 Heather VerValin Buck
Heather VerValin Buck Joseph Paul Stains
Joseph Paul Stains- School of Medicine, University of Maryland, Baltimore, MD, United States
Low bone mass is a pervasive global health concern, with implications for osteoporosis, frailty, disability, and mortality. Lifestyle factors, including sedentary habits, metabolic dysfunction, and an aging population, contribute to the escalating prevalence of osteopenia and osteoporosis. The application of mechanical load to bone through physical activity and exercise prevents bone loss, while sufficient mechanical load stimulates new bone mass acquisition. Osteocytes, cells embedded within the bone, receive mechanical signals and translate these mechanical cues into biological signals, termed mechano-transduction. Mechano-transduction signals regulate other bone resident cells, such as osteoblasts and osteoclasts, to orchestrate changes in bone mass. This review explores the mechanisms through which osteocyte-mediated response to mechanical loading regulates osteoblast differentiation and bone formation. An overview of bone cell biology and the impact of mechanical load will be provided, with emphasis on the mechanical cues, mechano-transduction pathways, and factors that direct progenitor cells toward the osteoblast lineage. While there are a wide range of clinically available treatments for osteoporosis, the majority act through manipulation of the osteoclast and may have significant disadvantages. Despite the central role of osteoblasts to the deposition of new bone, few therapies directly target osteoblasts for the preservation of bone mass. Improved understanding of the mechanisms leading to osteoblastogenesis may reveal novel targets for translational investigation.
1 Introduction
In the healthy skeleton, bone must respond to environmental cues to accommodate the organism’s needs. This is easily demonstrated in athletes, where the results of repetitive high-intensity loading are clear. Both male and female collegiate tennis players, for example, develop significantly increased bone mineral density in the dominant arm, as compared to the contralateral arm (McClanahan et al., 2002). Significant differences between an athlete’s dominant and non-dominant arm can also be observed in a range of sports, like baseball, golf, and volleyball. Through training and competition, athletes expose their skeletons to repeated and intense strain; their bones respond to increased strain by upregulating anabolic bone processes, like osteoblast differentiation, extracellular matrix deposition, and mineralization to strengthen the bone and resist fracture. This increase in bone mass is accomplished through the close interplay of various bone-resident cells, namely, osteoblasts, osteocytes, and osteoclasts. Generally, osteoblasts deposit and mineralize the collagen-rich extracellular matrix that will become new bone. Osteocytes receive mechanical and hormonal signals and transduce these cues among themselves and to osteoblasts and osteoclasts to coordinate bone deposition and resorption. Osteoclasts resorb mineralized bone and digest the collagenous extracellular matrix. Osteoclasts and osteoblasts are on-demand cells that form when they are needed and later become quiescent or undergo apoptosis. In contrast, osteocytes are long-lived cells that mediate the activation of osteoblast and osteoclasts. This review will discuss the bone anabolic response to mechanical load, with a focus on how osteocytes sense and respond to loading to affect osteoblast differentiation.
1.1 Osteoblasts
Osteoblasts are cuboidal cells found on bony surfaces, where they are the primary producer and depositor of the extracellular matrix that will become mineralized bone (Mohamed, 2008). They arise from mesenchymal progenitor cells, which can differentiate into a range of cells such as adipocytes, chondrocytes, and myocytes. They express mechanoresponsive calcium channels, like Piezo1, and have been shown in vitro to respond to tensile load with increased alkaline phosphatase expression and mineralization (Liu et al., 2022).
Following local bone resorption by osteoclasts, osteoblasts are recruited to the newly exposed bone surface where they begin to secrete collagen and other extracellular matrix proteins at their apical face (Blair et al., 2017). This newly formed extracellular matrix, termed osteoid, is an arrangement of primarily type I collagen trimers, which are highly interlinked, along with other extracellular matrix proteins like osteopontin, osteocalcin, bone sialoprotein, and osteonectin (Orgel et al., 2001). Osteoid also contains other organic components, like embedded latent growth factors, and provides the lattice into which mineralization will occur (Linkhart et al., 1996).
The inorganic component of bone, hydroxyapatite, is comprised of phosphate and calcium, vital minerals found in circulation. Following matrix deposition, osteoblasts support hydroxyapatite crystal nucleation by expressing proteins such as alkaline phosphatase, which provide the appropriate inorganic phosphate and hydrolyze the mineralization inhibitor, pyrophosphate (Andrade et al., 2019), and bone sialoprotein, which stimulates hydroxyapatite nucleation (Hunter et al., 1996). The presence of fibrillar collagen, alkaline phosphatase, and the absence of endogenous inhibitors of mineralization seem to be the key combination that permits this tissue to mineralize (Murshed et al., 2005). After mineralization, osteoblasts either become entrapped into the accumulating bone matrix and differentiate into osteocytes, become quiescent bone lining cells, or undergo apoptosis (Dallas and Bonewald, 2010).
1.2 Osteocytes
Osteocytes, which are found throughout mineralized bone in most vertebrates (Shahar and Dean, 2013), are the primary integrator of mechanical signaling (Schaffler et al., 2014), utilizing their distinctive location encased in mineralized bone and their neuron-like morphology to detect mechanical load in the surrounding bone. Osteocytic cell bodies are located within ovular chambers, called lacunae, and their long, thin processes reside in a geometrically complex, three-dimensional series of tunnels, called canaliculi. This series of chambers and tunnels, the lacunar-canalicular network, is highly interconnected and provides a conduit for direct cell-to-cell communication amongst osteocytes and between osteocytes and other bone resident cells, mediated by gap junctions (Moorer et al., 2017). Surrounding the osteocytes, the fluid-filled space within the lacunar-canalicular system contains extracellular matrix elements like collagen, glycocalyx (Burra et al., 2011), and perlecan (Thompson et al., 2011), which is believed to be a primary tethering protein, critical for load response (Wang et al., 2014).
When mechanically stimulated, osteocytes express many bone anabolic effectors, like WNT, nitric oxide, and PGE2, which act to upregulate the β-catenin pathway; unloaded osteocytes express catabolic signals like RANKL and the β-catenin pathway inhibitor sclerostin. Osteocytes are terminally differentiated osteoblasts (Mullen et al., 2013), and while the mechanism of their encapsulation within bone matrix has not been fully clarified (Franz-Odendaal et al., 2006; Robling and Bonewald, 2020), they are often distinguished from osteoblasts by their expression of markers such as DMP1, FGF23, podoplanin, and sclerostin (Delgado-Calle and Bellido, 2022).
1.3 Osteoclasts
Osteoclasts are large, multinucleated cells derived from macrophages and are the primary facilitator of bone resorption. They originate from hematopoietic stem cells and their differentiation is promoted by the binding of RANKL (receptor activator of NFκB ligand) (Yasuda et al., 1998) to its cognate receptor (RANK) on the surface of osteoclast progenitors. Osteocytic RANKL expression is increased during unloading, supporting increased osteoclastogenesis and bone resorption during disuse (Ono et al., 2020). Emerging data suggest that osteoclasts may also directly respond to mechanical stimulation (Dsouza and Komarova, 2023), but the way in which this response may participate in anabolic processes is not settled.
To remove mineralized bone, osteoclasts adhere to the bone surface, form an actin ring, and vectorially secrete acidic vesicles into the bone-facing extracellular milieu (Teitelbaum, 2011). The accumulation of hydrogen protons and proteolytic enzymes on the bone-facing surface dissolve mineralized bone and extracellular matrix, liberating calcium and phosphate ions and forming a resorption pit (Han et al., 2019).
1.4 Osteoprogenitors and the osteoblast cell lineage
A broad group of mesenchymal progenitor cells, commonly found in the bone marrow cavity, around vasculature, or in the fibrous periosteal and endocortical membrane surfaces that line cortical bone, give rise to skeletally associated cells including chondrocytes, adipocytes, and osteoblasts (Kurenkova et al., 2020; Matsushita et al., 2020). Lineage allocation of these progenitor cells is mediated by specific cues and activators. Canonically, the transcription factor Sox9 is required for the differentiation of chondrocytes (Wheatley et al., 1996), cells necessary for growth, limb development, and cartilage maintenance. Differentiation into adipocytes, cells that support metabolism and lipid storage, is supported by the transcription factor PPARγ (Lefterova et al., 2008), while osteoblast differentiation requires the transcription factors RUNX2 (Drissi et al., 2000) and Osterix (Nakashima et al., 2002).
There are several influences that can bias mesenchymal progenitor cells towards osteoblast or adipocyte differentiation. For example, consistent administration of a high-fat diet increases the relative differentiation of progenitor cells into adipocytes over osteoblasts (Parhami et al., 2001), while osteoblastogenesis is favored after mechanical load (Sen et al., 2008). The stiffness of the extracellular matrix surrounding bone-resident cells is also believed to influence differentiation and anabolic response. It has been demonstrated in vitro that mesenchymal progenitor cells are more likely to differentiate into the osteogenic lineage (Engler et al., 2006) and that expression of osteoblastic deposition markers (Zhang et al., 2017) increase in the presence of a relatively stiff extracellular matrix.
Even within the osteoblast lineage, varied cues can activate distinct progenitor populations to differentiate into bone-depositing osteoblasts. For example, discrete groups of skeletal progenitor cells may be recruited from the periosteal or endosteal compartments in response to fracture or mechanical loading (Atria and Castillo, 2023), though this is still an area of emerging research.
1.5 Mechanical load: Tissue and cellular responses
Like many other physiological systems, bone mass is regulated around a homeostatic set point. Mechanical loading, such as from customary physical activity to which an organism is acclimated, will be within the homeostatic range and insufficient to induce a net change in bone mass (Robling and Turner, 2009). This broad range of mechanical loading is referred to as the “adapted window” in Harold Frost’s mechano-stat theory (Frost, 2003) and is also known as the lazy zone. This level of load will be different for every organism and will adapt to the organism’s activity level and peak loading forces. For example, an individual who regularly strength trains will accumulate more skeletal mass to adapt to increased load, but may not receive additional anabolic benefit from exercising with a weight to which they have habituated, even if that weight would be sufficient to drive deposition in a novice weightlifter.
The mechanical stimulation experienced by a population of osteocytes (known as strain) is in part controlled by the strength and resistance of their surrounding bone. Relatively high loads cause shape changes in the bone, which shifts fluid within the lacunar-canalicular system, stimulating osteocytes (Price et al., 2011). These osteocytes locally induce osteoblastogenesis and increased bone mass. After mineralization, the same level of load may no longer be sufficient to cause deflection of the thicker, more resistant bone, resulting in reduced strain perceived by osteocytes. Consistent lack strain leads to resorption and thinner, more easily deflectable bones that will become more responsive to a given load. This process of continual readjustment tunes bone to the changing needs of the organism (Christen et al., 2014).
Load above the homeostatic range activates osteoblastogenesis (Turner et al., 1998) and leads to the deposition of new bone; this range is known as the bone overload zone. Deposition does not occur globally, however, and is generally proportional to the amount of experienced strain (Robling et al., 2002). In contrast to mechanical loading, unloading or disuse refers to a significant loss of mechanical stimuli and may be the result of local immobilization after fracture (Ceroni et al., 2012) or paralysis (Dionyssiotis et al., 2007), or may affect the total organism, as during bedrest (Leblanc et al., 1990) or microgravity (LeBlanc et al., 2000). Sustained unloading leads to an increase in osteoclast number and activity (Ishijima et al., 2001) and decreased osteoblastogenesis and deposition (Dufour et al., 2008), resulting in resorption and a net loss of bone mass. For either strenuous activity or disuse, bone adapts to the loading environment to which it is routinely exposed.
When sufficient force is applied to bone it flexes, causing fluid within the lacunar-canalicular system to be displaced (Piekarski and Munro, 1977). The passage of fluid over the surface of the osteocyte creates shear stress against the cell, while movement of the tethering proteins that link the osteocyte to the bone extracellular matrix is thought to amplify the mechanical stimulation of the osteocyte (You et al., 2001). Mechanical signals are transmitted into the osteocyte through factors such as force-gated ion channels like Piezo1 (Li et al., 2019) and TRPV4 (Lyons et al., 2017; Williams et al., 2020), which allow for rapid calcium influx and deformation of the cytoskeleton (Han et al., 2004; Lyons et al., 2017) and primary cilia (Lee et al., 2015), which engage a range of anabolic signaling elements in the local area, including nitric oxide (Klein-Nulend et al., 1995), reactive oxygen (Lyons et al., 2017), PGE2 (Jiang and Cheng, 2001), IGF1 (Lean et al., 1996), BMP-2 (da Silva Madaleno et al., 2020) and WNTs (Du et al., 2019). Additionally, loading causes osteocytes to reduce expression of proteins, like sclerostin and DKK1. Ultimately, many of these signaling factors participate in the β-catenin pathway.
In response to mechanical loading, osteocytes seem to respond in a binary manner with respect to calcium influx (Schaffler and Kennedy, 2012), with each osteocyte in either a ‘loaded’ or ‘non-loaded’ state. One loading event does not stimulate every osteocyte, however; osteocytes in regions of bone experiencing the highest mechanical strain are more likely to be stimulated, and osteocytes in low strain areas are less likely to be stimulated. The magnitude of the loading event can increase the percentage of osteocytes in a given anatomic region that go into the ‘loaded’ state, with strain frequency being the largest determinant of successful stimulation (Lewis et al., 2017). The type of force applied, like tensile, compressive, or stretch, can also influence the molecular response (Josephson and Morgan, 2023). The overall distribution and number of these ‘loaded’ and ‘non-loaded’ osteocytes contributes to the tuning of local bone mass.
2 Molecular signals mediate the cellular effects of mechanical loading
2.1 Sclerostin
Sclerostin is a glycoprotein secreted by mature osteocytes that can signal to a variety of cells and tissues (Weivoda et al., 2017) and also control deposition and resorption through regulation of bone-resident cells. Disruptive variants or genetic knockout of the Sost gene that encodes sclerostin are sufficient to produce a phenotype characterized by excessive skeletal deposition in both humans (Brunkow et al., 2001) and mice (Li et al., 2008).
An osteocyte in the lazy zone, or one experiencing disuse, will constitutively secrete sclerostin, signaling that bone formation is not needed. In response to mechanical loading and other bone anabolic cues, sclerostin protein is reduced and bone formation is unleashed.
When secreted by osteocytes, sclerostin suppresses osteoblast differentiation and promotes osteoclast number and activity (Suen and Qin, 2016). Additionally, sclerostin may bind to osteoblasts, preventing their differentiation into osteocytes (Atkins et al., 2011). Sclerostin also functions as an endocrine signal, perhaps facilitating glucose and fatty acid availability for new bone formation when sclerostin expression is low (Riddle, 2023), but the direct effect of these processes on bone mass have yet to be fully clarified (Fu et al., 2022; Gu et al., 2023).
Sclerostin suppresses the ability of mesenchymal progenitor cells, pre-osteoblasts, and bone lining cells to differentiate into osteoblasts via inhibition of the β-catenin pathway (Rutkovskiy et al., 2016). Sclerostin binds to LRP5/6, where it acts as a competitive inhibitor of WNT family members (Li et al., 2005), like WNT3 and WNT10 (Krishnan et al., 2006). Once sclerostin binds to LRP5/6, LRP5/6 cannot form a complex with Frizzled, and β-catenin becomes ubiquitinated and is degraded by the proteosome (Liao et al., 2022). Other mechanically regulated inhibitors of WNT binding, like DKK1, have been shown to suppress osteoblast differentiation in a similar way (Ke et al., 2012).
In contrast, a decrease in sclerotin abundance allows WNT to bind with LRP5/6, disrupting the β-catenin destruction complex (Stamos and Weis, 2013). Stabilized β-catenin translocates to the nucleus where it acts to support Runx2 and Osx (osterix) expression, leading to osteoblast differentiation and the expression of osteoid-associated genes (Felber et al., 2015). Thus, β-catenin signaling is fundamentally important to increased osteoblast number and subsequent matrix deposition.
In addition to bone formation, sclerostin may also regulate bone resorption. In vitro, treatment of osteocyte-like cells with exogenous sclerostin leads to increased RANKL expression (Wijenayaka et al., 2011). Overall, when sclerostin levels are high, osteoblast differentiation and bone formation are inhibited, while osteoclast formation and bone resorption are elevated. High levels of sclerostin, as is typically seen with disuse, is net catabolic to bone.
Loss of sclerostin abundance following mechanical load supports a powerful switch from catabolic to anabolic signaling. Short bouts of axial loading within the high-physiological range in vivo are sufficient to cause a temporary loss of sclerostin and induce osteoprotegerin (OPG) expression and an increase in bone mass in regions of high strain (Robling et al., 2008). Sclerostin protein undergoes tight, spatial regulation in regions of greatest mechanical strain, depressing bone formation where load is highest and bone is most likely to fail if appropriate adaptation does not occur. (Figure 1). This strain-dependent loss of sclerostin has been demonstrated following fluid shear stress in vitro as well (Gould et al., 2021a).
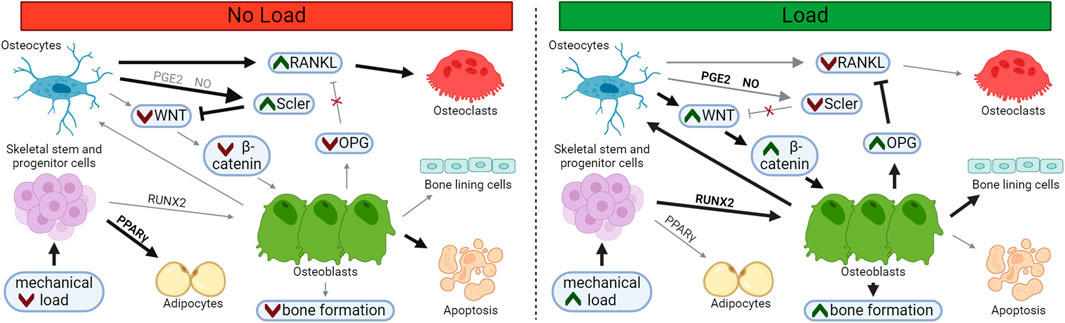
FIGURE 1. Loading Supports Osteoblast Differentiation and Suppresses Osteoclast Differentiation. In unloaded bone, sclerostin and RANKL expression is upregulated, supporting osteoclastogenesis and resorption. Following load, osteoblast and osteocyte differentiation increases, while osteoblast apoptosis and osteoclast differentiation decreases, leading to bone matrix deposition. Osteoblasts may embed and mature into osteocytes within the newly formed bone or may transition to quiescent bone lining cells on the newly formed bone surface. Bolded arrows and green chevrons indicate upregulated pathways, red chevrons indicate downregulated pathways.
2.2 Nitric oxide
Along with many other anabolic signaling molecules, osteocytes release nitric oxide in response to fluid shear stimulation (Klein-Nulend et al., 1995). Nitric oxide is produced by three isoforms (Nos1, Nos2, and Nos3), which are expressed in a wide range of cell types (Riancho et al., 1995), and is sufficient to drive the loss of sclerostin protein in vitro (Gould et al., 2021b). Global knockouts of the Nos3 gene have been shown to result in decreased trabecular bone volume basally in adult mice, while Nos2 knockout reduces bone mass recovery after tail suspension and reloading (Watanuki et al., 2002). Interestingly, Nos1 knockout produces increased murine bone mineral density in cortical and trabecular compartments (van’t Hof et al., 2004). While these isoforms convert the same substrates, they have diverse expression profiles, activators, and repressors, leading to disparate functions in bone. Nitric oxide has long been understood to play an intrinsic role in bone homeostasis, but the translational potential of nitric oxide for bone mass has not been clarified (Liu and Rosen, 2021).
2.3 PGE2
Like nitric oxide, prostaglandin E2 (PGE2) is also widely expressed across diverse tissues (Li et al., 2007), is produced by osteocytes in response to stimulation via pulsatile fluid flow (Ajubi et al., 1996), and is sufficient to reduce sclerostin protein abundance in vitro (Koide et al., 2017). PGE2 has been shown to support fracture healing (Wittenberg and Wittenberg, 1991) and bone formation (Li et al., 2007), and to also support osteoclastogenesis in vitro (Li et al., 2000). Despite increasing both resorption and deposition, treatment with PGE2 appears to result in a net increase in bone mass, though persistent, systemic side effects have prevented it from being utilized as a clinical therapy for low bone mass (Hartke and Lundy, 2001).
2.4 WNT
Removal of β-catenin pathway inhibitors (e.g., sclerostin, DKK1) is insufficient to stimulate the pathway unless the cognate ligands are also present during anabolic signaling (Tu et al., 2012). WNTs are canonical anabolic activators of the β-catenin pathway (Kim et al., 2013) and osteoblastic expression of WNT family genes increases after load (Holguin et al., 2016). Through WNT expression, additional osteoblasts may be recruited to support deposition following relatively high strain. WNT1 specifically has been demonstrated to be necessary for local anabolic bone response following axial loading (Lawson et al., 2022).
2.5 OPG
In addition to stimulating new bone formation, mechanical loading can also inhibit bone resorption. One mechanism of bone mass control is the regulation of osteoprotegerin following anabolic stimuli. Osteoprotegerin (OPG) is a cytokine receptor that is expressed by osteoblasts that can locally moderate osteoclastogenesis (Cawley et al., 2020). It is not membrane-bound, but rather secreted into extracellular space (Boyce and Xing, 2007) where it acts as a decoy RANKL receptor, preventing RANKL from stimulating osteoclastogenesis and resorption (Udagawa et al., 2000). OPG production increases following mechanical strain (Saunders et al., 2006) and is typically detectable in circulation as well as within the bone environment (Tsukasaki et al., 2020).
Conceptually, OPG expression could spatially restrain demineralization in loaded bone and prevent loss of bone mass in specific high-strain regions where new bone is being formed, even during periods of calcium deficit. Biologically active OPG is kept close to the osteoblast membrane, rather than diffused away, through association with osteoblastic surface proteins (Li and Xu, 2020), supporting OPG’s role in local control. (Figure 2). OPG has also been shown to support osteoblastic differentiation in vitro, increasing both osteoblast number and upregulation of markers associated with osteoblast maturity, like ALP (Yu et al., 2011).
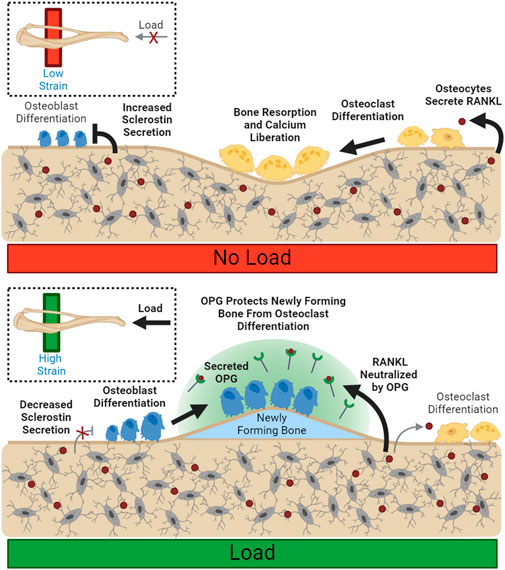
FIGURE 2. Load Spatially Controls Resorption and Deposition. Unloaded osteocytes secrete sclerostin, preventing osteoblast differentiation. Disinhibited RANKL expression allows for increased osteoclast differentiation and resorption. Load suppresses local sclerostin expression, allowing osteoblast differentiation. Local osteoclastic resorption and differentiation are downregulated through reduced RANKL availability. Bolded arrows indicate upregulated pathways for given conditions.
2.6 Embedded growth factors
The majority of organic bone matrix is comprised of type I collagen (Tzaphlidou, 2008), but other components are embedded during deposition, including growth factors such as BMPs, TGF-β and IGF-I (Solheim, 1998). Injury to bone can accumulate in the form of microscopic cracks due to intense, repetitive loading (O’Brien et al., 2003), or can occur acutely, as in fracture. Disruption of the bone results in osteoclastic dissolution of mineralized tissue, releasing these latent osteoid-embedded growth factors into the local environment. These factors may now stimulate osteoprogenitor recruitment and proliferation, osteoblast differentiation, and local bone deposition (Baylink et al., 1993) in order to restore and strengthen the bone.
3 Discussion
Osteoporosis, a clinical designation of severe bone loss with greatly increased fracture risk, can result in hospitalization or death, especially in the elderly and frail (Li et al., 2017). There are several classes of clinically available treatments for low bone mass and osteoporosis, some of which effectively increase BMD by targeting the molecular mediators of mechanical loading. Anti-RANKL (denosumab), acts like an OPG mimetic to suppress osteoclastogenesis, and anti-sclerostin (romosozumab) antibody treatments effectively reduce sclerostin bioavailability, reducing osteoclastogenesis and increasing osteoblastogenesis.
Another class of treatment, teriparatide (PTH1-34) and abaloparatide (PTHrP1-34) are synthetic analogs of parathyroid hormone, which has been shown to suppress sclerostin expression at the RNA (Bellido et al., 2005; Wein and Kronenberg, 2018) and protein (Drake et al., 2010; Gould et al., 2021b) level. Parathyroid hormone-analog treatments are highly effective at preserving and restoring bone mass through an increase in osteoblast number and matrix deposition, due in part to β-catenin pathway upregulation (Tian et al., 2011).
The balance of deposition and resorption is necessary for skeletal health, but can be altered in individuals with mineral deficiency, leading to low bone mineral density (BMD) and associated disorders. For example, prolonged vitamin D deficiency can contribute to low bone mass and relatively high proportions of unmineralized osteoid (Francis and Selby, 1997), ultimately leading to rickets or osteomalacia if left untreated. Exercise, as well as dietary vitamin supplementation, have been shown to improve BMD in these patients (Tønnesen et al., 2016).
Like many other tissues, the skeleton follows a ‘use it or lose it’ model, wherein continued disuse leads to a cycle of wasting, decreased capacity, and fragility, especially in aging (Leser et al., 2021), when the balance of deposition and resorption shifts towards net loss of mineralized bone. Bone mass and osteoblast number decrease with age (Almeida, 2012), but study after study (Layne and Nelson, 1999; Mosti et al., 2014; Watson et al., 2018; Holubiac et al., 2022) report the protective benefits of mechanical loading and exercise. Load increases expression of osteoblastogenesis-related signals, like WNTs and OPG, while suppressing sclerostin abundance and osteoclastogenesis-related signals like RANKL. Accordingly, exercise that produces sufficient mechanical load remains the best defense against bone loss.
Author contributions
HB: Writing–review and editing, Writing–original draft, Funding acquisition, Conceptualization. JS: Writing–review and editing, Supervision, Funding acquisition, Conceptualization.
Funding
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. Funding was provided by NIH R01-AR071614 and T32-GM008181.
Acknowledgments
Figures were created with BioRender.com.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ajubi N. E., Klein-Nulend J., Nijweide P. J., Vrijheid-Lammers T., Alblas M. J., Burger E. H. (1996). Pulsating fluid flow increases prostaglandin production by cultured chicken osteocytes-A cytoskeleton-dependent process. Biochem. Biophys. Res. Commun. 225 (1), 62–68. doi:10.1006/bbrc.1996.1131
Almeida M. (2012). Aging mechanisms in bone. Bonekey Rep. 1 (7), 102–107. doi:10.1038/bonekey.2012.102
Andrade M. A. R., Derradi R., Simão A. M. S., Millán J. L., Ramos A. P., Ciancaglini P., et al. (2019). Is alkaline phosphatase biomimeticaly immobilized on titanium able to propagate the biomineralization process? Arch. Biochem. Biophys. 663, 192–198. doi:10.1016/j.abb.2019.01.014
Atkins G. J., Rowe P. S., Lim H. P., Welldon K. J., Ormsby R., Wijenayaka A. R., et al. (2011). Sclerostin is a locally acting regulator of late-osteoblast/preosteocyte differentiation and regulates mineralization through a MEPE-ASARM-dependent mechanism. J. Bone Min. Res. 26 (7), 1425–1436. doi:10.1002/jbmr.345
Atria P. J., Castillo A. B. (2023). Skeletal adaptation to mechanical cues during homeostasis and repair: the niche, cells, and molecular signaling. Front. Physiol. 14, 1233920. doi:10.3389/fphys.2023.1233920
Baylink D. J., Finkelman R. D., Mohan S. (1993). Growth factors to stimulate bone formation. J. Bone Min. Res. 8, S565–S572. doi:10.1002/jbmr.5650081326
Bellido T., Ali A. A., Gubrij I., Plotkin L. I., Fu Q., O’Brien C. A., et al. (2005). Chronic elevation of parathyroid hormone in mice reduces expression of sclerostin by osteocytes: a novel mechanism for hormonal control of osteoblastogenesis. Endocrinology 146 (11), 4577–4583. doi:10.1210/en.2005-0239
Blair H. C., Larrouture Q. C., Li Y., Lin H., Beer-Stoltz D., Liu L., et al. (2017). Osteoblast differentiation and bone matrix formation in vivo and in vitro. Tissue Eng. Part B Rev. 23 (3), 268–280. doi:10.1089/ten.TEB.2016.0454
Boyce B. F., Xing L. (2007). The RANKL/RANK/OPG pathway. Curr. Osteoporos. Rep. 5 (3), 98–104. doi:10.1007/s11914-007-0024-y
Brunkow M. E., Gardner J. C., Van Ness J., Paeper B. W., Kovacevich B. R., Proll S., et al. (2001). Bone dysplasia sclerosteosis results from loss of the SOST gene product, a novel cystine knot-containing protein. Am. J. Hum. Genet. 68 (3), 577–589. doi:10.1086/318811
Burra S., Nicolella D. P., Jiang J. X. (2011). Dark horse in osteocyte biology: glycocalyx around the dendrites is critical for osteocyte mechanosensing. Commun. Integr. Biol. 4, 48–50. doi:10.4161/cib.4.1.13646
Cawley K. M., Bustamante-Gomez N. C., Guha A. G., MacLeod R. S., Xiong J., Gubrij I., et al. (2020). Local production of osteoprotegerin by osteoblasts suppresses bone resorption. Cell Rep. 32 (10), 108052. doi:10.1016/j.celrep.2020.108052
Ceroni D., Martin X., Delhumeau C., Rizzoli R., Kaelin A., Farpour-Lambert N. (2012). Effects of cast-mediated immobilization on bone mineral mass at various sites in adolescents with lower-extremity fracture. J. Bone Jt. Surg. Am. 94 (3), 208–216. doi:10.2106/JBJS.K.00420
Christen P., Ito K., Ellouz R., Boutroy S., Sornay-Rendu E., Chapurlat R. D., et al. (2014). Bone remodelling in humans is load-driven but not lazy. Nat. Commun. 5, 4855. doi:10.1038/ncomms5855
Dallas S. L., Bonewald L. F. (2010). Dynamics of the transition from osteoblast to osteocyte. Ann. N. Y. Acad. Sci. 1192 (1), 437–443. doi:10.1111/j.1749-6632.2009.05246.x
da Silva Madaleno C., Jatzlau J., Knaus P. (2020). BMP signalling in a mechanical context – implications for bone biology. Bone 137 (May), 115416. doi:10.1016/j.bone.2020.115416
Delgado-Calle J., Bellido T. (2022). The osteocyte as a signaling cell. Physiol. Rev. 102, 379–410. doi:10.1152/physrev.00043.2020
Dionyssiotis Y., Trovas G., Galanos A., Raptou P., Papaioannou N., Papagelopoulos P., et al. (2007). Bone loss and mechanical properties of tibia in spinal cord injured men. J. Musculoskelet. Neuronal Interact. 7 (1), 62–68.
Drake M. T., Srinivasan B., Mödder U. I., Peterson J. M., McCready L. K., Riggs B. L., et al. (2010). Effects of parathyroid hormone treatment on circulating sclerostin levels in postmenopausal women. J. Clin. Endocrinol. Metab. 95 (11), 5056–5062. doi:10.1210/jc.2010-0720
Drissi H., Luc Q., Shakoori R., De Sousa Lopes S. C., Choi J. Y., Terry A., et al. (2000). Transcriptional autoregulation of the bone related CBFA1/RUNX2 gene. J. Cell Physiol. 184 (3), 341–350. doi:10.1002/1097-4652(200009)184:3<341::AID-JCP8>3.0.CO;2-Z
Dsouza C., Komarova S. V. (2023). Mechanosensitivity and mechanotransductive properties of osteoclasts. Am. J. Physiol. Physiol. 326, 95–106. doi:10.1152/ajpcell.00347.2023
Du J. H., Lin S. X., Wu X. L., Yang S. M., Cao L. Y., Zheng A., et al. (2019). The function of wnt ligands on osteocyte and bone remodeling. J. Dent. Res. 98 (8), 930–938. doi:10.1177/0022034519854704
Dufour C., Holy X., Marie P. J. (2008). Transforming growth factor-beta prevents osteoblast apoptosis induced by skeletal unloading via PI3K/Akt, Bcl-2, and phospho-Bad signaling. Am. J. Physiol. - Endocrinol. Metab. 294 (4), 794–801. doi:10.1152/ajpendo.00791.2007
Engler A. J., Sen S., Sweeney H. L., Discher D. E. (2006). Matrix elasticity directs stem cell lineage specification. Cell 126 (4), 677–689. doi:10.1016/j.cell.2006.06.044
Felber K., Elks P. M., Lecca M., Roehl H. H. (2015). Expression of osterix is regulated by FGF and Wnt/β-catenin signalling during osteoblast differentiation. PLoS One 10 (12), 0144982. doi:10.1371/journal.pone.0144982
Francis R. M., Selby P. L. (1997). Osteomalacia. Baillieres Clin. Endocrinol. Metab. 11 (1), 145–163. doi:10.1016/s0950-351x(97)80569-1
Franz-Odendaal T. A., Hall B. K., Witten P. E. (2006). Buried alive: how osteoblasts become osteocytes. Dev. Dyn. 235 (1), 176–190. doi:10.1002/dvdy.20603
Frost H. M. (2003). Bone’s mechanostat: a 2003 update. Anat. Rec. - Part A Discov. Mol. Cell Evol. Biol. 275 (2), 1081–1101. doi:10.1002/ar.a.10119
Fu Y.-H., Liu W.-J., Lee C.-L., Wang J.-S. (2022). Associations of insulin resistance and insulin secretion with bone mineral density and osteoporosis in a general population. Front. Endocrinol. (Lausanne) 13, 971960. doi:10.3389/fendo.2022.971960
Gould N. R., Leser J. M., Torre O. M., Khairallah R. J., Ward C. W., Stains J. P. (2021a). In vitro fluid shear stress induced sclerostin degradation and CaMKII activation in osteocytes. Bio-protocol 11 (23), e4251–e4315. doi:10.21769/BioProtoc.4251
Gould N. R., Williams K. M., Joca H. C., Torre O. M., Lyons J. S., Leser J. M., et al. (2021b). Disparate bone anabolic cues activate bone formation by regulating the rapid lysosomal degradation of sclerostin protein. Elife 10, e64393–e64426. doi:10.7554/eLife.64393
Gu P., Pu B., Xin Q., Yue D., Luo L. L., Tao J. S., et al. (2023). The metabolic score of insulin resistance is positively correlated with bone mineral density in postmenopausal patients with type 2 diabetes mellitus. Sci. Rep. 13 (1), 8796. doi:10.1038/s41598-023-32931-8
Han G., Zuo J., Holliday L. S. (2019). Specialized roles for actin in osteoclasts: unanswered questions and therapeutic opportunities. Biomolecules 9, 17. MDPI AG. doi:10.3390/biom9010017
Han Y., Cowin S. C., Schaffler M. B., Weinbaum S. (2004). Mechanotransduction and strain amplification in osteocyte cell processes. Proc. Natl. Acad. Sci. U. S. A. 101 (47), 16689–16694. doi:10.1073/pnas.0407429101
Hartke J. R., Lundy M. W. (2001). Bone anabolic therapy with selective prostaglandin analogs. J. Musculoskelet. Neuronal Interact. 2 (1), 25–31. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15758474.
Holguin N., Brodt M. D., Silva M. J. (2016). Activation of wnt signaling by mechanical loading is impaired in the bone of old mice. J. Bone Min. Res. 31 (12), 2215–2226. doi:10.1002/jbmr.2900
Holubiac I. Ș., Leuciuc F. V., Crăciun D. M., Dobrescu T. (2022). Effect of strength training protocol on bone mineral density for postmenopausal women with osteopenia/osteoporosis assessed by dual-energy X-ray absorptiometry (DEXA). Sensors (Basel) 22 (5), 1904. doi:10.3390/s22051904
Hunter G. K., Hauschka P. V., Poole A. R., Rosenberg L. C., Goldberg H. A. (1996). Nucleation and inhibition of hydroxyapatite formation by mineralized tissue proteins. Biochem. J. 317, 59–64. doi:10.1042/bj3170059
Ishijima M., Rittling S. R., Yamashita T., Tsuji K., Kurosawa H., Nifuji A., et al. (2001). Enhancement of osteoclastic bone resorption and suppression of osteoblastic bone formation in response to reduced mechanical stress do not occur in the absence of osteopontin. J. Exp. Med. 193 (3), 399–404. doi:10.1084/jem.193.3.399
Jiang J. X., Cheng B. (2001). Mechanical stimulation of gap junctions in bone osteocytes is mediated by prostaglandin E2. Cell Commun. Adhes. 8 (4–6), 283–288. doi:10.3109/15419060109080738
Josephson T. O., Morgan E. F. (2023). Harnessing mechanical cues in the cellular microenvironment for bone regeneration. Front. Physiol. 14 (October), 1232698. doi:10.3389/fphys.2023.1232698
Ke H. Z., Richards W. G., Li X., Ominsky M. S. (2012). Sclerostin and dickkopf-1 as therapeutic targets in bone diseases. Endocr. Rev. 33 (5), 747–783. doi:10.1210/er.2011-1060
Kim J. H., Liu X., Wang J., Chen X., Zhang H., Kim S. H., et al. (2013). Wnt signaling in bone formation and its therapeutic potential for bone diseases. Ther. Adv. Musculoskelet. Dis. 5 (1), 13–31. doi:10.1177/1759720X12466608
Klein-Nulend J., Semeins C. M., Ajubi N. E., Nijweide P. J., Burger E. H. (1995). Pulsating fluid flow increases nitric oxide (NO) synthesis by osteocytes but not periosteal fibroblasts - correlation with prostaglandin upregulation. Biochem. Biophys. Res. Commun. 217 (2), 640–648. doi:10.1006/bbrc.1995.2822
Koide M., Kobayashi Y., Yamashita T., Uehara S., Nakamura M., Hiraoka B. Y., et al. (2017). Bone Formation is coupled to resorption via suppression of sclerostin expression by osteoclasts. J. Bone Min. Res. 32 (10), 2074–2086. doi:10.1002/jbmr.3175
Krishnan V., Bryant H. U., MacDougald O. A. (2006). Regulation of bone mass by Wnt signaling. J. Clin. Invest. 116 (5), 1202–1209. doi:10.1172/JCI28551
Kurenkova A. D., Medvedeva E. V., Newton P. T., Chagin A. S. (2020). Niches for skeletal stem cells of mesenchymal origin. Front. Cell Dev. Biol. 8 (July), 592. doi:10.3389/fcell.2020.00592
Lawson L. Y., Brodt M. D., Migotsky N., Chermside-Scabbo C. J., Palaniappan R., Silva M. J. (2022). Osteoblast-specific wnt secretion is required for skeletal homeostasis and loading-induced bone formation in adult mice. J. Bone Min. Res. 37 (1), 108–120. doi:10.1002/jbmr.4445
Layne J. E., Nelson M. E. (1999). The effects of progressive resistance training on bone density: a review. Med. Sci. Sports Exerc 31 (1), 25–30. doi:10.1097/00005768-199901000-00006
Lean J. M., Mackay A. G., Chow J. W. M., Chambers T. J. (1996). Osteocytic expression of mRNA for c-fos and IGF-I: an immediate early gene response to an osteogenic stimulus. Am. J. Physiol. - Endocrinol. Metab. 270 (6 33-6), 937–945. doi:10.1152/ajpendo.1996.270.6.E937
LeBlanc A., Schneider V., Shackelford L., West S., Oganov V., Bakulin A., et al. (2000). Bone mineral and lean tissue loss after long duration space flight. J. Musculoskelet. Neuronal Interact. 1 (2), 157–160. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15758512.
Leblanc A. D., Schneider V. S., Evans H. J., Engelbretson D. A., Krebs J. M. (1990). Bone mineral loss and recovery after 17 weeks of bed rest. J. Bone Min. Res. 5 (8), 843–850. doi:10.1002/jbmr.5650050807
Lee K. L., Guevarra M. D., Nguyen A. M., Chua M. C., Wang Y., Jacobs C. R. (2015). The primary cilium functions as a mechanical and calcium signaling nexus. Cilia 4 (1), 7. doi:10.1186/s13630-015-0016-y
Lefterova M. I., Zhang Y., Steger D. J., Schupp M., Schug J., Cristancho A., et al. (2008). PPARgamma and C/EBP factors orchestrate adipocyte biology via adjacent binding on a genome-wide scale. Genes Dev. 22 (21), 2941–2952. doi:10.1101/gad.1709008
Leser J. M., Harriot A., Buck H. V., Ward C. W., Stains J. P. (2021). Aging, osteo-sarcopenia, and musculoskeletal mechano-transduction. Front. Rehabil. Sci. 2, 782848. doi:10.3389/fresc.2021.782848
Lewis K. J., Frikha-Benayed D., Louie J., Stephen S., Spray D. C., Thi M. M., et al. (2017). Osteocyte calcium signals encode strain magnitude and loading frequency in vivo. Proc. Natl. Acad. Sci. U. S. A. 114 (44), 11775–11780. doi:10.1073/pnas.1707863114
Li G., Thabane L., Papaioannou A., Ioannidis G., Levine M. A. H., Adachi J. D. (2017). An overview of osteoporosis and frailty in the elderly. BMC Musculoskelet. Disord. 18 (1), 46–55. doi:10.1186/s12891-017-1403-x
Li M., Thompson D. D., Paralkar V. M. (2007). Prostaglandin E2 receptors in bone formation. Int. Orthop. 31 (6), 767–772. doi:10.1007/s00264-007-0406-x
Li M., Xu D. (2020). Antiresorptive activity of osteoprotegerin requires an intact heparan sulfate-binding site. Proc. Natl. Acad. Sci. U. S. A. 117 (29), 17187–17194. doi:10.1073/pnas.2005859117
Li X., Han L., Nookaew I., Mannen E., Silva M. J., Almeida M., et al. (2019). Stimulation of piezo1 by mechanical signals promotes bone anabolism. Elife 8, e49631–e49722. doi:10.7554/eLife.49631
Li X., Okada Y., Pilbeam C. C., Lorenzo J. A., Kennedy C. R. J., Breyer R. M., et al. (2000). Knockout of the murine prostaglandin EP2 receptor impairs Osteoclastogenesis in vitro. Endocrinology 141 (6), 2054–2061. doi:10.1210/endo.141.6.7518
Li X., Ominsky M. S., Niu Q. T., Sun N., Daugherty B., D’Agostin D., et al. (2008). Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J. Bone Min. Res. 23 (6), 860–869. doi:10.1359/jbmr.080216
Li X., Zhang Y., Kang H., Liu W., Liu P., Zhang J., et al. (2005). Sclerostin binds to LRP5/6 and antagonizes canonical Wnt signaling. J. Biol. Chem. 280 (20), 19883–19887. doi:10.1074/jbc.M413274200
Liao C., Liang S., Wang Y., Zhong T., Liu X. (2022). Sclerostin is a promising therapeutic target for oral inflammation and regenerative dentistry. J. Transl. Med. 20 (1), 221–313. doi:10.1186/s12967-022-03417-4
Linkhart T. A., Mohan S., Baylink D. J. (1996). Growth factors for bone growth and repair: IGF, TGFβ and BMP. Bone 19 (1), S1–S12. doi:10.1016/s8756-3282(96)00138-x
Liu H., Rosen C. J. (2021). Nitric oxide and bone: the phoenix rises again. J. Clin. Invest. 131 (5). doi:10.1172/jci147072
Liu P., Tu J., Wang W., Li Z., Li Y., Yu X., et al. (2022). Effects of mechanical stress stimulation on function and expression mechanism of osteoblasts. Front. Bioeng. Biotechnol. 10, 830722. doi:10.3389/fbioe.2022.830722
Lyons J. S., Joca H. C., Law R. A., Williams K. M., Kerr J. P., Shi G., et al. (2017). Microtubules tune mechanotransduction through NOX2 and TRPV4 to decrease sclerostin abundance in osteocytes. Sci. Signal 10 (506), eaan5748. doi:10.1126/scisignal.aan5748
Matsushita Y., Ono W., Ono N. (2020). Skeletal stem cells for bone development and repair: diversity matters. Curr. Osteoporos. Rep. 18 (3), 189–198. doi:10.1007/s11914-020-00572-9
McClanahan B. S., Harmon-Clayton K., Ward K. D., Klesges R. C., Vukadinovich C. M., Cantler E. D. (2002). Side-to-side comparisons of bone mineral density in upper and lower limbs of collegiate athletes. J. strength Cond. Res. 16 (4), 586–590. doi:10.1519/1533-4287(2002)016<0586:stscob>2.0.co;2
Mohamed AMFS (2008). An overview of bone cells and their regulating factors of differentiation. Malays. J. Med. Sci. 15 (1), 4–12.
Moorer M. C., Hebert C., Tomlinson R. E., Iyer S. R., Chason M., Stains J. P. (2017). Defective signaling, osteoblastogenesis and bone remodeling in a mouse model of connexin 43 C-terminal truncation. J. Cell Sci. 130 (3), 531–540. doi:10.1242/jcs.197285
Mosti M. P., Carlsen T., Aas E., Hoff J., Stunes A. K., Syversen U. (2014). Maximal strength training improves bone mineral density and neuromuscular performance in young adult women. J. strength Cond. Res. 28 (10), 2935–2945. doi:10.1519/JSC.0000000000000493
Mullen C. A., Haugh M. G., Schaffler M. B., Majeska R. J., McNamara L. M. (2013). Osteocyte differentiation is regulated by extracellular matrix stiffness and intercellular separation. J. Mech. Behav. Biomed. Mater 28, 183–194. doi:10.1016/j.jmbbm.2013.06.013
Murshed M., Harmey D., Millán J. L., McKee M. D., Karsenty G. (2005). Unique coexpression in osteoblasts of broadly expressed genes accounts for the spatial restriction of ECM mineralization to bone. Genes Dev. 19 (9), 1093–1104. doi:10.1101/gad.1276205
Nakashima K., Zhou X., Kunkel G., Zhang Z., Deng J. M., Behringer R. R., et al. (2002). The novel zinc finger-containing transcription factor osterix is required for osteoblast differentiation and bone formation. Cell 108 (1), 17–29. doi:10.1016/s0092-8674(01)00622-5
O’Brien F. J., Taylor D., Lee T. C. (2003). Microcrack accumulation at different intervals during fatigue testing of compact bone. J. Biomech. 36 (7), 973–980. doi:10.1016/s0021-9290(03)00066-6
Ono T., Hayashi M., Sasaki F., Nakashima T. (2020). RANKL biology: bone metabolism, the immune system, and beyond. Inflamm. Regen. 40 (1), 2. doi:10.1186/s41232-019-0111-3
Orgel J. P., Miller A., Irving T. C., Fischetti R. F., Hammersley A. P., Wess T. J. (2001). The in situ supermolecular structure of type I collagen. Structure 9 (11), 1061–1069. doi:10.1016/s0969-2126(01)00669-4
Parhami F., Tintut Y., Beamer W. G., Gharavi N., Goodman W., Demer L. L. (2001). Atherogenic high-fat diet reduces bone mineralization in mice. J. Bone Min. Res. 16 (1), 182–188. doi:10.1359/jbmr.2001.16.1.182
Piekarski K., Munro M. (1977). Transport mechanism operating between blood supply and osteocytes in long bones. Nature 269 (5623), 80–82. doi:10.1038/269080a0
Price C., Zhou X., Li W., Wang L. (2011). Real-time measurement of solute transport within the lacunar-canalicular system of mechanically loaded bone: direct evidence for load-induced fluid flow. J. Bone Min. Res. 26 (2), 277–285. doi:10.1002/jbmr.211
Riancho J. A., Zarrabeitia M. T., Fernandez-Luna J. L., Gonzalez-Macias J. (1995). Mechanisms controlling nitric oxide synthesis in osteoblasts. Mol. Cell Endocrinol. 107 (1), 87–92. doi:10.1016/0303-7207(94)03428-v
Riddle R. C. (2023). Endocrine functions of sclerostin. Curr. Opin. Endocr. Metab. Res. 28, 100433–100511. doi:10.1016/j.coemr.2022.100433
Robling A. G., Bonewald L. F. (2020). The osteocyte: new insights. Annu. Rev. Physiol. 82, 485–506. doi:10.1146/annurev-physiol-021119-034332
Robling A. G., Hinant F. M., Burr D. B., Turner C. H. (2002). Improved bone structure and strength after long-term mechanical loading is greatest if loading is separated into short bouts. J. Bone Min. Res. 17 (8), 1545–1554. doi:10.1359/jbmr.2002.17.8.1545
Robling A. G., Niziolek P. J., Baldridge L. A., Condon K. W., Allen M. R., Alam I., et al. (2008). Mechanical stimulation of bone in vivo reduces osteocyte expression of Sost/sclerostin. J. Biol. Chem. 283 (9), 5866–5875. doi:10.1074/jbc.M705092200
Robling A. G., Turner C. H. (2009). Mechanical signaling for bone modeling and remodeling. Crit. Rev. Eukaryot. Gene Expr. 19 (4), 319–338. doi:10.1615/critreveukargeneexpr.v19.i4.50
Rutkovskiy A., Stensløkken K.-O., Vaage I. J. (2016). Osteoblast differentiation at a glance. Med. Sci. Monit. Basic Res. 22, 95–106. doi:10.12659/msmbr.901142
Saunders M. M., Taylor A. F., Du C., Zhou Z., Pellegrini V. D., Donahue H. J. (2006). Mechanical stimulation effects on functional end effectors in osteoblastic MG-63 cells. J. Biomech. 39 (8), 1419–1427. doi:10.1016/j.jbiomech.2005.04.011
Schaffler M. B., Cheung W.-Y., Majeska R., Kennedy O. (2014). Osteocytes: master orchestrators of bone. Calcif. Tissue Int. 94 (1), 5–24. doi:10.1007/s00223-013-9790-y
Schaffler M. B., Kennedy O. D. (2012). Osteocyte signaling in bone. Curr. Osteoporos. Rep. 10 (2), 118–125. doi:10.1007/s11914-012-0105-4
Sen B., Xie Z., Case N., Ma M., Rubin C., Rubin J. (2008). Mechanical strain inhibits adipogenesis in mesenchymal stem cells by stimulating a durable beta-catenin signal. Endocrinology 149 (12), 6065–6075. doi:10.1210/en.2008-0687
Shahar R., Dean M. N. (2013). The enigmas of bone without osteocytes. Bonekey Rep. 2, 343. doi:10.1038/bonekey.2013.77
Stamos J. L., Weis W. I. (2013). The β-catenin destruction complex. Cold Spring Harb. Perspect. Biol. 5 (1), a007898–a7916. doi:10.1101/cshperspect.a007898
Suen P. K., Qin L. (2016). Sclerostin, an emerging therapeutic target for treating osteoporosis and osteoporotic fracture: a general review. J. Orthop. Transl. 4, 1–13. doi:10.1016/j.jot.2015.08.004
Teitelbaum S. L. (2011). The osteoclast and its unique cytoskeleton. Ann. N. Y. Acad. Sci. 1240 (1), 14–17. doi:10.1111/j.1749-6632.2011.06283.x
Thompson W. R., Modla S., Grindel B. J., Czymmek K. J., Kirn-Safran C. B., Wang L., et al. (2011). Perlecan/Hspg2 deficiency alters the pericellular space of the lacunocanalicular system surrounding osteocytic processes in cortical bone. J. Bone Min. Res. 26 (3), 618–629. doi:10.1002/jbmr.236
Tian Y., Xu Y., Fu Q., He M. (2011). Parathyroid hormone regulates osteoblast differentiation in a Wnt/β-catenin-dependent manner. Mol. Cell Biochem. 355 (1–2), 211–216. doi:10.1007/s11010-011-0856-8
Tønnesen R., Schwarz P., Hovind P. H., Jensen L. T. (2016). Physical exercise associated with improved BMD independently of sex and vitamin D levels in young adults. Eur. J. Appl. Physiol. 116 (7), 1297–1304. doi:10.1007/s00421-016-3383-1
Tsukasaki M., Asano T., Muro R., Huynh N. C. N., Komatsu N., Okamoto K., et al. (2020). OPG production matters where it happened. Cell Rep. 32 (10), 108124. doi:10.1016/j.celrep.2020.108124
Tu X., Rhee Y., Condon K. W., Bivi N., Allen M. R., Dwyer D., et al. (2012). Sost downregulation and local Wnt signaling are required for the osteogenic response to mechanical loading. Bone 50 (1), 209–217. doi:10.1016/j.bone.2011.10.025
Turner C. H., Owan I., Alvey T., Hulman J., Hock J. M. (1998). Recruitment and proliferative responses of osteoblasts after mechanical loading in vivo determined using sustained-release bromodeoxyuridine. Bone 22 (5), 463–469. doi:10.1016/s8756-3282(98)00041-6
Tzaphlidou M. (2008). Bone architecture: collagen structure and calcium/phosphorus maps. J. Biol. Phys. 34 (1–2), 39–49. doi:10.1007/s10867-008-9115-y
Udagawa N., Takahashi N., Yasuda H., Mizuno A., Itoh K., Ueno Y., et al. (2000). Osteoprotegerin produced by osteoblasts is an important regulator in osteoclast development and function. Endocrinology 141 (9), 3478–3484. doi:10.1210/endo.141.9.7634
van’t Hof R. J., Macphee J., Libouban H., Helfrich M. H., Ralston S. H. (2004). Regulation of bone mass and bone turnover by neuronal nitric oxide synthase. Endocrinology 145 (11), 5068–5074. doi:10.1210/en.2004-0205
Wang B., Lai X., Price C., Thompson W. R., Li W., Quabili T. R., et al. (2014). Perlecan-containing pericellular matrix regulates solute transport and mechanosensing within the osteocyte lacunar-canalicular system. J. Bone Min. Res. 29 (4), 878–891. doi:10.1002/jbmr.2105
Watanuki M., Sakai A., Sakata T., Tsurukami H., Miwa M., Uchida Y., et al. (2002). Role of inducible nitric oxide synthase in skeletal adaptation to acute increases in mechanical loading. J. Bone Min. Res. 17 (6), 1015–1025. doi:10.1359/jbmr.2002.17.6.1015
Watson S. L., Weeks B. K., Weis L. J., Harding A. T., Horan S. A., Beck B. R. (2018). High-intensity resistance and impact training improves bone mineral density and physical function in postmenopausal women with osteopenia and osteoporosis: the LIFTMOR randomized controlled trial. J. Bone Min. Res. 33 (2), 211–220. doi:10.1002/jbmr.3284
Wein M. N., Kronenberg H. M. (2018). Regulation of bone remodeling by parathyroid hormone. Cold Spring Harb. Perspect. Med. 8 (8), a031237–a31320. doi:10.1101/cshperspect.a031237
Weivoda M. M., Youssef S. J., Oursler M. J. (2017). Sclerostin expression and functions beyond the osteocyte. Bone 96 (507), 45–50. doi:10.1016/j.bone.2016.11.024
Wheatley S., Wright E., Jeske Y., Mccormack A., Bowles J., Koopman P. (1996). Aetiology of the skeletal dysmorphology syndrome campomelic dysplasia: expression of the Sox9 gene during chondrogenesis in mouse embryos. Ann. N. Y. Acad. Sci. 785, 350–352. doi:10.1111/j.1749-6632.1996.tb56306.x
Wijenayaka A. R., Kogawa M., Lim H. P., Bonewald L. F., Findlay D. M., Atkins G. J. (2011). Sclerostin stimulates osteocyte support of osteoclast activity by a RANKL-dependent pathway. PLoS One 6 (10), e25900. doi:10.1371/journal.pone.0025900
Williams K. M., Leser J. M., Gould N. R., Joca H. C., Lyons J. S., Khairallah R. J., et al. (2020). TRPV4 calcium influx controls sclerostin protein loss independent of purinergic calcium oscillations. Bone 136 (April), 115356. doi:10.1016/j.bone.2020.115356
Wittenberg J. M., Wittenberg R. H. (1991). Release of prostaglandins from bone and muscle after femoral osteotomy in rats. Acta Orthop. 62 (6), 577–581. doi:10.3109/17453679108994500
Yasuda H., Shima N., Nakagawa N., Yamaguchi K., Kinosaki M., Mochizuki S. I., et al. (1998). Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. U. S. A. 95 (7), 3597–3602. doi:10.1073/pnas.95.7.3597
You L., Cowin S. C., Schaffler M. B., Weinbaum S. (2001). A model for strain amplification in the actin cytoskeleton of osteocytes due to fluid drag on pericellular matrix. J. Biomech. 34 (11), 1375–1386. doi:10.1016/s0021-9290(01)00107-5
Yu H., de Vos P., Ren Y. (2011). Overexpression of osteoprotegerin promotes preosteoblast differentiation to mature osteoblasts. Angle Orthod. 81 (1), 100–106. doi:10.2319/050210-238.1
Keywords: osteoblast, differentiation, osteoblastogenesis, mechanical loading, sclerostin, Wnt, osteocyte
Citation: Buck HV and Stains JP (2024) Osteocyte-mediated mechanical response controls osteoblast differentiation and function. Front. Physiol. 15:1364694. doi: 10.3389/fphys.2024.1364694
Received: 02 January 2024; Accepted: 29 February 2024;
Published: 11 March 2024.
Edited by:
Claudine Blin-Wakkach, UMR7370 Laboratoire de Physio Médecine Moléculaire (LP2M), FranceReviewed by:
Katharina Jähn-Rickert, University Medical Center Hamburg-Eppendorf, GermanyAlain Guignandon, INSERM U1059 SAnté INgéniérie BIOlogie, France
Copyright © 2024 Buck and Stains. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Joseph Paul Stains, anN0YWluc0Bzb20udW1hcnlsYW5kLmVkdQ==