
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 17 February 2025
Sec. Skeletal Physiology
Volume 16 - 2025 | https://doi.org/10.3389/fphys.2025.1514302
 Xinda Zheng1†
Xinda Zheng1† Zhuqing Dong2†
Zhuqing Dong2† Xiaofei Ding1Qian Huang2Shengping Tang2Yuchen Zhang1Boxiang Li3*
Xiaofei Ding1Qian Huang2Shengping Tang2Yuchen Zhang1Boxiang Li3* Shijie Liao1*
Shijie Liao1*Legg–Calvé–Perthes disease (LCPD) is a hip disease caused by ischemia of the femoral epiphysis in children, which occurs in children aged 4–8 years (mean 6.5 years), with a male-to-female ratio of about 4:1. The disease has been reported for more than 100 years, but its etiology has not been elucidated. In recent years, a considerable amount of research has been carried out on the etiology of the disease, and the development of the disease is believed to involve a variety of molecular biological alterations, such as the COL2A1 mutation, which may be one of the causes of necrotic collapses of the epiphyseal cartilage matrix in LCPD. Tissue factor V Leiden mutation and insulin-like growth factor (IGF-1) abnormalities have also been reported in LCPD, but most theories need further confirmation. The in-depth study of LCPD cell biology has facilitated the suggestion regarding structural and/or functional abnormalities of microvascular endothelial cells in LCPD. This conjecture is supported by epidemiological and clinical evidence. Abnormal activation of osteoclasts, ischemic damage to epiphyseal cartilage, and activation of the bone marrow immune system all play important roles in the onset and progression of the disease. In this paper, we review the previous basic studies on LCPD and give an overview from the molecular biology and cell biology perspectives.
Legg–Calvé–Perthes disease (LCPD) was first described in 1910 by three researchers, Legg (Legg, 2006), Calvé (Calve, 2006), and Perthes (Perthes, 2012). LCPD is also referred to as ischemic necrosis of the femoral head epiphysis in children. The disease commonly occurs in children aged 4–8 years (average 6.5 years), with an incidence of 0.4/100,000 to 29/100,000, and a male-to-female ratio of about 4:1. Its incidence is associated with regional socioeconomic status, gender, and race (Loder and Skopelja, 2011; Kessler and Cannamela, 2018). The natural course of LCPD is approximately 34 months, and after healing of femoral head necrosis, varying degrees of deformity may persist, with patients at risk of early-onset arthritis after skeletal maturation (Catterall, 1971; Mose, 1980; Stulberg et al., 1981). Clinically, the etiology of LCPD is often not well understood, imposing a significant physical and psychological burden on children, making understanding the cause of this disease essential. Although this disease has been reported for over a century, extensive etiological research has been carried out by many scholars, but the exact cause remains uncertain. Traditional perspectives attribute the disease to factors such as low socioeconomic status, maternal or childhood tobacco exposure (Daniel et al., 2012; Perry et al., 2017), endocrine abnormalities like insulin-like growth factor 1 (IGF-1) dysfunction (Baş et al., 2015), and anatomical issues such as venous congestion in the femoral head, arterial occlusion, vascular anomalies, and congenital factors like in-utero effects (Perry et al., 2012a). Detailed discussions of these potential causes have been provided by scholars. For example, Gao et al. (2020) explained the relationship between passive smoking and LCPD from the perspective of different passive smoking types (e.g., paternal smoking, maternal smoking, tobacco exposure during pregnancy). The etiology of LCPD was fully elaborated by Rodríguez-Olivas et al. (2022), who suggested that LCPD may be correlated with factors such as the environment, race, and coagulation. Some scholars also found that some molecular markers related to the pathogenesis of LCPD, including VEGF, eNOS, and IL-6, may be involved in the disease progression of LCPD, potentially helping the treatment and diagnosis to some extent (Spasovski et al., 2023) (as shown in Figure 1).
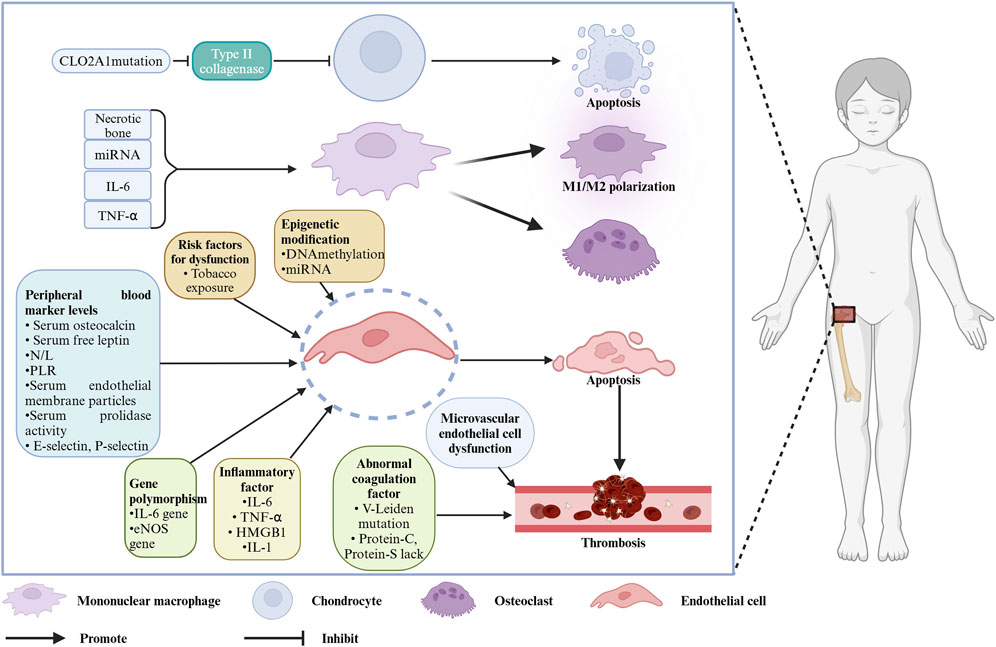
Figure 1. Molecules and cells associated with the pathogenesis of LCPD. Created with BioRender.com.
In recent years, significant progress has been made in etiological research on LCPD. Building on these reviews, we aim to summarize the current research status from a molecular biology perspective, focusing on aspects like gene mutations, epigenetic modifications, and gene polymorphisms. Molecular-level changes may further affect cell function, so we summarize the progress in pathogenesis research from a cellular biology perspective, particularly focusing on microvascular endothelial cell structural and/or functional abnormalities in the hip joint, abnormal osteoclast activation, ischemic damage to epiphyseal cartilage, and the bone marrow immune system. This deepens the understanding of the disease and provides a reference for further basic research.
The molecular level is the lowest level at which we currently understand the disease and may be the cause of the disease. Previous reports of changes in the molecular level of LCPD may include COL2A1 mutation, tissue factor V Leiden mutation, and IGF-1dysfunction, but these molecular changes as independent factors to cause the disease remain controversial, including sample selectivity bias, and small sample size. In recent years, significant progress has been made in the molecular biology of this disease. We therefore attempt to summarize the advances in molecular biological research from the perspectives of gene mutations, epigenetic modifications, and gene polymorphisms, to elucidate the molecular-level pathological changes in LCPD. The main findings of the included articles are summarized (Table 1).
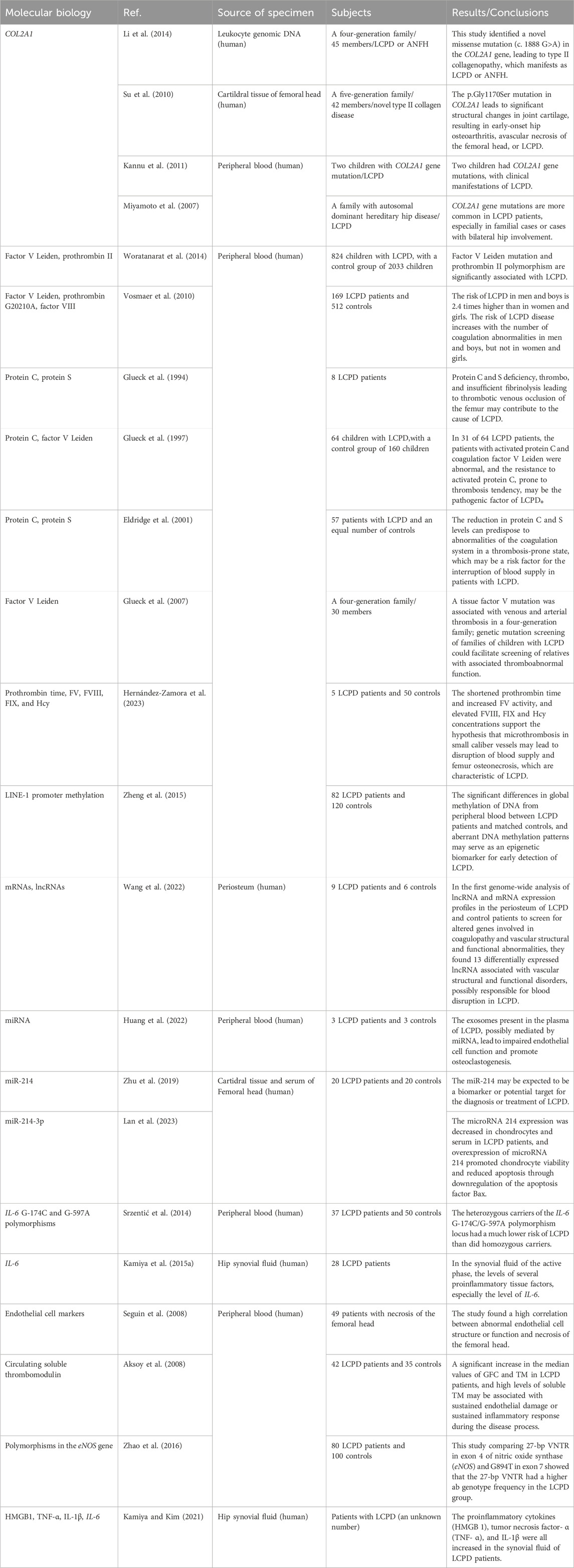
Table 1. Main findings of the included case-control studies.
Research on COL2A1 mutations in femoral head ischemic necrosis began with Liu et al. (2005), who conducted a four-generation pedigree analysis of two families with familial femoral head necrosis. They found that familial femoral head necrosis is dominantly inherited and identified a gene mutation in the 12q13 region (Chen et al., 2004), with further sequencing of the COL2A1 gene exon encoding collagenase II confirming a mutation at codon 1170 (Su et al., 2010). COL2A1 gene mutations are therefore the cause of familial femoral head ischemic necrosis. LCPD is idiopathic ischemic necrosis of the femoral head in children, and other scholars have also reported the same COL2A1 gene locus mutations in familial LCPD patients (Kannu et al., 2011; Miyamoto et al., 2007; Woratanarat et al., 2014; Su et al., 2008). Cartilage tissue consists of chondrocytes and cartilage matrix, with collagen II playing a key role in maintaining its homeostasis. COL2A1 encodes the precursor of the α-1 chain of collagenase II, and the aforementioned mutation results in a substitution of serine for glycine in the Gly-X-Y domain of collagenase II, leading to defects in the synthesis and structure of collagen II (Ibrahim and Little, 2016). This result explains the short stature observed in LCPD patients. This mutation also alters the cartilage matrix structure, reducing its strength. The hip joint is the main weight-bearing joint of the lower limbs, and the combination of load-bearing and reduced biomechanical properties of the epiphysis can lead to vascular occlusion of the epiphysis (Pinheiro et al., 2018). Therefore, a solid theoretical basis for COL2A1 mutations as a cause of LCPD is identified.
Some scholars have pointed out that although typical LCPD hip joint imaging changes are observed in these cases, it is difficult to distinguish whether these changes are caused by ischemia or by epiphyseal dysplasia. No sporadic cases of LCPD with COL2A1 mutations have been reported so far (Kim, 2011). COL2A1 gene mutations may be a cause of familial LCPD, but their role as an independent cause of the disease remains controversial. Chinese scholars recently conducted COL2A1 mutation testing on a familial LCPD family and identified a novel heterozygous mutation in exon 29 of COL2A1 (c.1888 G.A, p.Gly630Ser) (Li et al., 2014). This finding suggests the presence of a novel COL2A1 gene mutation site in LCPD patients, and whether this mutation exists in sporadic cases requires further research with a larger sample size.
Thrombophilia is a disease caused by abnormal coagulation-related factors (Crowther and Kelton, 2003; Baglin et al., 2010). Vosmaer et al. (2010) found that factor V Leiden, prothrombin G20210A mutations, Protein-S and Protein-C deficiencies, and elevated factor VIII are all associated with an increased risk of LCPD. As the number of abnormal coagulation factors increases, the risk also increases. Thrombophilia can potentially lead to venous thrombosis, which obstructs blood flow from the femoral head epiphysis, resulting in ischemia. Scholars have therefore conducted extensive studies on the relationship between thrombophilia and LCPD.
Protein C is a vitamin K-dependent plasma zymogen that plays a central role in the inhibition of the blood coagulation cascade. Human protein C is encoded by a gene on chromosome 2ql3-14 and spans approximately 10 kilobases; Protein S is essential for the anticoagulant role of protein C in blood coagulation (Hepner and Karlaftis, 2013; Griffin et al., 2007). Similarly, protein S is a vitamin K-dependent plasma glycoprotein with a molecular weight of approximately 75 kDa, consisting of 635 amino acid residues. Protein S is a potent cofactor in the regulation of the intrinsic pathway by activated protein C (APC) (Dahlbäck, 1991). The strong association between genetic or acquired protein C and protein S deficiencies and increased risk of venous thrombosis suggests that protein C and protein S play important and central roles in controlling the initiation and propagation stages of the coagulation cascade.
As early as 1994, a clinical controlled trial led by Glueck et al. (1994) first reported that among 8 LCPD patients, 3 had Protein-C deficiency and 1 had Protein-S deficiency. After expanding the sample size to 44 cases, they confirmed the presence of anticoagulant factor deficiencies in LCPD, with 19 cases of Protein-C deficiency and 4 cases of Protein-S deficiency. Another study by Glueck found that the factor V Leiden mutation increases resistance to Protein-C, suggesting that this result might explain the deficiency of anticoagulant factors (Glueck et al., 1997). These findings indicate a deficiency of anticoagulant factors in LCPD patients, leading Glueck to propose that thrombophilia is the cause of the disease. Other scholars reached similar conclusions. Eldridge et al. (2001) found that Protein-C and Protein-S levels in the disease group were significantly lower than in the control group. This finding suggests that the deficiency of Protein-C and Protein-S may contribute to the susceptibility to LCPD.
The factor V Leiden mutation is another cause of thrombophilia, and Glueck et al. (1997) initially reported that 8 out of 64 LCPD patients had the factor V Leiden mutation. Another study involving 61 LCPD patients found that the factor V Leiden mutation rate (4.9%) was significantly higher than in the control group (0.7%). Recently, a large-scale Dutch case-control study found that the incidence of LCPD was higher with the factor V Leiden mutation (16/166 vs. 16/509) (Vosmaer et al., 2010). These researchers believe that thrombophilia is a high-risk factor for LCPD. However, scholars from different regions have drawn different conclusions. For instance, Israeli researchers studied 119 LCPD patients and 276 normal children, finding no difference in the factor V Leiden mutation rate between the LCPD group (7/119) and the normal group (13/276) (Kenet et al., 2008).
Given the inconsistent findings, Thaveeratitharm et al. (Miyamoto et al., 2007) conducted a meta-analysis of the current literature to determine whether thrombophilia is related to LCPD. They found that the factor V Leiden mutation increased the risk of LCPD by three times compared with the control group. This study concluded that the factor V Leiden mutation is a risk factor for LCPD. The form of the factor V Leiden mutation involves a substitution of CGA with CAA at position 1691 of the gene (Glueck et al., 1997), and this mutation can be vertically transmitted. A study involving genetic screening of LCPD patients found the factor V Leiden mutation in first- and second-degree relatives, which was associated with thrombotic events within the family (Glueck et al., 2007). Additionally, researchers analyzed blood and plasma samples from 25 LCPD patients and 50 healthy controls, discovering reduced prothrombin time and elevated levels of factor V Leiden, factor VIII, factor IX, and Hcy in LCPD patients (Hernández-Zamora et al., 2023), potentially explaining the coagulation abnormalities in these patients.
In summary, coagulation factor-related abnormalities in blood coagulation may be a cause of LCPD. However, the likelihood of thrombosis in childhood is low, and no reports of coagulation abnormalities in children with LCPD have been documented. A possible explanation is that LCPD may be a hematological disorder, where the mutation potentially affects the coagulation state of the blood system, although the specific mechanism remains unclear.
Epigenetics suggests that acquired phenotypes can be inherited, and non-genetic changes influence the genotype. Epigenetic regulation includes selective transcriptional and post-transcriptional control, with DNA methylation being one of the modes of selective transcriptional regulation. In normal physiology, DNA methylation regulates the temporal expression of genes during embryogenesis. If methylation and demethylation are imbalanced, this can cause abnormal timing in gene expression. Considering that LCPD commonly occurs in children aged 4–8 and is self-limiting and self-healing (Herring et al., 2004), domestic scholars conducted research on DNA methylation levels in LCPD patients. LINE-1 is a class of non-long terminal repeat retrotransposons, accounting for approximately 18% of the human genome. Given their high frequency presence in the genome, LINE-1 methylation levels can be used as a marker for full DNA methylation levels (Zheng et al., 2015). The study analyzed LINE-1 promoter levels in 82 patients, finding a significant reduction compared with controls. Further subgroup analysis revealed that this difference existed only in male patients (Zheng et al., 2015). This finding correlates with the clinical tendency of LCPD to occur more frequently in men and boys. Furthermore, socioeconomic environment, maternal smoking, and childhood tobacco exposure are risk factors for LCPD (Perry et al., 2017), and these factors are thought to act through epigenetic mechanisms, with DNA methylation being one key regulatory pathway (Ladd-Acosta et al., 2016). Hence, DNA methylation levels could be a critical factor in LCPD pathogenesis.
Non-coding RNA regulation is an important part of epigenetic modifications and is considered a biomarker for clinical diagnosis, with its abnormal expression often involved in the pathogenesis of certain diseases. In LCPD patients, reduced expression of non-coding RNAs (such as lncRNA n335645) may downregulate the expression of related gene mRNAs (ILK, VCL, RRAS, or other genes), leading to vascular structural or functional damage and interruption of blood supply to the femoral head in LCPD patients (Wang et al., 2022). Exosomal miRNA extracted from the plasma of LCPD patients could promote endothelial cell dysfunction and osteoclastogenesis (Huang et al., 2022). In vitro experiments found that the expression of microRNA-214 in chondrocytes and serum of LCPD patients was reduced, while overexpression of microRNA-214 promoted chondrocyte viability and reduced apoptosis by downregulating the apoptotic factor Bax. This has potential as a biomarker or therapeutic target for the diagnosis or treatment of LCPD (Zhu et al., 2019; Lan et al., 2023).
In summary, analyzing global DNA methylation levels and non-coding RNA expression in patients provides the first epigenetic explanation for LCPD etiology. This study offers a reference for future etiological research on LCPD. Its limitation lies in only presenting a phenomenon. Whether the reduced DNA methylation and non-coding RNA expression share the same driving factors or originate from the same locus changes, and how these changes affect disease development, remain areas for further research.
Changes in DNA bases result in gene mutations, while natural variations in single nucleotides cause DNA sequence polymorphisms. DNA sequence polymorphism refers to the presence of two or more distinct genotypes or alleles, also known as genetic polymorphism. Genetic polymorphisms provide individual susceptibility, and environmental stimuli combined with this susceptibility lead to disease, which we refer to as the phenotype. Srzentić et al. (2014) analyzed IL-6 and TLR4 gene polymorphisms in 37 children with early-stage LCPD. They found that IL-6 G-174C/G-597A polymorphisms were in complete linkage disequilibrium, and the proportion of heterozygous carriers of the IL-6 G-174C/G-597A polymorphism was significantly lower in the disease group than in the control group. The risk of developing LCPD was therefore much lower in heterozygous carriers than in homozygous carriers. Theoretically, homozygous carriers have a genetic susceptibility to elevated IL-6. Clinically, Kim et al. (Kamiya et al., 2015a) found elevated levels of various pro-inflammatory factors in the synovial fluid of active LCPD patients, with IL-6 showing the most significant increase. They also found no TLR4 gene polymorphisms in either group of children, consistent with the theory that LCPD is a form of aseptic inflammation (Srzentić et al., 2014).
Recent studies suggest that endothelial cell structural or functional abnormalities exist in LCPD patients (Seguin et al., 2008; Aksoy et al., 2008; Perry et al., 2012b). Endothelial nitric oxide synthase (eNOS) regulates endothelial cell function by synthesizing nitric oxide. Zhao et al. (2016) compared 80 LCPD patients and 100 healthy children, examining the 27-bp VNTR in exon 4 and G894T in exon 7 of the eNOS gene. Results showed a higher frequency of the ab genotype for the 27-bp VNTR in the LCPD group. In exon 7, the heterozygous GT genotype for G894T was more frequent in the LCPD group than in the healthy control group. The results suggest that eNOS gene polymorphisms may be a risk factor for LCPD. The influence of IL-6 and eNOS gene polymorphisms on LCPD development can be explained by the theory of genetic susceptibility interacting with environmental stimuli (Glueck et al., 2007), though the specific mechanisms require further research.
Numerous researchers have investigated biomarkers associated with LCPD patients and discovered elevated expression of relevant molecules in the serum and synovial fluid of these patients. Pro-inflammatory cytokines (HMGB1), tumor necrosis factor-α (TNF-α), and IL-1 have been detected at elevated levels in the synovial fluid of LCPD patients (Kamiya and Kim, 2021). The elevation of these cytokines further upregulates IL-6 expression, exacerbating the inflammatory response in LCPD patients and leading to synovitis. Compared with healthy controls, LCPD patients have significantly elevated levels of osteocalcin (Wei et al., 2017) and free leptin (Lee et al., 2013) in their serum, which are associated with the staging and severity of LCPD. Undeniably, these molecules are involved in the development of LCPD to some extent, but whether they can serve as independent molecular markers of LCPD requires further research.
The balance of the femoral head epiphyseal bone depends on the equilibrium between osteoblastic differentiation, osteoclastic differentiation, and bone angiogenesis. Disruption of this balance can lead to disease. In recent years, scholars have studied whether these molecular changes affect cell function and subsequently cause disease. Current research suggests that the development of LCPD may be related to dysfunctions of microvascular endothelial cells and osteoclasts, ischemic damage to chondrocytes, and the recruitment of bone marrow immune cells. The main findings of the included articles are summarized (Table 2).
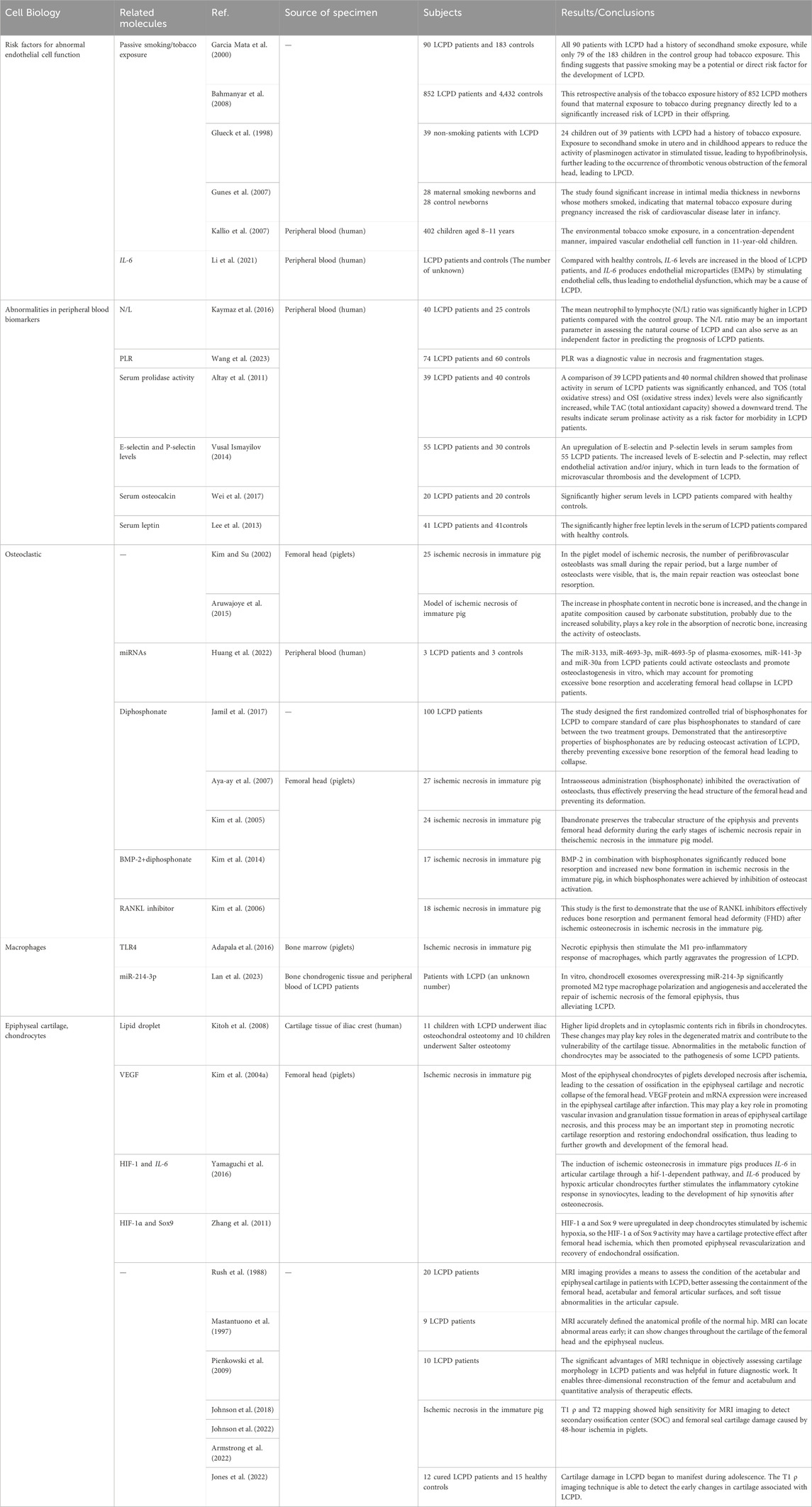
Table 2. Main findings of included cohort studies.
The physiological vascular changes in the femoral head of children have particular characteristics. In children aged 1–3 years, the femoral head epiphysis is supplied by the lateral circumflex femoral artery and the medial circumflex femoral artery. Between ages 4–7, the lateral circumflex artery to the epiphysis closes, and after age 7, the ligamentum teres artery grows in, the lateral circumflex artery reopens, and blood supply to the femoral head epiphysis is restored (Berthaume et al., 2016). LCPD commonly occurs in children aged 4–8 years (Herring et al., 2004), aligning with this physiological timeline. Theoretically, if the structure of the medial circumflex artery is abnormal or if angiogenesis is inhibited, leading to delayed reopening, disease may occur. Endothelial cells play an important role in angiogenesis, leading scholars to suggest that children with LCPD may have structural or functional abnormalities in their vascular endothelium (Seguin et al., 2008; Aksoy et al., 2008; Perry et al., 2012b; Hailer et al., 2010). This hypothesis is supported by epidemiological and clinical examinations.
The mainstream view holds that maternal passive smoking and/or postnatal infant tobacco exposure are closely related to the onset of LCPD. In 2000, Garcia Mata et al. (2000) reported that all 90 LCPD patients in the disease group had a history of secondhand smoke exposure, whereas only 79 of 183 control group children had tobacco exposure. Although no correlation was found between tobacco exposure and Catterall staging or Stulberg classification, the risk of LCPD in children exposed to passive smoking was more than five times higher than in normal children. In a retrospective analysis by Swedish scholars of 852 LCPD patients, maternal tobacco exposure during pregnancy was found to significantly increase the risk of LCPD in offspring (Bahmanyar et al., 2008). Subsequent studies by other scholars supported this view, with Daniel et al. (2012) finding that environmental tobacco exposure and smoke from indoor wood burning also increased the risk of the disease. Recently, Perry et al. (2017) suggested that environmental tobacco exposure and maternal tobacco exposure during pregnancy are risk factors for LCPD, potentially explaining the association between socioeconomic status and LCPD. Tobacco exposure is a risk factor for LCPD, and how it influences the occurrence of LCPD has attracted the interest of scholars. Glueck et al. (1998) reported that out of 39 LCPD patients, 24 children had a history of tobacco exposure, with 48% having low tissue plasminogen activator activity, significantly higher than the normal group. Thus, Glueck suggested that tobacco exposure leads to hypofibrinolysis, potentially further causing thrombotic venous occlusion in the femoral head, resulting in LCPD. Our research group previously found that serum endothelial microparticles (Li et al., 2021) and miRNA in plasma exosomes (Huang et al., 2022) from LCPD patients can promote endothelial dysfunction and inhibit angiogenesis in LCPD patients. Previous studies have confirmed that maternal tobacco exposure during pregnancy increases the risk of cardiovascular disease in infants, and tobacco exposure elevates oxidative stress levels in the body, damaging endothelial cell function in a dose-dependent manner (Gunes et al., 2007; Kallio et al., 2007). Some researchers therefore speculate that LCPD may not be caused by coagulation system abnormalities, but by vascular structural or functional issues, particularly endothelial dysfunction.
Some scholars have suggested that hip joint synovitis in LCPD is a chronic process and may be involved in the onset and progression of the disease. During the repair phase of femoral head ischemic necrosis, excessive resorption of necrotic bone, delayed new bone formation, and replacement of necrotic bone with fibrous vascular tissue occur. These processes persist over time. Using gadolinium-enhanced MRI, Kim et al. (Neal et al., 2015) detected increased synovitis signals in the affected hip, and these signals correlated with Waldenström staging. These findings suggest that synovitis is a chronic condition. To further explore the nature of hip synovitis, they performed cytokine quantification on joint fluid from active LCPD patients and found significant elevations in pro-inflammatory cytokines, with IL-6 being the most notably increased (Kamiya et al., 2015a). IL-6, as a pro-inflammatory cytokine, can directly damage endothelial cells and induce oxidative stress, leading to endothelial dysfunction, aligning with the findings of our research group (Li et al., 2021; Liu et al., 2024). Furthermore, Kim et al. (Kuroyanagi et al., 2018; Kamiya et al., 2019) demonstrated that IL-6 knockout mice showed restored angiogenesis following epiphyseal ischemic necrosis. The rise in pro-inflammatory cytokines in hip synovitis therefore acts as a risk factor for microvascular endothelial cell damage, potentially resulting in suppressed angiogenesis.
Vascular endothelial cells are important endocrine organs that regulate vascular tone, platelet aggregation, coagulation, and fibrinolysis. Biomarkers in the blood can reflect endothelial cell function, and in recent years, scholars have focused on how these biomarker levels present in LCPD patients. Researchers analyzing blood biomarkers in LCPD patients found that the neutrophil-to-lymphocyte ratio (N/L) (Kaymaz et al., 2016) and platelet-to-lymphocyte ratio (PLR) (Wang et al., 2023) were significantly higher in the LCPD group than in the control group, with the increase in N/L being most pronounced in the good prognosis Herring A/B group. These blood markers can reflect the body’s inflammation levels and are important for the diagnosis and treatment of LCPD. Reactive oxygen species (ROS) are present in the normal body, but excessive ROS can lead to endothelial cell damage. Altay et al. (2011) compared 39 LCPD patients with 40 normal children in terms of serum proline enzyme activity, total oxidant status (TOS), total antioxidant capacity (TAC), and oxidative stress index (OSI). They found that serum proline enzyme activity, TOS, and OSI were significantly elevated, while TAC was reduced in LCPD patients. These results suggest that elevated serum proline enzyme activity is a risk factor for LCPD. E-selectin and P-selectin shed from endothelial cells during activation or apoptosis, entering the bloodstream, and can reflect the functional status of microvascular endothelial cells. Vusal Ismayilov (2014) measured serum levels of E-selectin and P-selectin in 85 LCPD patients and found both to be upregulated in LCPD patients. Elevated E-selectin and P-selectin levels stimulate platelet and endothelial cell activation, leading to microvascular thrombosis and the development of LCPD. Additionally, endothelial nitric oxide synthase, the primary enzyme for nitric oxide production, exhibits increased gene polymorphisms in LCPD patients, elevating LCPD risk and potentially affecting endothelial cell relaxation function (Zhao et al., 2016). Despite the limited number of cases, these abnormal serum biomarkers reflect endothelial dysfunction in LCPD patients.
The interruption of blood supply to the femoral head epiphysis is an established mechanism of LCPD, but research has largely focused on extravascular factors, with little emphasis on the functional state of the vessels themselves. Swedish scholars conducted a retrospective analysis of 3,141 LCPD patients and performed regression analysis on the relationship with cardiovascular disease. They found that compared with non-LCPD patients, the risk ratio for cardiovascular disease was 1.70, and the risk of hypertension was statistically significantly higher (Hailer et al., 2010; Mörlin and Hailer, 2021). This result suggests that vascular dysfunction may be prevalent among LCPD patients. To further investigate the structural and functional state of microvascular endothelial cells in LCPD, the research team applied flow-mediated dilation (FMD) to directly assess endothelial function and recorded changes in arterial blood flow under ischemic stimuli. They found that the microvascular diameter supplying LCPD patients was smaller and blood flow velocity slower compared with the normal group, while the large vessels remained in a normal state (Perry et al., 2012b). The significance of this study lies in its direct revelation of structural or functional abnormalities in the microvascular endothelial cells of LCPD patients, providing direct evidence of endothelial dysfunction in LCPD.
The balance of bone remodeling depends on the balance between the number and function of osteoclasts, bone angiogenesis, and osteoblasts. Once this balance is disrupted, it can lead to disease. Clinical studies on the role of bone cells in the pathophysiology of LCPD are scarce, with most insights derived from basic research in animal models. The femoral head epiphysis of piglets has a similar anatomical structure to humans, and other scholars have conducted extensive studies using the piglet LCPD model (Kim and Su, 2002; Kim et al., 2001; Pringle et al., 2004; Koob et al., 2007; Kim et al., 2009).
In the early stages after ischemia, diffuse cell necrosis, apoptosis, and disruption of bone marrow stroma are observed in the femoral head epiphysis, followed by the formation of characteristic necrotic cavities, with osteoblast loss in the trabeculae (Kim et al., 2001). In adults, the repair phase of femoral head ischemic necrosis occurs through “creeping substitution,” where bone marrow mesenchymal stem cells differentiate into osteoblasts to repair necrotic bone. The repair process in LCPD differs from this process. During the repair phase, arteries from the femoral neck regenerate and extend into the secondary ossification center from the lateral side of the epiphyseal cartilage, restoring blood supply to the femoral head epiphysis (Kim et al., 2016). Osteoblasts around the fibrous vasculature are fewer, but a large number of osteoclasts are observed (Kim and Su, 2002). In early LCPD, hip joint synovitis is an aseptic inflammation, so why are osteoclasts activated? The current view is that the increase in necrotic bone and the chronic inflammatory state of the hip joint are two causes of this activation. Kim et al. (Aruwajoye et al., 2015) found that phosphate levels in necrotic bone were elevated, and phosphate replaced hydroxyapatite, increasing osteoclast activity. Elevated pro-inflammatory cytokines in the hip joint synovial fluid of patients can also activate osteoclasts. Our research group previously found that plasma exosomal miR-3133, miR-4693-3p, miR-4693-5p, miR-141-3p, and miR-30a from LCPD patients could activate osteoclasts and promote osteoclastogenesis in vitro. These effects may contribute to excessive bone resorption and accelerated femoral head collapse in LCPD patients (Huang et al., 2022).
TLRs are important immune response receptors in the human body and can recognize ligands through two pathways: pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs). Necrotic tissue or damaged cells release cytokines that are recognized by TLRs, activating downstream pathways, which is the mechanism of DAMPs. TLR4 belongs to the TLR family and is primarily expressed on the surface of monocytes and macrophages. Monocytes and macrophages can either promote or suppress inflammation depending on the environment, and pro-inflammatory monocytes can differentiate into osteoclasts, playing a role in bone resorption. Scholars found that in necrotic bone from the femoral head epiphysis in LCPD, phosphate replaces hydroxyapatite, leading to elevated phosphate levels (Aruwajoye et al., 2015). They concluded that necrotic bone is the source of high phosphate levels, which induces monocyte differentiation into osteoclasts through the DAMPs pathway, leading to enhanced bone resorption in the femoral head epiphysis of LCPD patients.
The repair process in LCPD is also accompanied by chronic inflammation. Kim et al. (Adapala et al., 2016) found that necrotic bone could activate the expression of pro-inflammatory cytokines. Through the DAMPs pathway, TLR4 activates monocytes, which then exhibit the pro-inflammatory M1 phenotype. Furthermore, cytokine quantification of joint fluid from active LCPD patients revealed elevated pro-inflammatory cytokines compared with normal controls, with the most significant difference seen in IL-6 levels (Kamiya et al., 2015a). IL-6 is part of the tumor necrosis factor (TNF) superfamily. Previous studies have shown that this family can induce osteoclast differentiation, with TNF-α and RANKL being key cytokines involved in osteoclast differentiation (Boyle et al., 2003). To determine whether IL-6 has a similar function in LCPD, Kim et al. constructed IL-6 knockout mice and found that osteoclast differentiation was inhibited in IL-6 knockout mice, femoral head epiphyseal ischemic necrosis deformities were alleviated, and femoral head bone mass increased (Kuroyanagi et al., 2018; Kamiya et al., 2019; Kim et al., 2018).
Osteoclasts are the only known cells responsible for bone resorption in the human body, and this discussion shows that their abnormal activation plays a role in the development of LCPD and contributes to deformity during healing. Studying osteoclasts can provide guidance for clinical treatment (Jamil et al., 2017). For example, the use of bisphosphonates (Aya-ay et al., 2007; Kim et al., 2005) combined with BMP2 (Kim et al., 2014) or RANKL (Kim et al., 2006) to treat ischemic bone necrosis improved femoral head deformities, proving the important role of osteoclast bone resorption in the development of femoral head deformities. Thus, from an etiological perspective and for guiding clinical treatment, the role of osteoclasts in LCPD is worthy of attention.
Unlike adult femoral head necrosis, LCPD is a self-limiting disease, and children with LCPD can heal without treatment, which may be related to the higher abundance of H-type vessels in young populations. H-type vessels are a new subtype of capillaries discovered by Kusumbe et al. (2014) in 2014 in the skeletal system of mice. These vessels couple angiogenesis and osteogenesis and play an important role in promoting bone repair. In human bone sections, the abundance of H-type vessels decreases with age, consistent with findings in mouse skeletal systems (Wang et al., 2017). Compared with other types of capillaries, H-type vessels express high levels of CD31 and endomucin. Endothelial cells in H-type vessels interact with bone cells (such as osteoblasts, osteoclasts, and chondrocytes) through cytokines or signaling pathways to maintain bone growth and homeostasis, playing a crucial role in bone diseases such as osteoporosis, osteoarthritis, and osteonecrosis (Liu et al., 2023; Qin et al., 2023; Xu et al., 2024; Ma et al., 2021).
With the introduction of the concept of osteoimmunology, the immune-inflammatory mechanisms between bone and the immune system have become a major focus in the study of bone metabolism-related diseases. In LCPD, necrotic epiphyses activate M1 pro-inflammatory responses in macrophages through Toll-like receptor 4, exacerbating the progression of LCPD to some extent (Adapala et al., 2016). However, in vitro, exosomes from chondrocytes overexpressing miR-214-3p can significantly promote M2 macrophage polarization and angiogenesis, thereby alleviating LCPD (Lan et al., 2023; Yu et al., 2023). In the early stages of LCPD bone necrosis, scholars have found that neutrophils and macrophages are recruited to the ischemic necrotic bone area, and their levels remain high throughout the bone repair phase (Phipps et al., 2016). This evidence is the most direct of immune cell involvement in bone necrosis, but how these immune cells contribute to the inflammatory response of bone necrosis and later promote bone repair is still unknown. Other immune cells in the bone marrow microenvironment (such as plasma cells, T cells, and B cells) have been shown to play important roles in bone metabolism-related diseases (Miron et al., 2024; Schlundt et al., 2023; Ma et al., 2022), but no studies on their role in LCPD have been reported. Therefore, exploring the etiology of LCPD from the perspective of osteoimmunity and H-type vessels, and even unraveling the mechanism of self-repair in this disease, may offer more possibilities for future clinical drug development.
The necrotic collapse of the femoral head in LCPD patients is related to epiphyseal cartilage degeneration. Ischemic injury to the epiphyseal cartilage leads to abnormal chondrocyte metabolism, resulting in disordered matrix calcification. Metabolic damage to ischemic chondrocytes in the epiphysis leads to fragility of the femoral epiphysis, which may be related to the development of LCPD in some patients (Kitoh et al., 2008). In a study on ischemia in immature piglets, Kim et al. (2004a) found that most epiphyseal chondrocytes underwent necrosis after ischemia, resulting in the cessation of endochondral ossification and necrotic collapse of the femoral head. Subsequently, deeper chondrocytes, under the stimulus of ischemia and hypoxia, secrete relevant cytokines that promote the reconstruction of epiphyseal blood flow and the restoration of endochondral ossification (Zhang et al., 2011). In the early stages of LCPD, metabolic dysfunction of superficial chondrocytes in the joint leads to necrotic collapse of the epiphyseal cartilage, after which the surviving deep chondrocytes initiate re-ossification and revascularization of the epiphysis. The KIM team also found that IL-6 levels are increased in the hip synovial fluid of LCPD patients, and articular chondrocytes are the main source of increased IL-6 levels, potentially contributing to the occurrence of hip synovitis in LCPD patients (Yamaguchi et al., 2016). Depression of IL-6 production in articular chondrocytes by using the IL-6 receptor inhibitor (tocilizumab) (Kamiya et al., 2019) or knockout of the IL-6 gene (Kuroyanagi et al., 2018) significantly promotes cartilage synthesis, bone formation, and revascularization after ischemic osteonecrosis. To more accurately identify the pathological changes in the femoral epiphysis in LCPD, imaging techniques can help diagnose the early pathological changes of the disease. As early as 1988, Rush et al. (1988) discovered through MRI imaging that it is possible to better assess the ischemic damage to the acetabulum and epiphyseal cartilage in LCPD patients. T1-weighted MRI images can clearly distinguish between the stages of vascular necrosis and fragmentation, and cartilage deformities on MRI are useful for differentiating these stages (Kumasaka et al., 1991). Imaging scans of the hip joints of nine children with LCPD at different stages revealed that MRI could identify abnormal areas earlier than conventional X-rays; it also showed changes in the evolution of the entire femoral head cartilage and epiphyseal nucleus, potentially distinguishing the evolution and occult patterns of osteochondropathy (Mastantuono et al., 1997; Pienkowski et al., 2009; Johnson et al., 2018). This is essential for studying the biomechanics of hip osteochondropathy and developing treatment plans. In adulthood, LCPD patients continue to experience cartilage degeneration of the femoral head, leaving varying degrees of femoral head deformities and even leading to early-onset hip osteoarthritis. However, with new MRI techniques, T1ρ and T2 mapping can sensitively detect ischemic damage to the femoral head SOC and epiphyseal cartilage within 48 h of ischemia induction (Johnson et al., 2022; Armstrong et al., 2022). T1ρ sequence scanning of the hip joint during the healing phase of LCPD can accurately detect early pathological changes in cartilage degeneration in LCPD patients, allowing for early prevention and therapeutic intervention (Jones et al., 2022). These techniques can be used clinically to assess the damage and repair of epiphyseal cartilage, providing better analysis of the ischemic pathological changes in LCPD.
In 22 LCPD patients, scholars obtained cylindrical biopsy specimens from the femoral metaphysis, and histopathological examination showed fat necrosis, vascular proliferation, and focal fibrosis, indicating previous ischemic episodes (Eckerwall et al., 1997). However, bone specimens from LCPD patients are difficult to obtain clinically, so using animal models is an important tool for studying the pathogenesis of Perthes disease and conducting in vivo experiments to more accurately observe the pathological changes of LCPD at the histological level. Current research uses animal models to simulate the pathological changes of LCPD. The main animals used include piglets, rabbits, rats, mice, and dogs, each with its own advantages and disadvantages. The advantage of the piglet LCPD model is that it better demonstrates the pathological changes of the femoral head in LCPD patients at the tissue level. Whether through imaging or histological staining, the model accurately presents the necrosis and repair process of LCPD, as well as the pathological changes in the acetabulum (Johnson et al., 2018; Kim et al., 2004b; Miashiro et al., 2023). However, piglets are large, have strict feeding requirements, high experimental costs, and are difficult to model, which are challenges many scholars face. Canine animal models share similar characteristics. Medium-sized animal models like rats (Boss et al., 2003; Little et al., 2005) and rabbits (Kamegaya et al., 1990) are easier to model, less expensive, and simpler to observe. Mouse models (Kamiya et al., 2015b) are better suited for subsequent genetic studies, aiding in the exploration of molecular or gene mutations in LCPD patients, as mentioned earlier in the article. In summary, clinical samples of LCPD are difficult to obtain, and animal model research helps us observe the histopathological changes of LCPD, offering the potential for cellular biotherapy and molecular genetic manipulation, providing more reliable theoretical support for elucidating the etiology and improving the diagnosis and treatment of LCPD.
Molecular-level changes create a predisposition to disruption of blood supply to the femoral head. For example, COL2A1 mutations weaken the strength of the epiphyseal cartilage matrix, mutations in Factor V Leiden, and deficiencies in Protein-C and Protein-S contribute to thrombophilia, while nitric oxide synthase and IL-6 gene polymorphisms potentially affect endothelial cell function. Pro-inflammatory factors in joint fluid, serum-free osteocalcin, and leptin levels all influence the progression of LCPD to some extent. These susceptibilities, when triggered by environmental factors, lead to the development of LCPD. This finding aligns with Kim’s (Glueck et al., 2007) view that the mechanism of LCPD involves genetic susceptibility combined with environmental factors. Molecular-level changes further impact cellular functions. Increasing reports suggest that structural or functional abnormalities of microvascular endothelial cells are common in LCPD patients, but whether endothelial dysfunction is the cause of LCPD or an intermediate consequence of other risk factors remains to be further investigated. Osteoclasts, the only cells responsible for bone resorption, are activated under LCPD pathological conditions, leading to increased bone resorption. This finding explains clinical manifestations like femoral head deformity and decreased bone density, but the molecular mechanisms require further exploration.
As research on LCPD progresses, the immune response mechanism between bones and immune cells is found to play an important role in LCPD. M1 polarization of bone macrophages promotes the inflammatory response in LCPD, while M2 polarization supports vascular regeneration in LCPD. Other immune cells in the bone marrow have also been shown to play important roles in bone metabolism and repair, but the exact molecular mechanisms in LCPD remain to be elucidated.
Currently, extensive research has been conducted on the development of LCPD animal models. Large animals (such as piglets and puppies) and medium-sized animals (such as rats and rabbits) are more suitable for observing pathological changes in the femoral head, joint cartilage, and epiphyseal cartilage, as well as vascular and connective tissue regeneration at the histopathological level. Mouse models are better suited for molecular-level studies. In juvenile mouse models of femoral epiphyseal ischemic necrosis, repair of epiphyseal necrosis is accompanied by vascular reconstruction and bone repair (Miashiro et al., 2023). This repair may occur through signaling pathways that stimulate endothelial cells to secrete angiogenic and osteogenic factors, consistent with findings by Kim et al. (Boyle et al., 2003; Kim et al., 2018) in piglet models. The self-repair mechanism of LCPD may be related to the osteogenesis-angiogenesis coupling function of H-type vessels. H-type vessels, which are bone-specific vessels that couple osteogenesis and angiogenesis, promote bone repair and vascular regeneration in bone-related diseases, and may become a potential target for treating LCPD. Overall, while the cause of LCPD remains unclear, advancements in molecular and cellular biology offer new perspectives for basic research into the disease.
XZ: Writing–original draft. ZD: Writing–original draft. XD: Project administration, Supervision, Writing–review and editing, Funding acquisition. QH: Methodology, Project administration, Supervision, Writing–review and editing. ST: Conceptualization, Investigation, Visualization, Writing–review and editing. YZ: Conceptualization, Investigation, Visualization, Writing–review and editing. BL: Methodology, Project administration, Supervision, Writing–review and editing, Funding acquisition. SL: Funding acquisition, Project administration, Supervision, Writing–review and editing, Methodology.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by the National Natural Science Foundation of China, grants 82060396 and 82160809, Natural Science Foundation of Guangxi, grant 2023GXNSFAA026342, “Medical Excellence Award” Funded by the Creative Research Development Grant from the First Affiliated Hospital of Guangxi Medical University (2022014),Youth Science and Technology Project of the First Affiliated Hospital of GuangxiMedical University (YYZS2022008),Guangxi Medical University Youth Science Foundation Program (GXMUYSF202233).
We apologize to all the scientists whose work could not be discussed in the pages of this review. The authors are grateful to all the members of the Dept.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declare that no Generative AI was used in the creation of this manuscript.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
LCPD, Legg-Calvé-Perthes disease; COL2A1, Collagen Type II Alpha 1 Chain; IGF-1, Insulin-like growth factor 1; IL-6, Interleukin-6; GFC, Global fibrinolytic capacity; TM, Thrombomodulin; VNTR, Variable Number of Tandem Repeats; eNOS, Endothelial nitric oxide synthase; HMGB1, Pro-inflammatory cytokines; TNF-α, Tumor necrosis factor-α; IL-1β, Interleukin-1β; LINE-1, Long Interspersed Nuclear Elements 1; TLR4, Toll-like receptors 4; TOS, Total oxidant status; TAC, Total antioxidant capacity; OSI, Oxidative stress index; VEGF, Vascular endothelial growth factor; HIF-1, Hypoxia-inducible factor-1; Sox9, SRY-box transcription factor 9; ROS, Reactive oxygen species.
Adapala N. S., Yamaguchi R., Phipps M., Aruwajoye O., Kim H. K. W. (2016). Necrotic bone stimulates proinflammatory responses in macrophages through the activation of toll-like receptor 4. Am. J. Pathol. 186, 2987–2999. doi:10.1016/j.ajpath.2016.06.024
Aksoy M. C., Aksoy D. Y., Haznedaroglu I. C., Sayinalp N., Kirazli S., Alpaslan M. (2008). Thrombomodulin and GFC levels in Legg-Calve-Perthes disease. Hematology 13, 324–328. doi:10.1179/102453308X343509
Altay M. A., Erturk C., Aksoy N., Taskin A., Bilge A., Celik H., et al. (2011). Serum prolidase activity and oxidative-antioxidative status in Legg-Calve-Perthes disease. J. Pediatr. Orthop. B 20, 222–226. doi:10.1097/BPB.0b013e32834493df
Armstrong A. R., Bhave S., Buko E. O., Chase K. L., Tóth F., Carlson C. S., et al. (2022). Quantitative T2 and T1ρ mapping are sensitive to ischemic injury to the epiphyseal cartilage in an in vivo piglet model of Legg-Calvé-Perthes disease. Osteoarthr. Cartil. 30, 1244–1253. doi:10.1016/j.joca.2022.05.009
Aruwajoye O. O., Kim H. K., Aswath P. B. (2015). Bone apatite composition of necrotic trabecular bone in the femoral head of immature piglets. Calcif. Tissue Int. 96, 324–334. doi:10.1007/s00223-015-9959-7
Aya-ay J., Athavale S., Morgan-Bagley S., Bian H., Bauss F., Kim H. K. (2007). Retention, distribution, and effects of intraosseously administered ibandronate in the infarcted femoral head. J. Bone Min. Res. 22, 93–100. doi:10.1359/jbmr.060817
Baglin T., Gray E., Greaves M., Hunt B. J., Keeling D., Machin S., et al. (2010). Clinical guidelines for testing for heritable thrombophilia. Br. J. Haematol. 149, 209–220. doi:10.1111/j.1365-2141.2009.08022.x
Bahmanyar S., Montgomery S. M., Weiss R. J., Ekbom A. (2008). Maternal smoking during pregnancy, other prenatal and perinatal factors, and the risk of Legg-Calve-Perthes disease. Pediatrics 122, e459–e464. doi:10.1542/peds.2008-0307
Baş V. N., Uytun S., Vurdem Ü E., Torun Y. A. (2015). Hypopituitarism and Legg-Calve-Perthes disease related to difficult delivery. Korean J. Pediatr. 58, 270–273. doi:10.3345/kjp.2015.58.7.270
Berthaume M. A., Perry D. C., Dobson C. A., Witzel U., Clarke N. M., Fagan M. J. (2016). Skeletal immaturity, rostral sparing, and disparate hip morphologies as biomechanical causes for Legg-Calvé-Perthes' disease. Clin. Anat. 29, 759–772. doi:10.1002/ca.22690
Boss J. H., Misselevich I., Peskin B., Zinman C., Levin D., Norman D., et al. (2003). Postosteonecrotic osteoarthritis-like disorder of the femoral head of rats. J. Comp. Pathol. 129, 235–239. doi:10.1016/s0021-9975(03)00031-8
Boyle W. J., Simonet W. S., Lacey D. L. (2003). Osteoclast differentiation and activation. Nature 423, 337–342. doi:10.1038/nature01658
Calve J. (2006). On a particular form of pseudo-coxalgia associated with a characteristic deformity of the upper end of the femur. 1910. Clin. Orthop. Relat. Res. 451, 14–16. doi:10.1097/01.blo.0000238799.05338.5a
Catterall A. (1971). The natural history of Perthes' disease. J. Bone Jt. Surg. Br. 53, 37–53. doi:10.1302/0301-620x.53b1.37
Chen W.-M., Liu Y.-F., Lin M.-W., Chen I.-C., Lin P.-Y., Lin G.-L., Jou Y.-S., et al. (2004). Autosomal dominant avascular necrosis of femoral head in two Taiwanese pedigrees and linkage to chromosome 12q13. Am. J. Hum. Genet. 75, 310–317. doi:10.1086/422702
Crowther M. A., Kelton J. G. (2003). Congenital thrombophilic states associated with venous thrombosis: a qualitative overview and proposed classification system. Ann. Intern Med. 138, 128–134. doi:10.7326/0003-4819-138-2-200301210-00014
Dahlbäck B. (1991). Protein S and C4b-binding protein: components involved in the regulation of the protein C anticoagulant system. Thromb. Haemost. 66, 049–061. doi:10.1055/s-0038-1646373
Daniel A. B., Shah H., Kamath A., Guddettu V., Joseph B. (2012). Environmental tobacco and wood smoke increase the risk of Legg-Calve-Perthes disease. Clin. Orthop. Relat. Res. 470, 2369–2375. doi:10.1007/s11999-011-2180-8
Eckerwall G., Hochbergs P., Simesen K., Willén H., Egund N., Wingstrand H. (1997). Metaphyseal histology and magnetic resonance imaging in Legg-Calvé-Perthes disease. J. Pediatr. Orthop. 17, 659–662. doi:10.1097/00004694-199709000-00016
Eldridge J., Dilley A., Austin H., M E.L.-J., Wolstein L., Doris J., et al. (2001). The role of protein C, protein S, and resistance to activated protein C in Legg-Perthes disease. Pediatrics 107, 1329–1334. doi:10.1542/peds.107.6.1329
Gao H., Huang Z., Jia Z., Ye H., Fu F., Song M., et al. (2020). Influence of passive smoking on the onset of Legg-Calvè-Perthes disease: a systematic review and meta-analysis. J. Pediatr. Orthop. B 29, 556–566. doi:10.1097/bpb.0000000000000725
Garcia Mata S., Ardanaz Aicua E., Hidalgo Ovejero A., Martinez Grande M. (2000). Legg-Calve-Perthes disease and passive smoking. J. Pediatr. Orthop. 20, 326–330. doi:10.1097/01241398-200005000-00011
Glueck C. J., Brandt G., Gruppo R., Crawford A., Roy D., Tracy T., et al. (1997). Resistance to activated protein C and Legg-Perthes disease. Clin. Orthop. Relat. Res. 338, 139–152. doi:10.1097/00003086-199705000-00021
Glueck C. J., Freiberg R. A., Crawford A., Gruppo R., Roy D., Tracy T., et al. (1998). Secondhand smoke, hypofibrinolysis, and Legg-Perthes disease. Clin. Orthop. Relat. Res. 352, 159–167. doi:10.1097/00003086-199807000-00019
Glueck C. J., Glueck H. I., Greenfield D., Freiberg R., Kahn A., Hamer T., et al. (1994). Protein C and S deficiency, thrombophilia, and hypofibrinolysis: pathophysiologic causes of Legg-Perthes disease. Pediatr. Res. 35, 383–388. doi:10.1203/00006450-199404000-00001
Glueck C. J., Tracy T., Wang P. (2007). Legg-Calve-Perthes disease, venous and arterial thrombi, and the factor V Leiden mutation in a four-generation kindred. J. Pediatr. Orthop. 27, 834–837. doi:10.1097/BPO.0b013e31815584bf
Griffin J. H., Fernández J. A., Gale A. J., Mosnier L. O. (2007). Activated protein C. J. Thromb. Haemost. 5 (Suppl. 1), 73–80. doi:10.1111/j.1538-7836.2007.02491.x
Gunes T., Koklu E., Yikilmaz A., Ozturk M. A., Akcakus M., Kurtoglu S., et al. (2007). Influence of maternal smoking on neonatal aortic intima-media thickness, serum IGF-I and IGFBP-3 levels. Eur. J. Pediatr. 166, 1039–1044. doi:10.1007/s00431-006-0376-9
Hailer Y. D., Montgomery S. M., Ekbom A., Nilsson O. S., Bahmanyar S. (2010). Legg-Calve-Perthes disease and risks for cardiovascular diseases and blood diseases. Pediatrics 125, e1308–e1315. doi:10.1542/peds.2009-2935
Hepner M., Karlaftis V. (2013). Protein C. Methods Mol. Biol. 992, 365–372. doi:10.1007/978-1-62703-339-8_29
Hernández-Zamora E., Rodríguez-Olivas A. O., Rosales-Cruz E., Galicia-Alvarado M. A., Zavala-Hernández C., Reyes-Maldonado E. (2023). Prothrombin time and coagulation factor IX as hemostatic risk markers for legg- calvé-perthes disease. Clin. Appl. Thromb. Hemost. 29, 10760296221151166. doi:10.1177/10760296221151166
Herring J. A., Kim H. T., Browne R. (2004). Legg-Calve-Perthes disease. Part II: prospective multicenter study of the effect of treatment on outcome. J. Bone Jt. Surg. Am. 86-A, 2121–2134.
Huang Q., Li B., Lin C., Chen X., Wang T., Liu J., et al. (2022). MicroRNA sequence analysis of plasma exosomes in early Legg-Calvé-Perthes disease. Cell Signal 91, 110184. doi:10.1016/j.cellsig.2021.110184
Ibrahim T., Little D. G. (2016). The pathogenesis and treatment of legg-calvé-perthes disease. JBJS Rev. 4, e4. doi:10.2106/jbjs.Rvw.15.00063
Jamil K., Zacharin M., Foster B., Donald G., Hassall T., Siafarikas A., et al. (2017). Protocol for a randomised control trial of bisphosphonate (zoledronic acid) treatment in childhood femoral head avascular necrosis due to Perthes disease. BMJ Paediatr. Open 1, e000084. doi:10.1136/bmjpo-2017-000084
Johnson C. P., Tóth F., Carlson C. S., Armstrong A. R., Zbýň Š., Wu B., et al. (2022). T1ρ and T2 mapping detect acute ischemic injury in a piglet model of Legg-Calvé-Perthes disease. J. Orthop. Res. 40, 484–494. doi:10.1002/jor.25044
Johnson C. P., Wang L., Tóth F., Aruwajoye O., Carlson C. S., Kim H. K. W., et al. (2018). Quantitative MRI helps to detect hip ischemia: preclinical model of legg-calvé-perthes disease. Radiology 289, 386–395. doi:10.1148/radiol.2018180497
Jones C. E., Mulpuri K., Teo T., Wilson D. R., d'Entremont A. G. (2022). T1ρ and T2 MRI show hip cartilage damage in adolescents with healed Legg-Calvé-Perthes disease. J. Pediatr. Orthop. B 31, 344–349. doi:10.1097/bpb.0000000000000892
Kallio K., Jokinen E., Raitakari O. T., Hamalainen M., Siltala M., Volanen I., et al. (2007). Tobacco smoke exposure is associated with attenuated endothelial function in 11-year-old healthy children. Circulation 115, 3205–3212. doi:10.1161/CIRCULATIONAHA.106.674804
Kamegaya M., Shinada Y., Akita T., Ogata S., Someya M., Tsuchiya K. (1990). Experimental avascular necrosis of the femoral capital epiphysis and induced subluxation of the hip in young rabbits. J. Pediatr. Orthop. 10, 1–5. doi:10.1097/01241398-199010010-00001
Kamiya N., Kim H. K. (2021). Elevation of proinflammatory cytokine HMGB1 in the synovial fluid of patients with legg-calvé-perthes disease and correlation with IL-6. JBMR Plus 5, e10429. doi:10.1002/jbm4.10429
Kamiya N., Kuroyanagi G., Aruwajoye O., Kim H. K. W. (2019). IL6 receptor blockade preserves articular cartilage and increases bone volume following ischemic osteonecrosis in immature mice. Osteoarthr. Cartil. 27, 326–335. doi:10.1016/j.joca.2018.10.010
Kamiya N., Yamaguchi R., Adapala N. S., Chen E., Neal D., Jack O., et al. (2015a). Legg-Calve-Perthes disease produces chronic hip synovitis and elevation of interleukin-6 in the synovial fluid. J. Bone Min. Res. 30, 1009–1013. doi:10.1002/jbmr.2435
Kamiya N., Yamaguchi R., Aruwajoye O., Adapala N. S., Kim H. K. (2015b). Development of a mouse model of ischemic osteonecrosis. Clin. Orthop. Relat. Res. 473, 1486–1498. doi:10.1007/s11999-015-4172-6
Kannu P., Irving M., Aftimos S., Savarirayan R. (2011). Two novel COL2A1 mutations associated with a Legg-Calve-Perthes disease-like presentation. Clin. Orthop. Relat. Res. 469, 1785–1790. doi:10.1007/s11999-011-1850-x
Kaymaz B., Büyükdogan K., Kaymaz N., Kömürcü E., Golge U. H., Goksel F., et al. (2016). Neutrophil to lymphocyte ratio may be a predictive marker of poor prognosis in Legg-Calvé-Perthes disease. Hip Int. 26, 598–601. doi:10.5301/hipint.5000381
Kenet G., Ezra E., Wientroub S., Steinberg D. M., Rosenberg N., Waldman D., et al. (2008). Perthes' disease and the search for genetic associations: collagen mutations, Gaucher's disease and thrombophilia. J. Bone Jt. Surg. Br. 90, 1507–1511. doi:10.1302/0301-620x.90b11.20318
Kessler J. I., Cannamela P. C. (2018). What are the demographics and epidemiology of legg-calve-perthes disease in a large southern California integrated health system? Clin. Orthop. Relat. Res. 476, 2344–2350. doi:10.1097/CORR.0000000000000490
Kim H. K. (2011). Legg-Calve-Perthes disease: etiology, pathogenesis, and biology. J. Pediatr. Orthop. 31, S141–S146. doi:10.1097/BPO.0b013e318223b4bd
Kim H. K., Aruwajoye O., Du J., Kamiya N. (2014). Local administration of bone morphogenetic protein-2 and bisphosphonate during non-weight-bearing treatment of ischemic osteonecrosis of the femoral head: an experimental investigation in immature pigs. J. Bone Jt. Surg. Am. 96, 1515–1524. doi:10.2106/JBJS.M.01361
Kim H. K., Bian H., Randall T., Garces A., Gerstenfeld L. C., Einhorn T. A. (2004a). Increased VEGF expression in the epiphyseal cartilage after ischemic necrosis of the capital femoral epiphysis. J. Bone Min. Res. 19, 2041–2048. doi:10.1359/jbmr.040911
Kim H. K., Burgess J., Thoveson A., Gudmundsson P., Dempsey M., Jo C. H. (2016). Assessment of femoral head revascularization in legg-calve-perthes disease using serial perfusion MRI. J. Bone Jt. Surg. Am. 98, 1897–1904. doi:10.2106/JBJS.15.01477
Kim H. K., Morgan-Bagley S., Kostenuik P. (2006). RANKL inhibition: a novel strategy to decrease femoral head deformity after ischemic osteonecrosis. J. Bone Min. Res. 21, 1946–1954. doi:10.1359/jbmr.060905
Kim H. K., Randall T. S., Bian H., Jenkins J., Garces A., Bauss F. (2005). Ibandronate for prevention of femoral head deformity after ischemic necrosis of the capital femoral epiphysis in immature pigs. J. Bone Jt. Surg. Am. 87, 550–557. doi:10.2106/JBJS.D.02192
Kim H. K., Skelton D. N., Quigley E. J. (2004b). Pathogenesis of metaphyseal radiolucent changes following ischemic necrosis of the capital femoral epiphysis in immature pigs. A preliminary report. J. Bone Jt. Surg. Am. 86, 129–135. doi:10.2106/00004623-200401000-00019
Kim H. K., Stephenson N., Garces A., Aya-ay J., Bian H. (2009). Effects of disruption of epiphyseal vasculature on the proximal femoral growth plate. J. Bone Jt. Surg. Am. 91, 1149–1158. doi:10.2106/JBJS.H.00654
Kim H. K., Su P. H. (2002). Development of flattening and apparent fragmentation following ischemic necrosis of the capital femoral epiphysis in a piglet model. J. Bone Jt. Surg. Am. 84-A, 1329–1334. doi:10.2106/00004623-200208000-00006
Kim H. K., Su P. H., Qiu Y. S. (2001). Histopathologic changes in growth-plate cartilage following ischemic necrosis of the capital femoral epiphysis. An experimental investigation in immature pigs. J. Bone Jt. Surg. Am. 83-A, 688–697. doi:10.2106/00004623-200105000-00007
Kim K. M., Wagle S., Moon Y. J., Wang S. I., Park B. H., Jang K. Y., et al. (2018). Interferon β protects against avascular osteonecrosis through interleukin 6 inhibition and silent information regulator transcript-1 upregulation. Oncotarget 9, 3562–3575. doi:10.18632/oncotarget.23337
Kitoh H., Kitakoji T., Kawasumi M., Ishiguro N. (2008). A histological and ultrastructural study of the iliac crest apophysis in Legg-Calve-Perthes disease. J. Pediatr. Orthop. 28, 435–439. doi:10.1097/BPO.0b013e318173ed54
Koob T. J., Pringle D., Gedbaw E., Meredith J., Berrios R., Kim H. K. (2007). Biomechanical properties of bone and cartilage in growing femoral head following ischemic osteonecrosis. J. Orthop. Res. 25, 750–757. doi:10.1002/jor.20350
Kumasaka Y., Watanabe H., Higashihara T., Kishimoto H., Harada K., Kozuka T. (1991). Changes in the cartilaginous contour of Legg-Calvé-Perthes disease: calculation on T1-weighted MR images. Nihon Igaku Hoshasen Gakkai Zasshi 51, 1232–1239.
Kuroyanagi G., Adapala N. S., Yamaguchi R., Kamiya N., Deng Z., Aruwajoye O., et al. (2018). Interleukin-6 deletion stimulates revascularization and new bone formation following ischemic osteonecrosis in a murine model. Bone 116, 221–231. doi:10.1016/j.bone.2018.08.011
Kusumbe A. P., Ramasamy S. K., Adams R. H. (2014). Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature 507, 323–328. doi:10.1038/nature13145
Ladd-Acosta C., Shu C., Lee B. K., Gidaya N., Singer A., Schieve L. A., et al. (2016). Presence of an epigenetic signature of prenatal cigarette smoke exposure in childhood. Environ. Res. 144, 139–148. doi:10.1016/j.envres.2015.11.014
Lan X., Yu R., Xu J., Jiang X. (2023). Exosomes from chondrocytes overexpressing miR-214-3p facilitate M2 macrophage polarization and angiogenesis to relieve Legg Calvé-Perthes disease. Cytokine 168, 156233. doi:10.1016/j.cyto.2023.156233
Lee J. H., Zhou L., Kwon K. S., Lee D., Park B. H., Kim J. R. (2013). Role of leptin in Legg-Calvé-Perthes disease. J. Orthop. Res. 31, 1605–1610. doi:10.1002/jor.22415
Legg A. T. (2006). An obscure affection of the hip joint. 1910. Clin. Orthop. Relat. Res. 451, 11–13. doi:10.1097/01.BLO.0000238798.05338.13
Li B., Huang Q., Lin C., Lu R., Wang T., Chen X., et al. (2021). Increased circulating CD31+/CD42b-EMPs in Perthes disease and inhibit HUVECs angiogenesis via endothelial dysfunction. Life Sci. 265, 118749. doi:10.1016/j.lfs.2020.118749
Li N., Yu J., Cao X., Wu Q. Y., Li W. W., Li T. F., et al. (2014). A novel p. Gly630Ser mutation of COL2A1 in a Chinese family with presentations of Legg-Calve-Perthes disease or avascular necrosis of the femoral head. PLoS One 9, e100505. doi:10.1371/journal.pone.0100505
Little D. G., McDonald M., Sharpe I. T., Peat R., Williams P., McEvoy T. (2005). Zoledronic acid improves femoral head sphericity in a rat model of perthes disease. J. Orthop. Res. 23, 862–868. doi:10.1016/j.orthres.2004.11.015
Liu J., Lin C., Li B., Huang Q., Chen X., Tang S., et al. (2024). Biochanin A inhibits endothelial dysfunction induced by IL-6-stimulated endothelial microparticles in Perthes disease via the NFκB pathway. Exp. Ther. Med. 27, 137. doi:10.3892/etm.2024.12425
Liu X., Zhang P., Gu Y., Guo Q., Liu Y. (2023). Type H vessels: functions in bone development and diseases. Front. Cell Dev. Biol. 11, 1236545. doi:10.3389/fcell.2023.1236545
Liu Y. F., Chen W. M., Lin Y. F., Yang R. C., Lin M. W., Li L. H., et al. (2005). Type II collagen gene variants and inherited osteonecrosis of the femoral head. N. Engl. J. Med. 352, 2294–2301. doi:10.1056/NEJMoa042480
Loder R. T., Skopelja E. N. (2011). The epidemiology and demographics of legg-calve-perthes' disease. ISRN Orthop. 2011, 504393. doi:10.5402/2011/504393
Ma J., Ren Y., Wang B., Sun W., Yue D., Wang W. (2021). Progress of developmental mechanism of subtype H vessels in osteonecrosis of the femoral head. Zhongguo Xiu Fu Chong Jian Wai Ke Za Zhi 35, 1486–1491. doi:10.7507/1002-1892.202103159
Ma M., Tan Z., Li W., Zhang H., Liu Y., Yue C. (2022). Osteoimmunology and osteonecrosis of the femoral head. Bone Jt. Res. 11, 26–28. doi:10.1302/2046-3758.111.Bjr-2021-0467.R1
Mastantuono M., Milella P. P., Della Rocca C., Nannerini M., De Paolis M., Larciprete M., et al. (1997). Role of magnetic resonance in the evaluation of the normal and osteochondrosis hip in early and late childhood. Radiol. Med. 94, 571–578.
Miashiro E. H., Zanella L. F., Cardoso G. S., Silva G. D. S., de Angelis K., de Almeida S. H. M. (2023). Animal model standardization for studying avascular necrosis of the femoral head in legg-calvé-perthes disease. Rev. Bras. Ortop. (Sao Paulo) 58, e771–e780. doi:10.1055/s-0042-1749418
Miron R. J., Bohner M., Zhang Y., Bosshardt D. D. (2024). Osteoinduction and osteoimmunology: emerging concepts. Periodontol. 2000 94, 9–26. doi:10.1111/prd.12519
Miyamoto Y., Matsuda T., Kitoh H., Haga N., Ohashi H., Nishimura G., et al. (2007). A recurrent mutation in type II collagen gene causes Legg-Calve-Perthes disease in a Japanese family. Hum. Genet. 121, 625–629. doi:10.1007/s00439-007-0354-y
Mörlin G. B., Hailer Y. D. (2021). High blood pressure and overweight in children with Legg-Calvé-Perthes disease: a nationwide population-based cohort study. BMC Musculoskelet. Disord. 22, 32. doi:10.1186/s12891-020-03889-9
Mose K. (1980). Methods of measuring in Legg-Calve-Perthes disease with special regard to the prognosis. Clin. Orthop. Relat. Res. 150, 103–109. doi:10.1097/00003086-198007000-00019
Neal D. C., O'Brien J. C., Burgess J., Jo C., Kim H. K., International Perthes Study G. (2015). Quantitative assessment of synovitis in Legg-Calve-Perthes disease using gadolinium-enhanced MRI. J. Pediatr. Orthop. B 24, 89–94. doi:10.1097/BPB.0000000000000107
Perry D. C., Bruce C. E., Pope D., Dangerfield P., Platt M. J., Hall A. J. (2012a). Comorbidities in Perthes' disease: a case control study using the General Practice Research database. J. Bone Jt. Surg. Br. 94, 1684–1689. doi:10.1302/0301-620X.94B12.29974
Perry D. C., Green D. J., Bruce C. E., Pope D., Dangerfield P., Platt M. J., et al. (2012b). Abnormalities of vascular structure and function in children with Perthes disease. Pediatrics 130, e126–e131. doi:10.1542/peds.2011-3269
Perry D. C., Thomson C., Pope D., Bruce C. E., Platt M. J. (2017). A case control study to determine the association between Perthes' disease and the recalled use of tobacco during pregnancy, and biological markers of current tobacco smoke exposure. Bone Jt. J. 99-B, 1102–1108. doi:10.1302/0301-620X.99B8.BJJ-2016-1282.R1
Perthes G. (2012). The classic: on juvenile arthritis deformans. 1910. Clin. Orthop. Relat. Res. 470, 2349–2368. doi:10.1007/s11999-012-2433-1
Phipps M. C., Huang Y., Yamaguchi R., Kamiya N., Adapala N. S., Tang L., et al. (2016). In vivo monitoring of activated macrophages and neutrophils in response to ischemic osteonecrosis in a mouse model. J. Orthop. Res. 34, 307–313. doi:10.1002/jor.22952
Pienkowski D., Resig J., Talwalkar V., Tylkowski C. (2009). Novel three-dimensional MRI technique for study of cartilaginous hip surfaces in Legg-Calvé-Perthes disease. J. Orthop. Res. 27, 981–988. doi:10.1002/jor.20909
Pinheiro M., Dobson C. A., Perry D., Fagan M. J. (2018). New insights into the biomechanics of legg-calve-perthes' disease: the role of epiphyseal skeletal immaturity in vascular obstruction. Bone Jt. Res. 7, 148–156. doi:10.1302/2046-3758.72.BJR-2017-0191.R1
Pringle D., Koob T. J., Kim H. K. (2004). Indentation properties of growing femoral head following ischemic necrosis. J. Orthop. Res. 22, 122–130. doi:10.1016/S0736-0266(03)00135-9
Qin X., Xi Y., Jiang Q., Chen C., Yang G. (2023). Type H vessels in osteogenesis, homeostasis, and related disorders. Differentiation 134, 20–30. doi:10.1016/j.diff.2023.09.005
Rodríguez-Olivas A. O., Hernández-Zamora E., Reyes-Maldonado E. (2022). Legg-Calvé-Perthes disease overview. Orphanet J. Rare Dis. 17, 125. doi:10.1186/s13023-022-02275-z
Rush B. H., Bramson R. T., Ogden J. A. (1988). Legg-Calvé-Perthes disease: detection of cartilaginous and synovial change with MR imaging. Radiology 167, 473–476. doi:10.1148/radiology.167.2.3357958
Schlundt C., Saß R. A., Bucher C. H., Bartosch S., Hauser A. E., Volk H. D., et al. (2023). Complex spatio-temporal interplay of distinct immune and bone cell subsets during bone fracture healing. Cells 13, 40. doi:10.3390/cells13010040
Seguin C., Kassis J., Busque L., Bestawros A., Theodoropoulos J., Alonso M. L., et al. (2008). Non-traumatic necrosis of bone (osteonecrosis) is associated with endothelial cell activation but not thrombophilia. Rheumatol. Oxf. 47, 1151–1155. doi:10.1093/rheumatology/ken206
Spasovski V., Srzentić Dražilov S., Nikčević G., Baščarević Z., Stojiljković M., Pavlović S., et al. (2023). Molecular biomarkers in perthes disease: a review. Diagn. (Basel) 13, 471. doi:10.3390/diagnostics13030471
Srzentić S., Spasovski V., Spasovski D., Zivković Z., Matanović D., Bascarević Z., et al. (2014). Association of gene variants in TLR4 and IL-6 genes with Perthes disease. Srp. Arh. Celok. Lek. 142, 450–456. doi:10.2298/SARH1408450S
Stulberg S. D., Cooperman D. R., Wallensten R. (1981). The natural history of Legg-Calve-Perthes disease. J. Bone Jt. Surg. Am. 63, 1095–1108. doi:10.2106/00004623-198163070-00006
Su P., Li R., Liu S., Zhou Y., Wang X., Patil N., et al. (2008). Age at onset-dependent presentations of premature hip osteoarthritis, avascular necrosis of the femoral head, or Legg-Calve-Perthes disease in a single family, consequent upon a p.Gly1170Ser mutation of COL2A1. Arthritis Rheum. 58, 1701–1706. doi:10.1002/art.23491
Su P., Zhang L., Peng Y., Liang A., Du K., Huang D. (2010). A histological and ultrastructural study of femoral head cartilage in a new type II collagenopathy. Int. Orthop. 34, 1333–1339. doi:10.1007/s00264-010-0985-9
Vosmaer A., Pereira R. R., Koenderman J. S., Rosendaal F. R., Cannegieter S. C. (2010). Coagulation abnormalities in Legg-Calve-Perthes disease. J. Bone Jt. Surg. Am. 92, 121–128. doi:10.2106/JBJS.I.00157
Vusal Ismayilov M. (2014). Increased soluble selectins as a ref source J pediatr hematol oncol. Pediatrics.
Wang L., Zhou F., Zhang P., Wang H., Qu Z., Jia P., et al. (2017). Human type H vessels are a sensitive biomarker of bone mass. Cell Death Dis. 8, e2760. doi:10.1038/cddis.2017.36
Wang S., Zhong H., Ze R., Hong P., Li J., Tang X. (2022). Microarray analysis of lncRNA and mRNA expression profiles in patients with Legg-Calve-Perthes disease. Front. Pediatr. 10, 974547. doi:10.3389/fped.2022.974547
Wang T., Luo X., Li B., Huang Q., Liu J., Tang S., et al. (2023). Platelet to lymphocyte ratio was a risk factor in Perthes disease. Sci. Rep. 13, 5052. doi:10.1038/s41598-023-32000-0
Wei W. B., Dang S. J., Li Y., Liu Z. Z. (2017). Alterations of serum osteocalcin levels in patients with Legg-Calvé-Perthes. Hip Int. 27, 92–95. doi:10.5301/hipint.5000431
Woratanarat P., Thaveeratitharm C., Woratanarat T., Angsanuntsukh C., Attia J., Thakkinstian A. (2014). Meta-analysis of hypercoagulability genetic polymorphisms in Perthes disease. J. Orthop. Res. 32, 1–7. doi:10.1002/jor.22473
Xu J., He S. J., Xia T. T., Shan Y., Wang L. (2024). Targeting type H vessels in bone-related diseases. J. Cell Mol. Med. 28, e18123. doi:10.1111/jcmm.18123
Yamaguchi R., Kamiya N., Adapala N. S., Drissi H., Kim H. K. (2016). HIF-1-Dependent IL-6 activation in articular chondrocytes initiating synovitis in femoral head ischemic osteonecrosis. J. Bone Jt. Surg. Am. 98, 1122–1131. doi:10.2106/jbjs.15.01209
Yu R., Ma C., Li G., Xu J., Feng D., Lan X. (2023). Inhibition of toll-like receptor 4 signaling pathway accelerates the repair of avascular necrosis of femoral epiphysis through regulating macrophage polarization in perthes disease. Tissue Eng. Regen. Med. 20, 489–501. doi:10.1007/s13770-023-00529-w
Zhang C., Yang F., Cornelia R., Tang W., Swisher S., Kim H. (2011). Hypoxia-inducible factor-1 is a positive regulator of Sox9 activity in femoral head osteonecrosis. Bone 48, 507–513. doi:10.1016/j.bone.2010.10.006
Zhao Y., Liao S., Lu R., Dang H., Zhao J., Ding X. (2016). Endothelial nitric oxide synthase gene polymorphism is associated with Legg-Calve-Perthes disease. Exp. Ther. Med. 11, 1913–1917. doi:10.3892/etm.2016.3111
Zheng P., Yang T., Ju L., Jiang B., Lou Y. (2015). Epigenetics in Legg–Calvé–Perthes disease: a study of global DNA methylation. J. Int. Med. Res. 43, 758–764. doi:10.1177/0300060515591062
Keywords: perthes disease, etiology, pathogenesis, molecular biology, cellular biology
Citation: Zheng X, Dong Z, Ding X, Huang Q, Tang S, Zhang Y, Li B and Liao S (2025) Progress in understanding Legg–Calvé–Perthes disease etiology from a molecular and cellular biology perspective. Front. Physiol. 16:1514302. doi: 10.3389/fphys.2025.1514302
Received: 21 October 2024; Accepted: 27 January 2025;
Published: 17 February 2025.
Edited by:
Rafael Scaf De Molon, São Paulo State University, BrazilReviewed by:
Nobuhiro Kamiya, Tenri University, JapanCopyright © 2025 Zheng, Dong, Ding, Huang, Tang, Zhang, Li and Liao. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Shijie Liao, Z3hsaWFvc2hpamllQDE2My5jb20=; Boxiang Li, bm5saWJveGlhbmdAMTYzLmNvbQ==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.