 Chenyu Zhuang1†
Chenyu Zhuang1† Hengyu Yan
Hengyu Yan Yan Li
Yan Li
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol., 24 March 2025
Sec. Integrative Physiology
Volume 16 - 2025 | https://doi.org/10.3389/fphys.2025.1529981
Background: Amyloid-β protein (Aβ) accumulation is a defining characteristic of Alzheimer’s disease (AD), resulting in neurodegeneration and a decline in cognitive function. Given orexin’s well-documented role in enhancing memory and cognition, this study investigates its potential to regulate Aβ-induced neurotoxicity, offering new perspectives into AD management.
Methods: This paper simulated Aβ accumulation in the hippocampus of AD patients by administering Aβ1-42 oligomers into the bilateral hippocampal dentate gyrus of ICR mice. Inflammatory cytokines (IL-6, TNF-α) and orexin-A levels were measured by ELISA. Additionally, the excitability of orexinergic neurons was assessed by IHC targeting c-Fos expression. These methodologies evaluated the Aβ-induced neuroinflammation, orexinergic system functionality, and dexamethasone’s (Dex) effects on these processes.
Results: Injection of Aβ1-42 oligomer resulted in elevated levels of IL-6, TNF-α, and orexin-A in the hippocampus, as well as increased excitability of orexinergic neurons in the lateral hypothalamus (LH). Dex treatment reduced neuroinflammation, causing a reduction in orexin-A levels and the excitability of orexinergic neurons.
Conclusion: Aβ-induced neuroinflammation is accompanied by enhanced levels of orexin-A and orexinergic neuron excitability. These findings suggest that the enhanced functionality of the orexinergic system may become a compensatory neuroprotective mechanism to counteract neuroinflammation and enhance cognitive function.
Alzheimer’s Disease (AD) is the most prevalent form of dementia, posing a significant challenge and having a profound impact on the health and quality of life of the elderly population. In recent years, China has witnessed an increase in the incidence, prevalence, and mortality rates of AD, imposing a considerable economic burden on patients’ families, society, and the healthcare system (Li et al., 2022; Ren et al., 2022; Ji et al., 2024). Despite advancements in diagnostic techniques and a deepening understanding of its pathology, the lack of a clearly defined underlying mechanism has hindered the development of effective treatments to slow the progression of AD. Pathologically, AD is featured by substantial atrophy and death of brain cells. The primary cause of this degeneration is the accumulation of extracellular amyloid-β protein (Aβ) plaques, coupled with intraneuronal neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau proteins (Braak and Braak, 1991; Winblad et al., 2016). The presence of Aβ plaques and NFTs disrupts neuronal communication, resulting in synaptic loss and ultimately causing widespread neuronal death (Selkoe, 2002; Mattson, 2004). Further pathological features include gliosis, neuroinflammation, and significant synaptic changes (Itagaki et al., 1989; Calsolaro and Edison, 2016; Tönnies and Trushina, 2017).
Orexin, also referred to as hypocretin, is a neuropeptide synthesized in the hypothalamus and consists of two isoforms: orexin-A and orexin-B (de Lecea et al., 1998; Sakurai et al., 1998). Orexinergic neurons are primarily located in the lateral hypothalamus (LH) and extensively project their fibers to different regions of the brain (Vraka et al., 2023). The orexinergic system is essential in regulating sleep-wake cycles, feeding behavior, energy homeostasis, as well as reward processing (Chemelli et al., 1999; Edwards et al., 1999; Volkoff et al., 1999; Willie et al., 2001; Carter et al., 2009; Tsujino and Sakurai, 2009; Leinninger et al., 2011). Extensive research has demonstrated that the orexinergic system exerts an influence on the pathophysiology of AD, particularly in relation to the generation and accumulation of Aβ. Kang et al. discovered that injecting orexin into the ventricles of APP/PS1 mice elevates the levels of Aβ in the brain ISF, while infusion of a dual orexin receptor antagonist (DORA) effectively decreases Aβ levels (Kang et al., 2009). Similarly, studies have indicated that knocking out the orexin gene in APP/PS1 mice results in a reduction of Aβ deposition in the brain (Roh et al., 2014). Human studies have revealed that patients with moderate to severe AD present higher levels of orexin in their CSF compared to the control individuals (Liguori et al., 2014). However, there remains some disagreement regarding whether the orexinergic system actually promotes the progression of AD. On one hand, focal overexpression of orexin in the hippocampus of APP/PS1/OR−/− mice did not alter the levels of Aβ accumulation (Roh et al., 2014). Additionally, postmortem analysis of hypothalamic tissue from patients with AD displayed a 40% reduction in orexin-immunoreactive neurons, accompanied by a slight decrease in the ventricular cerebrospinal fluid (CSF) levels of orexin-A. (Fronczek et al., 2012).
On the other hand, some physiological functions of orexin are inconsistent with a role in the development of AD. Orexin plays a crucial role in hippocampus-dependent social memory consolidation, and the introduction of exogenous orexin can ameliorate this deficit by modulating synaptic plasticity in the hippocampal region (Yang et al., 2013). By regulating the activity of various neurotransmitter systems, including cholinergic and dopaminergic pathways, orexin can enhance learning processes, facilitate the retrieval of memories, as well as support the consolidation of acquired information (Telegdy and Adamik, 2002; Palotai et al., 2014; Piantadosi et al., 2015). The administration of orexin-A via the nasal cavity or directly into the hippocampus has been shown to alleviate memory deficits in orexin/ataxin-3 transgenic mice (Yang et al., 2013; Mavanji et al., 2017). The impairment of memory caused by orexin deficiency has been confirmed in humans. For instance, patients with narcolepsy frequently exhibit memory and cognitive decline that is associated with the disease (Henry et al., 1993; Naumann et al., 2006; Witt et al., 2018). Other studies have demonstrated that orexin exerts neuroprotective effects. Specifically, orexin-A inhibits neuroinflammation by decreasing astrocyte proliferation, microglial activation, as well as the expression of chemokines and cytokines (Becquet et al., 2019). Orexin receptors hinder excessive autophagy through the MAPK/ERK/mTOR signaling pathway and confer neuroprotective benefits in AD via heterodimerization with GPR103 (Davies et al., 2015; Xu et al., 2021).
In summary, considering the diverse physiological functions of orexin, it is unreasonable to regard it as a direct causal factor in AD. The function of orexin in bolstering cognitive abilities, particularly memory and learning, fundamentally contradicts the underlying pathological mechanisms of AD. Although some studies suggest that orexin influences Aβ dynamics, its overall beneficial effects indicate a protective, rather than pathogenic, role in AD. Therefore, we hypothesize that the increased orexin levels observed in the brains of AD patients may represent a compensatory neuroprotective mechanism, rather than a causative factor. The accumulation of Aβ stimulates a compensatory increase in orexin, which, in turn, enhances a range of neuroprotective mechanisms, thereby mitigating the memory and cognitive impairments related to Aβ deposition. To certify this hypothesis, the pathological characteristics of Aβ accumulation in the hippocampus of AD patients were simulated in this study by injecting Aβ1-42 oligomers into the bilateral hippocampal dentate gyrus. Subsequently, we observed the impact of various concentrations of Aβ1-42 oligomers on neuroinflammation in the hippocampus and the functionality of the orexinergic system in the brain. Additionally, the intervention effects of dexamethasone (Dex) on these impacts were evaluated.
Human Amyloid Peptide (1–42) (P9001, Beyotime Biotechnology, Shanghai, China) was dissolved in dimethyl sulfoxide (DMSO; ST038, Beyotime Biotechnology, Shanghai, China) to prepare a 3 mM stock solution. This solution was then diluted in sterile phosphate-buffered saline (PBS; PYG0021, Boster Biological Technology, Wuhan, China) to achieve the desired concentration for Aβ oligomers formation. Dexamethasone sodium phosphate solution (Hefei Zhonglong Shenli Animal Pharmaceutical Co., Ltd., Hefei, China) was administered to specific mouse groups to investigate its effects on neuroinflammation and orexin-A expression. Injections were performed using a Mouse Brain Stereotaxic apparatus (STW-3, Chengdu Instrument Factory, Chengdu, China) and Microliter Syringes (PYG0021, Shanghai High Pigeon Industry & Trade, Shanghai, China). The injection rate was controlled by a Dual-channel Intelligent Syringe Pump (XMSP-2B, Ximai Nanotech, Shanghai, China). Quantification of IL-6, TNF-α, and orexin-A levels in hippocampal tissues was performed using Mouse IL-6, TNF-α, and orexin-A ELISA Kits (BYabscience, Nanjing, China; Catalog Numbers: BY-EM220188, BY-EM220852, BY-EM228148). Immunohistochemical analysis to assess orexinergic neuron excitability used an Anti-c-Fos antibody (ab222699, Abcam, Cambridge, United Kingdom) and the SABC-POD (F) rabbit IgG kit (SA1028, Boster Biological Technology, Wuhan, China) to detect c-Fos protein expression.
Drawing on prior reports on preparing Aβ oligomers (Kim et al., 2016), we made a 3 mM stock of Aβ1-42 in dimethyl sulfoxide (DMSO) and then diluted it 10-fold in phosphate-buffered saline (PBS) (300 μM Aβ, 90% PBS, 10% DMSO). The Aβ1-42 monomer solution was then incubated at 37°C for 24 h to promote the formation of oligomers. After incubation, the solution was dispensed and frozen at −80°C for later use.
Male ICR mice (8–10 weeks old, weight 25 ± 3 g) were sourced from the Comparative Medicine Center of Yangzhou University (Yangzhou, China). These mice were housed in standard laboratory conditions, with a temperature maintained at 25°C ± 1°C, humidity levels kept between 45% and 50%, and a controlled light cycle from 6 a.m. to 6 p.m. The animal protocol followed the ethical guidelines and scientific standards approved by the Institutional Animal Care and Use Committee of Yangzhou University (ethical approval code: YXYLL-2024-111, date: 21 May 2024). The animals were randomly assigned to seven groups: a Dex group, a sham group, a low-concentration Aβ1-42 group, a medium-concentration Aβ1-42 group, a high-concentration Aβ1-42 group, a high-concentration Aβ1-42 + Dex group, as well as sham + Dex group, with 6 animals in each group.
The sham group received an injection of 1 μL of 0.9% saline solution into the bilateral hippocampal dentate gyrus. The groups receiving low, medium, and high concentrations of Aβ1-42 oligomers were injected with 125 μM, 250 μM, and 375 μM of the oligomers, respectively, all at the same volume and injection site. For seven consecutive days, the Dex group, sham + Dex group and the high-concentration Aβ1-42 + Dex group underwent daily intraperitoneal injections of Dex (2 mg/kg). In contrast, the remaining groups received an equivalent volume of saline. On the seventh day, 4 hours following the intraperitoneal injection, the brains of the mice were collected for further analysis. For the purpose of conducting an enzyme-linked immunosorbent assay (ELISA), the brain was promptly excised, snap-frozen, and then stored at −80°C to ensure the preservation of its biological activity. For immunohistochemistry (IHC) analysis, the mice were anesthetized and underwent transcardial perfusion with 0.9% saline, followed by 4% paraformaldehyde (PFA). Subsequently, the brains were extracted and immersed in 4% PFA overnight for fixation. Following fixation, the brains were dehydrated, embedded in paraffin blocks, and then sectioned into 4 μm sagittal paramedian slices using a microtome. The sections were positioned between 0.7 and 0.74 mm lateral to the brain midline, which corresponds to the location of the LH. For each brain tissue, three consecutive sections were retained, and the average ratio of the positive area from these sections was computed to serve as the representative value for each individual sample (as illustrated in Figure 1).
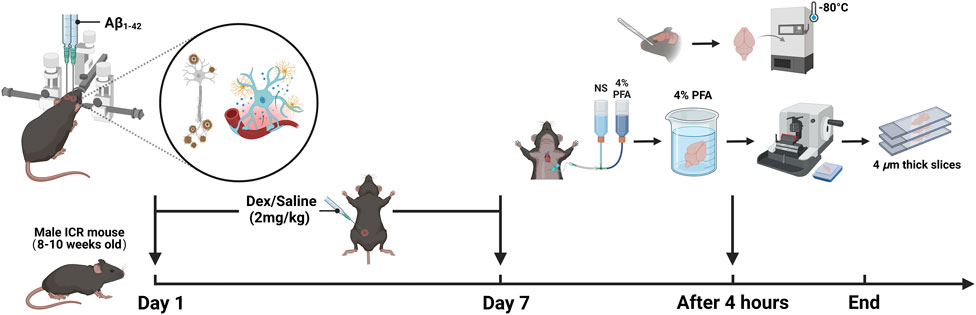
Figure 1. The design of experiments (created with BioRender.com).
The mice are first weighed and then anesthetized using 1% pentobarbital sodium (50 mg/kg) via intraperitoneal injection. Adequacy of anesthesia is confirmed by assessing the absence of a response to a pinch on the tail tip. Subsequently, the hair on the head of each mouse is cut off, and the mouse is fixed onto the ear bar of the brain stereotaxic apparatus. To prevent corneal dryness, sterile PBS is instilled into both eyes. The skin of the head is disinfected with iodophor, and a 1 cm incision is made in the center using ophthalmic scissors. The incision is then put open, and the mucosa covering the skull surface is peeled away using two injection needles, which help to dry the skull surface and facilitate the identification of the bregma. Once the bregma is identified, a point 2.0 mm posterior and 1.4 mm lateral to it is marked. At this marked location, a hole with a diameter of 1 mm is created using a grinder. Different concentrations of Aβ1−42 oligomers are drawn into a bilateral microsyringe and fixed onto the brain stereotaxic instrument. The needle tip is then moved to the drilled hole and inserted 2.5 mm from the skull surface to reach the hippocampus dentate gyrus. Aβ is injected simultaneously into both hippocampi over 5 min at a rate of 0.2 μL/min. Following the injection, the needle is left in place for an additional 5 min to aid in the diffusion of Aβ. The injection needle is then withdrawn, and the incision is sutured closed. After the surgery, each mouse is placed in a recovery box at 29°C until it has fully regained consciousness. During the first 3 days post-surgery, each mouse receives a daily subcutaneous injection of acetaminophen (200 mg/kg) for pain relief and enrofloxacin (5 mg/kg) for infection prevention, while closely monitoring the wound healing and overall health. If necessary, sterile PBS is applied to the wound area to maintain cleanliness. During the recovery period, the mice are housed individually in clean cages to prevent interference from other animals.
The mouse ELISA kit should be removed from the refrigerator and allowed to equilibrate at room temperature for 30 min. The steps outlined in the kit manual should be strictly adhered to, using a microplate reader to ascertain the optical density (OD) values for IL-6, TNF-α, and orexin in mouse hippocampal tissue samples. Subsequently, standard linear regression curves should be plotted, with the horizontal coordinate representing the standard concentration of IL-6, TNF-α, and orexin, and the vertical coordinate corresponding to the respective OD values. Based on the curve equation derived, the concentrations of these biomarkers in the mouse hippocampal samples can then be calculated.
The paraffin-embedded sections of brain tissue underwent deparaffinized in xylenes and were subsequently rehydrated through a graded series of alcohol solutions. After a 15-min pretreatment with 3% H2O2 to inactivate endogenous peroxidase activity, antigen retrieval was performed using citrate buffer. The sections were subsequently blocked with 5% BSA for 20 min and then incubated at 4°C overnight with the primary anti-c-Fos protein (1:500 dilution). The slices were then incubated with a secondary antibody for 30 min at 37°C, followed by the application of streptavidin-biotin complex (SABC) droplets. Between each step, the sections were thoroughly rinsed 3 times in Tris Buffered Saline (TBS) for 5 min each. The sections were then developed using diaminobenzidine (DAB) and counterstained with hematoxylin. Lastly, the sections underwent washing, dehydration, and clearing, as well as were mounted with cover slips.
Images of the sections were captured utilizing NIS Elements F 3.0 software (Nikon Corporation, Tokyo, Japan). Before photography, a blank site was evaluated with automatic white balancing. The immunohistochemical images were processed using ImageJ software (ImageJ 1.8.0, Rawak Software Inc., Stuttgart, Germany) to determine the ratio of positive areas.
Statistical analyses were conducted using GraphPad Prism version 10.1.2 (GraphPad Software, San Diego, CA, United States). The data for each group were presented as mean ± standard deviation (SD). One-way analysis of variance (ANOVA) was employed to compare data among multiple groups, followed by Tukey’s post hoc test for multiple comparisons. P < 0.05 was deemed statistically significant, whereas P > 0.05 was considered statistically non-significant (NS).
The levels of IL-6 and TNF-α in the hippocampus of mice were detected by ELISA to assess the expression of proinflammatory cytokines. Compared to the sham group, both IL-6 and TNF-α levels were significantly increased in the Aβ groups (p < 0.05, Figures 2A,B), indicating that injection of Aβ oligomers induces neuroinflammation. Dex treatment markedly decreased the Aβ-induced elevation of IL-6 and TNF-α levels (p < 0.05, Figures 2A,B), demonstrating its effectiveness in inhibiting pro-inflammatory cytokines and mitigating neuroinflammation in AD. No significant differences were observed in the levels of IL-6 and TNF-α among Dex, Sham, and Sham + Dex groups.
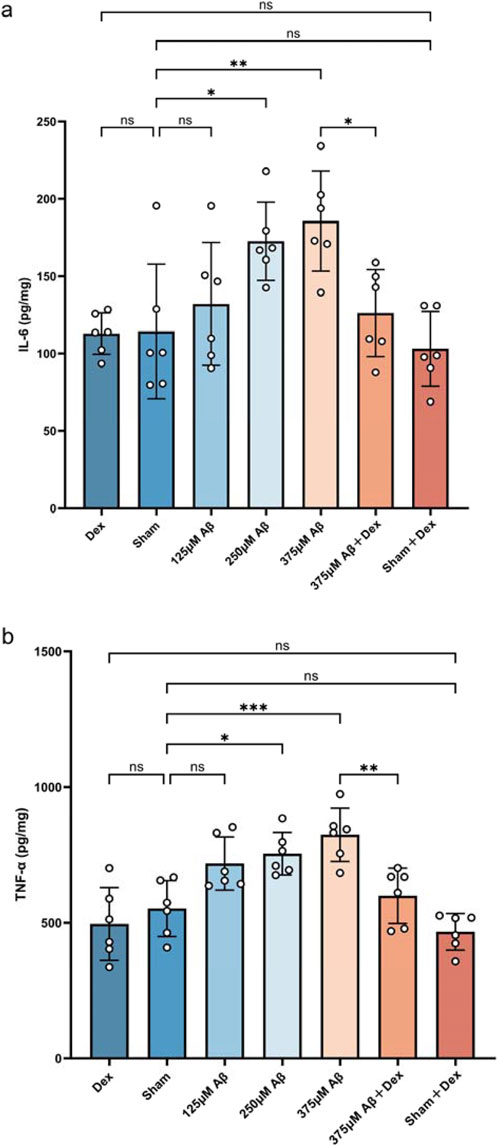
Figure 2. Aβ1-42 oligomers induce an increase in the secretion of pro-inflammatory cytokines. ELISA was utilized to measure the concentrations of inflammatory cytokines in hippocampal extracts obtained from each group. Injections of Aβ oligomers led to an upregulation in the expression of IL-6 (A) and TNF-α (B), while Dex treatment significantly decreased their levels. Statistical analysis was performed using one-way ANOVA followed by Tukey’s test. For IL-6, the ANOVA F = 6.250, with Tukey’s post hoc test showing p > 0.9999 for Dex (112.9 ± 13.39, n = 6) vs. Sham (114.3 ± 43.64, n = 6), p = 0.9977 for Dex vs. Sham + Dex (103.1 ± 24.17, n = 6), p = 0.9524 for Sham vs. 125 μM Aβ (132.1 ± 39.71, n = 6), p = 0.0365 for Sham vs. 250 μM Aβ (172.6 ± 25.24, n = 6), p = 0.0054 for Sham vs. 375 μM Aβ (185.7 ± 32.32, n = 6), p = 0.9954 for Sham vs Sham + Dex, and p = 0.0309 for 375 μM Aβ vs. 375 μM Aβ + Dex (126.2 ± 28.16, n = 6). For TNF-α, the ANOVA F = 11.44, with Tukey’s post hoc test showing p = 0.9532 for Dex (495.4 ± 133.8, n = 6) vs. Sham (552.1 ± 103.1, n = 6), p = 0.9986 for Dex vs. Sham + Dex (466.4 ± 67.31, n = 6), p = 0.0847 for Sham vs. 125 μM Aβ (717.9 ± 98.02, n = 6), p = 0.0185 for Sham vs. 250 μM Aβ (754.2 ± 78.35, n = 6), p = 0.0006 for Sham vs. 375 μM Aβ (824.1 ± 98.51, n = 6), p = 0.7457 for Sham vs. Sham + Dex, and p = 0.0066 for 375 μM Aβ vs. 375 μM Aβ + Dex (599.5 ± 102.0, n = 6). Data represent mean ± SD (n = 6); *: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001.
The levels of orexin-A in the hippocampus of mice continued to be monitored using the ELISA method. The results revealed that, compared to the sham group, orexin-A levels in the Aβ group were significantly evaluated (p < 0.05, Figure 3C), demonstrating a concentration-dependent relationship (p < 0.05, Figure 3C). Dex treatment significantly reduced orexin-A levels in the Aβ groups (p < 0.05, as displayed in Figure 3C), but the treatment had no significant effect on orexin-A levels in the sham + Dex group (p > 0.05, as shown in Figure 3C). No significant differences were observed in the levels of orexin-A among Dex, Sham, and Sham + Dex groups. This suggested that the increase of orexin-A was driven by the neuroinflammation induced by Aβ.
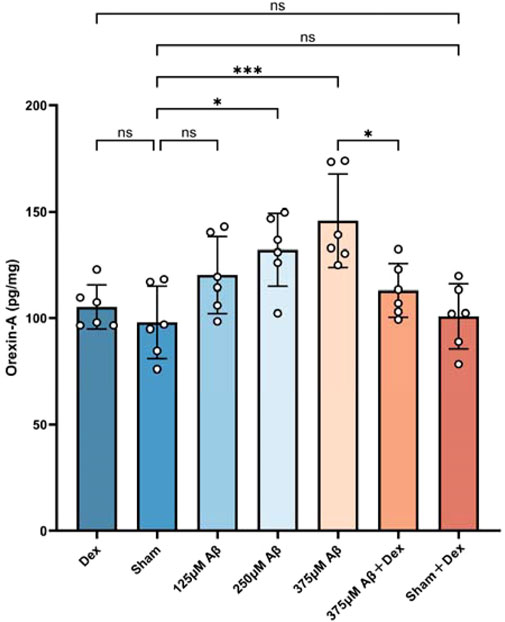
Figure 3. Elevated orexin-A levels induced by Aβ1-42 oligomer injection exhibited a clear concentration-dependency. The ELISA assay was used to evaluate the concentrations of orexin-A in hippocampal extracts obtained from different groups. The results demonstrated that injections of Aβ oligomers upregulated the expression of orexin-A (c), while Dex treatment reduced its level. Statistical analysis was performed using one-way ANOVA followed by Tukey’s test. The ANOVA F = 6.797, with Tukey’s post hoc test showing p = 0.9875 for Dex (105.2 ± 10.41, n = 6) vs. Sham (97.95 ± 16.99, n = 6), p = 0.9992 for Dex vs. Sham + Dex (100.8 ± 15.31, n = 6), p = 0.2528 for Sham vs. 125 μM Aβ (120.2 ± 18.12, n = 6), p = 0.0158 for Sham vs. 250 μM Aβ (132.1 ± 17.19, n = 6), p = 0.0003 for Sham vs. 375 μM Aβ (145.8 ± 22.05, n = 6), p > 0.9999 for Sham vs. Sham + Dex, and p = 0.0230 for 375 μM Aβ vs. 375 μM Aβ + Dex (113 ± 12.65, n = 6). Data are presented as mean ± SD (n = 6); *: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001.
Orexinergic neurons are predominantly located in the LH. To further assess functional changes in the orexinergic system, IHC was used to detect c-Fos positive neurons in the LH of mice. C-Fos gene, an immediate early gene transcribed in response to neuronal activation, is commonly used as a marker for identifying active neurons (Joo et al., 2016). The results demonstrated that compared to the sham group, injection of Aβ oligomer led to an increase in both c-Fos protein expression and the count of c-Fos-positive neurons (p < 0.05, Figures 4A,B), displaying a heightened excitability of orexinergic neurons. Additionally, the number of activated orexinergic neurons increased in correlation with higher concentrations of Aβ injection (p < 0.05, Figures 4A,B). Although Dex treatment significantly reduced c-Fos protein expression in the Aβ groups (p < 0.0001, Figures 4A,B), there was no measurable impact in the sham + Dex group (p > 0.05, Figures 4A,B), indicating that Dex does not affect the excitability of orexinergic neurons. No significant differences were observed in the count of c-Fos-positive neurons among the Dex, Sham, and Sham + Dex groups.

Figure 4. The excitability of orexinergic neurons in the lateral hypothalamus (LH) of AD mouse models induced by Aβ1-42 oligomers was increased. (a) Representative IHC staining images of c-Fos-positive neurons in the LH for each group, showing sagittal sections. Scale bar is indicated as 100 μm. The enlarged view is provided for better clarity. The 200 μm view can be viewed in the supplementary files link (https://www.frontiersin.org/articles/10.3389/fphys.2025.1529981/full#supplementary-material). The quantitative analysis of the area positive for c-Fos in neurons, conducted using ImageJ software, is presented in (b). The results demonstrated that the injection of Aβ oligomers increased c-Fos protein expression, resulting in an increased excitability of orexinergic neurons. Dex treatment specifically reduced the excitability of orexinergic neurons in the Aβ groups, without affecting the sham + Dex group. Statistical analysis was performed using one-way ANOVA followed by Tukey’s test. The ANOVA F = 62.8, with Tukey’s post hoc test showing p > 0.9999 for Dex (0.049 ± 0.01476, n = 6) vs. Sham (0.04633 ± 0.02271, n = 6), p = 0.9415 for Dex vs. Sham + Dex (0.06383 ± 0.009368, n = 6), p = 0.0189 for Sham vs. 125 μM Aβ (0.09667 ± 0.03789, n = 6), p < 0.0001 for Sham vs. 250 μM Aβ (0.2012 ± 0.02764, n = 6), p < 0.0001 for Sham vs. 375 μM Aβ (0.2533 ± 0.02904, n = 6), p = 0.8801 for Sham vs. Sham + Dex, and p < 0.0001 for 375 μM Aβ vs. 375 μM Aβ + Dex (0.1160 ± 0.02071, n = 6). Data are presented as mean ± SD (n = 6); *: p < 0.05, **: p < 0.01, ***: p < 0.001, ****: p < 0.0001.
Neuroinflammation is widely recognized as a central mechanism underlying the accumulation of amyloid-β protein (Aβ)-induced pathology in Alzheimer’s disease (AD). Research has shown that Aβ can stimulate microglia, causing them to shift towards the pro-inflammatory M1 phenotype and triggering the secretion of various pro-inflammatory factors, such as IL-6 and TNF-α, thereby initiating neuroinflammatory responses (Heneka et al., 2015; Tang and Le, 2016). This inflammatory response disrupts neuronal signal transmission, decreases synaptic plasticity, and enhances oxidative stress reactions, ultimately exacerbating neuronal damage (Block et al., 2007; Cai et al., 2019). Synaptic plasticity is a fundamental process that underlies learning and memory. The persistent release of pro-inflammatory factors can disrupt neuronal signaling pathways, causing synaptic loss and ultimately contributing to significant cognitive impairment in AD (De Strooper and Karran, 2016; Ransohoff, 2016). In this study, we successfully induced a significant upregulation of pro-inflammatory factors by injecting Aβ1-42 oligomers into the dentate gyrus of the mouse hippocampus, effectively simulating the neuroinflammatory pathological features of AD.
Previous studies have suggested that elevated orexin levels may lead to increased Aβ levels. For instance, Kang et al. found that injecting orexin into APP transgenic mice resulted in higher Aβ deposition, whereas administering a dual orexin receptor antagonist (DORA) reduced Aβ levels (Kang et al., 2009). Similarly, Roh et al. observed reduced Aβ deposition following the knockout of the orexin gene in APP/PS1 transgenic mice (Roh et al., 2014). These studies, along with our research, demonstrate a positive correlation between orexin and Aβ, although the causal relationships differ. It is important to note that the orexin system plays a key role in promoting and maintaining wakefulness (Arrigoni et al., 2010), facilitating voluntary activity (Kiwaki et al., 2004), and supporting energy metabolism (Tsujino and Sakurai, 2009). On one hand, during wakefulness, metabolic products such as adenosine and Aβ inevitably accumulate in the brain (Huang et al., 2011; Spira et al., 2014). On the other hand, there is literature showing that Aβ-degrading enzymes, such as neprilysin (NEP), exhibit increased activity during sleep when orexinergic neurons are inhibited, thereby enhancing Aβ degradation (Ogawa et al., 2005). However, previous studies seem to have overlooked the impact of metabolic product accumulation during wakefulness and the corresponding decrease in Aβ-degrading enzyme activity.
Recent research has demonstrated that the orexinergic system enhances cognitive functions. Specifically, orexin-A promotes the transformation of microglia into the anti-inflammatory M2 phenotype (Duffy et al., 2015), which exhibits anti-inflammatory and neuroprotective properties, improving cognitive deficits (Saijo and Glass, 2011; Wang et al., 2020). The role of the orexinergic system in regulating the body’s energy metabolism is well-established (Tsujino and Sakurai, 2009). From the survival perspective, it also fundamentally aids humans and animals in adapting to their environment and maintaining survival capabilities. For example, when animals are hungry or hypoglycemic, the excitability of orexinergic neurons increases (Risold et al., 1999), resulting in increased secretion of orexin-A (Ouedraogo et al., 2003). The enhanced functionality of the orexinergic system fosters and maintains wakefulness, augments autonomous activity, and boosts muscle energy consumption. Furthermore, it also enhances alertness and cognitive functions (Kiwaki et al., 2004; Yang et al., 2013). Collectively, these effects promote feeding behavior to fulfill energy requirements. This adaptive mechanism underscores the crucial role of the orexinergic system in ensuring survival by modulating physiological and behavioral responses according to the environment.
Neuroinflammation induced by the accumulation of Aβ results in cognitive impairments in both humans and animals, evidently compromising their adaptability and survival capabilities. To counteract the cognitive toxicity induced by Aβ, the body inevitably initiates compensatory responses. Our study demonstrates that intracerebral injection of Aβ led to a concentration-dependent increase in both orexin-A levels in the hippocampus, as well as an enhancement in the excitability of orexinergic neurons in the lateral hypothalamus (LH). This suggests that the accumulation of Aβ not only triggers neuroinflammation but also simultaneously augments the functionality of the orexinergic system.
Dexamethasone (Dex) is a glucocorticoid anti-inflammatory agent capable of penetrating the blood-brain barrier (BBB) (Vohra et al., 2021). In our study, intervention with Dex significantly mitigated Aβ-induced neuroinflammation, leading to decreased levels of orexin-A in the hippocampus and reduced excitability of orexinergic neurons in the LH. These findings further affirm that the elevation in orexin-A levels is a consequence of Aβ-induced neuroinflammation, and they bolster the hypothesis that the orexinergic system serves as a neuroprotective mechanism against Aβ-induced cognitive toxicity.
While our study provides valuable insights into the compensatory role of the orexinergic system in AD, there are several limitations that should be considered. First, although we focused on male mice to reduce variability caused by hormonal fluctuations, this choice limits our ability to fully assess potential sex-based differences in the impact of Aβ and the orexinergic system. Future studies should include both male and female animals to explore whether sex-based differences influence the compensatory mechanisms observed in AD. Furthermore, while our study utilized c-Fos as a marker of neuronal excitability, it is worth noting that more advanced techniques, such as electrophysiological recordings or calcium imaging, could offer a more comprehensive understanding of the dynamic activity of orexinergic neurons. This limitation could be addressed in future studies.
Besides, caution is needed when applying these findings to humans due to the inability of animal models to fully replicate the complexity of human AD, particularly in terms of genetics and the multifactorial nature of the disease. Further research is required to clarify the molecular mechanisms and signaling pathways through which the orexinergic system regulates AD pathology. Additionally, it is crucial to consider the implications of dosage, treatment duration, and long-term effects when exploring the orexinergic system as a therapeutic target. These considerations will help guide future research directions and their potential clinical applications.
The latest studies have explored various anti-inflammatory treatment strategies for AD. Both lupeol, a natural compound, and NB-02, a botanical therapeutic drug, have demonstrated neuroprotective effects by regulating neuroinflammation (Lee et al., 2021; Choe, 2024). Additionally, the activation of the receptor TREM2 on microglia has been proven to reduce neuroinflammation and improve cognitive outcomes in AD, while monoclonal antibodies targeting TREM2 further support its therapeutic potential (Fassler et al., 2021). Methylprednisolone and low-dose aspirin have also shown promise in regulating neuroinflammation and mitigating cognitive decline (Vallés et al., 2020; Sun et al., 2023). Furthermore, GLP-1 receptor agonists have been found to reduce neuroinflammation and amyloid precursor protein (APP) deposition, while improving memory and synaptic function (Nowell et al., 2022). In summary, these studies underscore the potential of targeting neuroinflammation as a therapeutic strategy for AD, and the findings of our study are in alignment with this growing body of work.
Neuroinflammation induced by Aβ is accompanied by an augmentation in the functionality of the orexinergic system, featured by an increase in the excitability of orexinergic neurons and an elevated secretion of orexin-A. The enhanced functionality of the orexinergic system acts as a compensatory response to counteract Aβ-induced neuroinflammation, ultimately aiming to improve cognition. This study contributes to our understanding of the pathophysiological mechanisms underlying AD.
The original contributions presented in the study are included in the article/Supplementary Material, further inquiries can be directed to the corresponding author.
The animal study was approved by the animal protocol was approved based on ethical procedures and scientific care by the Yangzhou University-Institutional Animal Care and Use Committee (ethical approval code: YXYLL-2024-111, date: 21 May 2024). The study was conducted in accordance with the local legislation and institutional requirements.
CZ: Funding acquisition, Methodology, Project administration, Validation, Writing–original draft, Writing–review and editing. HY: Investigation, Project administration, Validation, Writing–original draft. JL: Formal Analysis, Investigation, Visualization, Writing–original draft. YZ: Formal Analysis, Investigation, Software, Writing–original draft. YqL: Conceptualization, Resources, Writing–review and editing. GS: Data curation, Software, Writing–review and editing. YaL: Conceptualization, Funding acquisition, Resources, Supervision, Writing–review and editing.
The author(s) declare that financial support was received for the research and/or publication of this article. This research was funded by National Natural Science Foundation of China (82104724). This research was funded by Science and Technology Project of Guizhou Province [Qiankehe Foundation-ZK (2021) General 502].This research was funded by Undergraduate Students’ Innovation Training Program of Jiangsu Province (202411117052Z).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Gen AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2025.1529981/full#supplementary-material
Supplementary Data Sheet S1 | Representative IHC staining images of c-Fos-positive neurons in the LH for each experimental group, showing sagittal sections. The scale bars are indicated as 100 μm, 200 μm, and an enlarged view is provided for better clarity.
Arrigoni E., Mochizuki T., Scammell T. E. (2010). Activation of the basal forebrain by the orexin/hypocretin neurones. Acta Physiol. 198 (3), 223–235. doi:10.1111/j.1748-1716.2009.02036.x
Becquet L., Abad C., Leclercq M., Miel C., Jean L., Riou G., et al. (2019). Systemic administration of orexin A ameliorates established experimental autoimmune encephalomyelitis by diminishing neuroinflammation. J. Neuroinflammation 16 (1), 64. doi:10.1186/s12974-019-1447-y
Block M. L., Zecca L., Hong J. S. (2007). Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat. Rev. Neurosci. 8 (1), 57–69. doi:10.1038/nrn2038
Braak H., Braak E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82 (4), 239–259. doi:10.1007/bf00308809
Cai M., Lee J. H., Yang E. J. (2019). Electroacupuncture attenuates cognition impairment via anti-neuroinflammation in an Alzheimer's disease animal model. J. Neuroinflammation 16 (1), 264. doi:10.1186/s12974-019-1665-3
Calsolaro V., Edison P. (2016). Neuroinflammation in Alzheimer's disease: current evidence and future directions. Alzheimers Dement. 12 (6), 719–732. doi:10.1016/j.jalz.2016.02.010
Carter M. E., Adamantidis A., Ohtsu H., Deisseroth K., de Lecea L. (2009). Sleep homeostasis modulates hypocretin-mediated sleep-to-wake transitions. J. Neurosci. 29 (35), 10939–10949. doi:10.1523/jneurosci.1205-09.2009
Chemelli R. M., Willie J. T., Sinton C. M., Elmquist J. K., Scammell T., Lee C., et al. (1999). Narcolepsy in orexin knockout mice: molecular genetics of sleep regulation. Cell. 98 (4), 437–451. doi:10.1016/s0092-8674(00)81973-x
Choe K., Park J. S., Park H. Y., Tahir M., Park T. J., Kim M. O. (2024). Lupeol protect against LPS-induced neuroinflammation and amyloid beta in adult mouse Hippocampus. Front. Nutr. 11, 1414696. doi:10.3389/fnut.2024.1414696
Davies J., Chen J., Pink R., Carter D., Saunders N., Sotiriadis G., et al. (2015). Orexin receptors exert a neuroprotective effect in Alzheimer's disease (AD) via heterodimerization with GPR103. Sci. Rep. 5, 12584. doi:10.1038/srep12584
de Lecea L., Kilduff T. S., Peyron C., Gao X., Foye P. E., Danielson P. E., et al. (1998). The hypocretins: hypothalamus-specific peptides with neuroexcitatory activity. Proc. Natl. Acad. Sci. U. S. A. 95 (1), 322–327. doi:10.1073/pnas.95.1.322
De Strooper B., Karran E. (2016). The cellular phase of Alzheimer's disease. Cell. 164 (4), 603–615. doi:10.1016/j.cell.2015.12.056
Duffy C. M., Yuan C., Wisdorf L. E., Billington C. J., Kotz C. M., Nixon J. P., et al. (2015). Role of orexin A signaling in dietary palmitic acid-activated microglial cells. Neurosci. Lett. 606, 140–144. doi:10.1016/j.neulet.2015.08.033
Edwards C. M., Abusnana S., Sunter D., Murphy K. G., Ghatei M. A., Bloom S. R. (1999). The effect of the orexins on food intake: comparison with neuropeptide Y, melanin-concentrating hormone and galanin. J. Endocrinol. 160 (3), R7–R12. doi:10.1677/joe.0.160r007
Fassler M., Rappaport M., Cuño C. B., George J. (2021). Engagement of TREM2 by a novel monoclonal antibody induces activation of microglia and improves cognitive function in alzheimer’s disease models. J. Neuroinflammation 18 (1), 19. doi:10.1186/s12974-020-01980-5
Fronczek R., van Geest S., Frölich M., Overeem S., Roelandse F. W., Lammers G. J., et al. (2012). Hypocretin (orexin) loss in Alzheimer's disease. Neurobiol. Aging 33 (8), 1642–1650. doi:10.1016/j.neurobiolaging.2011.03.014
Heneka M. T., Carson M. J., El Khoury J., Landreth G. E., Brosseron F., Feinstein D. L., et al. (2015). Neuroinflammation in Alzheimer's disease. Lancet Neurol. 14 (4), 388–405. doi:10.1016/s1474-4422(15)70016-5
Henry G. K., Satz P., Heilbronner R. L. (1993). Evidence of a perceptual-encoding deficit in narcolepsy? Sleep 16 (2), 123–127. doi:10.1093/sleep/16.2.123
Huang Z. L., Urade Y., Hayaishi O. (2011). The role of adenosine in the regulation of sleep. Curr. Top. Med. Chem. 11 (8), 1047–1057. doi:10.2174/156802611795347654
Itagaki S., McGeer P. L., Akiyama H., Zhu S., Selkoe D. (1989). Relationship of microglia and astrocytes to amyloid deposits of Alzheimer disease. J. Neuroimmunol. 24 (3), 173–182. doi:10.1016/0165-5728(89)90115-x
Ji Q., Chen J., Li Y., Tao E., Zhan Y. (2024). Incidence and prevalence of Alzheimer's disease in China: a systematic review and meta-analysis. Eur. J. Epidemiol. 39 (7), 701–714. doi:10.1007/s10654-024-01144-2
Joo J. Y., Schaukowitch K., Farbiak L., Kilaru G., Kim T. K. (2016). Stimulus-specific combinatorial functionality of neuronal c-fos enhancers. Nat. Neurosci. 19 (1), 75–83. doi:10.1038/nn.4170
Kang J. E., Lim M. M., Bateman R. J., Lee J. J., Smyth L. P., Cirrito J. R., et al. (2009). Amyloid-beta dynamics are regulated by orexin and the sleep-wake cycle. Science 326 (5955), 1005–1007. doi:10.1126/science.1180962
Kim H. Y., Lee D. K., Chung B. R., Kim H. V., Kim Y. (2016). Intracerebroventricular injection of amyloid-β peptides in normal mice to acutely induce alzheimer-like cognitive deficits. J. Vis. Exp. 109, 53308. doi:10.3791/53308
Kiwaki K., Kotz C. M., Wang C., Lanningham-Foster L., Levine J. A. (2004). Orexin A (hypocretin 1) injected into hypothalamic paraventricular nucleus and spontaneous physical activity in rats. Am. J. Physiol. Endocrinol. Metab. 286 (4), E551–E559. doi:10.1152/ajpendo.00126.2003
Lee Y. F., Lariviere L., Russ A. N., Choi S.-Z., Bacskai B. J., Kastanenka K. V. (2021). Novel botanical therapeutic NB-02 effectively treats alzheimer’s neuropathophysiology in an APP/PS1 mouse model. Eneuro 8(3). doi:10.1523/eneuro.0389-20.2021
Leinninger G. M., Opland D. M., Jo Y. H., Faouzi M., Christensen L., Cappellucci L. A., et al. (2011). Leptin action via neurotensin neurons controls orexin, the mesolimbic dopamine system and energy balance. Cell. Metab. 14 (3), 313–323. doi:10.1016/j.cmet.2011.06.016
Li R., Qi J., Yang Y., Wu Y., Yin P., Zhou M., et al. (2022). Disease burden and attributable risk factors of Alzheimer's disease and dementia in China from 1990 to 2019. J. Prev. Alzheimers Dis. 9 (2), 306–314. doi:10.14283/jpad.2021.69
Liguori C., Romigi A., Nuccetelli M., Zannino S., Sancesario G., Martorana A., et al. (2014). Orexinergic system dysregulation, sleep impairment, and cognitive decline in Alzheimer disease. JAMA Neurol. 71 (12), 1498–1505. doi:10.1001/jamaneurol.2014.2510
Mattson M. P. (2004). Pathways towards and away from Alzheimer's disease. Nature 430 (7000), 631–639. doi:10.1038/nature02621
Mavanji V., Butterick T. A., Duffy C. M., Nixon J. P., Billington C. J., Kotz C. M. (2017). Orexin/hypocretin treatment restores hippocampal-dependent memory in orexin-deficient mice. Neurobiol. Learn Mem. 146, 21–30. doi:10.1016/j.nlm.2017.10.014
Naumann A., Bellebaum C., Daum I. (2006). Cognitive deficits in narcolepsy. J. Sleep. Res. 15 (3), 329–338. doi:10.1111/j.1365-2869.2006.00533.x
Nowell J., Blunt E., Edison P. (2022). Incretin and insulin signaling as novel therapeutic targets for alzheimer’s and Parkinson’s disease. Mol. Psychiatry 28 (1), 217–229. doi:10.1038/s41380-022-01792-4
Ogawa T., Kiryu-Seo S., Tanaka M., Konishi H., Iwata N., Saido T. C., et al. (2005). Altered expression of neprilysin family members in the pituitary gland of sleep-disturbed rats, an animal model of severe fatigue. J. Neurochem. 95 (4), 1156–1166. doi:10.1111/j.1471-4159.2005.03436.x
Ouedraogo R., Näslund E., Kirchgessner A. L. (2003). Glucose regulates the release of orexin-a from the endocrine pancreas. Diabetes 52 (1), 111–117. doi:10.2337/diabetes.52.1.111
Palotai M., Telegdy G., Ekwerike A., Jászberényi M. (2014). The action of orexin B on passive avoidance learning. Involvement of neurotransmitters. Behav. Brain Res. 272, 1–7. doi:10.1016/j.bbr.2014.06.016
Piantadosi P. T., Holmes A., Roberts B. M., Bailey A. M. (2015). Orexin receptor activity in the basal forebrain alters performance on an olfactory discrimination task. Brain Res. 1594, 215–222. doi:10.1016/j.brainres.2014.10.041
Ransohoff R. M. (2016). How neuroinflammation contributes to neurodegeneration. Science 353 (6301), 777–783. doi:10.1126/science.aag2590
Ren R., Qi J., Lin S., Liu X., Yin P., Wang Z., et al. (2022). The China alzheimer report 2022. Gen. Psychiatr. 35 (1), e100751. doi:10.1136/gpsych-2022-100751
Risold P. Y., Griffond B., Kilduff T. S., Sutcliffe J. G., Fellmann D. (1999). Preprohypocretin (orexin) and prolactin-like immunoreactivity are coexpressed by neurons of the rat lateral hypothalamic area. Neurosci. Lett. 259 (3), 153–156. doi:10.1016/s0304-3940(98)00906-9
Roh J. H., Jiang H., Finn M. B., Stewart F. R., Mahan T. E., Cirrito J. R., et al. (2014). Potential role of orexin and sleep modulation in the pathogenesis of Alzheimer's disease. J. Exp. Med. 211 (13), 2487–2496. doi:10.1084/jem.20141788
Saijo K., Glass C. K. (2011). Microglial cell origin and phenotypes in health and disease. Nat. Rev. Immunol. 11 (11), 775–787. doi:10.1038/nri3086
Sakurai T., Amemiya A., Ishii M., Matsuzaki I., Chemelli R. M., Tanaka H., et al. (1998). Orexins and orexin receptors: a family of hypothalamic neuropeptides and G protein-coupled receptors that regulate feeding behavior. Cell. 92 (4), 573–585. doi:10.1016/s0092-8674(00)80949-6
Selkoe D. J. (2002). Alzheimer's disease is a synaptic failure. Science 298 (5594), 789–791. doi:10.1126/science.1074069
Spira A. P., Chen-Edinboro L. P., Wu M. N., Yaffe K. (2014). Impact of sleep on the risk of cognitive decline and dementia. Curr. Opin. Psychiatry 27 (6), 478–483. doi:10.1097/yco.0000000000000106
Sun Y., Li J., A N., Li Z., Zhong W., Chen L., et al. (2023). Methylprednisolone alleviates cognitive functions through the regulation of neuroinflammation in alzheimer’s disease. Front. Immunol. 14, 1192940. doi:10.3389/fimmu.2023.1192940
Tang Y., Le W. (2016). Differential roles of M1 and M2 microglia in neurodegenerative diseases. Mol. Neurobiol. 53 (2), 1181–1194. doi:10.1007/s12035-014-9070-5
Telegdy G., Adamik A. (2002). The action of orexin A on passive avoidance learning. Involvement of transmitters. Regul. Pept. 104 (1-3), 105–110. doi:10.1016/s0167-0115(01)00341-x
Tönnies E., Trushina E. (2017). Oxidative stress, synaptic dysfunction, and Alzheimer's disease. J. Alzheimers Dis. 57 (4), 1105–1121. doi:10.3233/jad-161088
Tsujino N., Sakurai T. (2009). Orexin/hypocretin: a neuropeptide at the interface of sleep, energy homeostasis, and reward system. Pharmacol. Rev. 61 (2), 162–176. doi:10.1124/pr.109.001321
Vallés S. L., Aldasoro M. N., Aldasoro C., Guerra-Ojeda S., Iradi A., Vila J. M., et al. (2020). Action of low doses of aspirin in inflammation and oxidative stress induced by aβ1-42 on astrocytes in primary culture. Int. J. Med. Sci. 17 (6), 834–843. doi:10.7150/ijms.40959
Vohra M., Sharma A. R., Satyamoorthy K., Rai P. S. (2021). Pharmacogenomic considerations for repurposing of dexamethasone as a potential drug against SARS-CoV-2 infection. Per Med. 18 (4), 389–398. doi:10.2217/pme-2020-0183
Volkoff H., Bjorklund J. M., Peter R. E. (1999). Stimulation of feeding behavior and food consumption in the goldfish, Carassius auratus, by orexin-A and orexin-B. Brain Res. 846 (2), 204–209. doi:10.1016/s0006-8993(99)02052-1
Vraka K., Mytilinaios D., Katsenos A. P., Serbis A., Baloyiannis S., Bellos S., et al. (2023). Cellular localization of orexin 1 receptor in human hypothalamus and morphological analysis of neurons expressing the receptor. Biomolecules 13 (4), 592. doi:10.3390/biom13040592
Wang H., Peng G., Wang B., Yin H., Fang X., He F., et al. (2020). IL-1R(-/-) alleviates cognitive deficits through microglial M2 polarization in AD mice. Brain Res. Bull. 157, 10–17. doi:10.1016/j.brainresbull.2019.11.020
Willie J. T., Chemelli R. M., Sinton C. M., Yanagisawa M. (2001). To eat or to sleep? Orexin in the regulation of feeding and wakefulness. Annu. Rev. Neurosci. 24, 429–458. doi:10.1146/annurev.neuro.24.1.429
Winblad B., Amouyel P., Andrieu S., Ballard C., Brayne C., Brodaty H., et al. (2016). Defeating Alzheimer's disease and other dementias: a priority for European science and society. Lancet Neurol. 15 (5), 455–532. doi:10.1016/s1474-4422(16)00062-4
Witt S. T., Drissi N. M., Tapper S., Wretman A., Szakács A., Hallböök T., et al. (2018). Evidence for cognitive resource imbalance in adolescents with narcolepsy. Brain Imaging Behav. 12 (2), 411–424. doi:10.1007/s11682-017-9706-y
Xu D., Kong T., Zhang S., Cheng B., Chen J., Wang C. (2021). Orexin-A protects against cerebral ischemia-reperfusion injury by inhibiting excessive autophagy through OX1R-mediated MAPK/ERK/mTOR pathway. Cell. Signal 79, 109839. doi:10.1016/j.cellsig.2020.109839
Keywords: Alzheimer’s disease, orexin (hypocretin), amyloid-β protein, cognitive impairment, neuroprotection
Citation: Zhuang C, Yan H, Lu J, Zhou Y, Liu Y, Shi G and Li Y (2025) Compensatory enhancement of orexinergic system functionality induced by amyloid-β protein: a neuroprotective response in Alzheimer’s disease. Front. Physiol. 16:1529981. doi: 10.3389/fphys.2025.1529981
Received: 18 November 2024; Accepted: 05 March 2025;
Published: 24 March 2025.
Edited by:
Elizabeth S. Fernandes, Aarhus University, DenmarkReviewed by:
Itzel Escobedo, National Autonomous University of Mexico, MexicoCopyright © 2025 Zhuang, Yan, Lu, Zhou, Liu, Shi and Li. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Yan Li, bGl5YW56Z0B5enUuZWR1LmNu
†These authors have contributed equally to this work
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.