorylation state of eNOS modulates vascular reactivity and outcome of cerebral ischemia in vivo. J. Clin. Invest. 117 (7), 1961–1967. doi:10.1172/JCI29877
Bachetti T., Comini L., Pasini E., Cargnoni A., Curello S., Ferrari R. (2001). Ace-inhibition with quinapril modulates the nitric oxide pathway in normotensive rats. J. Mol. Cell Cardiol. 33 (3), 395–403. doi:10.1006/jmcc.2000.1311
Bahadoran Z., Mirmiran P., Kashfi K., Ghasemi A. (2021). Endogenous flux of nitric oxide: citrulline is preferred to Arginine. Acta Physiol. (Oxf) 231 (3), e13572. doi:10.1111/apha.13572
Balligand J. L., Feron O., Dessy C. (2009). eNOS activation by physical forces: from short-term regulation of contraction to chronic remodeling of cardiovascular tissues. Physiol. Rev. 89 (2), 481–534. doi:10.1152/physrev.00042.2007
Bauer P. M., Fulton D., Boo Y. C., Sorescu G. P., Kemp B. E., Jo H., et al. (2003). Compensatory phosphorylation and protein-protein interactions revealed by loss of function and gain of function mutants of multiple serine phosphorylation sites in endothelial nitric-oxide synthase. J. Biol. Chem. 278 (17), 14841–14849. doi:10.1074/jbc.M211926200
Bendall J. K., Alp N. J., Warrick N., Cai S., Adlam D., Rockett K., et al. (2005). Stoichiometric relationships between endothelial tetrahydrobiopterin, endothelial NO synthase (eNOS) activity, and eNOS coupling in vivo: insights from transgenic mice with endothelial-targeted GTP cyclohydrolase 1 and eNOS overexpression. Circ. Res. 97 (9), 864–871. doi:10.1161/01.RES.0000187447.03525.72
Bendall J. K., Douglas G., McNeill E., Channon K. M., Crabtree M. J. (2014). Tetrahydrobiopterin in cardiovascular health and disease. Antioxid. Redox Signal 20 (18), 3040–3077. doi:10.1089/ars.2013.5566
Benjamin N., O'Driscoll F., Dougall H., Duncan C., Smith L., Golden M., et al. (1994). Stomach NO synthesis. Nature 368 (6471), 502. doi:10.1038/368502a0
Benson M. A., Batchelor H., Chuaiphichai S., Bailey J., Zhu H., Stuehr D. J., et al. (2013). A pivotal role for tryptophan 447 in enzymatic coupling of human endothelial nitric oxide synthase (eNOS): effects on tetrahydrobiopterin-dependent catalysis and eNOS dimerization. J. Biol. Chem. 288 (41), 29836–29845. doi:10.1074/jbc.M113.493023
Bernatchez P., Sharma A., Bauer P. M., Marin E., Sessa W. C. (2011). A noninhibitory mutant of the caveolin-1 scaffolding domain enhances eNOS-derived NO synthesis and vasodilation in mice. J. Clin. Invest. 121 (9), 3747–3755. doi:10.1172/JCI44778
Bernatchez P. N., Bauer P. M., Yu J., Prendergast J. S., He P., Sessa W. C. (2005). Dissecting the molecular control of endothelial NO synthase by caveolin-1 using cell-permeable peptides. Proc. Natl. Acad. Sci. U. S. A. 102 (3), 761–766. doi:10.1073/pnas.0407224102
Bhatta A., Yao L., Xu Z., Toque H. A., Chen J., Atawia R. T., et al. (2017). Obesity-induced vascular dysfunction and arterial stiffening requires endothelial cell arginase 1. Cardiovasc Res. 113 (13), 1664–1676. doi:10.1093/cvr/cvx164
Black S. M., Fineman J. R., Steinhorn R. H., Bristow J., Soifer S. J. (1998). Increased endothelial NOS in lambs with increased pulmonary blood flow and pulmonary hypertension. Am. J. Physiol. 275 (5), H1643–H1651. doi:10.1152/ajpheart.1998.275.5.H1643
Blair A., Shaul P. W., Yuhanna I. S., Conrad P. A., Smart E. J. (1999). Oxidized low density lipoprotein displaces endothelial nitric-oxide synthase (eNOS) from plasmalemmal caveolae and impairs eNOS activation. J. Biol. Chem. 274 (45), 32512–32519. doi:10.1074/jbc.274.45.32512
Blau N., van Spronsen F. J., Levy H. L. (2010). Phenylketonuria. Lancet. 376 (9750), 1417–1427. doi:10.1016/S0140-6736(10)60961-0
Blekkenhorst L. C., Bondonno N. P., Liu A. H., Ward N. C., Prince R. L., Lewis J. R., et al. (2018). Nitrate, the oral microbiome, and cardiovascular health: a systematic literature review of human and animal studies. Am. J. Clin. Nutr. 107 (4), 504–522. doi:10.1093/ajcn/nqx046
Blom H. J., Smulders Y. (2011). Overview of homocysteine and folate metabolism. With special references to cardiovascular disease and neural tube defects. J. Inherit. Metab. Dis. 34 (1), 75–81. doi:10.1007/s10545-010-9177-4
Bode-Boger S. M., Boger R. H., Alfke H., Heinzel D., Tsikas D., Creutzig A., et al. (1996). L-arginine induces nitric oxide-dependent vasodilation in patients with critical limb ischemia. A randomized, controlled study. Circulation 93 (1), 85–90. doi:10.1161/01.cir.93.1.85
Bode-Boger S. M., Boger R. H., Galland A., Tsikas D., Frolich J. C. (1998). L-arginine-induced vasodilation in healthy humans: pharmacokinetic-pharmacodynamic relationship. Br. J. Clin. Pharmacol. 46 (5), 489–497. doi:10.1046/j.1365-2125.1998.00803.x
Bode-Boger S. M., Scalera F., Ignarro L. J. (2007). The L-arginine paradox: importance of the L-arginine/asymmetrical dimethylarginine ratio. Pharmacol. Ther. 114 (3), 295–306. doi:10.1016/j.pharmthera.2007.03.002
Boger R. H., Bode-Boger S. M., Mugge A., Kienke S., Brandes R., Dwenger A., et al. (1995). Supplementation of hypercholesterolaemic rabbits with L-arginine reduces the vascular release of superoxide anions and restores NO production. Atherosclerosis 117 (2), 273–284. doi:10.1016/0021-9150(95)05582-h
Boger R. H., Bode-Boger S. M., Phivthong-ngam L., Brandes R. P., Schwedhelm E., Mugge A., et al. (1998b). Dietary L-arginine and alpha-tocopherol reduce vascular oxidative stress and preserve endothelial function in hypercholesterolemic rabbits via different mechanisms. Atherosclerosis 141 (1), 31–43. doi:10.1016/s0021-9150(98)00145-2
Boger R. H., Bode-Boger S. M., Szuba A., Tsao P. S., Chan J. R., Tangphao O., et al. (1998c). Asymmetric dimethylarginine (ADMA): a novel risk factor for endothelial dysfunction: its role in hypercholesterolemia. Circulation 98 (18), 1842–1847. doi:10.1161/01.cir.98.18.1842
Boger R. H., Bode-Boger S. M., Thiele W., Creutzig A., Alexander K., Frolich J. C. (1998a). Restoring vascular nitric oxide formation by L-arginine improves the symptoms of intermittent claudication in patients with peripheral arterial occlusive disease. J. Am. Coll. Cardiol. 32 (5), 1336–1344. doi:10.1016/s0735-1097(98)00375-1
Bonauer A., Carmona G., Iwasaki M., Mione M., Koyanagi M., Fischer A., et al. (2009). MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science 324 (5935), 1710–1713. doi:10.1126/science.1174381
Bonderman D., Pretsch I., Steringer-Mascherbauer R., Jansa P., Rosenkranz S., Tufaro C., et al. (2014). Acute hemodynamic effects of riociguat in patients with pulmonary hypertension associated with diastolic heart failure (DILATE-1): a randomized, double-blind, placebo-controlled, single-dose study. Chest 146 (5), 1274–1285. doi:10.1378/chest.14-0106
Borek B., Nowicka J., Gzik A., Dziegielewski M., Jedrzejczak K., Brzezinska J., et al. (2023). Arginase 1/2 inhibitor OATD-02: from discovery to first-in-man setup in cancer immunotherapy. Mol. Cancer Ther. 22 (7), 807–817. doi:10.1158/1535-7163.MCT-22-0721
Bredt D. S., Hwang P. M., Glatt C. E., Lowenstein C., Reed R. R., Snyder S. H. (1991). Cloned and expressed nitric oxide synthase structurally resembles cytochrome P-450 reductase. Nature 351 (6329), 714–718. doi:10.1038/351714a0
Bredt D. S., Snyder S. H. (1990). Isolation of nitric oxide synthetase, a calmodulin-requiring enzyme. Proc. Natl. Acad. Sci. U. S. A. 87 (2), 682–685. doi:10.1073/pnas.87.2.682
Breuillard C., Cynober L., Moinard C. (2015). Citrulline and nitrogen homeostasis: an overview. Amino Acids 47 (4), 685–691. doi:10.1007/s00726-015-1932-2
Buga G. M., Singh R., Pervin S., Rogers N. E., Schmitz D. A., Jenkinson C. P., et al. (1996). Arginase activity in endothelial cells: inhibition by NG-hydroxy-L-arginine during high-output NO production. Am. J. Physiol. 271 (5 Pt 2), H1988–H1998. doi:10.1152/ajpheart.1996.271.5.H1988
Busnel O., Carreaux F., Carboni B., Pethe S., Goff S. V., Mansuy D., et al. (2005). Synthesis and evaluation of new omega-borono-alpha-amino acids as rat liver arginase inhibitors. Bioorg Med. Chem. 13 (7), 2373–2379. doi:10.1016/j.bmc.2005.01.053
Busse R., Mulsch A. (1990). Calcium-dependent nitric oxide synthesis in endothelial cytosol is mediated by calmodulin. FEBS Lett. 265 (1-2), 133–136. doi:10.1016/0014-5793(90)80902-u
Cai H., Harrison D. G. (2000). Endothelial dysfunction in cardiovascular diseases: the role of oxidant stress. Circ. Res. 87 (10), 840–844. doi:10.1161/01.res.87.10.840
Cai Z., Lu Q., Ding Y., Wang Q., Xiao L., Song P., et al. (2015). Endothelial nitric oxide synthase-derived nitric oxide prevents dihydrofolate reductase degradation via promoting S-nitrosylation. Arterioscler. Thromb. Vasc. Biol. 35 (11), 2366–2373. doi:10.1161/ATVBAHA.115.305796
Caldwell R. W., Rodriguez P. C., Toque H. A., Narayanan S. P., Caldwell R. B. (2018). Arginase: a multifaceted enzyme important in health and disease. Physiol. Rev. 98 (2), 641–665. doi:10.1152/physrev.00037.2016
Cao S., Yao J., McCabe T. J., Yao Q., Katusic Z. S., Sessa W. C., et al. (2001). Direct interaction between endothelial nitric-oxide synthase and dynamin-2. Implications for nitric-oxide synthase function. J. Biol. Chem. 276 (17), 14249–14256. doi:10.1074/jbc.M006258200
Castillo L., Chapman T. E., Yu Y. M., Ajami A., Burke J. F., Young V. R. (1993). Dietary arginine uptake by the splanchnic region in adult humans. Am. J. Physiol. 265 (4 Pt 1), E532–E539. doi:10.1152/ajpendo.1993.265.4.E532
Chalupsky K., Cai H. (2005). Endothelial dihydrofolate reductase: critical for nitric oxide bioavailability and role in angiotensin II uncoupling of endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. U. S. A. 102 (25), 9056–9061. doi:10.1073/pnas.0409594102
Cheang W. S., Wong W. T., Tian X. Y., Yang Q., Lee H. K., He G. W., et al. (2011). Endothelial nitric oxide synthase enhancer reduces oxidative stress and restores endothelial function in db/db mice. Cardiovasc Res. 92 (2), 267–275. doi:10.1093/cvr/cvr233
Chen C. A., De Pascali F., Basye A., Hemann C., Zweier J. L. (2013). Redox modulation of endothelial nitric oxide synthase by glutaredoxin-1 through reversible oxidative post-translational modification. Biochemistry 52 (38), 6712–6723. doi:10.1021/bi400404s
Chen C. A., Wang T. Y., Varadharaj S., Reyes L. A., Hemann C., Talukder M. A., et al. (2010). S-glutathionylation uncouples eNOS and regulates its cellular and vascular function. Nature 468 (7327), 1115–1118. doi:10.1038/nature09599
Chen P. F., Wu K. K. (2000). Characterization of the roles of the 594-645 region in human endothelial nitric-oxide synthase in regulating calmodulin binding and electron transfer. J. Biol. Chem. 275 (17), 13155–13163. doi:10.1074/jbc.275.17.13155
Chen Z., Zhang J., Stamler J. S. (2002). Identification of the enzymatic mechanism of nitroglycerin bioactivation. Proc. Natl. Acad. Sci. U. S. A. 99 (12), 8306–8311. doi:10.1073/pnas.122225199
Chen Z. P., Mitchelhill K. I., Michell B. J., Stapleton D., Rodriguez-Crespo I., Witters L. A., et al. (1999). AMP-activated protein kinase phosphorylation of endothelial NO synthase. FEBS Lett. 443 (3), 285–289. doi:10.1016/s0014-5793(98)01705-0
Chicoine L. G., Paffett M. L., Young T. L., Nelin L. D. (2004). Arginase inhibition increases nitric oxide production in bovine pulmonary arterial endothelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 287 (1), L60–L68. doi:10.1152/ajplung.00194.2003
Chuaiphichai S., Crabtree M. J., McNeill E., Hale A. B., Trelfa L., Channon K. M., et al. (2017). A key role for tetrahydrobiopterin-dependent endothelial NOS regulation in resistance arteries: studies in endothelial cell tetrahydrobiopterin-deficient mice. Br. J. Pharmacol. 174 (8), 657–671. doi:10.1111/bph.13728
Closs E. I., Ostad M. A., Simon A., Warnholtz A., Jabs A., Habermeier A., et al. (2012). Impairment of the extrusion transporter for asymmetric dimethyl-L-arginine: a novel mechanism underlying vasospastic angina. Biochem. Biophys. Res. Commun. 423 (2), 218–223. doi:10.1016/j.bbrc.2012.05.044
Colleluori D. M., Ash D. E. (2001). Classical and slow-binding inhibitors of human type II arginase. Biochemistry 40 (31), 9356–9362. doi:10.1021/bi010783g
Collet S., Carreau F., Boucher J. L., Pethe S., Lepoivre M., Danion-Bougot R., et al. (2000). Synthesis and evaluation of -borono- -amino acids † as active-site probes of arginase and nitric oxide synthases. J. Chem. Soc. Perkin Trans. 1 (2), 177–182. doi:10.1039/A908140B
Cooke J. P., Andon N. A., Girerd X. J., Hirsch A. T., Creager M. A. (1991). Arginine restores cholinergic relaxation of hypercholesterolemic rabbit thoracic aorta. Circulation 83 (3), 1057–1062. doi:10.1161/01.cir.83.3.1057
Cosentino F., Hurlimann D., Delli Gatti C., Chenevard R., Blau N., Alp N. J., et al. (2008). Chronic treatment with tetrahydrobiopterin reverses endothelial dysfunction and oxidative stress in hypercholesterolaemia. Heart 94 (4), 487–492. doi:10.1136/hrt.2007.122184
Costa T. J., Barros P. R., Arce C., Santos J. D., da Silva-Neto J., Egea G., et al. (2021). The homeostatic role of hydrogen peroxide, superoxide anion and nitric oxide in the vasculature. Free Radic. Biol. Med. 162, 615–635. doi:10.1016/j.freeradbiomed.2020.11.021
Crabtree M. J., Brixey R., Batchelor H., Hale A. B., Channon K. M. (2013). Integrated redox sensor and effector functions for tetrahydrobiopterin- and glutathionylation-dependent endothelial nitric-oxide synthase uncoupling. J. Biol. Chem. 288 (1), 561–569. doi:10.1074/jbc.M112.415992
Crabtree M. J., Channon K. M. (2011). Synthesis and recycling of tetrahydrobiopterin in endothelial function and vascular disease. Nitric Oxide 25 (2), 81–88. doi:10.1016/j.niox.2011.04.004
Crabtree M. J., Hale A. B., Channon K. M. (2011). Dihydrofolate reductase protects endothelial nitric oxide synthase from uncoupling in tetrahydrobiopterin deficiency. Free Radic. Biol. Med. 50 (11), 1639–1646. doi:10.1016/j.freeradbiomed.2011.03.010
Crabtree M. J., Tatham A. L., Al-Wakeel Y., Warrick N., Hale A. B., Cai S., et al. (2009a). Quantitative regulation of intracellular endothelial nitric-oxide synthase (eNOS) coupling by both tetrahydrobiopterin-eNOS stoichiometry and biopterin redox status: insights from cells with tet-regulated GTP cyclohydrolase I expression. J. Biol. Chem. 284 (2), 1136–1144. doi:10.1074/jbc.M805403200
Crabtree M. J., Tatham A. L., Hale A. B., Alp N. J., Channon K. M. (2009b). Critical role for tetrahydrobiopterin recycling by dihydrofolate reductase in regulation of endothelial nitric-oxide synthase coupling: relative importance of the de novo biopterin synthesis versus salvage pathways. J. Biol. Chem. 284 (41), 28128–28136. doi:10.1074/jbc.M109.041483
Cunnington C., Van Assche T., Shirodaria C., Kylintireas I., Lindsay A. C., Lee J. M., et al. (2012). Systemic and vascular oxidation limits the efficacy of oral tetrahydrobiopterin treatment in patients with coronary artery disease. Circulation 125 (11), 1356–1366. doi:10.1161/CIRCULATIONAHA.111.038919
Daiber A., Frein D., Namgaladze D., Ullrich V. (2002). Oxidation and nitrosation in the nitrogen monoxide/superoxide system. J. Biol. Chem. 277 (14), 11882–11888. doi:10.1074/jbc.M111988200
Dedio J., Konig P., Wohlfart P., Schroeder C., Kummer W., Muller-Esterl W. (2001). NOSIP, a novel modulator of endothelial nitric oxide synthase activity. Faseb J. 15 (1), 79–89. doi:10.1096/fj.00-0078com
Denton C. C., Shah P., Suriany S., Liu H., Thuptimdang W., Sunwoo J., et al. (2021). Loss of alpha-globin genes in human subjects is associated with improved nitric oxide-mediated vascular perfusion. Am. J. Hematol. 96 (3), 277–281. doi:10.1002/ajh.26058
Di Costanzo L., Ilies M., Thorn K. J., Christianson D. W. (2010). Inhibition of human arginase I by substrate and product analogues. Arch. Biochem. Biophys. 496 (2), 101–108. doi:10.1016/j.abb.2010.02.004
Di Leva F., Domi T., Fedrizzi L., Lim D., Carafoli E. (2008). The plasma membrane Ca2+ ATPase of animal cells: structure, function and regulation. Arch. Biochem. Biophys. 476 (1), 65–74. doi:10.1016/j.abb.2008.02.026
Dillon K. N., Kang Y., Maharaj A., Martinez M. A., Fischer S. M., Figueroa A. (2024). L-Citrulline supplementation attenuates aortic pressure and pressure waves during metaboreflex activation in postmenopausal women. Br. J. Nutr. 131 (3), 474–481. doi:10.1017/S000711452300199X
Di Lorenzo A., Lin M. I., Murata T., Landskroner-Eiger S., Schleicher M., Kothiya M., et al. (2013). eNOS-derived nitric oxide regulates endothelial barrier function through VE-cadherin and Rho GTPases. J. Cell Sci. 126 (Pt 24), 5541–5552. doi:10.1242/jcs.115972
Dimmeler S., Fleming I., Fisslthaler B., Hermann C., Busse R., Zeiher A. M. (1999). Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature 399 (6736), 601–605. doi:10.1038/21224
Donato A. J., Magerko K. A., Lawson B. R., Durrant J. R., Lesniewski L. A., Seals D. R. (2011). SIRT-1 and vascular endothelial dysfunction with ageing in mice and humans. J. Physiol. 589 (Pt 18), 4545–4554. doi:10.1113/jphysiol.2011.211219
Dong J. Y., Qin L. Q., Zhang Z., Zhao Y., Wang J., Arigoni F., et al. (2011). Effect of oral L-arginine supplementation on blood pressure: a meta-analysis of randomized, double-blind, placebo-controlled trials. Am. Heart J. 162 (6), 959–965. doi:10.1016/j.ahj.2011.09.012
Douglass M. S., Kaplowitz M. R., Zhang Y., Fike C. D. (2023). Impact of l-citrulline on nitric oxide signaling and arginase activity in hypoxic human pulmonary artery endothelial cells. Pulm. Circ. 13 (2), e12221. doi:10.1002/pul2.12221
Drab M., Verkade P., Elger M., Kasper M., Lohn M., Lauterbach B., et al. (2001). Loss of caveolae, vascular dysfunction, and pulmonary defects in caveolin-1 gene-disrupted mice. Science 293 (5539), 2449–2452. doi:10.1126/science.1062688
Drexler H., Zeiher A. M., Meinzer K., Just H. (1991). Correction of endothelial dysfunction in coronary microcirculation of hypercholesterolaemic patients by L-arginine. Lancet 338 (8782-8783), 1546–1550. doi:10.1016/0140-6736(91)92372-9
Dubois-Rande J. L., Zelinsky R., Roudot F., Chabrier P. E., Castaigne A., Geschwind H., et al. (1992). Effects of infusion of L-arginine into the left anterior descending coronary artery on acetylcholine-induced vasoconstriction of human atheromatous coronary arteries. Am. J. Cardiol. 70 (15), 1269–1275. doi:10.1016/0002-9149(92)90760-v
Eastman H. B., Swick A. G., Schmitt M. C., Azizkhan J. C. (1991). Stimulation of dihydrofolate reductase promoter activity by antimetabolic drugs. Proc. Natl. Acad. Sci. U. S. A. 88 (19), 8572–8576. doi:10.1073/pnas.88.19.8572
Erdmann E., Semigran M. J., Nieminen M. S., Gheorghiade M., Agrawal R., Mitrovic V., et al. (2013). Cinaciguat, a soluble guanylate cyclase activator, unloads the heart but also causes hypotension in acute decompensated heart failure. Eur. Heart J. 34 (1), 57–67. doi:10.1093/eurheartj/ehs196
Erez A., Nagamani S. C., Shchelochkov O. A., Premkumar M. H., Campeau P. M., Chen Y., et al. (2011). Requirement of argininosuccinate lyase for systemic nitric oxide production. Nat. Med. 17 (12), 1619–1626. doi:10.1038/nm.2544
Erwin P. A., Lin A. J., Golan D. E., Michel T. (2005). Receptor-regulated dynamic S-nitrosylation of endothelial nitric-oxide synthase in vascular endothelial cells. J. Biol. Chem. 280 (20), 19888–19894. doi:10.1074/jbc.M413058200
Erwin P. A., Mitchell D. A., Sartoretto J., Marletta M. A., Michel T. (2006). Subcellular targeting and differential S-nitrosylation of endothelial nitric-oxide synthase. J. Biol. Chem. 281 (1), 151–157. doi:10.1074/jbc.M510421200
Eshreif A., Al Batran R., Jamieson K. L., Darwesh A. M., Gopal K., Greenwell A. A., et al. (2020). l-Citrulline supplementation improves glucose and exercise tolerance in obese male mice. Exp. Physiol. 105 (2), 270–281. doi:10.1113/EP088109
Fernandez-Hernando C., Fukata M., Bernatchez P. N., Fukata Y., Lin M. I., Bredt D. S., et al. (2006). Identification of Golgi-localized acyl transferases that palmitoylate and regulate endothelial nitric oxide synthase. J. Cell Biol. 174 (3), 369–377. doi:10.1083/jcb.200601051
Feron O., Belhassen L., Kobzik L., Smith T. W., Kelly R. A., Michel T. (1996). Endothelial nitric oxide synthase targeting to caveolae. Specific interactions with caveolin isoforms in cardiac myocytes and endothelial cells. J. Biol. Chem. 271 (37), 22810–22814. doi:10.1074/jbc.271.37.22810
Feron O., Saldana F., Michel J. B., Michel T. (1998). The endothelial nitric-oxide synthase-caveolin regulatory cycle. J. Biol. Chem. 273 (6), 3125–3128. doi:10.1074/jbc.273.6.3125
Fish J. E., Marsden P. A. (2006). Endothelial nitric oxide synthase: insight into cell-specific gene regulation in the vascular endothelium. Cell Mol. Life Sci. 63 (2), 144–162. doi:10.1007/s00018-005-5421-8
Fish J. E., Yan M. S., Matouk C. C., St Bernard R., Ho J. J., Gavryushova A., et al. (2010). Hypoxic repression of endothelial nitric-oxide synthase transcription is coupled with eviction of promoter histones. J. Biol. Chem. 285 (2), 810–826. doi:10.1074/jbc.M109.067868
Fitzpatrick D. F., Bing B., Rohdewald P. (1998). Endothelium-dependent vascular effects of Pycnogenol. J. Cardiovasc Pharmacol. 32 (4), 509–515. doi:10.1097/00005344-199810000-00001
Fleming I., Bauersachs J., Busse R. (1997). Calcium-dependent and calcium-independent activation of the endothelial NO synthase. J. Vasc. Res. 34 (3), 165–174. doi:10.1159/000159220
Fleming I., Fisslthaler B., Dimmeler S., Kemp B. E., Busse R. (2001). Phosphorylation of Thr(495) regulates Ca(2+)/calmodulin-dependent endothelial nitric oxide synthase activity. Circ. Res. 88 (11), E68–E75. doi:10.1161/hh1101.092677
Fontana J., Fulton D., Chen Y., Fairchild T. A., McCabe T. J., Fujita N., et al. (2002). Domain mapping studies reveal that the M domain of hsp90 serves as a molecular scaffold to regulate Akt-dependent phosphorylation of endothelial nitric oxide synthase and NO release. Circ. Res. 90 (8), 866–873. doi:10.1161/01.res.0000016837.26733.be
Force USPST, Barry M. J., Nicholson W. K., Silverstein M., Chelmow D., Coker T. R., et al. (2023). Folic acid supplementation to prevent neural tube defects: US preventive Services Task force reaffirmation recommendation statement. JAMA 330 (5), 454–459. doi:10.1001/jama.2023.12876
Forstermann U., Closs E. I., Pollock J. S., Nakane M., Schwarz P., Gath I., et al. (1994). Nitric oxide synthase isozymes. Characterization, purification, molecular cloning, and functions. Hypertension 23 (6 Pt 2), 1121–1131. doi:10.1161/01.hyp.23.6.1121
Forstermann U., Mugge A., Alheid U., Bode S. M., Frolich J. C. (1989). Endothelium-derived relaxing factor (EDRF): a defence mechanism against platelet aggregation and vasospasm in human coronary arteries. Eur. Heart J. 10 (Suppl. F), 36–43. doi:10.1093/eurheartj/10.suppl_f.36
Forstermann U., Pollock J. S., Schmidt H. H., Heller M., Murad F. (1991). Calmodulin-dependent endothelium-derived relaxing factor/nitric oxide synthase activity is present in the particulate and cytosolic fractions of bovine aortic endothelial cells. Proc. Natl. Acad. Sci. U. S. A. 88 (5), 1788–1792. doi:10.1073/pnas.88.5.1788
Fraccarollo D., Widder J. D., Galuppo P., Thum T., Tsikas D., Hoffmann M., et al. (2008). Improvement in left ventricular remodeling by the endothelial nitric oxide synthase enhancer AVE9488 after experimental myocardial infarction. Circulation 118 (8), 818–827. doi:10.1161/CIRCULATIONAHA.107.717702
Frantz S., Adamek A., Fraccarollo D., Tillmanns J., Widder J. D., Dienesch C., et al. (2009). The eNOS enhancer AVE 9488: a novel cardioprotectant against ischemia reperfusion injury. Basic Res. Cardiol. 104 (6), 773–779. doi:10.1007/s00395-009-0041-3
Fredette N. C., Meyer M. R., Prossnitz E. R. (2017). Role of GPER in estrogen-dependent nitric oxide formation and vasodilation. J. steroid Biochem. Mol. Biol. 176, 65–72. doi:10.1016/j.jsbmb.2017.05.006
Fukuda Y., Teragawa H., Matsuda K., Yamagata T., Matsuura H., Chayama K. (2002). Tetrahydrobiopterin improves coronary endothelial function, but does not prevent coronary spasm in patients with vasospastic angina. Circ. J. 66 (1), 58–62. doi:10.1253/circj.66.58
Fukui T., Ishizaka N., Rajagopalan S., Laursen J. B., Capers Qt, Taylor W. R., et al. (1997). p22phox mRNA expression and NADPH oxidase activity are increased in aortas from hypertensive rats. Circ. Res. 80 (1), 45–51. doi:10.1161/01.res.80.1.45
Fulton D., Gratton J. P., McCabe T. J., Fontana J., Fujio Y., Walsh K., et al. (1999). Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature 399 (6736), 597–601. doi:10.1038/21218
Furchgott R. F., Zawadzki J. V. (1980). The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature 288 (5789), 373–376. doi:10.1038/288373a0
Gallis B., Corthals G. L., Goodlett D. R., Ueba H., Kim F., Presnell S. R., et al. (1999). Identification of flow-dependent endothelial nitric-oxide synthase phosphorylation sites by mass spectrometry and regulation of phosphorylation and nitric oxide production by the phosphatidylinositol 3-kinase inhibitor LY294002. J. Biol. Chem. 274 (42), 30101–30108. doi:10.1074/jbc.274.42.30101
Gao L., Chalupsky K., Stefani E., Cai H. (2009). Mechanistic insights into folic acid-dependent vascular protection: dihydrofolate reductase (DHFR)-mediated reduction in oxidant stress in endothelial cells and angiotensin II-infused mice: a novel HPLC-based fluorescent assay for DHFR activity. J. Mol. Cell Cardiol. 47 (6), 752–760. doi:10.1016/j.yjmcc.2009.07.025
Garcia-Cardena G., Fan R., Shah V., Sorrentino R., Cirino G., Papapetropoulos A., et al. (1998). Dynamic activation of endothelial nitric oxide synthase by Hsp90. Nature 392 (6678), 821–824. doi:10.1038/33934
Garcia-Cardena G., Fan R., Stern D. F., Liu J., Sessa W. C. (1996). Endothelial nitric oxide synthase is regulated by tyrosine phosphorylation and interacts with caveolin-1. J. Biol. Chem. 271 (44), 27237–27240. doi:10.1074/jbc.271.44.27237
Garcia-Cardena G., Martasek P., Masters B. S., Skidd P. M., Couet J., Li S., et al. (1997). Dissecting the interaction between nitric oxide synthase (NOS) and caveolin. Functional significance of the nos caveolin binding domain in vivo. J. Biol. Chem. 272 (41), 25437–25440. doi:10.1074/jbc.272.41.25437
Geller D. A., Lowenstein C. J., Shapiro R. A., Nussler A. K., Di Silvio M., Wang S. C., et al. (1993). Molecular cloning and expression of inducible nitric oxide synthase from human hepatocytes. Proc. Natl. Acad. Sci. U. S. A. 90 (8), 3491–3495. doi:10.1073/pnas.90.8.3491
Girerd X. J., Hirsch A. T., Cooke J. P., Dzau V. J., Creager M. A. (1990). L-arginine augments endothelium-dependent vasodilation in cholesterol-fed rabbits. Circ. Res. 67 (6), 1301–1308. doi:10.1161/01.res.67.6.1301
Giustiniani J., Couloubaly S., Baillet A., Pourci M. L., Cantaloube I., Fourniat C., et al. (2009). Basal endothelial nitric oxide synthase (eNOS) phosphorylation on Ser(1177) occurs in a stable microtubule- and tubulin acetylation-dependent manner. Exp. Cell Res. 315 (20), 3509–3520. doi:10.1016/j.yexcr.2009.07.018
Gray S. P., Di Marco E., Okabe J., Szyndralewiez C., Heitz F., Montezano A. C., et al. (2013). NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation 127 (18), 1888–1902. doi:10.1161/CIRCULATIONAHA.112.132159
Guzik T. J., Mussa S., Gastaldi D., Sadowski J., Ratnatunga C., Pillai R., et al. (2002). Mechanisms of increased vascular superoxide production in human diabetes mellitus: role of NAD(P)H oxidase and endothelial nitric oxide synthase. Circulation 105 (14), 1656–1662. doi:10.1161/01.cir.0000012748.58444.08
Hardy T. A., May J. M. (2002). Coordinate regulation of L-arginine uptake and nitric oxide synthase activity in cultured endothelial cells. Free Radic. Biol. Med. 32 (2), 122–131. doi:10.1016/s0891-5849(01)00781-x
Harris M. B., Ju H., Venema V. J., Liang H., Zou R., Michell B. J., et al. (2001). Reciprocal phosphorylation and regulation of endothelial nitric-oxide synthase in response to bradykinin stimulation. J. Biol. Chem. 276 (19), 16587–16591. doi:10.1074/jbc.M100229200
Hecker M., Sessa W. C., Harris H. J., Anggard E. E., Vane J. R. (1990). The metabolism of L-arginine and its significance for the biosynthesis of endothelium-derived relaxing factor: cultured endothelial cells recycle L-citrulline to L-arginine. Proc. Natl. Acad. Sci. U. S. A. 87 (21), 8612–8616. doi:10.1073/pnas.87.21.8612
Heidenreich P. A., Bozkurt B., Aguilar D., Allen L. A., Byun J. J., Colvin M. M., et al. (2022). 2022 AHA/ACC/HFSA guideline for the management of heart failure: executive summary: a report of the American College of cardiology/American heart association joint committee on clinical practice guidelines. J. Am. Coll. Cardiol. 79 (17), 1757–1780. doi:10.1016/j.jacc.2021.12.011
Heitzer T., Brockhoff C., Mayer B., Warnholtz A., Mollnau H., Henne S., et al. (2000a). Tetrahydrobiopterin improves endothelium-dependent vasodilation in chronic smokers: evidence for a dysfunctional nitric oxide synthase. Circ. Res. 86 (2), E36–E41. doi:10.1161/01.res.86.2.e36
Heitzer T., Krohn K., Albers S., Meinertz T. (2000b). Tetrahydrobiopterin improves endothelium-dependent vasodilation by increasing nitric oxide activity in patients with Type II diabetes mellitus. Diabetologia 43 (11), 1435–1438. doi:10.1007/s001250051551
Hellermann G. R., Solomonson L. P. (1997). Calmodulin promotes dimerization of the oxygenase domain of human endothelial nitric-oxide synthase. J. Biol. Chem. 272 (18), 12030–12034. doi:10.1074/jbc.272.18.12030
Higashi Y., Sasaki S., Nakagawa K., Fukuda Y., Matsuura H., Oshima T., et al. (2002). Tetrahydrobiopterin enhances forearm vascular response to acetylcholine in both normotensive and hypertensive individuals. Am. J. Hypertens. 15 (4 Pt 1), 326–332. doi:10.1016/s0895-7061(01)02317-2
Holowatz L. A., Kenney W. L. (2011). Acute localized administration of tetrahydrobiopterin and chronic systemic atorvastatin treatment restore cutaneous microvascular function in hypercholesterolaemic humans. J. Physiol. 589 (Pt 19), 4787–4797. doi:10.1113/jphysiol.2011.212100
Holton M., Mohamed T. M., Oceandy D., Wang W., Lamas S., Emerson M., et al. (2010). Endothelial nitric oxide synthase activity is inhibited by the plasma membrane calcium ATPase in human endothelial cells. Cardiovasc Res. 87 (3), 440–448. doi:10.1093/cvr/cvq077
Hong H. J., Hsiao G., Cheng T. H., Yen M. H. (2001). Supplemention with tetrahydrobiopterin suppresses the development of hypertension in spontaneously hypertensive rats. Hypertension 38 (5), 1044–1048. doi:10.1161/hy1101.095331
Huang P. L., Huang Z., Mashimo H., Bloch K. D., Moskowitz M. A., Bevan J. A., et al. (1995). Hypertension in mice lacking the gene for endothelial nitric oxide synthase. Nature 377 (6546), 239–242. doi:10.1038/377239a0
Icking A., Matt S., Opitz N., Wiesenthal A., Muller-Esterl W., Schilling K. (2005). NOSTRIN functions as a homotrimeric adaptor protein facilitating internalization of eNOS. J. Cell Sci. 118 (Pt 21), 5059–5069. doi:10.1242/jcs.02620
Ignarro L. J., Adams J. B., Horwitz P. M., Wood K. S. (1986a). Activation of soluble guanylate cyclase by NO-hemoproteins involves NO-heme exchange. Comparison of heme-containing and heme-deficient enzyme forms. J. Biol. Chem. 261 (11), 4997–5002. doi:10.1016/s0021-9258(19)89205-0
Ignarro L. J., Buga G. M., Wood K. S., Byrns R. E., Chaudhuri G. (1987). Endothelium-derived relaxing factor produced and released from artery and vein is nitric oxide. Proc. Natl. Acad. Sci. U. S. A. 84 (24), 9265–9269. doi:10.1073/pnas.84.24.9265
Ignarro L. J., Harbison R. G., Wood K. S., Kadowitz P. J. (1986b). Activation of purified soluble guanylate cyclase by endothelium-derived relaxing factor from intrapulmonary artery and vein: stimulation by acetylcholine, bradykinin and arachidonic acid. J. Pharmacol. Exp. Ther. 237 (3), 893–900.
Ignarro L. J., Wood K. S., Wolin M. S. (1982). Activation of purified soluble guanylate cyclase by protoporphyrin IX. Proc. Natl. Acad. Sci. U. S. A. 79 (9), 2870–2873. doi:10.1073/pnas.79.9.2870
Ilies M., Di Costanzo L., Dowling D. P., Thorn K. J., Christianson D. W. (2011). Binding of α,α-disubstituted amino acids to arginase suggests new avenues for inhibitor design. J. Med. Chem. 54 (15), 5432–5443. doi:10.1021/jm200443b
Ismaeel A., Papoutsi E., Miserlis D., Lavado R., Haynatzki G., Casale G. P., et al. (2020). The nitric oxide system in peripheral artery disease: connection with oxidative stress and biopterins. Antioxidants (Basel) 9 (7), 590. doi:10.3390/antiox9070590
Janaszak-Jasiecka A., Ploska A., Wieronska J. M., Dobrucki L. W., Kalinowski L. (2023). Endothelial dysfunction due to eNOS uncoupling: molecular mechanisms as potential therapeutic targets. Cell Mol. Biol. Lett. 28 (1), 21. doi:10.1186/s11658-023-00423-2
Janssens S. P., Simouchi A., Quertermous T., Bloch D. B., Bloch K. D. (1992). Cloning and expression of a cDNA encoding human endothelium-derived relating factor/nitric oxide synthase. J. Biol. Chem. 267 (31), 22694. doi:10.1016/s0021-9258(18)41728-0
Jiang F., Lim H. K., Morris M. J., Prior L., Velkoska E., Wu X., et al. (2011). Systemic upregulation of NADPH oxidase in diet-induced obesity in rats. Redox Rep. 16 (6), 223–229. doi:10.1179/174329211X13049558293713
Jo H., Otani H., Jo F., Shimazu T., Okazaki T., Yoshioka K., et al. (2011). Inhibition of nitric oxide synthase uncoupling by sepiapterin improves left ventricular function in streptozotocin-induced diabetic mice. Clin. Exp. Pharmacol. Physiol. 38 (8), 485–493. doi:10.1111/j.1440-1681.2011.05535.x
Ju H., Zou R., Venema V. J., Venema R. C. (1997). Direct interaction of endothelial nitric-oxide synthase and caveolin-1 inhibits synthase activity. J. Biol. Chem. 272 (30), 18522–18525. doi:10.1074/jbc.272.30.18522
Jung S. B., Kim C. S., Naqvi A., Yamamori T., Mattagajasingh I., Hoffman T. A., et al. (2010). Histone deacetylase 3 antagonizes aspirin-stimulated endothelial nitric oxide production by reversing aspirin-induced lysine acetylation of endothelial nitric oxide synthase. Circ. Res. 107 (7), 877–887. doi:10.1161/CIRCRESAHA.110.222968
Jurcau A., Ardelean A. I. (2022). Oxidative stress in ischemia/reperfusion injuries following acute ischemic stroke. Biomedicines 10 (3), 574. doi:10.3390/biomedicines10030574
Karantzoulis-Fegaras F., Antoniou H., Lai S. L., Kulkarni G., D'Abreo C., Wong G. K., et al. (1999). Characterization of the human endothelial nitric-oxide synthase promoter. J. Biol. Chem. 274 (5), 3076–3093. doi:10.1074/jbc.274.5.3076
Kashiwagi S., Atochin D. N., Li Q., Schleicher M., Pong T., Sessa W. C., et al. (2013). eNOS phosphorylation on serine 1176 affects insulin sensitivity and adiposity. Biochem. Biophys. Res. Commun. 431 (2), 284–290. doi:10.1016/j.bbrc.2012.12.110
Kassan M., Ait-Aissa K., Radwan E., Mali V., Haddox S., Gabani M., et al. (2016). Essential role of smooth muscle STIM1 in hypertension and cardiovascular dysfunction. Arterioscler. Thromb. Vasc. Biol. 36 (9), 1900–1909. doi:10.1161/ATVBAHA.116.307869
Katsuki S., Arnold W., Mittal C., Murad F. (1977). Stimulation of guanylate cyclase by sodium nitroprusside, nitroglycerin and nitric oxide in various tissue preparations and comparison to the effects of sodium azide and hydroxylamine. J. Cycl. Nucleotide Res. 3 (1), 23–35.
Kaye A. D., Jeha G. M., Pham A. D., Fuller M. C., Lerner Z. I., Sibley G. T., et al. (2020). Folic acid supplementation in patients with elevated homocysteine levels. Adv. Ther. 37 (10), 4149–4164. doi:10.1007/s12325-020-01474-z
Keller T. C. S, Lechauve C., Keller A. S., Broseghini-Filho G. B., Butcher J. T., Askew Page H. R., et al. (2022). Endothelial alpha globin is a nitrite reductase. Nat. Commun. 13 (1), 6405. doi:10.1038/s41467-022-34154-3
Keller T. C. S., Butcher J. T., Broseghini-Filho G. B., Marziano C., DeLalio L. J., Rogers S., et al. (2016). Modulating vascular hemodynamics with an alpha globin mimetic peptide (HbαX). Hypertension 68 (6), 1494–1503. doi:10.1161/HYPERTENSIONAHA.116.08171
Kim M., Han C. H., Lee M. Y. (2014). NADPH oxidase and the cardiovascular toxicity associated with smoking. Toxicol. Res. 30 (3), 149–157. doi:10.5487/TR.2014.30.3.149
Kim-Shapiro D. B., Gladwin M. T., Patel R. P., Hogg N. (2005). The reaction between nitrite and hemoglobin: the role of nitrite in hemoglobin-mediated hypoxic vasodilation. J. Inorg. Biochem. 99 (1), 237–246. doi:10.1016/j.jinorgbio.2004.10.034
Knorr M., Hausding M., Kroller-Schuhmacher S., Steven S., Oelze M., Heeren T., et al. (2011). Nitroglycerin-induced endothelial dysfunction and tolerance involve adverse phosphorylation and S-Glutathionylation of endothelial nitric oxide synthase: beneficial effects of therapy with the AT1 receptor blocker telmisartan. Arterioscler. Thromb. Vasc. Biol. 31 (10), 2223–2231. doi:10.1161/ATVBAHA.111.232058
Kondrikov D., Fonseca F. V., Elms S., Fulton D., Black S. M., Block E. R., et al. (2010). Beta-actin association with endothelial nitric-oxide synthase modulates nitric oxide and superoxide generation from the enzyme. J. Biol. Chem. 285 (7), 4319–4327. doi:10.1074/jbc.M109.063172
Konior A., Schramm A., Czesnikiewicz-Guzik M., Guzik T. J. (2014). NADPH oxidases in vascular pathology. Antioxid. Redox Signal 20 (17), 2794–2814. doi:10.1089/ars.2013.5607
Kossel A., Dakin H. D. (1904). Uber die Arginase. Z Physiol. Chem. 41, 321–331. doi:10.1515/bchm2.1904.41.4.321
Kou R., Greif D., Michel T. (2002). Dephosphorylation of endothelial nitric-oxide synthase by vascular endothelial growth factor. Implications for the vascular responses to cyclosporin A. J. Biol. Chem. 277 (33), 29669–29673. doi:10.1074/jbc.M204519200
Kovamees O., Shemyakin A., Checa A., Wheelock C. E., Lundberg J. O., Ostenson C. G., et al. (2016b). Arginase inhibition improves microvascular endothelial function in patients with type 2 diabetes mellitus. J. Clin. Endocrinol. Metab. 101 (11), 3952–3958. doi:10.1210/jc.2016-2007
Kovamees O., Shemyakin A., Eriksson M., Angelin B., Pernow J. (2016a). Arginase inhibition improves endothelial function in patients with familial hypercholesterolaemia irrespective of their cholesterol levels. J. Intern Med. 279 (5), 477–484. doi:10.1111/joim.12461
Kovamees O., Shemyakin A., Pernow J. (2014). Effect of arginase inhibition on ischemia-reperfusion injury in patients with coronary artery disease with and without diabetes mellitus. PLoS One 9 (7), e103260. doi:10.1371/journal.pone.0103260
Kraehling J. R., Sessa W. C. (2017). Contemporary approaches to modulating the nitric oxide-cGMP pathway in cardiovascular disease. Circ. Res. 120 (7), 1174–1182. doi:10.1161/CIRCRESAHA.117.303776
Landmesser U., Dikalov S., Price S. R., McCann L., Fukai T., Holland S. M., et al. (2003). Oxidation of tetrahydrobiopterin leads to uncoupling of endothelial cell nitric oxide synthase in hypertension. J. Clin. Invest. 111 (8), 1201–1209. doi:10.1172/JCI14172
Lass A., Suessenbacher A., Wolkart G., Mayer B., Brunner F. (2002). Functional and analytical evidence for scavenging of oxygen radicals by L-arginine. Mol. Pharmacol. 61 (5), 1081–1088. doi:10.1124/mol.61.5.1081
Laumonnier Y., Nadaud S., Agrapart M., Soubrier F. (2000). Characterization of an upstream enhancer region in the promoter of the human endothelial nitric-oxide synthase gene. J. Biol. Chem. 275 (52), 40732–40741. doi:10.1074/jbc.M004696200
Laursen J. B., Somers M., Kurz S., McCann L., Warnholtz A., Freeman B. A., et al. (2001). Endothelial regulation of vasomotion in apoE-deficient mice: implications for interactions between peroxynitrite and tetrahydrobiopterin. Circulation 103 (9), 1282–1288. doi:10.1161/01.cir.103.9.1282
Lee C. K., Han J. S., Won K. J., Jung S. H., Park H. J., Lee H. M., et al. (2009). Diminished expression of dihydropteridine reductase is a potent biomarker for hypertensive vessels. Proteomics 9 (21), 4851–4858. doi:10.1002/pmic.200800973
Levillain O. (2012). Expression and function of arginine-producing and consuming-enzymes in the kidney. Amino Acids 42 (4), 1237–1252. doi:10.1007/s00726-011-0897-z
Li C., Huang W., Harris M. B., Goolsby J. M., Venema R. C. (2005). Interaction of the endothelial nitric oxide synthase with the CAT-1 arginine transporter enhances NO release by a mechanism not involving arginine transport. Biochem. J. 386 (Pt 3), 567–574. doi:10.1042/BJ20041005
Li C., Ruan L., Sood S. G., Papapetropoulos A., Fulton D., Venema R. C. (2007). Role of eNOS phosphorylation at Ser-116 in regulation of eNOS activity in endothelial cells. Vasc. Pharmacol. 47 (5-6), 257–264. doi:10.1016/j.vph.2007.07.001
Li H., Horke S., Forstermann U. (2013). Oxidative stress in vascular disease and its pharmacological prevention. Trends Pharmacol. Sci. 34 (6), 313–319. doi:10.1016/j.tips.2013.03.007
Li H., Horke S., Forstermann U. (2014). Vascular oxidative stress, nitric oxide and atherosclerosis. Atherosclerosis 237 (1), 208–219. doi:10.1016/j.atherosclerosis.2014.09.001
Li H., Meininger C. J., Hawker J. R., Haynes T. E., Kepka-Lenhart D., Mistry S. K., et al. (2001). Regulatory role of arginase I and II in nitric oxide, polyamine, and proline syntheses in endothelial cells. Am. J. Physiol. Endocrinol. Metab. 280 (1), E75–E82. doi:10.1152/ajpendo.2001.280.1.E75
Li H., Witte K., August M., Brausch I., Godtel-Armbrust U., Habermeier A., et al. (2006). Reversal of endothelial nitric oxide synthase uncoupling and up-regulation of endothelial nitric oxide synthase expression lowers blood pressure in hypertensive rats. J. Am. Coll. Cardiol. 47 (12), 2536–2544. doi:10.1016/j.jacc.2006.01.071
Li J., Hou B., Tumova S., Muraki K., Bruns A., Ludlow M. J., et al. (2014). Piezo1 integration of vascular architecture with physiological force. Nature 515 (7526), 279–282. doi:10.1038/nature13701
Li Q., Atochin D., Kashiwagi S., Earle J., Wang A., Mandeville E., et al. (2013). Deficient eNOS phosphorylation is a mechanism for diabetic vascular dysfunction contributing to increased stroke size. Stroke 44 (11), 3183–3188. doi:10.1161/STROKEAHA.113.002073
Li X., Bazer F. W., Johnson G. A., Burghardt R. C., Wu G. (2023). Dietary supplementation with L-citrulline improves placental angiogenesis and embryonic survival in gilts. Exp. Biol. Med. (Maywood) 248 (8), 702–711. doi:10.1177/15353702231157943
Liao J. K., Shin W. S., Lee W. Y., Clark S. L. (1995). Oxidized low-density lipoprotein decreases the expression of endothelial nitric oxide synthase. J. Biol. Chem. 270 (1), 319–324. doi:10.1074/jbc.270.1.319
Lima B., Forrester M. T., Hess D. T., Stamler J. S. (2010). S-nitrosylation in cardiovascular signaling. Circ. Res. 106 (4), 633–646. doi:10.1161/CIRCRESAHA.109.207381
Lin Z., Kumar A., SenBanerjee S., Staniszewski K., Parmar K., Vaughan D. E., et al. (2005). Kruppel-like factor 2 (KLF2) regulates endothelial thrombotic function. Circ. Res. 96 (5), e48–e57. doi:10.1161/01.RES.0000159707.05637.a1
Lipton S. A., Choi Y. B., Pan Z. H., Lei S. Z., Chen H. S., Sucher N. J., et al. (1993). A redox-based mechanism for the neuroprotective and neurodestructive effects of nitric oxide and related nitroso-compounds. Nature 364 (6438), 626–632. doi:10.1038/364626a0
List B. M., Klosch B., Volker C., Gorren A. C., Sessa W. C., Werner E. R., et al. (1997). Characterization of bovine endothelial nitric oxide synthase as a homodimer with down-regulated uncoupled NADPH oxidase activity: tetrahydrobiopterin binding kinetics and role of haem in dimerization. Biochem. J. 323 (Pt 1), 159–165. doi:10.1042/bj3230159
Liu J., Garcia-Cardena G., Sessa W. C. (1995). Biosynthesis and palmitoylation of endothelial nitric oxide synthase: mutagenesis of palmitoylation sites, cysteines-15 and/or -26, argues against depalmitoylation-induced translocation of the enzyme. Biochemistry 34 (38), 12333–12340. doi:10.1021/bi00038a029
Lu J. L., Schmiege L. M., Kuo L., Liao J. C. (1996). Downregulation of endothelial constitutive nitric oxide synthase expression by lipopolysaccharide. Biochem. Biophys. Res. Commun. 225 (1), 1–5. doi:10.1006/bbrc.1996.1121
Lucotti P., Monti L., Setola E., La Canna G., Castiglioni A., Rossodivita A., et al. (2009). Oral L-arginine supplementation improves endothelial function and ameliorates insulin sensitivity and inflammation in cardiopathic nondiabetic patients after an aortocoronary bypass. Metabolism 58 (9), 1270–1276. doi:10.1016/j.metabol.2009.03.029
Lundberg J. O., Gladwin M. T., Weitzberg E. (2015). Strategies to increase nitric oxide signalling in cardiovascular disease. Nat. Rev. Drug Discov. 14 (9), 623–641. doi:10.1038/nrd4623
Lundberg J. O., Weitzberg E., Lundberg J. M., Alving K. (1994). Intragastric nitric oxide production in humans: measurements in expelled air. Gut 35 (11), 1543–1546. doi:10.1136/gut.35.11.1543
Maharaj A., Fischer S. M., Dillon K. N., Kang Y., Martinez M. A., Figueroa A. (2022). Effects of L-citrulline supplementation on endothelial function and blood pressure in hypertensive postmenopausal women. Nutrients 14 (20), 4396. doi:10.3390/nu14204396
Mahdi A., Kovamees O., Checa A., Wheelock C. E., von Heijne M., Alvarsson M., et al. (2018). Arginase inhibition improves endothelial function in patients with type 2 diabetes mellitus despite intensive glucose-lowering therapy. J. Intern Med. 284 (4), 388–398. doi:10.1111/joim.12785
Mahdi A., Pernow J., Kovamees O. (2019). Arginase inhibition improves endothelial function in an age-dependent manner in healthy elderly humans. Rejuvenation Res. 22 (5), 385–389. doi:10.1089/rej.2018.2135
Maier W., Cosentino F., Lutolf R. B., Fleisch M., Seiler C., Hess O. M., et al. (2000). Tetrahydrobiopterin improves endothelial function in patients with coronary artery disease. J. Cardiovasc Pharmacol. 35 (2), 173–178. doi:10.1097/00005344-200002000-00001
Man H. S. J., Sukumar A. N., Lam G. C., Turgeon P. J., Yan M. S., Ku K. H., et al. (2018). Angiogenic patterning by STEEL, an endothelial-enriched long noncoding RNA. Proc. Natl. Acad. Sci. U. S. A. 115 (10), 2401–2406. doi:10.1073/pnas.1715182115
Manea S. A., Antonescu M. L., Fenyo I. M., Raicu M., Simionescu M., Manea A. (2018). Epigenetic regulation of vascular NADPH oxidase expression and reactive oxygen species production by histone deacetylase-dependent mechanisms in experimental diabetes. Redox Biol. 16, 332–343. doi:10.1016/j.redox.2018.03.011
Marchi K. C., Ceron C. S., Muniz J. J., De Martinis B. S., Tanus-Santos J. E., Tirapelli C. R. (2016). NADPH oxidase plays a role on ethanol-induced hypertension and reactive oxygen species generation in the vasculature. Alcohol Alcohol 51 (5), 522–534. doi:10.1093/alcalc/agw043
Marrero M. B., Venema V. J., Ju H., He H., Liang H., Caldwell R. B., et al. (1999). Endothelial nitric oxide synthase interactions with G-protein-coupled receptors. Biochem. J. 343 (Pt 2), 335–340. doi:10.1042/0264-6021:3430335
Marsden P. A., Schappert K. T., Chen H. S., Flowers M., Sundell C. L., Wilcox J. N., et al. (1992). Molecular cloning and characterization of human endothelial nitric oxide synthase. FEBS Lett. 307 (3), 287–293. doi:10.1016/0014-5793(92)80697-f
Mattsson E. J., Kohler T. R., Vergel S. M., Clowes A. W. (1997). Increased blood flow induces regression of intimal hyperplasia. Arterioscler. Thromb. Vasc. Biol. 17 (10), 2245–2249. doi:10.1161/01.atv.17.10.2245
McCabe T. J., Fulton D., Roman L. J., Sessa W. C. (2000). Enhanced electron flux and reduced calmodulin dissociation may explain “calcium-independent” eNOS activation by phosphorylation. J. Biol. Chem. 275 (9), 6123–6128. doi:10.1074/jbc.275.9.6123
McClure S. J., Robinson P. J. (1996). Dynamin, endocytosis and intracellular signalling (review). Mol. Membr. Biol. 13 (4), 189–215. doi:10.3109/09687689609160598
McDonald K. K., Rouhani R., Handlogten M. E., Block E. R., Griffith O. W., Allison R. D., et al. (1997a). Inhibition of endothelial cell amino acid transport System y+ by arginine analogs that inhibit nitric oxide synthase. Biochim. Biophys. Acta 1324 (1), 133–141. doi:10.1016/s0005-2736(96)00226-x
McDonald K. K., Zharikov S., Block E. R., Kilberg M. S. (1997b). A caveolar complex between the cationic amino acid transporter 1 and endothelial nitric-oxide synthase may explain the arginine paradox. J. Biol. Chem. 272 (50), 31213–31216. doi:10.1074/jbc.272.50.31213
McQuillan L. P., Leung G. K., Marsden P. A., Kostyk S. K., Kourembanas S. (1994). Hypoxia inhibits expression of eNOS via transcriptional and posttranscriptional mechanisms. Am. J. Physiol. 267 (5 Pt 2), H1921–H1927. doi:10.1152/ajpheart.1994.267.5.H1921
Menzel D., Haller H., Wilhelm M., Robenek H. (2018). L-Arginine and B vitamins improve endothelial function in subjects with mild to moderate blood pressure elevation. Eur. J. Nutr. 57 (2), 557–568. doi:10.1007/s00394-016-1342-6
Miao Y., Ajami N. E., Huang T. S., Lin F. M., Lou C. H., Wang Y. T., et al. (2018). Enhancer-associated long non-coding RNA LEENE regulates endothelial nitric oxide synthase and endothelial function. Nat. Commun. 9 (1), 292. doi:10.1038/s41467-017-02113-y
Michel J. B., Feron O., Sase K., Prabhakar P., Michel T. (1997). Caveolin versus calmodulin. Counterbalancing allosteric modulators of endothelial nitric oxide synthase. J. Biol. Chem. 272 (41), 25907–25912. doi:10.1074/jbc.272.41.25907
Michell B. J., Griffiths J. E., Mitchelhill K. I., Rodriguez-Crespo I., Tiganis T., Bozinovski S., et al. (1999). The Akt kinase signals directly to endothelial nitric oxide synthase. Curr. Biol. 9 (15), 845–848. doi:10.1016/s0960-9822(99)80371-6
Michell B. J., Harris M. B., Chen Z. P., Ju H., Venema V. J., Blackstone M. A., et al. (2002). Identification of regulatory sites of phosphorylation of the bovine endothelial nitric-oxide synthase at serine 617 and serine 635. J. Biol. Chem. 277 (44), 42344–42351. doi:10.1074/jbc.M205144200
Milstien S., Katusic Z. (1999). Oxidation of tetrahydrobiopterin by peroxynitrite: implications for vascular endothelial function. Biochem. Biophys. Res. Commun. 263 (3), 681–684. doi:10.1006/bbrc.1999.1422
Mitchell J. A., Forstermann U., Warner T. D., Pollock J. S., Schmidt H. H., Heller M., et al. (1991). Endothelial cells have a particulate enzyme system responsible for EDRF formation: measurement by vascular relaxation. Biochem. Biophys. Res. Commun. 176 (3), 1417–1423. doi:10.1016/0006-291x(91)90444-c
Moens A. L., Takimoto E., Tocchetti C. G., Chakir K., Bedja D., Cormaci G., et al. (2008). Reversal of cardiac hypertrophy and fibrosis from pressure overload by tetrahydrobiopterin: efficacy of recoupling nitric oxide synthase as a therapeutic strategy. Circulation 117 (20), 2626–2636. doi:10.1161/CIRCULATIONAHA.107.737031
Morita M., Sakurada M., Watanabe F., Yamasaki T., Doi H., Ezaki H., et al. (2013). Effects of oral L-citrulline supplementation on lipoprotein oxidation and endothelial dysfunction in humans with vasospastic angina. Immunol. Endocr. Metab. Agents Med. Chem. 13 (3), 214–220. doi:10.2174/18715222113139990008
Mount P. F., Kemp B. E., Power D. A. (2007). Regulation of endothelial and myocardial NO synthesis by multi-site eNOS phosphorylation. J. Mol. Cell Cardiol. 42 (2), 271–279. doi:10.1016/j.yjmcc.2006.05.023
Muntau A. C., Longo N., Ezgu F., Schwartz I. V. D., Lah M., Bratkovic D., et al. (2024). Effects of oral sepiapterin on blood Phe concentration in a broad range of patients with phenylketonuria (APHENITY): results of an international, phase 3, randomised, double-blind, placebo-controlled trial. Lancet 404 (10460), 1333–1345. doi:10.1016/S0140-6736(24)01556-3
Munzel T., Daiber A. (2017). Does endothelial tetrahydrobiopterin control the endothelial NO synthase coupling state in arterial resistance arteries? Br. J. Pharmacol. 174 (14), 2422–2424. doi:10.1111/bph.13827
Munzel T., Daiber A., Gori T. (2013). More answers to the still unresolved question of nitrate tolerance. Eur. Heart J. 34 (34), 2666–2673. doi:10.1093/eurheartj/eht249
Munzel T., Daiber A., Ullrich V., Mulsch A. (2005). Vascular consequences of endothelial nitric oxide synthase uncoupling for the activity and expression of the soluble guanylyl cyclase and the cGMP-dependent protein kinase. Arterioscler. Thromb. Vasc. Biol. 25 (8), 1551–1557. doi:10.1161/01.ATV.0000168896.64927.bb
Munzel T., Giaid A., Kurz S., Stewart D. J., Harrison D. G. (1995b). Evidence for a role of endothelin 1 and protein kinase C in nitroglycerin tolerance. Proc. Natl. Acad. Sci. U. S. A. 92 (11), 5244–5248. doi:10.1073/pnas.92.11.5244
Munzel T., Meinertz T., Tebbe U., Schneider H. T., Stalleicken D., Wargenau M., et al. (2014). Efficacy of the long-acting nitro vasodilator pentaerithrityl tetranitrate in patients with chronic stable angina pectoris receiving anti-anginal background therapy with beta-blockers: a 12-week, randomized, double-blind, placebo-controlled trial. Eur. Heart J. 35 (14), 895–903. doi:10.1093/eurheartj/eht384
Munzel T., Sayegh H., Freeman B. A., Tarpey M. M., Harrison D. G. (1995a). Evidence for enhanced vascular superoxide anion production in nitrate tolerance. A novel mechanism underlying tolerance and cross-tolerance. J. Clin. Invest. 95 (1), 187–194. doi:10.1172/JCI117637
Murata T., Lin M. I., Huang Y., Yu J., Bauer P. M., Giordano F. J., et al. (2007). Reexpression of caveolin-1 in endothelium rescues the vascular, cardiac, and pulmonary defects in global caveolin-1 knockout mice. J. Exp. Med. 204 (10), 2373–2382. doi:10.1084/jem.20062340
Nadaud S., Philippe M., Arnal J. F., Michel J. B., Soubrier F. (1996). Sustained increase in aortic endothelial nitric oxide synthase expression in vivo in a model of chronic high blood flow. Circ. Res. 79 (4), 857–863. doi:10.1161/01.res.79.4.857
Naing A., Papadopoulos K. P., Pishvaian M. J., Rahma O., Hanna G. J., Garralda E., et al. (2024). First-in-human phase 1 study of the arginase inhibitor INCB001158 alone or combined with pembrolizumab in patients with advanced or metastatic solid tumours. BMJ Oncol. 3, e000249. doi:10.1136/bmjonc-2023-000249
Neumann P., Gertzberg N., Johnson A. (2004). TNF-alpha induces a decrease in eNOS promoter activity. Am. J. Physiol. Lung Cell Mol. Physiol. 286 (2), L452–L459. doi:10.1152/ajplung.00378.2002
Ng Y. Y. H., Dora K. A., Lemmey H. A. L., Lin J., Alden J., Wallis L., et al. (2024). Asymmetric dimethylarginine enables depolarizing spikes and vasospasm in mesenteric and coronary resistance arteries. Hypertension 81 (4), 764–775. doi:10.1161/HYPERTENSIONAHA.123.22454
Nishida C. R., Ortiz de Montellano P. R. (1999). Autoinhibition of endothelial nitric-oxide synthase. Identification of an electron transfer control element. J. Biol. Chem. 274 (21), 14692–14698. doi:10.1074/jbc.274.21.14692
Nishida K., Harrison D. G., Navas J. P., Fisher A. A., Dockery S. P., Uematsu M., et al. (1992). Molecular cloning and characterization of the constitutive bovine aortic endothelial cell nitric oxide synthase. J. Clin. Invest. 90 (5), 2092–2096. doi:10.1172/JCI116092
Nishimoto M., Mizuno R., Fujita T., Isshiki M. (2018). Stromal interaction molecule 1 modulates blood pressure via NO production in vascular endothelial cells. Hypertens. Res. 41 (7), 506–514. doi:10.1038/s41440-018-0045-1
Noreng S., Ota N., Sun Y., Ho H., Johnson M., Arthur C. P., et al. (2022). Structure of the core human NADPH oxidase NOX2. Nat. Commun. 13 (1), 6079. doi:10.1038/s41467-022-33711-0
Nystrom T., Nygren A., Sjoholm A. (2004). Tetrahydrobiopterin increases insulin sensitivity in patients with type 2 diabetes and coronary heart disease. Am. J. Physiol. Endocrinol. Metab. 287 (5), E919–E925. doi:10.1152/ajpendo.00046.2004
Oelze M., Knorr M., Kroller-Schon S., Kossmann S., Gottschlich A., Rummler R., et al. (2013). Chronic therapy with isosorbide-5-mononitrate causes endothelial dysfunction, oxidative stress, and a marked increase in vascular endothelin-1 expression. Eur. Heart J. 34 (41), 3206–3216. doi:10.1093/eurheartj/ehs100
Osowska S., Moinard C., Neveux N., Loi C., Cynober L. (2004). Citrulline increases arginine pools and restores nitrogen balance after massive intestinal resection. Gut 53 (12), 1781–1786. doi:10.1136/gut.2004.042317
Ou H., Shen Y. H., Utama B., Wang J., Wang X., Coselli J., et al. (2005). Effect of nuclear actin on endothelial nitric oxide synthase expression. Arterioscler. Thromb. Vasc. Biol. 25 (12), 2509–2514. doi:10.1161/01.ATV.0000189306.99112.4c
Paik W. K., Nochumson S., Kim S. (1978). Effect of modification on inhibitory amino acids on arginase activity. Biochem. Med. 19 (1), 39–46. doi:10.1016/0006-2944(78)90005-4
Palmer R. M., Ashton D. S., Moncada S. (1988). Vascular endothelial cells synthesize nitric oxide from L-arginine. Nature 333 (6174), 664–666. doi:10.1038/333664a0
Palmer R. M., Ferrige A. G., Moncada S. (1987). Nitric oxide release accounts for the biological activity of endothelium-derived relaxing factor. Nature 327 (6122), 524–526. doi:10.1038/327524a0
Park K., Mima A., Li Q., Rask-Madsen C., He P., Mizutani K., et al. (2016). Insulin decreases atherosclerosis by inducing endothelin receptor B expression. JCI Insight 1 (6), e86574. doi:10.1172/jci.insight.86574
Pernow J., Jung C. (2013). Arginase as a potential target in the treatment of cardiovascular disease: reversal of arginine steal? Cardiovasc Res. 98 (3), 334–343. doi:10.1093/cvr/cvt036
Pernow J., Kiss A., Tratsiakovich Y., Climent B. (2015). Tissue-specific up-regulation of arginase I and II induced by p38 MAPK mediates endothelial dysfunction in type 1 diabetes mellitus. Br. J. Pharmacol. 172 (19), 4684–4698. doi:10.1111/bph.13242
Peyton K. J., Liu X. M., Shebib A. R., Johnson F. K., Johnson R. A., Durante W. (2018). Arginase inhibition prevents the development of hypertension and improves insulin resistance in obese rats. Amino Acids 50 (6), 747–754. doi:10.1007/s00726-018-2567-x
Pollock J. S., Forstermann U., Mitchell J. A., Warner T. D., Schmidt H. H., Nakane M., et al. (1991). Purification and characterization of particulate endothelium-derived relaxing factor synthase from cultured and native bovine aortic endothelial cells. Proc. Natl. Acad. Sci. U. S. A. 88 (23), 10480–10484. doi:10.1073/pnas.88.23.10480
Porkert M., Sher S., Reddy U., Cheema F., Niessner C., Kolm P., et al. (2008). Tetrahydrobiopterin: a novel antihypertensive therapy. J. Hum. Hypertens. 22 (6), 401–407. doi:10.1038/sj.jhh.1002329
Pudlo M., Demougeot C., Girard-Thernier C. (2017). Arginase inhibitors: a rational approach over one century. Med. Res. Rev. 37 (3), 475–513. doi:10.1002/med.21419
Ranade S. S., Qiu Z., Woo S. H., Hur S. S., Murthy S. E., Cahalan S. M., et al. (2014). Piezo1, a mechanically activated ion channel, is required for vascular development in mice. Proc. Natl. Acad. Sci. U. S. A. 111 (28), 10347–10352. doi:10.1073/pnas.1409233111
Rapoport R. M., Waldman S. A., Ginsburg R., Molina C. R., Murad F. (1987). Effects of glyceryl trinitrate on endothelium-dependent and -independent relaxation and cyclic GMP levels in rat aorta and human coronary artery. J. Cardiovasc Pharmacol. 10 (1), 82–89. doi:10.1097/00005344-198707000-00012
Razani B., Engelman J. A., Wang X. B., Schubert W., Zhang X. L., Marks C. B., et al. (2001). Caveolin-1 null mice are viable but show evidence of hyperproliferative and vascular abnormalities. J. Biol. Chem. 276 (41), 38121–38138. doi:10.1074/jbc.M105408200
Rees D. D., Palmer R. M., Moncada S. (1989). Role of endothelium-derived nitric oxide in the regulation of blood pressure. Proc. Natl. Acad. Sci. U. S. A. 86 (9), 3375–3378. doi:10.1073/pnas.86.9.3375
Reule C. A., Goyvaerts B., Schoen C. (2017). Effects of an L-arginine-based multi ingredient product on endothelial function in subjects with mild to moderate hypertension and hyperhomocysteinemia - a randomized, double-blind, placebo-controlled, cross-over trial. BMC Complement. Altern. Med. 17 (1), 92. doi:10.1186/s12906-017-1603-9
Reyes L. A., Boslett J., Varadharaj S., De Pascali F., Hemann C., Druhan L. J., et al. (2015). Depletion of NADP(H) due to CD38 activation triggers endothelial dysfunction in the postischemic heart. Proc. Natl. Acad. Sci. U. S. A. 112 (37), 11648–11653. doi:10.1073/pnas.1505556112
Roos J., DiGregorio P. J., Yeromin A. V., Ohlsen K., Lioudyno M., Zhang S., et al. (2005). STIM1, an essential and conserved component of store-operated Ca2+ channel function. J. Cell Biol. 169 (3), 435–445. doi:10.1083/jcb.200502019
Rossitch E., Alexander E., Black P. M., Cooke J. P. (1991). L-arginine normalizes endothelial function in cerebral vessels from hypercholesterolemic rabbits. J. Clin. Invest. 87 (4), 1295–1299. doi:10.1172/JCI115132
Russell K. S., Haynes M. P., Caulin-Glaser T., Rosneck J., Sessa W. C., Bender J. R. (2000). Estrogen stimulates heat shock protein 90 binding to endothelial nitric oxide synthase in human vascular endothelial cells. Effects on calcium sensitivity and NO release. J. Biol. Chem. 275 (7), 5026–5030. doi:10.1074/jbc.275.7.5026
Ryoo S., Gupta G., Benjo A., Lim H. K., Camara A., Sikka G., et al. (2008). Endothelial arginase II: a novel target for the treatment of atherosclerosis. Circ. Res. 102 (8), 923–932. doi:10.1161/CIRCRESAHA.107.169573
Ryoo S., Lemmon C. A., Soucy K. G., Gupta G., White A. R., Nyhan D., et al. (2006). Oxidized low-density lipoprotein-dependent endothelial arginase II activation contributes to impaired nitric oxide signaling. Circ. Res. 99 (9), 951–960. doi:10.1161/01.RES.0000247034.24662.b4
Sasaki K., Heeschen C., Aicher A., Ziebart T., Honold J., Urbich C., et al. (2006). Ex vivo pretreatment of bone marrow mononuclear cells with endothelial NO synthase enhancer AVE9488 enhances their functional activity for cell therapy. Proc. Natl. Acad. Sci. U. S. A. 103 (39), 14537–14541. doi:10.1073/pnas.0604144103
Saura M., Zaragoza C., Cao W., Bao C., Rodriguez-Puyol M., Rodriguez-Puyol D., et al. (2002). Smad2 mediates transforming growth factor-beta induction of endothelial nitric oxide synthase expression. Circ. Res. 91 (9), 806–813. doi:10.1161/01.res.0000040397.23817.e5
Sawabe K., Yamamoto K., Harada Y., Ohashi A., Sugawara Y., Matsuoka H., et al. (2008). Cellular uptake of sepiapterin and push-pull accumulation of tetrahydrobiopterin. Mol. Genet. Metab. 94 (4), 410–416. doi:10.1016/j.ymgme.2008.04.007
Scalera F., Closs E. I., Flick E., Martens-Lobenhoffer J., Boissel J. P., Lendeckel U., et al. (2009). Paradoxical effect of L-arginine: acceleration of endothelial cell senescence. Biochem. Biophys. Res. Commun. 386 (4), 650–655. doi:10.1016/j.bbrc.2009.06.091
Schafer A., Fraccarollo D., Pfortsch S., Loch E., Neuser J., Vogt C., et al. (2011). Clopidogrel improves endothelial function and NO bioavailability by sensitizing adenylyl cyclase in rats with congestive heart failure. Basic Res. Cardiol. 106 (3), 485–494. doi:10.1007/s00395-011-0153-4
Schilling K., Opitz N., Wiesenthal A., Oess S., Tikkanen R., Muller-Esterl W., et al. (2006). Translocation of endothelial nitric-oxide synthase involves a ternary complex with caveolin-1 and NOSTRIN. Mol. Biol. Cell 17 (9), 3870–3880. doi:10.1091/mbc.e05-08-0709
Schulman S. P., Becker L. C., Kass D. A., Champion H. C., Terrin M. L., Forman S., et al. (2006). L-arginine therapy in acute myocardial infarction: the Vascular Interaction with Age in Myocardial Infarction (VINTAGE MI) randomized clinical trial. JAMA 295 (1), 58–64. doi:10.1001/jama.295.1.58
Scotland R. S., Morales-Ruiz M., Chen Y., Yu J., Rudic R. D., Fulton D., et al. (2002). Functional reconstitution of endothelial nitric oxide synthase reveals the importance of serine 1179 in endothelium-dependent vasomotion. Circ. Res. 90 (8), 904–910. doi:10.1161/01.res.0000016506.04193.96
Searles C. D. (2006). Transcriptional and posttranscriptional regulation of endothelial nitric oxide synthase expression. Am. J. Physiol. Cell Physiol. 291 (5), C803–C816. doi:10.1152/ajpcell.00457.2005
Searles C. D., Ide L., Davis M. E., Cai H., Weber M. (2004). Actin cytoskeleton organization and posttranscriptional regulation of endothelial nitric oxide synthase during cell growth. Circ. Res. 95 (5), 488–495. doi:10.1161/01.RES.0000138953.21377.80
Sena C. M., Nunes E., Louro T., Proenca T., Fernandes R., Boarder M. R., et al. (2008). Effects of alpha-lipoic acid on endothelial function in aged diabetic and high-fat fed rats. Br. J. Pharmacol. 153 (5), 894–906. doi:10.1038/sj.bjp.0707474
Sessa W. C., Garcia-Cardena G., Liu J., Keh A., Pollock J. S., Bradley J., et al. (1995). The Golgi association of endothelial nitric oxide synthase is necessary for the efficient synthesis of nitric oxide. J. Biol. Chem. 270 (30), 17641–17644. doi:10.1074/jbc.270.30.17641
Setoguchi S., Mohri M., Shimokawa H., Takeshita A. (2001). Tetrahydrobiopterin improves endothelial dysfunction in coronary microcirculation in patients without epicardial coronary artery disease. J. Am. Coll. Cardiol. 38 (2), 493–498. doi:10.1016/s0735-1097(01)01382-1
Shang Q., Bao L., Guo H., Hao F., Luo Q., Chen J., et al. (2017). Contribution of glutaredoxin-1 to S-glutathionylation of endothelial nitric oxide synthase for mesenteric nitric oxide generation in experimental necrotizing enterocolitis. Transl. Res. 188, 92–105. doi:10.1016/j.trsl.2016.01.004
Sharifi-Rad M., Anil Kumar N. V., Zucca P., Varoni E. M., Dini L., Panzarini E., et al. (2020). Lifestyle, oxidative stress, and antioxidants: back and forth in the pathophysiology of chronic diseases. Front. Physiol. 11, 694. doi:10.3389/fphys.2020.00694
Shatanawi A., Momani M. S., Al-Aqtash R., Hamdan M. H., Gharaibeh M. N. (2020). L-citrulline supplementation increases plasma nitric oxide levels and reduces arginase activity in patients with type 2 diabetes. Front. Pharmacol. 11, 584669. doi:10.3389/fphar.2020.584669
Shearer J. D., Richards J. R., Mills C. D., Caldwell M. D. (1997). Differential regulation of macrophage arginine metabolism: a proposed role in wound healing. Am. J. Physiol. 272 (2 Pt 1), E181–E190. doi:10.1152/ajpendo.1997.272.2.E181
Shemyakin A., Kovamees O., Rafnsson A., Bohm F., Svenarud P., Settergren M., et al. (2012). Arginase inhibition improves endothelial function in patients with coronary artery disease and type 2 diabetes mellitus. Circulation 126 (25), 2943–2950. doi:10.1161/CIRCULATIONAHA.112.140335
Shimazu T., Otani H., Yoshioka K., Fujita M., Okazaki T., Iwasaka T. (2011). Sepiapterin enhances angiogenesis and functional recovery in mice after myocardial infarction. Am. J. Physiol. Heart Circ. Physiol. 301 (5), H2061–H2072. doi:10.1152/ajpheart.00525.2011
Singer H. A., Peach M. J. (1982). Calcium- and endothelial-mediated vascular smooth muscle relaxation in rabbit aorta. Hypertension 4 (3 Pt 2), 19–25. doi:10.1161/01.hyp.4.3.19
Smith J. F., Lemmey H. A. L., Borysova L., Hiley C. R., Dora K. A., Garland C. J. (2020). Endothelial nitric oxide suppresses action-potential-like transient spikes and vasospasm in small resistance arteries. Hypertension 76 (3), 785–794. doi:10.1161/HYPERTENSIONAHA.120.15491
Soboloff J., Rothberg B. S., Madesh M., Gill D. L. (2012). STIM proteins: dynamic calcium signal transducers. Nat. Rev. Mol. cell Biol. 13 (9), 549–565. doi:10.1038/nrm3414
Soltis E. E., Cassis L. A. (1991). Influence of perivascular adipose tissue on rat aortic smooth muscle responsiveness. Clin. Exp. Hypertens. A 13 (2), 277–296. doi:10.3109/10641969109042063
Steggerda S. M., Bennett M. K., Chen J., Emberley E., Huang T., Janes J. R., et al. (2017). Inhibition of arginase by CB-1158 blocks myeloid cell-mediated immune suppression in the tumor microenvironment. J. Immunother. Cancer 5 (1), 101. doi:10.1186/s40425-017-0308-4
Straub A. C., Butcher J. T., Billaud M., Mutchler S. M., Artamonov M. V., Nguyen A. T., et al. (2014a). Hemoglobin α/eNOS coupling at myoendothelial junctions is required for nitric oxide scavenging during vasoconstriction. Arterioscler. Thromb. Vasc. Biol. 34 (12), 2594–2600. doi:10.1161/ATVBAHA.114.303974
Straub A. C., Lohman A. W., Billaud M., Johnstone S. R., Dwyer S. T., Lee M. Y., et al. (2012). Endothelial cell expression of haemoglobin α regulates nitric oxide signalling. Nature 491 (7424), 473–477. doi:10.1038/nature11626
Straub A. C., Zeigler A. C., Isakson B. E. (2014b). The myoendothelial junction: connections that deliver the message. Physiol. (Bethesda) 29 (4), 242–249. doi:10.1152/physiol.00042.2013
Stroes E., Kastelein J., Cosentino F., Erkelens W., Wever R., Koomans H., et al. (1997). Tetrahydrobiopterin restores endothelial function in hypercholesterolemia. J. Clin. Invest. 99 (1), 41–46. doi:10.1172/JCI119131
Stroes E. S., van Faassen E. E., Yo M., Martasek P., Boer P., Govers R., et al. (2000). Folic acid reverts dysfunction of endothelial nitric oxide synthase. Circ. Res. 86 (11), 1129–1134. doi:10.1161/01.res.86.11.1129
Stuehr D. J. (1997). Structure-function aspects in the nitric oxide synthases. Annu. Rev. Pharmacol. Toxicol. 37, 339–359. doi:10.1146/annurev.pharmtox.37.1.339
Stuehr D. J., Wei C. C., Wang Z., Hille R. (2005). Exploring the redox reactions between heme and tetrahydrobiopterin in the nitric oxide synthases. Dalton Trans. (21), 3427–3435. doi:10.1039/b506355h
Su Y., Edwards-Bennett S., Bubb M. R., Block E. R. (2003). Regulation of endothelial nitric oxide synthase by the actin cytoskeleton. Am. J. Physiol. Cell Physiol. 284 (6), C1542–C1549. doi:10.1152/ajpcell.00248.2002
Suarez Y., Fernandez-Hernando C., Pober J. S., Sessa W. C. (2007). Dicer dependent microRNAs regulate gene expression and functions in human endothelial cells. Circ. Res. 100 (8), 1164–1173. doi:10.1161/01.RES.0000265065.26744.17
Suvorava T., Nagy N., Pick S., Lieven O., Ruther U., Dao V. T., et al. (2015). Impact of eNOS-dependent oxidative stress on endothelial function and neointima formation. Antioxid. Redox Signal 23 (9), 711–723. doi:10.1089/ars.2014.6059
Suvorava T., Pick S., Kojda G. (2017). Selective impairment of blood pressure reduction by endothelial nitric oxide synthase dimer destabilization in mice. J. Hypertens. 35 (1), 76–88. doi:10.1097/HJH.0000000000001127
Takahashi S., Mendelsohn M. E. (2003). Calmodulin-dependent and -independent activation of endothelial nitric-oxide synthase by heat shock protein 90. J. Biol. Chem. 278 (11), 9339–9344. doi:10.1074/jbc.M212651200
Tang J. L., Zembowicz A., Xu X. M., Wu K. K. (1995). Role of Sp1 in transcriptional activation of human nitric oxide synthase type III gene. Biochem. Biophys. Res. Commun. 213 (2), 673–680. doi:10.1006/bbrc.1995.2184
Taubert D., Berkels R., Grosser N., Schroder H., Grundemann D., Schomig E. (2004). Aspirin induces nitric oxide release from vascular endothelium: a novel mechanism of action. Br. J. Pharmacol. 143 (1), 159–165. doi:10.1038/sj.bjp.0705907
Tengbom J., Kontidou E., Collado A., Yang J., Alvarsson M., Brinck J., et al. (2024). Differences in endothelial function between patients with Type 1 and Type 2 diabetes: effects of red blood cells and arginase. Clin. Sci. (Lond). 138 (15), 975–985. doi:10.1042/CS20240447
Terry L. E., VerMeer M., Giles J., Tran Q. K. (2017). Suppression of store-operated Ca2+ entry by activation of GPER: contribution to a clamping effect on endothelial Ca2+ signaling. Biochem. J. 474, 3627–3642. doi:10.1042/BCJ20170630
Thannickal V. J., Fanburg B. L. (2000). Reactive oxygen species in cell signaling. Am. J. Physiol. Lung Cell Mol. Physiol. 279 (6), L1005–L1028. doi:10.1152/ajplung.2000.279.6.L1005
Thomson L., Trujillo M., Telleri R., Radi R. (1995). Kinetics of cytochrome c2+ oxidation by peroxynitrite: implications for superoxide measurements in nitric oxide-producing biological systems. Arch. Biochem. Biophys. 319 (2), 491–497. doi:10.1006/abbi.1995.1321
Tomada I., Negrao R., Almeida H., Neves D. (2014). Long-term high-fat consumption leads to downregulation of Akt phosphorylation of eNOS at Ser1177 and upregulation of Sirtuin-1 expression in rat cavernous tissue. Age (Dordr). 36 (2), 597–611. doi:10.1007/s11357-013-9591-2
Totzeck M., Hendgen-Cotta U. B., Luedike P., Berenbrink M., Klare J. P., Steinhoff H. J., et al. (2012). Nitrite regulates hypoxic vasodilation via myoglobin-dependent nitric oxide generation. Circulation 126 (3), 325–334. doi:10.1161/CIRCULATIONAHA.111.087155
Tran Q. K. (2020). Reciprocality between estrogen biology and calcium signaling in the cardiovascular system. Front. Endocrinol. (Lausanne) 11, 568203. doi:10.3389/fendo.2020.568203
Tran Q. K., Black D. J., Persechini A. (2003). Intracellular coupling via limiting calmodulin. J. Biol. Chem. 278 (27), 24247–24250. doi:10.1074/jbc.C300165200
Tran Q. K., Black D. J., Persechini A. (2005). Dominant affectors in the calmodulin network shape the time courses of target responses in the cell. Cell Calcium 37 (6), 541–553. doi:10.1016/j.ceca.2005.02.001
Tran Q. K., Firkins R., Giles J., Francis S., Matnishian V., Tran P., et al. (2016). Estrogen enhances linkage in the vascular endothelial calmodulin network via a feedforward mechanism at the G protein-coupled estrogen receptor 1. J. Biol. Chem. 291 (20), 10805–10823. doi:10.1074/jbc.M115.697334
Tran Q. K., Leonard J., Black D. J., Nadeau O. W., Boulatnikov I. G., Persechini A. (2009). Effects of combined phosphorylation at Ser-617 and Ser-1179 in endothelial nitric-oxide synthase on EC50(Ca2+) values for calmodulin binding and enzyme activation. J. Biol. Chem. 284 (18), 11892–11899. doi:10.1074/jbc.M806205200
Tran Q. K., Leonard J., Black D. J., Persechini A. (2008). Phosphorylation within an autoinhibitory domain in endothelial nitric oxide synthase reduces the Ca(2+) concentrations required for calmodulin to bind and activate the enzyme. Biochemistry 47 (28), 7557–7566. doi:10.1021/bi8003186
Tran Q. K., VerMeer M., Burgard M. A., Hassan A. B., Giles J. (2015). Hetero-oligomeric complex between the G protein-coupled estrogen receptor 1 and the plasma membrane Ca2+-ATPase 4b. J. Biol. Chem. 290 (21), 13293–13307. doi:10.1074/jbc.M114.628743
Tsuboi T., Maeda M., Hayashi T. (2018). Administration of L-arginine plus L-citrulline or L-citrulline alone successfully retarded endothelial senescence. PLoS One 13 (2), e0192252. doi:10.1371/journal.pone.0192252
Tsukahara H., Gordienko D. V., Tonshoff B., Gelato M. C., Goligorsky M. S. (1994). Direct demonstration of insulin-like growth factor-I-induced nitric oxide production by endothelial cells. Kidney Int. 45 (2), 598–604. doi:10.1038/ki.1994.78
Uittenbogaard A., Shaul P. W., Yuhanna I. S., Blair A., Smart E. J. (2000). High density lipoprotein prevents oxidized low density lipoprotein-induced inhibition of endothelial nitric-oxide synthase localization and activation in caveolae. J. Biol. Chem. 275 (15), 11278–11283. doi:10.1074/jbc.275.15.11278
Umbrello M., Dyson A., Feelisch M., Singer M. (2013). The key role of nitric oxide in hypoxia: hypoxic vasodilation and energy supply-demand matching. Antioxid. Redox Signal 19 (14), 1690–1710. doi:10.1089/ars.2012.4979
Varadharaj S., Kelly O. J., Khayat R. N., Kumar P. S., Ahmed N., Zweier J. L. (2017). Role of dietary antioxidants in the preservation of vascular function and the modulation of health and disease. Front. Cardiovasc Med. 4, 64. doi:10.3389/fcvm.2017.00064
Vasquez-Vivar J., Kalyanaraman B., Martasek P., Hogg N., Masters B. S., Karoui H., et al. (1998). Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc. Natl. Acad. Sci. U. S. A. 95 (16), 9220–9225. doi:10.1073/pnas.95.16.9220
Venema R. C., Sayegh H. S., Kent J. D., Harrison D. G. (1996). Identification, characterization, and comparison of the calmodulin-binding domains of the endothelial and inducible nitric oxide synthases. J. Biol. Chem. 271 (11), 6435–6440. doi:10.1074/jbc.271.11.6435
Venema V. J., Zou R., Ju H., Marrero M. B., Venema R. C. (1997). Caveolin-1 detergent solubility and association with endothelial nitric oxide synthase is modulated by tyrosine phosphorylation. Biochem. Biophys. Res. Commun. 236 (1), 155–161. doi:10.1006/bbrc.1997.6921
Victor V. M., Rocha M., Sola E., Banuls C., Garcia-Malpartida K., Hernandez-Mijares A. (2009). Oxidative stress, endothelial dysfunction and atherosclerosis. Curr. Pharm. Des. 15 (26), 2988–3002. doi:10.2174/138161209789058093
Wang J., Sim A. S., Wang X. L., Wilcken D. E. (2006). L-arginine regulates asymmetric dimethylarginine metabolism by inhibiting dimethylarginine dimethylaminohydrolase activity in hepatic (HepG2) cells. Cell Mol. Life Sci. 63 (23), 2838–2846. doi:10.1007/s00018-006-6271-8
Wang J., Tokoro T., Matsui K., Higa S., Kitajima I. (2005). Pitavastatin at low dose activates endothelial nitric oxide synthase through PI3K-AKT pathway in endothelial cells. Life Sci. 76 (19), 2257–2268. doi:10.1016/j.lfs.2004.12.003
Wang S., Chennupati R., Kaur H., Iring A., Wettschureck N., Offermanns S. (2016). Endothelial cation channel PIEZO1 controls blood pressure by mediating flow-induced ATP release. J. Clin. Invest. 126 (12), 4527–4536. doi:10.1172/JCI87343
Wang W., Ha C. H., Jhun B. S., Wong C., Jain M. K., Jin Z. G. (2010). Fluid shear stress stimulates phosphorylation-dependent nuclear export of HDAC5 and mediates expression of KLF2 and eNOS. Blood 115 (14), 2971–2979. doi:10.1182/blood-2009-05-224824
Wang X., Reznick S., Li P., Liang W., van Breemen C. (2002). Ca(2+) removal mechanisms in freshly isolated rabbit aortic endothelial cells. Cell Calcium 31 (6), 265–277. doi:10.1016/s0143-4160(02)00075-1
Wariishi S., Miyahara K., Toda K., Ogoshi S., Doi Y., Ohnishi S., et al. (1995). A SP1 binding site in the GC-rich region is essential for a core promoter activity of the human endothelial nitric oxide synthase gene. Biochem. Biophys. Res. Commun. 216 (2), 729–735. doi:10.1006/bbrc.1995.2682
Watson C. P., Pazarentzos E., Fidanboylu M., Padilla B., Brown R., Thomas S. A. (2016). The transporter and permeability interactions of asymmetric dimethylarginine (ADMA) and L-arginine with the human blood-brain barrier in vitro. Brain Res. 1648 (Pt A)), 232–242. doi:10.1016/j.brainres.2016.07.026
Wever R. M., van Dam T., van Rijn H. J., de Groot F., Rabelink T. J. (1997). Tetrahydrobiopterin regulates superoxide and nitric oxide generation by recombinant endothelial nitric oxide synthase. Biochem. Biophys. Res. Commun. 237 (2), 340–344. doi:10.1006/bbrc.1997.7069
Whitsett J., Picklo M. J., Vasquez-Vivar J. (2007). 4-Hydroxy-2-nonenal increases superoxide anion radical in endothelial cells via stimulated GTP cyclohydrolase proteasomal degradation. Arterioscler. Thromb. Vasc. Biol. 27 (11), 2340–2347. doi:10.1161/ATVBAHA.107.153742
Wilson A. M., Harada R., Nair N., Balasubramanian N., Cooke J. P. (2007). L-arginine supplementation in peripheral arterial disease: no benefit and possible harm. Circulation 116 (2), 188–195. doi:10.1161/CIRCULATIONAHA.106.683656
Wohlfart P., Xu H., Endlich A., Habermeier A., Closs E. I., Hubschle T., et al. (2008). Antiatherosclerotic effects of small-molecular-weight compounds enhancing endothelial nitric-oxide synthase (eNOS) expression and preventing eNOS uncoupling. J. Pharmacol. Exp. Ther. 325 (2), 370–379. doi:10.1124/jpet.107.128009
Worthley M. I., Kanani R. S., Sun Y. H., Sun Y., Goodhart D. M., Curtis M. J., et al. (2007). Effects of tetrahydrobiopterin on coronary vascular reactivity in atherosclerotic human coronary arteries. Cardiovasc Res. 76 (3), 539–546. doi:10.1016/j.cardiores.2007.07.009
Wu G., Meininger C. J. (1993). Regulation of L-arginine synthesis from L-citrulline by L-glutamine in endothelial cells. Am. J. Physiol. 265 (6 Pt 2), H1965–H1971. doi:10.1152/ajpheart.1993.265.6.H1965
Wu G., Morris S. M. (1998). Arginine metabolism: nitric oxide and beyond. Biochem. J. 336 (Pt 1), 1–17. doi:10.1042/bj3360001
Wu G., Sharina I., Martin E. (2022). Soluble guanylyl cyclase: molecular basis for ligand selectivity and action in vitro and in vivo. Front. Mol. Biosci. 9, 1007768. doi:10.3389/fmolb.2022.1007768
Wu W., Geng P., Zhu J., Li J., Zhang L., Chen W., et al. (2019). KLF2 regulates eNOS uncoupling via Nrf2/HO-1 in endothelial cells under hypoxia and reoxygenation. Chem. Biol. Interact. 305, 105–111. doi:10.1016/j.cbi.2019.03.010
Xia Y., Roman L. J., Masters B. S., Zweier J. L. (1998b). Inducible nitric-oxide synthase generates superoxide from the reductase domain. J. Biol. Chem. 273 (35), 22635–22639. doi:10.1074/jbc.273.35.22635
Xia Y., Tsai A. L., Berka V., Zweier J. L. (1998a). Superoxide generation from endothelial nitric-oxide synthase. A Ca2+/calmodulin-dependent and tetrahydrobiopterin regulatory process. J. Biol. Chem. 273 (40), 25804–25808. doi:10.1074/jbc.273.40.25804
Xiang M., Lu Y., Xin L., Gao J., Shang C., Jiang Z., et al. (2021). Role of oxidative stress in reperfusion following myocardial ischemia and its treatments. Oxid. Med. Cell Longev. 2021, 6614009. doi:10.1155/2021/6614009
Xiong Y., Fru M. F., Yu Y., Montani J. P., Ming X. F., Yang Z. (2014). Long term exposure to L-arginine accelerates endothelial cell senescence through arginase-II and S6K1 signaling. Aging (Albany NY) 6 (5), 369–379. doi:10.18632/aging.100663
Xu H., Shi Y., Wang J., Jones D., Weilrauch D., Ying R., et al. (2007). A heat shock protein 90 binding domain in endothelial nitric-oxide synthase influences enzyme function. J. Biol. Chem. 282 (52), 37567–37574. doi:10.1074/jbc.M706464200
Xu J., Wang S., Wu Y., Song P., Zou M. H. (2009). Tyrosine nitration of PA700 activates the 26S proteasome to induce endothelial dysfunction in mice with angiotensin II-induced hypertension. Hypertension 54 (3), 625–632. doi:10.1161/HYPERTENSIONAHA.109.133736
Xu J., Wu Y., Song P., Zhang M., Wang S., Zou M. H. (2007). Proteasome-dependent degradation of guanosine 5'-triphosphate cyclohydrolase I causes tetrahydrobiopterin deficiency in diabetes mellitus. Circulation 116 (8), 944–953. doi:10.1161/CIRCULATIONAHA.106.684795
Yang F., Zhang L., Huo X. S., Yuan J. H., Xu D., Yuan S. X., et al. (2011). Long noncoding RNA high expression in hepatocellular carcinoma facilitates tumor growth through enhancer of zeste homolog 2 in humans. Hepatology 54 (5), 1679–1689. doi:10.1002/hep.24563
Yang Z., Ming X. F. (2013). Arginase: the emerging therapeutic target for vascular oxidative stress and inflammation. Front. Immunol. 4, 149. doi:10.3389/fimmu.2013.00149
Yeh D. C., Duncan J. A., Yamashita S., Michel T. (1999). Depalmitoylation of endothelial nitric-oxide synthase by acyl-protein thioesterase 1 is potentiated by Ca(2+)-calmodulin. J. Biol. Chem. 274 (46), 33148–33154. doi:10.1074/jbc.274.46.33148
Yoshioka K., Otani H., Shimazu T., Fujita M., Iwasaka T., Shiojima I. (2015). Sepiapterin prevents left ventricular hypertrophy and dilatory remodeling induced by pressure overload in rats. Am. J. Physiol. Heart Circ. Physiol. 309 (10), H1782–H1791. doi:10.1152/ajpheart.00417.2015
Yu J. G., O'Brien W. E., Lee T. J. (1997). Morphologic evidence for L-citrulline conversion to L-arginine via the argininosuccinate pathway in porcine cerebral perivascular nerves. J. Cereb. Blood Flow. Metab. 17 (8), 884–893. doi:10.1097/00004647-199708000-00007
Zamani M., Rezaiian F., Saadati S., Naseri K., Ashtary-Larky D., Yousefi M., et al. (2023). The effects of folic acid supplementation on endothelial function in adults: a systematic review and dose-response meta-analysis of randomized controlled trials. Nutr. J. 22 (1), 12. doi:10.1186/s12937-023-00843-y
Zhang Q., Church J. E., Jagnandan D., Catravas J. D., Sessa W. C., Fulton D. (2006). Functional relevance of Golgi- and plasma membrane-localized endothelial NO synthase in reconstituted endothelial cells. Arterioscler. Thromb. Vasc. Biol. 26 (5), 1015–1021. doi:10.1161/01.ATV.0000216044.49494.c4
Zhang Y., Murugesan P., Huang K., Cai H. (2020). NADPH oxidases and oxidase crosstalk in cardiovascular diseases: novel therapeutic targets. Nat. Rev. Cardiol. 17 (3), 170–194. doi:10.1038/s41569-019-0260-8
Zhao Y. Y., Liu Y., Stan R. V., Fan L., Gu Y., Dalton N., et al. (2002). Defects in caveolin-1 cause dilated cardiomyopathy and pulmonary hypertension in knockout mice. Proc. Natl. Acad. Sci. U. S. A. 99 (17), 11375–11380. doi:10.1073/pnas.172360799
Zimmermann K., Opitz N., Dedio J., Renne C., Muller-Esterl W., Oess S. (2002). NOSTRIN: a protein modulating nitric oxide release and subcellular distribution of endothelial nitric oxide synthase. Proc. Natl. Acad. Sci. U. S. A. 99 (26), 17167–17172. doi:10.1073/pnas.252345399
Zweier J. L., Chen C. A., Druhan L. J. (2011). S-glutathionylation reshapes our understanding of endothelial nitric oxide synthase uncoupling and nitric oxide/reactive oxygen species-mediated signaling. Antioxid. Redox Signal 14 (10), 1769–1775. doi:10.1089/ars.2011.3904
ABH 2(S)-amino-6-hexanoic acid
ACh acetylcholine
ADMA asymmetric dimethylarginine
Akt protein kinase B
AMPK AMP-activated protein kinase
ASL argininosuccinate lyase
ASS argininosuccinate synthase
BEC S-(2-boronoethyl)-l-cysteine
BH2 dihydrobiopterin
BH4 tetrahydrobiopterin
CAD Coronary Artery Disease
CaM calmodulin
CaMKII calcium/calmodulin-dependent protein kinase II
CAT-1 cationic amino acid transporter-1
Cav-1 caveolin-1
cGMP cyclic GMP
CSD caveolin-scaffolding domain
CVD cardiovascular disease
CytB5R3 cytochrome b5 reductase 3
DHFR dihydrofolate reductase
Dyn2 dynamin-2
ECs endothelial cells
EDD end-diastolic diameter
eNOS endothelial nitric oxide synthase
FAD flavin adenine dinucleotide
FMD% flow-mediated dilation percentage
FMN flavin mononucleotide
GPCR G-protein coupled receptor
Grx1 glutaredoxin
GSH reduced glutathione
GSSG oxidized glutathione
GTPCH guanosine triphosphate cyclohydrolase I
Hbα alpha subunit of hemoglobin
HDL high-density lipoprotein
HIF-1α hypoxia-inducible factor-1 alpha
Hsp90 heat shock protein 90
ICAM-1 intracellular adhesion molecule 1
iNOS inducible NOS
KLF2/KLF4 Krüppel-like factor 2/Krüppel-like factor 4
LAD left anterior descending coronary artery
lncRNAs long non-coding RNAs
LPS lipopolysaccharide
MEJ myoendothelial junctions
MI myocardial infarction
METHF 5-methyltetrahydrofolate
NADPH nicotinamide adenine dinucleotide phosphate
NF-κB nuclear factor kappa-light-chain-enhancer of activated B cells
nNOS neuronal NOS
NO nitric oxide
NOHA N-hydroxy-L-arginine
nor-NOHA N-hydroxy-nor-L-arginine
NOS NO synthases
NOSIP eNOS interacting protein
NOSTRIN eNOS trafficking inducer
NOX2 NADPH oxidase 2
Nrf2 nuclear factor erythroid 2-related factor
ox-LDL oxidized low-density lipoprotein
PAH pulmonary arterial hypertension
PDE phosphodiesterase
PKA protein kinase A
PKG protein kinase G
PMCA plasma membrane Ca2+-ATPase
PPIs protein-protein interactions
PTMs post-translational modifications
ROS reactive oxygen species
sGC soluble guanylate cyclase
SIRT1 sirtuin 1
SMAD2 mothers against decapentaplegic homolog-2
SNOs s-nitrosothiols
SOCE store-operated Ca2+ entry
Sp1 specificity protein 1
Sp3 specificity protein 3
STIM1 stromal interaction molecule 1
T1D type 1 diabetes
T2D type 2 diabetes
TAC transverse aortic constriction
TGF-β tumor growth factor-beta
TNF-α tumor necrosis factor-alpha
VEGF vascular endothelial growth factor
VSMCs vascular smooth muscle cells
Keywords: nitric oxide, eNOS, endothelium, preclinical evidence, clinical trials
Citation: Gonzalez M, Clayton S, Wauson E, Christian D and Tran Q-K (2025) Promotion of nitric oxide production: mechanisms, strategies, and possibilities. Front. Physiol. 16:1545044. doi: 10.3389/fphys.2025.1545044
Received: 13 December 2024; Accepted: 07 January 2025;
Published: 23 January 2025.
Edited by:
Christopher Garland, University of Oxford, United KingdomReviewed by:
Michael P. Massett, Texas Tech University, United StatesCopyright © 2025 Gonzalez, Clayton, Wauson, Christian and Tran. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Quang-Kim Tran, a2ltLnRyYW5AZG11LmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.