
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 02 July 2024
Sec. Mitochondrial Research
Volume 15 - 2024 | https://doi.org/10.3389/fphys.2024.1384966
This article is part of the Research TopicThe Role of Mitochondrial Genome in Healthy and Pathological AgingView all articles
 Indumathi Somasundaram1*Samatha M. Jain2Marcel Blot-Chabaud3
Indumathi Somasundaram1*Samatha M. Jain2Marcel Blot-Chabaud3 Surajit Pathak2*
Surajit Pathak2* Antara Banerjee2
Antara Banerjee2 Sonali Rawat4
Sonali Rawat4 Neeta Raj Sharma5
Neeta Raj Sharma5 Asim K. Duttaroy6*
Asim K. Duttaroy6*Aging is a complex process that features a functional decline in many organelles. Various factors influence the aging process, such as chromosomal abnormalities, epigenetic changes, telomere shortening, oxidative stress, and mitochondrial dysfunction. Mitochondrial dysfunction significantly impacts aging because mitochondria regulate cellular energy, oxidative balance, and calcium levels. Mitochondrial integrity is maintained by mitophagy, which helps maintain cellular homeostasis, prevents ROS production, and protects against mtDNA damage. However, increased calcium uptake and oxidative stress can disrupt mitochondrial membrane potential and permeability, leading to the apoptotic cascade. This disruption causes increased production of free radicals, leading to oxidative modification and accumulation of mitochondrial DNA mutations, which contribute to cellular dysfunction and aging. Mitochondrial dysfunction, resulting from structural and functional changes, is linked to age-related degenerative diseases. This review focuses on mitochondrial dysfunction, its implications in aging and age-related disorders, and potential anti-aging strategies through targeting mitochondrial dysfunction.
GRAPHICAL ABSTRACT | The figure shows the effect of mitochondrial dysfunction and mitophagy on stem cells leading to aging.
According to the United Nations (UN) in 2020 there were around 727 million people aged 65 years or over, and in the future, the number of aged people will increase compared to the younger population (United Nations Department of Economic and Social Affairs, P. D., 2020). This suggests a need to understand the aging process to improve individuals’ overall life expectancy and health. In humans, aging generally begins after thirty (Dziechciaz M and Filip, R., 2014) as a result of aggregation of physical, physiological, psychological, and social changes leading to an overall decline in physical and mental wellbeing and a reduction in mobility. Aging is a complex, multifactorial, and long-term process affecting individuals at various levels, including molecular, cellular, tissue, organ, and system (Cohen, 2018). Aging is associated with the onset of various age-related diseases and conditions, including cardiovascular diseases (Kizhakekuttu and Widlansky, 2010) such as atherosclerosis, hypertension, and heart problems, along with other diseases such as diabetes mellitus, arthritis, cataracts, hearing loss, weakened immune response, cancer, onset of neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease in the later stages of life (Liu, 2014; Balmik and Chinnathambi, 2018; Li et al., 2021). Studies addressing aging in humans and animal models identified various cellular processes and events associated with aging and age-associated diseases (Mitchell, S.J, et al., 2015; Casajus Pelegay et al., 2019). López-Otín et al. (2013) proposed nine hallmarks of aging: genomic instability, telomere attrition, epigenetic alterations, loss of proteostasis, deregulated nutrient sensing, cellular senescence, stem cell exhaustion, altered intracellular communication, and mitochondrial dysfunction (Liu et al., 2002). The crosstalk between these hallmarks of aging has been identified, which has shed more light on the molecular mechanism of the aging process. Among these hallmarks of aging, mitochondrial dysfunction has been comprehensively studied as its role in aging (Sun et al., 2016; Kauppila et al., 2017; Zhang et al., 2018; Bar-ziv et al., 2020). Moreover, the crosstalk of mitochondrial dysfunction with other hallmarks of aging suggests that mitochondrial dysfunction actively influences the aging process by contributing to other hallmarks of aging (Van der Rijt et al., 2020).
Mitochondria are unique double-membrane organelles that came into existence due to the engulfment of alpha-proteobacterium by a eukaryotic progenitor cell in an endosymbiosis process, demonstrating evolutionary importance in the advancement of eukaryotic life (Lane and Martin, 2010). Mitochondria, apart from the nucleus, comprises its genome, metabolome, transcriptome, and proteome (Cloonan et al., 2020). Mitochondria have been considered a cellular powerhouse, responsible for approximately 95% of cellular ATP production. They are responsible for significantly contributing to the maintenance of cellular homeostasis by contributing to metabolic processes such as the tricarboxylic acid cycle (TCA) and oxidative phosphorylation (OXPHOS) (Tzameli, 2012; Birsoy et al., 2015). Apart from regulating cellular energetics, mitochondria also play an essential role in intracellular calcium signaling thermogenesis, apoptosis, generation of reactive oxygen species (ROS), and regulation of oxidative stress response (Kowaltowski, 2000). Any defect or deficit in mitochondrial number and function might be responsible for cellular damage. Mitochondrial dysfunction has been reported to be associated with aging and almost all chronic aging-associated diseases (Nicolson, 2014; Nehlin, 2023) through reduced ATP production, alteration in the regulation of apoptosis, increased ROS production, and defective calcium signaling (Norat et al., 2020). Accumulation of mutations in mitochondrial DNA (mtDNA) is the primary cause of mitochondrial anomalies, further contributing to aging and associated diseases (Kujoth et al., 2007; Paraskevaidi et al., 2017). Here, we provide a detailed description of mitochondrial dysfunction, its implications in the aging process, the onset of aging-associated diseases, and potential therapeutic interventions targeting mitochondrial dysfunction to develop an effective strategy for treating age-related diseases.
The human mitochondrial genome comprises small double-stranded circular DNA spanning 16,569 bp, which contains 37 genes coding for 13 polypeptides, 2 rRNAs, and 22 tRNAs. All these 13 polypeptides encoded by mtDNA are active components of the oxidative phosphorylation system (Taanman, 1999). Mitochondria contain several copies of mtDNA, and its replication is a cell cycle-independent and relatively dynamic process (Bogenhagen, and Clayton, 1977). Replication of mtDNA is regulated by several specific mitochondrial proteins such as mitochondrial polymerase gamma A (PolγA), TWINKLE and the mitochondrial single-strand binding protein (mtSSB) (Bogenhagen, and Clayton, 1977). mtDNA has been organized into nucleoids, aggregates of one or more mtDNA copies linked to proteins that bind to mitochondrial DNA, like mitochondrial transcription factor (TFAM) (Bogenhagen, 2012). The Association of mtDNA with DNA binding proteins protects the DNA against ROS and reactive nitrogen species (RNS) produced during mitochondrial metabolism (Bogenhagen, 2012). mtDNA has up to a 15-fold higher mutation rate and a less efficient DNA repair mechanism compared to nuclear DNA (Short et al., 2005). Accumulation of mutations in mtDNA beyond a critical threshold may lead to significant adverse effects in mitochondrial functioning and mutations in components of OXPHOS complexes, resulting in mitochondrial dysfunction and increased ROS production (Wallace, 2010). According to a few studies, age-associated increases in point and deletion mutations in mtDNA have been observed in various human tissues, including the brain, heart, colon and skeletal muscle (Bua et al., 2006; Marzetti and Leeuwenburgh, 2006; Kennedy et al., 2013; Greaves et al., 2014). However, the role of these mutations in the aging phenotype and whether they are the cause or effect of aging is still unclear. A study (Bratic et al., 2015) in the mtDNA mutator mouse model gave more insights into the accumulation of mutations in mtDNA and its implications in aging phenotypes. This model was developed by homozygous knock-in of PolγA lacking exonuclease activity (Bratic et al., 2015). Another study (Edgar and Trifunovic, 200) suggested that an increase in the frequency of mutations in mtDNA resulted in a shortened lifespan and premature onset of aging phenotype, including reduced fertility, anemia, osteoporosis, hair loss, the curvature of the spine, reduced body weight and premature death in this mouse (Edgar and Trifunovic, 2009).
The accumulation of mutations in mtDNA and its implications in the aging process. More research (Ross et al., 2013) using mice with a nuclear genome of wild type and mtDNA mutations inherited from a mother heterozygous for mutator allele (PolγAwt/mut) showed that low levels of germline-transmitted mtDNA mutations may have long-term effects, including early aging and a decrease in lifespan (Ross et al., 2013; Ross et al., 2014).
Mitochondria hosts the OXPHOS and ATP production with assistance from the electron transport chain located in the inner membrane of mitochondria. The electron transport chain located in the inner mitochondrial membrane consists of four protein complexes and is coupled with ATP synthase, an ATP-producing enzyme. ROS are considered to be unwanted and toxic by-products of the mitochondrial electron transport system (Chistiakov et al., 2014). ROS are extremely reactive chemical species that include superoxide anion (O2−), hydroxyl radical (OH), and hydrogen peroxide (H2O2). Because they tend to donate or acquire another electron in order to achieve stability, these radicals have a single unpaired electron, which contributes to their high reactivity. Hydrogen peroxide (H2O2), hypochlorous acid (HOCl), hypobromous acid (HOBr), ozone (O3), singlet oxygen (1O2), nitrous acid (HNO2), nitrosyl cation (NO+), nitroxyl anion (NO−), dinitrogen trioxide (N2O3), dinitrogen tetraoxide (N2O4), nitronium (nitryl) cation (NO2+), organic peroxides (ROOH), aldehydes (HCOR), and peroxynitrite (ONOOH) are the non-radical species. Even while these non-radical species may readily trigger free radical reactions in organisms, they are not free radicals themselves.
The mitochondrial electron transport system produces almost 90% of total cellular ROS, making it a major ROS generator in cells (Bratic and Trifunovic, 2010). ROS are highly reactive molecules that can lead to oxidative deterioration of molecules like proteins, lipids, and DNA. ROS leads to various DNA lesions, including oxidized DNA bases and DNA strand breaks (Cui et al., 2012). Besides its harmful effects in inducing oxidative stress in cells, ROS also acts as a signaling molecule that influences various cellular pathways and enhances immunologic defence against pathogens (Yang and Lian, 2020). Many studies (Liu et al., 2002; Bowling and Beal, 1995) focused on aging demonstrate that ROS is one of the major mediators of age-related cellular damage and the pathogenesis of some age-associated neurodegenerative diseases. Furthermore, many theories explaining the aging process, including the free radical theory and the mitochondrial theory of aging, are based on the harmful effects of ROS and associated cellular damage (Chistiakov et al., 2014; Yan, 2014; Indo et al., 2015). Although elevated levels of ROS typically lead to cellular harm and accelerate aging, lower amounts might enhance overall defence mechanisms by triggering an adaptive reaction. This phenomenon has been termed mitochondrial hormesis. (Ristow and Schmeisser, 2014).
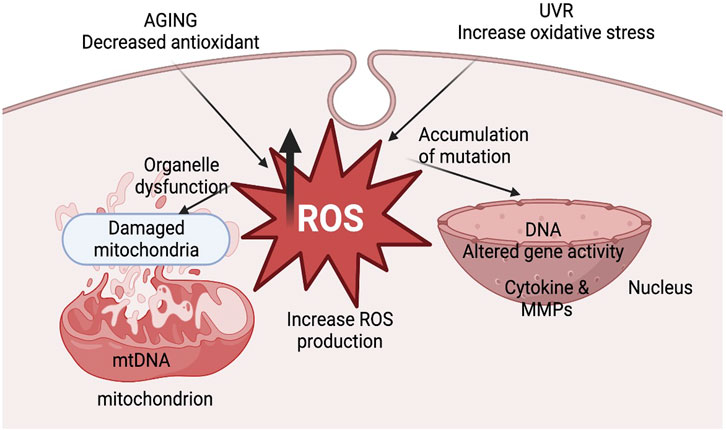
The cumulative damage to mitochondria and accumulation of mutations in mtDNA caused by ROS leads to deterioration in mitochondrial functions due to alteration in the expression of components of the electron transport chain. ROS-mediated damage in OXPHOS machinery alters the enzymatic activity of mitochondrial respiratory enzymes and reduces mitochondrial membrane potential, leading to compromised ATP production (Cedikova et al., 2016). There are two major sites of ROS production in the electron transport chain: Complex I and Complex III. Complex I is the major site affected by increased ROS production. Seven complex I components are encoded by mtDNA, which gets frequently mutated due to ROS, ultimately contributing to the aging process (Indo et al., 2015; Cedikova et al., 2016). According to one report by Nissanka and Moraes (2018), the activity of complex I is significantly hampered due to aging in rat brain and liver along with human platelets, leading to enhanced ROS generation (Nissanka and Moraes, 2018). This suggests that there might be a positive correlation between a defect in complex I activity and ROS generation and vice versa and their overall impact on the aging process. According to the free radical theory of aging, free radical production via mitochondria, oxidative damage of tissues, aging, and aging-related diseases are interlinked, eventually affecting an individual’s physiology and metabolism. These include -the absence of specific growth factors, hormones, DNA damage, chemotherapeutic agents, serum starvation, UV radiation, toxins, certain conditions like hyperthermia, hypoxia, viral infections, free radicals affecting mitochondrial membrane potential, increased levels of Ca+2, formation of reactive oxygen and nitrogen species and decline in the redox status as (i.e., glutathione, ATP, NADH) (Elmore, 2007; Pollack and Leeuwenburgh, 2001). Transgenic mice’s lifespans have been examined in another investigation to determine the impact of overexpressing antioxidant enzymes. The overexpression of either copper zinc superoxide dismutase (CuZnSOD) and catalase or CuZnSOD and manganese superoxide dismutase (MnSOD) may occur. Superoxide and hydrogen peroxide in the cytosolic and mitochondrial compartments are known to be scavenged by the overexpression of these important antioxidant enzymes. Therefore, the life span of mice is not increased by the overexpression of antioxidant enzymes (Pérez et al., 2009). This occurs because the increased levels of ROS can cause damage to mitochondrial DNA, as shown in Figure 1, leading to a decline in mitochondrial function. This decline in function, in turn, leads to increased oxidative stress that triggers a signaling cascade that contributes to skin structure and photoaging changes. Furthermore, through certain signaling genes like NF-kβ and AP-1, ROS can increase the expression of proinflammatory cytokines and induce inflammation. ROS can also directly damage the skin by increasing the expression of MMP (Benz and Yau, 2008; Rizwan et al., 2014). ROS-induced DNA mutations, lipid peroxidation, and protein oxidation are all implicated in skin aging and photoaging development (Rinnerthaler et al., 2015).
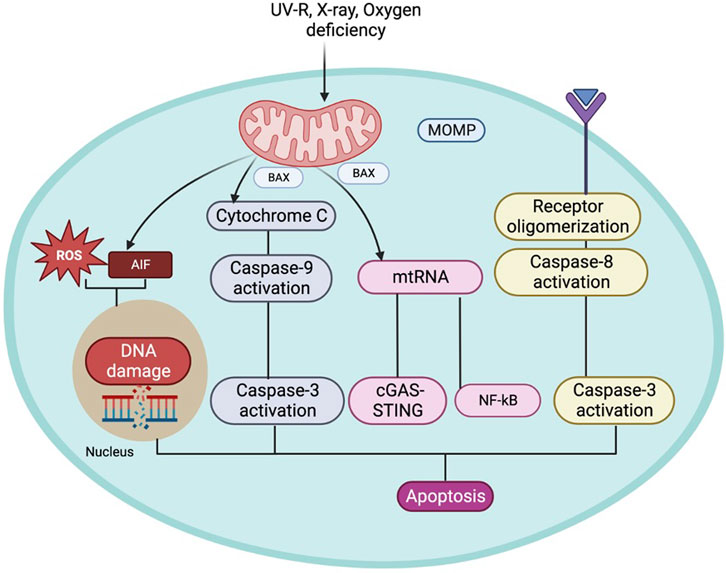
Figure 1. The illustration represents mitochondrial dysfunction and ROS production in aging.
Apoptosis is a homeostatic mechanism essential in controlling cell death, maintaining the average cell turnover, embryonic development, effective immune system functioning, maintaining cellular and tissue homeostasis, maintaining cell growth, differentiation, and tissue repair (Greenhalgh, 1998; Alberts et al., 2002; Mondello and Scovassi, 2010; Tower, 2015). This mechanism is executed without detrimental effects on the neighbouring cells as the process does not release cellular components into the surrounding tissue environment (Alberts et al., 2002; Kurosaka et al., 2003). The mitochondrial-mediated apoptosis pathway is initiated due to various death signals (Ravindran et al., 2011). The Bcl-2 family proteins are also critical in regulating the apoptosis progression by influencing the mitochondrial membrane permeability and Ca+2 influx, releasing cytochrome c, blocking electron transfer between complexes III and IV of the electron transport system, and preventing ATP generation. Cytochrome C is released from the mitochondria to the cytosol and activates Apaf-1, forming an “apoptosome” complex along with caspase-9, following cleavage and activation of the effector protease, caspase-3, leading to apoptosis. The pro-apoptotic and anti-apoptotic proteins trigger apoptosis or terminate the apoptosis progression by influencing the mitochondrial membrane permeability transition pores (mPTP), which is shown in (Figure 2) (Pollack and Leeuwenburgh, 2001; Eberle and Hossini, 2008). These observations (Srivastava, S., 2017) suggest that the mitochondrial apoptosis pathway plays a pivotal role in the aging process and pathology associated with age-related diseases. Additionally, mtRNA can cause apoptosis independently through the NF-κB and cGAS-STING pathways (Decout et al., 2021). If the permeability of the mitochondria increases, they will release apoptosis-inducing factors (AIF) which, when released into the cytoplasm or nucleus, can damage DNA and ROS, triggering apoptosis (Camplejohn et al., 2003; Sevrioukova, 2011).
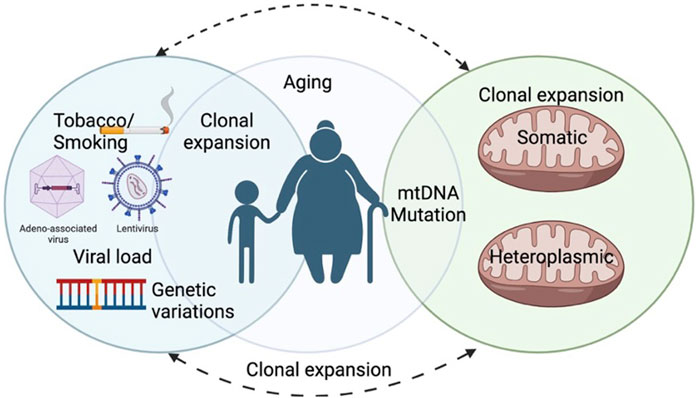
Figure 2. The figure represents the mitochondrial role leading to cell apoptosis and its association in aging.
Age-associated alterations in Mitochondrial Dynamics and the role of mitokines in aging: The mitochondrial network is regulated by the complex fission, fusion, and distribution events, which are essential for normal mitochondrial functions (Roy et al., 2015). A fine balance between mitochondrial fusion and fission events is maintained to optimize mitochondrial function. Mitochondrial fusion events are responsible for fusing mitochondrial contents to reduce the impact of damaged mtDNA and proteins (Silva Ramos et al., 2019). On the other hand, mitochondrial fission events are responsible for removing damaged mitochondrial contents by selective mitochondrial autophagy (Rubinsztein et al., 2011; Papackova, and Cahova, 2014). Mitochondrial biogenesis is a complex process regulated by the coordination of nuclear and mtDNA and triggered by factors such as hypoxia, caloric restriction, fasting, cold, physical exercise and hormonal stimulation. PGC-1α, PGC-1β, and PGC-1 are the family of PGC-1 proteins that activate mtDNA transcription; PGC-1α is regarded as the primary regulator of mitochondrial biogenesis.
Further, the nuclear transcription factors nuclear respiratory factor-1 (NRF-1), NRF-2, and estrogen-related receptor-α (ERR-α) are stimulated, and TFAM, the last effector of mtDNA transcription and replication, is expressed more frequently. The pathway is started by PGC-1α activation (either by phosphorylation or deacetylation). NRFs and ERRα modulate the expression of mitochondrial transcriptional factors and regulate the expression of mitochondrial respiratory subunits to initiate the process of biogenesis (Scarpulla, 2008; Zhu et al., 2013; Wang J. et al., 2020). There is an intricate balance between mitochondrial biogenesis and mitophagy, and defects in these mitochondrial dynamics may lead to alterations in mitochondrial and cellular functions. Altered mitochondrial dynamics have implications in the modulation of cellular processes that might be associated with aging (Terman et al., 2010).
Impaired mitochondrial dynamics have been implicated in age-associated changes due to functional and structural changes in mitochondria (Cedikova et al., 2016). According to a few studies, age-associated structural and functional mitochondrial defects have been observed in skeletal muscles, liver, kidney, heart, brain, diaphragm muscles, bone and fibroblasts (Yen et al., 1989; Shigenaga et al., 1994; Crane et al., 2010; Terman et al., 2010; Mahmoud and Hegazy, 2016; Marycz et al., 2016; Shum et al., 2016; Palomera-Avalos et al., 2017). In another study, a decline in mitochondrial density was observed in skeletal muscles with age progression (Crane et al., 2010). Mitochondrial fusion is mainly facilitated by two mitofusins, Mfn1 and Mfn2. These two mitofusins are on the outer membrane of mitochondria and play an important function in mitochondrial membrane fusion (koshiba, et al., 2004). According to one study, loss of Mfn1 and Mfn2 in mice increases mtDNA mutations in skeletal muscles, suggesting that mitofusins are important for mitochondrial integrity (Chen et al., 2010).
Moreover, decreased expression of Mfn2 protein and mRNA in the frontal cortex has been reported in patients with Alzheimer’s disease (Chen et al., 2010; Manczak et al., 2011). Age-associated defects in mitochondrial dynamics, along with impaired mitochondrial structure and functions, have been reported in various studies, indicating the importance of mitochondrial dynamics in health and aging.
Another important aspect of mitochondrial dynamics involves the cross-talk between mitochondria of distant cells under stress conditions through signalling molecules called mitokines. Mitokines include molecules expressed in the nucleus such as Fibroblast growth factor 21 (FGF21) and Growth Differentiation Factor-15 (GDF15) as well as mitochondrial derived peptides like humanin (HN) (Burtscher et al., 2023). Mitokines are essential factors to ensure cellular vitality and mitochondrial functions under stress conditions and other external stimulus in the form of exercise and diet (Burtscher J, et al., 2023). Mitochondria modifies its functions through metabolic reprogramming and epigenetic remodelling upon oxidative stress, impaired, ETC or proteotoxic stress generated by unfolded proteins (Mottis et al., 2019; Boos et al., 2020; Zhou et al., 2022). The dysregulation in mitokines signalling in the form of mitokines resistance or mitokines synthesis results in development of age-related decline in physiological functions and manifestation of metabolic, cardiovascular or neurological diseases. The plasma concentrations of FGF21 and GDF15 have increased with aging in humans and have been correlated to cardiovascular, metabolic and neurological diseases (Youm et al., 2016; Conte et al., 2019). Furthermore, mitokines such as HN and mitochondrial ORF of the 12S rRNA type-c (MOTS-c) play a protective role against cellular stress and inflammation. Additionally, HN has been shown to enhance insulin sensitivity and increase glucose transported 4 (GLUT4) expression in rodent model of diabetes (Lee et al., 2015; Wu et al., 2022). The levels of circulating HN are upregulated in aging individuals, suggesting its protective role against age-associated metabolic complications (Conte et al., 2019). Improved balance in mitokines level has been reported in individuals performing regular moderate to intensive exercise leading to improved cellular function and longevity (Taniguchi et al., 2016; Zhang et al., 2019). The role of mitokines in aging need to be characterized through intensive research to develop mitokines based therapeutic strategies against aging.
Autophagy is the controlled process associated with recycling components of the cell by delivering them to the lysosome for degradation under special circumstances such as nutrient starvation, dysfunction of particular cellular components, removal of cellular protein aggregates, control of cellular biomass, and elimination of intracellular pathogens (Ohsumi, 2014). Autophagy has been studied extensively concerning the aging process as a decrease in autophagy has been observed with aging (Rubinsztein et al., 2011; Madeo et al., 2010). Autophagy of mitochondria is known as mitophagy, which occurs during nutrient starvation, selective removal of mitochondria during differentiation of cells such as sperms, ocular lens cells, and red blood cells, as well as for the removal of dysfunctional mitochondria (Sato and Sato, 2013; Costello et al., 2013; Mortensen et al., 2010; Lemasters, 2005). Mitophagy has been associated with several conditions such as oxidative stress, hypoxia, defect in the electron transport chain, accumulation of protein aggregates, and iron starvation. Impairment in the mitophagy process has been associated with various pathological conditions, including aging and aging-associated diseases such as neurodegenerative diseases, cardiovascular diseases, and cancer (Chen et al., 2020; Evangelou et al., 2023).
The major regulator of mitophagy of dysfunctional mitochondria is PTEN-induced putative kinase 1 (PINK-1), and this process is also known as PINK1-mediated mitochondrial quality control (Wall et al., 2019). A defect in membrane potential dissipation prevents PINK-1 degradation followed by its autophosphorylation, which activates E3-ubiquitin ligase Parkin. Parkin promotes the ubiquitination of mitochondrial membrane proteins, further phosphorylated by PINK1. These phosphorylated polyubiquitination chains are recognized by adaptor proteins of core autophagy machinery such as p62 and OPTN, which further interacts with LC3 and initiates autophagosome formation around dysfunctional mitochondria, which is further fused with lysosome, leading to autolysosome formation and ultimately undergoes degradation (Palikaras et al., 2018). A widely studied pathway for mitophagy involves the PINK1/Parkin mechanism, wherein PINK1 recruits Parkin to dysfunctional mitochondria, triggering their degradation. However, aside from PINK1/Parkin-mediated mitophagy, alternative pathways exist that operate independently. One such pathway is SLR-independent mitophagy, wherein damaged mitochondria are identified by specific receptors like NIX/BNIP3L and FUNDC1. These receptors then engage with the autophagy machinery to facilitate degradation. In contrast, SLR-independent mitophagy pathways utilize different mechanisms for recognizing and targeting dysfunctional mitochondria for degradation. These pathways represent diverse strategies within cells for maintaining mitochondrial quality control and cellular homeostasis (Ganley and Simonsen, 2022).
Various studies involving model organisms such as Caenorhabditis elegans and Drosophila melanogaster have demonstrated defects in mitophagy and its implications for the health and lifespan of the organism (Bakula and Scheibye-Knudsen, 2020). Moreover, a study in transgenic mice expressing the fluorescent mitophagy reporter mt-Keima suggested that with age, there was a decrease in the mitophagy in the hippocampus region of the mouse brain (Sun et al., 2015). Also, decreased mitophagy has been observed in mouse hearts, and its implications in heart aging have been documented (Hoshino et al., 2013). Further association between defects in mitophagy and cardiovascular diseases has been reported, suggesting the role of decreased mitophagy in aging and age-associated diseases (Bravo-San Pedro et al., 2017). Decreased mitophagy has been reported to contribute to the pathogenesis of several age-associated disorders such as Parkinson’s disease, Alzheimer’s disease, cardiomyopathies, and cancer (Bernardini et al., 2017; Fivenson et al., 2017; Levine and Kroemer, 2019). The exact mechanism underlying decreased mitophagy in the aging process and age-associated diseases is still unclear, and more comprehensive studies are required to delineate detailed mechanisms.
Moreover, several researchers have attempted to target mitophagy as an effective strategy for developing anti-aging therapeutics (Bakula and Scheibye-Knudsen, 2020). Further analysis reveals that in the mito-QC reporter mouse, mitophagy in multiple organs appears to either increase or remain unchanged in older mice compared to younger ones. Transcriptomic analysis reveals a significant upregulation of the type I interferon response in the retina of older mice, which is associated with elevated levels of cytosolic mtDNA and activation of the cGAS/STING pathway (Boya, 2024; Jiménez-Loygorri et al., 2024).
Increased mitochondrial ROS production in aging cells contributes to another hallmark of aging apart from genomic instability and telomere attrition: epigenetic alterations. Increased levels of cellular ROS affect the DNA methylation status, contributing to age-associated modifications in epigenetic signatures (Liu L et al., 2002; Kietzmann et al., 2017). Increased ROS production due to age-associated mitochondrial dysfunction leads to 8-hydroxylamine (8-OHdG) DNA lesions, further inhibiting DNA methylation. These 8-OHdG lesions caused by increased ROS production are the major reason behind altered DNA methylation patterns observed during aging (Turk et al., 1995).
Proper mitochondrial and cytosolic proteostasis is important for healthy aging and longevity (Molenaars et al., 2020). mTOR is the key molecule of the nutrient-sensing pathway. It is a key regulator of mitochondrial activity, biogenesis, and dynamics (Morita et al., 2013; Morita et al., 2017). mTOR is an important serine/threonine kinase involved in cellular pathways regulating growth and proliferation. By stimulating the synthesis of mitochondria-related proteins encoded in the nucleus, mTOR regulates the energy generation of the mitochondria and the energy consumption of the mRNA translation machinery. Research shows that by promoting the synthesis of mitochondrial transcription factor A (TFAM), mitochondrial ribosomal proteins, and elements of complexes I and V, mTOR regulates mitochondrial activities. By inducing the synthesis of TFAM, mitochondrial ribosomal proteins, and elements of complexes I and V, mTOR regulates the activities of mitochondria (Morita. M., 2015). Furthermore, the nutrient-sensing mechanism/mammalian target of rapamycin complex 1 (mTORC1) plays a role in mitochondrial fission and apoptosis by stimulating the translation of mitochondrial fission process 1 (MTFP1) (Morita et al., 2015). The study (Zorov et al., 2014) highlights that both genetic interventions and pharmacological treatments targeting reactive oxygen species (ROS) resulting from dysfunctional mitochondria have demonstrated the normalization of several disease-related traits across different species. Additionally, dietary caloric restriction (CR) has proven effective and has recently been shown to decelerate the aging process in humans. Conversely, genetic modifications aimed at altering mitochondrial dynamics have yielded conflicting outcomes, potentially due to variations specific to cells, tissues, and the particular neurodegenerative diseases under investigation Figure 3 (Scott et al., 2019).
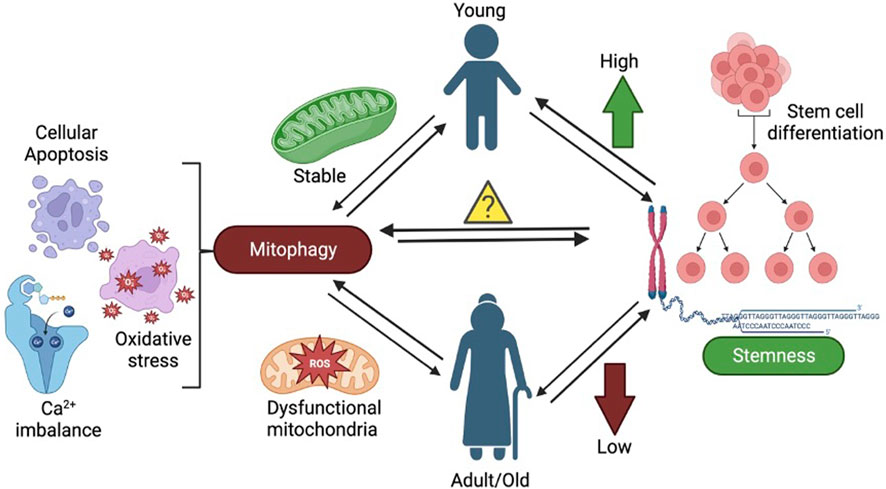
Figure 3. The representative illustration shows the Mutational burden and heteroplasmy in mitochondrial DNA that is linked to biological age, tobacco, and Viral infections.
Another aspect of aging affected by mitochondrial dysfunction is cellular senescence. Oxidative stress generated by a defect in mitochondrial components induces cellular senescence (Moiseeva et al., 2009). Cells become senescent when they enter a non-proliferative state while remaining active metabolically. High levels of ROS characterize the senescent state as a result of mitochondrial deregulation. Conversely, mitochondrial deregulation can drive cellular senescence (Yoon et al., 2003; Yoon et al., 2006; Byun et al., 2012). Abrupted energy transduction pathway, imbalance in mitochondrial dynamics, increased AMPK activity, and deregulation of mitochondrial calcium homeostasis may lead to cell cycle arrest (Ziegler et al., 2015). Increased ROS production has been shown to increase the rate of telomere shortening in cells and trigger the onset of cellular senescence through sustained DNA damage response (Passos et al., 2007; Chen et al., 2015; Davalli et al., 2018). Apart from this, defect in mitochondrial dynamics is also associated with the induction of cellular senescence (Lee et al., 2007).
Nicotinamide adenine dinucleotide (NAD) is a metabolite well implicated in senescence. NAD + acts as an electron carrier during the process of oxidative phosphorylation. The ratio of NAD+/NADH becomes low in cells undergoing senescence due to mitochondrial dysfunction (van der Veer et al., 2007; Lee et al., 2012). The lower NAD+/NADH ratio has been suggested to arise due to reduced cytosolic malate dehydrogenase levels, which utilize NADH to convert oxaloacetate to malate during oxidative phosphorylation. Low NAD+/NADH ratio also triggers senescence through p53 by activating 5′AMP-activated protein kinase (AMPK) (Jones et al., 2005). The cellular senescence driven via various factors of mitochondrial dysfunction has been collectively termed mitochondrial dysfunction-associated senescence (MiDAS) (Chapman J. et al., 2019). Cells undergoing MiDAS are characterized by lower NAD+/NADH ratios, leading to cell growth arrest and IL-1-associated senescence-associated secretory phenotype (SASP) (Wiley et al., 2016). Along with other aging-related hallmarks, stem cell exhaustion is associated with increased ROS production (Jang and Sharkis, 2007). A study in mitochondrial mutator mice demonstrated neuronal and hematopoietic stem cell dysfunction (Trifunovic et al., 2004).
There is a need to improve health and increase the life expectancy of the aged population. As a result, current biomedical research has been heavily focused on aging to identify potential targets for prolonging human health and lifespan (Madreiter-Sokolowski et al., 2018). Interventions like caloric restrictions, exercise, and pharmacological therapies have been studied, which can effectively improve health and slow down phenotypes of the aging process (Madreiter-Sokolowski et al., 2018; Nilsson and Tarnopolsky, 2019). Studies show that mitochondrial dysfunction is one of the most important aspects of aging and age-related disorders (Morita et al., 2013; Wu et al., 2022). Current research in developing effective therapeutic interventions and strategies for treating aging phenotypes and aging-associated diseases is inclined to utilize mitochondrial dysfunction as an effective target (Nicolson and Ash, 2017; Balcázar et al., 2020; Rai et al., 2020; Zimmermann et al., 2021).
Behavioral interventions such as caloric restriction and exercise are widely studied for delaying the aging process. Their association with mitochondrial biogenesis and function has been established in various model organisms (Madreiter-Sokolowski et al., 2018). Caloric restriction has been known to improve mitochondrial respiration, reducing ROS production (Madreiter-Sokolowski et al., 2018). Besides this, caloric restriction promotes mitochondrial biogenesis by activating AMPK signaling, activating SIRT1 and TFAM, and increasing mtDNA replication and transcription (Civitarese et al., 2007). However, further studies are required to determine the time of caloric restrictions in different individuals for optimum output related to improving health and delay in the cellular aging process. Besides caloric restriction, exercise is another behavioral intervention that has been reported to improve health and lead to delays in the aging process by influencing mitochondrial functions (Nicolson and Ash, 2017; Balcázar et al., 2020). According to a study in mice, PGC-1α and mitochondrial SIRT3 get downregulated with aging, which was further normalized by exercise, leading to an improved ROS defense mechanism (Gioscia-Ryan et al., 2016). These observations suggest that a combination of caloric restriction and regular exercise might improve health and delay aging by improving overall mitochondrial functions.
Various pharmacological molecules targeting mitochondrial dysfunction have been identified, with potential anti-aging properties. Polyphenols such as resveratrol and green tea polyphenols have been reported to extend lifespan in various model organisms, including mice (Rai et al., 2020). Resveratrol improves mitochondrial function by SIRT1-dependent activation of AMPK in mouse models (Price et al., 2012). Apart from polyphenols, natural and synthetic compounds are reported to improve mitochondrial functions. Natural compounds such as Urolithin A, a gut metabolite of ellagic acid, have been widely studied for their anti-aging activity in model organisms such as C. elegans and rodents (Price et al., 2012).
Moreover, Urolithin A has been reported to improve human mitochondrial and cellular health (Andreux et al., 2019). Apart from Urolithin A, the potential anti-aging activity of Actinonin and Polyamine spermidine via mitophagy induction has been reported (Bakula and Scheibye-Knudsen, 2020). N-3 fatty acids have also been studied to determine their anti-aging properties via improved mitochondrial ATP production and increased mitochondrial protein synthesis (Madreiter-Sokolowski et al., 2018). Melatonin has a multifaceted role as a free radical scavenger and a modulator of gene expression of antioxidant enzymes such as glutathione peroxidase and glutathione reductase, thus aiding in reducing oxidative damage (Ding et al., 2014; Kopustinskiene and Bernatoniene, 2021). It acts on mitochondrial uncoupling proteins (UCPs) and dissipates the proton gradient across the inner mitochondrial membrane (Agil et al., 2015; Tan, et al., 2016). The Chinese herb Scutellaria Baicalensis yields the flavonoid baicalein, which is a strong antioxidant and free radical scavenger while preventing the deposition of amyloid protein aggregates.
(Sonawane et al., 2019; Wang et al., 2021; Marković et al., 2011). More comprehensive studies are required to utilize these natural compounds as an effective anti-aging treatment to promote mitochondrial functions and reduce oxidative stress caused by mitochondrial dysfunction, as shown in Figure 4.
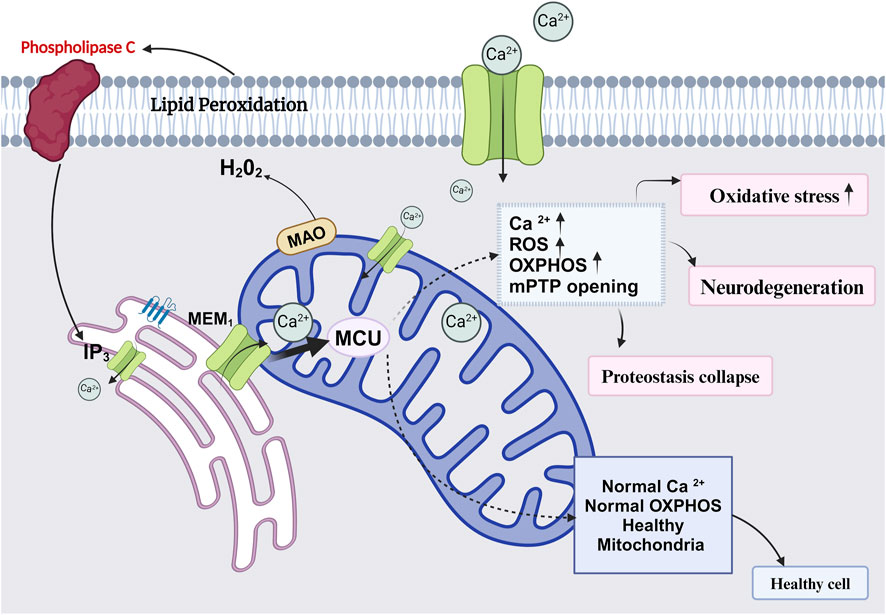
Figure 4. Diagram represents role of healthy and unhealthy mitochondria and its association in age associated diseases.
Apart from natural compounds, various synthetic compounds have been identified which demonstrate anti-aging activity by targeting mitochondrial functions. Randomized trials and population studies using natural antioxidants have produced poor results despite a substantial body of epidemiological and clinical data suggesting that antioxidant-rich diets lower blood pressure and cardiovascular risk (Kizhakekuttu and Widlansky, 2010). Apart from synthetic antioxidants, AMPK pathway activators, AICAR, and metformin have been identified as potential drugs for anti-aging therapy (Madreiter-Sokolowski et al., 2018). These AMPK pathway activators mimic the caloric restriction effects in the cells and lead to improved mitochondrial biogenesis, which further contributes to anti-aging properties (Toyama et al., 2016; Stancu, 2015). These studies suggest that targeting mitochondrial dysfunction and improving mitochondrial biogenesis could be a possible mode for developing anti-aging therapies. Furthermore, induction of mitophagy by behavioral interventions and treatment with natural and synthetic compounds might prove helpful in improving overall health and increasing life expectancy in the future.
Another approach to promote healthy aging in individuals has been directed towards cellular senescence. Therapeutics targeted against senescent cells are categorized into senolytics, i.e., killing senescent cells, and senomorphics/senostatics, i.e., targeted towards inhibition of SASP secretions (Atayik, and Çakatay, 2022; Nehlin, 2023; Van Deursen, 2014). Senolytic treatment has shown numerous beneficial effects in the form of improved genomic stability, alleviation of telomere attrition, mitochondrial dysfunction, and inflammatory response (Cho et al., 2023). Quercetin has been used as a senolyte which has been shown to induce AMPK-mediated apoptosis to eliminate senescent vascular smooth muscle cells and endothelial cells along with promoting mitophagy and NRF2-NFκB signaling (Wang L. et al., 2020; Jiang et al., 2020; Shao et al., 2022). Additionally, combined treatment of quercetin and Dasatinib is effective in improving overall cellular functions in aged hosts, decreasing the secretion of pro-inflammatory cytokines in human adipose tissues, and reducing senescent cell burden (Xu et al., 2018; Camell et al., 2021; Saccon et al., 2021). Fisetin is another flavonoid molecule present in various fruits and vegetables, known to exhibit senolytic effect (Khan et al., 2013). Additionally, fisetin facilitates the activation of Nfr2 in HUVECS, leading to hemeoxygenase one mediated antioxidant response (Yousefzadeh et al., 2018; Kwak et al., 2014). Piper longumine, an alkaloid in long peppers, inhibits platelet aggregation and thrombus formation (Chatterjee and Dutta, 1967; Iwashita et al., 2007). It has shown that the regulation of platelet-derived growth factor BB (PDGF-BB) mediated mitochondrial fission and mitochondrial protection in the case of hyperproliferation vascular phenomena (Salabei and Hill, 2013). Overall, the combination of various therapeutics interventions and senotherapeutic drugs can be effective in restoring mitochondrial functions and balancing mitochondrial dynamics, thereby targeting the mitochondria-associated aging phenotype to promote healthy aging.
Interestingly, TCA cycle metabolites can exhibit effects that oppose senescence. For example, α-ketoglutarate has demonstrated senomorphic properties by diminishing the SASP in senescent fibroblasts and extending the health span and lifespan in aged mice (Liao et al., 2021).
To begin, numerous senolytic medications presently under development exert their influence by focusing on anti-apoptotic proteins situated within mitochondria, which obstruct apoptosis driven by mitochondria. Senolytic drugs produce their senolytic impact by directing the anti-apoptotic B-cell lymphoma 2 (BCL-2) protein family within mitochondria (Basu, 2022; Martini, and Passos, 2023).
Mitochondria is a central organelle involved in the maintenance of cellular and physiological wellbeing, which contributes to cellular energetics and other important cellular processes such as cell signaling, calcium homeostasis, cell growth, and apoptosis. Systemic deterioration in mitochondrial components, functions, and dynamics has been associated with aging. Accumulation of mutations in mtDNA leads to increased production of defective mitochondrial proteins and increased ROS production. A further contribution of increased ROS production and activation of the mitochondrial apoptotic pathway in aged cells leads to systemic deterioration of various tissues, decreasing overall organ and tissue functions, including muscle, heart, liver, and brain function. Defective mitochondrial proteostasis has been associated with altered cellular functions in aging cells.
The decreased rate of mitophagy in aged cells further contributes to the accumulation of defective mitochondria and their effect on an organism’s cellular and overall health. The association of increased mitochondrial dysfunction with other hallmarks of aging makes it a possible target for developing therapies for treating aging phenotypes and associated disorders. Behavioral interventions and pharmacological compounds targeting mitochondrial dysfunction have increased hope in designing future therapeutic management of aging phenotypes and related diseases. Various therapeutic natural and synthetic molecules targeting mitochondrial functions have been studied for their implications on aging. There is still a need to identify the detailed molecular mechanism behind mitochondrial dysfunction, including the decrease in mitophagy in aging cells and their association with the overall aging process. Despite promising therapeutic interventions targeting mitochondrial dysfunction for treating pathologies associated with aging and age-related diseases, more comprehensive molecular and clinical studies are required to effectively utilize mitochondrial dysfunction as a target for anti-aging therapies.
IS: Conceptualization, Writing–original draft, Writing–review and editing. SJ: Writing–original draft, Writing–review and editing. MB-C: Writing–original draft, Writing–review and editing. SP: Writing–original draft, Writing–review and editing. AB: Writing–review and editing. SR: Writing–review and editing. NS: Writing–review and editing. AD: Writing–review and editing.
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
The authors of this paper are thankful to Chettinad Academy of Research and Education (CARE) for providing the support infrastructurally. Figures were created using Biorender.com.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
ROS, Reactive oxygen species; mtRNA, Mitochondrial RNA; DNA, Deoxyribonucleic acid; UN, United Nations; TCA, tricarboxylic acid cycle; OXPHOS, Oxidative phosphorylation; ATP, adenosine triphosphate; mtSSB, Mitochondrial single strand binding protein; TFAM, Mitochondrial transcriptional factor A; RNS, Réactive nitrogen species; MMP, Matrix metalloproteinases; AP-1, Activator Protein-1; CTL, cytolytic T lymphocytes; mPTP, membrane permeability transition pores; PGC-1a, Peroxisome proliferator-activated receptor-gamma coactivator (PGC)-1 alpha; PINK-1, PTEN induced putative kinase 1; OPTN, Plk1 phosphorylates Optineurin; 8-OHDG, 8- hydrooxylamine; mTOR, mammalian target of rapamycin; MTFP1, Translation of mitochondrial fission process 1; TFAM, Mitochondrial transcription factor A; AMPK, AMP-activated protein kinase; SIRT1, sirtuin (silent mating type information regulation two homolog) 1; UCPs, Mitochondrial uncoupling proteins; DAMPs, Damage-associated molecular patterns; CNS, Central nervous systems.
Agil A., El-Hammadi M., Jiménez-Aranda A., Tassi M., Abdo W., Fernández-Vázquez G., et al. (2015). Melatonin reduces hepatic mitochondrial dysfunction in diabetic obese rats. J. pineal Res. 59 (1), 70–79. doi:10.1111/jpi.12241
Alberts B., Johnson A., Lewis J., Raff M., Roberts K., Walter P. (2002). “Programmed cell death (apoptosis),” in Molecular biology of the cell. 4th edition (Germany: Garland Science).
Andreux P. A., Blanco-Bose W., Ryu D., Burdet F., Ibberson M., Aebischer P., et al. (2019). The mitophagy activator urolithin A is safe and induces a molecular signature of improved mitochondrial and cellular health in humans. Nat. Metab. 1 (6), 595–603. doi:10.1038/s42255-019-0073-4
Atayik M. C., Çakatay U. (2022). Mitochondria-targeted senotherapeutic interventions. Biogerontology 23 (4), 401–423. doi:10.1007/s10522-022-09973-y
Bakula D., Scheibye-Knudsen M. (2020). MitophAging: mitophagy in aging and disease. Front. Cell. Dev. Biol. 8, 239. doi:10.3389/fcell.2020.00239
Balcázar M., Cañizares S., Borja T., Pontón P., Bisiou S., Carabasse E., et al. (2020). Bases for treating skin aging with artificial mitochondrial transfer/transplant (AMT/T). Front. Bioeng. Biotechnol. 8, 919. doi:10.3389/fbioe.2020.00919
Balmik A. A., Chinnathambi S. (2018). Multi-faceted role of melatonin in neuroprotection and amelioration of Tau aggregates in Alzheimer’s disease. J. Alzheimer's Dis. 62 (4), 1481–1493. doi:10.3233/JAD-170900
Bar-Ziv R., Bolas T., Dillin A. (2020). Systemic effects of mitochondrial stress. EMBO Rep. 21 (6), e50094. doi:10.15252/embr.202050094
Basu A. (2022). The interplay between apoptosis and cellular senescence: bcl-2 family proteins as targets for cancer therapy. Pharmacol. Ther. 230, 107943. doi:10.1016/j.pharmthera.2021.107943
Benz C. C., Yau C. (2008). Ageing, oxidative stress and cancer: paradigms in parallax. Nat. Rev. Cancer 8 (11), 875–879. doi:10.1038/nrc2522
Bernardini J. P., Lazarou M., Dewson G. (2017). Parkin and mitophagy in cancer. Oncogene 36 (10), 1315–1327. doi:10.1038/onc.2016.302
Birsoy K., Wang T., Chen W. W., Freinkman E., Abu-Remaileh M., Sabatini D. M. (2015). An essential role of the mitochondrial electron transport chain in cell proliferation is to enable aspartate synthesis. Cell. 162 (3), 540–551. doi:10.1016/j.cell.2015.07.016
Bogenhagen D., Clayton D. A. (1977). Mouse L cell mitochondrial DNA molecules are selected randomly for replication throughout the cell cycle. Cell. 11 (4), 719–727. doi:10.1016/0092-8674(77)90286-0
Bogenhagen D. F. (2012). Mitochondrial DNA nucleoid structure. Biochimica Biophysica Acta (BBA)-Gene Regul. Mech. 1819 (9-10), 914–920. doi:10.1016/j.bbagrm.2011.11.005
Boos F., Labbadia J., Herrmann J. M. (2020). How the mitoprotein-induced stress response safeguards the cytosol: a unified view. Trends Cell. Biol. 30 (3), 241–254. doi:10.1016/j.tcb.2019.12.003
Bowling A. C., Beal M. F. (1995). Bioenergetic and oxidative stress in neurodegenerative diseases. Life Sci. 56 (14), 1151–1171. doi:10.1016/0024-3205(95)00055-b
Boya P. (2024). Mitophagy curtails cytosolic mtDNA-dependent activation of cGAS/STING inflammation during aging. Nat. Commun., 15 (1), 830.
Bratic A., Kauppila T. E., Macao B., Grönke S., Siibak T., Stewart J. B., et al. (2015). Complementation between polymerase-and exonuclease-deficient mitochondrial DNA polymerase mutants in genomically engineered flies. Nat. Commun. 6 (1), 8808. doi:10.1038/ncomms9808
Bratic I., Trifunovic A. (2010). Mitochondrial energy metabolism and ageing. Biochimica Biophysica Acta (BBA)-Bioenergetics 1797 (6-7), 961–967. doi:10.1016/j.bbabio.2010.01.004
Bravo-San Pedro J. M., Kroemer G., Galluzzi L. (2017). Autophagy and mitophagy in cardiovascular disease. Circulation Res. 120 (11), 1812–1824. doi:10.1161/CIRCRESAHA.117.311082
Bua E., Johnson J., Herbst A., Delong B., McKenzie D., Salamat S., et al. (2006). Mitochondrial DNA–deletion mutations accumulate intracellularly to detrimental levels in aged human skeletal muscle fibers. Am. J. Hum. Genet. 79 (3), 469–480. doi:10.1086/507132
Burtscher J., Soltany A., Visavadiya N. P., Burtscher M., Millet G. P., Khoramipour K., et al. (2023). Mitochondrial stress and mitokines in aging. Aging Cell. 22 (2), e13770. doi:10.1111/acel.13770
Byun H. O., Jung H. J., Seo Y. H., Lee Y. K., Hwang S. C., Hwang E. S., et al. (2012). GSK3 inactivation is involved in mitochondrial complex IV defect in transforming growth factor (TGF) β1-induced senescence. Exp. Cell. Res. 318 (15), 1808–1819. doi:10.1016/j.yexcr.2012.04.012
Camell C. D., Yousefzadeh M. J., Zhu Y., Prata L. G. L., Huggins M. A., Pierson M., et al. (2021). Senolytics reduce coronavirus-related mortality in old mice. Science 373 (6552), eabe4832. doi:10.1126/science.abe4832
Camplejohn R. S., Gilchrist R., Easton D., McKenzie-Edwards E., Barnes D. M., Eccles D. M., et al. (2003). Apoptosis, ageing and cancer susceptibility. Br. J. cancer 88 (4), 487–490. doi:10.1038/sj.bjc.6600767
Casajus Pelegay E., Puzzo F., Yilmazer A., Cagin U. (2019). Targeting mitochondrial defects to increase longevity in animal models of neurodegenerative diseases. Rev. Biomark. Stud. Metabolic Metabolism-Related Disord. 1134, 89–110. doi:10.1007/978-3-030-12668-1_5
Cedikova M., Pitule P., Kripnerova M., Markova M., Kuncova J. (2016). Multiple roles of mitochondria in aging processes. Physiological Res. 65, S519-S531. doi:10.33549/physiolres.933538
Chapman J., Fielder E., Passos J. F. (2019). Mitochondrial dysfunction and cell senescence: deciphering a complex relationship. FEBS Lett. 593 (13), 1566–1579. doi:10.1002/1873-3468.13498
Chatterjee A., Dutta C. P. (1967). Alkaloids of Piper longum Linn—I: structure and synthesis of piperlongumine and piperlonguminine. Tetrahedron 23 (4), 1769–1781. doi:10.1016/s0040-4020(01)82575-8
Chen G., Kroemer G., Kepp O. (2020). Mitophagy: an emerging role in aging and age-associated diseases. Front. Cell. Dev. Biol. 8, 200. doi:10.3389/fcell.2020.00200
Chen H., Ruiz P. D., McKimpson W. M., Novikov L., Kitsis R. N., Gamble M. J. (2015). MacroH2A1 and ATM play opposing roles in paracrine senescence and the senescence-associated secretory phenotype. Mol. Cell. 59 (5), 719–731. doi:10.1016/j.molcel.2015.07.011
Chen H., Vermulst M., Wang Y. E., Chomyn A., Prolla T. A., McCaffery J. M., et al. (2010). Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 141 (2), 280–289. doi:10.1016/j.cell.2010.02.026
Chistiakov D. A., Sobenin I. A., Revin V. V., Orekhov A. N., Bobryshev Y. V. (2014). Mitochondrial aging and age-related dysfunction of mitochondria. BioMed Res. Int. 2014, 238463. doi:10.1155/2014/238463
Cho S. J., Pronko A., Yang J., Stout-Delgado H. (2023). Impact of senolytic treatment on gene expression in aged lung. Int. J. Mol. Sci. 24 (8), 7628. doi:10.3390/ijms24087628
Civitarese A. E., Carling S., Heilbronn L. K., Hulver M. H., Ukropcova B., Deutsch W. A., et al. (2007). Calorie restriction increases muscle mitochondrial biogenesis in healthy humans. PLoS Med. 4 (3), e76. doi:10.1371/journal.pmed.0040076
Cloonan S. M., Kim K., Esteves P., Trian T., Barnes P. J. (2020). Mitochondrial dysfunction in lung ageing and disease. Eur. Respir. Rev. 29 (157), 200165. doi:10.1183/16000617.0165-2020
Cohen A. A. (2018). Aging across the tree of life: the importance of a comparative perspective for the use of animal models in aging. Biochimica Biophysica Acta (BBA)-Molecular Basis Dis. 1864 (9), 2680–2689. doi:10.1016/j.bbadis.2017.05.028
Conte M., Ostan R., Fabbri C., Santoro A., Guidarelli G., Vitale G., et al. (2019). Human aging and longevity are characterized by high levels of mitokines. Journals Gerontology Ser. A 74 (5), 600–607. doi:10.1093/gerona/gly153
Costello M. J., Brennan L. A., Basu S., Chauss D., Mohamed A., Gilliland K. O., et al. (2013). Autophagy and mitophagy participate in ocular lens organelle degradation. Exp. eye Res. 116, 141–150. doi:10.1016/j.exer.2013.08.017
Crane J. D., Devries M. C., Safdar A., Hamadeh M. J., Tarnopolsky M. A. (2010). The effect of aging on human skeletal muscle mitochondrial and intramyocellular lipid ultrastructure. Journals Gerontology Ser. A Biomed. Sci. Med. Sci. 65 (2), 119–128. doi:10.1093/gerona/glp179
Cui H., Kong Y., Zhang H. (2012). Oxidative stress, mitochondrial dysfunction, and aging. J. signal Transduct. 2012, 646354. doi:10.1155/2012/646354
Davalli P., Marverti G., Lauriola A., D’Arca D. (2018). Targeting oxidatively induced DNA damage response in cancer: opportunities for novel cancer therapies. Oxidative Med. Cell. Longev. 2018, 2389523. doi:10.1155/2018/2389523
Decout A., Katz J. D., Venkatraman S., Ablasser A. (2021). The cGAS–STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 21 (9), 548–569. doi:10.1038/s41577-021-00524-z
Ding K., Wang H., Xu J., Li T., Zhang L., Ding Y., et al. (2014). Melatonin stimulates antioxidant enzymes and reduces oxidative stress in experimental traumatic brain injury: the Nrf2–ARE signaling pathway as a potential mechanism. Free Radic. Biol. Med. 73, 1–11. doi:10.1016/j.freeradbiomed.2014.04.031
Dziechciaz M., Filip R. (2014). Biological psychological and social determinants of old age: bio-psycho-social aspects of human aging. Ann. Agric. Environ. Med. 21 (4), 835–838. doi:10.5604/12321966.1129943
Eberle J., Hossini A. M. (2008). Expression and function of bcl-2 proteins in melanoma. Curr. genomics 9 (6), 409–419. doi:10.2174/138920208785699571
Edgar D., Trifunovic A. (2009). The mtDNA mutator mouse: dissecting mitochondrial involvement in aging. Aging (Albany NY) 1 (12), 1028–1032. doi:10.18632/aging.100109
Evangelou K., Vasileiou P. V., Papaspyropoulos A., Hazapis O., Petty R., Demaria M., et al. (2023). Cellular senescence and cardiovascular diseases: moving to the “heart” of the problem. Physiol. Rev. 103 (1), 609–647. doi:10.1152/physrev.00007.2022
Fivenson E. M., Lautrup S., Sun N., Scheibye-Knudsen M., Stevnsner T., Nilsen H., et al. (2017). Mitophagy in neurodegeneration and aging. Neurochem. Int. 109, 202–209. doi:10.1016/j.neuint.2017.02.007
Ganley I. G., Simonsen A. (2022). Diversity of mitophagy pathways at a glance. J. Cell. Sci. 135 (23), jcs259748. doi:10.1242/jcs.259748
Gioscia-Ryan R. A., Battson M. L., Cuevas L. M., Zigler M. C., Sindler A. L., Seals D. R. (2016). Voluntary aerobic exercise increases arterial resilience and mitochondrial health with aging in mice. Aging (Albany NY) 8 (11), 2897–2914. doi:10.18632/aging.101099
Greaves L. C., Nooteboom M., Elson J. L., Tuppen H. A., Taylor G. A., Commane D. M., et al. (2014). Clonal expansion of early to mid-life mitochondrial DNA point mutations drives mitochondrial dysfunction during human ageing. PLoS Genet. 10 (9), e1004620. doi:10.1371/journal.pgen.1004620
Greenhalgh D. G. (1998). The role of apoptosis in wound healing. Int. J. Biochem. Cell. Biol. 30 (9), 1019–1030. doi:10.1016/s1357-2725(98)00058-2
Hoshino A., Mita Y., Okawa Y., Ariyoshi M., Iwai-Kanai E., Ueyama T., et al. (2013). Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat. Commun. 4 (1), 2308. doi:10.1038/ncomms3308
Indo H. P., Yen H. C., Nakanishi I., Matsumoto K. I., Tamura M., Nagano Y., et al. (2015). A mitochondrial superoxide theory for oxidative stress diseases and aging. J. Clin. Biochem. Nutr. 56 (1), 1–7. doi:10.3164/jcbn.14-42
Iwashita M., Oka N., Ohkubo S., Saito M., Nakahata N. (2007). Piperlongumine, a constituent of Piper longum L., inhibits rabbit platelet aggregation as a thromboxane A2 receptor antagonist. Eur. J. Pharmacol. 570 (1-3), 38–42. doi:10.1016/j.ejphar.2007.05.073
Jang Y. Y., Sharkis S. J. (2007). A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood, J. Am. Soc. Hematol. 110 (8), 3056–3063. doi:10.1182/blood-2007-05-087759
Jiang Y. H., Jiang L. Y., Wang Y. C., Ma D. F., Li X. (2020). Quercetin attenuates atherosclerosis via modulating oxidized LDL-induced endothelial cellular senescence. Front. Pharmacol. 11, 512. doi:10.3389/fphar.2020.00512
Jiménez-Loygorri J. I., Villarejo-Zori B., Viedma-Poyatos Á., Zapata-Muñoz J., Benítez-Fernández R., Frutos-Lisón M. D., et al. (2024). Mitophagy curtails cytosolic mtDNA-dependent activation of cGAS/STING inflammation during aging. Nat. Commun. 15 (1), 830. doi:10.1038/s41467-024-45044-1
Jones R. G., Plas D. R., Kubek S., Buzzai M., Mu J., Xu Y., et al. (2005). AMP-activated protein kinase induces a p53-dependent metabolic checkpoint. Mol. Cell. 18 (3), 283–293. doi:10.1016/j.molcel.2005.03.027
Kauppila T. E., Kauppila J. H., Larsson N. G. (2017). Mammalian mitochondria and aging: an update. Cell. metab. 25 (1), 57–71. doi:10.1016/j.cmet.2016.09.017
Kennedy S. R., Salk J. J., Schmitt M. W., Loeb L. A. (2013). Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet. 9 (9), e1003794. doi:10.1371/journal.pgen.1003794
Khan N., Syed D. N., Ahmad N., Mukhtar H. (2013). Fisetin: a dietary antioxidant for health promotion. Antioxidants redox Signal. 19 (2), 151–162. doi:10.1089/ars.2012.4901
Kietzmann T., Petry A., Shvetsova A., Gerhold J. M., Görlach A. (2017). The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. Br. J. Pharmacol. 174 (12), 1533–1554. doi:10.1111/bph.13792
Kizhakekuttu T. J., Widlansky M. E. (2010). Natural antioxidants and hypertension: promise and challenges. Cardiovasc. Ther., 28 (4), e20–e32.
Kopustinskiene D. M., Bernatoniene J. (2021). Molecular mechanisms of melatonin-mediated cell protection and signaling in health and disease. Pharmaceutics 13 (2), 129. doi:10.3390/pharmaceutics13020129
Koshiba T., Detmer S. A., Kaiser J. T., Chen H., McCaffery J. M., Chan D. C. (2004). Structural basis of mitochondrial tethering by mitofusin complexes. Science 305 (5685), 858–862. doi:10.1126/science.1099793
Kowaltowski A. J. (2000). Alternative mitochondrial functions in cell physiopathology: beyond ATP production. Braz. J. Med. Biol. Res. 33, 241–250. doi:10.1590/s0100-879x2000000200014
Kujoth G. C., Bradshaw P. C., Haroon S., Prolla T. A. (2007). The role of mitochondrial DNA mutations in mammalian aging. PLoS Genet. 3 (2), e24. doi:10.1371/journal.pgen.0030024
Kurosaka K., Takahashi M., Watanabe N., Kobayashi Y. (2003). Silent cleanup of very early apoptotic cells by macrophages. J. Immunol. 171 (9), 4672–4679. doi:10.4049/jimmunol.171.9.4672
Kwak S., Ku S. K., Bae J. S. (2014). Fisetin inhibits high-glucose-induced vascular inflammation in vitro and in vivo. Inflamm. Res. 63, 779–787. doi:10.1007/s00011-014-0750-4
Lane N., Martin W. (2010). The energetics of genome complexity. Nature 467 (7318), 929–934. doi:10.1038/nature09486
Lee C., Zeng J., Drew B. G., Sallam T., Martin-Montalvo A., Wan J., et al. (2015). The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell. metab. 21 (3), 443–454. doi:10.1016/j.cmet.2015.02.009
Lee S., Jeong S. Y., Lim W. C., Kim S., Park Y. Y., Sun X., et al. (2007). Mitochondrial fission and fusion mediators, hFis1 and OPA1, modulate cellular senescence. J. Biol. Chem. 282 (31), 22977–22983. doi:10.1074/jbc.M700679200
Lee S. M., Dho S. H., Ju S. K., Maeng J. S., Kim J. Y., Kwon K. S. (2012). Cytosolic malate dehydrogenase regulates senescence in human fibroblasts. Biogerontology, 13, pp.525–536.doi:10.1007/s10522-012-9397-0
Lemasters J. J. (2005). Dying a thousand deaths: redundant pathways from different organelles to apoptosis and necrosis. Gastroenterology, 129 (1). doi:10.1053/j.gastro.2005.06.006
Levine B., Kroemer G. (2019). Biological functions of autophagy genes: a disease perspective. Cell. 176 (1), 11–42. doi:10.1016/j.cell.2018.09.048
Li Z., Zhang Z., Ren Y., Wang Y., Fang J., Yue H., et al. (2021). Aging and age-related diseases: from mechanisms to therapeutic strategies. Biogerontology 22 (2), 165–187. doi:10.1007/s10522-021-09910-5
Liao Z., Yeo H. L., Wong S. W., Zhao Y. (2021). Cellular senescence: mechanisms and therapeutic potential. Biomedicines 9 (12), 1769. doi:10.3390/biomedicines9121769
Liu J. P. (2014). Molecular mechanisms of ageing and related diseases. Clin. Exp. Pharmacol. Physiology 41 (7), 445–458. doi:10.1111/1440-1681.12247
Liu L., Trimarchi J. R., Smith P. J., Keefe D. L. (2002). Mitochondrial dysfunction leads to telomere attrition and genomic instability. Aging Cell. 1 (1), 40–46. doi:10.1046/j.1474-9728.2002.00004.x
Liu Y., Fiskum G., Schubert D. (2002). Generation of reactive oxygen species by the mitochondrial electron transport chain. J. Neurochem. 80 (5), 780–787. doi:10.1046/j.0022-3042.2002.00744.x
López-Otín C., Blasco M. A., Partridge L., Serrano M., Kroemer G. (2013). The hallmarks of aging. Cell. 153, 1194–1217. doi:10.1016/j.cell.2013.05.039
Madeo F., Tavernarakis N., Kroemer G. (2010). Can autophagy promote longevity? Nat. Cell. Biol. 12 (9), 842–846. doi:10.1038/ncb0910-842
Madreiter-Sokolowski C. T., Sokolowski A. A., Waldeck-Weiermair M., Malli R., Graier W. F. (2018). Targeting mitochondria to counteract age-related cellular dysfunction. Genes. 9 (3), 165. doi:10.3390/genes9030165
Mahmoud Y. I., Hegazy H. G. (2016). Ginger and alpha lipoic acid ameliorate age-related ultrastructural changes in rat liver. Biotech. Histochem. 91 (2), 86–95. doi:10.3109/10520295.2015.1076578
Manczak M., Calkins M. J., Reddy P. H. (2011). Impaired mitochondrial dynamics and abnormal interaction of amyloid beta with mitochondrial protein Drp1 in neurons from patients with Alzheimer's disease: implications for neuronal damage. Hum. Mol. Genet. 20 (13), 2495–2509. doi:10.1093/hmg/ddr139
Marković Z. S., Dimitrić Marković J. M., Milenković D., Filipović N. (2011). Mechanistic study of the structure–activity relationship for the free radical scavenging activity of baicalein. J. Mol. Model. 17, 2575–2584. doi:10.1007/s00894-010-0942-y
Martini H., Passos J. F. (2023). Cellular senescence: all roads lead to mitochondria. FEBS J. 290 (5), 1186–1202. doi:10.1111/febs.16361
Marycz K., Kornicka K., Basinska K., Czyrek A. (2016). Equine metabolic syndrome affects viability, senescence, and stress factors of equine adipose-derived mesenchymal stromal stem cells: new insight into EqASCs isolated from EMS horses in the context of their aging. Oxidative Med. Cell. Longev. 2016, 1–17. doi:10.1155/2016/4710326
Marzetti E., Leeuwenburgh C. (2006). Skeletal muscle apoptosis, sarcopenia and frailty at old age. Exp. Gerontol. 41 (12), 1234–1238. doi:10.1016/j.exger.2006.08.011
Mitchell S. J., Scheibye-Knudsen M., Longo D. L., de Cabo R. (2015). Animal models of aging research: implications for human aging and age-related diseases. Annu. Rev. Anim. Biosci. 3 (1), 283–303. doi:10.1146/annurev-animal-022114-110829
Moiseeva O., Bourdeau V., Roux A., Deschênes-Simard X., Ferbeyre G. (2009). Mitochondrial dysfunction contributes to oncogene-induced senescence. Mol. Cell. Biol. 29 (16), 4495–4507. doi:10.1128/MCB.01868-08
Molenaars M., Janssens G. E., Williams E. G., Jongejan A., Lan J., Rabot S., et al. (2020). A conserved mito-cytosolic translational balance links two longevity pathways. Cell. metab. 31 (3), 549–563. doi:10.1016/j.cmet.2020.01.011
Mondello C., Scovassi A. I. (2010). Apoptosis: a way to maintain healthy individuals. Genome Stab. Hum. Dis. 50, 307–323. doi:10.1007/978-90-481-3471-7_16
Morita M., Gravel S. P., Chenard V., Sikström K., Zheng L., Alain T., et al. (2013). mTORC1 controls mitochondrial activity and biogenesis through 4E-BP-dependent translational regulation. Cell. metab. 18 (5), 698–711. doi:10.1016/j.cmet.2013.10.001
Morita M., Gravel S. P., Hulea L., Larsson O., Pollak M., St-Pierre J., et al. (2015). mTOR coordinates protein synthesis, mitochondrial activity and proliferation. Cell. cycle 14 (4), 473–480. doi:10.4161/15384101.2014.991572
Morita M., Prudent J., Basu K., Goyon V., Katsumura S., Hulea L., et al. (2017). mTOR controls mitochondrial dynamics and cell survival via MTFP1. Mol. Cell. 67 (6), 922–935. doi:10.1016/j.molcel.2017.08.013
Mortensen M., Ferguson D. J., Simon A. K. (2010). Mitochondrial clearance by autophagy in developing erythrocytes: clearly important, but just how much so? Cell. cycle 9 (10), 1901–1906. doi:10.4161/cc.9.10.11603
Mottis A., Herzig S., Auwerx J. (2019). Mitocellular communication: shaping health and disease. Science 366 (6467), 827–832. doi:10.1126/science.aax3768
Nehlin J. O. (2023). Senolytic and senomorphic interventions to defy senescence-associated mitochondrial dysfunction. Adv. protein Chem. Struct. Biol. 136, 217–247. doi:10.1016/bs.apcsb.2023.02.020
Nicolson G. L. (2014). Mitochondrial dysfunction and chronic disease: treatment with natural supplements. Integr. Med. A Clinician's J. 13 (4), 35–43.
Nicolson G. L., Ash M. E. (2017). Membrane Lipid Replacement for chronic illnesses, aging and cancer using oral glycerolphospholipid formulations with fructooligosaccharides to restore phospholipid function in cellular membranes, organelles, cells and tissues. Biochimica Biophysica Acta (BBA)-Biomembranes 1859 (9), 1704–1724. doi:10.1016/j.bbamem.2017.04.013
Nilsson M. I., Tarnopolsky M. A. (2019). Mitochondria and aging—the role of exercise as a countermeasure. Biology 8 (2), 40. doi:10.3390/biology8020040
Nissanka N., Moraes C. T. (2018). Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett. 592 (5), 728–742. doi:10.1002/1873-3468.12956
Norat P., Soldozy S., Sokolowski J. D., Gorick C. M., Kumar J. S., Chae Y., et al. (2020). Mitochondrial dysfunction in neurological disorders: exploring mitochondrial transplantation. NPJ Regen. Med. 5 (1), 22. doi:10.1038/s41536-020-00107-x
Ohsumi Y. (2014). Historical landmarks of autophagy research. Cell. Res. 24 (1), 9–23. doi:10.1038/cr.2013.169
Palikaras K., Lionaki E., Tavernarakis N. (2018). Mechanisms of mitophagy in cellular homeostasis, physiology and pathology. Nat. Cell. Biol. 20 (9), 1013–1022. doi:10.1038/s41556-018-0176-2
Palomera-Avalos V., Griñán-Ferré C., Puigoriol-Ilamola D., Camins A., Sanfeliu C., Canudas A. M., et al. (2017). Resveratrol protects SAMP8 brain under metabolic stress: focus on mitochondrial function and Wnt pathway. Mol. Neurobiol. 54, 1661–1676. doi:10.1007/s12035-016-9770-0
Papackova Z., Cahova M. (2014). Important role of autophagy in regulation of metabolic processes in health, disease and aging. Physiological Res. 63 (4), 409–420. doi:10.33549/physiolres.932684
Paraskevaidi M., Martin-Hirsch P. L., Kyrgiou M., Martin F. L., 2017. Underlying role of mitochondrial mutagenesis in the pathogenesis of a disease and current approaches for translational research. Mutagenesis, 32(3), pp.347–342. doi:10.1038/msb.2010.5
Passos J. F., Nelson G., Wang C., Richter T., Simillion C., Proctor C. J., et al. (2010). Feedback between p21 and reactive oxygen production is necessary for cell senescence. Mol. Syst. Biol., 6(1), p.347. doi:10.1038/msb.2010.5
Passos J. F., Saretzki G., Ahmed S., Nelson G., Richter T., Peters H., et al. (2007). Mitochondrial dysfunction accounts for the stochastic heterogeneity in telomere-dependent senescence. PLoS Biol. 5 (5), e110. doi:10.1371/journal.pbio.0050110
Pérez V. I., Van Remmen H., Bokov A., Epstein C. J., Vijg J., Richardson A. (2009). The overexpression of major antioxidant enzymes does not extend the lifespan of mice. Aging Cell. 8 (1), 73–75. doi:10.1111/j.1474-9726.2008.00449.x
Pollack M., Leeuwenburgh C. (2001). Apoptosis and aging: role of the mitochondria. Journals Gerontology Ser. A Biol. Sci. Med. Sci. 56 (11), B475–B482. doi:10.1093/gerona/56.11.b475
Price N. L., Gomes A. P., Ling A. J., Duarte F. V., Martin-Montalvo A., North B. J., et al. (2012). SIRT1 is required for AMPK activation and the beneficial effects of resveratrol on mitochondrial function. Cell. metab. 15 (5), 675–690. doi:10.1016/j.cmet.2012.04.003
Rai S. N., Singh C., Singh A., Singh M. P., Singh B. K. (2020). Mitochondrial dysfunction: a potential therapeutic target to treat Alzheimer’s disease. Mol. Neurobiol. 57, 3075–3088. doi:10.1007/s12035-020-01945-y
Ravindran J., Gupta N., Agrawal M., Bala Bhaskar A. S., Lakshmana Rao P. V. (2011). Modulation of ROS/MAPK signaling pathways by okadaic acid leads to cell death via, mitochondrial mediated caspase-dependent mechanism. Apoptosis 16, 145–161. doi:10.1007/s10495-010-0554-0
Rinnerthaler M., Bischof J., Streubel M. K., Trost A., Richter K. (2015). Oxidative stress in aging human skin. Biomolecules 5 (2), 545–589.
Ristow M., Schmeisser K. (2014). Mitohormesis: promoting health and lifespan by increased levels of reactive oxygen species (ROS). Dose-response 12 (2), 288–341. doi:10.2203/dose-response.13-035.Ristow
Rizwan S., ReddySekhar P., MalikAsrar B. (2014). Reactive oxygen species in inflammation and tissue injury. Antioxidants redox Signal. 20, 1126–1167. doi:10.1089/ars.2012.5149
Ross J. M., Coppotelli G., Hoffer B. J., Olson L. (2014). Maternally transmitted mitochondrial DNA mutations can reduce lifespan. Sci. Rep. 4 (1), 6569. doi:10.1038/srep06569
Ross J. M., Stewart J. B., Hagström E., Brené S., Mourier A., Coppotelli G., et al. (2013). Germline mitochondrial DNA mutations aggravate ageing and can impair brain development. Nature 501 (7467), 412–415. doi:10.1038/nature12474
Roy M., Reddy P. H., Iijima M., Sesaki H. (2015). Mitochondrial division and fusion in metabolism. Curr. Opin. Cell. Biol. 33, 111–118. doi:10.1016/j.ceb.2015.02.001
Rubinsztein D. C., Mariño G., Kroemer G. (2011). Autophagy and aging. Cell. 146 (5), 682–695. doi:10.1016/j.cell.2011.07.030
Saccon T. D., Nagpal R., Yadav H., Cavalcante M. B., Nunes A. D. D. C., Schneider A., et al. (2021). Senolytic combination of dasatinib and quercetin alleviates intestinal senescence and inflammation and modulates the gut microbiome in aged mice. Journals Gerontology Ser. A 76 (11), 1895–1905. doi:10.1093/gerona/glab002
Salabei J. K., Hill B. G. (2013). Mitochondrial fission induced by platelet-derived growth factor regulates vascular smooth muscle cell bioenergetics and cell proliferation. Redox Biol. 1 (1), 542–551. doi:10.1016/j.redox.2013.10.011
Sato M., Sato K. (2013). Maternal inheritance of mitochondrial DNA by diverse mechanisms to eliminate paternal mitochondrial DNA. Biochimica Biophysica Acta (BBA)-Molecular Cell. Res. 1833 (8), 1979–1984. doi:10.1016/j.bbamcr.2013.03.010
Scott A. C., Dündar F., Zumbo P., Chandran S. S., Klebanoff C. A., Shakiba M., et al. (2019). TOX is a critical regulator of tumour-specific T cell differentiation. Nature 571 (7764), 270–274.
Scarpulla R. C. (2008). Nuclear control of respiratory chain expression by nuclear respiratory factors and PGC-1-related coactivator. Ann. N. Y. Acad. Sci. 1147 (1), 321–334. doi:10.1196/annals.1427.006
Sevrioukova I. F. (2011). Apoptosis-inducing factor: structure, function, and redox regulation. Antioxidants redox Signal. 14 (12), 2545–2579. doi:10.1089/ars.2010.3445
Shao Y., Sun L., Yang G., Wang W., Liu X., Du T., et al. (2022). Icariin protects vertebral endplate chondrocytes against apoptosis and degeneration via activating Nrf-2/HO-1 pathway. Front. Pharmacol. 13, 937502. doi:10.3389/fphar.2022.937502
Shigenaga M. K., Hagen T. M., Ames B. N. (1994). Oxidative damage and mitochondrial decay in aging. Proc. Natl. Acad. Sci. 91 (23), 10771–10778. doi:10.1073/pnas.91.23.10771
Short K. R., Bigelow M. L., Kahl J., Singh R., Coenen-Schimke J., Raghavakaimal S., et al. (2005). Decline in skeletal muscle mitochondrial function with aging in humans. Proc. Natl. Acad. Sci. 102 (15), 5618–5623. doi:10.1073/pnas.0501559102
Shum L. C., White N. S., Nadtochiy S. M., Bentley K. L. D. M., Brookes P. S., Jonason J. H., et al. (2016). Cyclophilin D knock-out mice show enhanced resistance to osteoporosis and to metabolic changes observed in aging bone. PloS one 11 (5), e0155709. doi:10.1371/journal.pone.0155709
Silva Ramos E., Motori E., Brüser C., Kühl I., Yeroslaviz A., Ruzzenente B., et al. (2019). Mitochondrial fusion is required for regulation of mitochondrial DNA replication. PLoS Genet. 15 (6), e1008085. doi:10.1371/journal.pgen.1008085
Sonawane S. K., Balmik A. A., Boral D., Ramasamy S., Chinnathambi S. (2019). Baicalein suppresses Repeat Tau fibrillization by sequestering oligomers. Archives Biochem. biophysics 675, 108119. doi:10.1016/j.abb.2019.108119
Srivastava S. (2017). The mitochondrial basis of aging and age-related disorders. Genes. 8 (12), 398. doi:10.3390/genes8120398
Stancu A. L. (2015). AMPK activation can delay aging. Discoveries 3 (4), e53. doi:10.15190/d.2015.45
Sun N., Youle R. J., Finkel T. (2016). The mitochondrial basis of aging. Mol. Cell. 61 (5), 654–666. doi:10.1016/j.molcel.2016.01.028
Sun N., Yun J., Liu J., Malide D., Liu C., Rovira I. I., et al. (2015). Measuring in vivo mitophagy. Mol. Cell. 60 (4), 685–696. doi:10.1016/j.molcel.2015.10.009
Taanman J. W. (1999). The mitochondrial genome: structure, transcription, translation and replication. Biochimica Biophysica Acta (BBA)-Bioenergetics 1410 (2), 103–123. doi:10.1016/s0005-2728(98)00161-3
Tan D. X., Manchester L. C., Qin L., Reiter R. J. (2016). Melatonin: a mitochondrial targeting molecule involving mitochondrial protection and dynamics. Int. J. Mol. Sci. 17 (12), 2124. doi:10.3390/ijms17122124
Taniguchi H., Tanisawa K., Sun X., Kubo T., Higuchi M. (2016). Endurance exercise reduces hepatic fat content and serum fibroblast growth factor 21 levels in elderly men. J. Clin. Endocrinol. 101 (1), 191–198. doi:10.1210/jc.2015-3308
Terman A., Kurz T., Navratil M., Arriaga E. A., Brunk U. T. (2010). Mitochondrial turnover and aging of long-lived postmitotic cells: the mitochondrial–lysosomal axis theory of aging. Antioxidants redox Signal. 12 (4), 503–535. doi:10.1089/ars.2009.2598
Tower J. (2015). Programmed cell death in aging. Ageing Res. Rev. 23, 90–100. doi:10.1016/j.arr.2015.04.002
Toyama E. Q., Herzig S., Courchet J., Lewis Jr T. L., Losón O. C., Hellberg K., et al. (2016). Metabolism. AMP-activated protein kinase mediates mitochondrial fission in response to energy stress. Science 351 (6270), 275–281. doi:10.1126/science.aab4138
Trifunovic A., Wredenberg A., Falkenberg M., Spelbrink J. N., Rovio A. T., Bruder C. E., et al. (2004). Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429 (6990), 417–423. doi:10.1038/nature02517
Turk P. W., Laayoun A., Smith S. S., Weitzman S. A. (1995). DNA adduct 8-hydroxyl-2′-deoxyguanosine (8-hydroxyguanine) affects function of human DNA methyltransferase. Carcinogenesis 16 (5), 1253–1255. doi:10.1093/carcin/16.5.1253
Tzameli I. (2012). The evolving role of mitochondria in metabolism. Trends Endocrinol. Metabolism 23 (9), 417–419. doi:10.1016/j.tem.2012.07.008
United Nations Department of Economic and Social Affairs (2020). World population ageing 2020 highlights: living arrangements of older persons. USA: ST/ESA/SER.A/451.
Van der Rijt S., Molenaars M., McIntyre R. L., Janssens G. E., Houtkooper R. H. (2020). Integrating the hallmarks of aging throughout the tree of life: a focus on mitochondrial dysfunction. Front. Cell. Dev. Biol. 8, 594416. doi:10.3389/fcell.2020.594416
van der Veer E., Ho C., O'Neil C., Barbosa N., Scott R., Cregan S. P., et al. (2007). Extension of human cell lifespan by nicotinamide phosphoribosyltransferase. J. Biol. Chem. 282 (15), 10841–10845. doi:10.1074/jbc.C700018200
Van Deursen J. M. (2014). The role of senescent cells in ageing. Nature 509 (7501), 439–446. doi:10.1038/nature13193
Wall C. E., Rose C. M., Adrian M., Zeng Y. J., Kirkpatrick D. S., Bingol B. (2019). PPEF2 opposes PINK1-mediated mitochondrial quality control by dephosphorylating ubiquitin. Cell. Rep. 29 (10), 3280–3292. doi:10.1016/j.celrep.2019.10.130
Wallace D. C. (2010). Mitochondrial DNA mutations in disease and aging. Environ. Mol. Mutagen. 51 (5), 440–450. doi:10.1002/em.20586
Wang J., Toan S., Zhou H. (2020a). New insights into the role of mitochondria in cardiac microvascular ischemia/reperfusion injury. Angiogenesis 23, 299–314. doi:10.1007/s10456-020-09720-2
Wang L., Ma Y., Wei W., Wan P., Liu K., Xu M., et al. (2020b). Cadherin repeat 5 mutation associated with Bt resistance in a field-derived strain of pink bollworm. Sci. Rep. 10 (1), 16840. doi:10.1038/s41598-020-74102-z
Wang R., Li J., Niu D. B., Xu F. Y., Zeng X. A. (2021). Protective effect of baicalein on DNA oxidative damage and its binding mechanism with DNA: an in vitro and molecular docking study. Spectrochimica Acta Part A Mol. Biomol. Spectrosc. 253, 119605. doi:10.1016/j.saa.2021.119605
Wiley C. D., Velarde M. C., Lecot P., Liu S. U., Sarnoski E. A., Freund A., et al. (2016). Mitochondrial dysfunction induces senescence with a distinct secretory phenotype. Cell. metab. 23 (2), 303–314. doi:10.1016/j.cmet.2015.11.011
Wu Y., Sun L., Zhuang Z., Hu X., Dong D. (2022). Mitochondrial-derived peptides in diabetes and its complications. Front. Endocrinol. 12, 808120. doi:10.3389/fendo.2021.808120
Xu M., Pirtskhalava T., Farr J. N., Weigand B. M., Palmer A. K., Weivoda M. M., et al. (2018). Senolytics improve physical function and increase lifespan in old age. Nat. Med. 24 (8), 1246–1256. doi:10.1038/s41591-018-0092-9
Yan L. J. (2014). Positive oxidative stress in aging and aging-related disease tolerance. Redox Biol. 2, 165–169. doi:10.1016/j.redox.2014.01.002
Yang S., Lian G. (2020). ROS and diseases: role in metabolism and energy supply. Mol. Cell. Biochem. 467, 1–12. doi:10.1007/s11010-019-03667-9
Yen T. C., Chen Y. S., King K. L., Yeh S. H., Wei Y. H. (1989). Liver mitochondrial respiratory functions decline with age. Biochem. biophysical Res. Commun. 165 (3), 944–1003. doi:10.1016/0006-291x(89)92701-0
Yoon Y. S., Byun H. O., Cho H., Kim B. K., Yoon G. (2003). Complex II defect via down-regulation of iron-sulfur subunit induces mitochondrial dysfunction and cell cycle delay in iron chelation-induced senescence-associated growth arrest. J. Biol. Chem. 278 (51), 51577–51586. doi:10.1074/jbc.M308489200
Yoon Y. S., Yoon D. S., Lim I. K., Yoon S. H., Chung H. Y., Rojo M., et al. (2006). Formation of elongated giant mitochondria in DFO-induced cellular senescence: involvement of enhanced fusion process through modulation of Fis1. J. Cell. physiology 209 (2), 468–480. doi:10.1002/jcp.20753
Youm Y. H., Horvath T. L., Mangelsdorf D. J., Kliewer S. A., Dixit V. D. (2016). Prolongevity hormone FGF21 protects against immune senescence by delaying age-related thymic involution. Proc. Natl. Acad. Sci. 113 (4), 1026–1031. doi:10.1073/pnas.1514511113
Yousefzadeh M. J., Zhu Y. I., McGowan S. J., Angelini L., Fuhrmann-Stroissnigg H., Xu M., et al. (2018). Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 36, 18–28. doi:10.1016/j.ebiom.2018.09.015
Zhang H., Fealy C. E., Kirwan J. P. (2019). Exercise training promotes a GDF15-associated reduction in fat mass in older adults with obesity. Am. J. Physiology-Endocrinology Metabolism 316 (5), E829-E836–E836. doi:10.1152/ajpendo.00439.2018
Zhang H., Menzies K. J., Auwerx J. (2018). The role of mitochondria in stem cell fate and aging. Development 145 (8), dev143420. doi:10.1242/dev.143420
Zhou Z., Fan Y., Zong R., Tan K. (2022). The mitochondrial unfolded protein response: a multitasking giant in the fight against human diseases. Ageing Res. Rev. 81, 101702. doi:10.1016/j.arr.2022.101702
Zhu J., Wang K. Z., Chu C. T. (2013). After the banquet: mitochondrial biogenesis, mitophagy, and cell survival. Autophagy 9 (11), 1663–1676. doi:10.4161/auto.24135
Ziegler D. V., Wiley C. D., Velarde M. C. (2015). Mitochondrial effectors of cellular senescence: beyond the free radical theory of aging. Aging Cell. 14 (1), 1–7. doi:10.1111/acel.12287
Zimmermann A., Madreiter-Sokolowski C., Stryeck S., Abdellatif M. (2021). Targeting the mitochondria-proteostasis axis to delay aging. Front. Cell. Dev. Biol. 9, 656201. doi:10.3389/fcell.2021.656201
Keywords: mitochondrial dysfunction, mitochondrial DNA, ROS production, chromosomal aberrations, mitophagy
Citation: Somasundaram I, Jain SM, Blot-Chabaud M, Pathak S, Banerjee A, Rawat S, Sharma NR and Duttaroy AK (2024) Mitochondrial dysfunction and its association with age-related disorders. Front. Physiol. 15:1384966. doi: 10.3389/fphys.2024.1384966
Received: 12 February 2024; Accepted: 10 June 2024;
Published: 02 July 2024.
Edited by:
Martin Van Der Laan, Saarland University, GermanyReviewed by:
François Singh, University of Iceland, IcelandCopyright © 2024 Somasundaram, Jain, Blot-Chabaud, Pathak, Banerjee, Rawat, Sharma and Duttaroy. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Asim K. Duttaroy, YS5rLmR1dHRhcm95QG1lZGlzaW4udWlvLm5v; Indumathi Somasundaram, ZHJpbmR1bWF0aGlzb21hc3VuZGFyYW1AZ21haWwuY29t; Surajit Pathak, ZHJzdXJhaml0cGF0aGFrQGNhcmUuZWR1Lmlu
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.