
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
ORIGINAL RESEARCH article
Front. Physiol. , 01 July 2024
Sec. Integrative Physiology
Volume 15 - 2024 | https://doi.org/10.3389/fphys.2024.1397049
This article is part of the Research Topic Emerging Molecular Mechanisms in Cardiovascular Physiology and Pathology View all 7 articles
 Jade P. Marrow1,2†
Jade P. Marrow1,2† Razan Alshamali1,2†
Razan Alshamali1,2† Brittany A. Edgett1,2,3
Brittany A. Edgett1,2,3 Melissa A. Allwood1,2
Melissa A. Allwood1,2 Kyla L. S. Cochrane1,2
Kyla L. S. Cochrane1,2 Sara Al-Sabbag1
Sara Al-Sabbag1 Anmar Ayoub4
Anmar Ayoub4 Kjetil Ask4Gregory M. T. Hare2,5,6,7
Kjetil Ask4Gregory M. T. Hare2,5,6,7 Keith R. Brunt2,8
Keith R. Brunt2,8 Jeremy A. Simpson1,2*
Jeremy A. Simpson1,2*Erythropoietin (EPO) exerts non-canonical roles beyond erythropoiesis that are developmentally, structurally, and physiologically relevant for the heart as a paracrine factor. The role for paracrine EPO signalling and cellular crosstalk in the adult is uncertain. Here, we provided novel evidence showing cardiomyocyte restricted loss of function in Epo in adult mice induced hyper-compensatory increases in Epo expression by adjacent cardiac endothelial cells via HIF-2α independent mechanisms. These hearts showed concentric cellular hypertrophy, elevated contractility and relaxation, and greater resistance to ischemia-reperfusion injury. Voluntary exercise capacity compared to control hearts was improved independent of any changes to whole-body metabolism or blood O2 content or delivery (i.e., hematocrit). Our findings suggest cardiac EPO had a localized effect within the normoxic heart, which was regulated by cell-specific EPO-reciprocity between cardiomyocytes and endothelium. Within the heart, hyper-compensated endothelial Epo expression was accompanied by elevated Vegfr1 and Vegfb RNA, that upon pharmacological pan-inhibition of VEGF-VEGFR signaling, resulted in a paradoxical upregulation in whole-heart Epo. Thus, we provide the first evidence that a novel EPO-EPOR/VEGF-VEGFR axis exists to carefully mediate cardiac homeostasis via cardiomyocyte-endothelial EPO crosstalk.
Erythropoietin (EPO) is classically regarded as a renal-derived erythropoietic cytokine. In response to hypoxia, EPO stimulates the blast forming erythroid progenitor proliferation and survival to maintain hematocrit and/or restore oxygen delivery. Interestingly, non-renal sources of Epo are known to include the adult liver (Haidar et al., 1996), spleen (De Franciscis et al., 1979), ovaries (Masuda et al., 2000), uterus (Yasuda et al., 1998), testes (Magnanti et al., 2001), brain (Bernaudin et al., 1999; Weidemann et al., 2009; Urrutia et al., 2016), and heart (Wu et al., 1999; Kertesz et al., 2004; Chu et al., 2007; El Hasnaoui-Saadani et al., 2013). Within the heart, Epo production is generally ascribed to cardiomyocytes (Mengozzi et al., 2006; Miró-Murillo et al., 2011; El Hasnaoui-Saadani et al., 2013) and the endothelial lining (Mengozzi et al., 2006), yet the physiological significance of cardiac EPO is unclear. Since expression of the Epo receptor (Epor) occurs in multiple cardiac cell types (e.g., cardiomyocytes (Wright et al., 2004), endothelial cells (Beleslin-Cokic et al., 2004; Yang et al., 2014)), EPO likely elicits autocrine and/or paracrine cardiac-specific functional effects. While endothelial cells comprise ∼50% of the total number of cells in the murine heart, cardiomyocytes make up ∼30% (Pinto et al., 2016) and nearly 70% by total mass (Zak, 1973; Nag, 1980; Giordano et al., 2001). Thus, cardiomyocyte- and endothelial-derived factors as autocrine/paracrine effectors are vital for homeostasis. To date, the physiological role(s) of endogenously produced adult cardiomyocyte-derived EPO has not been investigated.
Recombinant human EPO (rhEPO) is neuroprotective (Bernaudin et al., 1999; Ponce et al., 2013; Bonnas et al., 2017; Wakhloo et al., 2020), cardioprotective (Cai et al., 2003; Calvillo et al., 2003; Parsa et al., 2003), and reportedly augments cardiac inotropy (Kaygisiz et al., 2006; Hefer et al., 2012) in prior pre-clinical studies. However, exogenous rhEPO is structurally distinct and differentially glycosylated compared to endogenous EPO. This can impact serum stability, receptor binding affinity, and bioactivity (Dube et al., 1988; Rahbek-Nielsen et al., 1997; Cheetham et al., 1998). Therefore, the beneficial effects of rhEPO cannot be inherently presumed to reflect endogenous, organ-specific functions of EPO. Instead, the pleiotropic roles of endogenous EPO should be assessed in vivo in a cell-specific context. Accordingly, we sought to investigate the impact of cardiomyocyte-restricted EPO signaling on cardiac autocrine/paracrine effects physiologically to contrast systemic or canonical erythropoietic functions determined using exogenous biology or receptor knock-out studies.
We previously generated a constitutive, cardiomyocyte specific Epo knockout mouse driven by the Mlc2v promoter (Allwood et al., 2024). When cardiomyocyte Epo is abolished during embryogenesis, cardiac cellular proliferation is reduced, leading to irreversible changes to overall morphology, function, and response to ischemic injury in the adult heart (Allwood et al., 2024). However, these mice also show a surprising transcriptional increase in endothelial cell derived Epo in compensation to loss of cardiomyocyte gene expression. This prohibits the distinction between developmental adaptations from physiological effects in the adult. To resolve whether cardiomyocyte-derived EPO signalling was physiologically relevant following a normal course of cardiogenic development, we used alpha-myosin heavy chain tamoxifen-induced Cre-LoxP loss of function in adult mice.
Herein we show that following the loss of cardiomyocyte derived Epo in the adult heart, compensatory hyperexpression persists by endothelial cells in a HIF2α-independent manner. This response was associated with concentric cellular hypertrophy, elevated contractile function, and a greater resistance to ischemia-reperfusion injury. Functionally, this phenotype translated to better voluntary exercise capacity, which was unrelated to changes in whole-body metabolism nor any change in hematocrit. Our findings suggest the overexpression of endogenous cardiac EPO acted locally, not systemically. In the absence of cardiomyocyte Epo, we observed concomitant upregulation of Epo, Vegfr1, and Vegfb RNA in the whole heart. When VEGF-VEGFR signaling was inhibited, a further increase in cardiac Epo could be observed. Collectively, our findings provide the first evidence for a paracrine cardioendothelial feedback loop by the EPO-EPOR/VEGF-VEGFR axis for maintaining cardiac homeostasis in the adult mouse.
Adult mice (C57Bl6 background) were bred and aged to 16 weeks of maturity for experiments and housed at 23°C–24°C with 45% humidity and maintained on a 12 h light/dark cycle with food and water provided ad libitum. This study was approved by the Animal Care Committee at the University of Guelph and all experiments were carried out in accordance with the guidelines from the Canadian Council on Animal Care.
Inducible CreLox transgenic mice expressing Epo LoxP (Zeigler et al., 2010) and Cre recombinase under the control of the cardiomyocyte-specific promoter, alpha-myosin heavy chain (αMHC-MerCreMer), were used in this study (Sohal et al., 2001) (Jackson Laboratory Strain # 005657, Supplementary Figure S1). Briefly, the 5′loxP site was inserted into intron 1 of the Epo gene (located 94 base pairs upstream from exon), and the 3’ loxP site was inserted into intron 4 (located 86 base pairs downstream of the exon 4), with the NEO cassette flanked by both loxP sites.
Experimental mice were bred using the following schemes: female EPO LoxP without Cre (EPOfl/fl: αMHC-MerCreMer−/−) crossed with male EPO LoxP mice expressing homozygous Cre Recombinase (EPOfl/fl: αMHC-MerCreMer+/+), which resulted in 100% heterozygosity; female EPOfl/fl: αMHC-MerCreMer+/+ crossed with male EPOfl/fl: αMHC-MerCreMer+/+ (100% homozygous), female EPOfl/fl: αMHC-MerCreMer+/− crossed with male EPOfl/fl: αMHC-MerCreMer+/− (expected Mendelian genetics were 25% homozygous, 50% heterozygous, 25% wildtype) and female EPOfl/fl: αMHC-MerCreMer+/− crossed with male EPOfl/fl: αMHC-MerCreMer+/+ (expected Mendelian genetics were 50% homozygous, 50% heterozygous). Genotyping used tail biopsies and PCR (REDExtract-N-Amp Tissue PCR Kit; Sigma-Aldrich, Oakville, ON, Canada) according to the manufacturer’s instructions. To confirm the presence of the floxed EPO alleles, the following primers sets were used: EPO-KO-F: 5′-AGTGAAGTTTGGCCGAGAAG-3’ (PCR Reaction A), EPO-KO-R: 5′- AGATCGAACTTGGCTCCTCA-3’ (PCR Reaction A), EPO-TAR-R: 5′-GTGGGACGTTCTGGAAGAAA-3’ (PCR Reaction A). PCR Reaction–Stage 1: 1 Cycle, 95°C for 2 min; Stage 2: 40 Cycles, 95°C for 45 s; Annealing: 59°C for 1 min; Extension: 72°C for 1 min; Stage 3 Additional Extension: 1 Cycle, 72°C for 5 min; Hold at 4°C. Gel Electrophoresis–load 18.5 uL sample/well and run gel for 45 min at 90V (lights OFF). Expected Results–Homozygous EPO flox sites = one band at 344 bp; Heterozygote EPO flox sites = band at 228 bp and 344 bp; Wildtype (i.e., no flox) = one band at 228 bp. Representative agarose gel (1.5%) characterizing EPO floxing is presented in Supplementary Figure S1B.
For expression of Cre recombinase under the αMHC-MerCreMer promoter, the following primer sets and conditions were used: MerCreMer Common (Fwd): 5′-TCTATTGCACACAGCAATCCA-3’ (PCR Reaction A and B), MerCreMer Reverse: 5′-CCAGCATTGTGAGAACAAGG-3’ (PCR Reaction A), Wild Type Reverse: 5′-CCAACTCTTGTGAGAGGAGCA-3’ (PCR Reaction B). PCR Protocol for Reaction A and B–Stage 1: 1 Cycle, 95°C for 2 min; Stage 2: 40 Cycles, 95°C for 30 s; Annealing: 60°C for 30 s; Extension: 72°C for 1 min; Stage 3 Additional Extension: 1 Cycle, 72°C for 5 min; Hold at 4°C. Gel Electrophoresis–load 16 uL sample/well and run gel for 35 min at 95 V (lights OFF). Expected Results–Transgenic allele (αMerCreMer) = ∼300 bp; Heterozygote allele = 295 bp and ∼300 bp; Wild type allele = 295 bp. Representative agarose gel (1.5%) characterizing αMerCreMer Cre recombinase is presented in Supplementary Figure S1C.
At 8 weeks of age, EPOfl/fl: αMHC-MerCreMer+/− mice were injected intraperitoneally (i.p.) with corn oil/tamoxifen mixture (25 mg/kg) once daily for 5 days to activate Cre recombinase and induce cardiomyocyte-specific deletion of Epo (denoted EPOΔ/Δ). Experiments were performed at 8 weeks post-injection (16 weeks old). Age-matched control mice (EPOfl/fl: αMHC-MerCreMer−/−, EPOfl/fl: αMHC-MerCreMer+/+ and EPOfl/fl: αMHC-MerCreMer+/− without tamoxifen) demonstrated no differences and were subsequently combined (denoted EPOfl/fl). Cre-null mice provided tamoxifen (EPOfl/fl: αMHC-MerCreMer−/− with tamoxifen) and mice without EPO floxed but with Cre (EPO+/+: αMHC-MerCreMer+/− with and without tamoxifen) showed no major phenotypical differences compared to wildtype mice (Supplementary Table S1).
EPOfl/fl and EPOΔ/Δ mice were subjected to a 3-day voluntary wheel running protocol. Mice were housed individually and allowed to run freely on an in-cage wheel (12 cm in diameter). Rotations were transmitted to a cycling computer (VDO M2.1 WR Cycling Computer) and distance, time, and pace were recorded. Data were presented as the averages from all 3 days of voluntary wheel running following a 24-h acclimation period.
The Comprehensive Laboratory Animal Monitoring System (CLAMS) metabolic caging apparatus (Columbus Instruments Oxymax) is a sealed indirect calorimeter used for the simultaneous measurement of multiple parameters, including oxygen consumption (VO2), carbon dioxide production (VCO2), and calculation of respiratory exchange ratio (RER) across 24-h. EPOfl/fl and EPOΔ/Δ mice were weighed and individually placed into the CLAMS caging for a 24-h acclimatization period followed by a subsequent 24-h data collection period. Mice were maintained on a 12-h light/dark cycle and provided food and water ad libitum. Data was recorded every 15 min for metabolic readings (VO2, VCO2) and total energy expenditure. The RER was calculated as the quotient of VCO2/VO2.
For the determination of hematocrit, blood was collected from the left ventricle and saphenous vein of EPOfl/fl and EPOΔ/Δ mice and centrifuged in heparinized microcapillary tubes (5000rpm at 23°C for 10 min). To calculate hematocrit, the length of red blood cells was divided by the length of total blood volume and expressed as a percentage. To measure hemoglobin (g/L), saphenous vein blood was collected by microcuvettes and measured by a HemoCue® Hb 201 (cat# 111716 Life Supply).
Mice were anesthetized using an isoflurane/oxygen mix (2%/100%). Echocardiography was performed using the Vevo2100 system (VisualSonics Inc., Toronto, ON, Canada) with the 40 MHz MS550D ultrasound transducer. Mice were maintained at 37.5°C throughout data collection, as confirmed with a TH-5 rectal probe thermometer (Physiotemp Instruments LLC, Clifton, NJ, United States). B-Mode images were captured from the parasternal long axis mid-papillary region to measure the left ventricle endocardial length (i.e., LV chamber length) from the aortic annulus to the apex during in diastole, as previously described (Vinhas et al., 2013). M-Mode images were collected from the parasternal long axis mid-papillary region and analyzed using the left ventricle trace function from the cardiac package (VisualSonics Inc., Toronto, ON, Canada) as previously described (Platt et al., 2017). Measurements represent the average data collected over three consecutive cardiac cycles. Data collection and analyses were performed in a blinded manner.
Cardiac function was investigated in vivo by invasive hemodynamics. Mice were anaesthetized using an isoflurane/oxygen mix (2%:100%). Animals were maintained at 37.5°C throughout data collection using a heated water pad. A 1.2F catheter (FTS-1211B-0018; Transonic Scisense Inc.) was inserted into the right carotid artery and advanced into the left ventricle to collect hemodynamics measurements. Hemodynamics were collected for 15 min. All hemodynamic signals were digitized at a sampling rate of 2000 Hz and recorded by computer using iWorx® analytic software (Labscribe2, Dover, NH, USA). Data collection and analyses were performed in a blinded manner. Tissues were collected, weighed, and randomly assigned to either histology, qPCR, or western blotting.
The ex vivo Langendorff preparation provides the direct assessment of systolic and diastolic cardiac function (and its susceptibility to ischemia-reperfusion injury), independent of preload, afterload, and heart rate, without the influence of neurohormonal effects or humoral factors in the blood. Accounting for these variables allows for conclusions to be made about purely intrinsic cardiac function. Mice were heparinised (200 IU/kg of body weight) for ex vivo cardiac assessment. After 20 minutes, mice were anesthetized using isoflurane, followed by a midline thoracic incision made to rapidly excise the heart. The heart was rinsed in ice-cold phosphate buffered saline and the aorta was cannulated onto a 21-gauge needle to allow for retrograde perfusion. Hearts were perfused with carbogenated (95% O2: 5% CO2) Krebs Henseleit buffer (pH of 7.4) at 70–75 mmHg. The Krebs Henseleit buffer contained the following compounds (in mM/L): 118 mM NaCl, 4.7 mM KCl, 1.2 mM MgSO4, 1.2 mM KH2PO4, 0.5 mM C3H3NaO3, 0.05 mM EDTA, 11 mM glucose, and 2 mM CaCl2. The left atrium was removed to allow the insertion of a deflated balloon attached to a pressure catheter into the left ventricle. The balloon was inflated to achieve an end diastolic pressure of 5–8 mmHg. Hearts were paced using a Grass SD9 Stimulator at a frequency of 7 Hz. A stabilization period of 20 min was followed by recording of baseline measurements, followed by 25 min of global no-flow ischemia. Afterwards, the perfusate line was reopened and hearts were re-perfused for 45 min. To assess cytoprotective function ex vivo, percent recovery of left ventricular pressure, and rate of change in pressure (dP/dtmax–index of contractility/inotropy, and dP/dtmin–index of relaxation) were calculated after 45 min of reperfusion. Data collection and analyses were performed in a blinded manner.
Blood was collected from the left ventricle via cardiac puncture and allowed to clot for 2 h on ice, then centrifuged in 1.5 mL Eppendorf tubes at (4,000 rpm at 4°C for 20 min). The supernatant was removed, snap frozen in liquid nitrogen, and stored at −80°C for quantification of serum EPO levels. EPO protein concentration was quantified from serum using a Quantikine Mouse EPO ELISA (MEP00B, R&D Systems) according to the manufacturer’s instructions.
Tissues selected for qPCR were snap frozen in liquid nitrogen and kept at −80°C (n = 8 per group for EPOfl/fl and EPOΔ/Δ, n = 7 EPOfl/- Cre+/−). Total RNA was isolated from the free left ventricle and kidneys using TRIzol (Invitrogen, Burlington, ON, Canada) with the Qiagen RNeasy kit (Qiagen, Hilden, Germany) according to the manufacturer’s instructions. RNA samples were treated with DNase (Qiagen), according to manufacturer’s instructions. Prior to cDNA synthesis, RNA concentrations were quantified (NanoDrop, ND1000; Thermo Fisher Scientific, Waltham, Massachusetts, USA). Protein contamination was assessed by measuring absorbance at 280 nm. Generation of cDNA was completed using a High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems by Thermo Fisher Scientific, Waltham, Massachusetts, USA) according to the manufacturer’s instructions, using 2000 ng of RNA per sample. Target gene RNA was quantified using the Platinum SYBR Green qPCR SuperMix-UDG with ROX (Invitrogen, Burlington, ON, Canada) using the primers listed in Table 1. Primers were designed to span an exon-exon region to eliminate any possibility of priming genomic contamination. qPCR was performed using 7,500 Real Time PCR detection system (Applied Biosystems, Foster City, CA, USA) with the following protocol: 1 cycle at 50°C for 2 min, 1 cycle at 95°C for 5 min, then 40 cycles at 95°C for 15 s, 1 min at 60°C for all genes (excluding Epo, where the annealing temperature was 58°C), followed by a dissociation curve to assess specificity of the reaction (Supplementary Figure S2). Samples were run in duplicate 25 uL reactions. Undetectable Epo RNA was assigned the value of the limit of detection of the assay (CT = 40). Results were analyzed according to the delta-delta CT method using reference genes (Table 1) and normalized to the EPOfl/fl group.
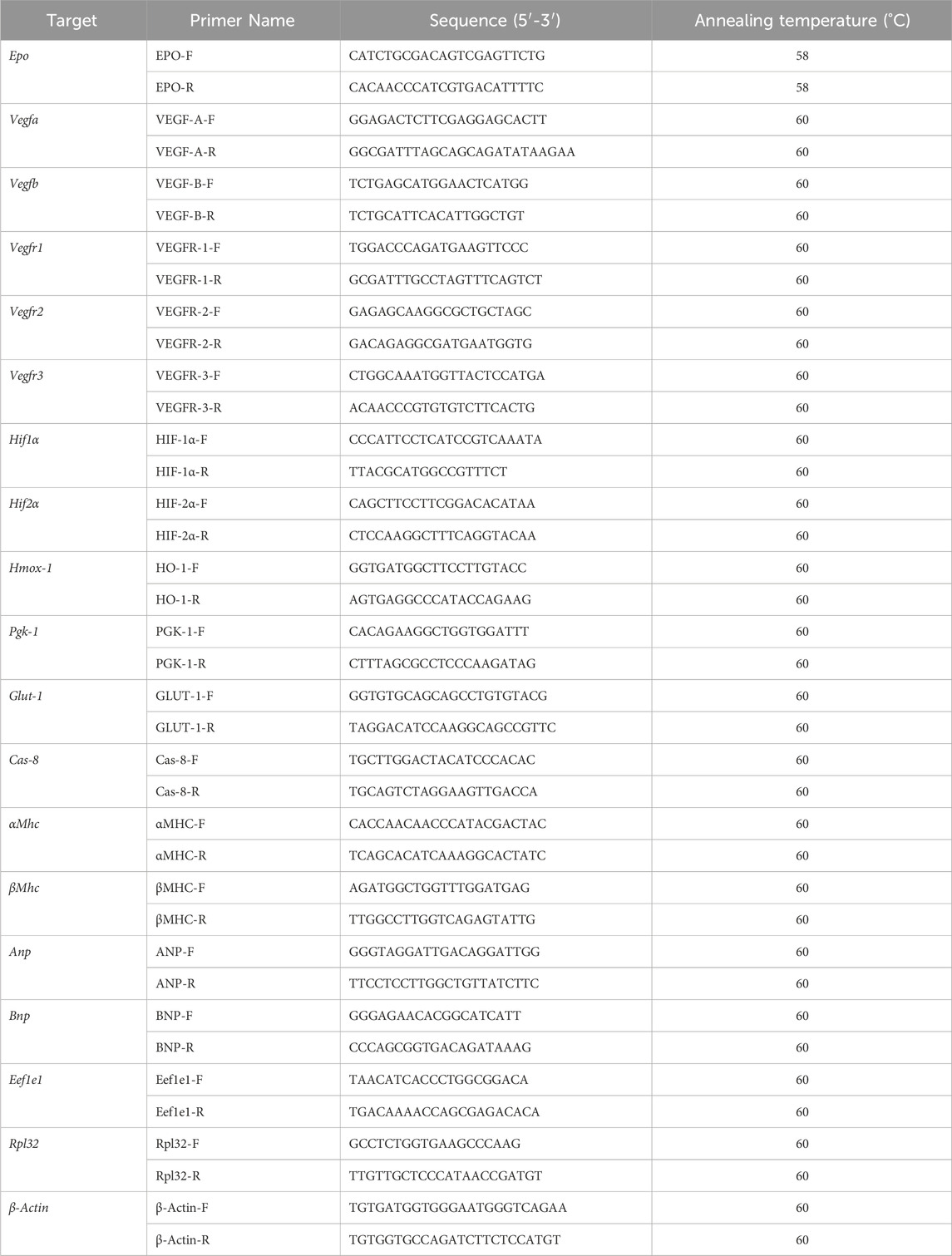
Table 1. List of mouse qPCR primers and sequences. Epo, erythropoietin; Vegf, vascular endothelial growth factor; Hif, hypoxia inducible factor; Hmox-1, heme oxygenase 1; Pgk-1, phosphoglycerate kinase 1; Glut-1, glucose transporter 1; Cas8, caspase 8; αMhc, alpha myosin heavy chain; βMhc, beta myosin heavy chain; Anp, atrial natriuretic peptide, Bnp, brain natriuretic peptide; Eef1e1, Eukaryotic Translation Elongation Factor 1 Epsilon 1; Rpl32, ribosomal protein L32; β-Actin, beta-actin.
The hearts were excised and block fixed in 10% buffered formalin for 24 h, followed by transfer into 70% ethanol for storage before processing and embedding in paraffin wax. Tissue microarrays were created using the tissue micro array (TMA) Master II instrument from 3D Histech Ltd. Regions of interest from the left ventricle were selected, cored, and added into a host paraffin block to create an array of 84 tissue cores, each measuring 2.0 mm in diameter (biological replicates n = 3).
To study Epo gene mRNA expression and localization from cardiac cell types, the microarray sections were stained using the following fluorophores: Epo with FITC (channel 2, accession # NM_007942.2) using RNAScope 2.5 LS Probe Mm-Epo-C2 Cat. No. 315508-C2, and with Cy5 (channel 1, accession # NM_007942.2) using RNAScope 2.5 LS Probe Mm-Epo-01 Cat. No. 444948. Then, kinase insert domain receptor (Kdr), an endothelial cell marker, was stained with Cy5 (channel 1, accession # NM_01612.2) using RNAScope 2.5 LS Probe-Mm-Kdr Cat. No. 414818. Myosin heavy chain 6 (Myh6), a cardiomyocyte marker, was stained with TRITC (channel 3, accession # NM_001164171.1) using RNAScope 2.5 LS Probe-Mm-Myh6 Cat. No. 506258. Finally, 4′,6-diamidino-2-phenylindole (Dapi) was used to stain nuclei. In-situ hybridization duplex staining was performed using commercially available assays and the ACD Bio program for the Leica Bond RX immunostainer. The slides were scanned using an Olympus VS 120 automated slide scanner, and images of 1 mm x 1 mm were acquired at ×40 magnification. The acquired images were analyzed using the HALO image analysis software from Indica Labs (v3.2.1851.229).
Mice were exsanguinated and 10 mL of 1x PBS, 10 mL of 0.5 mol/L KCl, and 10 mL of 10% buffered formalin (VWR, Mississauga, ON, Canada) were perfused through the right carotid artery to fix cardiomyocytes in diastole. Tissues were harvested, stored in 10% buffered formalin for 24 h, and then transferred into 70% ethanol. Hearts were processed and embedded in paraffin wax. Cross sections of the heart (5 μm) were mounted onto charged 1.2 mm Superfrost slides (Fisher Scientific). Paraffin embedded sections were then stained with either hematoxylin and eosin or Picrosirius Red for the determination of cardiomyocyte cross-sectional area (CSA) or percent fibrosis, respectively, within the left ventricle. Bright field images were acquired using an Olympus FSX 100 light microscope and analyzed blinded using ImageJ (for CSA) and cellSens (for interstitial fibrosis).
Cobalt chloride (CoCl2) is used as a hypoxia mimetic for its ability to chemically induce HIF-α stabilization and upregulate downstream HIF target genes under normoxic conditions (reviewed and discussed in detail (Muñoz-Sánchez and Chánez-Cárdenas, 2019; Packer, 2020)). The effects of CoCl2 are widespread, resulting in HIF-2α stabilization across multiple cell populations and tissues, including the myocardium (Wiesener et al., 2003). Mice were injected i. p. with either CoCl2 (30 mg/kg) dissolved in 1 mL sterile PBS (to simulate hypoxia) or vehicle control (PBS). Hearts were collected for HIF-2α western blotting at the following endpoints: 45 min, 1.5 h, or 3 h after injection.
For probing of HIF-2α, left ventricular samples were homogenized in a buffer with a phosphatase (PhosSTOP, cat. # 4906845001, Sigma) and protease inhibitor cocktail (cat. #P8340, Sigma), and separation of nuclear from cytoplasmic extracts was performed using the NE-PER kit as per the manufacturer’s instructions (cat. # 78833, Life Technologies). Nuclear protein extract concentrations were measured by bicinchoninic acid assay (cat. # 23277, Fisher Scientific). Samples were equally loaded (20ug/well) and separated by 10% SDS-PAGE, followed by immunoblotting. Nitrocellulose membranes were rinsed in ddH2O and then incubated in reversible Ponceau Stain for 7 min to confirm equal protein transfer. The stain was stripped using 200uM NaOH for 1 min and rinsed in ddH2O for 5 min x 3. Membranes were blocked (5% skim milk in 1x TBST (0.1% tween)) and incubated in a primary HIF-2α antibody (Sun et al., 2015) (1:1000; cat. # NB100-122SS, Bio-Techne; 5% bovine serum albumin in 1x TBST) overnight at 4°C. Membranes were washed for 5 min x 3 in TBST and then incubated with a goat anti-rabbit IgG horseradish peroxidase–conjugated secondary antibody (1:1000, cat. # HAF008, Bio-Techne; 1% skim milk for 1 h at 22°C). Membranes were washed for 5 min x 3 in 1x TBST. Signal was detected and quantified via enhanced chemiluminescence (cat. # 1705060, Bio-Rad) using a FluorChem HD imaging system (Alpha Innotech, Santa Clara, CA, USA). Values were obtained by measuring the target band (normalized to Ponceau) relative to the EPOfl/fl group.
Axitinib is an FDA-approved selective inhibitor of cellular phosphorylation of VEGF receptor tyrosine kinases (VEGFR-1, VEGFR-2, VEGFR-3) for the treatment of advanced renal cell carcinoma (Tyler and FCSHP, 2012; INLYTA, 2023). Axitinib (cat. #S1005, Selleck Chemicals) was prepared as previously described (Ma and Waxman, 2009). Briefly, axitinib was suspended at 5 mg/mL in polyethylene glycol 400 (cat. # PX1286B-2, Sigma) and sonicated at room temperature for 30 min until dissolved. Using 0.1N HCl, the pH was adjusted to 2.5, followed by a second round of 10-min sonication. To achieve a final ratio of 3:7 (v/v) of polyethylene glycol 400 to water, acidified water (pH 2.5) was added. The solution was prepared fresh and stored in the dark at 4°C. Axitinib was administered once a day through i. p. injection at a dose of 25 mg/kg body weight in a volume of 5 μL/g body weight (Ma and Waxman, 2009) for 4 days x 2 cycles (with 2 days rest). Data (invasive hemodynamics, saphenous vein hemoglobin levels, and tissues for qPCR) were collected 24 h after the last injection.
A one-step TUNEL in situ apoptosis kit (cat. # E-CK-A320, Elabscience Biotechnology Co.) was used to quantify differences in cell apoptosis between EPOfl/fl and EPOΔ/Δ hearts according to the manufacturer’s instructions. Fluorescent images were acquired using an Olympus FSX 100 microscope and the amount of green fluorescence of TUNEL-positive cells was analyzed blinded using ImageJ.
Graphing and statistical analyses of the data presented was performed using Prism version 9 software developed by (GraphPad Inc., La Jolla, CA, USA). Power calculations were used to determine the number of mice needed to detect a significant effect. Results were reported as mean ± SD (morphometrics, serum, hematocrit, qPCR, western blotting) or mean ± SEM (CLAMS, echocardiography, histology, invasive hemodynamics, Langendorff). To confirm whether data was normally distributed, a Shapiro-Wilk test was used. If the data was normally distributed, either a one-way ANOVA followed by Dunnett’s or Tukey’s post-hoc test or an unpaired Student’s t-test was performed. If the data was not normally distributed, a Kruskal–Wallis test with Dunn’s post-hoc test or Mann-Whitney U test was performed. Simple linear regression correlation analyses were run comparing Epo RNA expression to dP/dtmax, dP/dtmin, and dP/dt@LVP40 values and goodness-of-fit values were provided. p-value <0.05 was considered significant.
In the developing fetus, mice null for Epo and the Epor die by E13.5 due to impaired cardiogenesis and anemia (Wu et al., 1999; Kertesz et al., 2004). Our previous work using the Mlc2v promoter established cardiomyocyte specific Epo deletion during embryogenesis in mice induces a phenotype with less cardiomyocyte hypoplasia, compensatory cellular hypertrophy, and upregulation of Epo in the heart by the endothelial cell persisting into adulthood (Allwood et al., 2024). Therefore, the presence of cardiac EPO is critical for proper development of the heart, yet endogenous cardiac Epo expression, regulation, and physiological significance in the adult heart remains unknown. For this reason, in our follow up study, we hypothesized that the demand for compensatory cardiac EPO would be reduced in a fully, and normally, developed adult heart. Upon induced deletion of adult cardiomyocyte Epo in mice after an ordinary course of cardiogenesis, we expected whole-heart morphology to be normal and cardiac Epo production to be low. To verify this, mice were subjected to morphometric and quantitative tissue PCR analysis. In line with our previous reports (Allwood et al., 2024), the morphological measurements revealed no gross differences in body weight, heart weight, left ventricular weight, or heart weight/tibial length (HW/TL) ratio between groups (Supplementary Figure S3C; Table 2). Surprisingly, after tamoxifen induced MerCreMer deletion of Epo expression, there was a significant upregulation in cardiac Epo in EPOΔ/Δ mice compared to the EPOfl/fl group by qPCR (Figure 1A). This data was confirmed by a mildly graded response in mice with only one floxed EPO allele (i.e., EPOfl/- Cre+/−) (Figure 1A), suggesting this phenomenon does not exist in an all-or-nothing manner. Further, we sought to confirm our data was not specific to male mice. Indeed, female EPOΔ/Δ mice also showed a significant increase in whole-heart Epo upon targeted cardiomyocyte Epo deletion (Figure 1B) with no apparent sex effect (Supplementary Figure S4). To identify the specific cell type(s) responsible for Epo expression under normoxic conditions in EPOfl/fl and EPOΔ/Δ mice, we used RNA fluorescent in situ hybridization. As expected, we observed low basal Epo mRNA production by the cardiomyocyte (Figure 1Ci) and endothelial cell (Figure 1Cii) in EPOfl/fl mice. By contrast, in the EPOΔ/Δ mice, an over-abundance of Epo signal (shown using two regions of interest, Figure 1Ciii, iv) was co-localized with the endothelial cells, indicating that upon successful cardiomyocyte-Epo knockout, the endothelium compensated for the loss by increasing its own Epo expression. Therefore, not only have we verified this phenomenon in the adult mouse using a second independent CreLox line to support our earlier work (Allwood et al., 2024), but we also showed hyper-compensated cardiac Epo occurs regardless of floxed allele homozygosity (EPOΔ/Δ) or heterozygosity (EPOfl/-: Cre+/−), and in both sexes. Importantly, despite marked Epo overexpression in the adult EPOΔ/Δ mouse, on a whole-body and organ level, there were no visual abnormalities (Supplementary Figure S3A). There was also no change in renal Epo expression (Figure 2A), serum EPO (Figure 2B), or hematocrit (Figure 2C), suggesting that there would be no altered physiological consequences from compensatory cardiac endothelial overexpression.
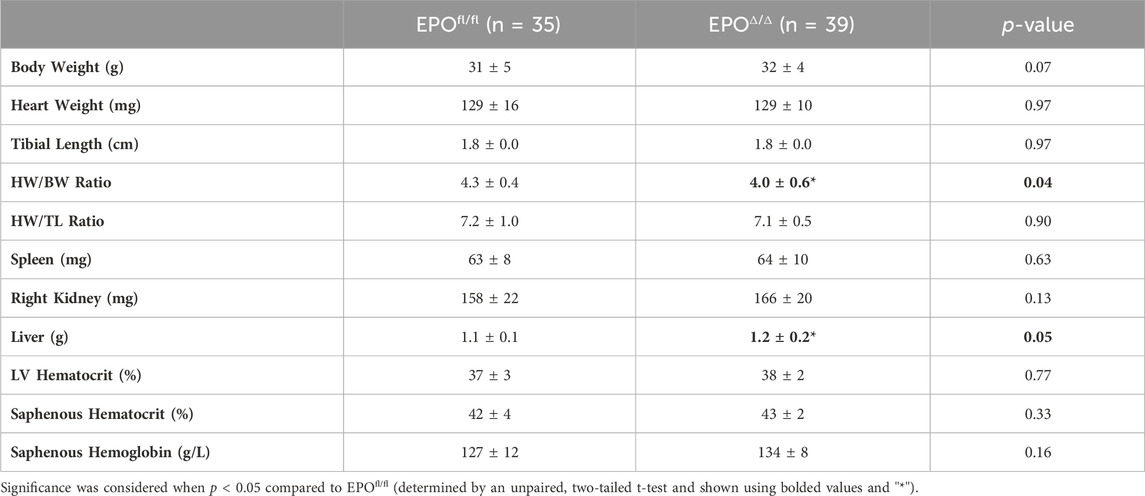
Table 2. Morphometrics. HW, heart weight; TL, tibial length; LV, left ventricle peak pressure. Data is presented as mean ± SD.
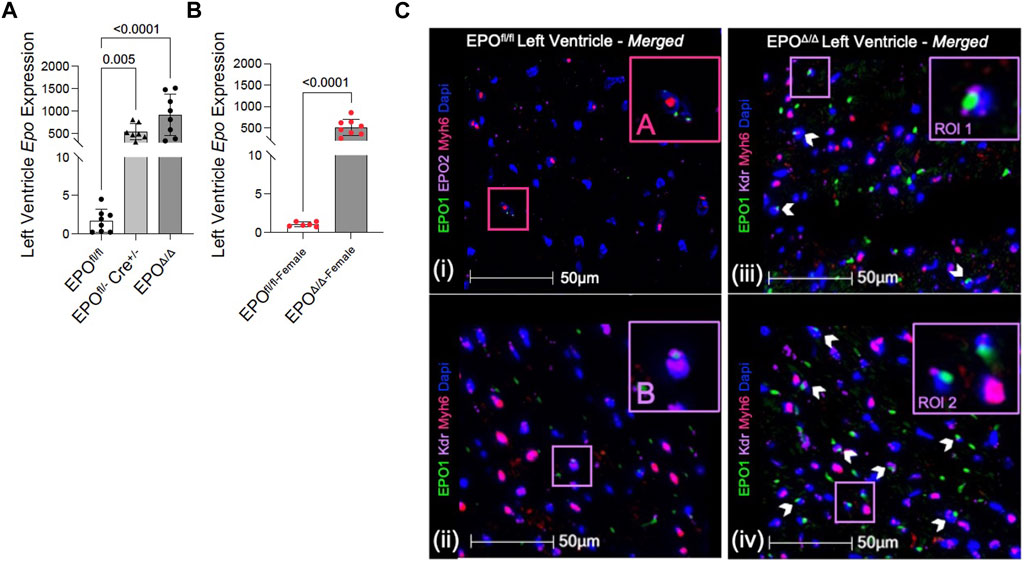
Figure 1. Epo RNA expression was significantly upregulated in male and female EPOΔ/Δ mice by qPCR. (A) Left ventricular expression of Epo RNA (normalized to Eef1e1) of EPOfl/fl, EPOfl/- Cre+/−+TAM, and EPOΔ/Δ mice. (B) Left ventricular expression of Epo RNA (normalized to Rpl32) of female EPOfl/fl and EPOΔ/Δ mice. Panels (Ci-iv): Cardiomyocytes and endothelial cells contributed to basal Epo expression in EPOfl/fl, which upon cardiomyocyte specific Epo deletion (EPOΔ/Δ), became overcompensated by the endothelial cells, leading to hyper-expression. (i) A cardiomyocyte (Myh6, TRITC) and (ii) endothelial cell (Kdr, Cy5) show co-localization with EPO (FITC, green) in EPOfl/fl. However, in EPOΔ/Δ mice, the cardiomyocyte does not appear to be a contributing source, confirming successful knockout of EPO. Rather, (iii, iv) show two regions of interests (RIO) to highlight intense endothelial-derived EPO signal. The pink box “(A)” is used to show cardiomyocyte-Epo colocalization in EPOfl/fl, purple box “(B)” is used to show endothelial-Epo colocalization in EPOfl/fl, purple boxes “ROI 1” and “ROI 2” show upregulated EPO signal from endothelial cells. A nuclei marker was used (Dapi, blue). Scale bar represents 50 µm. A one-way ANOVA followed by a Dunnett’s post-hoc test was used to detect differences in left ventricular expression between EPOfl/fl, EPOfl/- Cre+/−+TAM, and EPOΔ/Δ mice. An unpaired, two-tailed t-test was used to detect a difference between EPOfl/fl-female and EPOΔ/Δ-female. Data are expressed as mean ± SD and were considered significant when p < 0.05.
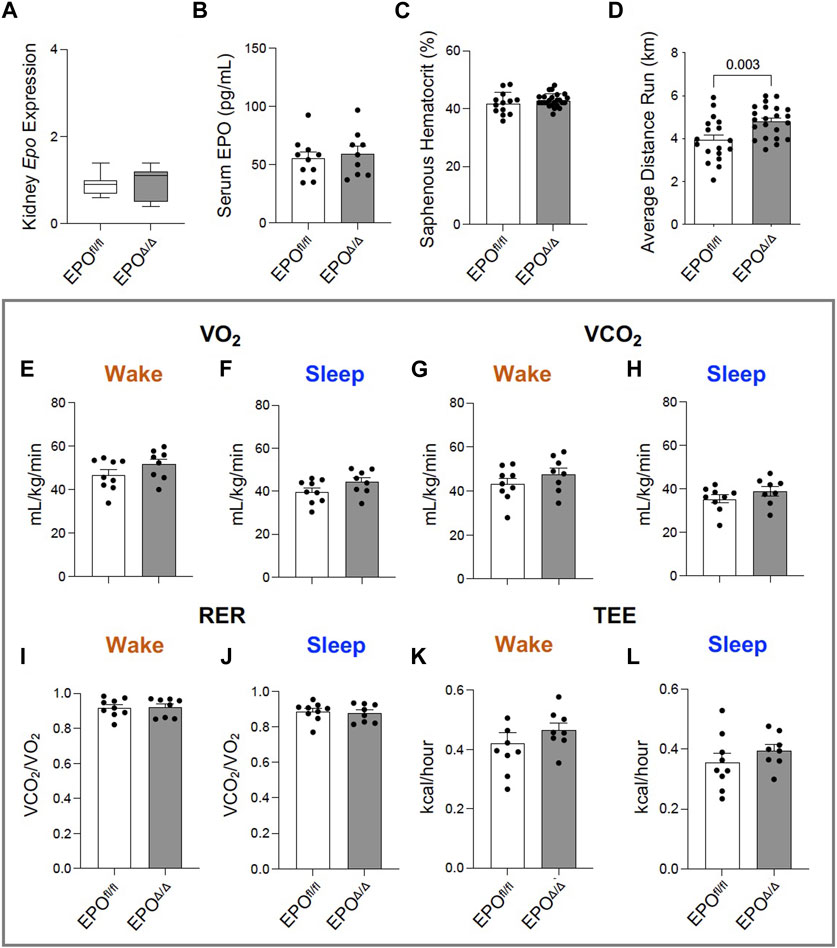
Figure 2. Greater voluntary wheel running capacity in EPOΔ/Δ compared to EPOfl/fl mice occurred independent from differences in hematocrit and whole-body metabolism. (A) Kidney Epo RNA expression (normalized to β-Actin) in EPOfl/fl and EPOΔ/Δ mice, (B) serum EPO, (C) hematocrit (%) collected from the saphenous vein of EPOfl/fl and EPOΔ/Δ mice, and (D) average voluntary wheel running distance (km) across 3 days. Comprehensive Laboratory Animal Monitoring System (CLAMS) measured (E, F) VO2 (mL/kg/min), (G, H) VCO2 (mL/kg/min), (I, J) RER (VCO2/VO2), and (K-L) total energy expenditure (TEE, kcal/hour) in EPOfl/fl and EPOΔ/Δ mice during their sleep and wake phases. An unpaired, two-tailed t-test was used to detect differences. Data are expressed as mean ± SD (for qPCR, serum EPO levels, and hematocrit) or mean ± SEM (for average running distance and whole-body metabolic readings). Data were considered significant when p < 0.05.
Exposing mice to either a physiological or pathological challenge could reveal additional insight to the innate compensatory mechanisms. We therefore subjected mice to an exercise test, and considering there was no change in hematocrit, we hypothesized no difference would be observed between groups. Yet, voluntary wheel running across three consecutive days revealed EPOΔ/Δ mice had increased running performance compared to control animals (Figure 2D). Since the difference in exercise capacity was not the result of erythropoiesis, we determined it could be linked to reported effects of EPO as a regulator of energy homeostasis as it increases metabolic activity, cellular respiratory capacity, and oxygen utilization shown previously in transgenic mice (Wang et al., 2013). However, using whole-body indirect calorimetry (CLAMS setup), our data showed there were no differences in VO2 (Figures 2E, F), VCO2 (Figures 2G, H), RER (Figures 2I, J), or total energy expenditure between groups (Figure 2K, L). We cannot exclude the possibility for differences in myocardial oxygen consumption at the level of the skeletal muscle by these data alone. It is plausible that EPOΔ/Δ mice were more efficient at extracting oxygen for mitochondrial cellular respiration (i.e., ATP production). Indeed, positive hypertrophic cardiac remodeling might account for the heightened exercise tolerance due to increased cardiac output.
Prior work showed that constitutive deletion of cardiomyocyte-specific EPO during embryogenesis causes early hypoplasia, and eventually, cardiac Epo over-expression led to hypertrophy, with no change in overall cardiac mass in adult hearts (Allwood et al., 2024). We sought to explore the physiological significance of cardiomyocyte deleted Epo in adult mice, independent of cardiogenesis. By echocardiography, there was a significant decrease in end systolic dimension (ESD) and a trending reduction in end diastolic dimension in EPOΔ/Δ mice (EDD, Figures 3A, B; Table 3). The left ventricle chamber length was decreased (Figures 3A, C) and posterior wall thickness was increased (Figures 3A, D). While heart rate (Figure 3E) and cardiac output (Figure 3F) were unchanged, EPOΔ/Δ mice had greater ejection fractions (Figure 3G), suggesting systolic function was being augmented. Together, these parameters indicated a concentric hypertrophy phenotype in the EPOΔ/Δ mice. To confirm these findings, we evaluated histologically cardiomyocytes in perfusion-fixed hearts (Figure 3H), identifying a significant increase in EPOΔ/Δ cross-sectional area compared to control (Figure 3I). These data combined suggest that as cells of the left ventricle wall widened, the length of the hearts shortened, resulting in no difference in global heart mass.
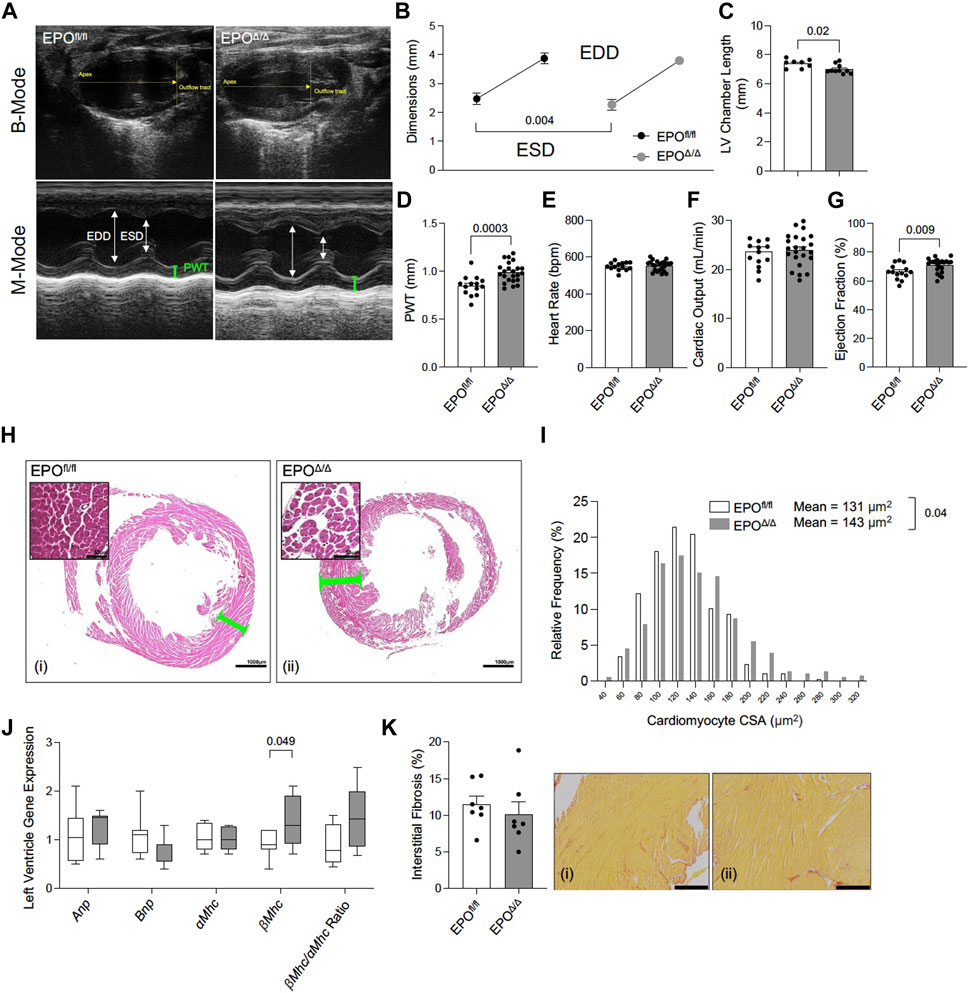
Figure 3. EPOΔ/Δ mice exhibited compensatory changes in cardiac structure and systolic function by echocardiography. (A) Representative B-Mode (top) and M-Mode (bottom) tracings from EPOfl/fl and EPOΔ/Δ mice. Echocardiography measurements included (B) end systolic dimension (ESD) and end diastolic dimension (EDD), (C) left ventricle (LV) chamber length, (D) posterior wall thickness (PWT), (E) heart rate, (F) cardiac output, (G) ejection fraction. (H) Perfusion fixed hearts (representative hematoxylin and eosin histological images from (i) EPOfl/fl and (ii) EPOΔ/Δ hearts–scale bars represent 50µm and 1000 µm) were used to measure differences in (I) cardiomyocyte cross-sectional area (CSA). (J) Expression of fetal genes (atrial natriuretic peptide (Anp), brain natriuretic peptide (Bnp), alpha myosin heavy chain (αMhc), beta myosin heavy chain (βMhc), and βMhc/αMhc ratio) was quantified. (K) Quantification of interstitial fibrosis (%) from (i) EPOfl/fl and (ii) EPOΔ/Δ hearts stained with Picrosirius Red. Scale bar represent 216 µm. An unpaired, two-tailed t-test was used to detect differences. Data are expressed as mean ± SEM (for echocardiography, CSA, and interstitial fibrosis) or mean ± SD (qPCR). Data were considered significant when p < 0.05.
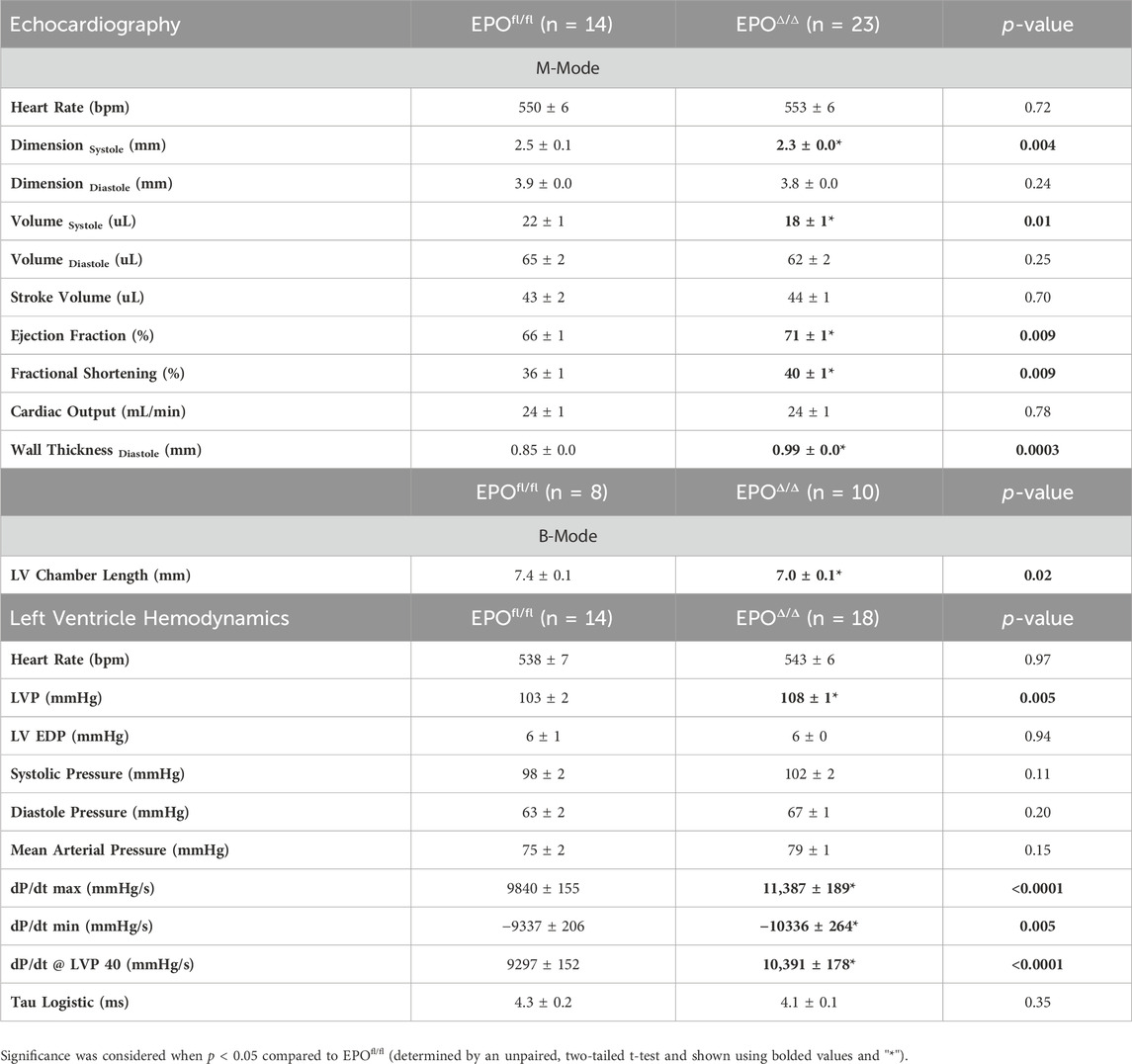
Table 3. In vivo cardiac assessment using echocardiography and invasive hemodynamics. LVP, left ventricle peak pressure; LV EDP, left ventricle end diastolic pressure. Data is presented as mean ± SEM.
Both pathological (e.g., hypertension) and physiological (e.g., exercise) conditions stimulate cellular and gross ventricular hypertrophy with inverse relationships to long-term health (Schoenfeld et al., 1998). However, a hallmark feature of pathological remodeling is the re-expression of certain fetal genes (Schoenfeld et al., 1998). Therefore, we quantified relative RNA levels of Anp, Bnp, α-Mhc, ß-Mhc, and the ratio of ß-Mhc/α-Mhc between groups by qPCR. No differences amongst Anp, Bnp, α-Mhc expression, or the ratio of ß-Mhc/α-Mhc (Figure 3J) were seen though ß-Mhc RNA was increased in EPOΔ/Δ mice. An incipient cause of ß-Mhc re-expression during cardiac hypertrophy and normal aging is linked to elevated fibrosis (Pandya et al., 2006). Accordingly, we quantified interstitial fibrosis of the left ventricle between groups (Figures 3Ki,ii), finding no difference. EPOΔ/Δ mice had compensatory cardiac function (i.e., increased ejection fraction) and the EPOΔ/Δ phenotype is inconsistent with pathological remodeling. Nonetheless, we reconciled this line of inquiry with a comprehensive assessment of cardiac function by invasive hemodynamics.
To gain a deeper understanding of the functional role of cardiac Epo overexpression and concentric cellular hypertrophy on cardiac function in our model, invasive hemodynamic analyses were performed (Figure 4A; Table 3). There were no differences in heart rate (Figure 4B). In terms of systolic function, EPOΔ/Δ mice demonstrated elevated left ventricular systolic pressures (LVP, Figure 4C) and an increase in cardiac contractility (dP/dtmax, dP/dt@LVP40, Figures 4D, E). To assess diastolic function, a multi-parameter approach was used (Ogilvie et al., 2020). For this reason, we reported indices of both relaxation and compliance: dP/dtmin (active relaxation phase), Tau Logistic (active relaxation phase), and end diastolic pressure (EDP, passive filling phase). dP/dtmin was significantly improved (Figure 4D), though this was not reflected by differences in EDP (Figure 4F) or Tau Logistic (Figure 4G). Taken together, both systolic and diastolic function were superior in the EPOΔ/Δ mice, suggesting endogenous cardiac EPO production was associated with better cardiac function. Supraphysiological levels of exogenous rhEPO confer inotropic and lusitropic effects ex vivo, therefore we were interested in the correlation between the levels of upregulated endogenous Epo RNA and these key hemodynamic parameters recorded in vivo. Simple linear regression analyses demonstrated a strong positive relationship between Epo RNA expression and dP/dtmax, dP/dtmin, and dP/dt@LVP40, with goodness-of-fits of R2 = 0.49, 0.48, and 0.67, respectively (Figures 4H–J). Therefore, our results indicate that in mice with higher endogenous cardiac Epo expression, inotropic and lusitropic function are greater.
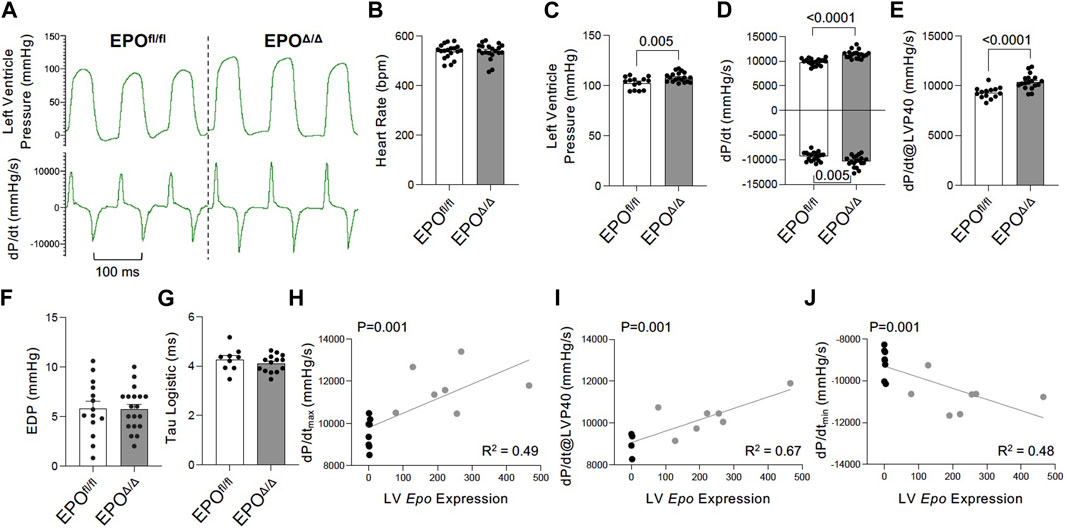
Figure 4. Improved inotropic and lusitropic cardiac function was observed in vivo by invasive hemodynamics in EPOΔ/Δ compared to EPOfl/fl mice. (A) Representative left ventricle pressure (LVP, top) and dP/dtmax and dP/dtmin (bottom) tracings from EPOfl/fl and EPOΔ/Δ mice. (B) Heart rate (bpm), (C) left ventricular systolic pressure (mmHg), (D) dP/dtmax and dP/dtmin (mmHg/s), (E) dP/dt@LVP40 (mmHg/s), (F) end diastolic pressure (EDP), and (G) Tau logistic (ms). Simple linear regression correlation analyses were run comparing Epo RNA expression to (H) dP/dtmax, (I) dP/dt@LVP40, and (J) dP/dtmin values and detected a positive correlation (p = 0.0014 for each) with moderate to strong goodness-of-fit values (R2 = 0.49, 0.67, 0.48, respectively). An unpaired two-tailed t-test was used to detect differences. Data are expressed as mean ± SEM and were considered significant when p < 0.05.
The in vivo assessment of cardiac function carries limitations–the data collected are dependent on preload, afterload, and heart rate. Conversely, the Langendorff preparation (i.e., ex vivo) allows for the isolated evaluation of systolic and diastolic cardiac function at a healthy baseline or after ischemic insult under controlled conditions without the influence of systemic neurohormonal or immunological factors. Since exogenous rhEPO has shown cardioprotective effects in vivo, investigating the physiological autocrine and/or paracrine effects of endogenous cardiac EPO on the heart was of particular interest. The EPOΔ/Δ group demonstrated a significant increase in baseline LVP (Figures 5A, B (top left)), developed pressure (Figure 5C), and dP/dtmax and dP/dtmin (Figure 5A (bottom left), D). While there were no significant differences in EDP or paced heart rate as they were manually set and controlled, intrinsic heart rate prior to pacing was elevated in EPOΔ/Δ mice (Table 4). After baseline recordings, mice were subjected to 25 min of global, no-flow ischemia, followed by 45 min of reperfusion (Figure 5A (right)). EPOΔ/Δ mice displayed better recovery post-ischemia: 71% of LVP (Figure 5E), 76% of dP/dtmax, (Figure 5F), and 61% of dP/dtmin (Figure 5G), whereas the EPOfl/fl mice recovered only 27% of LVP (Figure 5E), 28% of dP/dtmax (Figure 5F), and 25% of dP/dtmin (Figure 5G). Therefore, independent of heart rate, afterload, preload, and whole-body neurohormonal influence, hearts overexpressing endogenous cardiac EPO also displayed positive inotropic, lusitropic, chronotropic, and cardioprotective qualities. Having established the physiological impact of hyper-compensated cardiac Epo, we aimed to clarify some of the factors involved in regulating this phenomenon.
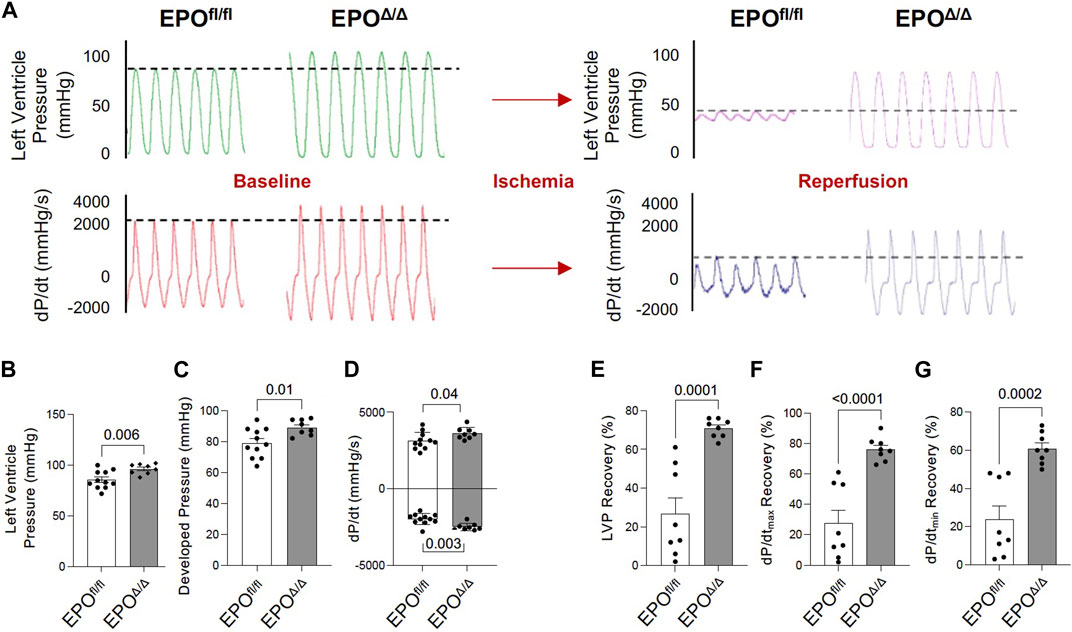
Figure 5. EPOΔ/Δ mice demonstrated greater inotropic and lusitropic cardiac function by the ex vivo Langendorff preparation, accompanied by superior cytoprotective abilities post-ischemia/reperfusion. (A) Representative isolated heart (Langendorff) function tracings at baseline (red and green) and post-ischemia reperfusion (pink and purple) for EPOfl/fl and EPOΔ/Δ mice. Baseline measurements included (B) left ventricle systolic pressure (mmHg), (C) developed pressure (mmHg), (D) dP/dtmax and min (mmHg/s). Baseline was followed by global no-flow ischemia and subsequent reperfusion, generating data for (E) % left ventricular pressure (LVP) recovery, (F) % dP/dtmax recovery, and (G) % dP/dtmin recovery. An unpaired two-tailed t-test was used to detect differences. Data are expressed as mean ± SEM and were considered significant when p < 0.05.
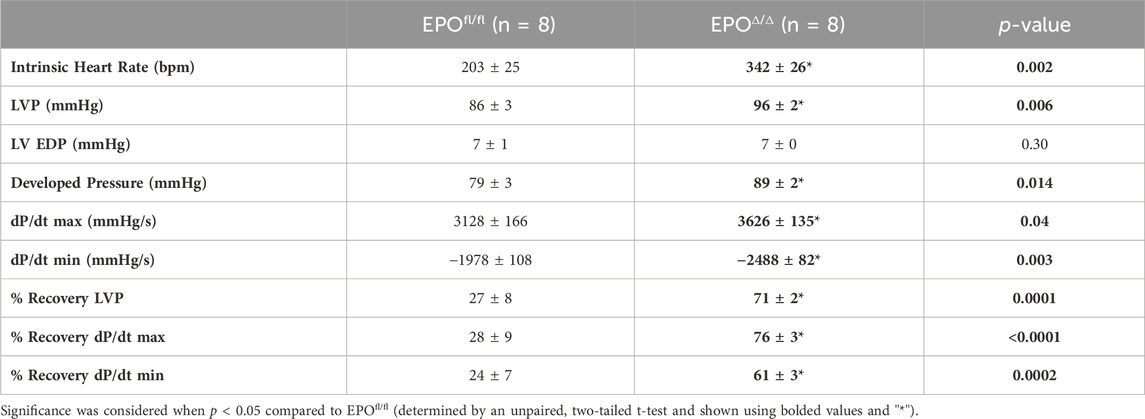
Table 4. Ex vivo cardiac assessment using Langendorff isolated heart preparation. LVP, left ventricle systolic pressure; LV EDP, left ventricle end diastolic pressure. Data is presented as mean ± SEM.
HIF-1α and HIF-2α are both primary transcription factors for regulating the hypoxic response. However, in terms of the transactivation of the Epo promoter, HIF-2α, and not HIF-1α, is the primary regulator in the adult kidney, liver, and brain (Warnecke et al., 2004; Gruber et al., 2007; Rankin et al., 2007; Yamashita et al., 2008; Yeo et al., 2008; Wang et al., 2015; Urrutia et al., 2016; Ghosh et al., 2021). Although there was no overt indication of hypoxia in this model (i.e., no differences in hematocrit or renal EPO expression), we aimed to rule out the canonical PHD2/VHL/HIF2α axis as the mechanism for mediating cardiac overexpression of Epo in EPOΔ/Δ mice. By immunoblotting, we showed no difference in the HIF-2α protein levels in left ventricular nuclear extracts of EPOfl/fl and EPOΔ/Δ mice (Figures 6A–C). This finding confirmed the overexpression of left ventricular Epo in EPOΔ/Δ mice was HIF2α-independent. Considering the unexpected nature of our findings, we wanted to conclusively exclude hypoxia as a stimulus for cardiac overexpression of Epo by investigating HIF1α-specific downstream target genes.
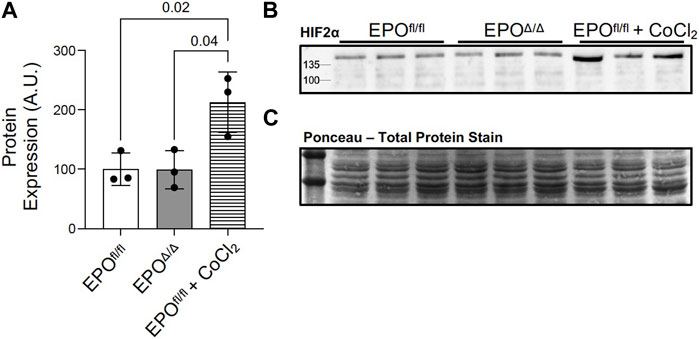
Figure 6. Marked overexpression of left ventricular Epo in EPOΔ/Δ mice was HIF2α-independent. (A) Western blot permitted the quantification of HIF-2α protein from nuclear extracts of left ventricle tissue of EPOfl/fl, EPOΔ/Δ, and EPOfl/fl mice treated with cobalt chloride (CoCl2) at 3 timepoints–3h post-injection, 1.5h post-injection, and 45min post-injection. (B) Representative western blot of HIF-2α and (C) ponceau stain, which demonstrates equal loading (20 µg per well). A one-way ANOVA followed by a Tukey’s post-hoc test was used to detect differences amongst the three groups. Data are expressed as mean ± SD and were considered significant when p < 0.05.
Under hypoxic conditions, HIF-1α escapes oxygen-dependent degradation, translocates to the nucleus, and complexes with HIF-1β (Wang et al., 1995). Working in concert with hepatocyte nuclear factor 4 (HNF-4) (Galson et al., 1995; Huang et al., 1997; Jelkmann, 2007) and the transcriptional co-activators, p300 and cAMP response element (CREB)-binding protein (Firth et al., 1995; Bunn et al., 1998), the HIF-1 complex binds the hypoxia response element to initiate the transcription of >500 downstream target genes (Mole et al., 2009), including Vegfa (Forsythe et al., 1996), Glut1 (Iyer et al., 1998), Ho-1 (Lee et al., 1997), Pgk-1 (Semenzas et al., 1994; Li et al., 1996a), Cas 8 (Zhao et al., 2018). Therefore, to rule out hypoxia and the involvement of HIF-1α in regulating cardiac Epo overexpression, we examined the expression levels of select HIF1α-specific target genes (e.g., Vegfa, Glut1, Hmox-1, Pgk-1, Cas 8) by qPCR. We measured no differences in their gene expression, and even downregulation of Cas 8 in EPOΔ/Δ mice (Figure 7). This indicated the upstream regulation of Epo in the EPOΔ/Δ mice by hypoxia and HIF-1α was unlikely. Epo is also regulated by HIF-independent mechanisms in a tissue-specific manner (Yasuda et al., 1998; Tam et al., 2006). One study demonstrates potent inhibition of VEGF induces hepatic synthesis of Epo and subsequent erythropoiesis through a HIF1α-independent mechanism (Tam et al., 2006). Therefore, we investigated the hypoxia-independent VEGF isoform, Vegfb, and its receptor, Vegfr1, by qPCR analyses. Interestingly, both genes were significantly upregulated in EPOΔ/Δ mice (Figure 7). Co-upregulation of endothelial-derived EPO and whole-heart Vegfb and Vegfr1 suggested a complex cellular interaction was governing this novel physiological concentric hypertrophy and cardioprotective phenotype. To identify a relationship between cardiac EPO and VEGF, we used the FDA-approved VEGF-specific tyrosine kinase inhibitor, axitinib, to interrupt VEGF signaling and observe the corresponding impact on Epo RNA levels in the heart.
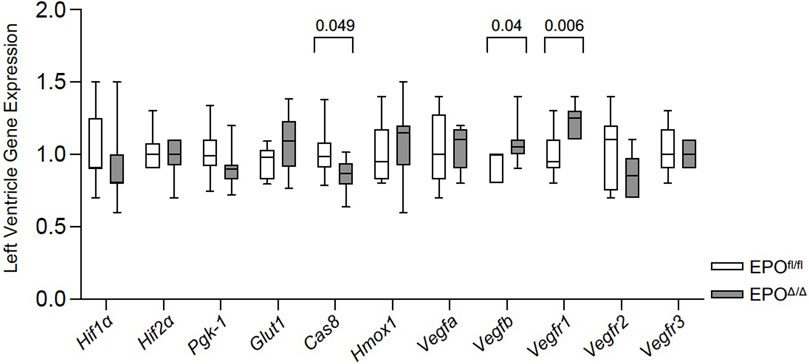
Figure 7. Vegfb and Vegfr1 RNA expression were upregulated in the left ventricle of EPOΔ/Δ mice. An unpaired two-tailed t-test was used to detect differences between EPOfl/fl and EPOΔ/Δ for individual genes of interest using qPCR. Data are expressed as mean ± SD and were considered significant when p < 0.05.
Several independent labs show active systemic crosstalk between EPO-EPOR and VEGF-VEGFR signal transduction pathways. rhEPO increases Vegf expression (Ribatti et al., 1999; Westenbrink et al., 2008; Lu et al., 2012; Oztas et al., 2020) resulting in angiogenesis and attenuated interstitial fibrosis, while VEGF-VEGFR blockade induces non-renal Epo expression (Tam et al., 2006) and erythropoiesis (Tam et al., 2006; Johnson et al., 2017). Whether these pathways interact similarly in the heart by endogenous forms of EPO and VEGF, is unknown. Therefore, to improve our understanding of the interplay between VEGF-VEGFR and EPO-EPOR in the heart, we interrupted VEGF-VEGFR signaling by inhibition using axitinib (Ma and Waxman, 2009) in EPOfl/fl mice. Preclinically, in a mouse model of prostate cancer, 24 days of axitinib treatment effectively arrests tumour growth by supressing tumour patency (i.e., blood perfusion) without disrupting blood perfusion to normal tissues (e.g., heart, kidney, liver, lung, muscle) (Ma and Waxman, 2009). In our study, axitinib was well-tolerated, and mice did not display any overt signs of distress during, or after, treatment. Short-term axitinib treatment did not elicit changes in erythropoiesis, as determined by no differences detected in hemoglobin levels measured between EPOfl/fl and EPOfl/fl+AXI groups (Figure 8A). When VEGF signaling was inhibited, cardiac expression of Epo RNA was significantly elevated by qPCR (Figure 8B). However, by invasive hemodynamics, we observed blunted heart rates and reduced left ventricle relaxation in axitinib-treated mice (Supplementary Table S2). These data were expected since tyrosine kinase inhibitors affect the cardiac conduction system, causing bradycardia and QTc prolongation (Kloth et al., 2015; Shopp et al., 2022), and are known to have cardiotoxic effects (Force and Kolaja, 2011; Dobbin et al., 2021). Our findings indicated cardiac Epo upregulation following VEGF inhibition does not rescue hearts from suspected axitinib-related bradycardia and cardiotoxicity. Importantly, however, we revealed the existence of a previously unrecognized link between VEGF and EPO in the heart. Future work is needed to fully define how the cardiac EPO-EPOR and VEGF-VEGFR axes are coordinated.
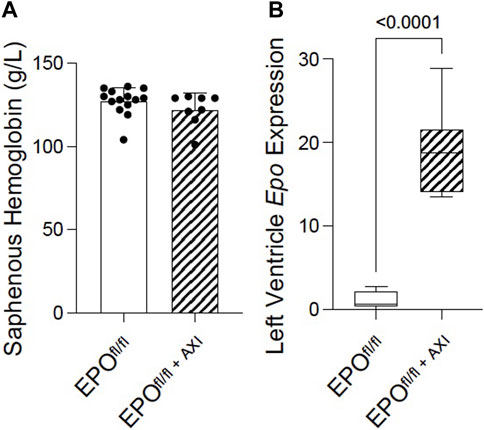
Figure 8. Pan-VEGFR inhibition via 8 days of axitinib treatment (i.p.) significantly upregulated left ventricular Epo RNA expression in EPOfl/fl mice independent of erythropoiesis. (A) Saphenous hemoglobin levels and (B) left ventricle Epo RNA expression by qPCR in EPOfl/fl compared to EPOfl/fl+AXI mice. Data are expressed as mean ± SD and were considered significant when p < 0.05 as determined by unpaired, two-tailed t-test.
Here we show that targeted deletion of the Epo gene in the adult cardiomyocyte results in compensatory overexpression by cardiac endothelial cells by HIF2α-independent conditions. The presence of excess Epo in the heart translated to notable changes in cardiac structure, and improvements in function and cytoprotection. Hyperproduction of Epo by the heart did not affect systemic erythropoiesis, suggesting EPO only acted locally via paracrine signaling between cardiomyocytes and endothelial cells. Pharmacological interruption of VEGF-VEGFR using the tyrosine kinase inhibitor, axitinib, allowed us to study how cardiac Epo expression could be modulated in the absence of VEGF signaling. Indeed, cardiac Epo RNA levels were upregulated in axitinib-treated mice. These data suggested a dynamic and interactive crosstalk between cardiomyocytes and endothelial cells involving the VEGF-VEGFR pathway that regulate the upstream production and subsequent physiological effects of cardiac EPO under hypoxia-independent conditions. Taken together, in the adult mouse heart, endothelial EPO is an important regulator of cell infrastructure, cardiac contractility, and ischemia-reperfusion susceptibility, such that excess Epo is associated with superior cardiac function directly independent of systemic red blood cell production.
Recent studies establish EPO as a pleiotropic cytokine. However, interpretation of these data does not always consider the form of EPO in question–exogenous vs. endogenous. rhEPO is differentially glycosylated compared to endogenous EPO (Rush et al., 1995; Skibeli et al., 2001; Lasne et al., 2002) and the oligo branching patterns and sialic acid content modify the pharmacodynamics and biological activity in vivo (i.e., stability and receptor binding) (Weikert et al., 1999; Skibeli et al., 2001). Therefore, high dose rhEPO may facilitate non-specific receptor binding, signal transduction, and physiological outcomes that do not apply to endogenous EPO. According to three ex vivo studies (Kaygisiz et al., 2006; Piuhola et al., 2008; Hefer et al., 2012), exogenous rhEPO is reportedly inotropic. The changes in contractility and relaxation are attributed to cAMP (Kaygisiz et al., 2006), increased calcium transients (rate and magnitude) and myofilament function via PI3-K and PKCε (Hefer et al., 2012), and endothelin-1 signaling (Piuhola et al., 2008). In another ex vivo study, hypoxia induces high endogenous plasma EPO levels, which correlate to increased atrial contraction (Sterin-Borda et al., 2003). Collectively, these studies suggest a positive primary effect of EPO on myocardial inotropic function.
Until now, the interpretation and application of these findings to an in vivo endogenous EPO system was limited. Therefore, using both ex vivo (i.e., Langendorff isolated hearts) and in vivo (i.e., invasive hemodynamics) preparations, our data revealed the improved cardiac functional effects conferred by endogenous EPO in EPOΔ/Δ mice for the first time. It is plausible endogenous EPO improved calcium handling by increasing cAMP-dependent protein kinase A (PKA) phosphorylation of phospholamban (PLB) (Simmerman and Jones, 1998), which stimulated subsequent sarcoplasmic reticulum calcium uptake (Kranias et al., 1985) and release by the ryanodine receptors (RyR) (Valdivia et al., 1995). Indeed, calcium transient assays and western blotting of key proteins (i.e., PLB, RyR) may clarify this line of inquiry. Alternatively, greater cardiac function in EPO overexpressing mice could be attributed to cardiomyocyte hypertrophy (i.e., CSA ∝ force output) (An et al., 1991). Investigating additional biochemical parameters (e.g., nitric oxide, endothelial or neuronal nitric oxide synthase, TGF-β, cyclic GMP) could clarify the mechanism(s) underlying this work, albeit they remain unclear. Since both cardiomyocytes (Wright et al., 2004) and endothelial cells (Anagnostou et al., 1994; Beleslin-Cokic et al., 2004; Yang et al., 2014) express the Epor, the EPO-mediated contractile effects could be the result of total activation of one, or both, cell receptors (Sterin-Borda et al., 2003). Thus, studies relying on transgenic knockout mice, radiolabeled EPO-receptor binding assays, immunoprecipitation, and western blotting, will ultimately improve our knowledge of the non-canonical EPO receptor activation mechanisms that govern EPO-mediated improvements on cardiac function.
We are the first to report that targeted deletion of Epo from the cardiomyocyte, both in the embryonic (Allwood et al., 2024) and now adult hearts (data presented here), results in a hyper-compensated response by the cardiac endothelial cell. This phenomenon suggests a critical physiological role for cardiac EPO, which must be conserved. The findings presented here were previously unrecognized. Using the same EPOfl/fl mice, Zeigler et al. characterized a conditional EPO-deficient model for chronic kidney disease (CKD) by tamoxifen-induced whole-body Epo knockout in the adult mouse (Zeigler et al., 2010). Considering 80%–95% efficiency of Cre recombination (Sohal et al., 2001; Hsieh et al., 2007; Bersell et al., 2013), their model achieves near ubiquitous Epo knockout from all cell types within the heart (Zeigler et al., 2010). Since the focus of their study was to establish the CKD model and prove EPO-deficient mice still participate in stress-induced erythropoiesis, no further physiological investigations were performed on the heart. Therefore, our study partially resolves this limitation and confirms a critical role for cardioendothelial derived Epo in modulating cardiac morphology, function, and susceptibility to ischemia. However, the complex cellular interplay we observed may not be limited to the cardiomyocytes and endothelial cells. For instance, decades of research indicate renal EPO synthesis is the sum of multiple EPO-producing cells (e.g., cells of the interstitial cortex and outer medulla (Maxwell et al., 1993), proximal convoluted tubule cells (Loya et al., 1994; Haidar et al., 1997), and peritubular fibroblasts (Koury et al., 1988; Lacombe et al., 1988))–therefore, it is plausible the novel regulatory network presented here extends to and includes other cardiac cells (e.g., smooth muscle cells, cardiac progenitors, monocytes/macrophages). While cardiac fibroblast cell isolation revealed no detectable Epo mRNA signal from either group (data not shown), other resident cardiac cells capable of Epo production in the heart could be identified using single cell RNA sequencing, fluorescent in situ hybridization, or Epo reporter mice should be used (Kragesteen et al., 2023). These findings would greatly inform the relevance of non-erythropoietic non-renal EPO, and open new avenues for EPO regulation to be explored.
Cardiac structure and function are governed by complex cellular interplay, that when disturbed, can cause pathology (Yin et al., 2017). Given their proximity, non-myocytes (e.g., fibroblasts, endothelial cells) regulate cardiomyocyte growth and development either through direct cell-to-cell contact or by the release of paracrine factors (Long et al., 1991; Gray et al., 1998; Yin et al., 2017). Cardiomyocytes, the force-producing cells of the heart, are unique from non-myocytes in that they very rarely self-replicate (Long et al., 1991; Li et al., 1996b; Soonpaa et al., 1996; Poolman and Brooks, 1998). Therefore, should cardiomyocyte-derived EPO be critical for global heart function, it is reasonable to speculate there is a feedforward mechanism involving other cell type(s) to compensate for any loss (proposed mechanism presented in Figure 9). Mice null for either Epo or the Epor die due to vascular abnormalities and anemia (Wu et al., 1999; Kertesz et al., 2004; Allwood et al., 2024). The data presented here emphasize that cardiac Epo production is also imperative for homeostatic function in the adult heart and is thus regulated by complex paracrine mechanisms that allow for redundancy to ensure adequate paracrine EPO is ever-present. Interestingly, this is not the case for VEGF-A. Upon cardiomyocyte or endothelial cell specific Vegfa deletion, no other VEGF isoforms or neighbouring cell types will increase their expression to compensate (Giordano et al., 2001). In these models, a lack of paracrine support leads to profound detriments in vascular homeostasis and cardiac function. The presence of redundancy for one system (i.e., EPO) and not the other (i.e., VEGF), is perhaps surprising considering the parallels between these growth factors and their upstream regulation by the HIFs, but could be explained, at least in part, by the isoform and/or transcription factors involved.
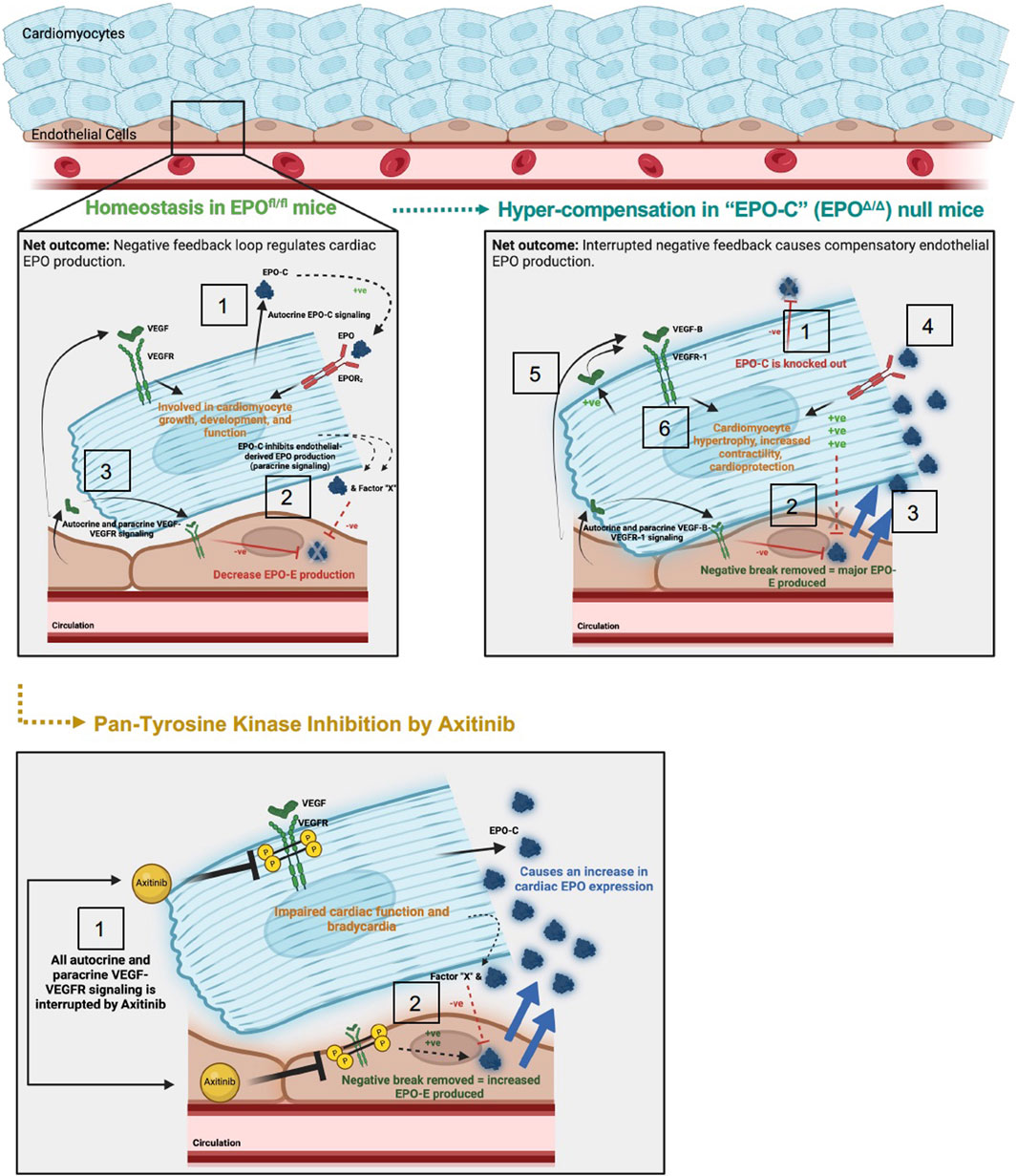
Figure 9. Schematic of proposed mechanism: cardiac structure and function rely on the homeostatic interaction between EPO-EPOR and VEGF-VEGFR signaling. Top left panel: (1) In adult wildtype mice (EPOfl/fl), the cardiomyocyte produces low levels of Epo at baseline (Figure 1). (2) Cardiomyocyte derived EPO (“EPO-C″) represses endothelial cell EPO (“EPO-E″) production in a paracrine fashion. (3) VEGF elicits paracrine stimulation of VEGFR on neighboring endothelial cells and cardiomyocytes, and activation of this pathway inhibits EPO-E production. The net outcome of these stimuli is reciprocal endothelial repression of EPO production. Top right panel: (1) In EPO-C null mice (EPOΔ/Δ) EPO is successfully knocked out of the cardiomyocyte. (2) The lack of EPO produced by the cardiomyocytes releases the inhibition of EPO-EPOR by the endothelial cell in a paracrine fashion. (3) This leads to overproduction of EPO by the endothelial cell. (4) The overproduction of EPO positively feeds back and binds the EPOR2 located on the cardiomyocyte (Wright et al., 2004). EPO binding to the EPOR2 on the cardiomyocyte may induce cardiomyocyte-production of VEGF. (5) Increased VEGF-B-VEGFR-1 signaling, along with (6) additional unknown intermediate factors, increase myocyte hypertrophy, contractile function, and cardioprotection (Karpanen et al., 2008b; Zentilin et al., 2010; Kivelä et al., 2014; Lal et al., 2017). Bottom panel: (1) Axitinib, a pan-tyrosine kinase inhibitor, prevents downstream VEGF-VEGFR signaling. (2) VEGF-induced inhibition of EPO is interrupted, resulting in an increase in EPO-E and EPO-C production. The net outcome of VEGF-VEGFR inhibition is impaired cardiac function and bradycardia, despite increased EPO production. The mechanisms underlying these physiological consequences require future work.
Dynamic cellular crosstalk between cardiomyocytes and endothelial cells involving EPO and VEGF signaling is likely as both the ligands and receptors have been localized to each (i.e., on cardiomyocytes: Epor (Wright et al., 2004), Vegfr (Zentilin et al., 2010), Epo (Mengozzi et al., 2006; El Hasnaoui-Saadani et al., 2013), and Vegf (Giordano et al., 2001); on endothelial cells: Epor2 (Anagnostou et al., 1994; Beleslin-Cokic et al., 2004; Yang et al., 2014), Vegfr (Lee et al., 2007), Epo (Mengozzi et al., 2006; Allwood et al., 2024), and Vegf (Lee et al., 2007)). On a whole-heart level, endogenous inactivation of the EPO-EPOR system reduces cardiomyocyte Vegf production and angiogenesis resulting in worse cardiac function in a model of pressure-overload (Asaumi et al., 2007). When cardiomyocyte derived Vegfa is knocked out during development, the adult heart demonstrates hypovascularity, cardiac dysfunction, and hypoxia induced Epo expression (Lee et al., 2007). Therefore, crosstalk amongst the EPO and VEGF axis appears to modulate morphology, contraction, and protection within the heart. At the molecular level, we observed elevated Epo RNA in EPOΔ/Δ mice alongside a significant increase in Vegfb gene expression (the hypoxia-independent form of VEGF predominantly found in the heart) and its receptor, Vegfr1. When VEGF-VEGFR signaling was neutralized in axitinib-treated mice, cardiac EPO expression (Figure 8B) was upregulated–a schematic representation of this new conceptual mechanism remains to be further defined (Figure 9). The uniform expression pattern of Epo, Vegfb, Vegfr1 in the EPOΔ/Δ heart may represent an intricate modulatory mechanism wherein VEGF-B indirectly drives cardiomyocyte hypertrophy, leading to better cardiac function, and lower susceptibility to ischemia-reperfusion injury (Shiojima et al., 2005; Tirziu et al., 2007; Karpanen et al., 2008a; Karpanen et al., 2008b; Zentilin et al., 2010; Kivelä et al., 2014; Lal et al., 2017; Lal et al., 2018). All in all, within the heart, the EPO-EPOR/VEGF-VEGFR axis exists to support cardiac homeostasis.
Hypoxia induced Epo synthesis in the kidney, liver, and brain is modulated by HIF-2α (Warnecke et al., 2004; Gruber et al., 2007; Rankin et al., 2007; Yamashita et al., 2008; Wang et al., 2015; Urrutia et al., 2016). However, the transcription factor responsible for cardiac Epo expression under hypoxic conditions (Chu et al., 2007; El Hasnaoui-Saadani et al., 2013; Deji et al., 2015) has received less attention. In one study, acute Vhl inactivation induces cardiac Epo production in a HIF1α-dependent manner. However, HIF-2α was not investigated under these conditions, therefore we cannot rule out its involvement in mediating the cardiac EPO response. Here, we provide evidence of the hypoxia mimetic, CoCl2, stabilized HIF-2α in the heart (Figure 6), which induced downstream local Epo production (data not shown). Yasuda et al., report 17β-estradiol- (E2) induced Epo production in the uterus is not regulated by hypoxia (Yasuda et al., 1998). In another study, potent VEGF inhibition increases hepatic Epo synthesis and modulates erythropoiesis in a HIF1α-independent manner (Tam et al., 2006). Our data show cardiac Epo overexpression in the EPOΔ/Δ heart was independent of HIF-2α stabilization (Figure 6) and investigations on HIF-1α target genes further suggest that Epo regulation was not related to a hypoxic stimulus (Figure 7). To our knowledge, this is the first report of cardiac Epo overexpression that occurs independent of HIF-1α and HIF-2α stabilization under normoxic conditions. The identity of the transcription factor(s)/repressors responsible for initiating this phenomenon remain to be solved. Using siRNA knock-down, chromatin immunoprecipitation assays that reveal DNA-protein interactions at the Epo enhancer binding sites, or transgenic knockout models, the mechanisms regulating hypoxia-independent cardiac Epo production may become apparent.
This study had limitations. We observed no gross organ hypertrophy, though cellular cross-sectional area and posterior wall thickness were increased in alignment with improvements in cardiac function. Considering cross-sectional area of a muscle is proportional to its force output, this seems reasonable. To maintain the same organ weight and size, a cardiomyocyte with increased cross-sectional area would either need to 1) shorten lengthwise or 2) undergo apoptosis. To address the former, in our previous study, we confirmed cardiogenesis is modulated by the presence of cardiac Epo–therefore, how long, and wide a cardiomyocyte grows is dependent upon endothelial cell (Colliva et al., 2020) and EPO signaling (Colliva et al., 2020; Allwood et al., 2024). Second, apoptosis is a hallmark feature of the transition from compensatory hypertrophy to heart failure (for review (Van Empel et al., 2005)). Accordingly, reduced contractile function of the heart and increased caspase 8 expression (Kruidering and Evan, 2000) would accompany cardiomyocyte apoptosis, which was not the case for the EPOΔ/Δ mice as confirmed by no difference in the amount of apoptotic cell death by TUNEL assay (Supplementary Figure S5A). Next, while we did not reconcile the transcription factor(s) responsible for regulating hypoxia-independent Epo overexpression, we established it was not via the canonical axis involving HIF-1 or HIF-2. Using next-generation chromatin immunoprecipitation assays/sequencing, the protein-DNA interactions that mediate this response might be uncovered. Further, by modifying the Epo gene construct, LacZ-tagged Epo reporter mice could reveal novel enhancer/promoter regions responsible for cardiac Epo expression. (i.e., 5′ kidney inducibility element, 3’ liver inducibility element).
We have uncovered a novel paradigm wherein adult cardiomyocyte Epo deletion induced endothelial cell derived Epo and subsequent Vegfb expression, which together appeared to stimulate cardiomyocyte hypertrophy in a feedforward manner. Along with more efficient cardiac force generation, EPOΔ/Δ mice demonstrated superior resistance to ischemic-reperfusion injury. Accordingly, there was a complex cellular interplay involving the EPO-EPOR and VEGF-VEGFR transduction pathways, which ultimately modulated cardiac structure and function, though future work is required to fully elucidate the mechanisms involved. Together in the heart, these pathways act in concert.
The raw data supporting the conclusion of this article will be made available by the authors, without undue reservation.
The animal study was approved by the Animal Care Committee at the University of Guelph and all experiments were carried out in accordance with the guidelines from the Canadian Council on Animal Care. The study was conducted in accordance with the local legislation and institutional requirements.
JM: Conceptualization, Data curation, Formal Analysis, Investigation, Writing–original draft, Writing–review and editing. RA: Conceptualization, Data curation, Formal Analysis, Investigation, Writing–review and editing. BE: Conceptualization, Formal Analysis, Writing–review and editing. MA: Conceptualization, Writing–review and editing. KC: Formal Analysis, Writing–review and editing. SA-S: Data curation, Formal Analysis, Writing–review and editing. AA: Data curation, Formal Analysis, Writing–review and editing. KA: Writing–review and editing. GH: Writing–review and editing. KB: Conceptualization, Funding acquisition, Resources, Supervision, Writing–review and editing. JAS: Conceptualization, Funding acquisition, Resources, Supervision, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. This work was supported by the Canadian Institutes of Health Research (CIHR) grants (JAS and KB); National Sciences Engineering Research Council (NSERC) grants (JAS and KB); with NSERC Alexander Graham Bell Canada Graduate Scholarship—Doctoral (JM); Killam Foundation Post-Doctoral Fellowship (BE); Ontario Graduate Scholarships (RA). We acknowledge the philanthropic support for cardiovascular research from David Southen and Betty and Jack Southen of London, ON, Canada, to the laboratory of JAS. The funding sources had no influence on study design, sample collection, data analysis and interpretation, or preparation of this manuscript.
Figure 9 was created using BioRender.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author(s) declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
The Supplementary Material for this article can be found online at: https://www.frontiersin.org/articles/10.3389/fphys.2024.1397049/full#supplementary-material
Allwood M. A., Edgett B. A., Platt M. J., Marrow J. P., Coyle-Asbil B., Holjak E. J. B., et al. (2024). Novel roles of cardiac-derived erythropoietin in cardiac development and function. J. Mol. Cell. Cardiol. 188, 90–104. doi:10.1016/j.yjmcc.2024.01.006
An K. N., Linscheid R. L., Brand P. W. (1991). Correlation of physiological cross-sectional areas of muscle and tendon. J. Hand Surg. Br. 16, 66–67. doi:10.1016/0266-7681(91)90130-g
Anagnostou A., Liu Z., Steiner M., Chin K., Lee E. S., Kessimian N., et al. (1994). Erythropoietin receptor mRNA expression in human endothelial cells. Proc. Natl. Acad. Sci. U. S. A. 91, 3974–3978. doi:10.1073/pnas.91.9.3974
Asaumi Y., Kagaya Y., Takeda M., Yamaguchi N., Tada H., Ito K., et al. (2007). Protective role of endogenous erythropoietin system in nonhematopoietic cells against pressure overload-induced left ventricular dysfunction in mice. Circulation 115, 2022–2032. doi:10.1161/CIRCULATIONAHA.106.659037
Beleslin-Cokic B. B., Cokic V. P., Yu X., Weksler B. B., Schechter A. N., Noguchi C. T. (2004). Erythropoietin and hypoxia stimulate erythropoietin receptor and nitric oxide production by endothelial cells. Blood 104, 2073–2080. doi:10.1182/blood-2004-02-0744
Bernaudin M., Marti H. H., Roussel S., Divoux D., Nouvelot A., MacKenzie E. T., et al. (1999). A potential role for erythropoietin in focal permanent cerebral ischemia in mice. J. Cereb. Blood Flow Metabolism 19, 643–651. doi:10.1097/00004647-199906000-00007
Bersell K., Choudhury S., Mollova M., Polizzotti B. D., Ganapathy B., Walsh S., et al. (2013). Moderate and high amounts of tamoxifen in αmHC-MerCreMer mice induce a DNA damage response, leading to heart failure and death. DMM Dis. Models Mech. 6, 1459–1469. doi:10.1242/dmm.010447
Bonnas C., Wüstefeld L., Winkler D., Kronstein-Wiedemann R., Dere E., Specht K., et al. (2017). EV-3, an endogenous human erythropoietin isoform with distinct functional relevance. Sci. Rep. 7, 3684. doi:10.1038/s41598-017-03167-0
Bunn H. F., Gu J., Huang L. E., Park J. W., Zhu H. E. (1998). Erythropoietin: a model system for studying oxygen-dependent gene regulation. J. Exp. Biol. 201, 1197–1201. doi:10.1242/jeb.201.8.1197
Cai Z., Manalo D. J., Wei G., Rodriguez E. R., Fox-Talbot K., Lu H., et al. (2003). Hearts from rodents exposed to intermittent hypoxia or erythropoietin are protected against ischemia-reperfusion injury. Circulation 108, 79–85. doi:10.1161/01.CIR.0000078635.89229.8A
Calvillo L., Latini R., Kajstura J., Leri A., Anversa P., Ghezzi P., et al. (2003). Recombinant human erythropoietin protects the myocardium from ischemia-reperfusion injury and promotes beneficial remodeling. Proc. Natl. Acad. Sci. U. S. A. 100, 4802–4806. doi:10.1073/pnas.0630444100
Cheetham J. C., Smith D. M., Aoki K. H., Stevenson J. L., Hoeffel T. J., Syed R. S., et al. (1998). NMR structure of human erythropoietin and a comparison with its receptor bound conformation. Nat. Struct. Biol. 5, 861–866. doi:10.1038/2302
Chu C. Y., Cheng C. H., Chen G. D., Chen Y. C., Hung C. C., Huang K. Y., et al. (2007). The zebrafish erythropoietin: functional identification and biochemical characterization. FEBS Lett. 581, 4265–4271. doi:10.1016/j.febslet.2007.07.073
Colliva A., Braga L., Giacca M., Zacchigna S. (2020). Endothelial cell–cardiomyocyte crosstalk in heart development and disease. J. Physiol. 598, 2923–2939. doi:10.1113/JP276758
De Franciscis P., De Bella G., Cifaldi S. (1979). Spleen as a production site for erythropoietin. Science 150, 1831–1833. doi:10.1126/science.150.3705.1831
Deji B. Z., Shang P., Danzeng W. J., Zhang H., Qiangba Y. Z. (2015). Expression and hypoxia adaptation analysis of the EPO gene in different tissues of plateau Tibetan pigs. Genet. Mol. Res. 14, 1700–1706. doi:10.4238/2015.March.6.16
Dobbin S. J. H., Petrie M. C., Myles R. C., Touyz R. M., Lang N. N. (2021). Cardiotoxic effects of angiogenesis inhibitors. Clin. Sci. (Lond) 135, 71–100. doi:10.1042/CS20200305
Dube S., Fishery J. W., Powell J. S. (1988). Glycosylation at specific sites of erythropoietin is essential for biosynthesis, secretion, and biological function. J. Biol. Chem. 263, 17516–17521. doi:10.1016/s0021-9258(19)77865-x
El Hasnaoui-Saadani R., Marchant D., Pichon A., Escoubet B., Pezet M., Hilfiker-Kleiner D., et al. (2013). Epo deficiency alters cardiac adaptation to chronic hypoxia. Respir. Physiol. Neurobiol. 186, 146–154. doi:10.1016/j.resp.2013.01.003
Firth J. D., Ebert B. L., Ratcliffe P. J. (1995). Hypoxic regulation of lactate dehydrogenase A. Interaction between hypoxia-inducible factor 1 and cAMP response elements. J. Biol. Chem. 270, 21021–21027. doi:10.1074/jbc.270.36.21021
Force T., Kolaja K. L. (2011). Cardiotoxicity of kinase inhibitors: the prediction and translation of preclinical models to clinical outcomes. Nat. Rev. Drug Discov. 10, 111–126. doi:10.1038/nrd3252
Forsythe J. A., Jiang B. H., Iyer N. V., Agani F., Leung S. W., Koos R. D., et al. (1996). Activation of vascular endothelial growth factor gene transcription by hypoxia-inducible factor 1. Mol. Cell. Biol. 16, 4604–4613. doi:10.1128/mcb.16.9.4604
Galson D. L., Tsuchiya T., Tendler D. S., Huang L. E., Ren Y., Ogura T., et al. (1995). The orphan receptor hepatic nuclear factor 4 functions as a transcriptional activator for tissue-specific and hypoxia-specific erythropoietin gene expression and is antagonized by EAR3/COUP-TF1. Mol. Cell. Biol. 15, 2135–2144. doi:10.1128/mcb.15.4.2135
Ghosh M. C., Zhang D. L., Ollivierre W. H., Noguchi A., Springer D. A., Linehan W. M., et al. (2021). Therapeutic inhibition of HIF-2α reverses polycythemia and pulmonary hypertension in murine models of human diseases. Blood 137, 2509–2519. doi:10.1182/blood.2020009138
Giordano F. J., Gerber H. P., Williams S. P., VanBruggen N., Bunting S., Ruiz-Lozano P., et al. (2001). A cardiac myocyte vascular endothelial growth factor paracrine pathway is required to maintain cardiac function. Proc. Natl. Acad. Sci. U. S. A. 98, 5780–5785. doi:10.1073/pnas.091415198
Gray M. O., Long C. S., Kalinyak J. E., Li H. T., Karliner J. S. (1998). Angiotensin II stimulates cardiac myocyte hypertrophy via paracrine release of TGF-beta 1 and endothelin-1 from fibroblasts. Cardiovasc Res. 40, 352–363. doi:10.1016/s0008-6363(98)00121-7
Gruber M., Hu C. J., Johnson R. S., Brown E. J., Keith B., Simon M. C. (2007). Acute postnatal ablation of Hif-2alpha results in anemia. Proc. Natl. Acad. Sci. U. S. A. 104, 2301–2306. doi:10.1073/pnas.0608382104
Haidar M. A., Loya F., Yang Y., Lin H., Glassman A., Goldwasser E., et al. (1997). Electron microscopic localization of LacZ expression in the proximal convoluted tubular cells of the kidney in transgenic mice carrying chimeric erythropoietin/lacZ gene constructs. J. Struct. Biol. 118, 220–225. doi:10.1006/jsbi.1997.3842
Haidar M. A., Loya F., Yang Y., Lin H., Glassman A., Keating M. J., et al. (1996). Differential expression of lacZ in the liver and kidney of transgenic mice carrying chimeric lacZ-erythropoietin gene constructs with or without its 1.2 kb 3’-flanking sequence. Nucleic Acids Res. 24, 3621–3628. doi:10.1093/nar/24.18.3621
Hefer D., Yi T., Selby D. E., Fishbaugher D. E., Tremble S. M., Begin K. J., et al. (2012). Erythropoietin induces positive inotropic and lusitropic effects in murine and human myocardium. J. Mol. Cell. Cardiol. 52, 256–263. doi:10.1016/j.yjmcc.2011.10.005
Hsieh P. C. H., Segers V. F. M., Davis M. E., MacGillivray C., Gannon J., Molkentin J. D., et al. (2007). Evidence from a genetic fate-mapping study that stem cells refresh adult mammalian cardiomyocytes after injury. Nat. Med. 13, 970–974. doi:10.1038/nm1618
Huang L. E., Ho V., Arany Z., Krainc D., Galson D., Tendler D., et al. (1997). Erythropoietin gene regulation depends on heme-dependent oxygen sensing and assembly of interacting transcription factors. Kidney Int. 51, 548–552. doi:10.1038/ki.1997.76
Iyer N. V., Kotch L. E., Agani F., Leung S. W., Laughner E., Wenger R. H., et al. (1998). Cellular and developmental control of O2 homeostasis by hypoxia-inducible factor 1 alpha. Genes. Dev. 12, 149–162. doi:10.1101/gad.12.2.149
Jelkmann W. (2007). Control of erythropoietin gene expression and its use in medicine. Methods Enzymol. 435, 179–197. doi:10.1016/S0076-6879(07)35010-6
Johnson A. C., Matias M., Boyle H., Escudier B., Molinier A., Laguerre B., et al. (2017). Haemoglobin level increase as an efficacy biomarker during axitinib treatment for metastatic renal cell carcinoma: a retrospective study. BMC Cancer 17, 355. doi:10.1186/s12885-017-3312-7
Karpanen T., Bry M., Ollila H. M., Seppänen-Laakso T., Liimatta E., Leskinen H., et al. (2008a). Overexpression of vascular endothelial growth factor-B in mouse heart alters cardiac lipid metabolism and induces myocardial hypertrophy. Circ. Res. 103, 1018–1026. doi:10.1161/CIRCRESAHA.108.178459
Karpanen T., Bry M., Ollila H. M., Seppänen-Laakso T., Liimatta E., Leskinen H., et al. (2008b). Overexpression of vascular endothelial growth factor-B in mouse heart alters cardiac lipid metabolism and induces myocardial hypertrophy. Circ. Res. 103, 1018–1026. doi:10.1161/CIRCRESAHA.108.178459
Kaygisiz Z., Erkasap N., Yazihan N., Sayar K., Ataoglu H., Uyar R., et al. (2006). Erythropoietin changes contractility, cAMP, and nitrite levels of isolated rat hearts. J. Physiological Sci. 56, 247–251. doi:10.2170/physiolsci.RP004806
Kertesz N., Wu J., Chen T. H. P., Sucov H. M., Wu H. (2004). The role of erythropoietin in regulating angiogenesis. Dev. Biol. 276, 101–110. doi:10.1016/j.ydbio.2004.08.025
Kivelä R., Bry M., Robciuc M. R., Räsänen M., Taavitsainen M., Silvola J. M. U., et al. (2014). VEGF-B-induced vascular growth leads to metabolic reprogramming and ischemia resistance in the heart. EMBO Mol. Med. 6, 307–321. doi:10.1002/emmm.201303147
Kloth J. S. L., Pagani A., Verboom M. C., Malovini A., Napolitano C., Kruit W. H. J., et al. (2015). Incidence and relevance of QTc-interval prolongation caused by tyrosine kinase inhibitors. Br. J. Cancer 112, 1011–1016. doi:10.1038/bjc.2015.82
Koury S., Bondurant M., Koury M. (1988). Localization of erythropoietin synthesizing cells in murine kidneys by in situ hybridization. Blood 71, 524–527. doi:10.1182/blood.v71.2.524.524
Kragesteen B. K., Giladi A., David E., Halevi S., Geirsdóttir L., Lempke O. M., et al. (2023). The transcriptional and regulatory identity of erythropoietin producing cells. Nat. Med. 29, 1191–1200. doi:10.1038/s41591-023-02314-7
Kranias E. G., Garvey J. L., Srivastava R. D., Solaro R. J. (1985). Phosphorylation and functional modifications of sarcoplasmic reticulum and myofibrils in isolated rabbit hearts stimulated with isoprenaline. Biochem. J. 226, 113–121. doi:10.1042/bj2260113
Kruidering M., Evan G. I. (2000). Caspase-8 in apoptosis: the beginning of “the end”. IUBMB Life 50, 85–90. doi:10.1080/713803693
Lacombe C., Da Silva J. L., Bruneval P., Fournier J. G., Wendling F., Casadevall N., et al. (1988). Peritubular cells are the site of erythropoietin synthesis in the murine hypoxic kidney. J. Clin. Investigation 81, 620–623. doi:10.1172/JCI113363
Lal N., Chiu A. P. L., Wang F., Zhang D., Jia J., Wan A., et al. (2017). Loss of VEGFB and its signaling in the diabetic heart is associated with increased cell death signaling. Am. J. Physiol. Heart Circ. Physiol. 312, H1163-H1175–H1175. doi:10.1152/ajpheart.00659.2016
Lal N., Puri K., Rodrigues B. (2018). Vascular endothelial growth factor B and its signaling. Front. Cardiovasc Med. 5, 39. doi:10.3389/fcvm.2018.00039
Lasne F., Martin L., Crepin N., De Ceaurriz J. (2002). Detection of isoelectric profiles of erythropoietin in urine: differentiation of natural and administered recombinant hormones. Anal. Biochem. 311, 119–126. doi:10.1016/s0003-2697(02)00407-4
Lee P. J., Jiang B. H., Chin B. Y., Iyer N. V., Alam J., Semenza G. L., et al. (1997). Hypoxia-inducible factor-1 mediates transcriptional activation of the heme oxygenase-1 gene in response to hypoxia. J. Biol. Chem. 272, 5375–5381. doi:10.1074/jbc.272.9.5375
Lee S., Chen T. T., Barber C. L., Jordan M. C., Murdock J., Desai S., et al. (2007). Autocrine VEGF signaling is required for vascular homeostasis. Cell. 130, 691–703. doi:10.1016/j.cell.2007.06.054
Li F., Wang X., Capasso J. M., Gerdes A. M. (1996b). Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J. Mol. Cell. Cardiol. 28, 1737–1746. doi:10.1006/jmcc.1996.0163
Li H., Ko H. P., Whitlock J. P. (1996a). Induction of phosphoglycerate kinase 1 gene expression by hypoxia: ROLES of arnt and HIF1α. J. Biol. Chem. 271, 21262–21267. doi:10.1074/jbc.271.35.21262
Long C. S., Henrich C. J., Simpson P. C. (1991). A growth factor for cardiac myocytes is produced by cardiac nonmyocytes. Cell. Regul. 2, 1081–1095. doi:10.1091/mbc.2.12.1081
Loya F., Yang Y., Lin H., Goldwasser E., Albitar M. (1994). Transgenic mice carrying the erythropoietin gene promoter linked to lacZ express the reporter in proximal convoluted tubule cells after hypoxia. Blood 84, 1831–1836. doi:10.1182/blood.v84.6.1831.1831
Lu J., Yao Y. y., Dai Q. m., Ma G. s., Zhang S. f., Cao L., et al. (2012). Erythropoietin attenuates cardiac dysfunction by increasing myocardial angiogenesis and inhibiting interstitial fibrosis in diabetic rats. Cardiovasc Diabetol. 11, 105. doi:10.1186/1475-2840-11-105
Ma J., Waxman D. J. (2009). Dominant effect of antiangiogenesis in combination therapy involving cyclophosphamide and axitinib. Clin. Cancer Res. 15, 578–588. doi:10.1158/1078-0432.CCR-08-1174
Magnanti M., Gandini O., Giuliani L., Gazzaniga P., Marti H. H., Gradilone A., et al. (2001). Erythropoietin expression in primary rat Sertoli and peritubular myoid cells. Blood 98, 2872–2874. doi:10.1182/blood.v98.9.2872
Masuda S., Kobayashi T., Chikuma M., Nagao M., Sasaki R. (2000). The oviduct produces erythropoietin in an estrogen- and oxygen-dependent manner. Am. J. Physiol. Endocrinol. Metab. 278, E1038–E1044. doi:10.1152/ajpendo.2000.278.6.E1038
Maxwell P. H., Osmond M. K., Pugh C. W., Heryet A., Nicholls L. G., Tan C. C., et al. (1993). Identification of the renal erythropoietin-producing cells using transgenic mice. Kidney Int. 44, 1149–1162. doi:10.1038/ki.1993.362
Mengozzi M., Latini R., Salio M., Sfacteria A., Piedimonte G., Gerwien J. G., et al. (2006). Increased erythropoietin production after myocardial infarction in mice. Heart 92, 838–839. doi:10.1136/hrt.2005.064105
Miró-Murillo M., Elorza A., Soro-Arnáiz I., Albacete-Albacete L., Ordoñez A., Balsa E., et al. (2011). Acute Vhl gene inactivation induces cardiac HIF-dependent erythropoietin gene expression. PLoS One 6, 22589. doi:10.1371/journal.pone.0022589
Mole D. R., Blancher C., Copley R. R., Pollard P. J., Gleadle J. M., Ragoussis J., et al. (2009). Genome-wide association of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha DNA binding with expression profiling of hypoxia-inducible transcripts. J. Biol. Chem. 284, 16767–16775. doi:10.1074/jbc.M901790200
Muñoz-Sánchez J., Chánez-Cárdenas M. E. (2019). The use of cobalt chloride as a chemical hypoxia model. J. Appl. Toxicol. 39, 556–570. doi:10.1002/jat.3749
Nag A. (1980). Study of non-muscle cells of the adult mammalian heart: a fine structural analysis and distribution. Cytobios 28, 41–61.
Ogilvie L. M., Edgett B. A., Huber J. S., Platt M. J., Eberl H. J., Lutchmedial S., et al. (2020). Hemodynamic assessment of diastolic function for experimental models. Am. J. Physiol. Heart Circ. Physiol. 318, H1139-H1158–H1158. doi:10.1152/ajpheart.00705.2019
Oztas Ý. M., Meric M., Beyaz M. O., Onalan M. A., Sonmez K., Oter K., et al. (2020). Cardioprotective effects of erythropoietin in diabetic rats determined by CD34 and vascular endothelial growth factor levels. Arch. Med. Sci. Atheroscler. Dis. 5, e1–e12. doi:10.5114/amsad.2020.92346
Packer M. (2020). Mutual antagonism of hypoxia-inducible factor isoforms in cardiac, vascular, and renal disorders. JACC Basic Transl. Sci. 5, 961–968. doi:10.1016/j.jacbts.2020.05.006
Pandya K., Kim H. S., Smithies O. (2006). Fibrosis, not cell size, delineates beta-myosin heavy chain reexpression during cardiac hypertrophy and normal aging in vivo. Proc. Natl. Acad. Sci. U. S. A. 103, 16864–16869. doi:10.1073/pnas.0607700103
Parsa C. J., Matsumoto A., Kim J., Riel R. U., Pascal L. S., Walton G. B., et al. (2003). A novel protective effect of erythropoietin in the infarcted heart. J. Clin. Investigation 112, 999–1007. doi:10.1172/JCI18200
Pinto A. R., Ilinykh A., Ivey M. J., Kuwabara J. T., D'Antoni M. L., Debuque R., et al. (2016). Revisiting cardiac cellular composition. Circ. Res. 118, 400–409. doi:10.1161/CIRCRESAHA.115.307778
Piuhola J., Kerkelä R., Keenan J. I., Hampton M. B., Richards A. M., Pemberton C. J. (2008). Direct cardiac actions of erythropoietin (EPO): effects on cardiac contractility, BNP secretion and ischaemia/reperfusion injury. Clin. Sci. 114, 293–304. doi:10.1042/CS20070229
Platt M. J., Huber J. S., Brunt K. R., Simpson J. A. (2017). Pulmonary flow as an improved method for determining cardiac output in mice after myocardial infarction. J. Am. Soc. Echocardiogr. 30, 612–623. doi:10.1016/j.echo.2017.02.008
Ponce L. L., Navarro J. C., Ahmed O., Robertson C. S. (2013). Erythropoietin neuroprotection with traumatic brain injury. Pathophysiology 20, 31–38. doi:10.1016/j.pathophys.2012.02.005
Poolman R. A., Brooks G. (1998). Expressions and activities of cell cycle regulatory molecules during the transition from myocyte hyperplasia to hypertrophy. J. Mol. Cell. Cardiol. 30, 2121–2135. doi:10.1006/jmcc.1998.0808
Rahbek-Nielsen H., Roepstorff P., Reischl H., Wozny M., Koll H., Haselbeck A. (1997). Glycopeptide profiling of human urinary erythropoietin by matrix-assisted laser desorption/ionization mass spectrometry. J. Mass Spectrom. 32, 948–958. doi:10.1002/(SICI)1096-9888(199709)32:9<948::AID-JMS551>3.0.CO;2-C
Rankin E. B., Biju M. P., Liu Q., Unger T. L., Rha J., Johnson R. S., et al. (2007). Hypoxia-inducible factor–2 (HIF-2) regulates hepatic erythropoietin in vivo. J. Clin. Investig. 117, 1068–1077. doi:10.1172/JCI30117
Ribatti D., Presta M., Vacca A., Ria R., Giuliani R., Dell'Era P., et al. (1999). Human erythropoietin induces a pro-angiogenic phenotype in cultured endothelial cells and stimulates neovascularization in vivo. Blood 93, 2627–2636. doi:10.1182/blood.v93.8.2627.408k21_2627_2636
Rush R. S., Derby P. L., Smith D. M., Merry C., Rogers G., Rohde M. F., et al. (1995). Microheterogeneity of erythropoietin carbohydrate structure. Anal. Chem. 67, 1442–1452. doi:10.1021/ac00104a022
Schoenfeld J. R., Vasser M., Jhurani P., Ng P., Hunter J. J., Ross J., et al. (1998). Distinct molecular phenotypes in murine cardiac muscle development, growth, and hypertrophy. J. Mol. Cell. Cardiol. 30, 2269–2280. doi:10.1006/jmcc.1998.0787
Semenzas G. L., Roth P. H., Fang H.-M., Wang G. L. (1994). Transcriptional regulation of genes encoding glycolytic enzymes by hypoxia-inducible factor 1. J. Biol. Chem. 269, 23757–23763. doi:10.1016/s0021-9258(17)31580-6
Shiojima I., Sato K., Izumiya Y., Schiekofer S., Ito M., Liao R., et al. (2005). Disruption of coordinated cardiac hypertrophy and angiogenesis contributes to the transition to heart failure. J. Clin. Investig. 115, 2108–2118. doi:10.1172/JCI24682
Shopp G. M., Helson L., Bouchard A., Salvail D., Majeed M. (2022) Liposomes ameliorate crizotinib-and nilotinib-induced inhibition of the cardiac I Kr channel and QTc prolongation.
Simmerman H. K. B., Jones L. R. (1998). Phospholamban: protein structure, mechanism of action, and role in cardiac function. Physiol. Rev. 78, 921–947. doi:10.1152/physrev.1998.78.4.921
Skibeli V., Nissen-Lie G., Torjesen P. (2001). Sugar profiling proves that human serum erythropoietin differs from recombinant human erythropoietin. Blood 98, 3626–3634. doi:10.1182/blood.v98.13.3626
Sohal D. S., Nghiem M., Crackower M. A., Witt S. A., Kimball T. R., Tymitz K. M., et al. (2001). Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circ. Res. 89, 20–25. doi:10.1161/hh1301.092687
Soonpaa M. H., Kim K. K., Pajak L., Franklin M., Field L. J. (1996). Cardiomyocyte DNA synthesis and binucleation during murine development. Am. J. Physiol. 271, H2183–H2189. doi:10.1152/ajpheart.1996.271.5.H2183
Sterin-Borda L., Barceló A. C., Bozzini C. E. (2003). Erythropoietin improves cardiac contractility in post-hypoxic mice. Br. J. Haematol. 121, 180–186. doi:10.1046/j.1365-2141.2003.04239.x
Sun W., Kosyna F. K., Jelkmann W., Depping R. (2015). Prolyl-4-Hydroxylase 2 potentially contributes to hepatocellular carcinoma-associated erythrocytosis by maintaining hepatocyte nuclear factor-4α expression. Cell. Physiology Biochem. 37, 2257–2264. doi:10.1159/000438581
Tam B. Y. Y., Wei K., Rudge J. S., Hoffman J., Holash J., Park S. k., et al. (2006). VEGF modulates erythropoiesis through regulation of adult hepatic erythropoietin synthesis. Nat. Med. 12, 793–800. doi:10.1038/nm1428
Tirziu D., Chorianopoulos E., Moodie K. L., Palac R. T., Zhuang Z. W., Tjwa M., et al. (2007). Myocardial hypertrophy in the absence of external stimuli is induced by angiogenesis in mice. J. Clin. Investig. 117, 3188–3197. doi:10.1172/JCI32024
Tyler P. D., Fcshp T. (2012). Axitinib: newly approved for renal cell carcinoma. J. Adv. Pract. Oncol. 3, 333–335. doi:10.6004/jadpro.2012.3.5.7
Urrutia A. A., Afzal A., Nelson J., Davidoff O., Gross K. W., Haase V. H. (2016). Prolyl-4-hydroxylase 2 and 3 coregulate murine erythropoietin in brain pericytes. Blood 128, 2550–2560. doi:10.1182/blood-2016-05-713545
Valdivia H. H., Kaplan J. H., Ellis-Davies G. C. R., Lederer W. J. (1995). Rapid adaptation of cardiac ryanodine receptors: modulation by Mg2+ and phosphorylation. Science 267, 1997–2000. doi:10.1126/science.7701323
Van Empel V. P. M., Bertrand A. T. A., Hofstra L., Crijns H. J., Doevendans P. A., De Windt L. J. (2005). Myocyte apoptosis in heart failure. Cardiovasc Res. 67, 21–29. doi:10.1016/j.cardiores.2005.04.012
Vinhas M., Araújo A. C., Ribeiro S., Rosário L. B., Belo J. A. (2013). Transthoracic echocardiography reference values in juvenile and adult 129/Sv mice. Cardiovasc Ultrasound 11, 12. doi:10.1186/1476-7120-11-12
Wakhloo D., Scharkowski F., Curto Y., Javed Butt U., Bansal V., Steixner-Kumar A. A., et al. (2020). Functional hypoxia drives neuroplasticity and neurogenesis via brain erythropoietin. Nat. Commun. 11, 1313. doi:10.1038/s41467-020-15041-1
Wang G. L., Jiang B. H., Rue E. A., Semenza G. L. (1995). Hypoxia-inducible factor 1 is a basic-helix-loop-helix-PAS heterodimer regulated by cellular O2 tension. Proc. Natl. Acad. Sci. U. S. A. 92, 5510–5514. doi:10.1073/pnas.92.12.5510
Wang J., Hayashi Y., Xu Z., Wu L., Yan X., Kalfa T. A., et al. (2015). Activation of HIF-2a-EPO Axis in kidney or liver is sufficient to drive erythrocytosis in a novel inducible HIF-2a transgenic mouse model. Blood 126, 931. doi:10.1182/blood.v126.23.931.931
Wang L., Teng R., Rogers H., Wu H., Kopp J. B., et al. (2013). PPARa and sirt1 mediate erythropoietin action in increasing metabolic activity and browning of white adipocytes to protect against obesity and metabolic disorders. Diabetes 62, 4122–4131. doi:10.2337/db13-0518
Warnecke C., Zaborowska Z., Kurreck J., Erdmann V. A., Frei U., Wiesener M., et al. (2004). Differentiating the functional role of hypoxia-inducible factor (HIF)-1alpha and HIF-2alpha (EPAS-1) by the use of RNA interference: erythropoietin is a HIF-2alpha target gene in Hep3B and Kelly cells. FASEB J. 18, 1462–1464. doi:10.1096/fj.04-1640fje
Weidemann A., Kerdiles Y. M., Knaup K. X., Rafie C. A., Boutin A. T., Stockmann C., et al. (2009). The glial cell response is an essential component of hypoxia-induced erythropoiesis in mice. J. Clin. Investig. 119, 3373–3383. doi:10.1172/JCI39378
Weikert S., Papac D., Briggs J., Cowfer D., Gawlitzek M., et al. (1999). Engineering Chinese hamster ovary cells to maximize sialic acid content of recombinant glycoproteins. Nat. Biotechnol. 17, 1116–1121. doi:10.1038/15104
Westenbrink B. D., Oeseburg H., Kleijn L., van der Harst P., Belonje A. M. S., Voors A. A., et al. (2008). Erythropoietin stimulates normal endothelial progenitor cell-mediated endothelial turnover, but attributes to neovascularization only in the presence of local ischemia. Cardiovasc Drugs Ther. 22, 265–274. doi:10.1007/s10557-008-6094-y
Wiesener M. S., Jürgensen J. S., Rosenberger C., Scholze C. K., Hörstrup J. H., Warnecke C., et al. (2003). Widespread hypoxia-inducible expression of HIF-2alpha in distinct cell populations of different organs. FASEB J. 17, 271–273. doi:10.1096/fj.02-0445fje
Wright G. L., Hanlon P., Amin K., Steenbergen C., Murphy E., Arcasoy M. O. (2004). Erythropoietin receptor expression in adult rat cardiomyocytes is associated with an acute cardioprotective effect for recombinant erythropoietin during ischemia-reperfusion injury. FASEB J. 18, 1031–1033. doi:10.1096/fj.03-1289fje
Wu H., Lee S. H., Gao J., Liu X., Iruela-Arispe M. L. (1999). Inactivation of erythropoietin leads to defects in cardiac morphogenesis. Development 126, 3597–3605. doi:10.1242/dev.126.16.3597
Yamashita T., Ohneda O., Sakiyama A., Iwata F., Ohneda K., Fujii-Kuriyama Y. (2008). The microenvironment for erythropoiesis is regulated by HIF-2alpha through VCAM-1 in endothelial cells. Blood 112, 1482–1492. doi:10.1182/blood-2007-11-122648
Yang Z., Wang H., Jiang Y., Hartnett M. E. (2014). VEGFA activates erythropoietin receptor and enhances VEGFR2-mediated pathological angiogenesis. Am. J. Pathol. 184, 1230–1239. doi:10.1016/j.ajpath.2013.12.023
Yasuda Y., Masuda S., Chikuma M., Inoue K., Nagao M., Sasaki R. (1998). Estrogen-dependent production of erythropoietin in uterus and its implication in uterine angiogenesis. J. Biol. Chem. 273, 25381–25387. doi:10.1074/jbc.273.39.25381
Yeo E. J., Cho Y. S., Kim M. S., Park J. W. (2008). Contribution of HIF-1alpha or HIF-2alpha to erythropoietin expression: in vivo evidence based on chromatin immunoprecipitation. Ann. Hematol. 87, 11–17. doi:10.1007/s00277-007-0359-6
Yin J. C., Platt M. J., Tian X., Wu X., Backx P. H., Simpson J. A., et al. (2017). Cellular interplay via cytokine hierarchy causes pathological cardiac hypertrophy in RAF1-mutant Noonan syndrome. Nat. Commun. 8, 15518–15611. doi:10.1038/ncomms15518
Zak R. (1973). Cell proliferation during cardiac growth. Am. J. Cardiol. 31, 211–219. doi:10.1016/0002-9149(73)91034-5
Zeigler B. M., Vajdos J., Qin W., Loverro L., Niss K. (2010). A mouse model for an erythropoietin-deficiency anemia. Dis. Model. Mech. 3, 763–772. doi:10.1242/dmm.004788
Zentilin L., Puligadda U., Lionetti V., Zacchigna S., Collesi C., Pattarini L., et al. (2010). Cardiomyocyte VEGFR-1 activation by VEGF-B induces compensatory hypertrophy and preserves cardiac function after myocardial infarction. FASEB J. 24, 1467–1478. doi:10.1096/fj.09-143180
Zhao X., Liu L., Wei X., Luan W., Liu P., et al. (2018). Hypoxia-inducible factor 1-α (HIF-1α) induces apoptosis of human uterosacral ligament fibroblasts through the death receptor and mitochondrial pathways. Med. Sci. Monit. 24, 8722–8733. doi:10.12659/MSM.913384

Keywords: erythropoietin, Cre-Lox, EPAS1 gene, hemodynamics, compensation, vascular endothelial growth factor, erythropoiesis
Citation: Marrow JP, Alshamali R, Edgett BA, Allwood MA, Cochrane KLS, Al-Sabbag S, Ayoub A, Ask K, Hare GMT, Brunt KR and Simpson JA (2024) Cardiomyocyte crosstalk with endothelium modulates cardiac structure, function, and ischemia-reperfusion injury susceptibility through erythropoietin. Front. Physiol. 15:1397049. doi: 10.3389/fphys.2024.1397049
Received: 06 March 2024; Accepted: 03 June 2024;
Published: 01 July 2024.
Edited by:
Prabhu Mathiyalagan, Benthos Prime Central, United StatesReviewed by:
Francisco O. Silva, University of Texas Southwestern Medical Center, United StatesCopyright © 2024 Marrow, Alshamali, Edgett, Allwood, Cochrane, Al-Sabbag, Ayoub, Ask, Hare, Brunt and Simpson. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jeremy A. Simpson, amVyZW15c0B1b2d1ZWxwaC5jYQ==
†These authors have contributed equally to this work and share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.