 Kevin L. Shores
Kevin L. Shores George A. Truskey
George A. Truskey- Department of Biomedical Engineering, Duke University, Durham, NC, United States
Hutchinson-Gilford Progeria Syndrome (HGPS) is a premature aging disorder that causes severe cardiovascular disease, resulting in the death of patients in their teenage years. The disease pathology is caused by the accumulation of progerin, a mutated form of the nuclear lamina protein, lamin A. Progerin binds to the inner nuclear membrane, disrupting nuclear integrity, and causes severe nuclear abnormalities and changes in gene expression. This results in increased cellular inflammation, senescence, and overall dysfunction. The molecular mechanisms by which progerin induces the disease pathology are not fully understood. Progerin’s detrimental impact on nuclear mechanics and the role of the nucleus as a mechanosensor suggests dysfunctional mechanotransduction could play a role in HGPS. This is especially relevant in cells exposed to dynamic, continuous mechanical stimuli, like those of the vasculature. The endothelial (ECs) and smooth muscle cells (SMCs) within arteries rely on physical forces produced by blood flow to maintain function and homeostasis. Certain regions within arteries produce disturbed flow, leading to an impaired transduction of mechanical signals, and a reduction in cellular function, which also occurs in HGPS. In this review, we discuss the mechanics of nuclear mechanotransduction, how this is disrupted in HGPS, and what effect this has on cell health and function. We also address healthy responses of ECs and SMCs to physiological mechanical stimuli and how these responses are impaired by progerin accumulation.
Introduction
Mechanotransduction is the translation of mechanical stimuli into biochemical signals through numerous mechanosensitive cellular components such as stretch-activated ion channels, G protein-coupled receptors (GPCRs), integrins, and cytoskeletal filaments. The cellular response to mechanical forces is critical for tissue development and homeostasis (Humphrey et al., 2014; Davis et al., 2023). By decoupling nuclear and cytoplasmic responses to external mechanical stresses, a role for the nucleus as a mechanosensor has been identified (Kirby and Lammerding, 2018). Nuclear mechanotransduction involves the generation and regulation of signals transmitted through the cytoskeleton or modulation of gene expression by nuclear deformation. How the nucleus senses mechanical stimuli and what effect that has on cellular function is of particular interest in the context of various diseases, specifically those that affect nuclear architecture like laminopathies. These are genetic disorders that cause mutations in proteins of the nuclear lamina, leading to structural disruption of the nucleus or gene mis-regulation (Maraldi et al., 2011). Some of the most studied laminopathies include Emery-Dreifuss muscular dystrophy, dilated cardiomyopathy, and the accelerated aging disorder, Hutchinson-Gilford Progeria Syndrome (HGPS) (Maurer and Lammerding, 2019). Of these, HGPS is the most severe, resulting in the premature death of patients in their early teens due to extreme cardiovascular disease caused by progerin, a truncated and farnesylated form of the lamin A protein (Gordon et al., 2014). Cells that are exposed to high, continuous mechanical stresses are often the most affected in HGPS. These include bone (Schmidt et al., 2012), skin (Sagelius et al., 2008), and vascular cells, particularly the smooth muscle cells (SMCs) within larger arteries (Murtada et al., 2023; Olive et al., 2010). Vascular cells are constantly exposed to varying mechanical stresses from blood flow and intraluminal pressure, and thus are highly sensitive to perturbations in these forces (Davis et al., 2023). Disturbances in mechanical loads within the vasculature, which occur within curved or branched arteries, can promote cellular dysfunction, leading to manifestation of disease (Chiu and Chien, 2011). Impairment of cellular responsiveness to physiological mechanical signaling may contribute to cellular dysfunction and disease in HGPS (Hennekam, 2006). Here, we discuss the influence of progerin expression on cellular mechanotransduction to fluid and solid stresses acting on vascular cells and evaluate how this might contribute to the vascular pathology seen in HGPS.
Pathophysiology of Hutchinson-Gilford Progeria Syndrome
HGPS is a rare and fatal disease that results in premature aging of children. Common symptoms of the disease include delayed growth, alopecia, bone dysplasia, joint contracture, scleroderma, lipodystrophy, and severe atherosclerosis (Olive et al., 2010; Gordon et al., 2007; Merideth et al., 2008; Ullrich and Gordon, 2015). The atherosclerosis, manifested as vascular stiffening, calcification, and fibrotic thickening of the vessel wall, leads to myocardial infarction or stroke. The disease develops rapidly with patients only surviving into their mid-teens (Olive et al., 2010; Hennekam, 2006; Gordon et al., 2014).
In over 90% of the affected population, the disease pathology is caused by a point mutation in the LMNA gene (c. 1824C>T) (Eriksson et al., 2003; De Sandre-Giovannoli et al., 2003). This mutation disrupts the post-translational processing of pre-lamin A, a major component of the nuclear lamina, by activating a cryptic splice site, causing an in-frame deletion of 50 amino acids near the C-terminus. Under healthy conditions, prelamin A is post-translationally modified by two transfer reactions, adding a farnesyl and carboxymethyl group, and two proteolytic cleavages that remove these groups to form mature lamin A (Musich and Zou, 2009). The mutated protein in HGPS, called progerin, lacks sites for endoproteolytic cleavage and remains farnesylated and carboxymethylated (Ahmed et al., 2018; Eriksson et al., 2003). At the cellular level, this leads to increased inflammation (Osorio et al., 2012), accelerated senescence (Cao et al., 2011; Wheaton et al., 2017; Benson et al., 2010), and stem cell dysfunction (Scaffidi and Misteli, 2008; Espada et al., 2008; Rosengardten et al., 2011). Progerin accumulates in the nuclear lamina causing abnormal nuclear shapes and protrusions (“blebs”) (Goldman et al., 2004), defects in DNA repair mechanisms (Gonzalo and Kreienkamp, 2015), epigenetic alterations (Shumaker et al., 2006), loss of heterochromatin (Goldman et al., 2004; McCord et al., 2013), and nuclear stiffening (Booth et al., 2015). While it is not yet fully understood how progerin causes cellular and nuclear dysfunction, a disruption to mechanotransduction appears to play a significant role.
Nuclear mechanotransduction and the impact of laminopathies
Mechanotransduction within the nucleus occurs through the nuclear envelope. This structure is comprised of the outer nuclear membrane (ONM), the inner nuclear membrane (INM), the perinuclear space between the ONM and INM, nuclear pore complexes (NPCs), and the nuclear lamina (Kalukula et al., 2022). The nuclear envelope is connected to the cytoskeleton via the linker of nucleoskeleton and cytoskeleton (LINC) complex. Actin filaments, microtubules, or intermediate filaments in the cytoplasm bind directly or indirectly to nesprin proteins that are localized to the ONM (Figure 1). Within the perinuclear space, these nesprins bind SUN (Sad1p, UNC-84) domain-containing proteins that span the INM and connect to the lamins of the nuclear lamina, NPCs, or directly to chromatin (Crisp et al., 2006; Kalukula et al., 2022). The nuclear lamina, comprised of A-type lamins (lamin A and C) and B-type lamins (lamin B1 and B2) bind with NPCs, LINC complex proteins, and chromatin (Burke and Stewart, 2013; de Leeuw et al., 2018). These elements provide a direct link from the cytoskeleton to DNA and facilitate changes in gene expression that occur in response to mechanical stimuli. The role of the LINC complex in vascular mechanotransduction was recently reviewed (Bougaran and Bautch, 2024) and only key features will be noted here.
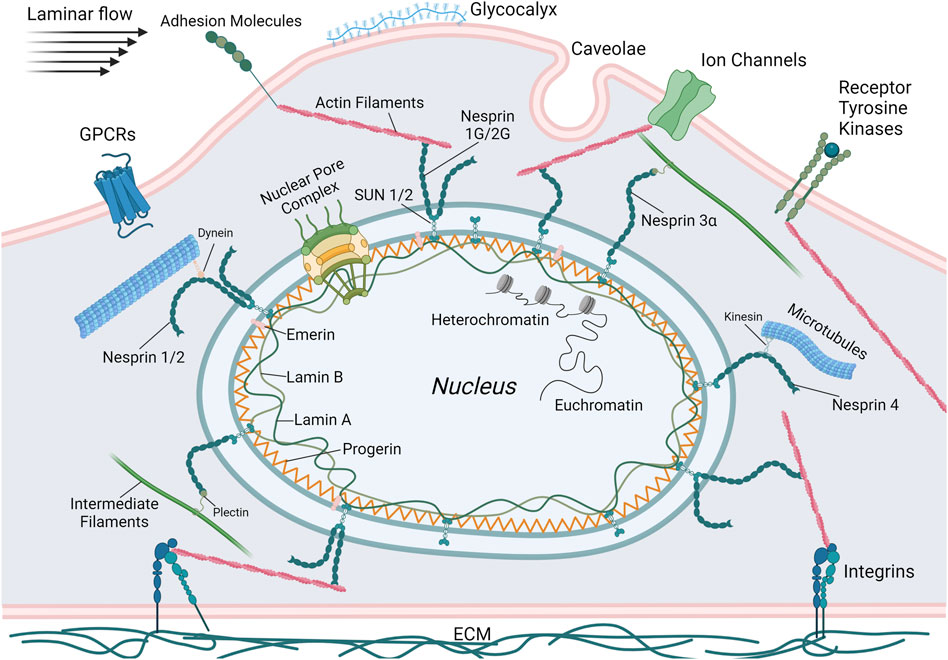
Figure 1. Schematic illustrating the primary mechanosensing elements of ECs including G-protein coupled receptors (GPCRs), adhesion molecules, ion channels, receptor tyrosine kinases, integrins, caveolae, and the glycocalyx. Also depicted are the components of the LINC complex. Lamins and, in the case of HGPS, progerin, are localized to the nuclear periphery and bind DNA or transcription factors. SUN proteins bridge the nuclear membrane, connecting intranuclear lamins with extranuclear KASH proteins (nesprins 1-4). These nesprins then bind to the cytoskeletal elements actin, microtubules, and intermediate filaments that are connected to or influenced by the various mechanosensors. Thus, the complete mechanotransductive pathway that regulates gene expression in response to external mechanical stresses can be visualized. This figure was generated using BioRender.
The nuclear lamina is essential for maintaining nuclear structure and organization of the genome (Gruenbaum and Foisner, 2015). Lamins provide a structural scaffold, anchoring chromatin and transcription factors to the nuclear periphery. This anchoring facilitates the compartmentalization of the genome, which is essential for accurate DNA transcription, replication, and repair. Furthermore, lamin expression levels are directly associated with nuclear mechanical stability, as well as tissue rigidity and plasticity. Higher amounts of lamin A and C correlate with increased nuclear stiffness, while lower amounts result in more deformable and fragile nuclei (Maurer and Lammerding, 2019). Lamins recruit DNA damage response machinery and regulate transcription factor binding. They also modulate heterochromatin, domains of densely packed DNA that are transcriptionally repressed and anchor it to the nuclear periphery (Camozzi et al., 2014). Specifically, lamin A interacts with INM proteins to regulate the location of heterochromatin based on extracellular mechanical stresses (Capanni et al., 2009; Maraldi et al., 2011; Solovei et al., 2013). Lamins also bind and anchor transcription factors to the nuclear periphery, regulating expression of the genes associated with these factors (Gruenbaum and Foisner, 2015). Dysfunctional lamin A, like that seen in HGPS, can detrimentally alter its ability to maintain appropriate nuclear mechanical properties and regulate gene expression.
In addition to lamins regulating gene expression through the organization of heterochromatin and transcription factors, there are several other ways that mechanical forces influence the activation or repression of genes. Mechanical stresses can induce nuclear deformations that result in immediate stretching of chromatin through the connections of the cytoskeleton and LINC complex (Tajik et al., 2016). While not completely understood, the presiding theory is that chromatin opening reveals previously inaccessible areas of the genome, resulting in rapid transcription of mechanosensitive genes (Dupont and Wickström, 2022; Kirby and Lammerding, 2018). In addition to the direct stretching of chromatin, these nuclear deformations can also stretch the NPCs localized within the nuclear envelope. NPCs allow the exchange of macromolecules between the nucleus and cytoplasm. NPCs are governed by the binding of soluble nuclear transport factors and transport signals (e.g., nuclear localization sequences or nuclear export sequences) to macromolecules that then allow docking to and transport through the NPC (Strambio-De-Castillia et al., 2010). Stretching of the nuclear membrane increases the diameter of the NPC opening, allowing import of mechanoresponsive transcriptional regulators like YAP (Yes-associated protein) (Elosegui-Artola et al., 2017). Depending on the nature of the mechanical force and the resultant deformation of the nuclear membrane, NPC stretching can promote nuclear export, or the NPC opening can constrict, inhibiting exchange of macromolecules (Kalukula et al., 2022). The nuclear membrane also controls stretch-sensitive ion channels that can be opened during nuclear deformations induced by mechanical stresses. These channels allow increased uptake of calcium into the nucleus, altering chromatin organization (Nava et al., 2020). Finally, external forces can induce conformational changes and increase phosphorylation of lamins (Buxboim et al., 2014b; Swift et al., 2013). Lamin conformational changes can expose residues, altering interactions with other proteins and downstream signaling (Ihalainen et al., 2015), while lamin phosphorylation influences their nucleo-cytoskeletal coupling (Guilluy et al., 2014), chromatin binding (Liu and Ikegami, 2020), and regulation of nuclear stiffness through lamin disassembly (Buxboim et al., 2014a). Due to truncation of amino acids, progerin lacks some of the major phosphorylation sites found on lamin A (Ao et al., 2023). This reduces its rate of phosphorylation in response to mechanical stimuli (Cho et al., 2018) and promotes cellular senescence (Ao et al., 2023).
Laminopathies like HGPS result from mutations that alter lamin function or expression. There are two prevailing hypotheses as to how these dysfunctional lamins cause disease: (1) mutant lamins make the nucleus more fragile, resulting in greater nuclear ruptures and cell death, particularly in tissues exposed to high mechanical stresses, and (2) lamin mutations alter gene expression through impaired chromatin interactions or inhibition of transcription factor binding (Maurer and Lammerding, 2019). Nuclear mechanotransduction can influence both proposed mechanisms. Cells with lamin A mutations or deletions exhibit increases in nuclear ruptures following mechanical loading that leads to greater cell death (Lammerding et al., 2004; Kim et al., 2021; Earle et al., 2020; Kim et al., 2024; Sears and Roux, 2022). Of note, decreased levels of lamin A correspond to more deformable nuclei with increased fragility, making them more susceptible to rupture (Lammerding et al., 2004). In contrast, some lamin A mutations, like those that produce progerin, increase nuclear stiffness (Dahl et al., 2006), reducing its compliance and ability to deform under mechanical loading, resulting in a greater susceptibility to rupture (Kim et al., 2021; Yue et al., 2023; Verstraeten et al., 2008). Preceding cell death, nuclear rupture can also promote senescence and the production of pro-inflammatory cytokines known as the senescence-associated secretory phenotype (SASP). Genomic DNA released into the cytosol following nuclear rupture binds to cyclic GMP-AMP synthase (cGAS), which triggers SASP production (Dou et al., 2017; Glück et al., 2017). HGPS and other laminopathies also cause DNA damage. This can occur through dysfunctional nuclear-cytoskeletal coupling, in the form of nuclear membrane rupture (Kalukula et al., 2022), that causes impaired recruitment (Liu et al., 2005) or cytoplasmic mislocalization (Xia et al., 2018) of DNA damage response factors.
Progerin can change the epigenetic landscape by altering the state of histone methylation, specifically reducing methylation of histone 3 on lysine 9 (H3K9) and H3K27, while increasing methylation of H4K20 (Shumaker et al., 2006; Columbaro et al., 2005). Progerin is also involved in the regulation of histone deacetylases (HDACs), with HGPS cells showing a loss of HDAC1 that correlated with heterochromatin defects (Pegoraro et al., 2009). While these epigenetic changes seen with HGPS have yet to be directly correlated with aberrant nuclear mechanotransduction, there is evidence for their regulation in response to mechanical stimuli under other conditions (Kalukula et al., 2022). Changes to nuclear mechanics induced by progerin accumulation may contribute to the apparent epigenetic alterations. H3K9me3 is suppressed in cardiomyocytes and dissociates from the nuclear periphery during environmental stiffening that reduces nuclear deformations, presumably increasing nuclear stiffness (Seelbinder et al., 2021). A similar mechanism of epigenetic alteration could be imagined in HGPS, where progerin accumulation leads to nuclear stiffening and an impairment of its response to mechanical stimuli (Booth et al., 2015), leading to a reduction in histone methylation. These detriments to nuclear structure and genome stability seen in HGPS are especially prevalent in cells under constant biomechanical stress, like those of the vasculature. Moreover, progerin accumulation within the nucleus can alter mechanotransduction pathways leading to cellular dysfunction.
Mechanotransduction in healthy endothelial cells
The arterial endothelium is exposed to two primary stresses (force per unit area) in the body: shear stress from the flow of blood and circumferential stress due to stretching of arteries from increases in blood pressure (Hahn and Schwartz, 2009). Responses to these forces modulate critical homeostatic functions such as inflammation, vasomotor tone, and vascular remodeling (Chiu and Chien, 2011). ECs sense these forces in a variety of ways such as ion channels (e.g., Piezo1, TRPV4, inner-rectifier K+ channel), G protein-coupled receptors (GPCRs), tyrosine kinase receptors (e.g., VEGFR2), caveolae, the glycocalyx, and integrins (Figure 1). (See (Davis et al., 2023) for an extensive review). Increases in laminar shear stress, brought on by increasing blood flow due to altered physical activity such as exercise, induce ECs to generate prostacyclin and nitric oxide (NO). These molecules promote relaxation of smooth muscle cells (SMCs) in the vascular media, providing control over vasomotor tone and ultimately blood pressure (Davies, 1995). Prostacyclin, additionally, promotes SMC health and function by increasing transcription of transgelin (SM22α), which results in a more contractile SMC phenotype (Tsai et al., 2009). NO is generated in response to shear stress through the platelet/endothelial cell adhesion molecule 1 (PECAM1)/vascular endothelium (VE)-cadherin/vascular endothelial growth factor receptor 2 (VEGFR2) complex. Initially, PECAM1 is phosphorylated in response to flow and activates a Src family kinase. VE-cadherin associates with VEGFR2 (also known as FLK1) and brings it into proximity with PECAM1. VEGFR2 is then activated by the Src family kinase and proceeds to recruit and activate phosphoinositide 3-kinase (PI3K). PI3K then phosphorylates endothelial nitric oxide synthase (eNOS) in an Akt-dependent manner. This phosphorylated eNOS, through a series of redox reactions, produces NO to relax SMCs and dilate the vessel (Tzima et al., 2005). ECs can also modulate vascular tone by releasing potassium through calcium-sensitive potassium channels that transport the potassium to the SMCs, triggering hyperpolarization and relaxation (Garland and Dora, 2017).
In addition to generating NO, shear forces also cause ECs to align in the direction of flow. This reorganization of the cytoskeleton is mediated by the PECAM1/VE-cadherin/VEGFR2 complex as well. Specifically, PI3K promotes conformational activation of integrins that, in turn, activate small GTPases (Rac, Rho, and CDC42), which regulate cell-cell junctions and remodeling of the actin cytoskeleton to elongate and align ECs in the flow direction (Osborn et al., 2006; Hahn and Schwartz, 2009). The cytoskeleton is critical for EC sensing and reacting to external stimuli as inhibiting actin, microtubules, or intermediate filaments block many EC responses to flow (Hahn and Schwartz, 2009).
Shear stresses have a direct effect on the nucleus and LINC complex in ECs. Cytoskeletal reorganization under flow leads to protrusion of the nucleus, exposing it to higher shear stresses than the rest of the cell (Tkachenko et al., 2013; Barbee et al., 1994; Barbee et al., 1995). Like the cell body cytoskeleton, the nucleus elongates in the direction of flow, providing a visual confirmation for its mechanosensing capacity (Danielsson et al., 2022b; Nava et al., 2020; Deguchi et al., 2005). Within the nuclear envelope, the LINC complex proteins exhibit essential functions in regulating many EC mechanotransductive pathways. SUN1 and 2 regulate nesprin-1 interactions with microtubules, which influences signaling of the small GTPase Ras homology family (Rho) proteins that alter cell-cell junctions or induce expression of EC tight junction proteins, leading to increased barrier function (Yang et al., 2020; Buglak et al., 2023).
In ECs, many transcription factors and transcriptional coactivators are upregulated in response to laminar shear stresses. These, along with the alteration of histone acetylation and phosphorylation induced by flow (Chen et al., 2008; Wang et al., 2010; Chen et al., 2010), modulate gene expression. Krüppel-like Factor (KLF) 2 and 4 and nuclear factor erythroid 2-related factor 2 (NRF2) are transcription factors critical to maintaining EC homeostasis. KLF2 expression is upregulated by phosphorylation and nuclear export of histone deacetylase 5 (HDAC5) in response to shear. Specifically, KLF2 induces eNOS expression and inhibits pro-inflammatory signaling through suppression of vascular cell adhesion molecule 1 (VCAM1) and E-selectin. KLF2 recruits the histone acetyltransferase p300 and outcompetes nuclear factor-κB (NF-κB), which promotes the expression of inflammatory genes (SenBanerjee et al., 2004; Dekker et al., 2002). In addition to increased recruitment, hemodynamic shear stress increases the activity of the transcriptional coactivator p300, which leads to increased eNOS transcription (Chen et al., 2008). YAP is another transcriptional coactivator that regulates many functions in ECs. Its phosphorylation leads to its proteasomal degradation in the cytosol, while dephosphorylation results in nuclear localization and activation of genes (Dupont et al., 2011). YAP nuclear localization is increased by the resultant NPC opening from mechanically stretching cells (Elosegui-Artola et al., 2017). Its translocation is also dependent on nuclear compression. Nuclei that became softer after silencing of lamin A/C experienced greater compression and elevated YAP nuclear localization (Koushki et al., 2023). Laminar flow promotes YAP phosphorylation in ECs, inhibiting its nuclear localization, through activation of G-protein-integrin interactions and RhoA inhibition (Wang et al., 2016a). YAP suppression downregulates pro-inflammatory gene expression such as monocyte chemoattractant protein-1 (MCP1), intercellular adhesion molecule-1 (ICAM1), and vascular cell adhesion molecule-1 (VCAM1) (Liu et al., 2019; Xu et al., 2016; Wang et al., 2016b). In contrast, YAP activation promotes inflammation and proliferation in ECs (Wang et al., 2016a). How the shear stress response of ECs is disrupted in HGPS is not completely understood, but several studies have started to reveal possible mechanisms that relate EC dysfunction with an impaired mechanosensing ability.
Disruption of mechanotransduction in HGPS endothelial cells
Many of the homeostatic functions of ECs discussed in the previous section are diminished or even reversed under disturbed flow that generates low, oscillatory shear stresses around vessel branches or in curved arteries (Andueza et al., 2020; Jiang et al., 2014; Lee et al., 2012) (Table 1). When exposed to disturbed flow, ECs lose their ability to regulate vasomotor tone by inducing SMC relaxation and vessel dilation (Chiu and Chien, 2011). This is caused by a reduction in KLF2/4, which activates eNOS and NO production under physiological laminar shear (Andueza et al., 2020; Jiang et al., 2014). Phosphorylation of eNOS is also reduced due to inhibition of the mechanosensitive adenosine monophosphate-activated protein kinase (AMPK) pathway (Guo et al., 2007). Disturbed flow increases EC inflammation due to reduced KLF2 expression (SenBanerjee et al., 2004), increased YAP/TAZ nuclear localization (Wang et al., 2016a; Wang et al., 2016b), and enhanced NF-κB activity (Nagel et al., 1999). It also reduces autophagic flux (Vion et al., 2017) and increases endoplasmic reticulum stress (Civelek et al., 2009). Additionally, ECs lose their migratory capacity and exhibit impaired alignment with the flow direction due to, in part, a disruption of nuclear-cytoskeletal coupling by loss of nesprin-1 (Chancellor et al., 2010; Denis et al., 2021; King et al., 2014). Moreover, low shear stresses (5 dynes/cm2) decrease nesprin −2 and lamin A in ECs, which leads to an increase in apoptosis (Han et al., 2015).
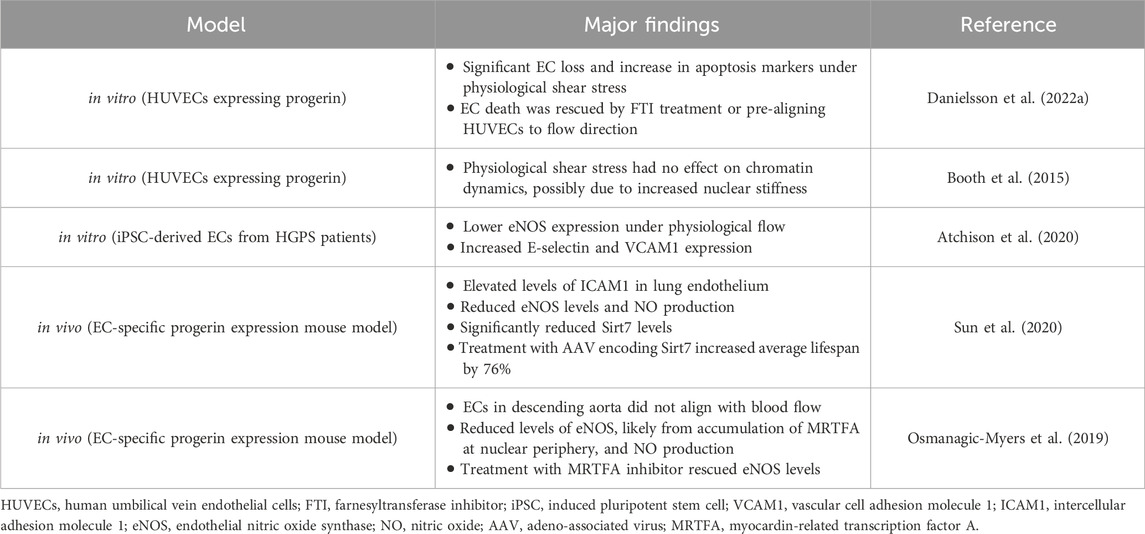
Table 1. Summary of the major findings from studies showing a disrupted mechanotransduction pathway in ECs expressing progerin.
When exposed to the more uniform laminar flow conditions in straight regions of blood vessels, HGPS ECs mirror the dysfunction seen under disturbed flow, indicating an inability to respond to external mechanical stimuli, presumably because of progerin accumulation within the nucleus. Several in vitro studies have shown HGPS EC dysfunction in response to laminar shear stress. Danielsson and colleagues expressed progerin in human umbilical vein endothelial cells (HUVECs) and exposed them to physiological shear (12 dynes/cm2) for 3 days. They found significant cell loss and increased markers of apoptosis compared to control HUVECs (Danielsson et al., 2022a). These responses were rescued by treatment with a farnesyltransferase inhibitor (FTI), that prevents progerin accumulation at the nuclear periphery (Yang et al., 2006), or, interestingly, by pre-aligning the HUVECs to flow prior to inducing progerin expression (Danielsson et al., 2022a). This suggests that progerin accumulation inhibits ECs ability to respond to flow, and that this impairment can result in increased EC apoptosis. HGPS ECs have lower eNOS expression after exposure to steady laminar shear stress for 24 h (Atchison et al., 2020; Gete et al., 2021), increased inflammation (Atchison et al., 2020; Osmanagic-Myers et al., 2019; Sun et al., 2020), and an inability to properly align under physiological flow (Osmanagic-Myers et al., 2019; Danielsson et al., 2022a). Treatment of HGPS ECs with the FTI lonafarnib or the rapamycin analog Everolimus restored eNOS and KLF2 expression (Abutaleb et al., 2023).
Another study measured chromatin dynamics under 20 dynes/cm2 shear stress in HUVECs treated with exogenously expressed progerin (Booth et al., 2015). External forces had no effect on chromatin dynamics in HUVECS expressing progerin compared to untreated HUVECs, possibly due to the increased nuclear stiffness caused by progerin accumulation (Verstraeten et al., 2008). This suggests a disruption in the nuclear mechanotransduction pathway that can alter chromatin organization, and possibly inhibit the regulation of critical mechanoresponsive genes.
Tissue-engineered blood vessels (TEBVs) prepared using induced pluripotent stem cell (iPSC)-derived ECs and SMCs from HGPS patients (viECs) (Atchison et al., 2020) showed suppressed dilation in response to acetylcholine (Ach) and increases in E-selectin and VCAM1 expression (Atchison et al., 2020). These results suggest a disruption in the mechanosensing pathway, inhibiting eNOS upregulation and promoting a pro-inflammatory response under laminar flow conditions.
Two in vivo studies using HGPS mouse models corroborated some of the findings from in vitro experiments. In both models, mice solely express progerin in ECs, allowing the investigation of their effects on HGPS vascular pathology without the influence from other vascular cells. Using single-cell transcriptomics analysis, Sun and colleagues found significant increases in the inflammatory response in the lung endothelium of EC-specific progerin expressing mice, with elevated levels of ICAM1 (Sun et al., 2020). They supported these findings by overexpressing progerin in HUVECs and showing significantly elevated levels of inflammatory genes. Relative to controls, progerin-expressing ECs exhibited significantly attenuated relaxation of the thoracic aorta in response to Ach and reduced eNOS levels. Sodium nitroprusside treatment, which induces SMC relaxation in an EC-independent manner (Bonaventura et al., 2008), was unaffected, suggesting the dysfunction is due to progerin-expressing ECs. In progerin-expressing ECs, levels of Sirtuin 7 (Sirt7), a nicotinamide adenine dinucleotide (NAD+)-dependent deacylase were reduced significantly. Further, enhanced interactions of Sirt7 and progerin compared to lamin A resulted in increased polyubiquitination of the Sirt7. Treatment with an adeno-associated viral vector encoding Sirt7 resulted in a 76% increase in average lifespan (Sun et al., 2020). While EC response to mechanical stimuli was not directly measured in this study, the detrimental effects of progerin expression, such as increased inflammation and reduced eNOS and NO production, suggest dysfunctional mechanosensing.
In another EC-specific progerin expressing mouse model (Osmanagic-Myers et al., 2019), ECs within the descending aorta were not aligned with the direction of blood flow. Progerin-expressing ECs exposed to 12 dynes/cm2 shear stress in vitro did not align with flow or exhibit nuclear elongation. Progerin-expressing ECs also showed reduced levels of eNOS, which resulted in lower systemic NO levels. To determine possible mechanisms for this dysfunctional response to shear stresses, the authors found upregulation of both SUN1 and 2 as well as an increase in actin polymerization, which may have been caused by a mislocalization of emerin (a nuclear envelope protein that regulates expression of mechanosensitive genes) into discrete aggregates (Osmanagic-Myers et al., 2019).
SUN1 expression also increased in another HGPS mouse model and human HGPS fibroblasts (Chen et al., 2012). This led to increases in nuclear defects and cellular senescence, with the reduction in heterochromatin due to increased SUN1 expression appearing to play a role. SUN1 knockdown significantly decreased cellular senescence in HGPS fibroblasts and more than doubled the lifespan of progeroid mice (Chen et al., 2012). The upregulation of SUN1 in progerin-expressing ECs, seen by Osmanagic-Myers and colleagues (Osmanagic-Myers et al., 2019), may have contributed to the dysfunctional cell response to mechanical stimuli. Additionally, a recent study evaluating the effects of cyclic stretch on mesenchymal stromal cells from a Zmpste24−/− mouse model supports the contribution of increased SUN2 expression and actin polymerization in promoting cellular dysfunction (Yue et al., 2023). The Zmpste24−/− model lacks the metalloprotease needed to cleave the farnesyl group from prelamin A during posttranslational processing. This results in a permanently farnesylated prelamin A that accumulates at the nuclear envelope, similarly to progerin, leading to misshapen nuclei, increased nuclear stiffness, and elevated cellular senescence (Mu et al., 2020). This model exhibited increases in SUN2 expression, actin polymerization, and nuclear stiffness that correlated with increased DNA damage and cellular senescence. SUN2 suppression reduced DNA damage and senescence following mechanical loading. This response was potentially mediated by nuclear decoupling and softening as polymerized actin levels and nuclear stiffness also decreased with SUN2 knockdown (Yue et al., 2023). Though not directly measured, an increased nuclear stiffness due to elevated SUN2 expression and subsequent actin polymerization may have influenced the impaired shear response of ECs (Booth et al., 2015; Danielsson et al., 2022a) seen by Osmanagic-Myers and colleagues (Osmanagic-Myers et al., 2019). These authors also found increased accumulation of myocardin-related transcription factor A (MRTFA) at the nuclear periphery, associated with progerin. MRTFA binds to the eNOS promoter region and reduces its expression (Fang et al., 2011). The enhanced localization of MRTFA at the nuclear periphery in progerin-expressing ECs appeared to increase its association with eNOS leading to eNOS suppression. Treating ECs with a small molecule MRTFA inhibitor (CCG-203971) rescued eNOS levels. Importantly, MRTFA is a mechanosensitive transcription factor whose nuclear localization is increased by emerin expression and subsequent actin polymerization (Osmanagic-Myers et al., 2019). Collectively, this study provides evidence for the impact of progerin expression on the ability of ECs to properly respond to mechanical stimuli. Understanding what other mechanotransduction pathways might be affected in ECs will be an important next step for determining how to mitigate their dysfunctional response.
Mechanotransduction in healthy vascular smooth muscle cells
Vascular SMCs are exposed to intraluminal stresses caused by changes in blood pressure that lead to stretch that depends on the extent of vessel dilation and cell orientation (Davis et al., 2023). A phenomenon known as the myogenic response causes SMCs in arterioles to constrict in response to increases in luminal pressure. This response does not require an intact endothelium or innervation, although it can be influenced by them (Davis and Hill, 1999). The myogenic response has several physiological functions, primarily establishing a basal vascular tone and responding to changes in blood flow. Ion channels are most likely involved in the force sensing mechanism (see Ref (Davis et al., 2023) for an extensive review). Ion channels are activated by mechanosensing GPCRs such as angiotensin II type 1 receptor (AT1R). Cell stretching changes GPCR conformation, allowing it to activate an associated G protein (Zou et al., 2004). The activated ion channels cause a depolarization of the SMC plasma membrane, which stimulates voltage-gated calcium channels, leading to an increase in intracellular calcium. The increased calcium activates myosin light chain 20 (MLC20), resulting in vasoconstriction (Davis et al., 2023). In addition to ion channels, GPCRs activate enzymes such as Ras homology family A (RhoA). This enzyme controls the activity of several downstream effector proteins that impact SMC proliferation, migration, differentiation, and contractility. For example, the effector protein, Rho associated coiled-coil containing protein kinase 2 (ROCK2) alters contractility by promoting phosphorylation of MLC20 (de Godoy and Rattan, 2011).
Mechanical forces significantly influence gene expression and phenotype changes in SMCs, particularly under pathological conditions (Owens et al., 2004). In vitro, physiological stretching of cells (≤10%) results in maintenance of a contractile phenotype (Mao et al., 2012; Reusch et al., 1996; Yao et al., 2014; Huang et al., 2015), while supraphysiological stretching decreases the expression of contractile genes encoding proteins such as calponin, transgelin, α-smooth muscle actin, and myosin heavy chain 11 (Wang et al., 2018; Wan et al., 2015; Hu et al., 2014). Upregulation of contractile proteins under physiological loading appears to be matrix-dependent, with SMCs cultured on basement membrane laminin showing the greatest upregulation and those cultured on fibronectin showing no upregulation (Reusch et al., 1996). Correspondingly, cyclic stretching regulates integrin expression to influence interactions with extracellular matrix (ECM) and downstream mechanotransduction pathways (Mao et al., 2012).
Transforming growth factor β and sirtuin pathways are implicated in the regulation and maintenance of a contractile SMC phenotype (Yao et al., 2014; Huang et al., 2015). HDACs also contribute to SMC phenotype regulation, with HDAC7 being upregulated and HDACs 3 and 4 being downregulated in a more contractile, quiescent phenotype (Yan et al., 2009). Changes to the SMC phenotype were influenced by both matrix stiffness and stretching through regulation of RhoA-ROCK2 and calcium influx pathways, respectively (Swiatlowska et al., 2022). Discoidin domain receptor-1 (DDR1) is a membrane protein that acts as a mechanosensor after binding to collagen (Ngai et al., 2020). Increased stiffness of the ECM, which occurs as HGPS progresses, leads to increased expression and activation of DDR1. This leads to YAP activation (Ngai et al., 2022) and stress fiber formation and expression of genes involved in calcification. Further, YAP activation leads to further DDR1 expression creating a positive feedback loop promoting vessel stiffness (Ngai et al., 2022).
Disruption of mechanotransduction in HGPS smooth muscle cells
While HGPS ECs clearly show some level of dysfunction in response to physiological shear stresses, the substantial decrease in the number of SMCs within the medial layer of large arteries (Varga et al., 2006) is believed to be the driver of the cardiovascular dysfunction in HGPS. As such, many studies have attempted to determine the mechanism behind this significant reduction in cellular content unique to the vascular media. Although the loss of SMCs is the predominant feature associated with HGPS pathology, there is also an SMC dysfunction that appears to be attributed, at least in part, to altered mechanotransduction, including increased inflammation (Ribas et al., 2017), elevated DNA damage (Kim et al., 2021), and reduced contractile response (de Godoy and Rattan, 2011; Murtada et al., 2020) (Table 2). While HGPS SMCs do manifest a dysfunctional contractile response (Del Campo et al., 2020; Murtada et al., 2020), the myogenic response and SMC phenotype switching in HGPS have not been examined.

Table 2. Summary of the major findings from studies showing a disrupted mechanotransduction pathway in SMCs expressing progerin.
Exposing SMCs derived from iPSCs of HGPS donors to 16% cyclic stretch for 24 h resulted in an increased inflammatory response, through upregulation of IL-1β and IL-6, as well as elevated levels of DNA damage (Ribas et al., 2017). While 16% stretch is generally considered pathological (Jensen et al., 2021), these deleterious results were not seen in non-HGPS controls, indicating the HGPS SMCs have a comparatively dysfunctional mechanoresponse to cyclic stretching. Treatment of these SMCs with an FTI or statin attenuated the inflammatory response to mechanical forces, suggesting progerin expression plays a role in this response. While no direct mechanism was determined for the increased inflammation, HGPS cells cultured under static conditions did not express the inflammatory markers (Ribas et al., 2017). Several in vivo studies using HGPS mouse models have also shown increased SMC dysfunction, though not always in the context of impaired mechanotransduction. One study that focused on the biomechanical changes of the vasculature with disease progression found, in addition to significant SMC loss, there was a total loss of vessel constriction capacity (Murtada et al., 2020) in response to phenylephrine (PE), as opposed to increased luminal pressure. However, PE mediates contraction through the RhoA-ROCK pathway, which is reduced in HGPS models (Hale et al., 2008), and, as noted, RhoA-ROCK suppression induces a more synthetic phenotype in SMCs (de Godoy and Rattan, 2011). In addition, mice in which only SMCs express progerin, exhibited significantly lower vessel constriction by PE or KCl compared with control mice that did not express progerin (Del Campo et al., 2020). Such a response was not observed in mice that expressed progerin in ECs alone.
In vivo studies observed significant SMC loss and vascular remodeling in the ascending aorta of a mouse model expressing human progerin after 12 months (Song et al., 2014). Interestingly, SMC loss did not occur in the descending aorta. The authors hypothesized this was due to the increased mechanical stresses within the ascending aorta, determining that the reduction in lumen diameter due to vascular remodeling of the ascending aorta could lead to significantly higher mechanical stresses not experienced by the descending aorta or within the ascending aorta of healthy controls (Song et al., 2014). Others have also seen differences in disease severity between different vascular regions, with the areas exposed to the highest mechanical forces being most affected (Murtada et al., 2023). In addition to significant SMC loss, there was also a reduction in intermediate filament proteins, particularly vimentin (Song et al., 2014) (Figure 2). These are important to many mechanotransduction pathways and help disperse mechanical stresses in cells (Hu et al., 2019). The descending aorta did not show the same vimentin reduction. In an ex vivo experiment using a microfluidic system, the descending aortas of WT and HGPS mice were exposed to high levels of shear stress (75 dynes/cm2). The WT samples did not show any changes in vimentin expression, but HGPS samples had a substantial reduction (Song et al., 2014). This study suggests that SMC loss seen in HGPS is dependent on the magnitude of pathological mechanical forces and that a potential mechanism involves the reduction in mechanotransduction related proteins.
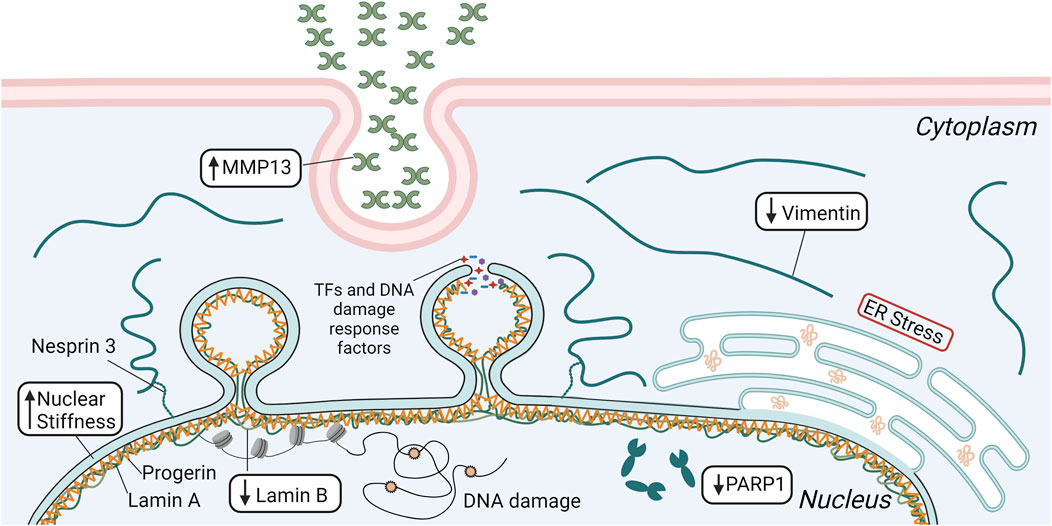
Figure 2. Schematic depicting several mechanisms that are involved in the significant SMC loss seen in HGPS, including elevated matrix metalloproteinase 13 (MMP13) secretion and endoplasmic reticulum (ER) stress, reduced vimentin and poly [ADP-ribose] polymerase 1 (PARP1) expression, and decreased levels of lamin B. Lower lamin B in concert with progerin accumulation increases nuclear stiffness, leading to greater incidence of nuclear rupture and DNA damage that ultimately results in increased SMC apoptosis. This figure was generated using BioRender.
Another in vivo study found a connection among components of the nucleoskeleton, pathological biomechanical forces, and loss of SMCs in an HGPS mouse model (Kim et al., 2018). The HGPS phenotype (e.g., greater ECM deposition and adventitial thickening with fewer SMCs) was more severe at regions of disturbed flow within the aorta. To determine if HGPS led to increased sensitivity to irregular mechanical stresses, progerin-expressing SMCs were stretched in vitro for 24 h. Stretching caused 40% of the SMCs to detach from the flexible membrane and these were determined non-viable, whereas WT SMCs were unaffected by the same mechanical regimen. The authors hypothesized that this response to mechanical forces may be due to the low levels of lamin B1 in SMCs combined with progerin expression, while WT SMCs are protected by their high levels of lamin A. They disrupted the connection between the nuclear and cytoskeleton by overexpressing the KASH domain of nesprin-2 (a component of the LINC complex). Since nesprins interact with SUN proteins through KASH domains, overexpressing the KASH domain of nesrpin2 occupies SUN protein binding sites, preventing the connection between the nuclear and cytoskeleton and reducing the transmission of external forces to the nucleus (Kim et al., 2018). This disruption of the LINC complex greatly improved progerin-expressing SMC survival after stretching. In vivo, KASH overexpression increased SMC content to 65% of WT mice in the outer curvature of the ascending aorta, and, interestingly, adventitial fibrosis was eliminated. Improvements were also seen in the inner curvature of the ascending aorta, albeit, not as pronounced, where the pathology is more severe (Kim et al., 2018). This study suggests that the resilience of the nucleus to external mechanical forces is impaired by progerin expression, leading to nuclear damage and cell death. While the exact mechanism is unknown, the authors speculate it is due to the increased nuclear rigidity imparted by progerin accumulation in conjunction with the low levels of lamin B1 seen in SMCs (Kim et al., 2018).
Increasing lamin B1 in progerin-expressing SMCs leads to an 80% reduction in nuclear membrane ruptures, lower amounts of DNA damage, and increased cell survival after 2 hours of stretching. Elevated lamin B1 decreases nuclear stiffness by 23% (Kim et al., 2021). This suggests that in HGPS SMCs, lower nuclear compliance due to reduced lamin B1 and elevated progerin leads to greater incidence of nuclear membrane ruptures and cell death. In vivo, progerin expression increases with age, while lamin B1 decreases, and this correlates with increased nuclear membrane ruptures in the aortic SMCs of older HGPS mice (Kim et al., 2021). In healthy SMCs, lamin A forms an organized network on the nuclear membrane with openings of mean size around 0.085 µm2 (Kim et al., 2024). However, when the SMCs express progerin, the network is less organized with larger openings through which blebs formed (Figure 2). The blebs were associated with low levels of lamin B1. Overexpressing lamin B1 normalized the meshwork and reduced bleb formation. Collectively, these in vivo studies suggest that progerin accumulation leads to a dysfunction in the components of the nucleoskeleton of SMCs that impairs their ability to resist damage caused by elevated or disturbed external mechanical forces, leading to nuclear/DNA damage and cell death.
Future directions and conclusion
It is clear from in vitro and in vivo studies that the progerin accumulation negatively impacts mechanotransduction pathways in both ECs and SMCs. To what extent this impaired mechanoresponse influences the cellular dysfunction seen in HGPS needs additional study.
For ECs, it would be interesting to investigate the effect of modulating LINC complex proteins on their response to physiological flow, such as cell alignment, inflammation, and elevated expression of homeostatic proteins like eNOS, KLF2, and NRF2. Additionally, other mechanotransduction pathways important to EC homeostasis are disrupted in non-endothelial HGPS cells (Son et al., 2024; Kubben et al., 2016). As mentioned, in ECs, the mechanosensitive AMPK pathway activates eNOS to produce NO (Guo et al., 2007). This pathway was inhibited in HGPS fibroblasts and activating it rescued the HGPS phenotype, reducing progerin expression and DNA damage, while restoring heterochromatin marks (Son et al., 2024). Similarly, NRF2, which provides antioxidant protection and suppresses inflammation in ECs (Chen et al., 2006), is mislocalized in HGPS fibroblasts to the nuclear periphery by progerin, preventing its activation. Providing activated NRF2 prevented nuclear defects, lowered progerin expression, and reduced oxidative stress (Kubben et al., 2016). These pathways should be investigated in the context of EC mechanotransduction as they may also be inhibited by progerin expression. This could provide additional explanation for the impaired mechanoresponse of HGPS ECs. Also, understanding how nuclear mechanics change in ECs with progerin accumulation and how this might differ from SMCs could provide insight into ECs ability to survive the high shear stresses in larger arteries while SMCs cannot.
While it is unclear what causes the characteristic loss of SMCs in HGPS, progerin accumulation may decrease nuclear compliance leading to increased nuclear ruptures and cell death. Song and colleagues attributed this to the loss of vimentin in SMCs of the ascending aorta that are exposed to high shear stresses (Song et al., 2014). In support of this hypothesis, others have found vimentin to be necessary for nuclear resistance to deformation under mechanical strains (Neelam et al., 2015; Patteson et al., 2019). The nuclear elasticity imparted by vimentin reduces the incidence of nuclear rupture and DNA damage (Patteson et al., 2019). Studies with cultured cells indicate that loss of vimentin is compensated by increased ECM synthesis and stiffening of the ECM (Grolleman et al., 2023), which could then explain changes in the vessel wall composition and stiffness in HGPS. Moreover, increased endoplasmic reticulum stress (Hamczyk et al., 2019), elevated matrix metalloprotease 13 expression (Pitrez et al., 2020), and increased poly (ADP-ribose) polymerase 1 expression (Zhang et al., 2014) influence SMC loss and treatment of the specific condition resulted in improved SMC survival.
Vascular dysfunction in HGPS is likely caused by multiple factors, with impaired mechanotransduction having an influence. Progerin accumulation not only affects the mechanosensing of the cell through disruption of the nuclear-cytoskeletal coupling, but can sequester transcription factors at the nuclear periphery, affecting their activity, and alter the epigenome. The long lifetime of progerin in large arteries can amplify any of these pathways (Hasper et al., 2023). While some studies involving HGPS SMCs suggest it is mainly the change in nuclear stiffness that promotes the pathological features, ones focused on ECs show protein mislocalization (e.g., MRTFA) or destabilization (e.g., Sirt7) having a prominent influence. Understanding how these two mechanisms of progerin accumulation alter cellular function in each of these vascular cells can influence what treatment methods may be required for improving the cardiovascular disease caused by HGPS.
Author contributions
KS: Investigation, Writing–original draft, Writing–review and editing. GT: Conceptualization, Funding acquisition, Investigation, Resources, Supervision, Writing–review and editing.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. Supported by NIH grant HL138252 (GAT) and an NIH predoctoral fellowship to KS (F31HL172589).
Acknowledgments
Figures were created using BioRender.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abutaleb N. O., Atchison L., Choi L., Bedapudi A., Shores K., Gete Y., et al. (2023). Lonafarnib and everolimus reduce pathology in iPSC-derived tissue engineered blood vessel model of Hutchinson-Gilford Progeria Syndrome. Sci. Rep. 13, 5032. doi:10.1038/s41598-023-32035-3
Ahmed M. S., Ikram S., Bibi N., Mir A. (2018). Hutchinson-0067ilford progeria syndrome: a premature aging disease. Mol. Neurobiol. 55, 4417–4427. doi:10.1007/s12035-017-0610-7
Andueza A., Kumar S., Kim J., Kang D. W., Mumme H. L., Perez J. I., et al. (2020). Endothelial reprogramming by disturbed flow revealed by single-cell RNA and chromatin accessibility study. Cell Rep. 33, 108491. doi:10.1016/j.celrep.2020.108491
Ao Y., Wu Z., Liao Z., Lan J., Zhang J., Sun P., et al. (2023). Role of C-terminal phosphorylation of lamin A in DNA damage and cellular senescence. Cells 12, 639. doi:10.3390/cells12040639
Atchison L., Abutaleb N. O., Snyder-Mounts E., Gete Y., Ladha A., Ribar T., et al. (2020). iPSC-derived endothelial cells affect vascular function in a tissue-engineered blood vessel model of hutchinson-gilford progeria syndrome. Stem Cell Rep. 14, 325–337. doi:10.1016/j.stemcr.2020.01.005
Barbee K. A., Davies P. F., Lal R. (1994). Shear stress-induced reorganization of the surface topography of living endothelial cells imaged by atomic force microscopy. Circulation Res. 74, 163–171. doi:10.1161/01.res.74.1.163
Barbee K. A., Mundel T., Lal R., Davies P. F. (1995). Subcellular distribution of shear stress at the surface of flow-aligned and nonaligned endothelial monolayers. Am. J. Physiology-Heart Circulatory Physiology 268, H1765–H1772. doi:10.1152/ajpheart.1995.268.4.H1765
Benson E. K., Lee S. W., Aaronson S. A. (2010). Role of progerin-induced telomere dysfunction in HGPS premature cellular senescence. J. Cell Sci. 123, 2605–2612. doi:10.1242/jcs.067306
Bonaventura D., Lunardi C. N., Rodrigues G. J., Neto M. A., Bendhack L. M. (2008). A novel mechanism of vascular relaxation induced by sodium nitroprusside in the isolated rat aorta. Nitric Oxide 18, 287–295. doi:10.1016/j.niox.2008.02.004
Booth E. A., Spagnol S. T., Alcoser T. A., Dahl K. N. (2015). Nuclear stiffening and chromatin softening with progerin expression leads to an attenuated nuclear response to force. Soft Matter 11, 6412–6418. doi:10.1039/c5sm00521c
Bougaran P., Bautch V. L. (2024). Life at the crossroads: the nuclear LINC complex and vascular mechanotransduction. Front. Physiol. 15, 1411995. doi:10.3389/fphys.2024.1411995
Buglak D. B., Bougaran P., Kulikauskas M. R., Liu Z., Monaghan-Benson E., Gold A. L., et al. (2023). Nuclear SUN1 stabilizes endothelial cell junctions via microtubules to regulate blood vessel formation. Elife 12, e83652. doi:10.7554/eLife.83652
Burke B., Stewart C. L. (2013). The nuclear lamins: flexibility in function. Nat. Rev. Mol. Cell Biol. 14, 13–24. doi:10.1038/nrm3488
Buxboim A., Swift J., Irianto J., Spinler K. R., Dingal P. C., Athirasala A., et al. (2014a). Matrix elasticity regulates lamin-A,C phosphorylation and turnover with feedback to actomyosin. Curr. Biol. 24, 1909–1917. doi:10.1016/j.cub.2014.07.001
Buxboim A., Swift J., Irianto J., Spinler K. R., Dingal P. D. P., Athirasala A., et al. (2014b). Matrix elasticity regulates lamin-A, C phosphorylation and turnover with feedback to actomyosin. Curr. Biol. 24, 1909–1917. doi:10.1016/j.cub.2014.07.001
Camozzi D., Capanni C., Cenni V., Mattioli E., Columbaro M., Squarzoni S., et al. (2014). Diverse lamin-dependent mechanisms interact to control chromatin dynamics. Focus on laminopathies. Nucleus 5, 427–440. doi:10.4161/nucl.36289
Cao K., Blair C. D., Faddah D. A., Kieckhaefer J. E., Olive M., Erdos M. R., et al. (2011). Progerin and telomere dysfunction collaborate to trigger cellular senescence in normal human fibroblasts. J. Clin. Invest 121, 2833–2844. doi:10.1172/JCI43578
Capanni C., Del Coco R., Mattioli E., Camozzi D., Columbaro M., Schena E., et al. (2009). Emerin-prelamin A interplay in human fibroblasts. Biol. Cell 101, 541–554. doi:10.1042/BC20080175
Chancellor T. J., Lee J., Thodeti C. K., Lele T. (2010). Actomyosin tension exerted on the nucleus through nesprin-1 connections influences endothelial cell adhesion, migration, and cyclic strain-induced reorientation. Biophys. J. 99, 115–123. doi:10.1016/j.bpj.2010.04.011
Chen C. Y., Chi Y. H., Mutalif R. A., Starost M. F., Myers T. G., Anderson S. A., et al. (2012). Accumulation of the inner nuclear envelope protein Sun1 is pathogenic in progeric and dystrophic laminopathies. Cell 149, 565–577. doi:10.1016/j.cell.2012.01.059
Chen X. L., Dodd G., Thomas S., Zhang X., Wasserman M. A., Rovin B. H., et al. (2006). Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am. J. Physiol. Heart Circ. Physiol. 290, H1862–H1870. doi:10.1152/ajpheart.00651.2005
Chen W., Bacanamwo M., Harrison D. G. (2008). Activation of p300 histone acetyltransferase activity is an early endothelial response to laminar shear stress and is essential for stimulation of endothelial nitric-oxide synthase mRNA transcription. J. Biol. Chem. 283, 16293–16298. doi:10.1074/jbc.M801803200
Chen Z., Peng I. C., Cui X., Li Y. S., Chien S., Shyy J. Y. (2010). Shear stress, SIRT1, and vascular homeostasis. Proc. Natl. Acad. Sci. U. S. A. 107, 10268–10273. doi:10.1073/pnas.1003833107
Chiu J. J., Chien S. (2011). Effects of disturbed flow on vascular endothelium: pathophysiological basis and clinical perspectives. Physiol. Rev. 91, 327–387. doi:10.1152/physrev.00047.2009
Cho S., Abbas A., Irianto J., Ivanovska I. L., Xia Y., Tewari M., et al. (2018). Progerin phosphorylation in interphase is lower and less mechanosensitive than lamin-A,C in iPS-derived mesenchymal stem cells. Nucleus 9, 230–245. doi:10.1080/19491034.2018.1460185
Civelek M., Manduchi E., Riley R. J., Stoeckert C. J., Davies P. F. (2009). Chronic endoplasmic reticulum stress activates unfolded protein response in arterial endothelium in regions of susceptibility to atherosclerosis. Circ. Res. 105, 453–461. doi:10.1161/CIRCRESAHA.109.203711
Columbaro M., Capanni C., Mattioli E., Novelli G., Parnaik V., Squarzoni S., et al. (2005). Rescue of heterochromatin organization in Hutchinson-Gilford progeria by drug treatment. Cell. Mol. life Sci. 62, 2669–2678. doi:10.1007/s00018-005-5318-6
Crisp M., Liu Q., Roux K., Rattner J. B., Shanahan C., Burke B., et al. (2006). Coupling of the nucleus and cytoplasm: role of the LINC complex. J. Cell Biol. 172, 41–53. doi:10.1083/jcb.200509124
Dahl K. N., Scaffidi P., Islam M. F., Yodh A. G., Wilson K. L., Misteli T. 2006. Distinct structural and mechanical properties of the nuclear lamina in Hutchinson-Gilford progeria syndrome, Proc. Natl. Acad. Sci., 103, 10271–10276. doi:10.1073/pnas.0601058103
Danielsson B. E., Peters H. C., Bathula K., Spear L. M., Noll N. A., Dahl K. N., et al. (2022a). Progerin-expressing endothelial cells are unable to adapt to shear stress. Biophys. J. 121, 620–628. doi:10.1016/j.bpj.2022.01.004
Danielsson B. E., Tieu K. V., Spagnol S. T., Vu K. K., Cabe J. I., Raisch T. B., et al. (2022b). Chromatin condensation regulates endothelial cell adaptation to shear stress. Mol. Biol. Cell 33, ar101. doi:10.1091/mbc.E22-02-0064
Davies P. F. (1995). Flow-mediated endothelial mechanotransduction. Physiol. Rev. 75, 519–560. doi:10.1152/physrev.1995.75.3.519
Davis M. J., Earley S., Li Y.-S., Chien S. (2023). Vascular mechanotransduction. Physiol. Rev. 103, 1247–1421. doi:10.1152/physrev.00053.2021
Davis M. J., Hill M. A. (1999). Signaling mechanisms underlying the vascular myogenic response. Physiol. Rev. 79, 387–423. doi:10.1152/physrev.1999.79.2.387
DE Godoy M. A., Rattan S. (2011). Role of rho kinase in the functional and dysfunctional tonic smooth muscles. Trends Pharmacol. Sci. 32, 384–393. doi:10.1016/j.tips.2011.03.005
Deguchi S., Maeda K., Ohashi T., Sato M. (2005). Flow-induced hardening of endothelial nucleus as an intracellular stress-bearing organelle. J. Biomechanics 38, 1751–1759. doi:10.1016/j.jbiomech.2005.06.003
Dekker R. J., VAN Soest S., Fontijn R. D., Salamanca S., DE Groot P. G., Vanbavel E., et al. (2002). Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Krüppel-like factor (KLF2). Blood 100, 1689–1698. doi:10.1182/blood-2002-01-0046
Del Campo L., Sánchez-López A., González-Gómez C., Andrés-Manzano M. J., Dorado B., Andrés V. (2020). Vascular smooth muscle cell-specific progerin expression provokes contractile impairment in a mouse model of hutchinson-gilford progeria syndrome that is ameliorated by nitrite treatment. Cells 9, 656. doi:10.3390/cells9030656
DE Leeuw R., Gruenbaum Y., Medalia O. (2018). Nuclear lamins: thin filaments with major functions. Trends Cell Biol. 28, 34–45. doi:10.1016/j.tcb.2017.08.004
Denis K. B., Cabe J. I., Danielsson B. E., Tieu K. V., Mayer C. R., Conway D. E. (2021). The LINC complex is required for endothelial cell adhesion and adaptation to shear stress and cyclic stretch. Mol. Biol. Cell 32, 1654–1663. doi:10.1091/mbc.E20-11-0698
DE Sandre-Giovannoli A., Bernard R., Cau P., Navarro C., Amiel J., Boccaccio I., et al. (2003). Lamin a truncation in Hutchinson-Gilford progeria. Science 300, 2055. doi:10.1126/science.1084125
Dou Z., Ghosh K., Vizioli M. G., Zhu J., Sen P., Wangensteen K. J., et al. (2017). Cytoplasmic chromatin triggers inflammation in senescence and cancer. Nature 550, 402–406. doi:10.1038/nature24050
Dupont S., Morsut L., Aragona M., Enzo E., Giulitti S., Cordenonsi M., et al. (2011). Role of YAP/TAZ in mechanotransduction. Nature 474, 179–183. doi:10.1038/nature10137
Dupont S., Wickström S. A. (2022). Mechanical regulation of chromatin and transcription. Nat. Rev. Genet. 23, 624–643. doi:10.1038/s41576-022-00493-6
Earle A. J., Kirby T. J., Fedorchak G. R., Isermann P., Patel J., Iruvanti S., et al. (2020). Mutant lamins cause nuclear envelope rupture and DNA damage in skeletal muscle cells. Nat. Mater. 19, 464–473. doi:10.1038/s41563-019-0563-5
Elosegui-Artola A., Andreu I., Beedle A. E. M., Lezamiz A., Uroz M., Kosmalska A. J., et al. (2017). Force triggers YAP nuclear entry by regulating transport across nuclear pores. Cell 171, 1397–1410. doi:10.1016/j.cell.2017.10.008
Eriksson M., Brown W. T., Gordon L. B., Glynn M. W., Singer J., Scott L., et al. (2003). Recurrent de novo point mutations in lamin A cause Hutchinson–Gilford progeria syndrome. Nature 423, 293–298. doi:10.1038/nature01629
Espada J., Varela I., Flores I., Ugalde A. P., Cadiñanos J., Pendás A. M., et al. (2008). Nuclear envelope defects cause stem cell dysfunction in premature-aging mice. J. Cell Biol. 181, 27–35. doi:10.1083/jcb.200801096
Fang F., Yang Y., Yuan Z., Gao Y., Zhou J., Chen Q., et al. (2011). Myocardin-related transcription factor A mediates OxLDL-induced endothelial injury. Circ. Res. 108, 797–807. doi:10.1161/CIRCRESAHA.111.240655
Garland C. J., Dora K. A. (2017). EDH: endothelium-dependent hyperpolarization and microvascular signalling. Acta Physiol. 219, 152–161. doi:10.1111/apha.12649
Gete Y. G., Koblan L. W., Mao X., Trappio M., Mahadik B., Fisher J. P., et al. (2021). Mechanisms of angiogenic incompetence in Hutchinson-Gilford progeria via downregulation of endothelial NOS. Aging Cell 20, e13388. doi:10.1111/acel.13388
Glück S., Guey B., Gulen M. F., Wolter K., Kang T.-W., Schmacke N. A., et al. (2017). Innate immune sensing of cytosolic chromatin fragments through cGAS promotes senescence. Nat. cell Biol. 19, 1061–1070. doi:10.1038/ncb3586
Goldman R. D., Shumaker D. K., Erdos M. R., Eriksson M., Goldman A. E., Gordon L. B., et al. (2004). Accumulation of mutant lamin A causes progressive changes in nuclear architecture in Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. U. S. A. 101, 8963–8968. doi:10.1073/pnas.0402943101
Gonzalo S., Kreienkamp R. (2015). DNA repair defects and genome instability in Hutchinson–Gilford Progeria Syndrome. Curr. Opin. Cell Biol. 34, 75–83. doi:10.1016/j.ceb.2015.05.007
Gordon L. B., Massaro J., D'Agostino R. B. S. R., Campbell S. E., Brazier J., Brown W. T., et al. (2014). Impact of farnesylation inhibitors on survival in Hutchinson-Gilford progeria syndrome. Circulation 130, 27–34. doi:10.1161/CIRCULATIONAHA.113.008285
Gordon L. B., Mccarten K. M., Giobbie-Hurder A., Machan J. T., Campbell S. E., Berns S. D., et al. (2007). Disease progression in Hutchinson-Gilford progeria syndrome: impact on growth and development. Pediatrics 120, 824–833. doi:10.1542/peds.2007-1357
Grolleman J., VAN Engeland N. C. A., Raza M., Azimi S., Conte V., Sahlgren C. M., et al. (2023). Environmental stiffness restores mechanical homeostasis in vimentin-depleted cells. Sci. Rep. 13, 18374. doi:10.1038/s41598-023-44835-8
Gruenbaum Y., Foisner R. (2015). Lamins: nuclear intermediate filament proteins with fundamental functions in nuclear mechanics and genome regulation. Annu. Rev. Biochem. 84, 131–164. doi:10.1146/annurev-biochem-060614-034115
Guilluy C., Osborne L. D., VAN Landeghem L., Sharek L., Superfine R., Garcia-Mata R., et al. (2014). Isolated nuclei adapt to force and reveal a mechanotransduction pathway in the nucleus. Nat. Cell Biol. 16, 376–381. doi:10.1038/ncb2927
Guo D., Chien S., Shyy J. Y. (2007). Regulation of endothelial cell cycle by laminar versus oscillatory flow: distinct modes of interactions of AMP-activated protein kinase and Akt pathways. Circ. Res. 100, 564–571. doi:10.1161/01.RES.0000259561.23876.c5
Hahn C., Schwartz M. A. (2009). Mechanotransduction in vascular physiology and atherogenesis. Nat. Rev. Mol. Cell Biol. 10, 53–62. doi:10.1038/nrm2596
Hale C. M., Shrestha A. L., Khatau S. B., Stewart-Hutchinson P. J., Hernandez L., Stewart C. L., et al. (2008). Dysfunctional connections between the nucleus and the actin and microtubule networks in laminopathic models. Biophys. J. 95, 5462–5475. doi:10.1529/biophysj.108.139428
Hamczyk M. R., Villa-Bellosta R., Quesada V., Gonzalo P., Vidak S., Nevado R. M., et al. (2019). Progerin accelerates atherosclerosis by inducing endoplasmic reticulum stress in vascular smooth muscle cells. EMBO Mol. Med. 11, e9736. doi:10.15252/emmm.201809736
Han Y., Wang L., Yao Q. P., Zhang P., Liu B., Wang G. L., et al. (2015). Nuclear envelope proteins Nesprin2 and LaminA regulate proliferation and apoptosis of vascular endothelial cells in response to shear stress. Biochim. Biophys. Acta 1853, 1165–1173. doi:10.1016/j.bbamcr.2015.02.013
Hasper J., Welle K., Swovick K., Hryhorenko J., Ghaemmaghami S., Buchwalter A. (2023). Long lifetime and tissue-specific accumulation of lamin A/C in Hutchinson–Gilford progeria syndrome. J. Cell Biol. 223, e202307049. doi:10.1083/jcb.202307049
Hennekam R. C. M. (2006). Hutchinson–Gilford progeria syndrome: review of the phenotype. Am. J. Med. Genet. Part A 140A, 2603–2624. doi:10.1002/ajmg.a.31346
Huang K., Yan Z. Q., Zhao D., Chen S. G., Gao L. Z., Zhang P., et al. (2015). SIRT1 and FOXO mediate contractile differentiation of vascular smooth muscle cells under cyclic stretch. Cell Physiol. Biochem. 37, 1817–1829. doi:10.1159/000438544
Hu B., Song J. T., Qu H. Y., Bi C. L., Huang X. Z., Liu X. X., et al. (2014). Mechanical stretch suppresses microRNA-145 expression by activating extracellular signal-regulated kinase 1/2 and upregulating angiotensin-converting enzyme to alter vascular smooth muscle cell phenotype. PLoS One 9, e96338. doi:10.1371/journal.pone.0096338
Hu J., Li Y., Hao Y., Zheng T., Gupta S. K., Parada G. A., et al. (2019). High stretchability, strength, and toughness of living cells enabled by hyperelastic vimentin intermediate filaments. Proc. Natl. Acad. Sci. U. S. A. 116, 17175–17180. doi:10.1073/pnas.1903890116
Humphrey J. D., Dufresne E. R., Schwartz M. A. (2014). Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 15, 802–812. doi:10.1038/nrm3896
Ihalainen T. O., Aires L., Herzog F. A., Schwartlander R., Moeller J., Vogel V. (2015). Differential basal-to-apical accessibility of lamin A/C epitopes in the nuclear lamina regulated by changes in cytoskeletal tension. Nat. Mater. 14, 1252–1261. doi:10.1038/nmat4389
Jensen L. F., Bentzon J. F., Albarrán-Juárez J. (2021). The phenotypic responses of vascular smooth muscle cells exposed to mechanical cues. Cells 10, 2209. doi:10.3390/cells10092209
Jiang Y. Z., Jiménez J. M., Ou K., Mccormick M. E., Zhang L. D., Davies P. F. (2014). Hemodynamic disturbed flow induces differential DNA methylation of endothelial Kruppel-Like Factor 4 promoter in vitro and in vivo. Circ. Res. 115, 32–43. doi:10.1161/CIRCRESAHA.115.303883
Kalukula Y., Stephens A. D., Lammerding J., Gabriele S. (2022). Mechanics and functional consequences of nuclear deformations. Nat. Rev. Mol. Cell Biol. 23, 583–602. doi:10.1038/s41580-022-00480-z
Kim P. H., Chen N. Y., Heizer P. J., Tu Y., Weston T. A., Fong J. L., et al. (2021). Nuclear membrane ruptures underlie the vascular pathology in a mouse model of Hutchinson-Gilford progeria syndrome. JCI Insight 6, e151515. doi:10.1172/jci.insight.151515
Kim P. H., Kim J. R., Tu Y., Jung H., Jeong J. Y. B., Tran A. P., et al. (2024). Progerin forms an abnormal meshwork and has a dominant-negative effect on the nuclear lamina. Proc. Natl. Acad. Sci. 121, e2406946121. doi:10.1073/pnas.2406946121
Kim P. H., Luu J., Heizer P., Tu Y., Weston T. A., Chen N., et al. (2018). Disrupting the LINC complex in smooth muscle cells reduces aortic disease in a mouse model of Hutchinson-Gilford progeria syndrome. Sci. Transl. Med. 10, eaat7163. doi:10.1126/scitranslmed.aat7163
King S. J., Nowak K., Suryavanshi N., Holt I., Shanahan C. M., Ridley A. J. (2014). Nesprin-1 and nesprin-2 regulate endothelial cell shape and migration. Cytoskelet. Hob. 71, 423–434. doi:10.1002/cm.21182
Kirby T. J., Lammerding J. (2018). Emerging views of the nucleus as a cellular mechanosensor. Nat. Cell Biol. 20, 373–381. doi:10.1038/s41556-018-0038-y
Koushki N., Ghagre A., Srivastava L. K., Molter C., Ehrlicher A. J. (2023). Nuclear compression regulates YAP spatiotemporal fluctuations in living cells. Proc. Natl. Acad. Sci. U. S. A. 120, e2301285120. doi:10.1073/pnas.2301285120
Kubben N., Zhang W., Wang L., Voss T. C., Yang J., Qu J., et al. (2016). Repression of the antioxidant NRF2 pathway in premature aging. Cell 165, 1361–1374. doi:10.1016/j.cell.2016.05.017
Lammerding J., Schulze P. C., Takahashi T., Kozlov S., Sullivan T., Kamm R. D., et al. (2004). Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. J. Clin. Invest 113, 370–378. doi:10.1172/JCI19670
Lee D. Y., Lee C. I., Lin T. E., Lim S. H., Zhou J., Tseng Y. C., et al. (2012). Role of histone deacetylases in transcription factor regulation and cell cycle modulation in endothelial cells in response to disturbed flow. Proc. Natl. Acad. Sci. U. S. A. 109, 1967–1972. doi:10.1073/pnas.1121214109
Liu S. Y., Ikegami K. (2020). Nuclear lamin phosphorylation: an emerging role in gene regulation and pathogenesis of laminopathies. Nucleus 11, 299–314. doi:10.1080/19491034.2020.1832734
Liu B., Wang J., Chan K. M., Tjia W. M., Deng W., Guan X., et al. (2005). Genomic instability in laminopathy-based premature aging. Nat. Med. 11, 780–785. doi:10.1038/nm1266
Liu D., Lv H., Liu Q., Sun Y., Hou S., Zhang L., et al. (2019). Atheroprotective effects of methotrexate via the inhibition of YAP/TAZ under disturbed flow. J. Transl. Med. 17, 378. doi:10.1186/s12967-019-02135-8
Mao X., Said R., Louis H., Max J. P., Bourhim M., Challande P., et al. (2012). Cyclic stretch-induced thrombin generation by rat vascular smooth muscle cells is mediated by the integrin αvβ3 pathway. Cardiovasc Res. 96, 513–523. doi:10.1093/cvr/cvs274
Maraldi N. M., Capanni C., Cenni V., Fini M., Lattanzi G. (2011). Laminopathies and lamin-associated signaling pathways. J. Cell Biochem. 112, 979–992. doi:10.1002/jcb.22992
Maurer M., Lammerding J. (2019). The driving force: nuclear mechanotransduction in cellular function, fate, and disease. Annu. Rev. Biomed. Eng. 21, 443–468. doi:10.1146/annurev-bioeng-060418-052139
Mccord R. P., Nazario-Toole A., Zhang H., Chines P. S., Zhan Y., Erdos M. R., et al. (2013). Correlated alterations in genome organization, histone methylation, and DNA-lamin A/C interactions in Hutchinson-Gilford progeria syndrome. Genome Res. 23, 260–269. doi:10.1101/gr.138032.112
Merideth M. A., Gordon L. B., Clauss S., Sachdev V., Smith A. C., Perry M. B., et al. (2008). Phenotype and course of Hutchinson-Gilford progeria syndrome. N. Engl. J. Med. 358, 592–604. doi:10.1056/NEJMoa0706898
Murtada S.-I., Kawamura Y., Caulk A. W., Ahmadzadeh H., Mikush N., Zimmerman K., et al. (2020). Paradoxical aortic stiffening and subsequent cardiac dysfunction in Hutchinson–Gilford progeria syndrome. J. R. Soc. Interface 17, 20200066. doi:10.1098/rsif.2020.0066
Murtada S. I., Kawamura Y., Cavinato C., Wang M., Ramachandra A. B., Spronck B., et al. (2023). Biomechanical and transcriptional evidence that smooth muscle cell death drives an osteochondrogenic phenotype and severe proximal vascular disease in progeria. Biomech. Model Mechanobiol. 22, 1333–1347. doi:10.1007/s10237-023-01722-5
Musich P. R., Zou Y. (2009). Genomic instability and DNA damage responses in progeria arising from defective maturation of prelamin A. Aging (Albany NY) 1, 28–37. doi:10.18632/aging.100012
Mu X., Tseng C., Hambright W. S., Matre P., Lin C. Y., Chanda P., et al. (2020). Cytoskeleton stiffness regulates cellular senescence and innate immune response in Hutchinson-Gilford Progeria Syndrome. Aging Cell 19, e13152. doi:10.1111/acel.13152
Nagel T., Resnick N., Dewey C. F., Gimbrone M. A. (1999). Vascular endothelial cells respond to spatial gradients in fluid shear stress by enhanced activation of transcription factors. Arterioscler. Thromb. Vasc. Biol. 19, 1825–1834. doi:10.1161/01.atv.19.8.1825
Nava M. M., Miroshnikova Y. A., Biggs L. C., Whitefield D. B., Metge F., Boucas J., et al. (2020). Heterochromatin-driven nuclear softening protects the genome against mechanical stress-induced damage. Cell 181, 800–817. doi:10.1016/j.cell.2020.03.052
Neelam S., Chancellor T. J., Li Y., Nickerson J. A., Roux K. J., Dickinson R. B., et al. (2015). Direct force probe reveals the mechanics of nuclear homeostasis in the mammalian cell. Proc. Natl. Acad. Sci. U. S. A. 112, 5720–5725. doi:10.1073/pnas.1502111112
Ngai D., Lino M., Rothenberg K. E., Simmons C. A., Fernandez-Gonzalez R., Bendeck M. P. (2020). DDR1 (discoidin domain receptor-1)-RhoA (Ras homolog family member A) Axis senses matrix stiffness to promote vascular calcification. Arteriosclerosis, Thrombosis, Vasc. Biol. 40, 1763–1776. doi:10.1161/ATVBAHA.120.314697
Ngai D., Mohabeer A. L., Mao A., Lino M., Bendeck M. P. (2022). Stiffness-responsive feedback autoregulation of DDR1 expression is mediated by a DDR1-YAP/TAZ axis. Matrix Biol. 110, 129–140. doi:10.1016/j.matbio.2022.05.004
Olive M., Harten I., Mitchell R., Beers J. K., Djabali K., Cao K., et al. (2010). Cardiovascular pathology in hutchinson-gilford progeria: correlation with the vascular pathology of aging. Arteriosclerosis, Thrombosis, Vasc. Biol. 30, 2301–2309. doi:10.1161/ATVBAHA.110.209460
Osborn E. A., Rabodzey A., Dewey C. F., Hartwig J. H. (2006). Endothelial actin cytoskeleton remodeling during mechanostimulation with fluid shear stress. Am. J. Physiol. Cell Physiol. 290, C444–C452. doi:10.1152/ajpcell.00218.2005
Osmanagic-Myers S., Kiss A., Manakanatas C., Hamza O., Sedlmayer F., Szabo P. L., et al. (2019). Endothelial progerin expression causes cardiovascular pathology through an impaired mechanoresponse. J. Clin. Invest 129, 531–545. doi:10.1172/JCI121297
Osorio F. G., Bárcena C., Soria-Valles C., Ramsay A. J., DE Carlos F., Cobo J., et al. (2012). Nuclear lamina defects cause ATM-dependent NF-κB activation and link accelerated aging to a systemic inflammatory response. Genes Dev. 26, 2311–2324. doi:10.1101/gad.197954.112
Owens G. K., Kumar M. S., Wamhoff B. R. (2004). Molecular regulation of vascular smooth muscle cell differentiation in development and disease. Physiol. Rev. 84, 767–801. doi:10.1152/physrev.00041.2003
Patteson A. E., Vahabikashi A., Pogoda K., Adam S. A., Mandal K., Kittisopikul M., et al. (2019). Vimentin protects cells against nuclear rupture and DNA damage during migration. J. Cell Biol. 218, 4079–4092. doi:10.1083/jcb.201902046
Pegoraro G., Kubben N., Wickert U., Göhler H., Hoffmann K., Misteli T. (2009). Ageing-related chromatin defects through loss of the NURD complex. Nat. cell Biol. 11, 1261–1267. doi:10.1038/ncb1971
Pitrez P. R., Estronca L., Monteiro L. M., Colell G., Vazão H., Santinha D., et al. (2020). Vulnerability of progeroid smooth muscle cells to biomechanical forces is mediated by MMP13. Nat. Commun. 11, 4110. doi:10.1038/s41467-020-17901-2
Reusch P., Wagdy H., Reusch R., Wilson E., Ives H. E. (1996). Mechanical strain increases smooth muscle and decreases nonmuscle myosin expression in rat vascular smooth muscle cells. Circ. Res. 79, 1046–1053. doi:10.1161/01.res.79.5.1046
Ribas J., Zhang Y. S., Pitrez P. R., Leijten J., Miscuglio M., Rouwkema J., et al. (2017). Biomechanical strain exacerbates inflammation on a progeria-on-a-chip model. Small 13. doi:10.1002/smll.201603737
Rosengardten Y., Mckenna T., Grochová D., Eriksson M. (2011). Stem cell depletion in Hutchinson-Gilford progeria syndrome. Aging Cell 10, 1011–1020. doi:10.1111/j.1474-9726.2011.00743.x
Sagelius H., Rosengardten Y., Hanif M., Erdos M. R., Rozell B., Collins F. S., et al. (2008). Targeted transgenic expression of the mutation causing Hutchinson-Gilford progeria syndrome leads to proliferative and degenerative epidermal disease. J. Cell Sci. 121, 969–978. doi:10.1242/jcs.022913
Scaffidi P., Misteli T. (2008). Lamin A-dependent misregulation of adult stem cells associated with accelerated ageing. Nat. Cell Biol. 10, 452–459. doi:10.1038/ncb1708
Schmidt E., Nilsson O., Koskela A., Tuukkanen J., Ohlsson C., Rozell B., et al. (2012). Expression of the Hutchinson-Gilford progeria mutation during osteoblast development results in loss of osteocytes, irregular mineralization, and poor biomechanical properties. J. Biol. Chem. 287, 33512–33522. doi:10.1074/jbc.M112.366450
Sears R. M., Roux K. J. (2022). Mechanisms of A-type lamin targeting to nuclear ruptures are disrupted in LMNA- and BANF1-associated progerias. Cells 11, 865. doi:10.3390/cells11050865
Seelbinder B., Ghosh S., Schneider S. E., Scott A. K., Berman A. G., Goergen C. J., et al. (2021). Nuclear deformation guides chromatin reorganization in cardiac development and disease. Nat. Biomed. Eng. 5, 1500–1516. doi:10.1038/s41551-021-00823-9
Senbanerjee S., Lin Z., Atkins G. B., Greif D. M., Rao R. M., Kumar A., et al. (2004). KLF2 Is a novel transcriptional regulator of endothelial proinflammatory activation. J. Exp. Med. 199, 1305–1315. doi:10.1084/jem.20031132
Shumaker D. K., Dechat T., Kohlmaier A., Adam S. A., Bozovsky M. R., Erdos M. R., et al. 2006. Mutant nuclear lamin A leads to progressive alterations of epigenetic control in premature aging, Proc. Natl. Acad. Sci., 103, 8703–8708. doi:10.1073/pnas.0602569103
Solovei I., Wang A. S., Thanisch K., Schmidt C. S., Krebs S., Zwerger M., et al. (2013). LBR and lamin A/C sequentially tether peripheral heterochromatin and inversely regulate differentiation. Cell 152, 584–598. doi:10.1016/j.cell.2013.01.009
Son S. M., Park S. J., Breusegem S. Y., Larrieu D., Rubinsztein D. C. (2024). p300 nucleocytoplasmic shuttling underlies mTORC1 hyperactivation in Hutchinson–Gilford progeria syndrome. Nat. Cell Biol. 26, 235–249. doi:10.1038/s41556-023-01338-y
Song M., San H., Anderson S. A., Cannon R. O., Orlic D. (2014). Shear stress-induced mechanotransduction protein deregulation and vasculopathy in a mouse model of progeria. Stem Cell Res. Ther. 5, 41. doi:10.1186/scrt429
Strambio-DE-Castillia C., Niepel M., Rout M. P. (2010). The nuclear pore complex: bridging nuclear transport and gene regulation. Nat. Rev. Mol. Cell Biol. 11, 490–501. doi:10.1038/nrm2928
Sun S., Qin W., Tang X., Meng Y., Hu W., Zhang S., et al. (2020). Vascular endothelium-targeted Sirt7 gene therapy rejuvenates blood vessels and extends life span in a Hutchinson-Gilford progeria model. Sci. Adv. 6, eaay5556. doi:10.1126/sciadv.aay5556
Swiatlowska P., Sit B., Feng Z., Marhuenda E., Xanthis I., Zingaro S., et al. (2022). Pressure and stiffness sensing together regulate vascular smooth muscle cell phenotype switching. Sci. Adv. 8, eabm3471. doi:10.1126/sciadv.abm3471
Swift J., Ivanovska I. L., Buxboim A., Harada T., Dingal P. D. P., Pinter J., et al. (2013). Nuclear lamin-A scales with tissue stiffness and enhances matrix-directed differentiation. Science 341, 1240104. doi:10.1126/science.1240104
Tajik A., Zhang Y., Wei F., Sun J., Jia Q., Zhou W., et al. (2016). Transcription upregulation via force-induced direct stretching of chromatin. Nat. Mater. 15, 1287–1296. doi:10.1038/nmat4729
Tkachenko E., Gutierrez E., Saikin S. K., Fogelstrand P., Kim C., Groisman A., et al. (2013). The nucleus of endothelial cell as a sensor of blood flow direction. Biol. Open 2, 1007–1012. doi:10.1242/bio.20134622
Tsai M.-C., Chen L., Zhou J., Tang Z., Hsu T.-F., Wang Y., et al. (2009). Shear stress induces synthetic-to-contractile phenotypic modulation in smooth muscle cells via peroxisome proliferator-activated receptor alpha/delta activations by prostacyclin released by sheared endothelial cells. Circulation Res. 105, 471–480. doi:10.1161/CIRCRESAHA.109.193656
Tzima E., Irani-Tehrani M., Kiosses W. B., Dejana E., Schultz D. A., Engelhardt B., et al. (2005). A mechanosensory complex that mediates the endothelial cell response to fluid shear stress. Nature 437, 426–431. doi:10.1038/nature03952
Ullrich N. J., Gordon L. B. (2015). Hutchinson-Gilford progeria syndrome. Handb. Clin. Neurol. 132, 249–264. doi:10.1016/B978-0-444-62702-5.00018-4
Varga R., Eriksson M., Erdos M. R., Olive M., Harten I., Kolodgie F., et al. (2006). Progressive vascular smooth muscle cell defects in a mouse model of Hutchinson-Gilford progeria syndrome. Proc. Natl. Acad. Sci. U. S. A. 103, 3250–3255. doi:10.1073/pnas.0600012103
Verstraeten V. L. R. M., Ji J. Y., Cummings K. S., Lee R. T., Lammerding J. (2008). Increased mechanosensitivity and nuclear stiffness in Hutchinson–Gilford progeria cells: effects of farnesyltransferase inhibitors. Aging Cell 7, 383–393. doi:10.1111/j.1474-9726.2008.00382.x
Vion A.-C., Kheloufi M., Hammoutene A., Poisson J., Lasselin J., Devue C., et al. 2017. Autophagy is required for endothelial cell alignment and atheroprotection under physiological blood flow, Proc. Natl. Acad. Sci., 114, E8675–E8684. doi:10.1073/pnas.1702223114
Wan X. J., Zhao H. C., Zhang P., Huo B., Shen B. R., Yan Z. Q., et al. (2015). Involvement of BK channel in differentiation of vascular smooth muscle cells induced by mechanical stretch. Int. J. Biochem. Cell Biol. 59, 21–29. doi:10.1016/j.biocel.2014.11.011
Wang K.-C., Yeh Y.-T., Nguyen P., Limqueco E., Lopez J., Thorossian S., et al. (2016a). Flow-dependent YAP/TAZ activities regulate endothelial phenotypes and atherosclerosis. Proc. Natl. Acad. Sci. 113, 11525–11530. doi:10.1073/pnas.1613121113
Wang L., Luo J. Y., Li B., Tian X. Y., Chen L. J., Huang Y., et al. (2016b). Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature 540, 579–582. doi:10.1038/nature20602
Wang W., Ha C. H., Jhun B. S., Wong C., Jain M. K., Jin Z. G. (2010). Fluid shear stress stimulates phosphorylation-dependent nuclear export of HDAC5 and mediates expression of KLF2 and eNOS. Blood 115, 2971–2979. doi:10.1182/blood-2009-05-224824
Wang Y., Cao W., Cui J., Yu Y., Zhao Y., Shi J., et al. (2018). Arterial wall stress induces phenotypic switching of arterial smooth muscle cells in vascular remodeling by activating the YAP/TAZ signaling pathway. Cell Physiol. Biochem. 51, 842–853. doi:10.1159/000495376
Wheaton K., Campuzano D., Ma W., Sheinis M., Ho B., Brown G. W., et al. (2017). Progerin-induced replication stress facilitates premature senescence in hutchinson-gilford progeria syndrome. Mol. Cell Biol. 37, e716-006599. doi:10.1128/MCB.00659-16
Xia Y., Ivanovska I. L., Zhu K., Smith L., Irianto J., Pfeifer C. R., et al. (2018). Nuclear rupture at sites of high curvature compromises retention of DNA repair factors. J. Cell Biol. 217, 3796–3808. doi:10.1083/jcb.201711161
Xu S., Koroleva M., Yin M., Jin Z. G. (2016). Atheroprotective laminar flow inhibits Hippo pathway effector YAP in endothelial cells. Transl. Res. 176, 18–28. doi:10.1016/j.trsl.2016.05.003
Yan Z. Q., Yao Q. P., Zhang M. L., Qi Y. X., Guo Z. Y., Shen B. R., et al. (2009). Histone deacetylases modulate vascular smooth muscle cell migration induced by cyclic mechanical strain. J. Biomech. 42, 945–948. doi:10.1016/j.jbiomech.2009.01.012
Yang S. H., Meta M., Qiao X., Frost D., Bauch J., Coffinier C., et al. (2006). A farnesyltransferase inhibitor improves disease phenotypes in mice with a Hutchinson-Gilford progeria syndrome mutation. J. Clin. Invest 116, 2115–2121. doi:10.1172/JCI28968
Yang F., Zhang Y., Zhu J., Wang J., Jiang Z., Zhao C., et al. (2020). Laminar flow protects vascular endothelial tight junctions and barrier function via maintaining the expression of long non-coding RNA MALAT1. Front. Bioeng. Biotechnol. 8, 647. doi:10.3389/fbioe.2020.00647
Yao Q. P., Zhang P., Qi Y. X., Chen S. G., Shen B. R., Han Y., et al. (2014). The role of SIRT6 in the differentiation of vascular smooth muscle cells in response to cyclic strain. Int. J. Biochem. Cell Biol. 49, 98–104. doi:10.1016/j.biocel.2014.01.016
Yue X., Cui J., Sun Z., Liu L., Li Y., Shao L., et al. (2023). Nuclear softening mediated by Sun2 suppression delays mechanical stress-induced cellular senescence. Cell Death Discov. 9, 167. doi:10.1038/s41420-023-01467-1
Zhang H., Xiong Z. M., Cao K. (2014). Mechanisms controlling the smooth muscle cell death in progeria via down-regulation of poly(ADP-ribose) polymerase 1. Proc. Natl. Acad. Sci. U. S. A. 111, E2261–E2270. doi:10.1073/pnas.1320843111
Keywords: progeria, progerin, lamin a, mechanotransduction, endothelial cell, vascular smooth muscle cell, atherosclerosis
Citation: Shores KL and Truskey GA (2024) Mechanotransduction of the vasculature in Hutchinson-Gilford Progeria Syndrome. Front. Physiol. 15:1464678. doi: 10.3389/fphys.2024.1464678
Received: 14 July 2024; Accepted: 13 August 2024;
Published: 22 August 2024.
Edited by:
Julia J Mack, UCLA Health System, United StatesReviewed by:
Vicente Andres, Spanish National Centre for Cardiovascular Research, SpainG.W. Gant Luxton, University of California, United States
Copyright © 2024 Shores and Truskey. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: George A. Truskey, Z3RydXNrZXlAZHVrZS5lZHU=