 Yamei Wu1,2†
Yamei Wu1,2† Hao Ying
Hao Ying Lu Huang
Lu Huang
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 08 February 2024
Sec. Mitochondrial Research
Volume 15 - 2024 | https://doi.org/10.3389/fphys.2024.1344951
Mitochondria are ubiquitous in eukaryotic cells. Normal maintenance of function is the premise and basis for various physiological activities. Mitochondrial dysfunction is commonly observed in a wide range of pathological conditions, such as neurodegenerative, metabolic, cardiovascular, and various diseases related to foetal growth and development. The placenta is a highly energy-dependent organ that acts as an intermediary between the mother and foetus and functions to maintain foetal growth and development. Recent studies have demonstrated that mitochondrial dysfunction is associated with placental disorders. Defects in mitochondrial quality control mechanisms may lead to preeclampsia and foetal growth restriction. In this review, we address the quality control mechanisms of mitochondria and the relevant pathologies of mitochondrial dysfunction in placenta-related diseases, such as preeclampsia and foetal growth restriction. This review also investigates the relation between mitochondrial dysfunction and placental disorders.
Mitochondria are double-membrane organelles consisting of dynamic inner and outer membranes. Mitochondria are the main sites of biological oxidation and energy conversion in eukaryotic cells. Mitochondria are involved in the regulation of several physiological mechanisms. These include energy metabolism (Zhang et al., 2021), biosynthesis (Ryoo and Kwak, 2018; Fuchs et al., 2020), calcium signal transduction (Jouaville et al., 1998), production of reactive oxygen species (ROS) (Hernansanz-Agustin and Enriquez, 2021; Kuznetsov et al., 2022), and apoptosis (Wang et al., 2021; Tian et al., 2022). Therefore, mitochondrial dysfunction may lead to various diseases, such as cardiovascular diseases, skeletal muscle disorders, and neurodegenerative diseases (Mani et al., 2021; Sadoshima et al., 2021; Chen et al., 2022; Vodickova et al., 2022). Mitochondrial homeostasis is maintained at the organelle level through dynamically regulated quality-control mechanisms, including mitochondrial fusion and fission, biogenesis, and mitophagy (Green and Van Houten, 2011; Kotiadis et al., 2014; Suliman and Piantadosi, 2016). At the molecular level, homeostasis is mediated by the mitochondrial unfolded protein response, mitochondrial molecular chaperones, and proteases which constitute the mitochondrial protein quality control system that maintains the dynamic balance of the mitochondrial proteome (Vazquez-Calvo et al., 2020).
The placenta consists of the amniotic membrane, chorionic villous membrane, and decidua basalis, which are important organs for maintaining foetal growth and development. Therefore, it has a high energy demand. The placenta performs material exchange, defence, synthesis, and immunity (Shao et al., 2022). Mitochondria consume oxygen via oxidative phosphorylation to produce adenosine triphosphate (ATP). When mitochondrial dysfunction occurs, ATP synthesis is reduced and placental function is impaired, resulting in various pregnancy complications, such as preeclampsia (Marin et al., 2020; Smith et al., 2021) and foetal growth restriction (Kiyokoba et al., 2022). Furthermore, mitochondrial dysfunction amplifies oxidative stress and the production of ROS, leading to a self-perpetuating cycle of mitochondrial damage. This, in turn, contributes to placental dysfunction (Manna et al., 2019). In this study, we explored the intricate relation between mitochondrial dysfunction and placenta-related diseases, focusing on their role in conditions, such as preeclampsia and foetal growth restriction.
The morphology, size, and number of mitochondria are controlled by constant fusion and fission (Zemirli et al., 2018). In mammalian cells, mitochondrial outer membrane (MOM) fusion is mainly regulated by the dynamin-related GTPases mitofusin 1 and mitofusin 2 (MFN2) (Sloat et al., 2019), whereas mitochondrial inner membrane fusion is mainly regulated by optic atrophy 1, which is also a dynamin-related GTPase (Youle and van der Bliek, 2012). Moreover, Misato protein (MSTO1) is a soluble cytoplasmic protein. MSTO1 translocates to the MOM and interacts with mitochondrial fusion proteins at the MOM–cytoplasm interface (Al Ojaimi et al., 2022). F-Box and Leucine-rich repeat protein 4 is a nuclear-encoded mitochondrial protein that is in the intermembrane space. F-Box and Leucine-rich repeat protein 4 may also play a role in mitochondrial fusion by interacting with and regulating mitochondrial fusion proteins (Wang et al., 2020). Proteins involved in mediating mitochondrial fission include dynamin-related protein 1 (Drp1), fission protein 1, and mitochondrial dynamic proteins MiD49 and MiD51 (Serasinghe and Chipuk, 2017). The phosphorylation of Drp1 at Ser637 controls the mitochondrial fission-promoting activity of Drp1 (Kanamaru et al., 2012). cAMP-dependent protein kinase phosphorylates Drp1 and inhibits its translocation to mitochondrial fission sites. In contrast, calcineurin-mediated dephosphorylation of Drp1 causes Drp1 to accumulate in the mitochondria and promotes mitochondrial fission (Suarez-Rivero et al., 2016). Mitochondrial dynamics are complex and are related to mitochondrial function through fusion and fission. Research has indicated that mitochondrial dynamics play a crucial role in various pathological manifestations, including neurodegenerative diseases, cancer, cardiomyopathy, and metabolic disorders (Serasinghe and Chipuk, 2017).
Mitophagy refers to the process by which cells selectively remove aging and damaged mitochondria through autophagy, thereby controlling mitochondrial quality and maintaining mitochondrial homeostasis (Tian et al., 2022). There are two main mitophagy pathways, the ubiquitin-mediated and the receptor-mediated pathway (Chen et al., 2022). The ubiquitin-mediated pathway includes the PINK1-Parkin pathway. PINK1 is localised to the MOM and is introduced into the intermembrane space by the translocase outer membrane and into the mitochondrial inner membrane by the translocase of the inner membrane 23. At the mitochondrial inner membrane, PINK1 is processed with mitochondrial processing peptidase and PGAM5-associated rhomboid-like protease, and eventually degraded (Figure 1). Under pathological conditions, damaged mitochondria undergo mitochondrial membrane depolarisation. In the PINK1-Parkin pathway, when the mitochondria are damaged and undergo membrane depolarisation, PINK1 accumulates in the MOM. PINK1 is then activated through autophosphorylation, leading to the direct or indirect phosphorylation of Ser65 in the parkin Ubl domain. Subsequently, p-parkin ubiquitinates mitochondrial outer membrane proteins, forming complexes with sequestosome1 (p62) and microtubule-associated protein 1 light chain 3 II. These events ultimately result in mitophagy (Figure 1) (Eldeeb et al., 2022; Wu et al., 2022; Yildirim et al., 2022).
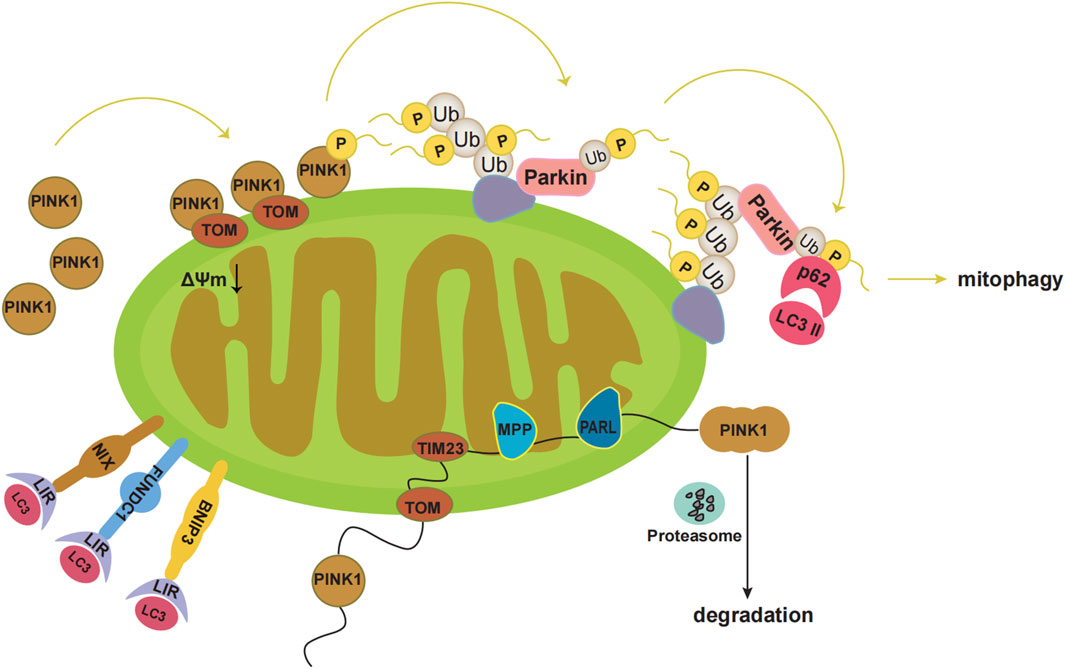
FIGURE 1. Pathways of mitophagy. Under physiological conditions, PINK1 is localized to the mitochondrial outer membrane, which is introduced into the intermembrane space by translocase outer membrane (TOM). Then PINK1 is introduced into the mitochondrial inner membrane by translocase of the inner membrane 23 (TIM23). At the mitochondrial inner membrane, the mitochondrial processing peptidase (MPP) removes PINK1’s N-terminal MTS. Subsequently, PGAM5-associated rhomboid-like protease (PARL) cleaves PINK1. Finally, PINK1 in the double-cleavage form is released into the cytoplasm and is degraded rapidly by the ubiquitin proteasome system. Under pathological conditions, the damaged mitochondria undergo mitochondrial membrane depolarization. PINK1 accumulates at the mitochondrial outer membrane. Then PINK1 kinase is activated by autophosphorylation, which directly phosphorylates Ser65 in the parkin Ubl domain, or first phosphorylates Ser65 in ubiquitin and leads to conformational changes in parkin, then causes the release of the Ubl domain from the parkin core, and finally phosphorylates Ser65 in the parkin Ubl domain. Subsequently, p-Parkin ubiquitinates mitochondrial outer membrane proteins, which bind sequestosome1 (p62) and microtubule-associated protein 1 light chain 3 II (LC3II). Eventually mitophagy occurs. Additionally, BNIP3, NIX, and FUNDC1 can interact with the autophagosome membrane protein LC3 through the LC3 interaction region (LIR).
In addition, various mitochondrial proteins, such as NIP-3-like protein X (NIX), FUN14 domain-containing 1 (FUNDC1), and BNIP3 can act as mitophagy receptors to guide the clearance of damaged mitochondria (Xie et al., 2020; Linqing et al., 2021; Xie et al., 2021; Liu et al., 2022; Ning et al., 2022). BNIP3, NIX, and FUNDC1 interact with the autophagosomal membrane protein LC3 via the LC3 interaction region (Figure 1) (Wei et al., 2015). Phosphorylation of Ser34/35 near the LC3 interaction region of NIX enhances its binding to LC3 (Rogov et al., 2017). In addition, NIX may induce mitophagy by promoting the production of ROS (Li et al., 2015; Li et al., 2022). Both NIX and BNIP3 are considerably upregulated by HIF-1α under hypoxic conditions, facilitating the clearance of damaged mitochondria and preventing ROS accumulation (Mazure and Pouyssegur, 2010). NIX may act as a substrate for Parkin in the PINK1-Parkin pathway (Gao et al., 2015). BNIP3 may interact with PINK1 to prevent its proteolytic activity and support its accumulation on the mitochondrial membrane, thereby promoting mitophagy (Zhang et al., 2016). FUNDC1 mediated mitophagy is primarily regulated through phosphorylation and ubiquitination. In the presence of oxygen, FUNDC1 is phosphorylated by Src kinase at Tyr18 and by CK2 at Ser13 (Zhang et al., 2022). Under hypoxic conditions, FUNDC1 is ubiquitinated by lysine 119 of the mitochondrial E3 ligase membrane-associated RING-CH5, which protects mitochondria from degradation by mitophagy. During severe hypoxia, CK2 and Src kinases are inhibited, dephosphorylating FUNDC1 and increasing its binding affinity to LC3, thereby promoting mitophagy (Chen et al., 2017a).
Mitochondria have a bilayer membrane structure with approximately 140 proteins in the outer membrane, 130 soluble proteins in the intermembrane space, and 800 and 500 proteins in the inner membrane and matrix, respectively (Jadiya and Tomar, 2020). Molecular chaperones, proteases, the ubiquitin-proteasome system, mitophagy, mitochondria-derived vesicles, and the mitochondrial unfolded protein response are involved in mitochondrial protein quality control (Friedlander et al., 2021; Song et al., 2021).
Molecular chaperones, such as Hsp60 (Fan et al., 2020) and Hsp70 (Havalova et al., 2021), can promote and maintain the correct folding of proteins to ensure their normal function. Mitochondrial proteases play crucial roles in controlling the quantity of mitochondrial proteins. They are responsible for protein turnover and processing within mitochondria (Steele and Glynn, 2019; Szczepanowska and Trifunovic, 2022). The ubiquitin-proteasome system is involved in the degradation of more than 80% of endogenous proteins in eukaryotes, and ubiquitin molecules ubiquitinate and degrade substrate proteins mainly through the formation of multiubiquitin chains (Alsayyah et al., 2020; Kodron et al., 2021). Currently, the specific mechanism by which mitochondria-derived vesicles are involved in mitochondrial protein quality control is not clear. Several studies have pointed out that vesicles contain damaged MOMs, inner membranes, and matrix proteins that are transported to lysosomes for degradation (Soubannier et al., 2012), suggesting that vesicles may be a complementary mechanism that controls mitochondrial quality prior to mitochondrial degradation at the organelle level (Baixauli et al., 2014). The mitochondrial unfolded protein response is a stress response. Under stress signalling conditions, such as decreased mitochondrial membrane potential, mitochondrial DNA (mtDNA) clearance, mitochondrial accumulation of misfolded proteins, or an imbalance between nuclear and mitochondrial-encoding proteins, the gene transcription of nuclear-encoded mitochondrial chaperones and proteases is activated. This activation enhances protein homeostasis (Svagusa et al., 2020). Tight-coupled molecular interactions between multiple players in the mitochondrial protein quality control system ensures protein balance and mitochondrial function to maintain overall cellular fitness.
Preeclampsia is a multisystem disorder that occurs during pregnancy, characterised by new-onset hypertension (systolic pressure/diastolic pressure, ≥140/90 mmHg) and proteinuria (≥300 mg/24 h) after 20 weeks of gestation. In 2013, the American Conference of Obstetricians and Gynecologists noted that proteinuria was not necessary for the diagnosis of preeclampsia. Preeclampsia can also be diagnosed as hypertension associated with thrombocytopenia, renal insufficiency, liver impairment, pulmonary oedema, or brain or visual impairment.
The association between mitochondrial dysfunction and preeclampsia was first explored in 1989, and several members of a family with mitochondrial dysfunction were found to have a high incidence of preeclampsia and eclampsia (Torbergsen et al., 1989). This was followed by a steady stream of studies linking mitochondrial dysfunction to preeclampsia. One study reported morphological data showing degeneration and apoptotic changes in the mitochondria of the preeclamptic placenta (Shi et al., 2013). The study also found that compared with those in normal placentas, preeclampsia placentas had four upregulated mitochondrial proteins and 22 downregulated mitochondrial proteins that are involved in many key processes in the development of preeclampsia, such as apoptosis, fatty acid oxidation, respiratory chain, ROS generation, tricarboxylic acid cycle, and oxidative stress. Thus, mitochondrial dysfunction is closely associated with the occurrence and development.
As research has progressed in recent years, our understanding of the specific mechanisms underlying preeclampsia has deepened. It has become increasingly clear that mitochondrial dysfunction plays a central role in the molecular mechanism of preeclampsia, particularly when it is caused by dysfunction in placental 11β-hydroxysteroid dehydrogenase type 2 (Long et al., 2022). Their study revealed that dysfunction in 11β-hydroxysteroid dehydrogenase type 2 resulted in mtDNA instability and impaired mitochondrial dynamics, contributing to the development of preeclampsia, and the mitochondrial-targeted antioxidant MitoTEMPO has significantly alleviated the symptoms of preeclampsia.
Interestingly, conflicting conclusions exist in the literature regarding the expression of mitochondrial fusion and fission proteins in preeclamptic placentas. These discrepancies in protein expression may be attributed to variations in the severity of preeclampsia and differences in study populations. Fusion is a process that helps preserve mitochondrial function by mixing the contents of damaged mitochondria, whereas fission is the first step in mitophagy. The mitochondrial fusion proteins mitofusin 1, MFN2, and optic atrophy 1 are downregulated in the placentas of women with preeclampsia (Zhou et al., 2017). Another study also showed that MFN2 and ATP expression was significantly reduced in the preeclamptic placenta compared with that in normal placenta (Yu et al., 2016). However, maternal serum MFN2 levels were higher in patients with preeclampsia (Aydogan Mathyk et al., 2020). In conclusion, despite conflicting findings from various studies, it is evident that there is a disruption in the balance between mitochondrial fusion and fission in the preeclamptic placenta.
Mitophagy serves the function of eliminating damaged mitochondria. Defects in the autophagic pathway may contribute to the onset and progression of preeclampsia. A previous study found that the inhibition of BNIP3 expression reduced autophagic activity, leading to the accumulation of damaged mitochondria in vivo, thus participating in the development of preeclampsia (Zhou et al., 2021). The downregulation of BNIP3 expression in preeclamptic placentae has also been confirmed in other studies (Tong et al., 2018; Ma et al., 2019). These results suggest that decreased autophagy is closely associated with preeclampsia. However, another study found contradictory results. Low levels of FUNDC1 ubiquitination have been found in hypoxic trophoblast cells and the placentas of pregnant women with preeclampsia (Chen et al., 2022). Mitochondrial E3 ligase membrane-associated RING-CH5 has been reported to inhibit hypoxia-induced mitochondrial autophagy through ubiquitination and degradation of FUNDC1, and inhibition of FUNDC1 ubiquitination and degradation increases mitochondrial sensitivity to autophagy-induced stress (Chen et al., 2017a; Chen et al., 2017b). Therefore, FUNDC1 ubiquitination is a process of mitochondrial desensitisation that prevents functional mitochondria from being removed via autophagy. The decrease in FUNDC1 ubiquitination promotes autophagy, resulting in excessive enhancement of autophagy and destruction of normal mitochondria, which are related to the occurrence and development of preeclampsia. In summary, changes in autophagic activity are associated with preeclampsia.
Foetal growth restriction is defined as foetal body mass or abdominal circumference less than the 10th percentile of body mass for gestational age and is also known as intrauterine growth restriction (IUGR). According to the 2021 guidelines issued by the American College of Obstetricians and Gynecologists, the aetiology of FGR can be divided into maternal, foetal, and placental factors.
Many recent studies have explored the relation between mitochondrial dysfunction and foetal growth restriction. Excessive oxidative stress and perturbed mitochondrial antioxidant capacity can interfere with mtDNA replication, leading to a decrease in mtDNA content in the peripheral blood (Knez et al., 2017). A study found that the mtDNA copy number (mtDNAcn) in the peripheral blood of women with preeclampsia associated with IUGR was significantly lower than that in women with preeclampsia associated with appropriate for gestational age intrauterine foetal growth (Busnelli et al., 2019). Moreover, several studies have reported a reduction in mtDNA copy number levels in the placenta of patients with IUGR (Diaz et al., 2014; Mando et al., 2014; Poidatz et al., 2015; Luo et al., 2018). However, another study found that an increase in mtDNA copy number was an indicator of mitochondrial functional impairment, and the mtDNA content in the IUGR pregnancy group was significantly increased (Naha et al., 2020). Therefore, we speculate that the change in mtDNA copy number is closely related to foetal growth restriction. It was also found that mitochondrial Sirtuin-3 (Sirt3) protein levels were significantly decreased, and succinate dehydrogenase activity was decreased in the IUGR pregnancy group (Naha et al., 2020). Sirt3 is translated as a 44 kDa inactivated protein that is cleaved by 142 amino acids to produce an active 28 kDa protein that acts as a deacetylase on several mitochondrial maintenance proteins (Sundaresan et al., 2008). In addition, mitochondrial Sirt3 has been shown to be associated with ATP synthase subunits to regulate ATP synthesis and maintain membrane potential (Yang et al., 2016). Therefore, decreased Sirt3 protein expression and succinate dehydrogenase activity suggest that mitochondrial function is impaired in growth-restricted placentas.
The placenta is crucial for the growth and development of the foetus, and because of its high energy demand, the maintenance of mitochondrial function is a necessary condition for the growth and development of the foetus. Studies have shown that abnormalities in mitochondrial fusion and fission, mitophagy, and various mechanisms in the mitochondrial protein quantity control system may lead to mitochondrial dysfunction and placenta-related diseases. Recent studies have shown that exogenous supplementation with antioxidants, such as vitamin D, coenzyme Q10, and melatonin, can improve mitochondrial function, inhibit oxidative stress, reduce inflammatory responses, and protect mitochondria from oxidative damage (Reiter et al., 2020; Phillips et al., 2022). One study has shown that mitochondrial transfer can improve embryo quality and resolve pregnancy complications caused by abnormal mtDNA (Morimoto et al., 2023). However, the specific mechanisms by which mitochondrial dysfunction leads to placental disorders remain unclear. It is expected that the specific molecular mechanisms will be further clarified using animal models and human placental samples to provide new ideas for the prevention and treatment of placenta-related diseases.
YW: Writing–original draft, Writing–review and editing. ML: Writing–original draft, Writing–review and editing. HY: Writing–review and editing. YiG: Writing–review and editing. YZ: Writing–review and editing. YaG: Writing–review and editing. LH: Writing–review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. This work was supported by grants from Top Talent Project of Wuxi Health Commission (BJ2020081) and China Scholarship Council, Maternal and Child Health Research Project of Wuxi Health Commission (FYKY202106). Wuxi Science and Technology Bureau project “Light of Taihu Lake” science and technology research (basic research) (K20221035).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Al Ojaimi M., Salah A., El-Hattab A. W. (2022). Mitochondrial fission and fusion: molecular mechanisms, biological functions, and related disorders. Membranes 12 (9), 893. doi:10.3390/membranes12090893
Alsayyah C., Ozturk O., Cavellini L., Belgareh-Touze N., Cohen M. M. (2020). The regulation of mitochondrial homeostasis by the ubiquitin proteasome system. Biochimica Biophysica Acta-Bioenergetics 1861 (12), 148302. doi:10.1016/j.bbabio.2020.148302
Aydogan Mathyk B., Temel Yuksel I., Tayyar A., Aslan Cetin B., Tayyar A. T., Koroglu N. (2020). Maternal serum mitofusin-2 levels in patients with preeclampsia: the possible role of mitochondrial dysfunction in preeclampsia. J. Matern. Fetal Neonatal Med. 33 (11), 1861–1866. doi:10.1080/14767058.2018.1532497
Baixauli F., Lopez-Otin C., Mittelbrunn M. (2014). Exosomes and autophagy: coordinated mechanisms for the maintenance of cellular fitness. Front. Immunol. 5, 403. doi:10.3389/fimmu.2014.00403
Busnelli A., Lattuada D., Ferrari S., Reschini M., Colciaghi B., Somigliana E., et al. (2019). Mitochondrial DNA copy number in peripheral blood in the first trimester of pregnancy and different preeclampsia clinical phenotypes development: a pilot study. Reprod. Sci. 26 (8), 1054–1061. doi:10.1177/1933719118804410
Chen G., Chen L., Huang Y., Zhu X., Yu Y. (2022a). Increased FUN14 domain containing 1 (FUNDC1) ubiquitination level inhibits mitophagy and alleviates the injury in hypoxia-induced trophoblast cells. Bioengineered 13 (2), 3620–3633. doi:10.1080/21655979.2021.1997132
Chen T.-H., Koh K.-Y., Lin K.M.-C., Chou C.-K. (2022b). Mitochondrial dysfunction as an underlying cause of skeletal muscle disorders. Int. J. Mol. Sci. 23 (21), 12926. doi:10.3390/ijms232112926
Chen Y., Qin W., Li L., Wu P., Wei D. (2022c). Mitophagy: critical role in atherosclerosis progression. DNA Cell Biol. 41 (10), 851–860. doi:10.1089/dna.2022.0249
Chen Z., Liu L., Cheng Q., Li Y., Wu H., Zhang W., et al. (2017a). Mitochondrial E3 ligase MARCH5 regulates FUNDC1 to fine-tune hypoxic mitophagy. Embo Rep. 18 (3), 495–509. doi:10.15252/embr.201643309
Chen Z., Siraj S., Liu L., Chen Q. (2017b). MARCH5-FUNDC1 axis fine-tunes hypoxia-induced mitophagy. Autophagy 13 (7), 1244–1245. doi:10.1080/15548627.2017.1310789
Diaz M., Aragones G., Sanchez-Infantes D., Bassols J., Perez-Cruz M., de Zegher F., et al. (2014). Mitochondrial DNA in placenta: associations with fetal growth and superoxide dismutase activity. Horm. Res. Paediatr. 82 (5), 303–309. doi:10.1159/000366079
Eldeeb M. A., Esmaili M., Hassan M., Ragheb M. A. (2022). The role of PTEN-L in modulating PINK1-parkin-mediated mitophagy. Neurotox. Res. 40, 1103–1114. doi:10.1007/s12640-022-00475-w
Fan F., Duan Y., Yang F., Trexler C., Wang H., Huang L., et al. (2020). Deletion of heat shock protein 60 in adult mouse cardiomyocytes perturbs mitochondrial protein homeostasis and causes heart failure. Cell Death Differ. 27 (2), 587–600. doi:10.1038/s41418-019-0374-x
Friedlander J. E., Shen N., Zeng A., Korm S., Feng H. (2021). Failure to guard: mitochondrial protein quality control in cancer. Int. J. Mol. Sci. 22 (15), 8306. doi:10.3390/ijms22158306
Fuchs P., Rugen N., Carrie C., Elsaesser M., Finkemeier I., Giese J., et al. (2020). Single organelle function and organization as estimated from Arabidopsis mitochondrial proteomics. Plant J. 101 (2), 420–441. doi:10.1111/tpj.14534
Gao F., Chen D., Si J., Hu Q., Qin Z., Fang M., et al. (2015). The mitochondrial protein BNIP3L is the substrate of PARK2 and mediates mitophagy in PINK1/PARK2 pathway. Hum. Mol. Genet. 24 (9), 2528–2538. doi:10.1093/hmg/ddv017
Green D. R., Van Houten B. (2011). SnapShot: mitochondrial quality control. Cell 147 (4), 950. doi:10.1016/j.cell.2011.10.036
Havalova H., Ondrovicova G., Keresztesova B., Bauer J. A., Pevala V., Kutejova E., et al. (2021). Mitochondrial HSP70 chaperone system-the influence of post-translational modifications and involvement in human diseases. Int. J. Mol. Sci. 22 (15), 8077. doi:10.3390/ijms22158077
Hernansanz-Agustin P., Enriquez J. A. (2021). Generation of reactive oxygen species by mitochondria. Antioxidants 10 (3), 415. doi:10.3390/antiox10030415
Jadiya P., Tomar D. (2020). Mitochondrial protein quality control mechanisms. Genes (Basel) 11 (5), 563. doi:10.3390/genes11050563
Jouaville L. S., Ichas F., Mazat J. P. (1998). Modulation of cell calcium signals by mitochondria. Mol. Cell Biochem. 184 (1-2), 371–376. doi:10.1023/a:1006850121769
Kanamaru Y., Sekine S., Ichijo H., Takeda K. (2012). The phosphorylation-dependent regulation of mitochondrial proteins in stress responses. J. signal Transduct. 2012, 931215. doi:10.1155/2012/931215
Kiyokoba R., Uchiumi T., Yagi M., Toshima T., Tsukahara S., Fujita Y., et al. (2022). Mitochondrial dysfunction-induced high hCG associated with development of fetal growth restriction and pre-eclampsia with fetal growth restriction. Sci. Rep. 12 (1), 4056. doi:10.1038/s41598-022-07893-y
Knez J., Marrachelli V. G., Cauwenberghs N., Winckelmans E., Zhang Z., Thijs L., et al. (2017). Peripheral blood mitochondrial DNA content in relation to circulating metabolites and inflammatory markers: a population study. Plos One 12 (7), e0181036. doi:10.1371/journal.pone.0181036
Kodron A., Mussulini B. H., Pilecka I., Chacinska A. (2021). The ubiquitin-proteasome system and its crosstalk with mitochondria as therapeutic targets in medicine. Pharmacol. Res. 163, 105248. doi:10.1016/j.phrs.2020.105248
Kotiadis V. N., Duchen M. R., Osellame L. D. (2014). Mitochondrial quality control and communications with the nucleus are important in maintaining mitochondrial function and cell health. Biochimica Biophysica Acta-General Subj. 1840 (4), 1254–1265. doi:10.1016/j.bbagen.2013.10.041
Kuznetsov A. V., Margreiter R., Ausserlechner M. J., Hagenbuchner J. (2022). The complex interplay between mitochondria, ROS and entire cellular metabolism. Antioxidants 11 (10), 1995. doi:10.3390/antiox11101995
Li L., Tan J., Miao Y., Lei P., Zhang Q. (2015). ROS and autophagy: interactions and molecular regulatory mechanisms. Cell. Mol. Neurobiol. 35 (5), 615–621. doi:10.1007/s10571-015-0166-x
Li Y., Shang C., Liu Z., Han J., Li W., Xiao P., et al. (2022). Apoptin mediates mitophagy and endogenous apoptosis by regulating the level of ROS in hepatocellular carcinoma. Cell Commun. Signal. 20 (1), 134. doi:10.1186/s12964-022-00940-1
Linqing L., Yuhan Q., Erfei L., Yong Q., Dong W., Chengchun T., et al. (2021). Hypoxia-induced PINK1/Parkin-mediated mitophagy promotes pulmonary vascular remodeling. Biochem. Biophys. Res. Commun. 534, 568–575. doi:10.1016/j.bbrc.2020.11.040
Liu X., Hussain R., Mehmood K., Tang Z., Zhang H., Li Y. (2022). Mitochondrial-endoplasmic reticulum communication-mediated oxidative stress and autophagy. Biomed. Res. Int. 2022. doi:10.1155/2022/6459585
Long J., Huang Y., Tang Z., Shan Y., Feng D., Wang W., et al. (2022). Mitochondria targeted antioxidant significantly alleviates preeclampsia caused by 11β-HSD2 dysfunction via OPA1 and MtDNA maintenance. Antioxidants 11 (8), 1505. doi:10.3390/antiox11081505
Luo Z., Luo W., Li S., Zhao S., Sho T., Xu X., et al. (2018). Reactive oxygen species mediated placental oxidative stress, mitochondrial content, and cell cycle progression through mitogen-activated protein kinases in intrauterine growth restricted pigs. Reprod. Biol. 18 (4), 422–431. doi:10.1016/j.repbio.2018.09.002
Ma J., Yang J., Lv S., Gao M., Sun Y., Chen Z. J., et al. (2019). Dysfunction of B-cell lymphoma 2/adenovirus E1B 19KD interacting protein 3 in decidua is involved in the pathogenesis of preeclampsia. J. Hypertens. 37 (10), 2048–2060. doi:10.1097/HJH.0000000000002139
Mando C., De Palma C., Stampalija T., Anelli G. M., Figus M., Novielli C., et al. (2014). Placental mitochondrial content and function in intrauterine growth restriction and preeclampsia. Am. J. Physiol. Endocrinol. Metab. 306 (4), E404–E413. doi:10.1152/ajpendo.00426.2013
Mani S., Sevanan M., Krishnamoorthy A., Sekar S. (2021). A systematic review of molecular approaches that link mitochondrial dysfunction and neuroinflammation in Parkinson’s disease. Neurol. Sci. 42 (11), 4459–4469. doi:10.1007/s10072-021-05551-1
Manna S., McCarthy C., McCarthy F. P. (2019). Placental ageing in adverse pregnancy outcomes: telomere shortening, cell senescence, and mitochondrial dysfunction. Oxidative Med. Cell. Longev. 2019, 3095383. doi:10.1155/2019/3095383
Marin R., Chiarello D. I., Abad C., Rojas D., Toledo F., Sobrevia L. (2020). Oxidative stress and mitochondrial dysfunction in early-onset and late-onset preeclampsia. Biochimica Biophysica Acta-Molecular Basis Dis. 1866 (12), 165961. doi:10.1016/j.bbadis.2020.165961
Mazure N. M., Pouyssegur J. (2010). Hypoxia-induced autophagy: cell death or cell survival? Curr. Opin. Cell Biol. 22 (2), 177–180. doi:10.1016/j.ceb.2009.11.015
Morimoto Y., Gamage U. S. K., Yamochi T., Saeki N., Morimoto N., Yamanaka M., et al. (2023). Mitochondrial transfer into human oocytes improved embryo quality and clinical outcomes in recurrent pregnancy failure cases. Int. J. Mol. Sci. 24 (3), 2738. doi:10.3390/ijms24032738
Naha R., Anees A., Chakrabarty S., Naik P. S., Pandove M., Pandey D., et al. (2020). Placental mitochondrial DNA mutations and copy numbers in intrauterine growth restricted (IUGR) pregnancy. Mitochondrion 55, 85–94. doi:10.1016/j.mito.2020.08.008
Ning P., Jiang X., Yang J., Zhang J., Yang F., Cao H. (2022). Mitophagy: a potential therapeutic target for insulin resistance. Front. physiol. 13. doi:10.3389/fphys.2022.957968
Phillips E. A., Hendricks N., Bucher M., Maloyan A. (2022). Vitamin D supplementation improves mitochondrial function and reduces inflammation in placentae of obese women. Front. Endocrinol. 13, 893848. doi:10.3389/fendo.2022.893848
Poidatz D., Dos Santos E., Duval F., Moindjie H., Serazin V., Vialard F., et al. (2015). Involvement of estrogen-related receptor-gamma and mitochondrial content in intrauterine growth restriction and preeclampsia. Fertil. Steril. 104 (2), 483–490. doi:10.1016/j.fertnstert.2015.05.005
Reiter R. J., Ma Q., Sharma R. (2020). Melatonin in mitochondria: mitigating clear and present dangers. Physiology 35 (2), 86–95. doi:10.1152/physiol.00034.2019
Rogov V. V., Suzuki H., Marinkovic M., Lang V., Kato R., Kawasaki M., et al. (2017). Phosphorylation of the mitochondrial autophagy receptor Nix enhances its interaction with LC3 proteins. Sci. Rep. 7 (1), 1131. doi:10.1038/s41598-017-01258-6
Ryoo I.-g., Kwak M.-K. (2018). Regulatory crosstalk between the oxidative stress-related transcription factor Nfe2l2/Nrf2 and mitochondria. Toxicol. Appl. Pharmacol. 359, 24–33. doi:10.1016/j.taap.2018.09.014
Sadoshima J., Kitsis R. N., Sciarretta S. (2021). Editorial: mitochondrial dysfunction and cardiovascular diseases. Front. Cardiovasc. Med. 8, 645986. doi:10.3389/fcvm.2021.645986
Serasinghe M. N., Chipuk J. E. (2017). Mitochondrial fission in human diseases. Handb. Exp. Pharmacol. 240, 159–188. doi:10.1007/164_2016_38
Shao X., Yu W., Yang Y., Wang F., Yu X., Wu H., et al. (2022). The mystery of the life tree: the placentas†. Biol. Reproduction 107 (1), 301–316. doi:10.1093/biolre/ioac095
Shi Z., Long W., Zhao C., Guo X., Shen R., Ding H. (2013). Comparative proteomics analysis suggests that placental mitochondria are involved in the development of pre-eclampsia. Plos One 8 (5), e64351. doi:10.1371/journal.pone.0064351
Sloat S. R., Whitley B. N., Engelhart E. A., Hoppins S. (2019). Identification of a mitofusin specificity region that confers unique activities to Mfn1 and Mfn2. Mol. Biol. Cell 30 (17), 2309–2319. doi:10.1091/mbc.E19-05-0291
Smith A. N., Wang X., Thomas D. G., Tatum R. E., Booz G. W., Cunningham M. W. (2021). The role of mitochondrial dysfunction in preeclampsia: causative factor or collateral damage? Am. J. Hypertens. 34 (5), 442–452. doi:10.1093/ajh/hpab003
Song J., Herrmann J. M., Becker T. (2021). Quality control of the mitochondrial proteome. Nat. Rev. Mol. Cell Biol. 22 (1), 54–70. doi:10.1038/s41580-020-00300-2
Soubannier V., McLelland G. L., Zunino R., Braschi E., Rippstein P., Fon E. A., et al. (2012). A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr. Biol. 22 (2), 135–141. doi:10.1016/j.cub.2011.11.057
Steele T. E., Glynn S. E. (2019). Mitochondrial AAA proteases: a stairway to degradation. Mitochondrion 49, 121–127. doi:10.1016/j.mito.2019.07.012
Suarez-Rivero J. M., Villanueva-Paz M., de la Cruz-Ojeda P., de la Mata M., Cotan D., Oropesa-Avila M., et al. (2016). Mitochondrial dynamics in mitochondrial diseases. Dis. (Basel, Switz. 5 (1), 1. doi:10.3390/diseases5010001
Suliman H. B., Piantadosi C. A. (2016). Mitochondrial quality control as a therapeutic target. Pharmacol. Rev. 68 (1), 20–48. doi:10.1124/pr.115.011502
Sundaresan N. R., Samant S. A., Pillai V. B., Rajamohan S. B., Gupta M. P. (2008). SIRT3 is a stress-responsive deacetylase in cardiomyocytes that protects cells from stress-mediated cell death by deacetylation of Ku70. Mol. Cell. Biol. 28 (20), 6384–6401. doi:10.1128/mcb.00426-08
Svagusa T., Martinic M., Martinic M., Kovacevic L., Sepac A., Milicic D., et al. (2020). Mitochondrial unfolded protein response, mitophagy and other mitochondrial quality control mechanisms in heart disease and aged heart. Croat. Med. J. 61 (2), 126–138. doi:10.3325/cmj.2020.61.126
Szczepanowska K., Trifunovic A. (2022). Mitochondrial matrix proteases: quality control and beyond. Febs J. 289 (22), 7128–7146. doi:10.1111/febs.15964
Tian C., Liu Y., Li Z., Zhu P., Zhao M. (2022a). Mitochondria related cell death modalities and disease. Front. Cell Dev. Biol. 10, 832356. doi:10.3389/fcell.2022.832356
Tian Y., Liu X., Pei X., Gao H., Pan P., Yang Y. (2022b). Mechanism of mitochondrial homeostasis controlling ovarian Physiology. Endocrinology 164 (1), bqac189. doi:10.1210/endocr/bqac189
Tong J., Zhao W., Lv H., Li W. P., Chen Z. J., Zhang C. (2018). Transcriptomic profiling in human decidua of severe preeclampsia detected by RNA sequencing. J. Cell Biochem. 119 (1), 607–615. doi:10.1002/jcb.26221
Torbergsen T., Oian P., Mathiesen E., Borud O. (1989). Pre-eclampsia--a mitochondrial disease? Acta obstetricia Gynecol. Scand. 68 (2), 145–148. doi:10.3109/00016348909009902
Vazquez-Calvo C., Suhm T., Buettner S., Ott M. (2020). The basic machineries for mitochondrial protein quality control. Mitochondrion 50, 121–131. doi:10.1016/j.mito.2019.10.003
Vodickova A., Koren S. A., Wojtovich A. P. (2022). Site-specific mitochondrial dysfunction in neurodegeneration. Mitochondrion 64, 1–18. doi:10.1016/j.mito.2022.02.004
Wang N., Wang C., Zhao H., He Y., Lan B., Sun L., et al. (2021). The MAMs structure and its role in cell death. Cells 10 (3), 657. doi:10.3390/cells10030657
Wang S., Lin L., Wang Y., Wang A., Liu Z., Wu S., et al. (2020). Novel homozygous mutation in the FBXL4 gene is associated with mitochondria DNA depletion syndrome-13. J. Neurological Sci. 416, 116948. doi:10.1016/j.jns.2020.116948
Wei H., Liu L., Chen Q. (2015). Selective removal of mitochondria via mitophagy: distinct pathways for different mitochondrial stresses. Biochimica Biophysica Acta-Molecular Cell Res. 1853 (10), 2784–2790. doi:10.1016/j.bbamcr.2015.03.013
Wu Y., Jiang T., Hua J., Xiong Z., Dai K., Chen H., et al. (2022). PINK1/Parkin-mediated mitophagy in cardiovascular disease: from pathogenesis to novel therapy. Int. J. Cardiol. 361, 61–69. doi:10.1016/j.ijcard.2022.05.025
Xie Y., Liu J., Kang R., Tang D. (2020). Mitophagy receptors in tumor biology. Front. Cell Dev. Biol. 8. doi:10.3389/fcell.2020.594203
Xie Y., Liu J., Kang R., Tang D. (2021). Mitophagy in pancreatic cancer. Front. Oncol. 11. doi:10.3389/fonc.2021.616079
Yang W., Nagasawa K., Munch C., Xu Y., Satterstrom K., Jeong S., et al. (2016). Mitochondrial Sirtuin network reveals dynamic SIRT3-dependent deacetylation in response to membrane depolarization. Cell 167 (4), 985–1000. doi:10.1016/j.cell.2016.10.016
Yildirim R. M., Ergun Y., Basar M. (2022). Mitochondrial dysfunction, mitophagy and their correlation with perinatal complications: preeclampsia and low birth weight. Biomedicines 10 (10), 2539. doi:10.3390/biomedicines10102539
Youle R. J., van der Bliek A. M. (2012). Mitochondrial fission, fusion, and stress. Science 337 (6098), 1062–1065. doi:10.1126/science.1219855
Yu J., Guo X., Chen R., Feng L. (2016). Downregulation of mitofusin 2 in placenta is related to preeclampsia. Biomed Res. Int. 2016, 6323086. doi:10.1155/2016/6323086
Zemirli N., Morel E., Molino D. (2018). Mitochondrial dynamics in basal and stressful conditions. Int. J. Mol. Sci. 19 (2), 564. doi:10.3390/ijms19020564
Zhang P., Konja D., Zhang Y., Wang Y. (2021). Communications between mitochondria and endoplasmic reticulum in the regulation of metabolic homeostasis. Cells 10 (9), 2195. doi:10.3390/cells10092195
Zhang T., Xue L., Li L., Tang C., Wan Z., Wang R., et al. (2016). BNIP3 protein suppresses PINK1 kinase proteolytic cleavage to promote mitophagy. J. Biol. Chem. 291 (41), 21616–21629. doi:10.1074/jbc.M116.733410
Zhang Y., Lian Y., Lian X., Zhang H., Chen Y., Sheng H., et al. (2022). FUNDC1 mediated mitophagy in epileptic hippocampal neuronal injury induced by magnesium-free fluid. Neurochem. Res. 48, 284–294. doi:10.1007/s11064-022-03749-z
Zhou X., Han T. L., Chen H., Baker P. N., Qi H., Zhang H. (2017). Impaired mitochondrial fusion, autophagy, biogenesis and dysregulated lipid metabolism is associated with preeclampsia. Exp. Cell Res. 359 (1), 195–204. doi:10.1016/j.yexcr.2017.07.029
Keywords: mitochondrial dysfunction, preeclampsia, foetal growth restriction, placenta, mitochondria
Citation: Wu Y, Li M, Ying H, Gu Y, Zhu Y, Gu Y and Huang L (2024) Mitochondrial quality control alterations and placenta-related disorders. Front. Physiol. 15:1344951. doi: 10.3389/fphys.2024.1344951
Received: 27 November 2023; Accepted: 17 January 2024;
Published: 08 February 2024.
Edited by:
Diego De Stefani, University of Padua, ItalyReviewed by:
Yang Yang, First Affiliated Hospital of Zhengzhou University, ChinaCopyright © 2024 Wu, Li, Ying, Gu, Zhu, Gu and Huang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Lu Huang, OTg2MjAyMzM3M0BqaWFuZ25hbi5lZHUuY24=; Yanfang Gu, ODQyOTIzMDU4QHFxLmNvbQ==
†These authors share first authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.