 Md Monirul Hoque1,2
Md Monirul Hoque1,2 Joy Olaoluwa Gbadegoye1,2
Joy Olaoluwa Gbadegoye1,2 Fasilat Oluwakemi Hassan1,2
Fasilat Oluwakemi Hassan1,2 Amr Raafat1
Amr Raafat1 Djamel Lebeche1,2,3,4*
Djamel Lebeche1,2,3,4*- 1Departments of Physiology, The University of Tennessee Health Science Center, Memphis, TN, United States
- 2College of Graduate Health Sciences, The University of Tennessee Health Science Center, Memphis, TN, United States
- 3Medicine-Cardiology, College of Medicine, The University of Tennessee Health Science Center, Memphis, TN, United States
- 4Pharmaceutical Sciences, College of Pharmacy, The University of Tennessee Health Science Center, Memphis, TN, United States
Cardiac fibrosis is a major and complex pathophysiological process that ultimately culminates in cardiac dysfunction and heart failure. This phenomenon includes not only the replacement of the damaged tissue by a fibrotic scar produced by activated fibroblasts/myofibroblasts but also a spatiotemporal alteration of the structural, biochemical, and biomechanical parameters in the ventricular wall, eliciting a reactive remodeling process. Though mechanical stress, post-infarct homeostatic imbalances, and neurohormonal activation are classically attributed to cardiac fibrosis, emerging evidence that supports the roles of immune system modulation, inflammation, and metabolic dysregulation in the initiation and progression of cardiac fibrogenesis has been reported. Adaptive changes, immune cell phenoconversions, and metabolic shifts in the cardiac nonmyocyte population provide initial protection, but persistent altered metabolic demand eventually contributes to adverse remodeling of the heart. Altered energy metabolism, mitochondrial dysfunction, various immune cells, immune mediators, and cross-talks between the immune cells and cardiomyocytes play crucial roles in orchestrating the transdifferentiation of fibroblasts and ensuing fibrotic remodeling of the heart. Manipulation of the metabolic plasticity, fibroblast–myofibroblast transition, and modulation of the immune response may hold promise for favorably modulating the fibrotic response following different cardiovascular pathological processes. Although the immunologic and metabolic perspectives of fibrosis in the heart are being reported in the literature, they lack a comprehensive sketch bridging these two arenas and illustrating the synchrony between them. This review aims to provide a comprehensive overview of the intricate relationship between different cardiac immune cells and metabolic pathways as well as summarizes the current understanding of the involvement of immune–metabolic pathways in cardiac fibrosis and attempts to identify some of the previously unaddressed questions that require further investigation. Moreover, the potential therapeutic strategies and emerging pharmacological interventions, including immune and metabolic modulators, that show promise in preventing or attenuating cardiac fibrosis and restoring cardiac function will be discussed.
1 Introduction
Myocardial fibrosis is a common pathophysiologic companion of many different myocardial conditions, where the cardiac interstitium expands through the deposition of extracellular matrix (ECM) proteins (Frangogiannis, 2021). Unlike other organs, the adult mammalian heart has limited regenerative potential. In response to ischemic insults, systemic diseases, or any other harmful stimulus to the circulatory system or the heart itself, the damaged cardiomyocytes are replaced by a fibrotic scar (Gyöngyösi et al., 2017). Though this event is crucial for the preservation of ventricular rupture (Travers et al., 2016), over time, excessive and continuous ECM deposition leads to irreversible ventricular remodeling and distorted organ geometry and significantly impairs the function of the heart (Li et al., 2018; Maruyama and Imanaka-Yoshida, 2022; Majid et al., 2023).
A wide repertoire of cell populations, including cardiomyocytes, fibroblasts, endothelial cells, smooth muscle cells, and pericytes; different types of immune cells (myeloid and lymphoid); adipocytes; mesothelial cells; and neuronal cells synchronously maintain cardiac function (Meilhac and Buckingham, 2018; Litviňuková et al., 2020; Tucker et al., 2020; Marín-Sedeño et al., 2021). Sustenance of cardiac homeostasis depends on the integrity of individual cells and the cellular interactions mediated by juxtacrine, paracrine, and endocrine signals. Any pathogenic stimuli disrupting the cardiac microenvironment, metabolic demand, hemodynamic stability, or these crosstalk networks strain the cardiomyocytes and lead to counter-responses (Pogontke et al., 2019; Marín-Sedeño et al., 2021; Liu et al., 2023) and initiation of an inflammatory cascade (Hara and Tallquist, 2023). Activation of the resident immune cells as well as recruitment of innate and adaptive immune cells attempt to adapt to the insult. However, the activation of pattern recognition receptors (PRRs) by damage-associated molecular patterns (DAMPs) or pathogen-associated molecular patterns (PAMPs) initiates downstream signaling cascades that might upregulate the expression of genes encoding pro-inflammatory cytokines and chemokines (Mann, 2011; Adamo et al., 2020a; Silvis et al., 2020). Persistent low-grade inflammation activates tissue-resident macrophages and mast cells to recruit and activate B and T cells, which trigger plasma protein infiltration. The temporal imbalance between the activity of matrix metalloproteinases (MMPs) and tissue inhibitor of MMPs (TIMPs) with a collateral increase in transforming growth factor-β (TGF-β) signaling and NLR family pyrin domain-containing 3 (NLRP3) inflammasome formation leads to aberrant fibrotic deposition and left ventricular (LV) remodeling (Mezzaroma et al., 2011; Zhang et al., 2011; Adamo et al., 2020a). Changes in cardiac performances resulting from altered intercellular signaling, loss of cell activity, or cell death can then result in further changes in cell-to-cell communication (Buckingham et al., 2005; Marín-Sedeño et al., 2021) (Figure 1).
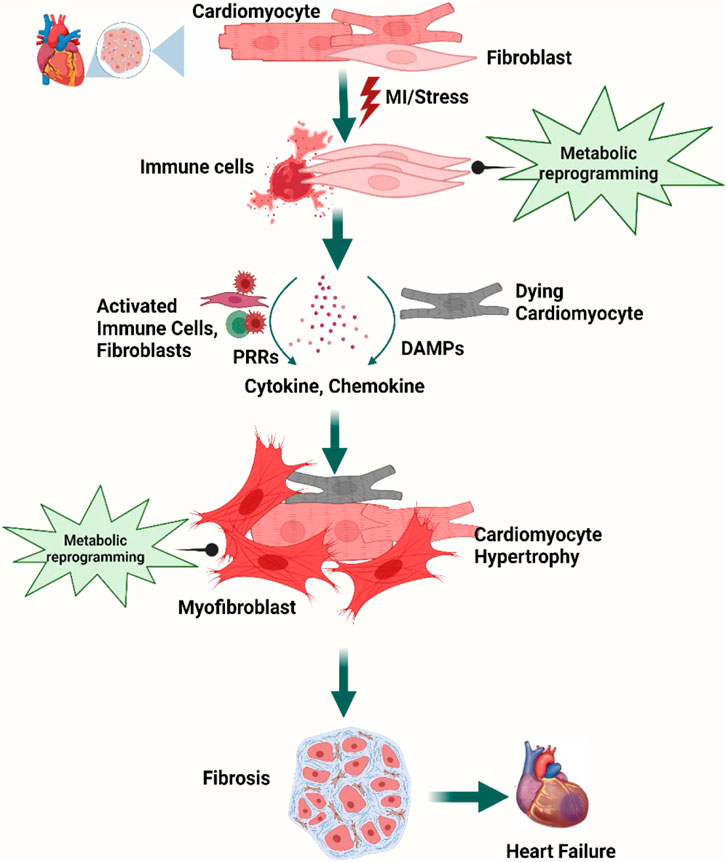
Figure 1. Overview of cellular steps leading to cardiac fibrosis. Following injury, cardiac fibroblasts are activated and transdifferentiated into myofibroblasts to replace dying cardiomyocytes. Dying myocytes release damage-associated molecular patterns (DAMPs) which activate immune cells and trigger downstream signaling cascades of pro-inflammatory cytokines and chemokines. A plethora of cytokines and chemokines in addition to myofibroblasts undergo metabolic alterations that drive the development and progression of myocardial fibrosis and heart failure. Figure created using BioRender.
Along with immune activation, metabolic reprogramming, supply–demand mismatch, and disruption of the equilibrium in local metabolites in the affected cardiac tissue contribute to progression of cardiac fibrosis. Metabolic reprogramming allows the cells to adopt dynamic changes in cellular metabolic pathways and biological functions in response to various stimuli or environmental conditions, for example, cellular metabolism switches from oxidative metabolism (i.e., oxidative phosphorylation, OXPHOS) to the more oxygen-sparing carbohydrate metabolism (i.e., glycolysis) and utilization of glutamine and fatty acid increases to meet the energy and biosynthetic demands during acute and chronic cardiac stress (Sun et al., 2020a; Sabogal-Guáqueta et al., 2023; Ritterhoff and Tian, 2017; Rosano and Vitale, 2018; Yoganathan et al., 2023; Razeghi et al., 2001; Akki et al., 2008; Aubert et al., 2016; Bedi et al., 2016; Tran and Wang, 2019; Lin et al., 2023). With progressive low-grade inflammation in the background of cardiac insult, cytokines and nutrient metabolites activate inflammatory programs through shared pathways, and resident and recruited immune cells undergo metabolic shifts on their own as well during fibrotic events (Sun et al., 2020a; Sabogal-Guáqueta et al., 2023). The energy demand and metabolic intermediates for immune cell activation and differentiation are dependent more on glycolysis than on the tricarboxylic acid (TCA) cycle and OXPHOS (Srivastava et al., 2018; Cho et al., 2020; Farah et al., 2021; Wenzl et al., 2021). Moreover, the fibroblasts adopt a metabolic phenotype using glycolysis and glutamine-derived α-ketoglutarate to adapt to the altered microenvironment (Kawaguchi et al., 2011; Mouton and Hall, 2020; Farah et al., 2021; Francisco et al., 2021).
Immunometabolism is still a burgeoning field, and much of the existing knowledge on cardiac immune cell metabolism is based on myocardial infarction (MI) models (Mouton and Hall, 2020). In the past few years, technological advances and highly sensitive metabolomics approaches have redefined the inextricable relationships between immune activation, molecular signaling, and metabolism and discovered their association to immune cell functions in the course of the disease. However, there are knowledge gaps in the complete sketch of immunometabolism from the perspective of cardiac fibrosis that require further exploration to use immune modulation strategies to prevent cardiac remodeling.
In this review, we provide an update on the current understanding of the involvement of immune and metabolic systems in cardiac fibrosis and the potential of immune–metabolic reprogramming in the management of cardiac fibrosis and restoration of cardiac function.
2 Contribution of the immune system to cardiac homeostasis
A large number of innate and adaptive immune cells are found in the heart. Following infiltration of the cardiac tissue at gestation, the immune cells persist in the myocardium and engage in essential housekeeping functions; defend against pathogens, toxic insults, hypoxia, or other injury; and maintain normal cardiac function throughout life. Though the resident and recruited immune cell populations change in different stages of life as well as over the course of injurious stimuli, the major subtypes of inflammatory cells include leukocytes, mononuclear phagocytes, neutrophils, B cells, and T cells. The complex interactions and crosstalk between different subsets of immune cells that either reside or infiltrate the cardiac tissue and the resident cardiac and non-cardiac cells comprising cardiomyocytes, fibroblasts, and endothelial cells maintain the physiological microenvironment in the heart (Pinto et al., 2012; Ramos et al., 2017; Swirski and Nahrendorf, 2018; Marelli-Berg and Aksentijevic, 2019; Steffens et al., 2022).
Architecturally, the heart is heterogeneous, as is the distribution of immune cells: various macrophage subsets are non-uniformly distributed in distinct niches of the heart. Dendritic cells are found abundantly in the cardiac valves and aortic sinus (Choi et al., 2009), whereas the atrioventricular node contains a high concentration of tissue-resident macrophages (Hulsmans et al., 2017). On the other hand, the coronary vasculature is rich in CCR2- (CC chemokine receptor 2) macrophages and fetal monocyte-derived macrophages are concentrated adjacent to the endocardial trabeculae (Leid et al., 2016). These findings suggest that biochemical, neurohormonal, nutritional, or metabolic alterations of different niches of the heart involve different populations of inflammatory cells as principal responders and lead to different pathophysiological courses. Moreover, the organized chambers, vessels, and myocardium are immersed in the serosal fluid within the pericardium, which contains leukocytes, macrophages, and B cells and provides tissue-infiltrating leukocytes during challenges (Butts et al., 2017). The pericardial adipose tissue supplies lymphocytes and coordinates granulopoiesis and the activation of immune cells (Horckmans et al., 2018), whereas white adipose tissue synchronizes the supply and mast cell accumulation in the heart following MI (Ngkelo et al., 2016). We lack a complete picture of the spatiotemporal distribution of different immune cell populations. A summary of the immune cells along with their distribution and role in the heart is enlisted in Table 1.
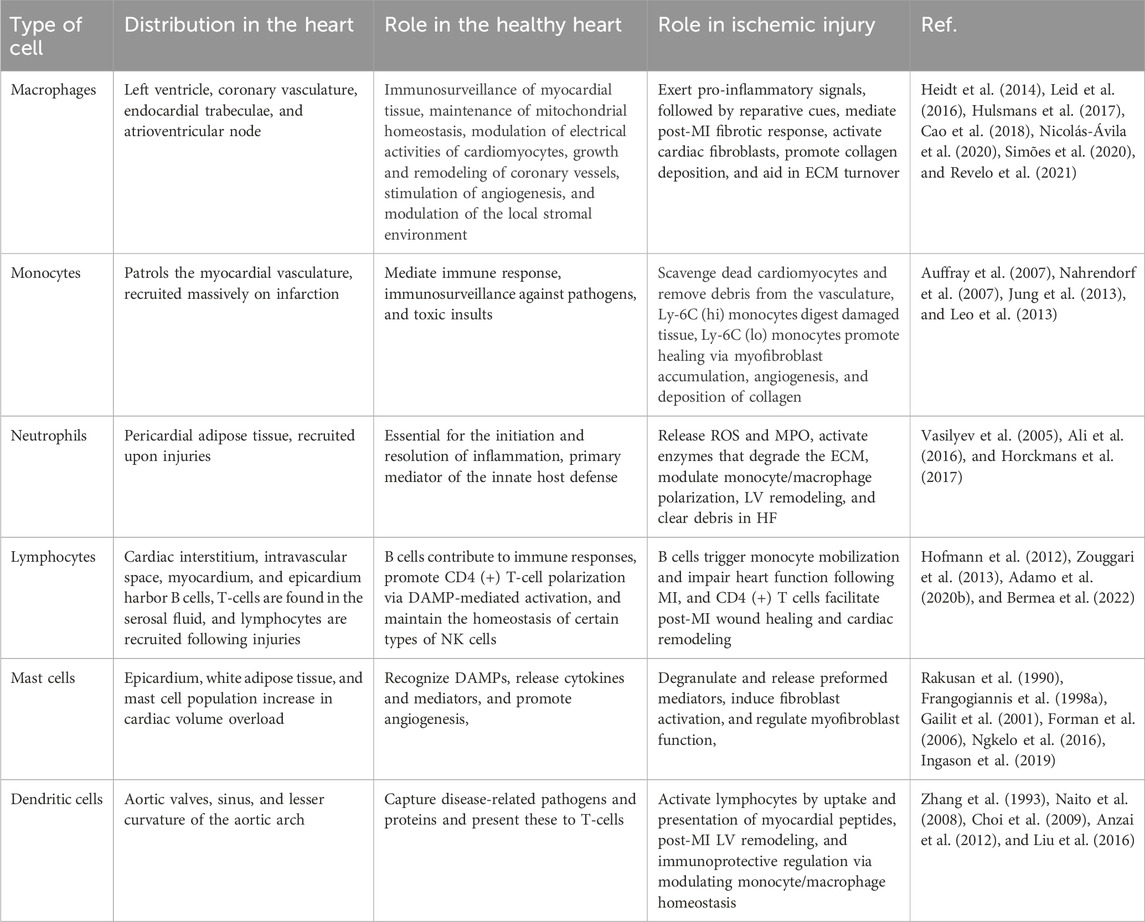
Table 1. Role of different immune cells in healthy and ischemic heart.
Specific localization of different immune cells in the heart suggests that they have specific interactions with resident cardiac cells. Cardiomyocytes, fibroblasts, and endothelial cells not only express receptors that recognize inflammatory mediators from inflammatory cells but also produce growth factors, cytokines, and chemokines to which leukocytes respond. Mast cell-derived tumor necrosis factor (TNF) activates endothelial cells (Frangogiannis et al., 1998a), IL-6 from cardiomyocytes activates neutrophils via intercellular adhesion molecule 1 (ICAM1) expression (Gwechenberger et al., 1999), IL-17 from T cells stimulates cardiac fibroblasts (Wu et al., 2014), and fibroblast-derived granulocyte–macrophage colony-stimulating factor (GM-CSF) induces the production and recruitment of myeloid cells (Anzai et al., 2017). On the other hand, macrophage-derived TGFβ, vascular endothelial growth factor (VEGF), and IL-10 promote collagen production, neo-angiogenesis, and resolution of inflammation (Nahrendorf et al., 2007; Hilgendorf et al., 2014) (Figure 2).
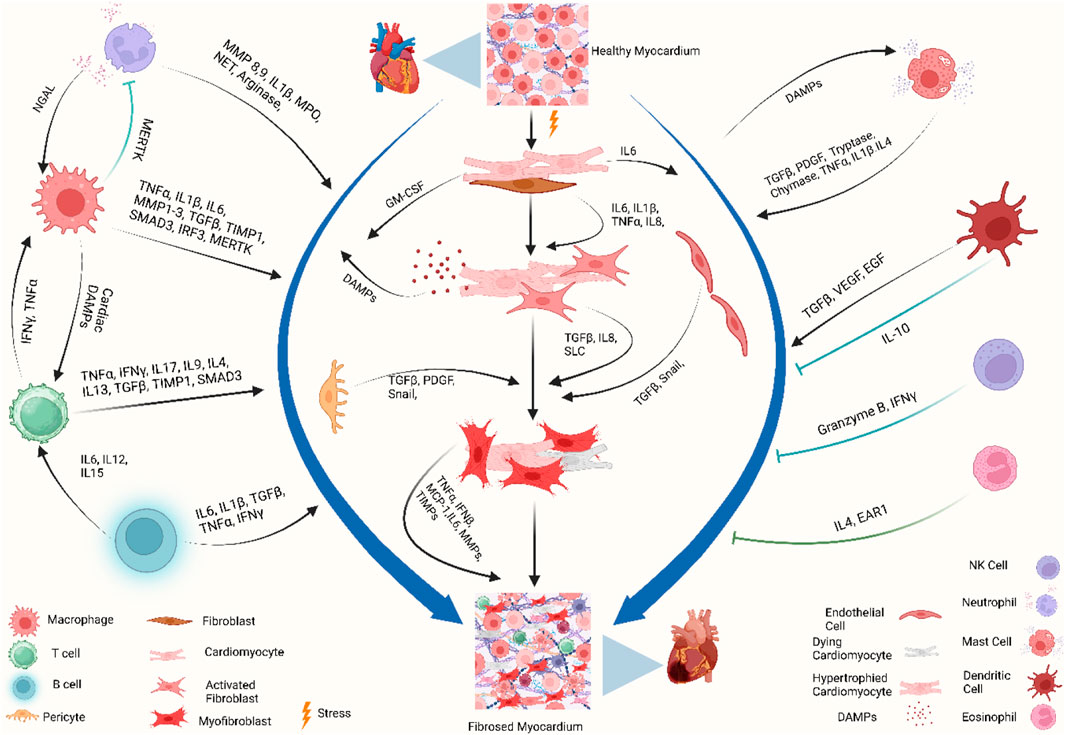
Figure 2. Immune crosstalk between inflammatory cells and cardiac cells during the transformation of the cardiac microenvironment toward fibrosis. The injured myocardium alters the cardiac microenvironment and releases different growth factors and inflammatory mediators, i.e., IL-6, IL-1β, GM-CSF, TNF-α, PDGF, and DAMPs, from injured cardiomyocytes, cardiac fibroblasts, endothelial cells, and pericytes (Gwechenberger et al., 1999; MeléNdez et al., 2010a; Anzai et al., 2017). Upon activation, cardiac fibroblasts further promote the cascades and release MMPs, TIMPs, MCP-1, and TNF-α, which play a role in ECM remodeling and fibrogenesis (Yoshimura et al., 2014; Pluijmert et al., 2021). During these events, the recruited and activated immune cells release another wave of cytokines and chemokines which further promote cardiac remodeling. Recruited monocytes and macrophages trigger inflammatory responses by releasing DAMPs, detect extracellular DNA from dying cells, and release type I interferons (King et al., 2017). Resident macrophages, IL-10 signaling, and MERTK modulate inflammatory cells, promote reparative events, inhibit hyperactivation of fibroblasts, and mitigate fibrosis (DeBerge et al., 2017; Jung et al., 2017; Revelo et al., 2021). Simultaneously, neutrophils, lymphocytes, and mast cells crosstalk with macrophages and fibroblasts to stage the background of ECM turnover and deposition of collagen by releasing inflammatory cytokines. Dendritic cells and NK cells contribute to improving fibrosis by modulating adverse inflammatory events (Ong et al., 2015; Choo et al., 2017). When these profibrotic interplay and crosstalk between different resident and non-resident immune and non-immune cells in the stressed heart outweigh the reparatory mechanisms, the consequential events ultimately lead to cardiac fibrosis. Abbreviations: MERTK, proto-oncogene tyrosine-protein kinase MER; NGAL, neutrophil gelatinase-associated lipocalin; IFNγ, interferon gamma; TNF-α, tumor necrosis factor-alpha; IL1β, interleukin-1 beta; IL6, interleukin 6; IL15, interleukin 15; IL12, interleukin 12; IL8, interleukin 8; IL17, interleukin 17; IL9, interleukin 9; IL4, interleukin 4; IL13, interleukin 13; MMP, matrix metalloproteinase; TIMP, tissue inhibitors of metalloproteinases; SMAD, suppressor of mothers against decapentaplegic; IRF3, interferon regulatory factor 3; MPO, myeloperoxidase; NET, neutrophil extracellular traps; GM-CSF, granulocyte–macrophage colony-stimulating factor; DAMP, damage-associated molecular pattern; Snail, zinc finger protein SNAIL1; SLC, solute carrier; MCP-1, monocyte chemoattractant protein-1; VEGF, vascular endothelial growth factor; EGF, epidermal growth factor; EAR1, eosinophil-associated ribonuclease 1. Figure created using BioRender.
Newer subsets of immune cells and novel roles of different inflammatory cells are being revealed with ongoing research. Leukocytes and their products are gaining focus in the context of normal physiological as well as pathological fibrotic events. A brief discussion of the major contributors to cardiac fibrosis is provided in the following section.
2.1 Monocytes and macrophages in cardiac fibrosis
Cardiac macrophages are part of a steady-state cell network that contributes not only to forming a repertoire of immune cells but also to maintaining the mechanically strenuous and energy-intense pumping function of the heart. They electrically couple to cardiomyocytes through connexin 43 (CX43, also known as GJA1)-containing gap junctions in normal mouse and human hearts (Fernández-Ruiz, 2017). Macrophage ablation results in cardiac pathologies such as progressive atrioventricular block and conduction abnormalities in the atria and ventricles (Hulsmans et al., 2017). Moreover, macrophages help in cell and matrix turnover and regulate angiogenesis and matrix deposition and removal in cardiac pathologies (Guo et al., 2018; Simões et al., 2020; Li et al., 2023a). Hence, spatiotemporal alteration and phenoconversion of macrophage populations in cardiac fibrosis not only remodel the structure but also cardiac rhythmicity (Figure 2).
2.1.1 Subsets of cardiac macrophages
Macrophages and monocytes regulate fibrotic responses across many tissues (Wynn and Ramalingam, 2012). The myocardium of adult mammals typically has a restricted number of resident macrophages (Gersch et al., 2002; Heidt et al., 2014; Mylonas et al., 2015) which play a role in maintaining cardiac homeostasis (Hulsmans et al., 2017). Moreover, the development of sophisticated tools, such as single-cell RNA sequencing, has found evidence of heterogeneous and ontogenetically diverse macrophages in the heart. At least seven subsets of cardiac and pericardial macrophage populations have been identified in the infarcted heart with unique spatiotemporal dynamics and morphologic characteristics (Bajpai et al., 2018; Wei et al., 2023). Tissue-resident cardiac macrophages are principally divided based on the expression of CC-chemokine receptor 2 (CCR2). CCR2- macrophages are of embryonic origin, and they seed the cardiac tissue during embryonic and early postnatal development. They support the development of coronary vasculature (Leid et al., 2016), cardiac regeneration, and electrical conduction within the atrioventricular (AV) node (Aurora et al., 2014; Lavine et al., 2014; Hulsmans et al., 2017). On the other hand, adult hematopoietic lineages produce CCR2+ macrophages, which facilitate neutrophil extravasation and monocyte recruitment (Li et al., 2016) and initiation of inflammation within the diseased heart (Epelman et al., 2014b; Heidt et al., 2014; Epelman et al., 2015; Bajpai et al., 2019). Another recent study reported the existence of a GATA6+ population of macrophages with an anti-fibrotic role within the pericardial cavity (Deniset et al., 2019). The dynamics of macrophage lineages change over time depending on the injurious stimuli and try to adopt changes to cope with the altered microenvironment. Thus, different subsets of macrophages contribute to the maintenance as well as pathological events of the heart.
2.1.2 Macrophage plays an integral role in cardiac fibrosis
The macrophage heterogeneity influences the outcome of myocardial injuries in neonatal and adult hearts. The neonatal heart contains only CCR2- cardiac resident macrophages, while the adult heart contains both CCR2- and CCR2+ macrophage populations (Aurora et al., 2014; Lavine et al., 2014). Following an injury, for example, after an MI, the neonatal heart expands CCR2- macrophages, while the adult heart expands CCR2+ macrophages. In the adult heart, monocyte-derived CCR2+ macrophages are recruited by the inflammatory signal transduction pathways, DAMPs, and other chemokine-driven pathways and replace the CCR2- population (Dewald et al., 2005; Frangogiannis et al., 2007). Upon recruitment, the CCR2+ population continues the phagocytic activities and promotes inflammation by activating the inflammatory bodies, by pattern recognition receptor (PRR) signaling, and by producing cytokines (Epelman et al., 2014a; Heidt et al., 2014; Sager et al., 2016; Bajpai et al., 2019; Chen et al., 2023; Isidoro and Deniset, 2023). These macrophages play a role in regulating inflammation, promoting fibrosis, facilitating matrix remodeling, supporting angiogenesis, and contributing to the process of regeneration. They release different cytokines, such as interleukin-1β, tumor necrosis factor-alpha (TNF-α), interleukin-6 (IL-6), interleukin-16 (IL-16), interleukin-18 (IL-18), transforming growth factor-β (TGF-β1), platelet-derived growth factor (PDGF), matrix metalloprotease-9 (MMP-9), and tissue inhibitor of metalloproteinase 1 (TIMP 1), and induce alpha-smooth muscle actin (αSMA), lysyl oxidase (LOX), and collagen type 1 alpha 2 (Col1a2) expression in fibroblasts, thus exhibiting profibrotic activity (Lindsey et al., 2006; MeléNdez et al., 2010a; Tamaki et al., 2013; de Couto et al., 2015; Frangogiannis, 2015; Chen and Frangogiannis, 2017; Honold and Nahrendorf, 2018; Liao et al., 2020; Moskalik et al., 2022). On the other hand, the macrophages with a reparatory phenotype (CCR2-) release IL-10 after the acute phase and attempt to repair cardiac function (Krishnamurthy et al., 2009; Jung et al., 2017; Moskalik et al., 2022). Metabolic reprogramming further polarizes macrophages into pro-inflammatory (M1) or anti-inflammatory (M2) type and, in turn, this phenoconversion orchestrates the background of scarring or healing of the injured myocardium (Kim et al., 2021; Kang et al., 2023) (Figure 2). Moreover, subsets of myeloid cells can engage in the fibrosis process by being converted to fibroblast-like cells or differentiating fibroblasts into myofibroblasts (Meng et al., 2016; Sinha et al., 2018; Haider et al., 2019; Simões and Riley, 2022; Wu et al., 2022).
2.2 Granulocytes in cardiac fibrosis
2.2.1 Direct role of neutrophils in cardiac remodeling
The role of neutrophils in the regulation of fibrosis is context-dependent. Reperfusion of the post-ischemic myocardium promotes neutrophil infiltration, which exacerbates the pro-inflammatory response and contributes to the ischemia–reperfusion (I/R) injury of the ischemic border zone. They produce and release reactive oxygen species (ROS) and myeloperoxidase (MPO), resulting in the generation of cytotoxic aldehydes, oxidative stress, activation of enzymes degrading the ECM and causing cardiomyocyte apoptosis, and maladaptive remodeling (Vinten-Johansen, 2004; Vasilyev et al., 2005; Fröhlich et al., 2013; Koeth et al., 2013; Puhl and Steffens, 2019; Okyere and Tilley, 2020). However, neutrophils exhibit a strong association with acute inflammatory reactions in the myocardium and play a role in tissue repair by influencing the behavior of macrophages (Horckmans et al., 2017). In a mouse model, neutrophils have been shown to direct macrophages toward a reparative phenotype through the secretion of neutrophil gelatinase-associated lipocalin (NGAL) which mediates efficient clearance of debris. Moreover, antibody-mediated depletion of neutrophils increases cardiac fibrosis and worsens cardiac function (Horckmans et al., 2017) (Figure 2). Therefore, in regulation of fibrosis and for favorable cardiac remodeling, a balanced neutrophil response plays a crucial role.
Studies have found a high plasticity potential of neutrophils and their roles in modulating the outcome of inflammatory events. Ma et al. (2016) reported that the temporal polarization of neutrophils toward pro-inflammatory N1 exacerbates left ventricle (LV) remodeling, whereas the anti-inflammatory N2 phenotype attenuates adverse LV remodeling. DAMPs released from necrosed myocytes stimulate N1 polarization by stimulating TLR4, whereas TGF-β1 inhibition attenuates pro-inflammatory neutrophils (Ma et al., 2016).
The presence of neutrophils in the wounded region is transient since they are rapidly eliminated. Replacement of neutrophils with Ly6Clow macrophages is aligned with the transition from the inflammatory phase to the reparatory phase and decreased production of inflammatory cytokines, growth factors, and chemokines (Nahrendorf et al., 2007). Therefore, the involvement of neutrophils in chronic cardiac fibrosis is restricted to the initial phases of fibroblast activation. According to a recent study, a noteworthy mechanism depends on neutrophils and can potentially contribute to age-related cardiac fibrosis. Within the core of an aging organism, the stimulation of ROS in neutrophils can initiate the creation of neutrophil extracellular traps (NETs). This process is facilitated by activation of the peptidyl arginine deiminase 4 enzyme (PAD4). In vivo, tests indicate that the production of NETs through the involvement of PAD4 plays a role in the development of interstitial fibrotic alterations and the onset of left ventricular diastolic dysfunction (Martinod et al., 2017) (Figure 2).
2.2.2 Contribution of basophils to cardiac fibrosis
Because of their rarity, basophils have long been overlooked in immunological research. Basophils circulate in the bloodstream under homeostatic conditions, but they infiltrate tissues during inflammation (Miyake et al., 2022). Despite their low numbers, basophils affect the accumulation of myeloid cells and influence cardiac remodeling. Sicklinger et al. (2021) showed that basophil depletion promoted a shift from Ly6Clow macrophages with reparatory phenotype toward inflammatory Ly6Chi monocytes in the infarcted myocardium. Induction of IL-4 and IL-13 by glycoprotein IPSE/α-1 in basophils improves cardiac functions and post-MI cardiac healing (Sicklinger et al., 2021). The role of basophils in cardiac fibrosis is further corroborated by the finding that low blood basophil counts are associated with increased scar size and poor outcomes in patients with acute MI (Miyake et al., 2022). Moreover, IL-4 released from the infiltrating basophils acts on resident fibroblasts, triggers myofibroblast expansion, and leads to the production of connective tissue elements from myofibroblasts (Schiechl et al., 2016).
2.3 Lymphocytes in cardiac fibrosis
2.3.1 B cells in cardiac fibrosis
The adaptive immune cells, B and T lymphocytes, are found in small numbers in a normal physiological heart but increase following injury (Zouggari et al., 2013; Wang et al., 2019). The B-cell population in the human heart is divided between the intravascular space and the interstitial space (Bermea et al., 2022) and further divided into subgroups. B1 and B2 cells contribute to the innate immune response through secretion of IFN-γ, IL-6, and IL-17; promote CD4+ T-cell polarization; and regulate the mobilization of monocytes through the production of CCL7, whereas Bregs or B10 cells support immunologic tolerance, resolve the acute inflammatory response, and maintain the homeostasis of certain types of natural killer cells through the secretion of IL-10, IL-35, and TGF-β (Zouggari et al., 2013; Chimen et al., 2015; Shen and Fillatreau, 2015) (Figure 2).
Cardiac B cells in neonatal mice promote cardiomyocyte proliferation, angiogenesis, and regeneration of the heart and inhibit inflammatory responses, while adult B cells promote inflammation and impair cardiac function following myocardial injury (Zouggari et al., 2013; Tan et al., 2023). Depletion of neonatal B cells reduces cardiac regeneration and promotes fibrotic scarring in the post-MI heart, whereas B-cell depletion in adult mice inhibits myocardial fibrosis and improves cardiac function (Tan et al., 2023). Moreover, activated B cells contribute to sustained immune system activation and myocardial inflammation, promote the synthesis of myocardial collagen types I and III, and damage the left ventricular ejection fraction (Mo et al., 2021). Studies have found that B cells promote fibrosis through releasing inflammatory cytokines like IL-1β, IL-6, and TNFα, whereas depletion of B cells results in attenuation of collagen deposition following MI, transverse aortic constriction, and nonischemic cardiomyopathy (Yu et al., 2013; Cordero-Reyes et al., 2016; Ma et al., 2018).
2.3.2 Heterogeneity of T-cell populations in cardiac fibrosis
T-cell receptor engagement, antigenic stimuli, tissue microenvironment, and metabolic reprogramming shape the repertoire of T cells into that of T helper cells (Th1, Th2, Th9, Th17, and Th22), cytotoxic T lymphocytes (CTLs), regulatory T (Treg) cells, and natural killer T (NKT) cells (Zhang and Zhang, 2020). Induction of T cells by cardiac DAMPs processed by antigen-presenting cells results in cardiotropism, transformation of cardiac fibroblasts, and maladaptive cardiac remodeling (Bayer et al., 2023) (Figure 2). An increasing body of literature indicates that distinct subpopulations of T lymphocytes play a substantial role in the direct stimulation of fibroblasts and progression of cardiac fibrosis (Nevers et al., 2015; Li et al., 2017; Abdullah and Jin, 2018). One study proposed the presence of a binding interaction between activated Th1 cells and cardiac fibroblasts in the left ventricular pressure overload model. The interaction has the potential to trigger the synthesis of TGF-β by fibroblasts, resulting in the differentiation of fibroblasts into myofibroblasts (Nevers et al., 2017). Furthermore, an increased number of Th2 cells in cardiac tissues are afflicted with fibrosis (Duerrschmid et al., 2015). The behavior under observation can be attributed to the increased regulation of profibrotic cytokines, such as IL-4 and IL-13, which effectively enhance collagen synthesis by fibroblasts. The infiltration of Th17 cells into the fibrous myocardium has been suggested to be involved in the pathogenesis of autoimmune myocarditis, contributing to the progression of a fibrous myocardium (Baldeviano et al., 2010). Moreover, T cells can stimulate fibroblast activation via their fibrogenic activity. The profibrotic impact produced by T lymphocytes suggests their involvement in the survival of cardiac elastocytes, resulting in the replacement of dead cells by fibrous tissues (Kallikourdis et al., 2017).
2.3.3 Emerging insights regarding the role of T cells in cardiac fibrosis
Although the precise influence of different T-cell subpopulations on the development of fibrosis remains unclear, a growing body of evidence indicates that the use of regulatory T cells (Tregs) in cellular therapy holds promise in reduction of the incidence of myocardial infarction and consequently the ensuing fibrotic response (Kvakan et al., 2009; Tang et al., 2012). Tregs could reduce the fibrogenic activity of macrophages and CD8+ T-cell depletion after experimental MI in mouse models, which reduces inflammation and preserves ventricular function (Weirather et al., 2014; Santos-Zas et al., 2021). Furthermore, Saxena et al. (2014) have reported that Tregs could impact the phenotype of fibroblasts. Moreover, they have been observed to secrete signals that inhibit fibrosis (Tang et al., 2012); however, the precise characteristics of these signals have yet to be elucidated. Notably, Tregs can secrete fibrous mediators, such as TGF-β (Taylor et al., 2006; Saxena et al., 2014; Wang et al., 2023). Hence, the regulatory role of these cells in the fibrotic response is likely dependent on the specific circumstances and the balance between fibrotic and anti-fibrotic cellular processes.
2.3.4 Emerging role of NK cells in cardiac fibrosis
Natural killer cells (NK cells) are type-I innate lymphoid cells known for their role in the recognition and elimination of virus-infected and malignant cells and in limiting their spread (Chiossone et al., 2018). NK cells also modulate immune responses by reciprocally interacting with macrophages, dendritic cells, T cells, and endothelial cells and contribute to the regulation of cardiac diseases (Vivier et al., 2008; Ong et al., 2017). Sustained NK cell deficit in patients with coronary artery disease was associated with persistent low-grade inflammation in the heart (Backteman et al., 2013). As an unabated chronic inflammation plays a role in eventual fibrotic remodeling of the cardiac tissue, NK cells might play a potential role in repairing and maintaining tissue homeostasis by modulating the inflammatory cascades (Tosello-Trampont et al., 2017). Activated NK cells aggregate in the heart, release granzyme B and IFN-γ, and exhibit increased expression of activation markers such as CD69, TRAIL (tumor necrosis factor-related apoptosis-inducing ligand treatment), and CD27. Hyperactivation of NK cells suppresses eosinophil activation and causes eosinophil apoptosis in an anti-inflammatory milieu, leading to decreased cardiac fibrosis (Ong et al., 2015). Moreover, NK cells lower the expression of eosinophil-related chemokines, eotaxin 1 (CCL11), eotaxin 2 (CCL24), CXCL9, and CXCL10 by resident cardiac fibroblasts (Ong et al., 2015). In post-myocardial injuries, NK cells lower cardiomyocyte apoptosis, collagen deposition, and consequent fibrosis and promote neovascularization (Ayach et al., 2006; Boukouaci et al., 2014). However, the role of NK cells in MI still lacks full characterization, and more research studies are required to explore the potential of NK cells in limiting the deposition of collagen and development of cardiac fibrosis.
2.4 Mast cells in cardiac fibrosis
2.4.1 Mast cells do more than allergic reaction
The myocardium of adult animals harbors a limited population of mast cells. Notably, the abundance of mast cells in big mammals, such as dogs, surpasses that observed in mice (Frangogiannis et al., 1999). Cardiac mast cells are multifaceted resident immune sentinel cells playing a pivotal role in fibrotic remodeling in response to various myocardial injuries (Legere et al., 2020; Jin et al., 2022). Heart failure has been found to correlate with elevated quantities of mast cells (Shiota et al., 2003; Wei et al., 2003; Somasundaram et al., 2005; Luitel et al., 2017; Guimbal et al., 2021). Mast cells recognize DAMPs through TLR and ST2 (interleukin 1 receptor-like 1 receptor) and release preformed mediators and synthesize and secrete cytokines, chemokines, and lipid mediators (Janicki et al., 2015; Shao et al., 2015; Jin et al., 2022). The precise mechanisms responsible for increasing mast cells in the fibrotic cardiac area have yet to be fully elucidated. The growth factor known as stem cell factor (SCF) is crucial in the recruitment, development, and proliferation of fully developed mast cells. Additionally, it has been suggested that SCF may contribute to the localized increase in mast cells in the heart, thereby influencing cardiac pathology (Frangogiannis et al., 1998a). The origin of mast cells that infiltrate the diseased myocardium could be derived from adipose tissue (Ngkelo et al., 2016). Though the role of mast cells in the angiogenic responses in hypoxic tissues and cardiac fibrosis is reported, it lacks a complete picture. The pathophysiological role of mast cells depends on the tissue microenvironment and can be both pro- or anti-fibrotic in nature (Legere et al., 2019) (Figure 2).
2.4.2 Mast cells contribute to experimental cardiac fibrosis
Experimental data show that mast cell growth significantly impacts cardiac fibrosis progression (Levick et al., 2011; Levick and Widiapradja, 2018; Widiapradja et al., 2019). In a mouse model of left ventricular pressure overload, mice without mast cells showed decreased perivascular fibrosis, which is associated with decreased progression to decompensated heart failure (Hara et al., 2002). After pressure overload, mast cells accumulate in the artery, contributing to ventricular fibrillation through platelet-derived growth factor-A (PDGF-A) expression (Liao et al., 2010). In hypertensive rat models, the stabilization of mast cells reduces fibrotic cardiac remodeling by preventing myocardial infiltration by macrophages (Levick et al., 2009). On the other hand, cardiac fibroblasts showed a profibrotic phenotype in response to mast cell mediators in mice with cardiac-specific overexpression of TNF (tumor necrosis factor) (Zhang et al., 2011). The significant profibrotic effects of mediators derived from mast cells are supported by the findings that left ventricular diastolic dysfunctions are present in many patients with systemic activation disorders of mast cells (Kolck et al., 2007).
2.4.3 Mast cell derivatives activate fibroblasts and promote fibrosis
Mast cells can store a diverse array of preformed fibrogenic mediators within granules alongside their capacity to generate cytokines and growth factors (Grützkau et al., 1998; Patella et al., 1998). These synthetically generated bioactive chemicals exhibit notable effectiveness in stimulating fibroblasts upon exposure to external stimuli. Following an injury, mast cell degranulation can be initiated through a range of mechanisms, such as the activation of the complement system, the formation of ROS, the activation of adenosine receptors, or the stimulation of cytokines (Frangogiannis et al., 1998a; Murray et al., 2004; Meléndez et al., 2010b). The process of degranulation results in the release of substantial quantities of fibrous agents, which have the potential to initiate or intensify the fibrous response, such as TNF-α (Frangogiannis et al., 1998a), TGF-β (Shiota et al., 2003), IL-4 (Kanellakis et al., 2012), and PDGF (Nazari et al., 2016). Nevertheless, it is important to acknowledge that fibrinogen production is not limited solely to mast cells. Various additional cell types, such as macrophages, lymphocytes, vascular cells, and myocytes, collectively contribute to the pathogenesis of cardiac fibrosis, as discussed earlier. The precise involvement of mast cells in this pathway has yet to be determined, although histamines, tryptases, and chymases can substantially influence the fibrotic process due to their distinct localization within mast cells (de Almeida et al., 2002) (Figure 2).
Chymases can generate angiotensin II (Urata et al., 1990), potentially making it a key mast cell-derived mediator in cardiac fibrosis. It has been proposed that over 75% of cardiac-specific angiotensin II in failing hearts may come from the chymase pathway, independent of ACE (angiotensin-converting enzyme) (Urata et al., 1990). This pathway remains unaffected by ACE inhibitors, potentially offering a mechanism for cardiac fibrosis progression despite ACE inhibition. Chymase might also participate in the fibrotic response by activating MMPs (Fang et al., 1997; Stewart et al., 2003). Both rodent and large animal studies on cardiac fibrosis emphasize the significance of mast cell chymases, suggesting potential therapeutic avenues. For instance, chymase inhibition reduced fibrosis and diastolic dysfunction in a dog model of tachycardia-induced heart failure (Matsumoto et al., 2003), decreased cardiac fibrosis and MMP expression in a porcine reperfusion infarction model (Oyamada et al., 2011), and attenuated interstitial fibrosis while preventing diastolic dysfunction in a rat non-reperfused myocardial infarction model (Kanemitsu et al., 2006). Tryptase, the most abundant product of human mast cells, effectively activates fibroblasts by promoting proliferation (Ruoss et al., 1991) and collagen I synthesis (Cairns and Walls, 1997) through the protease-activated receptor (PAR)-2, leading to ERK-MAPK signaling activation (McLarty et al., 2011). Despite in vitro evidence supporting the fibrogenic effects of tryptase and its expansion in fibrotic hearts (Frangogiannis et al., 1998b; Somasundaram et al., 2005), there is a lack of studies investigating the in vivo role of tryptases in fibrotic cardiac remodeling.
Although the available data generally indicate that mediators derived from mast cells play a role in the accumulation of fibrous tissue, certain experimental studies have proposed that mast cells might possess features that counteract fibrosis (Joseph et al., 2005). The specific processes responsible for this protective phenomenon are still not fully understood. A potential association may exist between the indirect effects of mast cells on the viability of cardiomyocytes or the expression pattern of growth factors (Ngkelo et al., 2016). Like macrophages, mast cells can modify the expression profile of growth factors and proteins in reaction to signals from the microenvironment and metabolic demand (Xiong et al., 2022). The data indicate a possible transition from a state that promotes fibrosis to one that inhibits fibrosis.
2.5 Dendritic cells in cardiac fibrosis
Dendritic cells (DCs) are novel players in various fibrotic diseases where they possess a central role as antigen-presenting cells to regulate the immune system and inflammatory response. Studies have reported an immunoprotective role of the infiltrated DCs in experimental post-MI healing (Nagai et al., 2014; Carvalheiro et al., 2020; Sun et al., 2021). A decreased number of DCs, associated with increased infiltration of macrophages, impaired reparative fibrosis, and cardiac rupture following MI, suggests a protective role of DCs in the cardiac healing process (Nagai et al., 2014). The myocardium harbors both conventional dendritic cells (cDC) and plasmacytoid dendritic cells (pDCs), which maintain the infiltration of other leukocytes in the injury site area. Infiltration of activated DCs into the injured myocardium mediates the regulation of monocyte and macrophage homeostasis in the infarct area (Naito et al., 2008). Depletion of bone marrow-derived CD11c+ cells resulted in sustained release of IL-1β, IL-18, TNF-α; a high level of MMP-9 activity; and a decreased level of IL-10 in a mouse model, which resulted in enhanced fibrosis (Anzai et al., 2012) (Figure 2).
Cardiac cDCs recruited by chemokine receptor CCR2 cause upregulation of cardiomyocyte hypertrophy and inflammation by advanced glycation end products (Cao et al., 2015). Santos et al. (2020) showed that tolerogenic DCs attenuated lymphoproliferation and increased the FOXP3+ CD4+ Treg cells in a mouse model. These events led to diminished IFN-γ, IL-12, and Col1a2 and increased IL-10, resulting in improved cardiac remodeling (Santos et al., 2020). These studies support the role of DCs in post-injury cardiac remodeling; however, more studies are needed to provide a clearer mechanistic insight into how DCs influence fibrosis.
3 Metabolic regulation of cardiac homeostasis
3.1 Metabolic flexibility of the homeostatic heart
The normal adult heart derives approximately 70%–90% of ATP from the oxidation of fatty acids (FAs) and the remaining from the oxidation of glucose, lactate, ketone bodies, and certain amino acids. Mitochondrial oxidative phosphorylation generates most of the ATP required, whereas glycolysis and GTP formation in the TCA cycle provide only around 5% (Lopaschuk et al., 2010; Doenst et al., 2013). The coordinated FA oxidation enables the heart to maintain its ability to switch between available substrates. Moreover, FA produces the greatest ATP yield per 2-carbon energy substrates with the highest O2 consumption (Fillmore et al., 2014). On the other hand, glucose works as a flexible substrate as it provides ATP by cytoplasmic glycolysis and mitochondrial oxidation of the pyruvate derived from glucose metabolism. More importantly, glucose supplies ATP most efficiently in the ischemic and stressed myocardium (Tian et al., 2023). Glucose metabolism generates pyruvate, lactate, and acetyl-CoA, which are further used in the pentose phosphate pathway or replenish TCA cycle intermediates in the mitochondria. Thus, glucose plays a dynamic role in the overall cardiac metabolic balance.
Ketone bodies and amino acids have a minor contribution to the overall cardiac oxidative metabolism in a normal heart, but prolonged fasting, ketogenic diet, and poorly controlled diabetes increase the ketone body utilization by the heart in vivo, while lactate or ketone supplementation in the perfusate reduces the glucose and FA oxidation in isolated perfused hearts ex vivo (Jeffrey et al., 1995; Stanley et al., 2003; Wentz et al., 2010; Papazafiropoulou et al., 2021). Ketone bodies are readily metabolized by the heart, and β-hydroxybutyrate is predominantly oxidized in the heart. Ketones produce ATP with a median efficiency compared to FAs and glucose and hence are a major fuel for the heart with increased circulating ketone levels (Ho et al., 2021). On the other hand, the carbon skeleton produced on branched-chain amino acid (BCAA) metabolism, including acetyl-CoA, α-ketoglutarate, acetoacetyl CoA, succinyl CoA, pyruvate, fumarate, and oxaloacetate, is used in the processes of the TCA cycle to offset the energy demand (Newgard, 2012). The TCA cycle enzymes produce the reducing equivalents NADH and FADH2 within the mitochondrial matrix (Tymoczko et al., 2002) and act as a pivotal mechanism to interconnect the glycolytic, beta-oxidation, and amino acid oxidation pathways (Brandt U and Heinrich Pc, 2014).
Mitochondria not only generate energy but also contribute to cellular signaling, maintain redox equilibrium, and act as a hub for the interconnected metabolic pathways of different substrates (Schaper et al., 1985). Fatty acyl-coenzyme A (CoA) and pyruvate from FA and glucose metabolism, respectively, feed mitochondria, whereas lactate, ketone bodies, and amino acids get oxidized directly in the mitochondria. All the energy-yielding substrates converge on acetyl-CoA production via specific catabolic pathways, which ultimately enter the TCA cycle and accomplish the energy transfer through oxidative phosphorylation (Kolwicz et al., 2013; Passarella and Schurr, 2018). In short, the healthy adult heart is metabolically flexible, with FAs being the predominant substrate, followed by lactate, ketone bodies, glucose, and BCAAs (Ng et al., 2023). To meet its ATP demand for continual cardiac contraction and pumping activities, the heart can readily shift between different energy substrates.
3.2 Metabolic reprogramming in the stressed heart
During the early events of fibrogenesis, mitochondrial and cellular homeostatic signaling and metabolic balance experience both qualitative and quantitative derangements. A shift in substrate preference away from FAs toward more anaerobic substrates leads the energy-compromised organ to suffer from a progressive burnout, which causes further functional deterioration (He et al., 2022). Perfused hypertrophic rat hearts showed a decrease in FA oxidation and increased glucose utilization consistently when subjected to regional myocardial infarction (Remondino et al., 2000), pressure overload (Allard et al., 1994; Christe and Rodgers, 1994), or volume overload (el Alaoui-Talibi et al., 1992; El Alaoui-Talibi et al., 1997) ex vivo. During stressful conditions, substrate versatility and complex regulatory mechanisms contribute to metabolic flexibility by transcriptional regulation and post-translational modification of crucial proteins in different metabolic pathways.
Limited oxygen supply during ischemia suppresses aerobic glucose and FA oxidation. The activation of the oxygen-sensing pathway and the HIF-1α leads to the transcriptional upregulation of glycolytic enzymes (Del Rey et al., 2017; Taylor and Scholz, 2022; Woods et al., 2022). Along with the shift of cardiac substrate metabolism from FA oxidation to glycolysis, GLUT4 gets translocated toward the sarcolemma, FA transporter FAT/CD36 moves away from the sarcolemma, and the glycogen content decreases (Heather et al., 2013). On the other hand, import of glucose into the cells by GLUT-4 depends on insulin, while glycogenolysis, triggered by an increase in AMP and inorganic phosphate and a decrease in ATP levels, generates glucose as an alternative source. Though glucose is not the major metabolic substrate for cardiac tissue, its utilization increases during ischemia, increased workload, and pressure overload hypertrophy (Stanley et al., 1992; Leong et al., 2002; Nascimben et al., 2004; He et al., 2012; Bertrand et al., 2020). This evidence supports metabolic reprogramming in cardiac cells during altered cardiac states as well as a shift in the metabolic axis, which is dependent not only on the energy demand but also on endocrine and neurohormonal homeostasis. Moreover, the heart relies more on glucose as its primary energy source in the context of disorders like diabetes and metabolic syndrome. This metabolic alteration has been found to have consequential effects on fibrosis-related cellular processes (Doenst et al., 2013; Hartupee and Mann, 2017).
Upregulation of ketone body utilization is another feature of the ischemic and hypertrophied failing heart to cope with the injurious event. 3-Hydroxybutyrate (3-OHB) enhances the bioenergetic thermodynamics of isolated mitochondria in the context of low FA levels. Moreover, a mouse model lacking 3-OHB oxidation is less adaptive to ischemic insult and pressure overload and culminated in worsened heart failure and remodeling (Aubert et al., 2016; Bedi et al., 2016; Horton et al., 2019; Manolis et al., 2023) However, more research is needed on cardiac ketone body metabolism in the context of pathological fibrosis and heart failure to elucidate the role of ketone bodies in the altered cardiac microenvironment.
Though amino acids have little contribution as oxidative fuel, myocardial uptake of several amino acids increases as a consequence of metabolic remodeling in pathological conditions. Amino acids are used in oxidative stress due to their potential non-oxidative metabolism and low contribution to cellular acidification. Glutamate and glutamine have been found to prolong cellular function when converted to α-ketoglutarate, while asparagine and aspartate remove amine groups and excess TCA cycle intermediates (Wischmeyer et al., 2003; Liu et al., 2007; Alkan and Bogner-Strauss, 2019). Moreover, glutamine and glutamate are used in ischemic and hypertrophied hearts to produce ATP directly through substrate-level phosphorylation and safeguard the cardiac tissue from being damaged by free radicals and low pH (Drake et al., 2012). Though the heart shows metabolic flexibility in terms of substrate preference governed by plasma substrate and hormone levels in normal physiological conditions, this phenomenon is significantly affected during pathology and failure (Jiang et al., 2021; Shi and Qiu, 2022). All these events are brought about as a result of the injury, metabolic adaptation to the altered microenvironment, and energy supply–demand mismatch and lead to changes in the ECM and architectural organization of the cardiac tissue and culminate in fibrosis.
3.3 How metabolism dyshomeostasis contributes to cardiac fibrosis?
3.3.1 Direct effect of metabolism on the heart: cardiac fibroblasts and cardiomyocytes
Highly regulated and interconnected networks of metabolic pathways not only provide the energy currency for the functional integrity of the heart but also maintain the structural and spatiotemporal homeostasis of the cardiac tissue. Different metabolic pathways perform predominant roles and orchestrate the background of metabolic reprogramming while adapting to different stages of physiological development or pathological conditions. Cardiac fibrosis is the endpoint of multifarious cardiovascular pathologies, such as ischemic and nonischemic heart failure, pressure and volume overloads, genetic cardiomyopathies, diabetes, and aging (Gibb et al., 2020).
The myocardium contains a complex and intricate consortium of cardiomyocytes, endothelium, fibroblasts, pericytes, and immune cells. Upon injury, these cells acquire a fibrogenic phenotype by upregulating the expression of fibrosis-related genes and exhibit matrix synthetic and remodeling profiles. Moreover, DAMPs from dead cardiomyocytes activate inflammation, and collectively with the inflammatory cytokines, TGF-β, and other mediators, these events contribute to cardiac fibrosis (Zeisberg et al., 2007; Zhang et al., 2015; Alex et al., 2023). However, correlative expression studies of ECM proteins attribute the cardiac fibroblasts as the primary cell type responsible for myocardial fibrogenesis. Cardiac fibroblasts comprise around 10%–20% of the total cell population in the heart (Pinto et al., 2016). The nature and activities of fibroblasts, their response to the altered microenvironment, and metabolic reprogramming define the course of cardiac remodeling following any insult (Ali et al., 2014; Kanisicak et al., 2016; Kaur et al., 2016).
Though quiescent cardiac fibroblasts derive energy from mitochondrial oxidative phosphorylation, activation of fibroblasts and their differentiation into myofibroblasts display a stark increase in aerobic glycolysis and lactate production (Gibb et al., 2020). Moreover, the early stages of cardiac fibroblast activation align with altered mitochondrial morphology (Xin et al., 2019), produce mitochondrial reactive oxygen species (mtROS) (Jain et al., 2013), and show features of mitochondrial Ca2+ uptake (Lombardi et al., 2019). Cardiac fibroblasts display remarkable plasticity in response to injurious stimuli, change their own behaviors, and adapt quickly to the altered environment by transitioning between differentiation states (Fu et al., 2020). Metabolic patterns are remodeled between the initiation of cardiac fibroblast activation and their full differentiation into myofibroblasts. Quiescent cardiac fibroblasts require ATP to acquire contractile phenotypes, while fully activated cardiac fibroblasts use amino acid synthesis for collagen production. Moreover, lactate, succinate, and other amino acids serve as stimulators of myofibroblast differentiation (Tian and Ren, 2023).
Altered glycolysis along with increased glycolytic enzymes, such as hexokinase, phosphofructokinase-1 (PFK1), pyruvate kinase, and lactate dehydrogenase (LDH), have been reported in activated cardiac fibroblasts and fibrotic diseases of different organs (Kottmann et al., 2012; Xie et al., 2015; Ding et al., 2017; Selvarajah et al., 2019). Following MI or angiotensin II administration, cardiac fibroblasts showed enhanced glycolysis and glutaminolysis, promoted the myofibroblast phenotype, and exacerbated myocardial injury (Lombardi et al., 2019). On the other hand, inhibition of glucose oxidation during enhanced glucose breakdown enables cardiac fibroblast activation and leads to the accumulation of lactate in myofibroblasts, which ultimately promotes histone lactylation following MI to express genes with a reparatory phenotype (Wang et al., 2022). Furthermore, increased levels of lactate enhance cardiac fibrosis and worsen cardiac dysfunction by promoting endothelial-to-mesenchymal transition via the transcription factor Snail1 lactylation after MI (Fan et al., 2023). Myofibroblasts undergo a shift away from FA oxidation toward glutamine utilization and α-ketoglutarate (α-KG) production. Enhanced glutaminolysis augments α-KG biosynthesis and cardiac fibroblast activation and contributes to de novo collagen synthesis from differentiated myofibroblasts (Gibb et al., 2022a).
3.3.2 Direct effect of metabolism on nonmyocytes: immune cells
Although the cardiomyocyte is the heart’s parenchymal cell, the healthy heart also contains large amounts of nonmyocyte cells that help the organ contract (Pinto et al., 2016). Through paracrine factor secretion, modifications to the ECM, gap junction coupling, and nitric oxide (NO) generation (Travers et al., 2016; He et al., 2017; DeBerge et al., 2019; Humeres and Frangogiannis, 2019; Mouton and Hall, 2020), nonmyocytes directly contribute to cardiac contractility. According to Pinto et al. (2016), a significant proportion of nonmyocytes in the heart, approximately 10%, consist of cardiac immune cells, with the majority being macrophages. These resident immune cells play important roles in the maintenance of normal cardiac homeostasis, and their metabolic state, immune phenotype, and overall disease progression are inextricably interwoven with the cardiac microenvironment. Immune cells, immune effectors, and their interactions with the parenchyma and stromal components are critical for tissue homeostasis and response to acute and chronic stressful conditions. The expression of multiple pro and anti-inflammatory cytokines and the presence of activated macrophages, immunomodulatory regulatory T-cells (Tregs), and other cell types in the cardiac tissue may have adaptive roles in tissue health and maintenance (Zaidi et al., 2021). In the following section, the role of metabolic reprogramming and phenoconversion of immune cells in the context of cardiac fibrosis is discussed.
3.3.3 Metabolism of cardiac immune cells during cardiac fibrogenesis
Cardiac metabolism gets reprogrammed in pathology, evidenced by an augmented dependence on glucose metabolism and a reduced level of FA oxidation (Young et al., 2001; Akki et al., 2008; Kolwicz et al., 2013), and so do the immune cells in the heart. While myocytes exhibit metabolic adaptations in response to changes in ATP demand and substrate availability, immune cells primarily undergo metabolic reprogramming to facilitate phenotypic transition between distinct subsets (Zhang et al., 2019a). M1 macrophages and neutrophils depend on glycolytic metabolism to drive pro-inflammatory processes, including increased pentose phosphate pathway (PPP) activity to produce NADPH to synthesize pro-inflammatory lipid mediators (Zhang et al., 2019a) (Figure 3). The diminished activity and truncation of the TCA cycle with the succinate dehydrogenase enzyme leads to increased succinate dehydrogenase levels. This increase in succinate dehydrogenase levels subsequently triggers the activation of HIF-1α, which promotes the expression of genes associated with inflammation and glycolysis in macrophages. Upregulation of HIF-1α and glycolysis in macrophages following MI causes them to initially acquire a pro-inflammatory phenotype, generally referred to as the M1 phenotype, which plays a crucial role in myocardial remodeling (Mouton et al., 2018). In contrast, polarized anti-inflammatory M2-type macrophages, expressing high levels of genes with a reparatory phenotype (Yan et al., 2013), rely on oxidative phosphorylation, a complete TCA cycle, and the inhibition of glycolysis and the PPP (Zhang et al., 2019a). T lymphocytes inside the injured tissue undergo a metabolic reprogramming characterized by an increase in glycolytic metabolism and the presence of pro-inflammatory T-helper 17 (Th-17) cells (Wu et al., 2019). The scope of promoting M2 macrophage and anti-inflammatory regulatory T-cell phenotype by metabolic intervention might nurture the promise of metabolic modulation in the progression of cardiac fibrosis (Zhang et al., 2019b; Zhao et al., 2020; Pérez and Rius-Pérez, 2022; Taylor and Scholz, 2022).
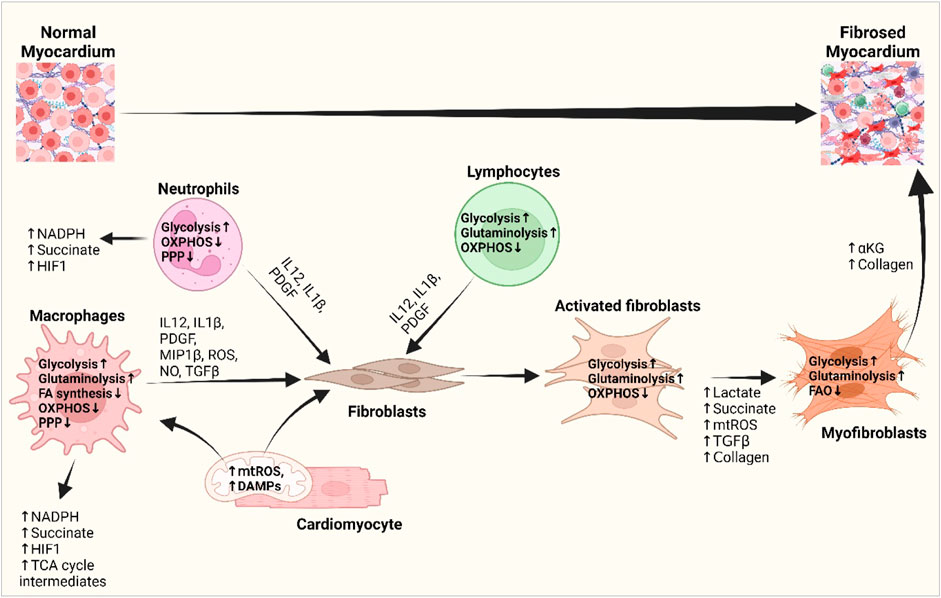
Figure 3. Interplay of inflammatory cells with fibroblasts and cardiomyocytes and their metabolic reprogramming in cardiac fibrogenesis. DAMPs from the damaged cardiomyocytes and mitochondrial ROS activate macrophages and fibroblasts and bring about metabolic changes to meet the altered metabolic demand. Increased glycolysis and amino acid utilization and decreased oxidative phosphorylation in macrophages, neutrophils, and lymphocytes promote the release of different growth and inflammatory factors, i.e., IL-1β, IL-12, PDGF, and ROS, which subsequently activate fibroblasts (Wang et al., 2008; Yoshikawa et al., 2022). Metabolic shifts promote the production of lactate, succinate, HIF1-α, and TCA cycle intermediates and stimulate myofibroblast differentiation. Myofibroblasts shift from fatty acid oxidation to glutaminolysis and promote αKG and collagen biosynthesis and fibrotic deposition in the heart (Gibb et al., 2022a; Fan et al., 2023). Abbreviations: DAMP, damage-associated molecular pattern; FA synthesis, fatty acid synthesis; HIF1, hypoxia-inducible factor 1; IL-1β, interleukin-1 beta; IL-12, interleukin-4; MIP1β, macrophage inflammatory protein-1 beta; mtROS, mitochondrial reactive oxygen species; NADPH, nicotinamide adenine dinucleotide phosphate; NO, nitric oxide; OXPHOS, oxidative phosphorylation; PDGF, platelet-derived growth factor; PPP, pentose phosphate pathway; TCA cycle, tricarboxylic acid cycle; TGF-β, transforming growth factor-beta. Figure created using BioRender.
3.3.3.1 Reprogramming in glucose metabolism
The mounting of an immune response and functional reprogramming within a cell is associated with innate metabolic changes (O'Neill and Hardie, 2013). One of the most well-recognized changes is the activation of anerobic glycolysis, which is a common feature of inflammatory activation of activated macrophages (Rodríguez-Prados et al., 2010); dendritic cells (DCs) (Krawczyk et al., 2010); natural killer (NK) cells (Donnelly et al., 2014); B cells (Doughty et al., 2006); effector subsets of T cells: Th1, Th2, and Th17 (Michalek et al., 2011; Shi et al., 2011); and memory T cells (Gubser et al., 2013). Upregulation of glycolysis forces them toward the pro-inflammatory phenotype (Andrew et al., 2014; Freemerman et al., 2014), while inhibition of glycolysis inhibits immune cell activation and drives them to polarize toward macrophages with a reparatory phenotype and T cells (Soto-Heredero et al., 2020).
The glycolytic phenotype shows increased expression of glycolytic enzymes and offers the immune cell a survival advantage in hypoxic environments as well as provides the Kreb’s cycle intermediates to produce inflammatory cytokines (Abhishek et al., 2015). For example, the glycolytic enzyme hexokinase 1 has been shown to directly interact with and activate the NLRP3 inflammasome (Finucane et al., 2019; Baik et al., 2023). Activation of NLRP3 is strongly linked with fibrosis, which drives the differentiation of fibroblasts into myofibroblasts by chronic upregulation of IL-1β and IL-18 and subsequent autocrine signaling that maintains an activated inflammasome (Artlett, 2022). Moreover, PDGF-BB, IL-12, IL-1β, and MIP-1β (macrophage inflammatory protein-1 beta) from immune cells promote the glycolytic process in fibroblasts which provide more ATP and biosynthetic intermediates for excessive production of the ECM, while inhibition of glycolysis attenuates fibroblast activation and cardiac fibrosis (Chen et al., 2021a; Feng et al., 2023). Furthermore, enhanced glycolysis in immune cells can lead to increased production of ROS, contributing to oxidative stress and tissue injury, both of which are implicated in cardiac fibrosis (Harvey et al., 2014; Dhalla et al., 2022). Hence, increased glycolysis is considered a hallmark of metabolic reprogramming in most immune cells undergoing rapid activation in response to stimulation of pattern recognition receptors, cytokine receptors, or antigen receptors which amplify the production of inflammatory cytokines (Figure 3). Thus, the complex interplay between the immune system and fibrotic processes in the heart could be used to modulate the fibrogenic cascades in the heart.
3.3.3.2 Reprogramming in the TCA cycle
The TCA cycle and oxidative phosphorylation are intact in M2 macrophages and most T-cell subsets, whereas in effector T cells, there is a shift away from the TCA cycle, and in M1 macrophages, the TCA cycle breaks down at two sites: after citrate and after succinate (Abhishek et al., 2015; Liu et al., 2021). As the TCA cycle disrupts in activated macrophages, the mitochondrial accumulation of citrate and succinate feeds into oxidative pathways to generate key effector molecules and metabolites in macrophages. Production of nitric oxide (NO), prostaglandins, IL-1β, and stabilization of HIF1α promote immune function (Infantino et al., 2011; Tannahill et al., 2013). The chronicity of ongoing residual inflammation in cardiac tissue and activated immune cells play crucial roles in the fibrotic deposition of the ECM in the heart. The intermediates of the TCA cycle contribute to the modulation of the metabolic phenotype of immune cells, which consequently promotes fibroblast activity and ultimately exerts a fibrogenic effect on the heart. As the TCA cycle occurs in the mitochondrial matrix, the integrity of mitochondria as well as mitochondrial substrates during stressful conditions play roles in immune regulation as well (Martínez-Reyes and Chandel, 2020).
TCA cycle metabolites such as citrate, succinate, fumarate, oxaloacetate, α-KG, and L-malate accumulate in the cells with mitochondrial stress and link cellular metabolism to innate leukocyte responses and fibrosis (Patil et al., 2019; Ryan et al., 2019; Wu et al., 2023). Transport of mitochondrial citrate by the citrate carrier SLC25A1 (solute carrier family 25 member 1) to the cytosol is upregulated in M1 macrophages in an NF-κB- or signal transducer and transcription (STAT)-dependent manner, which promotes NO, ROS, and prostaglandin E2 (PGE2) production, while inhibition of the citrate carrier reduces the inflammation (Infantino et al., 2011; Infantino et al., 2014; Williams and O'Neill, 2018). Increased glycolysis leads to increased production of α-KG from the TCA cycle to support collagen synthesis and might promote fibrosis (Henderson and O'Reilly, 2021) (Figure 3).
A key link between the TCA cycle (mitochondria) and fibrosis is oxidative injury, where increased ROS and mitochondrial DAMPs induce TGF-β expression in macrophages and fibroblasts and consequently induce myofibroblast differentiation, NLRP3 inflammasome activation, alter MMP/TIMP balances, and set off signaling cascades triggering fibrosis (Liu and Gaston Pravia, 2010; Ung et al., 2021). Furthermore, mitochondrial dysfunction combined with energy deficiency-driven activation of CPI (carnitine palmitoyltransferase I) results in a substantially greater content of long-chain (LC) acylcarnitines. At high concentrations, LC acylcarnitines impede oxidative phosphorylation, causing mitochondrial membrane hyperpolarization and increasing the formation of ROS in cardiac mitochondria (Tominaga et al., 2008; Liepinsh et al., 2016). Metabolic reprogramming of the TCA cycle in immune cells might provide mechanistic insights to modulate the fibrogenic events in cardiac remodeling.
3.3.3.3 Reprogramming in lipid metabolism
FA oxidation and FA synthesis have opposing roles in the immune system. Inflammatory signals drive FA synthesis, immune cell proliferation, and inflammatory cytokine production, whereas non-inflammatory and tolerogenic immune cells prefer FA oxidation and the production of suppressive cytokines, leading to inhibition of inflammation. Effector T cells and activated macrophages show enhanced lipid synthesis, whereas M2 macrophages, regulatory T cells, and memory T cells show FA oxidation, which limits their growth and allows them to persist (Posokhova et al., 2008; Ecker et al., 2010; Michalek et al., 2011; Feingold et al., 2012) (Figure 3).
Accumulation of intracellular FAs stimulates IL-1α production in foam cells, leading to increased inflammation. M1 polarization is induced by IFNγ and LPS and acquires an inflammatory phenotype via glycolytic metabolism, whereas M2 polarization is promoted by IL-4, which induces FA oxidation by STAT6 and PPARγ-co-activator 1β (PGC1β). Overexpression of PGC1β attenuates M1 polarization even in the presence of IFNγ and LPS (Vats et al., 2006; Huang et al., 2014; Nomura et al., 2016). Moreover, intracellular accumulation of lipids and lipid intermediates leads to lipotoxicity, culminating in alterations in the structural morphology and impaired cardiac function. Lipotoxicity has the potential to induce apoptosis in cardiomyocytes by the augmentation of ROS production and endoplasmic reticulum stress, ultimately leading to the development of heart failure (Zlobine et al., 2016).
Activation of NF-κB through TLR4 signaling induces SREBP (sterol receptor element-binding protein) expression and promotes lipid synthesis, which induces the cleavage and maturation of pro-IL-1 and pro-IL-18 and promotes the M1 macrophage phenotype (Im et al., 2011). On the other hand, liver X receptor (LXR) upregulation and AMPK-enhanced β-oxidation of lipids induce the anti-inflammatory phenotype of immune cells (Zelcer and Tontonoz, 2006). Moreover, altered lipid availability contributes to mitochondrial dysfunction and changes the macrophage phenotype. Proliferation of T cells depends on glycolysis and β-oxidation, whereas their activation depends on de novo lipogenesis. Lipogenesis also contributes to differentiation of Th17 cells to Tregs. Activated T cells shift their dependence from lipid metabolism to glucose breakdown (Shyer et al., 2020). As phenoconversion of immune cells essentially influences fibroblasts and myofibroblasts in the heart, modulation of FA metabolism by means of IL-10, AMPK, mTOR, or TLR signaling holds promise in intervening in cardiac fibrosis (Sag et al., 2008).
3.3.3.4 Reprogramming in amino acid metabolism
The availability and metabolism of various amino acids play important roles in immune function, of which glutamine, arginine, and tryptophan are the most important. Adequate glutamine is used for the induction of IL-1 and NO production through feeding into arginine synthesis. Inadequate glutamine supply impedes cytotoxic macrophages from producing NO in vitro. Glutaminolysis promotes glycolysis via the α-KG/mTOR/HIF-1α pathway as well as contributes to amino acid synthesis and lipid metabolism and promotes ECM production in fibroblast and myofibroblast persistence (Gibb et al., 2022b).
α-KG produced via glutaminolysis feeds OXPHOS and FA oxidation and promotes M2 polarization of the macrophages through Jmjd3 (Jumonji domain-containing 3)-dependent demethylation of H3K27 and attenuates the M1 phenotype by inhibiting IKK activation through PKH (prolyl hydroxylase domain) (Bai et al., 2019; Jia et al., 2019). Moreover, glutamine supports the M2 phenotype by UDP-GlcNAc (glutamine–UDP-N-acetylglucosamine) synthesis via the hexosamine biosynthetic pathway. On the other hand, succinate synthesized via glutamine-dependent anaplerosis or the γ-aminobutyric acid (GABA) shunt promotes polarization of M1 macrophages (Ren et al., 2019). This evidence suggests that the cytotoxic function of macrophages could be modified by modulating the amino acid metabolism. Both T-cell and B-cell activation as well as the balance between effector T cells and Treg cells depend markedly on glutamine usage. Loss of transporter protein ASCT2 (alanine–serine–cysteine transporter type-2) was found to reduce glutamine level in the cells and cause a defect in effector T-cell function by reducing mTORC1 activity in T cells (Crawford and Cohen, 1985; Carr et al., 2010; Le et al., 2012; Nakaya et al., 2014).
Arginine plays a dual role in immune activation. The flux of arginine into the NO synthesis pathway produces NO by inducible nitric oxide synthase (iNOS) and promotes inflammatory M1 macrophages, whereas the arginase pathway promotes tolerant immune responses and often is associated with wound healing. mTOR signaling regulates numerous events that are crucial for T-cell and monocyte differentiation (Weichhart et al., 2015). mTORC1 activity in T cells is suppressed in arginine-depleted in vitro cultures (Cobbold et al., 2009). Arginase 1 expression in macrophages limits the inflammatory potential of effector Th2 cells and suppresses fibrosis. Moreover, macrophage-specific expression of arginase-1 promotes inflammation and fibrosis by enhancing L-proline, polyamine, and Th2 cytokine production (Pesce et al., 2009). The metabolic fates of the products of arginase and arginine deaminase in the immune cells suggest that arginine metabolism plays a key role in inflammation.
Extracellular amino acids support the energy-intensive T-cell activation process and contribute to immune regulation. Reduced extracellular amino acids, i.e., leucine, during ischemia, impair T-cell mobilization and mTOR-dependent Th1 and Th17 differentiation (Sinclair et al., 2013). Moreover, an excessive amount of BCAA impairs mitochondrial function. This impairment is characterized by the disruption of the mitochondrial membrane potential and the opening of the mitochondrial permeability transition pore. The accumulation of branched-chain keto acids (BCKAs) from the degraded BCAA facilitates ROS generation (Zhao et al., 2023). The metabolites, ROS, and oxidative stress crosstalk with fibroblasts activate profibrotic cascades, alter the turnover of the ECM, and ultimately shift the balance toward fibrosis. Manipulation of the cooperativity among cells for production of substrates for collagen synthesis gives us an insight into treating cardiac fibrosis (Cowling et al., 2019; Molek et al., 2021; Raziyeva et al., 2022).
3.3.3.5 Reprogramming in the pentose phosphate pathway (PPP)
Glycolysis feeds the PPP, which allows the diversion of intermediates from the glycolytic pathway toward the production of nucleotide and amino acid precursors as well as generates reducing equivalents of NADPH, which has an important role in the maintenance of a favorable cellular redox environment. Macrophages and neutrophils use NADPH for rapid ROS production to clear the insulting agent as well as for the induction of antioxidants to prevent excessive tissue damage (O'Neill et al., 2016). The role of the PPP in immune cell activation, ROS production, and cell polarization has been found crucial in the study of sedoheptulose kinase carbohydrate kinase-like protein (CARKL) on macrophages. CARKL limits the flux through the PPP, and its suppression directs macrophages toward the M1 phenotype (Haschemi et al., 2012; Sun et al., 2020b). NADPH generated by the PPP is used in the regeneration of antioxidants such as glutathione, which plays a crucial role in protecting cells from oxidative damage associated with inflammation and fibrosis.
4 The interplay between the immune system and metabolism in cardiac fibrosis
Is cardiac fibrosis an endpoint of the derangement of the normal immune physiology and metabolism of the heart? Various factors can contribute to the development of fibrosis, with one notable factor being the interplay between immune cells and metabolic pathways. However, the relationship between metabolic changes and immune responses during fibroblast-to-myofibroblast transition remains unclear. The crosstalk between immune cells and metabolic pathways is a complex and dynamic interaction that plays a crucial role in various physiological and pathological processes. Immune cell activation and function are intimately linked to metabolic pathways (Chi, 2022), allowing cells to meet the energy demands required for their activities. The metabolic processes initiated by immune cells have the potential to impact the activation of fibroblasts and the subsequent buildup of the ECM through their influence on cytokine production and tissue healing mechanisms, hence facilitating development of fibrosis (Czubryt, 2019; Mouton and Hall, 2020; Eming et al., 2021; DeBerge et al., 2023; Hara and Tallquist, 2023). Macrophages undergo phenotypic shifts from an M1 state to an M2 state. These transitions are accompanied by distinct metabolic profiles, including glycolysis and oxidative phosphorylation, which contribute to the respective activities of the macrophages (Wynn and Ramalingam, 2012) (Figure 3). Similarly, activated T cells undergo a metabolic shift from oxidative phosphorylation to glycolysis, facilitating the prompt energy generation and biosynthesis required for proliferation and effective functioning (Figure 3).
The impact of metabolic pathways, including the TCA cycle and FA oxidation, on the development and function of immune cells and fibroblasts provides newer perspectives on different metabolites in fibrotic diseases (MacIver et al., 2013). An illustration of this phenomenon involves the utilization of metabolites, such as succinate, fumarate, and itaconate, as important signaling molecules that modulate physiology and pathology and regulate intercellular communication within the immune system (MacIver et al., 2013; Tannahill et al., 2013; Mills et al., 2016; Mills et al., 2018; Hooftman et al., 2023). These metabolites can influence various immune cell populations, including regulatory T cells. The influence of metabolic pathways on the differentiation and function of Treg has been reported. Maintenance of Treg suppressive function relies on FA oxidation, underscoring the significance of metabolic signals in regulating the immune system (MacIver et al., 2013). The phenomenon of metabolic reprogramming enables immune cells to adjust their energy and biosynthetic requirements in response to varying activation conditions and leads the cells of the surrounding microenvironment to initiate fibrosis. The role of mTOR in integrating nutritional availability and metabolic conditions to govern the differentiation and responsiveness of T cells suggests a strong interconnection between the activation and functionality of immune cells and several metabolic pathways (van der Windt et al., 2012) (Figure 3).
The interaction between metabolism and the fibrotic response is bidirectional, with metabolism playing a causal role in dictating cellular signaling and the effector functions of fibroblasts and immune cells. For example, TGF-β1, a profibrotic molecule expressed by activated fibroblasts that contributes to ECM remodeling (Kawaguchi et al., 2011; Mouton and Hall, 2020; Farah et al., 2021; Francisco et al., 2021), is a strong activator of glycolysis (Jiang et al., 2015) and immune response (Li et al., 2006). Conversely, glycolysis is involved in the activation of fibroblasts and is a critical regulator of TGF-β1 and collagen synthesis in cardiac fibroblasts (Singh et al., 2008)), suggesting that glycolysis upregulation is important in ECM remodeling, with TGF-β1 playing a potent role as a metabolic regulator of cardiac fibrosis (Figure 4). Similarly, lipid metabolism is also a key player in regulating ECM homeostasis and fibrosis. Peroxisome proliferator-activated receptor (PPAR), a major facilitator of FA oxidation, promotes fibroblast-mediated as well as macrophage-mediated degradation of the ECM, reflecting the potential interconnection between lipid metabolism and ECM remodeling (Figure 4).
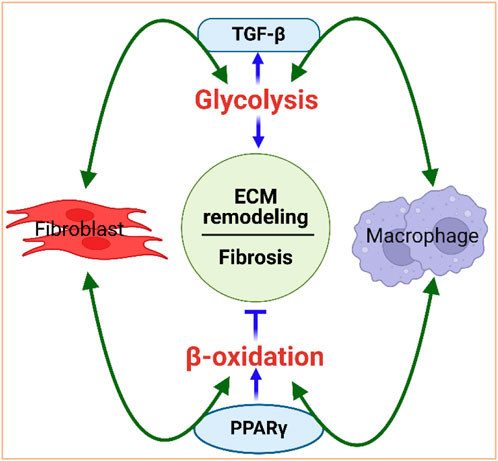
Figure 4. Interdependence of metabolism and ECM remodeling. Fibroblasts and macrophages work in concert to regulate the ECM. They are the primary cell types that mediate collagen internalization and degradation. For instance, fibroblasts and macrophages activate glycolysis, via TGF-β signaling, and promote fibrosis. Glycolysis, in turn, can increase TGF-β, further activating fibroblasts and macrophages. Fibroblasts and macrophages also activate beta-oxidation via PPAR signaling to promote degradation of the ECM. PPARγ can control macrophage polarization to either pro-inflammatory M1 or to anti-inflammatory M2 macrophages. Figure created using BioRender.
5 Targeting metabolism and immune response in cardiac fibrosis
Basic research has detailed the cellular and molecular mechanisms and signaling pathways driving this lesion; however, there is a clear lack of personalized anti-fibrotic strategies permissible for its effective treatment. Recent key findings implicating the innate and adaptive immune response and metabolic changes during the pathological transition of cardiac fibroblasts have tremendous potential and may offer opportunities to facilitate novel therapeutic strategies for the regulation of the treatment of fibrotic remodeling (Figure 5). For instance, given the remarkable success of immunotherapy in cancer treatment, the use of chimeric antigen receptor T cells (CAR T-cells) or modified T-cell receptors would be an ambitious approach. CAR T cells have been successfully used in the treatment of certain leukemias and solid tumors (Petrausch et al., 2012; Jogalekar et al., 2022). Similarly, studies have shown that modified CAR T cells designed against activated fibroblasts improve myocardial fibrosis and cardiac function in mouse models of heart failure (Aghajanian et al., 2019; Rurik et al., 2022).
Figure 5. Targeting metabolism and immunity in cardiac fibrosis. Cellular map showing the sites of possible potential interventions in immune and metabolic pathways to improve the outcome of cardiac fibrosis. Figure created using BioRender.
Exploitation of metabolic pathways as possible targets for anti-fibrotic therapy should be considered, although it might be challenging to really determine the specific role of immune cell metabolism and its effect in cardiac fibrosis due to several factors acting in pathological condition; however, the influence of metabolism on immune cells remains quite unclear due to the spatiotemporal distribution of various immunological cells at various phases of cardiac fibrosis. Certain cell types which prove effective at a certain time might not have similar effects after some time. However, the combination of metabolic approaches could improve the prognosis of heart failure when combined with other treatment regimens such as ACE inhibitors, beta-blockers, and mineralocorticoid blockers (Stanley et al., 2005; McMurray et al., 2012; Yurista et al., 2022). As such, the use of metabolic agents in cardiac fibrosis and heart failure could be beneficial. Some common metabolic agents that could be used target various metabolic processes, including FA/lipid metabolism and glucose/pyruvate metabolism (Rosano et al., 2015). For instance, niacin, also known as nicotinic acid, is involved in the lipid metabolism pathway and could alter the energy metabolism of cardiac fibroblasts by inhibiting β-oxidation and increasing glucose oxidation (Rosano et al., 2015). This increased glycolysis induction serves as a compensatory response to reduce mitochondrial oxidative metabolism and ATP production in heart failure (Lopaschuk et al., 2021; Ng et al., 2023) (Figure 5).
Glucose metabolism tightly regulates extracellular matrix production, and targeting glycolysis has shown exciting results in tackling fibrosis and other diseases. Glycolysis inhibitors have been reported as emerging therapeutic candidates in some cancer treatments (Akins et al., 2018; Chelakkot et al., 2023), while glycolytic reprogramming has been implicated as a regulator of pulmonary fibrosis (Xie et al., 2015; Li et al., 2022). This leads to the question of whether these inhibitors could be beneficial as well in the reversal of myofibroblasts in cardiac fibrosis (Figure 5). Although fibrosis is important for the normal healing process, excessive and continuous deposition of myofibroblasts in the injured myocardium is rather deleterious. It is therefore imperative that both immune responses and metabolic control should be adequately used in the arrest of fibroblast deposition at the adequate point in time during fibrosis. Chen et al. (2021a) reported that 2 deoxy-D-glucose, a glycolysis inhibitor, could alleviate cardiac fibrosis by reducing collagen volume and α smooth muscle actin expression. They also reported the reversal of fibroblast activation in vivo with glycolytic inhibitors. However, specific mechanisms through which these effects were achieved have not been well-documented. Another inhibitor of glycolysis, Kallistatin/Serpina3c, which inhibits the transcription of enolase, a key enzyme in glycolysis, has also been reported to inhibit cardiac fibrosis after MI, hence preventing fibroblast proliferation (Ji et al., 2022). Several attempts have been made to use immunomodulators in the therapy of cardiac fibrosis, yet without much success, such as the use of TNF-α (Baumgarten et al., 2000; Rurik et al., 2021) or TGF-β neutralizing antibodies (Kuwahara et al., 2002) (Figure 5). This has been suggested to be associated with reduced efficacy of TGF-β inhibitors when used as monotherapy (Parichatikanond et al., 2020). However, some other TGF-β inhibitors such as pirfenidone have proven effective in experimental conditions in the treatment of fibrosis, but the mechanisms are not totally understood (Nguyen et al., 2010; Park et al., 2019). It might be beneficial if TGF-β inhibitors with proven effects are used with other metabolic regulators of fibrosis to achieve better treatment outcomes. A combined use of immune modulators or some existing glycolytic inhibitors together with off-market anti-fibrotic medications could proffer better treatment options for cardiac fibrosis.
Pharmacological interventions have been shown to improve cardiac function through inhibition of FA oxidation and improved glucose oxidation (Lopaschuk et al., 1988; Lopaschuk et al., 2010), suggesting that metabolic rewiring may serve as an essential strategy. For instance, a recent study showed that deletion of a key enzyme in the FA oxidation pathway, CPT1b, not only reduced cardiac fibrosis but also stimulated cardiomyocyte proliferation post ischemia–reperfusion injury (Li et al., 2023b). Thus, it is possible, in principle, to pharmacologically block the activity of the CPT1b and open the door for the development of an inhibitor that can be used to effectively control the activity of the enzyme. This finding highlights the potential of metabolic reprogramming of other key enzymes and/or metabolites in the development of an effective anti-fibrotic therapy that may eventually be used in humans. Along these lines, we have recently reported that gene modulation and alteration make a substantial difference in the reaction of cells to the presence of pathological conditions or insults to the cells. We have shown that the deletion of delta-like 1 homolog (Dlk1) accelerates fibroblast–myofibroblast differentiation and that Dlk1-null mice have marked myocardial fibrosis (Rodriguez et al., 2019). It is interesting to know that Dlk1-null mice also exhibit accelerated adiposity (Moon et al., 2002), likely via increased preadipocyte replication/adipogenesis (Mortensen et al., 2012). Increased visceral adiposity contributes to fibroblast activation and cardiac fibrosis (Sawaki et al., 2018).
Further research would be important to understand the role of various gene alterations or manipulations on immune cell metabolism and changes that this metabolism could cause (Hinz et al., 2001; Van Den Borne et al., 2010; Hecker et al., 2011). Although the mechanisms of myofibroblast deactivation/dedifferentiation are being investigated, many details still remain unknown (Fortier et al., 2021). Efforts should be invested in strategies based on the manipulation of immune–metabolic pathways governing the function and phenotyping changes in myofibroblasts. As discussed earlier, macrophages can aid in the process of myocardial healing and fibrosis and may acquire multiple roles at different stages during wound resolution (Haider et al., 2019). In this regard, it is conceivable to assume that macrophages can unexpectedly participate in the modulation of myofibroblast dedifferentiation and myofibroblast removal/apoptosis or can undergo conversion into fibroblast-like cells, boosting interstitial collagen production to optimize healing (Haider et al., 2019). Furthermore, therapies targeting receptor tyrosine kinase, such as anlotinib, have been shown to extract some anti-fibrotic effects in endometrial cancer and non-small cell lung cancer and to inhibit glycolysis in myofibroblasts to reverse pulmonary fibrosis (Chen et al., 2021b). It will be interesting to know if it also confers the same benefits in cardiac fibrosis. Our rapidly expanding knowledge of CRISPR/Cas9 and RNA technologies in targeting specific mediators of myofibroblast dedifferentiation should possibly bring forward novel critical anti-fibrotic approaches (Figure 4).
6 Conclusion
Metabolic alterations in the myocardium associated with maladaptive hypertrophy and myocardial fibrosis induce a prolonged inflammatory response and energetic imbalance, which perturb cellular function and stimulate fibrotic events. An increasing number of studies have found that altered glycolysis, amino acid, and lipid metabolism in cardiac fibroblasts and immune cells induce an inflammatory environment and an extensive interdependent signaling contributes to the deposition of ECM during fibrosis. Simultaneously, these cellular and functional changes alter cellular metabolism, setting up a positive feedback chain of events that drive fibrosis. Hence, therapies targeting metabolic pathways in the immune cells or cardiac fibroblasts represent a very promising area of research for the treatment of cardiac fibrosis. However, the complexity of the immuno-metabolic networks in myocardial fibrosis creates challenges to therapeutic translation. Nevertheless, the similarities in metabolic derangements in glycolysis and β-oxidation across multiple fibrotic tissues should provide opportunities to target key metabolic pathways in cardiac fibrosis. Fibroblasts and macrophages are the primary cell types that mediate collagen internalization and degradation; therefore, reversing metabolic alterations must target both cells to effectively reduce fibrosis. Although the field of immunometabolism is burgeoning every day, there remain knowledge gaps about the contributions of in vivo immunometabolism directly within the myocardium. Moreover, much of our current insights into pathological myocardial remodeling originate from studies of myocardial ischemia, and there is a much larger knowledge gap between immune cell metabolism and nonischemic, cardiometabolic heart disease. While some anti-fibrotic drugs targeting metabolic dysregulation have shown promising results, more rigorous clinical studies are needed to test their therapeutic efficacy and adverse effects. In this respect, drug repurposing strategies in parallel with systematic, large-scale drug screening should be highly considered (Zhao et al., 2019). Moreover, further research is still required to elucidate how the interplay of inflammation and metabolic rewiring promotes fibrosis and what is the difference in the immunometabolism during physiological tissue repair and pathophysiological fibrotic response.
Author contributions
MH: Data curation, Writing–original draft, Writing–review and editing. JG: Writing–original draft. FH: Writing–original draft. AR: Writing–original draft. DL: Conceptualization, Funding acquisition, Investigation, Resources, Supervision, Visualization, Writing–review and editing, Data curation, Project administration.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Abdullah C. S., Jin Z. Q. (2018). Targeted deletion of T-cell S1P receptor 1 ameliorates cardiac fibrosis in streptozotocin-induced diabetic mice. Faseb J. 32 (10), 5426–5435. doi:10.1096/fj.201800231R
Abhishek K. J., Huang S. C. C., Sergushichev A., Lampropoulou V., Ivanova Y., Loginicheva E., et al. (2015). Network integration of parallel metabolic and transcriptional data reveals metabolic modules that regulate macrophage polarization. Immunity 42 (3), 419–430. doi:10.1016/j.immuni.2015.02.005
Adamo L., Rocha-Resende C., Lin C. Y., Evans S., Williams J., Dun H., et al. (2020b). Myocardial B cells are a subset of circulating lymphocytes with delayed transit through the heart. JCI Insight 5 (3), e134700. doi:10.1172/jci.insight.134700
Adamo L., Rocha-Resende C., Prabhu S. D., Mann D. L. (2020a). Reappraising the role of inflammation in heart failure. Nat. Rev. Cardiol. 17 (5), 269–285. doi:10.1038/s41569-019-0315-x
Aghajanian H., Kimura T., Rurik J. G., Hancock A. S., Leibowitz M. S., Li L., et al. (2019). Targeting cardiac fibrosis with engineered T cells. Nature 573 (7774), 430–433. doi:10.1038/s41586-019-1546-z
Akins N. S., Nielson T. C., Le H. V. (2018). Inhibition of glycolysis and glutaminolysis: an emerging drug discovery approach to combat cancer. Curr. Top. Med. Chem. 18 (6), 494–504. doi:10.2174/1568026618666180523111351
Akki A., Smith K., Seymour A.-M. L. (2008). Compensated cardiac hypertrophy is characterised by a decline in palmitate oxidation. Mol. Cell. Biochem. 311 (1-2), 215–224. doi:10.1007/s11010-008-9711-y
Alex L., Tuleta I., Hernandez S. C., Hanna A., Venugopal H., Astorkia M., et al. (2023). Cardiac pericytes acquire a fibrogenic phenotype and contribute to vascular maturation after myocardial infarction. Circulation 148 (11), 882–898. doi:10.1161/CIRCULATIONAHA.123.064155
Ali M., Pulli B., Courties G., Tricot B., Sebas M., Iwamoto Y., et al. (2016). Myeloperoxidase inhibition improves ventricular function and remodeling after experimental myocardial infarction. JACC Basic Transl. Sci. 1 (7), 633–643. doi:10.1016/j.jacbts.2016.09.004
Ali S. R., Ranjbarvaziri S., Talkhabi M., Zhao P., Subat A., Hojjat A., et al. (2014). Developmental heterogeneity of cardiac fibroblasts does not predict pathological proliferation and activation. Circ. Res. 115 (7), 625–635. doi:10.1161/CIRCRESAHA.115.303794
Alkan H. F., Bogner-Strauss J. G. (2019). Maintaining cytosolic aspartate levels is a major function of the TCA cycle in proliferating cells. Mol. Cell Oncol. 6 (5), e1536843. doi:10.1080/23723556.2018.1536843
Allard M. F., Schönekess B. O., Henning S. L., English D. R., Lopaschuk G. D. (1994). Contribution of oxidative metabolism and glycolysis to ATP production in hypertrophied hearts. Am. J. Physiol. 267 (2), H742–H750. doi:10.1152/ajpheart.1994.267.2.H742
Andrew N. M., Gerriets V. A., Nichols A. G., Michalek R. D., Rudolph M. C., Deoliveira D., et al. (2014). The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 20 (1), 61–72. doi:10.1016/j.cmet.2014.05.004
Anzai A., Anzai T., Nagai S., Maekawa Y., Naito K., Kaneko H., et al. (2012). Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling. Circulation 125 (10), 1234–1245. doi:10.1161/CIRCULATIONAHA.111.052126
Anzai A., Choi J. L., He S., Fenn A. M., Nairz M., Rattik S., et al. (2017). The infarcted myocardium solicits GM-CSF for the detrimental oversupply of inflammatory leukocytes. J. Exp. Med. 214 (11), 3293–3310. doi:10.1084/jem.20170689
Artlett C. M. (2022). The mechanism and regulation of the NLRP3 inflammasome during fibrosis. Biomolecules 12 (5), 634. doi:10.3390/biom12050634
Aubert G., Martin O. J., Horton J. L., Lai L., Vega R. B., Leone T. C., et al. (2016). The failing heart relies on ketone bodies as a fuel. Circulation 133 (8), 698–705. doi:10.1161/CIRCULATIONAHA.115.017355
Auffray C., Fogg D., Garfa M., Elain G., Join-Lambert O., Kayal S., et al. (2007). Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 317 (5838), 666–670. doi:10.1126/science.1142883
Aurora A. B., Porrello E. R., Tan W., Mahmoud A. I., Hill J. A., Bassel-Duby R., et al. (2014). Macrophages are required for neonatal heart regeneration. J. Clin. Investigation 124 (3), 1382–1392. doi:10.1172/JCI72181
Ayach B. B., Yoshimitsu M., Dawood F., Sun M., Arab S., Chen M., et al. (2006). Stem cell factor receptor induces progenitor and natural killer cell-mediated cardiac survival and repair after myocardial infarction. Proc. Natl. Acad. Sci. 103 (7), 2304–2309. doi:10.1073/pnas.0510997103
Backteman K., Ernerudh J., Jonasson L. (2013). Natural killer (NK) cell deficit in coronary artery disease: no aberrations in phenotype but sustained reduction of NK cells is associated with low-grade inflammation. Clin. Exp. Immunol. 175 (1), 104–112. doi:10.1111/cei.12210
Bai L., Bernard K., Tang X., Hu M., Horowitz J. C., Thannickal V. J., et al. (2019). Glutaminolysis epigenetically regulates antiapoptotic gene expression in idiopathic pulmonary fibrosis fibroblasts. Am. J. Respir. Cell Mol. Biol. 60 (1), 49–57. doi:10.1165/rcmb.2018-0180OC
Baik S. H., Ramanujan V. K., Becker C., Fett S., Underhill D. M., Wolf A. J. (2023). Hexokinase dissociation from mitochondria promotes oligomerization of VDAC that facilitates NLRP3 inflammasome assembly and activation. Sci. Immunol. 8 (84), eade7652. doi:10.1126/sciimmunol.ade7652
Bajpai G., Bredemeyer A., Li W., Zaitsev K., Koenig A. L., Lokshina I., et al. (2019). Tissue resident CCR2− and CCR2+ cardiac macrophages differentially orchestrate monocyte recruitment and fate specification following myocardial injury. Circulation Res. 124 (2), 263–278. doi:10.1161/CIRCRESAHA.118.314028
Bajpai G., Schneider C., Wong N., Bredemeyer A., Hulsmans M., Nahrendorf M., et al. (2018). The human heart contains distinct macrophage subsets with divergent origins and functions. Nat. Med. 24 (8), 1234–1245. doi:10.1038/s41591-018-0059-x
Baldeviano G. C., Barin J. G., Talor M. V., Srinivasan S., Bedja D., Zheng D., et al. (2010). Interleukin-17A is dispensable for myocarditis but essential for the progression to dilated cardiomyopathy. Circ. Res. 106 (10), 1646–1655. doi:10.1161/CIRCRESAHA.109.213157
Baumgarten G., Knuefermann P., Mann D. L. (2000). Cytokines as emerging targets in the treatment of heart failure. Trends Cardiovasc Med. 10 (5), 216–223. doi:10.1016/s1050-1738(00)00063-3
Bayer A. L., Smolgovsky S., Ngwenyama N., Hernández-Martínez A., Kaur K., Sulka K., et al. (2023). T-cell MyD88 is a novel regulator of cardiac fibrosis through modulation of T-cell activation. Circ. Res. 133 (5), 412–429. doi:10.1161/CIRCRESAHA.123.323030
Bedi K. C., Snyder N. W., Brandimarto J., Aziz M., Mesaros C., Worth A. J., et al. (2016). Evidence for intramyocardial disruption of lipid metabolism and increased myocardial ketone utilization in advanced human heart failure. Circulation 133 (8), 706–716. doi:10.1161/CIRCULATIONAHA.115.017545
Bermea K. C., Kostelecky N., Rousseau S. T., Lin C. Y., Adamo L. (2022). The human myocardium harbors a population of naive B-cells with a distinctive gene expression signature conserved across species. Front. Immunol. 13, 973211. doi:10.3389/fimmu.2022.973211
Bertrand L., Auquier J., Renguet E., Angé M., Cumps J., Horman S., et al. (2020). Glucose transporters in cardiovascular system in health and disease. Pflügers Archiv - Eur. J. Physiology 472 (9), 1385–1399. doi:10.1007/s00424-020-02444-8
Boukouaci W., Lauden L., Siewiera J., Dam N., Hocine H. R., Khaznadar Z., et al. (2014). Natural killer cell crosstalk with allogeneic human cardiac-derived stem/progenitor cells controls persistence. Cardiovasc. Res. 104 (2), 290–302. doi:10.1093/cvr/cvu208
Brandt U D. C. (2014). “Abbau von Acetyl-CoA zu CO2 und H2O,” in Löffler/Petrides Biochemie und Pathobiochemie. Editor M. M. G. L. Heinrich Pc (Heidelberg, Berlin: Springer), 226–234.
Buckingham M., Meilhac S., Zaffran S. (2005). Building the mammalian heart from two sources of myocardial cells. Nat. Rev. Genet. 6 (11), 826–835. doi:10.1038/nrg1710
Butts B., Goeddel L. A., George D. J., Steele C., Davies J. E., Wei C. C., et al. (2017). Increased inflammation in pericardial fluid persists 48 hours after cardiac surgery. Circulation 136 (23), 2284–2286. doi:10.1161/CIRCULATIONAHA.117.029589
Cairns J. A., Walls A. F. (1997). Mast cell tryptase stimulates the synthesis of type I collagen in human lung fibroblasts. J. Clin. Invest. 99 (6), 1313–1321. doi:10.1172/JCI119290
Cao D. J., Schiattarella G. G., Villalobos E., Jiang N., May H. I., Li T., et al. (2018). Cytosolic DNA sensing promotes macrophage transformation and governs myocardial ischemic injury. Circulation 137 (24), 2613–2634. doi:10.1161/CIRCULATIONAHA.117.031046
Cao W., Chen J., Chen Y., Chen S., Chen X., Huang H., et al. (2015). Advanced glycation end products induced immune maturation of dendritic cells controls heart failure through NF-κB signaling pathway. Arch. Biochem. Biophys. 580, 112–120. doi:10.1016/j.abb.2015.07.003
Carr E. L., Kelman A., Wu G. S., Gopaul R., Senkevitch E., Aghvanyan A., et al. (2010). Glutamine uptake and metabolism are coordinately regulated by ERK/MAPK during T lymphocyte activation. J. Immunol. 185 (2), 1037–1044. doi:10.4049/jimmunol.0903586
Carvalheiro T., Zimmermann M., Radstake T. R. D. J., Marut W. (2020). Novel insights into dendritic cells in the pathogenesis of systemic sclerosis. Clin. Exp. Immunol. 201 (1), 25–33. doi:10.1111/cei.13417
Chelakkot C., Chelakkot V. S., Shin Y., Song K. (2023). Modulating glycolysis to improve cancer therapy. Int. J. Mol. Sci. 24 (3), 2606. doi:10.3390/ijms24032606
Chen B., Frangogiannis N. G. (2017). Immune cells in repair of the infarcted myocardium. Microcirculation 24 (1). doi:10.1111/micc.12305
Chen C., Wang J., Liu C., Hu J. (2023). Cardiac resident macrophages: key regulatory mediators in the aftermath of myocardial infarction. Front. Immunol. 14, 1207100. doi:10.3389/fimmu.2023.1207100
Chen W., Zhang J., Zhong W., Liu Y., Lu Y., Zeng Z., et al. (2021b). Anlotinib inhibits PFKFB3-driven glycolysis in myofibroblasts to reverse pulmonary fibrosis. Front. Pharmacol. 12, 744826. doi:10.3389/fphar.2021.744826
Chen Z. T., Gao Q. Y., Wu M. X., Wang M., Sun R. L., Jiang Y., et al. (2021a). Glycolysis inhibition alleviates cardiac fibrosis after myocardial infarction by suppressing cardiac fibroblast activation. Front. Cardiovasc Med. 8, 701745. doi:10.3389/fcvm.2021.701745
Chi H. (2022). Immunometabolism at the intersection of metabolic signaling, cell fate, and systems immunology. Cell. Mol. Immunol. 19 (3), 299–302. doi:10.1038/s41423-022-00840-x
Chimen M., McGettrick H. M., Apta B., Kuravi S. J., Yates C. M., Kennedy A., et al. (2015). Homeostatic regulation of T cell trafficking by a B cell–derived peptide is impaired in autoimmune and chronic inflammatory disease. Nat. Med. 21 (5), 467–475. doi:10.1038/nm.3842
Chiossone L., Dumas P. Y., Vienne M., Vivier E. (2018). Natural killer cells and other innate lymphoid cells in cancer. Nat. Rev. Immunol. 18 (11), 671–688. doi:10.1038/s41577-018-0061-z
Cho S. J., Moon J. S., Nikahira K., Yun H. S., Harris R., Hong K. S., et al. (2020). GLUT1-dependent glycolysis regulates exacerbation of fibrosis via AIM2 inflammasome activation. Thorax 75 (3), 227–236. doi:10.1136/thoraxjnl-2019-213571
Choi J.-H., Do Y., Cheong C., Koh H., Boscardin S. B., Oh Y. S., et al. (2009). Identification of antigen-presenting dendritic cells in mouse aorta and cardiac valves. J. Exp. Med. 206 (3), 497–505. doi:10.1084/jem.20082129
Choo E. H., Lee J. H., Park E. H., Park H. E., Jung N. C., Kim T. H., et al. (2017). Infarcted myocardium-primed dendritic cells improve remodeling and cardiac function after myocardial infarction by modulating the regulatory T cell and macrophage polarization. Circulation 135 (15), 1444–1457. doi:10.1161/CIRCULATIONAHA.116.023106
Christe M. E., Rodgers R. L. (1994). Altered glucose and fatty acid oxidation in hearts of the spontaneously hypertensive rat. J. Mol. Cell Cardiol. 26 (10), 1371–1375. doi:10.1006/jmcc.1994.1155
Cobbold S. P., Adams E., Farquhar C. A., Nolan K. F., Howie D., Lui K. O., et al. (2009). Infectious tolerance via the consumption of essential amino acids and mTOR signaling. Proc. Natl. Acad. Sci. 106 (29), 12055–12060. doi:10.1073/pnas.0903919106
Cordero-Reyes A. M., Youker K. A., Trevino A. R., Celis R., Hamilton D. J., Flores-Arredondo J. H., et al. (2016). Full expression of cardiomyopathy is partly dependent on B-cells: a pathway that involves cytokine activation, immunoglobulin deposition, and activation of apoptosis. J. Am. Heart Assoc. 5 (1), e002484. doi:10.1161/JAHA.115.002484
Cowling R. T., Kupsky D., Kahn A. M., Daniels L. B., Greenberg B. H. (2019). Mechanisms of cardiac collagen deposition in experimental models and human disease. Transl. Res. 209, 138–155. doi:10.1016/j.trsl.2019.03.004
Crawford J., Cohen H. J. (1985). The essential role of L-glutamine in lymphocyte differentiation in vitro. J. Cell Physiol. 124 (2), 275–282. doi:10.1002/jcp.1041240216
Czubryt M. P. (2019). Cardiac fibroblast to myofibroblast phenotype conversion-an unexploited therapeutic target. J. Cardiovasc Dev. Dis. 6 (3), 28. doi:10.3390/jcdd6030028
de Almeida A., Mustin D., Forman M. F., Brower G. L., Janicki J. S., Carver W. (2002). Effects of mast cells on the behavior of isolated heart fibroblasts: modulation of collagen remodeling and gene expression. J. Cell Physiol. 191 (1), 51–59. doi:10.1002/jcp.10071
DeBerge M., Chaudhary R., Schroth S., Thorp E. B. (2023). Immunometabolism at the heart of cardiovascular disease. JACC Basic Transl. Sci. 8 (7), 884–904. doi:10.1016/j.jacbts.2022.12.010
DeBerge M., Shah S. J., Wilsbacher L., Thorp E. B. (2019). Macrophages in heart failure with reduced versus preserved ejection fraction. Trends Mol. Med. 25 (4), 328–340. doi:10.1016/j.molmed.2019.01.002
DeBerge M., Yeap X. Y., Dehn S., Zhang S., Grigoryeva L., Misener S., et al. (2017). MerTK cleavage on resident cardiac macrophages compromises repair after myocardial ischemia reperfusion injury. Circ. Res. 121 (8), 930–940. doi:10.1161/CIRCRESAHA.117.311327
de Couto G., Liu W., Tseliou E., Sun B., Makkar N., Kanazawa H., et al. (2015). Macrophages mediate cardioprotective cellular postconditioning in acute myocardial infarction. J. Clin. Invest. 125 (8), 3147–3162. doi:10.1172/JCI81321
Del Rey M. J., Valín Á., Usategui A., García-Herrero C. M., Sánchez-Aragó M., Cuezva J. M., et al. (2017). Hif-1α knockdown reduces glycolytic metabolism and induces cell death of human synovial fibroblasts under normoxic conditions. Sci. Rep. 7 (1), 3644. doi:10.1038/s41598-017-03921-4
Deniset J. F., Belke D., Lee W. Y., Jorch S. K., Deppermann C., Hassanabad A. F., et al. (2019). Gata6(+) pericardial cavity macrophages relocate to the injured heart and prevent cardiac fibrosis. Immunity 51 (1), 131–140. doi:10.1016/j.immuni.2019.06.010
Dewald O., Zymek P., Winkelmann K., Koerting A., Ren G., Abou-Khamis T., et al. (2005). CCL2/Monocyte Chemoattractant Protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ. Res. 96 (8), 881–889. doi:10.1161/01.RES.0000163017.13772.3a
Dhalla N. S., Elimban V., Bartekova M., Adameova A. (2022). Involvement of oxidative stress in the development of subcellular defects and heart disease. Biomedicines 10 (2), 393. doi:10.3390/biomedicines10020393
Ding H., Jiang L., Xu J., Bai F., Zhou Y., Yuan Q., et al. (2017). Inhibiting aerobic glycolysis suppresses renal interstitial fibroblast activation and renal fibrosis. Am. J. Physiology-Renal Physiology 313 (3), F561–F575. doi:10.1152/ajprenal.00036.2017
Doenst T., Nguyen T. D., Abel E. D. (2013). Cardiac metabolism in heart failure: implications beyond ATP production. Circulation Res. 113 (6), 709–724. doi:10.1161/CIRCRESAHA.113.300376
Donnelly R. P., Loftus R. M., Keating S. E., Liou K. T., Biron C. A., Gardiner C. M., et al. (2014). mTORC1-dependent metabolic reprogramming is a prerequisite for NK cell effector function. J. Immunol. 193 (9), 4477–4484. doi:10.4049/jimmunol.1401558
Doughty C. A., Bleiman B. F., Wagner D. J., Dufort F. J., Mataraza J. M., Roberts M. F., et al. (2006). Antigen receptor-mediated changes in glucose metabolism in B lymphocytes: role of phosphatidylinositol 3-kinase signaling in the glycolytic control of growth. Blood 107 (11), 4458–4465. doi:10.1182/blood-2005-12-4788
Drake K. J., Sidorov V. Y., McGuinness O. P., Wasserman D. H., Wikswo J. P. (2012). Amino acids as metabolic substrates during cardiac ischemia. Exp. Biol. Med. (Maywood) 237 (12), 1369–1378. doi:10.1258/ebm.2012.012025
Duerrschmid C., Trial J., Wang Y., Entman M. L., Haudek S. B. (2015). Tumor necrosis factor: a mechanistic link between angiotensin-II-induced cardiac inflammation and fibrosis. Circ. Heart Fail 8 (2), 352–361. doi:10.1161/CIRCHEARTFAILURE.114.001893
Ecker J., Liebisch G., Englmaier M., Grandl M., Robenek H., Schmitz G. (2010). Induction of fatty acid synthesis is a key requirement for phagocytic differentiation of human monocytes. Proc. Natl. Acad. Sci. 107 (17), 7817–7822. doi:10.1073/pnas.0912059107
El Alaoui-Talibi Z., Guendouz A., Moravec M., Moravec J. (1997). Control of oxidative metabolism in volume-overloaded rat hearts: effect of propionyl-L-carnitine. Am. J. Physiol. 272 (4), H1615–H1624. doi:10.1152/ajpheart.1997.272.4.H1615
el Alaoui-Talibi Z., Landormy S., Loireau A., Moravec J. (1992). Fatty acid oxidation and mechanical performance of volume-overloaded rat hearts. Am. J. Physiol. 262 (4), H1068–H1074. doi:10.1152/ajpheart.1992.262.4.H1068
Eming S. A., Murray P. J., Pearce E. J. (2021). Metabolic orchestration of the wound healing response. Cell Metab. 33 (9), 1726–1743. doi:10.1016/j.cmet.2021.07.017
Epelman S., Kory J. L., Gwendalyn J. R. (2014a). Origin and functions of tissue macrophages. Immunity 41 (1), 21–35. doi:10.1016/j.immuni.2014.06.013
Epelman S., Lavine K. J., Beaudin A. E., Sojka D. K., Carrero J. A., Calderon B., et al. (2014b). Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 40 (1), 91–104. doi:10.1016/j.immuni.2013.11.019
Epelman S., Liu P. P., Mann D. L. (2015). Role of innate and adaptive immune mechanisms in cardiac injury and repair. Nat. Rev. Immunol. 15 (2), 117–129. doi:10.1038/nri3800
Fan M., Yang K., Wang X., Chen L., Gill P. S., Ha T., et al. (2023). Lactate promotes endothelial-to-mesenchymal transition via Snail1 lactylation after myocardial infarction. Sci. Adv. 9 (5), eadc9465. doi:10.1126/sciadv.adc9465
Fang K. C., Raymond W. W., Blount J. L., Caughey G. H. (1997). Dog mast cell alpha-chymase activates progelatinase B by cleaving the Phe88-Gln89 and Phe91-Glu92 bonds of the catalytic domain. J. Biol. Chem. 272 (41), 25628–25635. doi:10.1074/jbc.272.41.25628
Farah H., Young S. P., Mauro C., Jones S. W. (2021). Metabolic dysfunction and inflammatory disease: the role of stromal fibroblasts. FEBS J. 288 (19), 5555–5568. doi:10.1111/febs.15644
Feingold K. R., Shigenaga J. K., Kazemi M. R., McDonald C. M., Patzek S. M., Cross A. S., et al. (2012). Mechanisms of triglyceride accumulation in activated macrophages. J. Leukoc. Biol. 92 (4), 829–839. doi:10.1189/jlb.1111537
Feng L., Chen X., Huang Y., Zhang X., Zheng S., Xie N. (2023). Immunometabolism changes in fibrosis: from mechanisms to therapeutic strategies. Front. Pharmacol. 14, 1243675. doi:10.3389/fphar.2023.1243675
Fernández-Ruiz I. (2017). Immunology: surprising role of cardiac macrophages in heart electrical conduction. Nat. Rev. Cardiol. 14 (6), 315. doi:10.1038/nrcardio.2017.72
Fillmore N., Mori J., Lopaschuk G. D. (2014). Mitochondrial fatty acid oxidation alterations in heart failure, ischaemic heart disease and diabetic cardiomyopathy. Br. J. Pharmacol. 171 (8), 2080–2090. doi:10.1111/bph.12475
Finucane O. M., Sugrue J., Rubio-Araiz A., Guillot-Sestier M. V., Lynch M. A. (2019). The NLRP3 inflammasome modulates glycolysis by increasing PFKFB3 in an IL-1β-dependent manner in macrophages. Sci. Rep. 9 (1), 4034. doi:10.1038/s41598-019-40619-1
Forman M. F., Brower G. L., Janicki J. S. (2006). Rat cardiac mast cell maturation and differentiation following acute ventricular volume overload. Inflamm. Res. 55 (10), 408–415. doi:10.1007/s00011-006-6016-z
Fortier S. M., Penke L. R., King D., Pham T. X., Ligresti G., Peters-Golden M. (2021). Myofibroblast dedifferentiation proceeds via distinct transcriptomic and phenotypic transitions. JCI Insight 6 (6), e144799. doi:10.1172/jci.insight.144799
Francisco J., Zhang Y., Nakada Y., Jeong J. I., Huang C. Y., Ivessa A., et al. (2021). AAV-mediated YAP expression in cardiac fibroblasts promotes inflammation and increases fibrosis. Sci. Rep. 11 (1), 10553. doi:10.1038/s41598-021-89989-5
Frangogiannis N. G. (2015). Emerging roles for macrophages in cardiac injury: cytoprotection, repair, and regeneration. J. Clin. Invest. 125 (8), 2927–2930. doi:10.1172/JCI83191
Frangogiannis N. G. (2021). Cardiac fibrosis. Cardiovasc. Res. 117 (6), 1450–1488. doi:10.1093/cvr/cvaa324
Frangogiannis N. G., Burns A. R., Michael L. H., Entman M. L. (1999). Histochemical and morphological characteristics of canine cardiac mast cells. Histochem J. 31 (4), 221–229. doi:10.1023/a:1003541332070
Frangogiannis N. G., Dewald O., Xia Y., Ren G., Haudek S., Leucker T., et al. (2007). Critical role of monocyte chemoattractant protein-1/CC chemokine ligand 2 in the pathogenesis of ischemic cardiomyopathy. Circulation 115 (5), 584–592. doi:10.1161/CIRCULATIONAHA.106.646091
Frangogiannis N. G., Lindsey M. L., Michael L. H., Youker K. A., Bressler R. B., Mendoza L. H., et al. (1998a). Resident cardiac mast cells degranulate and release preformed TNF-alpha, initiating the cytokine cascade in experimental canine myocardial ischemia/reperfusion. Circulation 98 (7), 699–710. doi:10.1161/01.cir.98.7.699
Frangogiannis N. G., Perrard J. L., Mendoza L. H., Burns A. R., Lindsey M. L., Ballantyne C. M., et al. (1998b). Stem cell factor induction is associated with mast cell accumulation after canine myocardial ischemia and reperfusion. Circulation 98 (7), 687–698. doi:10.1161/01.cir.98.7.687
Freemerman A. J., Johnson A. R., Sacks G. N., Milner J. J., Kirk E. L., Troester M. A., et al. (2014). Metabolic reprogramming of macrophages: glucose transporter 1 (GLUT1)-mediated glucose metabolism drives a proinflammatory phenotype. J. Biol. Chem. 289 (11), 7884–7896. doi:10.1074/jbc.M113.522037
Fröhlich G. M., Meier P., White S. K., Yellon D. M., Hausenloy D. J. (2013). Myocardial reperfusion injury: looking beyond primary PCI. Eur. Heart J. 34 (23), 1714–1722. doi:10.1093/eurheartj/eht090
Fu X., Liu Q., Li C., Li Y., Wang L. (2020). Cardiac fibrosis and cardiac fibroblast lineage-tracing: recent advances. Front. Physiology 11, 416. doi:10.3389/fphys.2020.00416
Gailit J., Marchese M. J., Kew R. R., Gruber B. L. (2001). The differentiation and function of myofibroblasts is regulated by mast cell mediators. J. Investigative Dermatology 117 (5), 1113–1119. doi:10.1046/j.1523-1747.2001.15211.x
Gersch C., Dewald O., Zoerlein M., Michael L. H., Entman M. L., Frangogiannis N. G. (2002). Mast cells and macrophages in normal C57/BL/6 mice. Histochem Cell Biol. 118 (1), 41–49. doi:10.1007/s00418-002-0425-z
Gibb A. A., Huynh A. T., Gaspar R. B., Ploesch T. L., Lombardi A. A., Lorkiewicz P. K., et al. (2022a). Glutamine uptake and catabolism is required for myofibroblast formation and persistence. J. Mol. Cell Cardiol. 172, 78–89. doi:10.1016/j.yjmcc.2022.08.002
Gibb A. A., Lazaropoulos M. P., Elrod J. W. (2020). Myofibroblasts and fibrosis: mitochondrial and metabolic control of cellular differentiation. Circulation Res. 127 (3), 427–447. doi:10.1161/CIRCRESAHA.120.316958
Gibb A. A., Murray E. K., Huynh A. T., Gaspar R. B., Ploesch T. L., Bedi K., et al. (2022b). Glutaminolysis is essential for myofibroblast persistence and in vivo targeting reverses fibrosis and cardiac dysfunction in heart failure. Circulation 145 (21), 1625–1628. doi:10.1161/CIRCULATIONAHA.121.057879
Grützkau A., Krüger-Krasagakes S., Baumeister H., Schwarz C., Kögel H., Welker P., et al. (1998). Synthesis, storage, and release of vascular endothelial growth factor/vascular permeability factor (VEGF/VPF) by human mast cells: implications for the biological significance of VEGF206. Mol. Biol. Cell 9 (4), 875–884. doi:10.1091/mbc.9.4.875
Gubser P. M., Bantug G. R., Razik L., Fischer M., Dimeloe S., Hoenger G., et al. (2013). Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat. Immunol. 14 (10), 1064–1072. doi:10.1038/ni.2687
Guimbal S., Cornuault L., Rouault P., Hollier P. L., Chapouly C., Bats M. L., et al. (2021). Mast cells are the trigger of small vessel disease and diastolic dysfunction in diabetic obese mice. Arteriosclerosis, Thrombosis, Vasc. Biol. 41 (4), e193–e207. doi:10.1161/ATVBAHA.121.315900
Guo L., Akahori H., Harari E., Smith S. L., Polavarapu R., Karmali V., et al. (2018). CD163+ macrophages promote angiogenesis and vascular permeability accompanied by inflammation in atherosclerosis. J. Clin. Investigation 128 (3), 1106–1124. doi:10.1172/JCI93025
Gwechenberger M., Mendoza L. H., Youker K. A., Frangogiannis N. G., Smith C. W., Michael L. H., et al. (1999). Cardiac myocytes produce interleukin-6 in culture and in viable border zone of reperfused infarctions. Circulation 99 (4), 546–551. doi:10.1161/01.cir.99.4.546
Gyöngyösi M., Winkler J., Ramos I., Do Q. T., Firat H., McDonald K., et al. (2017). Myocardial fibrosis: biomedical research from bench to bedside. Eur. J. Heart Fail. 19 (2), 177–191. doi:10.1002/ejhf.696
Haider N., Boscá L., Zandbergen H. R., Kovacic J. C., Narula N., González-Ramos S., et al. (2019). Transition of macrophages to fibroblast-like cells in healing myocardial infarction. J. Am. Coll. Cardiol. 74 (25), 3124–3135. doi:10.1016/j.jacc.2019.10.036
Hara A., Tallquist M. D. (2023). Fibroblast and immune cell cross-talk in cardiac fibrosis. Curr. Cardiol. Rep. 25 (6), 485–493. doi:10.1007/s11886-023-01877-8
Hara M., Ono K., Hwang M. W., Iwasaki A., Okada M., Nakatani K., et al. (2002). Evidence for a role of mast cells in the evolution to congestive heart failure. J. Exp. Med. 195 (3), 375–381. doi:10.1084/jem.20002036
Hartupee J., Mann D. L. (2017). Neurohormonal activation in heart failure with reduced ejection fraction. Nat. Rev. Cardiol. 14 (1), 30–38. doi:10.1038/nrcardio.2016.163
Harvey A. P., Grieve D. J. (2014). “Reactive oxygen species (ROS) signaling in cardiac remodeling and failure,” in Systems biology of free radicals and antioxidants. Editor I. Laher (Berlin, Heidelberg: Springer Berlin Heidelberg), 951–992.
Haschemi A., Kosma P., Gille L., Evans C. R., Burant C. F., Starkl P., et al. (2012). The sedoheptulose kinase CARKL directs macrophage polarization through control of glucose metabolism. Cell Metab. 15 (6), 813–826. doi:10.1016/j.cmet.2012.04.023
He J., Liu Q., Wang J., Xu F., Fan Y., He R., et al. (2022). Identification of the metabolic remodeling profile in the early-stage of myocardial ischemia and the contributory role of mitochondrion. Bioengineered 13 (4), 11106–11121. doi:10.1080/21655979.2022.2068882
He L., Kim T., Long Q., Liu J., Wang P., Zhou Y., et al. (2012). Carnitine palmitoyltransferase-1b deficiency aggravates pressure overload-induced cardiac hypertrophy caused by lipotoxicity. Circulation 126 (14), 1705–1716. doi:10.1161/CIRCULATIONAHA.111.075978
He X., Zeng H., Chen S. T., Roman R. J., Aschner J. L., Didion S., et al. (2017). Endothelial specific SIRT3 deletion impairs glycolysis and angiogenesis and causes diastolic dysfunction. J. Mol. Cell Cardiol. 112, 104–113. doi:10.1016/j.yjmcc.2017.09.007
Heather L. C., Pates K. M., Atherton H. J., Cole M. A., Ball D. R., Evans R. D., et al. (2013). Differential translocation of the fatty acid transporter, FAT/CD36, and the glucose transporter, GLUT4, coordinates changes in cardiac substrate metabolism during ischemia and reperfusion. Circ. Heart Fail 6 (5), 1058–1066. doi:10.1161/CIRCHEARTFAILURE.112.000342
Hecker L., Jagirdar R., Jin T., Thannickal V. J. (2011). Reversible differentiation of myofibroblasts by MyoD. Exp. Cell Res. 317 (13), 1914–1921. doi:10.1016/j.yexcr.2011.03.016
Heidt T., Courties G., Dutta P., Sager H. B., Sebas M., Iwamoto Y., et al. (2014). Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circulation Res. 115 (2), 284–295. doi:10.1161/CIRCRESAHA.115.303567
Henderson J., O'Reilly S. (2021). The emerging role of metabolism in fibrosis. Trends Endocrinol. Metab. 32 (8), 639–653. doi:10.1016/j.tem.2021.05.003
Hilgendorf I., Gerhardt L. M. S., Tan T. C., Winter C., Holderried T. A. W., Chousterman B. G., et al. (2014). Ly-6C high monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circulation Res. 114 (10), 1611–1622. doi:10.1161/CIRCRESAHA.114.303204
Hinz B., Mastrangelo D., Iselin C. E., Chaponnier C., Gabbiani G. (2001). Mechanical tension controls granulation tissue contractile activity and myofibroblast differentiation. Am. J. Pathol. 159 (3), 1009–1020. doi:10.1016/S0002-9440(10)61776-2
Ho K. L., Karwi Q. G., Wagg C., Zhang L., Vo K., Altamimi T., et al. (2021). Ketones can become the major fuel source for the heart but do not increase cardiac efficiency. Cardiovasc Res. 117 (4), 1178–1187. doi:10.1093/cvr/cvaa143
Hofmann U., Beyersdorf N., Weirather J., Podolskaya A., Bauersachs J., Ertl G., et al. (2012). Activation of CD4 + T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation 125 (13), 1652–1663. doi:10.1161/CIRCULATIONAHA.111.044164
Honold L., Nahrendorf M. (2018). Resident and monocyte-derived macrophages in cardiovascular disease. Circ. Res. 122 (1), 113–127. doi:10.1161/CIRCRESAHA.117.311071
Hooftman A., Peace C. G., Ryan D. G., Day E. A., Yang M., McGettrick A. F., et al. (2023). Macrophage fumarate hydratase restrains mtRNA-mediated interferon production. Nature 615 (7952), 490–498. doi:10.1038/s41586-023-05720-6
Horckmans M., Bianchini M., Santovito D., Megens R. T. A., Springael J. Y., Negri I., et al. (2018). Pericardial adipose tissue regulates granulopoiesis, fibrosis, and cardiac function after myocardial infarction. Circulation 137 (9), 948–960. doi:10.1161/CIRCULATIONAHA.117.028833
Horckmans M., Ring L., Duchene J., Santovito D., Schloss M. J., Drechsler M., et al. (2017). Neutrophils orchestrate post-myocardial infarction healing by polarizing macrophages towards a reparative phenotype. Eur. Heart J. 38 (3), 187–197. doi:10.1093/eurheartj/ehw002
Horton J. L., Davidson M. T., Kurishima C., Vega R. B., Powers J. C., Matsuura T. R., et al. (2019). The failing heart utilizes 3-hydroxybutyrate as a metabolic stress defense. JCI Insight 4 (4), e124079. doi:10.1172/jci.insight.124079
Huang S.C.-C., Everts B., Ivanova Y., O'Sullivan D., Nascimento M., Smith A. M., et al. (2014). Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat. Immunol. 15 (9), 846–855. doi:10.1038/ni.2956
Hulsmans M., Clauss S., Xiao L., Aguirre A. D., King K. R., Hanley A., et al. (2017). Macrophages facilitate electrical conduction in the heart. Cell 169 (3), 510–522. doi:10.1016/j.cell.2017.03.050
Humeres C., Frangogiannis N. G. (2019). Fibroblasts in the infarcted, remodeling, and failing heart. JACC Basic Transl. Sci. 4 (3), 449–467. doi:10.1016/j.jacbts.2019.02.006
Im S. S., Yousef L., Blaschitz C., Liu J. Z., Edwards R. A., Young S. G., et al. (2011). Linking lipid metabolism to the innate immune response in macrophages through sterol regulatory element binding protein-1a. Cell Metab. 13 (5), 540–549. doi:10.1016/j.cmet.2011.04.001
Infantino V., Convertini P., Cucci L., Panaro M. A., Di Noia M. A., Calvello R., et al. (2011). The mitochondrial citrate carrier: a new player in inflammation. Biochem. J. 438 (3), 433–436. doi:10.1042/BJ20111275
Infantino V., Iacobazzi V., Menga A., Avantaggiati M. L., Palmieri F. (2014). A key role of the mitochondrial citrate carrier (SLC25A1) in TNFα- and IFNγ-triggered inflammation. Biochim. Biophys. Acta 1839 (11), 1217–1225. doi:10.1016/j.bbagrm.2014.07.013
Ingason A. B., Mechmet F., Atacho D. A. M., Steingrímsson E., Petersen P. H. (2019). Distribution of mast cells within the mouse heart and its dependency on Mitf. Mol. Immunol. 105, 9–15. doi:10.1016/j.molimm.2018.11.009
Isidoro C. A., Deniset J. F. (2023). The role of macrophage subsets in and around the heart in modulating cardiac homeostasis and pathophysiology. Front. Immunol. 14, 1111819. doi:10.3389/fimmu.2023.1111819
Jain M., Rivera S., Monclus E. A., Synenki L., Zirk A., Eisenbart J., et al. (2013). Mitochondrial reactive oxygen species regulate transforming growth factor-β signaling. J. Biol. Chem. 288 (2), 770–777. doi:10.1074/jbc.M112.431973
Janicki J. S., Brower G. L., Levick S. P. (2015). The emerging prominence of the cardiac mast cell as a potent mediator of adverse myocardial remodeling. New York: Springer, 121–139.
Jeffrey F. M. H., Diczku V., Sherry A. D., Malloy C. R. (1995). Substrate selection in the isolated working rat heart: effects of reperfusion, afterload, and concentration. Basic Res. Cardiol. 90 (5), 388–396. doi:10.1007/BF00788500
Ji J. J., Qian L. L., Zhu Y., Jiang Y., Guo J. Q., Wu Y., et al. (2022). Kallistatin/Serpina3c inhibits cardiac fibrosis after myocardial infarction by regulating glycolysis via Nr4a1 activation. Biochim. Biophys. Acta Mol. Basis Dis. 1868 (9), 166441. doi:10.1016/j.bbadis.2022.166441
Jia Y., Reddy M. A., Das S., Oh H. J., Abdollahi M., Yuan H., et al. (2019). Dysregulation of histone H3 lysine 27 trimethylation in transforming growth factor-β1-induced gene expression in mesangial cells and diabetic kidney. J. Biol. Chem. 294 (34), 12695–12707. doi:10.1074/jbc.RA119.007575
Jiang L., Xiao L., Sugiura H., Huang X., Ali A., Kuro-o M., et al. (2015). Metabolic reprogramming during TGFβ1-induced epithelial-to-mesenchymal transition. Oncogene 34 (30), 3908–3916. doi:10.1038/onc.2014.321
Jiang M., Xie X., Cao F., Wang Y. (2021). Mitochondrial metabolism in myocardial remodeling and mechanical unloading: implications for ischemic heart disease. Front. Cardiovasc. Med. 8, 789267. doi:10.3389/fcvm.2021.789267
Jin J., Jiang Y., Chakrabarti S., Su Z. (2022). Cardiac mast cells: a two-head regulator in cardiac homeostasis and pathogenesis following injury. Front. Immunol. 13, 963444. doi:10.3389/fimmu.2022.963444
Jogalekar M. P., Rajendran R. L., Khan F., Dmello C., Gangadaran P., Ahn B. C. (2022). CAR T-Cell-Based gene therapy for cancers: new perspectives, challenges, and clinical developments. Front. Immunol. 13, 925985. doi:10.3389/fimmu.2022.925985
Joseph J., Kennedy R. H., Devi S., Wang J., Joseph L., Hauer-Jensen M. (2005). Protective role of mast cells in homocysteine-induced cardiac remodeling. Am. J. Physiol. Heart Circ. Physiol. 288 (5), H2541–H2545. doi:10.1152/ajpheart.00806.2004
Jung K., Kim P., Leuschner F., Gorbatov R., Kim J. K., Ueno T., et al. (2013). Endoscopic time-lapse imaging of immune cells in infarcted mouse hearts. Circulation Res. 112 (6), 891–899. doi:10.1161/CIRCRESAHA.111.300484
Jung M., Ma Y., Iyer R. P., DeLeon-Pennell K. Y., Yabluchanskiy A., Garrett M. R., et al. (2017). IL-10 improves cardiac remodeling after myocardial infarction by stimulating M2 macrophage polarization and fibroblast activation. Basic Res. Cardiol. 112 (3), 33. doi:10.1007/s00395-017-0622-5
Kallikourdis M., Martini E., Carullo P., Sardi C., Roselli G., Greco C. M., et al. (2017). T cell costimulation blockade blunts pressure overload-induced heart failure. Nat. Commun. 8, 14680. doi:10.1038/ncomms14680
Kanellakis P., Ditiatkovski M., Kostolias G., Bobik A. (2012). A pro-fibrotic role for interleukin-4 in cardiac pressure overload. Cardiovasc Res. 95 (1), 77–85. doi:10.1093/cvr/cvs142
Kanemitsu H., Takai S., Tsuneyoshi H., Nishina T., Yoshikawa K., Miyazaki M., et al. (2006). Chymase inhibition prevents cardiac fibrosis and dysfunction after myocardial infarction in rats. Hypertens. Res. 29 (1), 57–64. doi:10.1291/hypres.29.57
Kang S., Wang B., Xie Y., Cao X., Wang M. (2023). The role of M1 and M2 myocardial macrophages in promoting proliferation and healing via activating epithelial-to-mesenchymal transition. Biomedicines 11 (10), 2666. doi:10.3390/biomedicines11102666
Kanisicak O., Khalil H., Ivey M. J., Karch J., Maliken B. D., Correll R. N., et al. (2016). Genetic lineage tracing defines myofibroblast origin and function in the injured heart. Nat. Commun. 7, 12260. doi:10.1038/ncomms12260
Kaur H., Takefuji M., Ngai C. Y., Carvalho J., Bayer J., Wietelmann A., et al. (2016). Targeted ablation of periostin-expressing activated fibroblasts prevents adverse cardiac remodeling in mice. Circ. Res. 118 (12), 1906–1917. doi:10.1161/CIRCRESAHA.116.308643
Kawaguchi M., Takahashi M., Hata T., Kashima Y., Usui F., Morimoto H., et al. (2011). Inflammasome activation of cardiac fibroblasts is essential for myocardial ischemia/reperfusion injury. Circulation 123 (6), 594–604. doi:10.1161/CIRCULATIONAHA.110.982777
Kim Y., Nurakhayev S., Nurkesh A., Zharkinbekov Z., Saparov A. (2021). Macrophage polarization in cardiac tissue repair following myocardial infarction. Int. J. Mol. Sci. 22 (5), 2715. doi:10.3390/ijms22052715
King K. R., Aguirre A. D., Ye Y. X., Sun Y., Roh J. D., Ng R. P., et al. (2017). IRF3 and type I interferons fuel a fatal response to myocardial infarction. Nat. Med. 23 (12), 1481–1487. doi:10.1038/nm.4428
Koeth R. A., Haselden V., Tang W. H. W. (2013). “Chapter one - myeloperoxidase in cardiovascular disease,” in Advances in clinical chemistry. Editor G. S. Makowski (Elsevier), 1–32.
Kolck U. W., Alfter K., Homann J., von Kügelgen I., Molderings G. J. (2007). Cardiac mast cells: implications for heart failure. J. Am. Coll. Cardiol. 49 (10), 1107–1108. doi:10.1016/j.jacc.2006.12.018
Kolwicz S. C., Purohit S., Tian R. (2013). Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circulation Res. 113 (5), 603–616. doi:10.1161/CIRCRESAHA.113.302095
Kottmann R. M., Kulkarni A. A., Smolnycki K. A., Lyda E., Dahanayake T., Salibi R., et al. (2012). Lactic acid is elevated in idiopathic pulmonary fibrosis and induces myofibroblast differentiation via pH-dependent activation of transforming growth factor-β. Am. J. Respir. Crit. Care Med. 186 (8), 740–751. doi:10.1164/rccm.201201-0084OC
Krawczyk C. M., Holowka T., Sun J., Blagih J., Amiel E., DeBerardinis R. J., et al. (2010). Toll-like receptor-induced changes in glycolytic metabolism regulate dendritic cell activation. Blood 115 (23), 4742–4749. doi:10.1182/blood-2009-10-249540
Krishnamurthy P., Rajasingh J., Lambers E., Qin G., Losordo D. W., Kishore R. (2009). IL-10 inhibits inflammation and attenuates left ventricular remodeling after myocardial infarction via activation of STAT3 and suppression of HuR. Circulation Res. 104 (2), e9–e18. doi:10.1161/CIRCRESAHA.108.188243
Kuwahara F., Kai H., Tokuda K., Kai M., Takeshita A., Egashira K., et al. (2002). Transforming growth factor-beta function blocking prevents myocardial fibrosis and diastolic dysfunction in pressure-overloaded rats. Circulation 106 (1), 130–135. doi:10.1161/01.cir.0000020689.12472.e0
Kvakan H., Kleinewietfeld M., Qadri F., Park J. K., Fischer R., Schwarz I., et al. (2009). Regulatory T cells ameliorate angiotensin II-induced cardiac damage. Circulation 119 (22), 2904–2912. doi:10.1161/CIRCULATIONAHA.108.832782
Lavine K. J., Epelman S., Uchida K., Weber K. J., Nichols C. G., Schilling J. D., et al. (2014). Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc. Natl. Acad. Sci. 111 (45), 16029–16034. doi:10.1073/pnas.1406508111
Le A., Lane A. N., Hamaker M., Bose S., Gouw A., Barbi J., et al. (2012). Glucose-independent glutamine metabolism via TCA cycling for proliferation and survival in B cells. Cell Metab. 15 (1), 110–121. doi:10.1016/j.cmet.2011.12.009
Legere S. A., Haidl I. D., Castonguay M. C., Brunt K. R., Légaré J. F., Marshall J. S., et al. (2020). Increased mast cell density is associated with decreased fibrosis in human atrial tissue. J. Mol. Cell. Cardiol. 149, 15–26. doi:10.1016/j.yjmcc.2020.09.001
Legere S. A., Haidl I. D., Légaré J. F., Marshall J. S. (2019). Mast cells in cardiac fibrosis: new insights suggest opportunities for intervention. Front. Immunol. 10, 580. doi:10.3389/fimmu.2019.00580
Leid J., Carrelha J., Boukarabila H., Epelman S., Jacobsen S. E. W., Lavine K. J. (2016). Primitive embryonic macrophages are required for coronary development and maturation. Circulation Res. 118 (10), 1498–1511. doi:10.1161/CIRCRESAHA.115.308270
Leong H. S., Grist M., Parsons H., Wambolt R. B., Lopaschuk G. D., Brownsey R., et al. (2002). Accelerated rates of glycolysis in the hypertrophied heart: are they a methodological artifact? Am. J. Physiol. Endocrinol. Metab. 282 (5), E1039–E1045. doi:10.1152/ajpendo.00507.2001
Leo M. C., Stamatiades E. G., Auffray C., Hanna R. N., Glover L., Vizcay-Barrena G., et al. (2013). Nr4a1-Dependent Ly6Clow monocytes monitor endothelial cells and orchestrate their disposal. Cell 153 (2), 362–375. doi:10.1016/j.cell.2013.03.010
Levick S. P., McLarty J. L., Murray D. B., Freeman R. M., Carver W. E., Brower G. L. (2009). Cardiac mast cells mediate left ventricular fibrosis in the hypertensive rat heart. Hypertension 53 (6), 1041–1047. doi:10.1161/HYPERTENSIONAHA.108.123158
Levick S. P., Meléndez G. C., Plante E., McLarty J. L., Brower G. L., Janicki J. S. (2011). Cardiac mast cells: the centrepiece in adverse myocardial remodelling. Cardiovasc Res. 89 (1), 12–19. doi:10.1093/cvr/cvq272
Levick S. P., Widiapradja A. (2018). Mast cells: key contributors to cardiac fibrosis. Int. J. Mol. Sci. 19 (1), 231. doi:10.3390/ijms19010231
Li C., Sun X. N., Zeng M. R., Zheng X. J., Zhang Y. Y., Wan Q., et al. (2017). Mineralocorticoid receptor deficiency in T cells attenuates pressure overload-induced cardiac hypertrophy and dysfunction through modulating T-cell activation. Hypertension 70 (1), 137–147. doi:10.1161/HYPERTENSIONAHA.117.09070
Li J., Zhai X., Sun X., Cao S., Yuan Q., Wang J. (2022). Metabolic reprogramming of pulmonary fibrosis. Front. Pharmacol. 13, 1031890. doi:10.3389/fphar.2022.1031890
Li L., Zhao Q., Kong W. (2018). Extracellular matrix remodeling and cardiac fibrosis. Matrix Biol. 68-69, 490–506. doi:10.1016/j.matbio.2018.01.013
Li M. O., Wan Y. Y., Sanjabi S., Robertson A. K. L., Flavell R. A. (2006). Transforming growth factor-beta regulation of immune responses. Annu. Rev. Immunol. 24 (1), 99–146. doi:10.1146/annurev.immunol.24.021605.090737
Li R., Chen B., Kubota A., Hanna A., Humeres C., Hernandez S. C., et al. (2023a). Protective effects of macrophage-specific integrin α5 in myocardial infarction are associated with accentuated angiogenesis. Nat. Commun. 14 (1), 7555. doi:10.1038/s41467-023-43369-x
Li W., Hsiao H. M., Higashikubo R., Saunders B. T., Bharat A., Goldstein D. R., et al. (2016). Heart-resident CCR2+ macrophages promote neutrophil extravasation through TLR9/MyD88/CXCL5 signaling. JCI Insight 1 (12), e87315. doi:10.1172/jci.insight.87315
Li X., Wu F., Günther S., Looso M., Kuenne C., Zhang T., et al. (2023b). Inhibition of fatty acid oxidation enables heart regeneration in adult mice. Nature 622 (7983), 619–626. doi:10.1038/s41586-023-06585-5
Liao C. H., Akazawa H., Tamagawa M., Ito K., Yasuda N., Kudo Y., et al. (2010). Cardiac mast cells cause atrial fibrillation through PDGF-A-mediated fibrosis in pressure-overloaded mouse hearts. J. Clin. Invest. 120 (1), 242–253. doi:10.1172/JCI39942
Liao C. W., Chou C. H., Wu X. M., Chen Z. W., Chen Y. H., Chang Y. Y., et al. (2020). Interleukin-6 plays a critical role in aldosterone-induced macrophage recruitment and infiltration in the myocardium. Biochim. Biophys. Acta Mol. Basis Dis. 1866 (3), 165627. doi:10.1016/j.bbadis.2019.165627
Liepinsh E., Makrecka-Kuka M., Volska K., Kuka J., Makarova E., Antone U., et al. (2016). Long-chain acylcarnitines determine ischaemia/reperfusion-induced damage in heart mitochondria. Biochem. J. 473 (9), 1191–1202. doi:10.1042/BCJ20160164
Lin L.-C., Liu Z. Y., Yang J. J., Zhao J. Y., Tao H. (2023). Lipid metabolism reprogramming in cardiac fibrosis. Trends Endocrinol. Metabolism 35, 164–175. doi:10.1016/j.tem.2023.10.004
Lindsey M. L., Escobar G. P., Dobrucki L. W., Goshorn D. K., Bouges S., Mingoia J. T., et al. (2006). Matrix metalloproteinase-9 gene deletion facilitates angiogenesis after myocardial infarction. Am. J. Physiology-Heart Circulatory Physiology 290 (1), H232–H239. doi:10.1152/ajpheart.00457.2005
Litviňuková M., Talavera-López C., Maatz H., Reichart D., Worth C. L., Lindberg E. L., et al. (2020). Cells of the adult human heart. Nature 588 (7838), 466–472. doi:10.1038/s41586-020-2797-4
Liu C., Liu Y., Chen H., Yang X., Lu C., Wang L., et al. (2023). Myocardial injury: where inflammation and autophagy meet. Burns Trauma 11, 11. doi:10.1093/burnst/tkac062
Liu H., Gao W., Yuan J., Wu C., Yao K., Zhang L., et al. (2016). Exosomes derived from dendritic cells improve cardiac function via activation of CD4+ T lymphocytes after myocardial infarction. J. Mol. Cell. Cardiol. 91, 123–133. doi:10.1016/j.yjmcc.2015.12.028
Liu J., Marchase R. B., Chatham J. C. (2007). Glutamine-induced protection of isolated rat heart from ischemia/reperfusion injury is mediated via the hexosamine biosynthesis pathway and increased protein O-GlcNAc levels. J. Mol. Cell Cardiol. 42 (1), 177–185. doi:10.1016/j.yjmcc.2006.09.015
Liu R. M., Gaston Pravia K. A. (2010). Oxidative stress and glutathione in TGF-beta-mediated fibrogenesis. Free Radic. Biol. Med. 48 (1), 1–15. doi:10.1016/j.freeradbiomed.2009.09.026
Liu Y., Xu R., Gu H., Zhang E., Qu J., Cao W., et al. (2021). Metabolic reprogramming in macrophage responses. Biomark. Res. 9 (1), 1. doi:10.1186/s40364-020-00251-y
Lombardi A. A., Gibb A. A., Arif E., Kolmetzky D. W., Tomar D., Luongo T. S., et al. (2019). Mitochondrial calcium exchange links metabolism with the epigenome to control cellular differentiation. Nat. Commun. 10 (1), 4509. doi:10.1038/s41467-019-12103-x
Lopaschuk G. D., Karwi Q. G., Tian R., Wende A. R., Abel E. D. (2021). Cardiac energy metabolism in heart failure. Circulation Res. 128 (10), 1487–1513. doi:10.1161/CIRCRESAHA.121.318241
Lopaschuk G. D., Ussher J. R., Folmes C. D. L., Jaswal J. S., Stanley W. C. (2010). Myocardial fatty acid metabolism in health and disease. Physiol. Rev. 90 (1), 207–258. doi:10.1152/physrev.00015.2009
Lopaschuk G. D., Wall S. R., Olley P. M., Davies N. J. (1988). Etomoxir, a carnitine palmitoyltransferase I inhibitor, protects hearts from fatty acid-induced ischemic injury independent of changes in long chain acylcarnitine. Circulation Res. 63 (6), 1036–1043. doi:10.1161/01.res.63.6.1036
Luitel H., Sydykov A., Schymura Y., Mamazhakypov A., Janssen W., Pradhan K., et al. (2017). Pressure overload leads to an increased accumulation and activity of mast cells in the right ventricle. Physiol. Rep. 5 (6), e13146. doi:10.14814/phy2.13146
Ma Y., Mouton A. J., Lindsey M. L. (2018). Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl. Res. 191, 15–28. doi:10.1016/j.trsl.2017.10.001
Ma Y., Yabluchanskiy A., Iyer R. P., Cannon P. L., Flynn E. R., Jung M., et al. (2016). Temporal neutrophil polarization following myocardial infarction. Cardiovasc Res. 110 (1), 51–61. doi:10.1093/cvr/cvw024
MacIver N. J., Michalek R. D., Rathmell J. C. (2013). Metabolic regulation of T lymphocytes. Annu. Rev. Immunol. 31, 259–283. doi:10.1146/annurev-immunol-032712-095956
Majid A., Hassan F. O., Hoque M. M., Gbadegoye J. O., Lebeche D. (2023). Bioactive compounds and cardiac fibrosis: current insight and future prospect. J. Cardiovasc. Dev. Dis. 10 (7), 313. doi:10.3390/jcdd10070313
Mann D. L. (2011). The emerging role of innate immunity in the heart and vascular system: for whom the cell tolls. Circulation Res. 108 (9), 1133–1145. doi:10.1161/CIRCRESAHA.110.226936
Manolis A. S., Manolis T. A., Manolis A. A. (2023). Ketone bodies and cardiovascular disease: an alternate fuel source to the rescue. Int. J. Mol. Sci. 24 (4), 3534. doi:10.3390/ijms24043534
Marelli-Berg F. M., Aksentijevic D. (2019). Immunometabolic cross-talk in the inflamed heart. Cell Stress 3 (8), 240–266. doi:10.15698/cst2019.08.194
Marín-Sedeño E., de Morentin X. M., Pérez-Pomares J. M., Gómez-Cabrero D., Ruiz-Villalba A. (2021). Understanding the adult mammalian heart at single-cell RNA-seq resolution. Front. Cell Dev. Biol. 9, 645276. doi:10.3389/fcell.2021.645276
Martínez-Reyes I., Chandel N. S. (2020). Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 11 (1), 102. doi:10.1038/s41467-019-13668-3
Martinod K., Witsch T., Erpenbeck L., Savchenko A., Hayashi H., Cherpokova D., et al. (2017). Peptidylarginine deiminase 4 promotes age-related organ fibrosis. J. Exp. Med. 214 (2), 439–458. doi:10.1084/jem.20160530
Maruyama K., Imanaka-Yoshida K. (2022). The pathogenesis of cardiac fibrosis: a review of recent progress. Int. J. Mol. Sci. 23 (5), 2617. doi:10.3390/ijms23052617
Matsumoto T., Wada A., Tsutamoto T., Ohnishi M., Isono T., Kinoshita M. (2003). Chymase inhibition prevents cardiac fibrosis and improves diastolic dysfunction in the progression of heart failure. Circulation 107 (20), 2555–2558. doi:10.1161/01.CIR.0000074041.81728.79
McLarty J. L., Meléndez G. C., Brower G. L., Janicki J. S., Levick S. P. (2011). Tryptase/Protease-activated receptor 2 interactions induce selective mitogen-activated protein kinase signaling and collagen synthesis by cardiac fibroblasts. Hypertension 58 (2), 264–270. doi:10.1161/HYPERTENSIONAHA.111.169417
McMurray J. J., Adamopoulos S., Anker S. D., Auricchio A., Böhm M., Dickstein K., et al. (2012). ESC guidelines for the diagnosis and treatment of acute and chronic heart failure 2012: the task force for the diagnosis and treatment of acute and chronic heart failure 2012 of the European society of cardiology. Developed in collaboration with the heart failure association (HFA) of the ESC. Eur. Heart J. 33 (14), 1787–1847. doi:10.1093/eurheartj/ehs104
Meilhac S. M., Buckingham M. E. (2018). The deployment of cell lineages that form the mammalian heart. Nat. Rev. Cardiol. 15 (11), 705–724. doi:10.1038/s41569-018-0086-9
MeléNdez G. C., McLarty J. L., Levick S. P., Du Y., Janicki J. S., Brower G. L. (2010a). Interleukin 6 mediates myocardial fibrosis, concentric hypertrophy, and diastolic dysfunction in rats. Hypertension 56 (2), 225–231. doi:10.1161/HYPERTENSIONAHA.109.148635
Meléndez G. C., Voloshenyuk T. G., McLarty J. L., Levick S. P., Brower G. L. (2010b). Oxidative stress-mediated cardiac mast cell degranulation. Toxicol. Environ. Chem. 92 (7), 1293–1301. doi:10.1080/02772240903306409
Meng X.-M., Wang S., Huang X. R., Yang C., Xiao J., Zhang Y., et al. (2016). Inflammatory macrophages can transdifferentiate into myofibroblasts during renal fibrosis. Cell Death Dis. 7 (12), e2495. doi:10.1038/cddis.2016.402
Mezzaroma E., Toldo S., Farkas D., Seropian I. M., Van Tassell B. W., Salloum F. N., et al. (2011). The inflammasome promotes adverse cardiac remodeling following acute myocardial infarction in the mouse. Proc. Natl. Acad. Sci. 108 (49), 19725–19730. doi:10.1073/pnas.1108586108
Michalek R. D., Gerriets V. A., Jacobs S. R., Macintyre A. N., MacIver N. J., Mason E. F., et al. (2011). Cutting edge: distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 186 (6), 3299–3303. doi:10.4049/jimmunol.1003613
Mills E. L., Kelly B., Logan A., Costa A. S. H., Varma M., Bryant C. E., et al. (2016). Succinate dehydrogenase supports metabolic repurposing of mitochondria to drive inflammatory macrophages. Cell 167 (2), 457–470. doi:10.1016/j.cell.2016.08.064
Mills E. L., Ryan D. G., Prag H. A., Dikovskaya D., Menon D., Zaslona Z., et al. (2018). Itaconate is an anti-inflammatory metabolite that activates Nrf2 via alkylation of KEAP1. Nature 556 (7699), 113–117. doi:10.1038/nature25986
Miyake K., Ito J., Karasuyama H., Sato R., Saitoh S. I., Murakami Y. (2022). Nucleic acid sensing by toll-like receptors in the endosomal compartment. Front. Immunol. 13, 941931. doi:10.3389/fimmu.2022.941931
Mo F., Luo Y., Yan Y., Li J., Lai S., Wu W. (2021). Are activated B cells involved in the process of myocardial fibrosis after acute myocardial infarction? An in vivo experiment. BMC Cardiovasc. Disord. 21 (1), 5. doi:10.1186/s12872-020-01775-9
Molek P., Zmudzki P., Wlodarczyk A., Nessler J., Zalewski J. (2021). The shifted balance of arginine metabolites in acute myocardial infarction patients and its clinical relevance. Sci. Rep. 11 (1), 83. doi:10.1038/s41598-020-80230-3
Moon Y. S., Smas C. M., Lee K., Villena J. A., Kim K. H., Yun E. J., et al. (2002). Mice lacking paternally expressed Pref-1/Dlk1 display growth retardation and accelerated adiposity. Mol. Cell Biol. 22 (15), 5585–5592. doi:10.1128/mcb.22.15.5585-5592.2002
Mortensen S. B., Jensen C. H., Schneider M., Thomassen M., Kruse T. A., Laborda J., et al. (2012). Membrane-tethered delta-like 1 homolog (DLK1) restricts adipose tissue size by inhibiting preadipocyte proliferation. Diabetes 61 (11), 2814–2822. doi:10.2337/db12-0176
Moskalik A., Niderla-Bielińska J., Ratajska A. (2022). Multiple roles of cardiac macrophages in heart homeostasis and failure. Heart Fail. Rev. 27 (4), 1413–1430. doi:10.1007/s10741-021-10156-z
Mouton A. J., DeLeon-Pennell K. Y., Rivera Gonzalez O. J., Flynn E. R., Freeman T. C., Saucerman J. J., et al. (2018). Mapping macrophage polarization over the myocardial infarction time continuum. Basic Res. Cardiol. 113 (4), 26. doi:10.1007/s00395-018-0686-x
Mouton A. J., Hall J. E. (2020). Novel roles of immunometabolism and nonmyocyte metabolism in cardiac remodeling and injury. Am. J. Physiol. Regul. Integr. Comp. Physiol. 319 (4), R476–R484. doi:10.1152/ajpregu.00188.2020
Murray D. B., Gardner J. D., Brower G. L., Janicki J. S. (2004). Endothelin-1 mediates cardiac mast cell degranulation, matrix metalloproteinase activation, and myocardial remodeling in rats. Am. J. Physiology-Heart Circulatory Physiology 287 (5), H2295–H2299. doi:10.1152/ajpheart.00048.2004
Mylonas K. J., Jenkins S. J., Castellan R. F. P., Ruckerl D., McGregor K., Phythian-Adams A. T., et al. (2015). The adult murine heart has a sparse, phagocytically active macrophage population that expands through monocyte recruitment and adopts an ‘M2’phenotype in response to Th2 immunologic challenge. Immunobiology 220 (7), 924–933. doi:10.1016/j.imbio.2015.01.013
Nagai T., Honda S., Sugano Y., Matsuyama T. a., Ohta-Ogo K., Asaumi Y., et al. (2014). Decreased myocardial dendritic cells is associated with impaired reparative fibrosis and development of cardiac rupture after myocardial infarction in humans. J. Am. Heart Assoc. 3 (3), e000839. doi:10.1161/JAHA.114.000839
Nahrendorf M., Swirski F. K., Aikawa E., Stangenberg L., Wurdinger T., Figueiredo J. L., et al. (2007). The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 204 (12), 3037–3047. doi:10.1084/jem.20070885
Naito K., Anzai T., Sugano Y., Maekawa Y., Kohno T., Yoshikawa T., et al. (2008). Differential effects of GM-CSF and G-CSF on infiltration of dendritic cells during early left ventricular remodeling after myocardial infarction. J. Immunol. 181 (8), 5691–5701. doi:10.4049/jimmunol.181.8.5691
Nakaya M., Xiao Y., Zhou X., Chang J. H., Chang M., Cheng X., et al. (2014). Inflammatory T cell responses rely on amino acid transporter ASCT2 facilitation of glutamine uptake and mTORC1 kinase activation. Immunity 40 (5), 692–705. doi:10.1016/j.immuni.2014.04.007
Nascimben L., Ingwall J. S., Lorell B. H., Pinz I., Schultz V., Tornheim K., et al. (2004). Mechanisms for increased glycolysis in the hypertrophied rat heart. Hypertension 44 (5), 662–667. doi:10.1161/01.HYP.0000144292.69599.0c
Nazari M., Ni N. C., Lüdke A., Li S. H., Guo J., Weisel R. D., et al. (2016). Mast cells promote proliferation and migration and inhibit differentiation of mesenchymal stem cells through PDGF. J. Mol. Cell Cardiol. 94, 32–42. doi:10.1016/j.yjmcc.2016.03.007
Nevers T., Salvador A. M., Grodecki-Pena A., Knapp A., Velázquez F., Aronovitz M., et al. (2015). Left ventricular T-cell recruitment contributes to the pathogenesis of heart failure. Circ. Heart Fail 8 (4), 776–787. doi:10.1161/CIRCHEARTFAILURE.115.002225
Nevers T., Salvador A. M., Velazquez F., Ngwenyama N., Carrillo-Salinas F. J., Aronovitz M., et al. (2017). Th1 effector T cells selectively orchestrate cardiac fibrosis in nonischemic heart failure. J. Exp. Med. 214 (11), 3311–3329. doi:10.1084/jem.20161791
Newgard C. B. (2012). Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab. 15 (5), 606–614. doi:10.1016/j.cmet.2012.01.024
Ng S. M., Neubauer S., Rider O. J. (2023). Myocardial metabolism in heart failure. Curr. Heart Fail. Rep. 20 (1), 63–75. doi:10.1007/s11897-023-00589-y
Ngkelo A., Richart A., Kirk J. A., Bonnin P., Vilar J., Lemitre M., et al. (2016). Mast cells regulate myofilament calcium sensitization and heart function after myocardial infarction. J. Exp. Med. 213 (7), 1353–1374. doi:10.1084/jem.20160081
Nguyen D. T., Ding C., Wilson E., Marcus G. M., Olgin J. E. (2010). Pirfenidone mitigates left ventricular fibrosis and dysfunction after myocardial infarction and reduces arrhythmias. Heart rhythm. 7 (10), 1438–1445. doi:10.1016/j.hrthm.2010.04.030
Nicolás-Ávila J. A., Lechuga-Vieco A. V., Esteban-Martínez L., Sánchez-Díaz M., Díaz-García E., Santiago D. J., et al. (2020). A network of macrophages supports mitochondrial homeostasis in the heart. Cell 183 (1), 94–109. doi:10.1016/j.cell.2020.08.031
Nomura M., Liu J., Rovira I. I., Gonzalez-Hurtado E., Lee J., Wolfgang M. J., et al. (2016). Fatty acid oxidation in macrophage polarization. Nat. Immunol. 17 (3), 216–217. doi:10.1038/ni.3366
Okyere A. D., Tilley D. G. (2020). Leukocyte-dependent regulation of cardiac fibrosis. Front. Physiol. 11, 301. doi:10.3389/fphys.2020.00301
O'Neill L. A. J., Hardie D. G. (2013). Metabolism of inflammation limited by AMPK and pseudo-starvation. Nature 493 (7432), 346–355. doi:10.1038/nature11862
O'Neill L. A. J., Kishton R. J., Rathmell J. (2016). A guide to immunometabolism for immunologists. Nat. Rev. Immunol. 16 (9), 553–565. doi:10.1038/nri.2016.70
Ong S., Ligons D. L., Barin J. G., Wu L., Talor M. V., Diny N., et al. (2015). Natural killer cells limit cardiac inflammation and fibrosis by halting eosinophil infiltration. Am. J. Pathol. 185 (3), 847–861. doi:10.1016/j.ajpath.2014.11.023
Ong S., Rose N. R., Čiháková D. (2017). Natural killer cells in inflammatory heart disease. Clin. Immunol. 175, 26–33. doi:10.1016/j.clim.2016.11.010
Oyamada S., Bianchi C., Takai S., Chu L. M., Sellke F. W. (2011). Chymase inhibition reduces infarction and matrix metalloproteinase-9 activation and attenuates inflammation and fibrosis after acute myocardial ischemia/reperfusion. J. Pharmacol. Exp. Ther. 339 (1), 143–151. doi:10.1124/jpet.111.179697
Papazafiropoulou A., Georgopoulos M., Katsilambros N. (2021). Ketone bodies and the heart. Archives Med. Sci. – Atheroscler. Dis. 6 (1), 209–214. doi:10.5114/amsad.2021.112475
Parichatikanond W., Luangmonkong T., Mangmool S., Kurose H. (2020). Therapeutic targets for the treatment of cardiac fibrosis and cancer: focusing on TGF-β signaling. Front. Cardiovasc Med. 7, 34. doi:10.3389/fcvm.2020.00034
Park S., Nguyen N. B., Pezhouman A., Ardehali R. (2019). Cardiac fibrosis: potential therapeutic targets. Transl. Res. 209, 121–137. doi:10.1016/j.trsl.2019.03.001
Passarella S., Schurr A. (2018). l-Lactate transport and metabolism in mitochondria of hep G2 cells-the cori cycle revisited. Front. Oncol. 8, 120. doi:10.3389/fonc.2018.00120
Patella V., Marinò I., Arbustini E., Lamparter-Schummert B., Verga L., Adt M., et al. (1998). Stem cell factor in mast cells and increased mast cell density in idiopathic and ischemic cardiomyopathy. Circulation 97 (10), 971–978. doi:10.1161/01.cir.97.10.971
Patil N. K., Bohannon J. K., Hernandez A., Patil T. K., Sherwood E. R. (2019). Regulation of leukocyte function by citric acid cycle intermediates. J. Leukoc. Biol. 106 (1), 105–117. doi:10.1002/JLB.3MIR1118-415R
Pérez S., Rius-Pérez S. (2022). Macrophage polarization and reprogramming in acute inflammation: a redox perspective. Antioxidants 11 (7), 1394. doi:10.3390/antiox11071394
Pesce J. T., Ramalingam T. R., Mentink-Kane M. M., Wilson M. S., El Kasmi K. C., Smith A. M., et al. (2009). Arginase-1-expressing macrophages suppress Th2 cytokine-driven inflammation and fibrosis. PLoS Pathog. 5 (4), e1000371. doi:10.1371/journal.ppat.1000371
Petrausch U., Schuberth P. C., Hagedorn C., Soltermann A., Tomaszek S., Stahel R., et al. (2012). Re-directed T cells for the treatment of fibroblast activation protein (FAP)-positive malignant pleural mesothelioma (FAPME-1). BMC Cancer 12, 615. doi:10.1186/1471-2407-12-615
Pinto A. R., Ilinykh A., Ivey M. J., Kuwabara J. T., D'Antoni M. L., Debuque R., et al. (2016). Revisiting cardiac cellular composition. Circ. Res. 118 (3), 400–409. doi:10.1161/CIRCRESAHA.115.307778
Pinto A. R., Paolicelli R., Salimova E., Gospocic J., Slonimsky E., Bilbao-Cortes D., et al. (2012). An abundant tissue macrophage population in the adult murine heart with a distinct alternatively-activated macrophage profile. PLoS ONE 7 (5), e36814. doi:10.1371/journal.pone.0036814
Pluijmert N. J., Atsma D. E., Quax P. H. A. (2021). Post-ischemic myocardial inflammatory response: a complex and dynamic process susceptible to immunomodulatory therapies. Front. Cardiovasc. Med. 8, 647785. doi:10.3389/fcvm.2021.647785
Pogontke C., Guadix J. A., Ruiz-Villalba A., Pérez-Pomares J. M. (2019). Development of the myocardial interstitium. Anatomical Rec. 302 (1), 58–68. doi:10.1002/ar.23915
Posokhova E. N., Khoshchenko O. M., Chasovskikh M. I., Pivovarova E. N., Dushkin M. I. (2008). Lipid synthesis in macrophages during inflammation in vivo: effect of agonists of peroxisome proliferator activated receptors alpha and gamma and of retinoid X receptors. Biochem. Mosc. 73 (3), 296–304. doi:10.1134/s0006297908030097
Puhl S.-L., Steffens S. (2019). Neutrophils in post-myocardial infarction inflammation: damage vs. Resolution? Front. Cardiovasc. Med. 6, 25. doi:10.3389/fcvm.2019.00025
Rakusan K., Sarkar K., Turek Z., Wicker P. (1990). Mast cells in the rat heart during normal growth and in cardiac hypertrophy. Circulation Res. 66 (2), 511–516. doi:10.1161/01.res.66.2.511
Ramos G. C., van den Berg A., Nunes-Silva V., Weirather J., Peters L., Burkard M., et al. (2017). Myocardial aging as a T-cell–mediated phenomenon. Proc. Natl. Acad. Sci. 114 (12), E2420–E2429. doi:10.1073/pnas.1621047114
Razeghi P., Young M. E., Alcorn J. L., Moravec C. S., Frazier O. H., Taegtmeyer H. (2001). Metabolic gene expression in fetal and failing human heart. Circulation 104 (24), 2923–2931. doi:10.1161/hc4901.100526
Raziyeva K., Kim Y., Zharkinbekov Z., Temirkhanova K., Saparov A. (2022). Novel therapies for the treatment of cardiac fibrosis following myocardial infarction. Biomedicines 10 (9), 2178. doi:10.3390/biomedicines10092178
Remondino A., Rosenblatt-Velin N., Montessuit C., Tardy I., Papageorgiou I., Dorsaz P. A., et al. (2000). Altered expression of proteins of metabolic regulation during remodeling of the left ventricle after myocardial infarction. J. Mol. Cell Cardiol. 32 (11), 2025–2034. doi:10.1006/jmcc.2000.1234
Ren W., Xia Y., Chen S., Wu G., Bazer F. W., Zhou B., et al. (2019). Glutamine metabolism in macrophages: a novel target for obesity/type 2 diabetes. Adv. Nutr. 10 (2), 321–330. doi:10.1093/advances/nmy084
Revelo X. S., Parthiban P., Chen C., Barrow F., Fredrickson G., Wang H., et al. (2021). Cardiac resident macrophages prevent fibrosis and stimulate angiogenesis. Circulation Res. 129 (12), 1086–1101. doi:10.1161/CIRCRESAHA.121.319737
Ritterhoff J., Tian R. (2017). Metabolism in cardiomyopathy: every substrate matters. Cardiovasc. Res. 113 (4), 411–421. doi:10.1093/cvr/cvx017
Rodriguez P., Sassi Y., Troncone L., Benard L., Ishikawa K., Gordon R. E., et al. (2019). Deletion of delta-like 1 homologue accelerates fibroblast-myofibroblast differentiation and induces myocardial fibrosis. Eur. Heart J. 40 (12), 967–978. doi:10.1093/eurheartj/ehy188
Rodríguez-Prados J.-C., Través P. G., Cuenca J., Rico D., Aragonés J., Martín-Sanz P., et al. (2010). Substrate fate in activated macrophages: a comparison between innate, classic, and alternative activation. J. Immunol. 185 (1), 605–614. doi:10.4049/jimmunol.0901698
Rosano G. M., Vitale C. (2018). Metabolic modulation of cardiac metabolism in heart failure. Card. Fail Rev. 4 (2), 99–103. doi:10.15420/cfr.2018.18.2
Rosano G. M. C., Vitale C., Spoletini I. (2015). Metabolic approach to heart failure: the role of metabolic modulators. Egypt. Heart J. 67 (3), 177–181. doi:10.1016/j.ehj.2015.03.004
Ruoss S. J., Hartmann T., Caughey G. (1991). Mast cell tryptase is a mitogen for cultured fibroblasts. J. Clin. investigation 88 (2), 493–499. doi:10.1172/JCI115330
Rurik J. G., Aghajanian H., Epstein J. A. (2021). Immune cells and immunotherapy for cardiac injury and repair. Circ. Res. 128 (11), 1766–1779. doi:10.1161/CIRCRESAHA.121.318005
Rurik J. G., Tombácz I., Yadegari A., Méndez Fernández P. O., Shewale S. V., Li L., et al. (2022). CAR T cells produced in vivo to treat cardiac injury. Science 375 (6576), 91–96. doi:10.1126/science.abm0594
Ryan D. G., Murphy M. P., Frezza C., Prag H. A., Chouchani E. T., O'Neill L. A., et al. (2019). Coupling Krebs cycle metabolites to signalling in immunity and cancer. Nat. Metab. 1, 16–33. doi:10.1038/s42255-018-0014-7
Sabogal-Guáqueta A. M., Marmolejo-Garza A., Trombetta-Lima M., Oun A., Hunneman J., Chen T., et al. (2023). Species-specific metabolic reprogramming in human and mouse microglia during inflammatory pathway induction. Nat. Commun. 14 (1), 6454. doi:10.1038/s41467-023-42096-7
Sag D., Carling D., Stout R. D., Suttles J. (2008). Adenosine 5'-monophosphate-activated protein kinase promotes macrophage polarization to an anti-inflammatory functional phenotype. J. Immunol. 181 (12), 8633–8641. doi:10.4049/jimmunol.181.12.8633
Sager H. B., Hulsmans M., Lavine K. J., Moreira M. B., Heidt T., Courties G., et al. (2016). Proliferation and recruitment contribute to myocardial macrophage expansion in chronic heart failure. Circulation Res. 119 (7), 853–864. doi:10.1161/CIRCRESAHA.116.309001
Santos E. S., de Aragão-França L. S., Meira C. S., Cerqueira J. V., Vasconcelos J. F., Nonaka C. K. V., et al. (2020). Tolerogenic dendritic cells reduce cardiac inflammation and fibrosis in chronic chagas disease. Front. Immunol. 11, 488. doi:10.3389/fimmu.2020.00488
Santos-Zas I., Lemarié J., Zlatanova I., Cachanado M., Seghezzi J. C., Benamer H., et al. (2021). Cytotoxic CD8+ T cells promote granzyme B-dependent adverse post-ischemic cardiac remodeling. Nat. Commun. 12 (1), 1483. doi:10.1038/s41467-021-21737-9
Sawaki D., Czibik G., Pini M., Ternacle J., Suffee N., Mercedes R., et al. (2018). Visceral adipose tissue drives cardiac aging through modulation of fibroblast senescence by osteopontin production. Circulation 138 (8), 809–822. doi:10.1161/CIRCULATIONAHA.117.031358
Saxena A., Dobaczewski M., Rai V., Haque Z., Chen W., et al. (2014). Regulatory T cells are recruited in the infarcted mouse myocardium and may modulate fibroblast phenotype and function. Am. J. Physiol. Heart Circ. Physiol. 307 (8), H1233–H1242. doi:10.1152/ajpheart.00328.2014
Schaper J., Meiser E., Stämmler G. (1985). Ultrastructural morphometric analysis of myocardium from dogs, rats, hamsters, mice, and from human hearts. Circulation Res. 56 (3), 377–391. doi:10.1161/01.res.56.3.377
Schiechl G., Hermann F. J., Rodriguez Gomez M., Kutzi S., Schmidbauer K., Talke Y., et al. (2016). Basophils trigger fibroblast activation in cardiac allograft fibrosis development. Am. J. Transplant. 16 (9), 2574–2588. doi:10.1111/ajt.13764
Selvarajah B., Azuelos I., Platé M., Guillotin D., Forty E. J., Contento G., et al. (2019). mTORC1 amplifies the ATF4-dependent de novo serine-glycine pathway to supply glycine during TGF-β1-induced collagen biosynthesis. Sci. Signal. 12 (582), eaav3048. doi:10.1126/scisignal.aav3048
Shao Z., Nazari M., Guo L., Li S. H., Sun J., Liu S. M., et al. (2015). The cardiac repair benefits of inflammation do not persist: evidence from mast cell implantation. J. Cell. Mol. Med. 19 (12), 2751–2762. doi:10.1111/jcmm.12703
Shen P., Fillatreau S. (2015). Antibody-independent functions of B cells: a focus on cytokines. Nat. Rev. Immunol. 15 (7), 441–451. doi:10.1038/nri3857
Shi L. Z., Wang R., Huang G., Vogel P., Neale G., Green D. R., et al. (2011). HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J. Exp. Med. 208 (7), 1367–1376. doi:10.1084/jem.20110278
Shi X., Qiu H. (2022). New insights into energy substrate utilization and metabolic remodeling in cardiac physiological adaption. Front. Physiology 13, 831829. doi:10.3389/fphys.2022.831829
Shiota N., Rysä J., Kovanen P. T., Ruskoaho H., Kokkonen J. O., Lindstedt K. A. (2003). A role for cardiac mast cells in the pathogenesis of hypertensive heart disease. J. Hypertens. 21 (10), 1935–1944. doi:10.1097/00004872-200310000-00022
Shyer J. A., Flavell R. A., Bailis W. (2020). Metabolic signaling in T cells. Cell Res. 30 (8), 649–659. doi:10.1038/s41422-020-0379-5
Sicklinger F., Meyer I. S., Li X., Radtke D., Dicks S., Kornadt M. P., et al. (2021). Basophils balance healing after myocardial infarction via IL-4/IL-13. J. Clin. Investigation 131 (13), e136778. doi:10.1172/JCI136778
Silvis M. J. M., Kaffka Genaamd Dengler S. E., Odille C. A., Mishra M., van der Kaaij N. P., Doevendans P. A., et al. (2020). Damage-associated molecular patterns in myocardial infarction and heart transplantation: the road to translational success. Front. Immunol. 11, 599511. doi:10.3389/fimmu.2020.599511
Simões F. C., Cahill T. J., Kenyon A., Gavriouchkina D., Vieira J. M., Sun X., et al. (2020). Macrophages directly contribute collagen to scar formation during zebrafish heart regeneration and mouse heart repair. Nat. Commun. 11 (1), 600. doi:10.1038/s41467-019-14263-2
Simões F. C., Riley P. R. (2022). Immune cells in cardiac repair and regeneration. Development 149 (8), dev199906. doi:10.1242/dev.199906
Sinclair L. V., Rolf J., Emslie E., Shi Y. B., Taylor P. M., Cantrell D. A. (2013). Control of amino-acid transport by antigen receptors coordinates the metabolic reprogramming essential for T cell differentiation. Nat. Immunol. 14 (5), 500–508. doi:10.1038/ni.2556
Singh V. P., Baker K. M., Kumar R. (2008). Activation of the intracellular renin-angiotensin system in cardiac fibroblasts by high glucose: role in extracellular matrix production. Am. J. Physiol. Heart Circ. Physiol. 294 (4), H1675–H1684. doi:10.1152/ajpheart.91493.2007
Sinha M., Sen C. K., Singh K., Das A., Ghatak S., Rhea B., et al. (2018). Direct conversion of injury-site myeloid cells to fibroblast-like cells of granulation tissue. Nat. Commun. 9 (1), 936. doi:10.1038/s41467-018-03208-w
Somasundaram P., Ren G., Nagar H., Kraemer D., Mendoza L., Michael L. H., et al. (2005). Mast cell tryptase may modulate endothelial cell phenotype in healing myocardial infarcts. J. Pathol. 205 (1), 102–111. doi:10.1002/path.1690
Soto-Heredero G., Gómez de Las Heras M. M., Gabandé-Rodríguez E., Oller J., Mittelbrunn M. (2020). Glycolysis – a key player in the inflammatory response. FEBS J. 287 (16), 3350–3369. doi:10.1111/febs.15327
Srivastava S. P., Li J., Kitada M., Fujita H., Yamada Y., Goodwin J. E., et al. (2018). SIRT3 deficiency leads to induction of abnormal glycolysis in diabetic kidney with fibrosis. Cell Death Dis. 9 (10), 997. doi:10.1038/s41419-018-1057-0
Stanley W. C., Hall J. L., Stone C. K., Hacker T. A. (1992). Acute myocardial ischemia causes a transmural gradient in glucose extraction but not glucose uptake. Am. J. Physiology-Heart Circulatory Physiology 262 (1), H91–H96. doi:10.1152/ajpheart.1992.262.1.H91
Stanley W. C., Meadows S. R., Kivilo K. M., Roth B. A., Lopaschuk G. D. (2003). beta-Hydroxybutyrate inhibits myocardial fatty acid oxidation in vivo independent of changes in malonyl-CoA content. Am. J. Physiology-Heart Circulatory Physiology 285 (4), H1626–H1631. doi:10.1152/ajpheart.00332.2003
Stanley W. C., Recchia F. A., Lopaschuk G. D. (2005). Myocardial substrate metabolism in the normal and failing heart. Physiol. Rev. 85 (3), 1093–1129. doi:10.1152/physrev.00006.2004
Steffens S., Nahrendorf M., Madonna R. (2022). Immune cells in cardiac homeostasis and disease: emerging insights from novel technologies. Eur. Heart J. 43 (16), 1533–1541. doi:10.1093/eurheartj/ehab842
Stewart J. A., Wei C. C., Brower G. L., Rynders P. E., Hankes G. H., Dillon A. R., et al. (2003). Cardiac mast cell- and chymase-mediated matrix metalloproteinase activity and left ventricular remodeling in mitral regurgitation in the dog. J. Mol. Cell Cardiol. 35 (3), 311–319. doi:10.1016/s0022-2828(03)00013-0
Sun K., Li Y.-y., Jin J. (2021). A double-edged sword of immuno-microenvironment in cardiac homeostasis and injury repair. Signal Transduct. Target. Ther. 6 (1), 79. doi:10.1038/s41392-020-00455-6
Sun L., Yang X., Yuan Z., Wang H. (2020a). Metabolic reprogramming in immune response and tissue inflammation. Arteriosclerosis, Thrombosis, Vasc. Biol. 40 (9), 1990–2001. doi:10.1161/ATVBAHA.120.314037
Sun L., Zhou F., Shao Y., Lv Z., Li C. (2020b). Sedoheptulose kinase bridges the pentose phosphate pathway and immune responses in pathogen-challenged sea cucumber Apostichopus japonicus. Dev. Comp. Immunol. 109, 103694. doi:10.1016/j.dci.2020.103694
Swirski F. K., Nahrendorf M. (2018). Cardioimmunology: the immune system in cardiac homeostasis and disease. Nat. Rev. Immunol. 18 (12), 733–744. doi:10.1038/s41577-018-0065-8
Tamaki S., Mano T., Sakata Y., Ohtani T., Takeda Y., Kamimura D., et al. (2013). Interleukin-16 promotes cardiac fibrosis and myocardial stiffening in heart failure with preserved ejection fraction. PLoS ONE 8 (7), e68893. doi:10.1371/journal.pone.0068893
Tan Y., Duan X., Wang B., Liu X., Zhan Z. (2023). Murine neonatal cardiac B cells promote cardiomyocyte proliferation and heart regeneration. npj Regen. Med. 8 (1), 7. doi:10.1038/s41536-023-00282-7
Tang T. T., Yuan J., Zhu Z. F., Zhang W. C., Xiao H., Xia N., et al. (2012). Regulatory hīs ameliorate cardiac remodeling after myocardial infarction. Basic Res. Cardiol. 107 (1), 232. doi:10.1007/s00395-011-0232-6
Tannahill G. M., Curtis A. M., Adamik J., Palsson-McDermott E. M., McGettrick A. F., Goel G., et al. (2013). Succinate is an inflammatory signal that induces IL-1β through HIF-1α. Nature 496 (7444), 238–242. doi:10.1038/nature11986
Taylor A., Verhagen J., Blaser K., Akdis M., Akdis C. A. (2006). Mechanisms of immune suppression by interleukin-10 and transforming growth factor-beta: the role of T regulatory cells. Immunology 117 (4), 433–442. doi:10.1111/j.1365-2567.2006.02321.x
Taylor C. T., Scholz C. C. (2022). The effect of HIF on metabolism and immunity. Nat. Rev. Nephrol. 18 (9), 573–587. doi:10.1038/s41581-022-00587-8
Tian G., Ren T. (2023). Mechanical stress regulates the mechanotransduction and metabolism of cardiac fibroblasts in fibrotic cardiac diseases. Eur. J. Cell Biol. 102 (2), 151288. doi:10.1016/j.ejcb.2023.151288
Tian H., Zhao X., Zhang Y., Xia Z. (2023). Abnormalities of glucose and lipid metabolism in myocardial ischemia-reperfusion injury. Biomed. Pharmacother. 163, 114827. doi:10.1016/j.biopha.2023.114827
Tominaga H., Katoh H., Odagiri K., Takeuchi Y., Kawashima H., Saotome M., et al. (2008). Different effects of palmitoyl-L-carnitine and palmitoyl-CoA on mitochondrial function in rat ventricular myocytes. Am. J. Physiol. Heart Circ. Physiol. 295 (1), H105–H112. doi:10.1152/ajpheart.01307.2007
Tosello-Trampont A., Surette F. A., Ewald S. E., Hahn Y. S. (2017). Immunoregulatory role of NK cells in tissue inflammation and regeneration. Front. Immunol. 8, 301. doi:10.3389/fimmu.2017.00301
Tran D. H., Wang Z. V. (2019). Glucose metabolism in cardiac hypertrophy and heart failure. J. Am. Heart Assoc. 8 (12), e012673. doi:10.1161/JAHA.119.012673
Travers J. G., Kamal F. A., Robbins J., Yutzey K. E., Blaxall B. C. (2016). Cardiac fibrosis: the fibroblast awakens. Circulation Res. 118 (6), 1021–1040. doi:10.1161/CIRCRESAHA.115.306565
Tucker N. R., Chaffin M., Fleming S. J., Hall A. W., Parsons V. A., Bedi K. C., et al. (2020). Transcriptional and cellular diversity of the human heart. Circulation 142 (5), 466–482. doi:10.1161/CIRCULATIONAHA.119.045401
Tymoczko J. L., Berg J. M., Stryer L. (2002). Biochemistry: W H freeman, carbon atoms of degraded amino acids emerge as major metabolic intermediates, ed. T. ed. New York: W. H. Freeman.
Ung C. Y., Onoufriadis A., Parsons M., McGrath J. A., Shaw T. J. (2021). Metabolic perturbations in fibrosis disease. Int. J. Biochem. Cell Biol. 139, 106073. doi:10.1016/j.biocel.2021.106073
Urata H., Kinoshita A., Misono K. S., Bumpus F. M., Husain A. (1990). Identification of a highly specific chymase as the major angiotensin II-forming enzyme in the human heart. J. Biol. Chem. 265 (36), 22348–22357. doi:10.1016/s0021-9258(18)45712-2
Van Den Borne S. W. M., Diez J., Blankesteijn W. M., Verjans J., Hofstra L., Narula J. (2010). Myocardial remodeling after infarction: the role of myofibroblasts. Nat. Rev. Cardiol. 7 (1), 30–37. doi:10.1038/nrcardio.2009.199
van der Windt G. J., Everts B., Chang C. H., Curtis J. D., Freitas T. C., Amiel E., et al. (2012). Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 36 (1), 68–78. doi:10.1016/j.immuni.2011.12.007
Vasilyev N., Williams T., Brennan M. L., Unzek S., Zhou X., Heinecke J. W., et al. (2005). Myeloperoxidase-generated oxidants modulate left ventricular remodeling but not infarct size after myocardial infarction. Circulation 112 (18), 2812–2820. doi:10.1161/CIRCULATIONAHA.105.542340
Vats D., Mukundan L., Odegaard J. I., Zhang L., Smith K. L., Morel C. R., et al. (2006). Oxidative metabolism and PGC-1beta attenuate macrophage-mediated inflammation. Cell Metab. 4 (1), 13–24. doi:10.1016/j.cmet.2006.05.011
Vinten-Johansen J. (2004). Involvement of neutrophils in the pathogenesis of lethal myocardial reperfusion injury. Cardiovasc. Res. 61 (3), 481–497. doi:10.1016/j.cardiores.2003.10.011
Vivier E., Tomasello E., Baratin M., Walzer T., Ugolini S. (2008). Functions of natural killer cells. Nat. Immunol. 9 (5), 503–510. doi:10.1038/ni1582
Wang J., Duan Y., Sluijter J. P., Xiao J. (2019). Lymphocytic subsets play distinct roles in heart diseases. Theranostics 9 (14), 4030–4046. doi:10.7150/thno.33112
Wang N., Wang W., Wang X., Mang G., Chen J., Yan X., et al. (2022). Histone lactylation boosts reparative gene activation post-myocardial infarction. Circ. Res. 131 (11), 893–908. doi:10.1161/CIRCRESAHA.122.320488
Wang Q., Donthi R. V., Wang J., Lange A. J., Watson L. J., Jones S. P., et al. (2008). Cardiac phosphatase-deficient 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase increases glycolysis, hypertrophy, and myocyte resistance to hypoxia. Am. J. Physiol. Heart Circ. Physiol. 294 (6), H2889–H2897. doi:10.1152/ajpheart.91501.2007
Wang X., Zhou H., Liu Q., Cheng P., Zhao T., Yang T., et al. (2023). Targeting regulatory T cells for cardiovascular diseases. Front. Immunol. 14. doi:10.3389/fimmu.2023.1126761
Wei C. C., Lucchesi P. A., Tallaj J., Bradley W. E., Powell P. C., Dell'Italia L. J. (2003). Cardiac interstitial bradykinin and mast cells modulate pattern of LV remodeling in volume overload in rats. Am. J. Physiol. Heart Circ. Physiol. 285 (2), H784–H792. doi:10.1152/ajpheart.00793.2001
Wei K.-H., Chowdhury K., Lim K. L., Liu K. T., Ko T. M., et al. (2023). Comparative single-cell profiling reveals distinct cardiac resident macrophages essential for zebrafish heart regeneration. eLife 12, e84679. doi:10.7554/eLife.84679
Weichhart T., Hengstschläger M., Linke M. (2015). Regulation of innate immune cell function by mTOR. Nat. Rev. Immunol. 15 (10), 599–614. doi:10.1038/nri3901
Weirather J., Hofmann U. D. W., Beyersdorf N., Ramos G. C., Vogel B., Frey A., et al. (2014). Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ. Res. 115 (1), 55–67. doi:10.1161/CIRCRESAHA.115.303895
Wentz A. E., d'Avignon D. A., Weber M. L., Cotter D. G., Doherty J. M., Kerns R., et al. (2010). Adaptation of myocardial substrate metabolism to a ketogenic nutrient environment. J. Biol. Chem. 285 (32), 24447–24456. doi:10.1074/jbc.M110.100651
Wenzl F. A., Ambrosini S., Mohammed S. A., Kraler S., Lüscher T. F., Costantino S., et al. (2021). Inflammation in metabolic cardiomyopathy. Front. Cardiovasc. Med. 8, 742178. doi:10.3389/fcvm.2021.742178
Widiapradja A., Manteufel E. J., Dehlin H. M., Pena J., Goldspink P. H., Sharma A., et al. (2019). Regulation of cardiac mast cell maturation and function by the neurokinin-1 receptor in the fibrotic heart. Sci. Rep. 9 (1), 11004. doi:10.1038/s41598-019-47369-0
Williams N. C., O'Neill L. A. J. (2018). A role for the krebs cycle intermediate citrate in metabolic reprogramming in innate immunity and inflammation. Front. Immunol. 9, 141. doi:10.3389/fimmu.2018.00141
Wischmeyer P. E., Jayakar D., Williams U., Singleton K. D., Riehm J., Bacha E. A., et al. (2003). Single dose of glutamine enhances myocardial tissue metabolism, glutathione content, and improves myocardial function after ischemia-reperfusion injury. JPEN J. Parenter. Enter. Nutr. 27 (6), 396–403. doi:10.1177/0148607103027006396
Woods P. S., Kimmig L. M., Sun K. A., Meliton A. Y., Shamaa O. R., Tian Y., et al. (2022). HIF-1α induces glycolytic reprograming in tissue-resident alveolar macrophages to promote cell survival during acute lung injury. eLife 11, e77457. doi:10.7554/eLife.77457
Wu J., Sun P., Chen Q., Sun Y., Shi M., Mang G., et al. (2019). Metabolic reprogramming orchestrates CD4(+) T-cell immunological status and restores cardiac dysfunction in autoimmune induced-dilated cardiomyopathy mice. J. Mol. Cell Cardiol. 135, 134–148. doi:10.1016/j.yjmcc.2019.08.002
Wu J.-J., Sun Z. L., Liu S. Y., Chen Z. H., Yuan Z. D., Zou M. L., et al. (2022). The ASIC3-M-CSF-M2 macrophage-positive feedback loop modulates fibroblast-to-myofibroblast differentiation in skin fibrosis pathogenesis. Cell Death Dis. 13 (6), 527. doi:10.1038/s41419-022-04981-9
Wu L., Ong S., Talor M. V., Barin J. G., Baldeviano G. C., Kass D. A., et al. (2014). Cardiac fibroblasts mediate IL-17A–driven inflammatory dilated cardiomyopathy. J. Exp. Med. 211 (7), 1449–1464. doi:10.1084/jem.20132126
Wu L., Yan Z., Jiang Y., Chen Y., Du J., Guo L., et al. (2023). Metabolic regulation of dendritic cell activation and immune function during inflammation. Front. Immunol. 14. doi:10.3389/fimmu.2023.1140749
Wynn T. A., Ramalingam T. R. (2012). Mechanisms of fibrosis: therapeutic translation for fibrotic disease. Nat. Med. 18 (7), 1028–1040. doi:10.1038/nm.2807
Xie N., Tan Z., Banerjee S., Cui H., Ge J., Liu R. M., et al. (2015). Glycolytic reprogramming in myofibroblast differentiation and lung fibrosis. Am. J. Respir. Crit. Care Med. 192 (12), 1462–1474. doi:10.1164/rccm.201504-0780OC
Xin Y., Wu W., Qu J., Wang X., Lei S., Yuan L., et al. (2019). Inhibition of mitofusin-2 promotes cardiac fibroblast activation via the PERK/ATF4 pathway and reactive oxygen species. Oxidative Med. Cell. Longev. 2019, 3649808. doi:10.1155/2019/3649808
Xiong X., Li J., Zhang S., Jia X., Xiao C. (2022). Involvement of polyamines from cardiac mast cells in myocardial remodeling induced by pressure overload through mitochondrial permeability transition pore opening. Front. Cardiovasc. Med. 9, 850688. doi:10.3389/fcvm.2022.850688
Yan X., Anzai A., Katsumata Y., Matsuhashi T., Ito K., Endo J., et al. (2013). Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J. Mol. Cell. Cardiol. 62, 24–35. doi:10.1016/j.yjmcc.2013.04.023
Yoganathan T., Perez-Liva M., Balvay D., Le Gall M., Lallemand A., Certain A., et al. (2023). Author Correction: acute stress induces long-term metabolic, functional, and structural remodeling of the heart. Nat. Commun. 14 (1), 4143. doi:10.1038/s41467-023-39910-7
Yoshikawa S., Nagao M., Toh R., Shinohara M., Iino T., Irino Y., et al. (2022). Inhibition of glutaminase 1-mediated glutaminolysis improves pathological cardiac remodeling. Am. J. Physiol. Heart Circ. Physiol. 322 (5), H749–H761. doi:10.1152/ajpheart.00692.2021
Yoshimura T., Galligan C., Takahashi M., Chen K., Liu M., Tessarollo L., et al. (2014). Non-myeloid cells are major contributors to innate immune responses via production of monocyte chemoattractant protein-1/CCL2. Front. Immunol. 4, 482. doi:10.3389/fimmu.2013.00482
Young M. E., Laws F. A., Goodwin G. W., Taegtmeyer H. (2001). Reactivation of peroxisome proliferator-activated receptor alpha is associated with contractile dysfunction in hypertrophied rat heart. J. Biol. Chem. 276 (48), 44390–44395. doi:10.1074/jbc.M103826200
Yu M., Wen S., Wang M., Liang W., Li H. H., Long Q., et al. (2013). TNF-α-Secreting B cells contribute to myocardial fibrosis in dilated cardiomyopathy. J. Clin. Immunol. 33 (5), 1002–1008. doi:10.1007/s10875-013-9889-y
Yurista S. R., Chen S., Welsh A., Tang W. H. W., Nguyen C. T. (2022). Targeting myocardial substrate metabolism in the failing heart: ready for prime time? Curr. Heart Fail. Rep. 19 (4), 180–190. doi:10.1007/s11897-022-00554-1
Zaidi Y., Aguilar E. G., Troncoso M., Ilatovskaya D. V., DeLeon-Pennell K. Y. (2021). Immune regulation of cardiac fibrosis post myocardial infarction. Cell. Signal. 77, 109837. doi:10.1016/j.cellsig.2020.109837
Zeisberg E. M., Tarnavski O., Zeisberg M., Dorfman A. L., McMullen J. R., Gustafsson E., et al. (2007). Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 13 (8), 952–961. doi:10.1038/nm1613
Zelcer N., Tontonoz P. (2006). Liver X receptors as integrators of metabolic and inflammatory signaling. J. Clin. Investigation 116 (3), 607–614. doi:10.1172/JCI27883
Zhang J., Yu Z. X., Fujita S., Yamaguchi M. L., Ferrans V. J. (1993). Interstitial dendritic cells of the rat heart. Quantitative and ultrastructural changes in experimental myocardial infarction. Circulation 87 (3), 909–920. doi:10.1161/01.cir.87.3.909
Zhang M., Zhang S. (2020). T cells in fibrosis and fibrotic diseases. Front. Immunol. 11, 1142. doi:10.3389/fimmu.2020.01142
Zhang S., Bories G., Lantz C., Emmons R., Becker A., Liu E., et al. (2019a). Immunometabolism of phagocytes and relationships to cardiac repair. Front. Cardiovasc Med. 6, 42. doi:10.3389/fcvm.2019.00042
Zhang S., Weinberg S., DeBerge M., Gainullina A., Schipma M., Kinchen J. M., et al. (2019b). Efferocytosis fuels requirements of fatty acid oxidation and the electron transport chain to polarize macrophages for tissue repair. Cell Metab. 29 (2), 443–456. doi:10.1016/j.cmet.2018.12.004
Zhang W., Chancey A. L., Tzeng H. P., Zhou Z., Lavine K. J., Gao F., et al. (2011). The development of myocardial fibrosis in transgenic mice with targeted overexpression of tumor necrosis factor requires mast cell–fibroblast interactions. Circulation 124 (19), 2106–2116. doi:10.1161/CIRCULATIONAHA.111.052399
Zhang W., Lavine K. J., Epelman S., Evans S. A., Weinheimer C. J., Barger P. M., et al. (2015). Necrotic myocardial cells release damage-associated molecular patterns that provoke fibroblast activation in vitro and trigger myocardial inflammation and fibrosis in vivo. J. Am. Heart Assoc. 4 (6), e001993. doi:10.1161/JAHA.115.001993
Zhao P., Zhou W., Zhang Y., Li J., Zhao Y., Pan L., et al. (2020). Aminooxyacetic acid attenuates post-infarct cardiac dysfunction by balancing macrophage polarization through modulating macrophage metabolism in mice. J. Cell. Mol. Med. 24 (4), 2593–2609. doi:10.1111/jcmm.14972
Zhao S., Zhou L., Wang Q., Cao J. H., Chen Y., Wang W., et al. (2023). Elevated branched-chain amino acid promotes atherosclerosis progression by enhancing mitochondrial-to-nuclear H(2)O(2)-disulfide HMGB1 in macrophages. Redox Biol. 62, 102696. doi:10.1016/j.redox.2023.102696
Zhao X., Psarianos P., Ghoraie L. S., Yip K., Goldstein D., Gilbert R., et al. (2019). Metabolic regulation of dermal fibroblasts contributes to skin extracellular matrix homeostasis and fibrosis. Nat. Metab. 1 (1), 147–157. doi:10.1038/s42255-018-0008-5
Zlobine I., Gopal K., Ussher J. R. (2016). Lipotoxicity in obesity and diabetes-related cardiac dysfunction. Biochim. Biophys. Acta 1861 (10), 1555–1568. doi:10.1016/j.bbalip.2016.02.011
Keywords: cardiac fibrosis, immune cells, immunometabolism, cardiometabolism, inflammation, metabolic reprogramming
Citation: Hoque MM, Gbadegoye JO, Hassan FO, Raafat A and Lebeche D (2024) Cardiac fibrogenesis: an immuno-metabolic perspective. Front. Physiol. 15:1336551. doi: 10.3389/fphys.2024.1336551
Received: 16 November 2023; Accepted: 07 March 2024;
Published: 21 March 2024.
Edited by:
Daniel M. Johnson, The Open University, United KingdomReviewed by:
Joshua Travers, University of Colorado Anschutz Medical Campus, United StatesMelissa Reichelt, The University of Queensland, Australia
Copyright © 2024 Hoque, Gbadegoye, Hassan, Raafat and Lebeche. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Djamel Lebeche, ZGxlYmVjaGVAdXRoc2MuZWR1