 Tiantian Wang
Tiantian Wang Ying Huang
Ying Huang Xiangcheng Zhang
Xiangcheng Zhang- Department of Critical Care Medicine, The Affiliated Huaian No 1 People’s Hospital of Nanjing Medical University, Huaian, Jiangsu, China
Sepsis-associated acute kidney injury presents as a critical condition characterized by prolonged hospital stays, elevated mortality rates, and an increased likelihood of transition to chronic kidney disease. Sepsis-associated acute kidney injury suppresses fatty acid oxidation and oxidative phosphorylation in the mitochondria of renal tubular epithelial cells, thus favoring a metabolic shift towards glycolysis for energy production. This shift acts as a protective mechanism for the kidneys. However, an extended reliance on glycolysis may contribute to tubular atrophy, fibrosis, and subsequent chronic kidney disease progression. Metabolic reprogramming interventions have emerged as prospective strategies to counteract sepsis-associated acute kidney injury by restoring normal metabolic function, offering potential therapeutic and preventive modalities. This review delves into the metabolic alterations of tubular epithelial cells associated with sepsis-associated acute kidney injury, stressing the importance of metabolic reprogramming for the immune response and the urgency of metabolic normalization. We present various intervention targets that could facilitate the recovery of oxidative phosphorylation-centric metabolism. These novel insights and strategies aim to transform the clinical prevention and treatment landscape of sepsis-associated acute kidney injury, with a focus on metabolic mechanisms. This investigation could provide valuable insights for clinicians aiming to enhance patient outcomes in the context of sepsis-associated acute kidney injury.
Background
Sepsis-associated acute kidney injury (SA-AKI) is characterized by the presence of both consensus sepsis criteria (as defined by Sepsis-3 recommendations) and acute kidney injury (AKI) criteria [as defined by Kidney Disease: Improving Global Outcomes (KDIGO) recommendations] when AKI occurs within 7 days from diagnosis of sepsis (Kidney Int Suppl, 2011; Singer et al., 2016; Zarbock et al., 2023). The prevalence of SA-AKI is associated with increased mortality rates, lengthy stays in intensive care units, extended hospitalizations, greater long-term impairments, and reduced quality of life (Schuler et al., 2018; Poston and Koyner, 2019; Stanski et al., 2020; Zarbock et al., 2023).
While the precise pathophysiological mechanisms underlying SA-AKI remain incompletely delineated, contributing factors are acknowledged to encompass systemic and renal inflammation, complement activation, dysregulation of the renin-angiotensin-aldosterone system, mitochondrial dysfunction, metabolic reprogramming, and vascular anomalies that span both microcirculatory dysfunction and macrocirculatory alteration (Zarbock et al., 2023).
During the early phase of sepsis, renal tubular epithelial cells (TECs) switch from oxidative phosphorylation (OXPHOS), a highly efficient energy production pathway, to aerobic glycolysis, which is substantially less efficient (Gómez et al., 2017). This metabolic shift is thought to be an essential protective response to infection. This review highlights various therapeutic targets that have been identified to modulate this metabolic adaptation, potentially pioneering new clinical approaches for managing SA-AKI.
Unveiling TECs’ metabolic machinery: exploring normal metabolism
The kidney plays a pivotal role in reabsorbing glucose, ions, and nutrients via ion channels and transporters located in the proximal tubules. The energy requisite for this selective reabsorption is primarily supplied by the kidney’s mitochondria, which produce ATP to fuel the Na+-K+ ATPase pump, a process driven by the ionic gradient this pump establishes. The study suggests that this reabsorption process is responsible for up to 70% of the kidney’s total energy consumption (Bhargava and Schnellmann, 2017).
To fulfill these high energy requirements, renal TECs mainly depend on fatty acid oxidation (FAO) and OXPHOS for efficient energy production, as depicted in Figure 1 (Han et al., 2017; Harzandi et al., 2021). Specifically, TECs uptake long-chain fatty acids via transport proteins and subsequently facilitate their oxidation within the mitochondria (Yang et al., 2017). This process yields acetyl-CoA, reduced flavin adenine dinucleotide (FADH2), and reduced nicotinamide adenine dinucleotide (NADH) (Houten et al., 2016). Acetyl-CoA is further channeled into the tricarboxylic acid (TCA) cycle, creating additional FADH2 and NADH, which then feed into the electron transport chain to ultimately generate ATP (Gao and Chen, 2022). Given that the FAO-driven OXPHOS pathway can yield between 106 and 129 molecules of ATP per cycle, it is recognized as one of the most efficient pathways for cellular energy production (Jang et al., 2020).
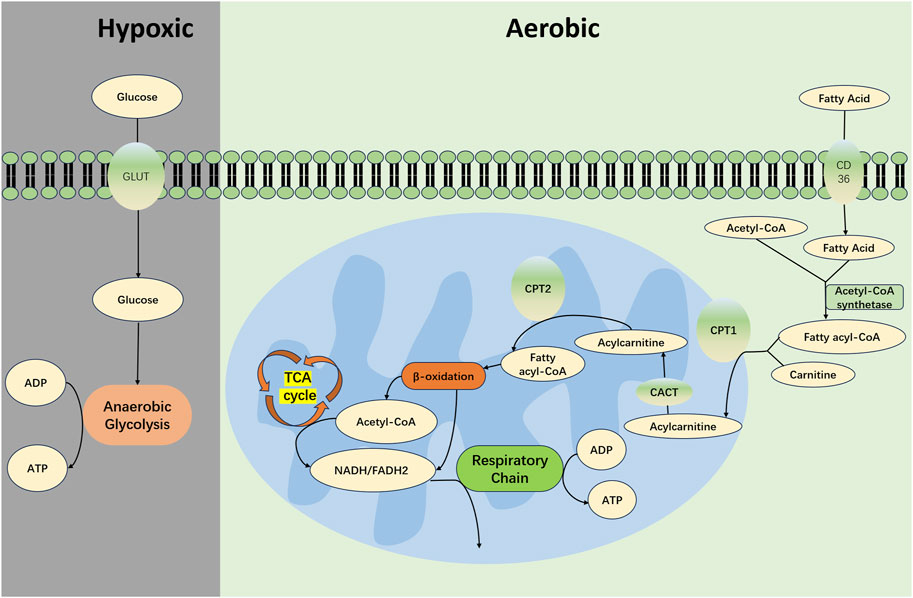
FIGURE 1. Metabolism in Normal Renal TECs. In the proximal tubules of the kidney, long-chain fatty acids are absorbed via transport proteins such as CD36 and fatty acid binding protein (FABP). These fatty acids are catalyzed by acyl-CoA synthetase on the outer mitochondrial membrane to form acyl-CoA in conjunction with coenzyme A (CoA). Acyl-CoA is then shuttled through a channel into the mitochondrial intermembrane space where it encounters carnitine palmitoyltransferase 1 (CPT1) that is embedded in the membrane. Under the catalysis of CPT1, acyl-CoA is conjugated with carnitine to form acylcarnitine. This acylcarnitine is then transported across the mitochondrial inner membrane by carnitine-acylcarnitine translocase (CACT). Subsequently, acylcarnitine is converted back to acyl-CoA by carnitine acyltransferase 2 (CPT2), which is associated with the mitochondrial inner membrane. Acyl-CoA then undergoes a series of reactions including dehydrogenation, hydration, dehydrogenation, and thiolytic cleavage, constituting the β-oxidation process. This leads to the production of acetyl-CoA, while concomitantly generating FADH2 and NADH. The acetyl-CoA enters the TCA cycle, resulting in additional molecules of FADH2 and NADH. These electron carriers—FADH2 and NADH—from both the β-oxidation and the TCA cycle partake in the respiratory chain, transferring electrons to oxygen atoms; this electron transfer event is coupled with the phosphorylation of ADP to create ATP. Under conditions of oxygen deprivation, cells primarily generate energy through anaerobic glycolysis.CD36, cluster of differentiation 36; TEC, Tubular Epithelial Cell; CPT-1, carnitine O-palmitoyltransferase 1; CACT, carnitine-acylcarnitine translocase; CPT-2, carnitine O-palmitoyltransferase 2; TCA, tricarboxylic acid cycle; FADH2, reduced flavin adenine dinucleotide; NADH, reduced nicotinamide adenine dinucleotide; ATP, adenosine triphosphate; ADP, adenosine diphosphate.
Moreover, with renal TECs representing primary sites for FAO and OXPHOS, it is noteworthy that the kidneys contain the second-highest concentration of mitochondria in the body, following only the myocardium (Pagliarini et al., 2008). This substantial mitochondrial endowment is essential in meeting the rigorous energy demands of kidney function.
Building resilience: exploring TECs’ resistance and tolerance mechanisms
Upon pathogen invasion, a host organism activates two cardinal defense strategies to combat the infection: resistance and tolerance (Schneider and Ayres, 2008).
Host resistance reduces the pathogen burden by blocking the entry of pathogens or eliminating them (Schneider and Ayres, 2008). The immune system is pivotal for resistance. While the innate immune system often effectively clears pathogens, it can sometimes be overwhelmed, leading to harmful host reactions. In sepsis, the immune response may become dysregulated, manifesting as excessive inflammation or suppression, both of which are harmful (van der Poll et al., 2017). Sepsis may trigger both inflammation and immune suppression sequentially or simultaneously (Liu D. et al., 2022). Upon invasion, immune cells, particularly renal TECs expressing Toll-like receptors (TLRs), identify pathogen-associated (PAMPs) and damage-associated molecular patterns (DAMPs). This interaction activates immune cascades (Jang and Rabb, 2015). Specifically, TLR2 and TLR4 initiate upregulation of specific genes to produce inflammatory cytokines (e.g., TNF-α, MCP-1) through NF-κB activation, leading to further signaling events (Naito et al., 2008; Fry, 2012). The cytokines produced promote the inflammatory response. TECs also sense PAMPs and DAMPs through TLR-4 on their basolateral membranes in the peritubular capillaries (Peerapornratana et al., 2019). This detection and subsequent activation of TECs’ innate immunity can cause oxidative stress, reactive oxygen species (ROS) production, and mitochondrial dysfunction, contributing to TEC injury (Kalakeche et al., 2011; Fry, 2012; Dellepiane et al., 2016). Hence, resistance during inflammation involves trade-offs and can result in immunopathology, compromising the host’s health (Toro et al., 2021).
Alongside resistance, the body also employs tolerance as a defense mechanism to minimize infection-induced tissue damage through multiple pathways (Medzhitov et al., 2012). The concept of tolerance, first introduced by Schafer (Schafer, 1971) and later expanded on by Schneider and Ayres (Schneider and Ayres, 2008) in 2018, applies to both animals and humans. Ganeshan, Nikkanen (Ganeshan et al., 2019) showed that in a mouse model of lipopolysaccharide (LPS)-induced sepsis, hepatic metabolism shifted from fatty acid oxidation toward acetyl-CoA and amino acid catabolism, resulting in a hypometabolic state that enables tissue tolerance and protection against bacterial infections. We hypothesize that sepsis-induced AKI may lead to metabolic reprogramming in the host, enhancing the state of tolerance to counteract damage not just from pathogens, but also from the host’s immune response.
Mitochondrial dysfunction in SA-AKI: implications for TECs’ energy metabolism
Mitochondria, the sites of FAO and OXPHOS, are integral to the energy metabolism of TECs. In the context of SA-AKI, metabolic reprogramming is intimately associated with mitochondrial dysfunction. Research has demonstrated increases in mitochondrial DNA (mtDNA) oxidation and damage, alongside reductions in mitochondrial abundance in the kidneys of SA-AKI patients (van der Slikke et al., 2021). Mitochondrial dysfunction in the setting of sepsis precipitates ATP depletion and an upsurge in reactive oxygen species (ROS), both of which are contributory factors to cellular instability and subsequent renal organ dysfunction (Brealey et al., 2002). Yet, the exact molecular pathways through which mitochondria contribute to the pathogenesis of SA-AKI remain to be fully elucidated.
Mitochondrial damage within TECs during SA-AKI manifests as diminished mitochondrial mass, fragmentation, cristae disruption, and diverse extents of mitochondrial swelling (Parikh et al., 2015). Additionally, the challenges posed by sepsis extend to compromising the vital quality control mechanisms of mitochondria such as fission/fusion dynamics, biogenesis, and mitophagy—all of which are essential in preserving mitochondrial integrity and function.
For instance, dysfunctional mitochondria can initiate fission, resulting in two daughter mitochondria which may then fuse with healthy counterparts, aiming to ameliorate the damage (Sun et al., 2019). Nonetheless, this intricate process of mitochondrial fission and fusion becomes compromised during septic acute kidney injury (Liu et al., 2019).
Mitochondrial biogenesis, the process that elevates mitochondrial mass and enhances glycolytic and OXPHOS enzyme levels, ultimately increases mitochondrial energy production capabilities (Sun et al., 2019). Insufficient or delayed induction of mitochondrial biogenesis during critical illness magnifies the susceptibility to oxidative and nitrosative mitochondrial damage, impairs the clearance of various cellular organelles, and contributes to a net reduction in mitochondrial content, culminating in cellular energy deficits. Conversely, early initiation of mitochondrial biogenesis and prompt response to oxidative stress are associated with higher survival rates in patients with critical illnesses (Carré et al., 2010). Promoting biogenesis through diverse strategies in septic mouse models has proven effective in preserving renal function and improving survival outcomes (Jin et al., 2020; Li et al., 2023). Furthermore, in vitro analyses demonstrate that the NRF2 pathway enhances mitophagy in renal cells and tubules, stimulates mitochondrial biogenesis under septic conditions, increases the abundance of functional mitochondria, and promotes a more robust mitochondrial network, which collectively mitigate inflammation, oxidative stress, and apoptosis (Chen et al., 2022). These findings collectively underscore the significance of mitochondrial biogenesis in the recovery process of TECs post-injury.
Mitochondrial autophagy, otherwise designated as mitophagy, serves as an essential process for the selective identification, encapsulation, and breakdown of defective mitochondria, which is vital in the context of SA-AKI (Parikh et al., 2015). Therapeutic strategies that enhance mitophagy, such as the administration of melatonin which augments mitochondrial autophagy by enabling SIRT3-dependent deacetylation of transcription factor A (TFAM), have shown promise in mitigating the effects of SA-AKI (Deng et al., 2023).
Metabolic shifts and reprogramming: unraveling changes and tolerance mechanisms in TECs during SA-AKI
After sepsis-induced AKI, renal TECs experience metabolic shifts, transitioning from FAO-driven OXPHOS to a glycolytic state, as depicted in Figure 2.
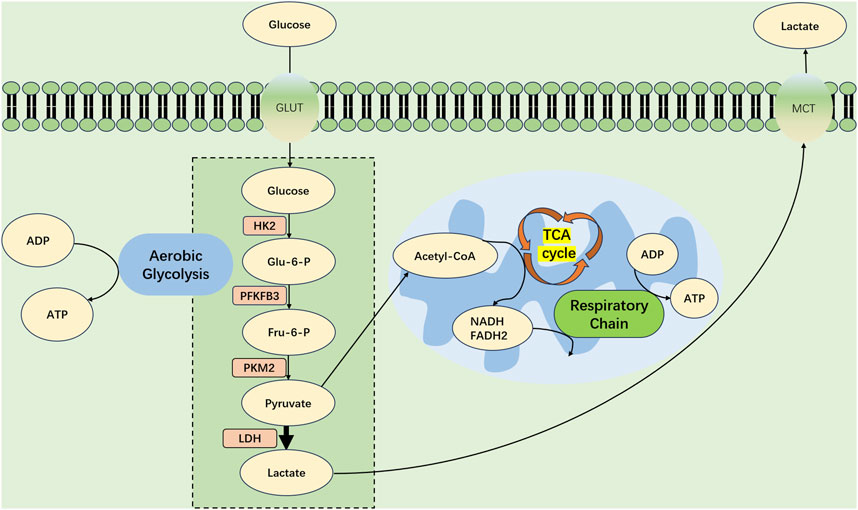
FIGURE 2. Metabolic Shift in Tubular Epithelial Cells During Septic Acute Kidney Injury. At the advent of SA-AKI, renal tubular epithelial cells (TECs) experience a profound metabolic transformation, migrating from OXPHOS toward a predominance of aerobic glycolysis. Within this metabolic realignment, the bulk of pyruvate forged by glycolytic processes eschews mitochondrial entry, opting instead for conversion into lactate—a process catalyzed by lactate dehydrogenase (LDH). This strategic metabolic adaptation is key, supporting an increased production of ATP through glycolysis to meet the amplified energy requirements imposed by the septic challenge. GLUT, glucose transporter protein; PFKFB3, fructose-2,6-bisphosphatase 3; HK2, hexokinase 2; PKM2, pyruvate kinase M2; Glu-6-P glucose 6-phosphate; Fru6-P Fructose 6-phosphate; LDH, Lactate Dehydrogenase; MCT, Monocarboxylate TransporterCD36, cluster of differentiation 36; TCA, tricarboxylic acid cycle; FADH2, reduced flavin adenine dinucleotide; NADH, reduced nicotinamide adenine dinucleotide; ATP, adenosine triphosphate; ADP, adenosine diphosphate.
Researchers, utilizing a mouse model of cecal ligation and puncture (CLP)-induced sepsis, noted diminished renal fatty acid oxidase expression and a pronounced upregulation of glycolytic enzymes (Li et al., 2020).
Moreover, a dedicated metabolic and osmolyte study revealed significant post-CLP alterations within renal TECs; renal biopsies, obtained 8 hours after the procedure, demonstrated elevated glycolytic intermediates and reduced tricarboxylic acid (TCA) cycle intermediates (Waltz et al., 2016). Comparable metabolic shifts were observed in atrophic TECs associated with polycystic kidney disease and ischemia-reperfusion injury, processes mediated by activation of the Akt/phosphatidylinositol 3-kinase/mammalian target of rapamycin complex 1 (Akt/PI3K/mTORC1) signaling pathway (Lan et al., 2016; Podrini et al., 2020).
Notably, mTORC1 activation serves to stabilize the transcription factor hypoxia-inducible factor 1α (HIF-1α), thereby steering cellular metabolism toward aerobic glycolysis. Given these insights, it is plausible that TECs adapt their metabolism through similar mechanisms to other cell types under septic conditions.
This metabolic shift represents a protective, adaptive response that allows for enhanced infection tolerance, reduced cellular damage, and maintained viability (Liu J. et al., 2022). In their pivotal study, Katherine et al. discovered that glucose administration to malaria-infected mice bolstered glycolysis, leading to increased disease tolerance and enhanced survival rates (Cumnock et al., 2018).
During the early stages of LPS-induced AKI, the metabolic switch to glycolysis proves integral for establishing trained immunity (Cheng et al., 2014). Specifically, in LPS-induced SA-AKI mouse models, hexokinase (HK) activity, critical to the glycolytic pathway, markedly increased.
This HK surge facilitates activation of the pentose phosphate pathway (PPP), essential for maintaining glutathione (GSH) in a reduced state and upholding renal antioxidant defense mechanisms (Smith et al., 2014). In addition, aerobic glycolysis provides both sufficient energy and vital structural components, such as fatty acids, amino acids, and nucleotides, needed for cellular functions including mitosis (Vander Heiden et al., 2009).
Restoring OXPHOS: rebalancing TECs’ metabolism
Initially, the organism’s metabolic shift toward glycolysis at the onset of sepsis may enhance tolerance and facilitate cellular survival; however, prolonged dependence on this pathway is ultimately deleterious. It becomes imperative for metabolism to transition back to OXPHOS. This necessity arises from several key findings.
First, upon SA-AKI onset, metabolic reprogramming in TECs leads to decreased utilization of free fatty acids and subsequent lipid accumulation (Jang et al., 2020). Such accumulation contributes to renal tubular fibrosis and glomerulosclerosis development (Gai et al., 2019). A recent investigation demonstrated that stimulating uncoupling protein 1 (UCP1) lessens lipid buildup, thereby inhibiting AKI progression through the AMPK/ULK1/autophagy pathway (Xiong et al., 2021).
Moreover, evidence suggests that timely metabolic restoration to OXPHOS and FAO can effectively rescue renal tubular ion transport and renal function (Toro et al., 2021). Activation of the farnesol X receptor (FXR) promotes FAO-related gene expression in TECs; this activation mitigates the effects of cisplatin-induced AKI and reduces lipid accumulation, protecting against FAO-mediated renal damage (Xu et al., 2022). These findings accentuate the therapeutic potential of targeting FXR and enhancing FAO in AKI treatment.
Furthermore, continuous aerobic glycolysis in renal TECs is linked to persistent local inflammation and compounded injury, reminiscent of the metabolic activity observed in immune cells (Toro et al., 2021; Zhu et al., 2022). Pyruvate kinase M2 (PKM2) plays a vital role in glycolysis’ final rate-limiting step, while high mobility group box 1 (HMGB1) acts as a powerful cytokine in the sepsis context. Elevated PKM2 levels have been shown to prompt HMGB1 release from macrophages, leading to inflammation. Remarkably, using a PKM2 inhibitor like picrotoxin can diminish HMGB1 levels, offering protection from sepsis (Yang et al., 2014).
Prolonged glycolysis during AKI also adversely influences renal tubular regeneration, progressing to tubular atrophy, fibrosis, and potential chronic kidney disease (CKD). Interventions enhancing FAO, activating PGC-1α with ZLN-005, or inhibiting glycolysis with 2-DG, a hexokinase 2 inhibitor, have proven to forestall the transition from AKI to CKD by preventing pericyte-to-myofibroblast transformation (Lan et al., 2016; Li et al., 2018; Xu et al., 2023).
While these findings illuminate the critical role of metabolic reprogramming in the prognosis of AKI to CKD, research on the appropriate timing for metabolic recovery from glycolysis to OXPHOS remains limited.
Targeting treatment prospects: identifying potential therapeutic avenues
The metabolic functions of the kidneys are pivotal in the progression of SA-AKI. Accordingly, targeting renal energy metabolism emerges as a pivotal strategy for the prevention and treatment of SA-AKI (Figure 3). FAO and OXPHOS are the primary sources of renal energy, and the role of peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α) in regulating mitochondrial biogenesis is crucial. PGC-1α, which is principally expressed in the renal proximal tubular cells, becomes activated by AMPK phosphorylation and sirtuin1 (SIRT1) deacetylation, enhancing its regulatory functions (Fontecha-Barriuso et al., 2020).
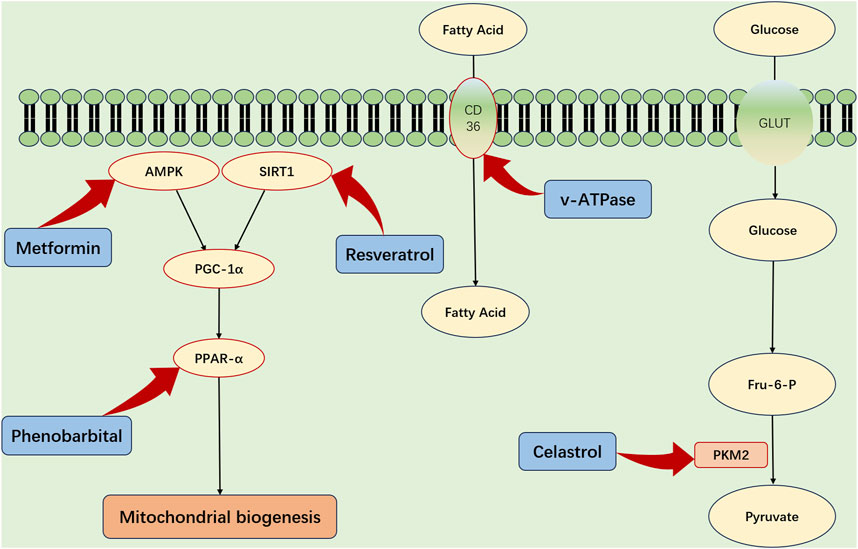
FIGURE 3. Strategic Metabolic Interventions for Renal Protection in SA-AKI. The refined activation of critical metabolic influencers—specifically PGC-1α, AMPK, SIRT1, and PPAR-α—and the enhancement of fatty acid absorption through CD36 mediation offer viable strategies for defending renal integrity amidst sepsis-associated acute kidney injury (SA-AKI). Additionally, strategically modulating PKM2, a key enzyme in the process of aerobic glycolysis, could provide a supplementary pathway to diminish the metabolic imbalances that are hallmark features of SA-AKI. AMPK, adenosine monophosphate kinase; PGC-1α, peroxisome proliferator-activated receptor gamma; Sirt1, sirtuin1; PPAR-α, peroxisome proliferator-activated receptor-α; TFAM, transcription factor A; GLUT, glucose transporter protein; PKM2, pyruvate kinase M2; Glu-6-P glucose 6-phosphate; Fru6-P Fructose 6-phosphate.
During SA-AKI, a notable decline in PGC-1α expression impairs energy production, underscoring the significance of PGC-1α (Tran et al., 2011; Smith et al., 2015). Therefore, enhancing PGC-1α levels—possibly through the administration of recombinant human erythropoietin (rhEPO) or by pharmacologically stimulating the AMPK pathway—may help mitigate the detrimental effects of sepsis (Cantó and Auwerx, 2009; Jin et al., 2020; Li et al., 2023).
In addition, Peroxisome proliferator-activated receptor-α (PPAR-α) and CD36 are key modulators of renal metabolism. PPAR-α enhances FAO enzyme expression and aids fatty acid uptake via CD36 (Iwaki et al., 2019). Preclinical models indicate that PPAR-α agonists, such as fenofibrate, could offer protective benefits for renal health (Kaur et al., 2021). CD36 plays a critical role in allowing proximal tubular cells to obtain fatty acids attached to albumin from the bloodstream, which is essential for maintaining adequate energy levels (Yang et al., 2017). Targeted interventions aimed at CD36 might influence renal metabolism, though additional investigations are warranted to confirm this premise.
Additionally, targeting glycolysis in TECs may be considered a promising therapeutic strategy. PKM2, an enzyme crucial to the glycolytic pathway, and its inhibitors, notably celastrol and shikonin, are proposed to attenuate renal injury associated with SA-AKI (Wu et al., 2021; Luo et al., 2022).
In conclusion, manipulating metabolic pathways within the kidneys opens avenues for novel SA-AKI treatments. The strategic activation of PGC-1α pathways, stimulating the PPAR-α axis, and curtailing PKM2 activity present viable options for renal recovery. However, although these preliminary findings are hopeful, they require further exploration and verification through rigorous research.
Discussion
Metabolic reprogramming may offer renal protection by enabling a tolerance mechanism during the initial stages of SA-AKI. However, a prolonged dependence on aerobic glycolysis may result in adverse outcomes, such as tubular atrophy and fibrosis. In contrast, transitioning to OXPHOS metabolism has the potential to decelerate AKI progression and assist in the recovery of renal function.
While evidence underscores the crucial role of OXPHOS in kidney repair, the research landscape regarding the timing and regulation of this metabolic shift is limited in scope. Specifically, determining the optimal timing to initiate this metabolic shift—whether it should occur in the hyperacute phase, the days following the onset of AKI, or during the chronic phase—remains largely unexplored. Furthermore, although resistance and tolerance mechanisms are postulated in existing studies, there is a dearth of solid empirical evidence to support these claims, especially in the context of SA-AKI. Consequently, this highlights the necessity for comprehensive research to elucidate their specific roles and interactions. Additionally, despite the extensive research on various types of acute kidney injury, focused studies on SA-AKI are notably lacking. Given the potential variations in pathogenesis across different AKI types, further investigation is essential to bridge this knowledge gap.
As of now, there are no clinically approved therapeutic agents designed to modulate metabolic reprogramming, and the pharmacological safety profiles of such interventions are yet to be determined, calling for broad-ranging research. Nonetheless, the profound impact of metabolic reprogramming in TECs on SA-AKI progression emphasizes its potential as a focal point for future research. Therefore, targeting metabolic changes in TECs shows promise as a strategy for both preventing and treating SA-AKI.
Author contributions
TW: Conceptualization, Resources, Writing–original draft, Writing–review and editing. YH: Conceptualization, Formal Analysis, Investigation, Resources, Supervision, Writing–review and editing. XiZ: Conceptualization, Data curation, Investigation, Resources, Software, Writing–review and editing. YZ: Conceptualization, Investigation, Methodology, Project administration, Resources, Software, Writing–review and editing. XgZ: Conceptualization, Funding acquisition, Investigation, Resources, Supervision, Validation, Writing–review and editing.
Funding
The author(s) declare that no financial support was received for the research, authorship, and/or publication of this article.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Bhargava P., Schnellmann R. G. (2017). Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 13 (10), 629–646. doi:10.1038/nrneph.2017.107
Brealey D., Brand M., Hargreaves I., Heales S., Land J., Smolenski R., et al. (2002). Association between mitochondrial dysfunction and severity and outcome of septic shock. Lancet 360 (9328), 219–223. doi:10.1016/S0140-6736(02)09459-X
Cantó C., Auwerx J. (2009). PGC-1alpha, SIRT1 and AMPK, an energy sensing network that controls energy expenditure. Curr. Opin. Lipidol. 20 (2), 98–105. doi:10.1097/MOL.0b013e328328d0a4
Carré J. E., Orban J. C., Re L., Felsmann K., Iffert W., Bauer M., et al. (2010). Survival in critical illness is associated with early activation of mitochondrial biogenesis. Am. J. Respir. Crit. Care Med. 182 (6), 745–751. doi:10.1164/rccm.201003-0326OC
Chen Z., Wang H., Hu B., Chen X., Zheng M., Liang L., et al. (2022). Transcription factor nuclear factor erythroid 2 p45-related factor 2 (NRF2) ameliorates sepsis-associated acute kidney injury by maintaining mitochondrial homeostasis and improving the mitochondrial function. Eur. J. Histochem 66 (3), 3412. doi:10.4081/ejh.2022.3412
Cheng S. C., Quintin J., Cramer R. A., Shepardson K. M., Saeed S., Kumar V., et al. (2014). mTOR- and HIF-1α-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 345 (6204), 1250684. doi:10.1126/science.1250684
Cumnock K., Gupta A. S., Lissner M., Chevee V., Davis N. M., Schneider D. S. (2018). Host energy source is important for disease tolerance to malaria. Curr. Biol. 28 (10), 1635–1642. doi:10.1016/j.cub.2018.04.009
Dellepiane S., Marengo M., Cantaluppi V. (2016). Detrimental cross-talk between sepsis and acute kidney injury: new pathogenic mechanisms, early biomarkers and targeted therapies. Crit. Care 20, 61. doi:10.1186/s13054-016-1219-3
Deng Z., He M., Hu H., Zhang W., Zhang Y., Ge Y., et al. (2023). Melatonin attenuates sepsis-induced acute kidney injury by promoting mitophagy through SIRT3-mediated TFAM deacetylation. Autophagy 20, 151–165. doi:10.1080/15548627.2023.2252265
Fontecha-Barriuso M., Martin-Sanchez D., Martinez-Moreno J. M., Monsalve M., Ramos A. M., Sanchez-Niño M. D., et al. (2020). The role of PGC-1α and mitochondrial biogenesis in kidney diseases. Biomolecules 10 (2), 347. doi:10.3390/biom10020347
Fry D. E. (2012). Article commentary: sepsis, systemic inflammatory response, and multiple organ dysfunction: the mystery continues. Am. Surg. 78 (1), 1–8. doi:10.1177/000313481207800102
Gai Z., Wang T., Visentin M., Kullak-Ublick G. A., Fu X., Wang Z. (2019). Lipid accumulation and chronic kidney disease. Nutrients 11 (4), 722. doi:10.3390/nu11040722
Ganeshan K., Nikkanen J., Man K., Leong Y. A., Sogawa Y., Maschek J. A., et al. (2019). Energetic trade-offs and hypometabolic states promote disease tolerance. Cell 177 (2), 399–413. doi:10.1016/j.cell.2019.01.050
Gao Z., Chen X. (2022). Fatty acid β-oxidation in kidney diseases: perspectives on pathophysiological mechanisms and therapeutic opportunities. Front. Pharmacol. 13, 805281. doi:10.3389/fphar.2022.805281
Gómez H., Kellum J. A., Ronco C. (2017). Metabolic reprogramming and tolerance during sepsis-induced AKI. Nat. Rev. Nephrol. 13 (3), 143–151. doi:10.1038/nrneph.2016.186
Han S. H., Wu M. Y., Nam B. Y., Park J. T., Yoo T. H., Kang S. W., et al. (2017). PGC-1α protects from notch-induced kidney fibrosis development. J. Am. Soc. Nephrol. 28 (11), 3312–3322. doi:10.1681/ASN.2017020130
Harzandi A., Lee S., Bidkhori G., Saha S., Hendry B. M., Mardinoglu A., et al. (2021). Acute kidney injury leading to CKD is associated with a persistence of metabolic dysfunction and hypertriglyceridemia. iScience 24 (2), 102046. doi:10.1016/j.isci.2021.102046
Houten S. M., Violante S., Ventura F. V., Wanders R. J. (2016). The biochemistry and Physiology of mitochondrial fatty acid β-oxidation and its genetic disorders. Annu. Rev. Physiol. 78, 23–44. doi:10.1146/annurev-physiol-021115-105045
Iwaki T., Bennion B. G., Stenson E. K., Lynn J. C., Otinga C., Djukovic D., et al. (2019). PPARα contributes to protection against metabolic and inflammatory derangements associated with acute kidney injury in experimental sepsis. Physiol. Rep. 7 (10), e14078. doi:10.14814/phy2.14078
Jang H. R., Rabb H. (2015). Immune cells in experimental acute kidney injury. Nat. Rev. Nephrol. 11 (2), 88–101. doi:10.1038/nrneph.2014.180
Jang H. S., Noh M. R., Kim J., Padanilam B. J. (2020). Defective mitochondrial fatty acid oxidation and lipotoxicity in kidney diseases. Front. Med. (Lausanne). 7, 65. doi:10.3389/fmed.2020.00065
Jin K., Ma Y., Manrique-Caballero C. L., Li H., Emlet D. R., Li S., et al. (2020). Activation of AMP-activated protein kinase during sepsis/inflammation improves survival by preserving cellular metabolic fitness. Faseb J. 34 (5), 7036–7057. doi:10.1096/fj.201901900R
Kalakeche R., Hato T., Rhodes G., Dunn K. W., El-Achkar T. M., Plotkin Z., et al. (2011). Endotoxin uptake by S1 proximal tubular segment causes oxidative stress in the downstream S2 segment. J. Am. Soc. Nephrol. 22 (8), 1505–1516. doi:10.1681/ASN.2011020203
Kaur J., Kaur T., Sharma A. K., Kaur J., Yadav H. N., Pathak D., et al. (2021). Fenofibrate attenuates ischemia reperfusion-induced acute kidney injury and associated liver dysfunction in rats. Drug Dev. Res. 82 (3), 412–421. doi:10.1002/ddr.21764
Kidney Int Suppl (2011). Section 2: AKI definition. Kidney Int. Suppl. 2 (1), 19–36. doi:10.1038/kisup.2011.32
Lan R., Geng H., Singha P. K., Saikumar P., Bottinger E. P., Weinberg J. M., et al. (2016). Mitochondrial pathology and glycolytic shift during proximal tubule atrophy after ischemic AKI. J. Am. Soc. Nephrol. 27 (11), 3356–3367. doi:10.1681/ASN.2015020177
Li K., Gao L., Zhou S., Ma Y. R., Xiao X., Jiang Q., et al. (2023). Erythropoietin promotes energy metabolism to improve LPS-induced injury in HK-2 cells via SIRT1/PGC1-α pathway. Mol. Cell Biochem. 478 (3), 651–663. doi:10.1007/s11010-022-04540-y
Li M., Jia F., Zhou H., Di J., Yang M. (2018). Elevated aerobic glycolysis in renal tubular epithelial cells influences the proliferation and differentiation of podocytes and promotes renal interstitial fibrosis. Eur. Rev. Med. Pharmacol. Sci. 22 (16), 5082–5090. doi:10.26355/eurrev_201808_15701
Li Y., Nourbakhsh N., Pham H., Tham R., Zuckerman J. E., Singh P. (2020). Evolution of altered tubular metabolism and mitochondrial function in sepsis-associated acute kidney injury. Am. J. Physiol. Ren. Physiol. 319 (2), F229–f44. doi:10.1152/ajprenal.00390.2019
Liu D., Huang S. Y., Sun J. H., Zhang H. C., Cai Q. L., Gao C., et al. (2022a). Sepsis-induced immunosuppression: mechanisms, diagnosis and current treatment options. Mil. Med. Res. 9 (1), 56. doi:10.1186/s40779-022-00422-y
Liu J., Zhou G., Wang X., Liu D. (2022b). Metabolic reprogramming consequences of sepsis: adaptations and contradictions. Cell Mol. Life Sci. 79 (8), 456. doi:10.1007/s00018-022-04490-0
Liu J. X., Yang C., Zhang W. H., Su H. Y., Liu Z. J., Pan Q., et al. (2019). Disturbance of mitochondrial dynamics and mitophagy in sepsis-induced acute kidney injury. Life Sci. 235, 116828. doi:10.1016/j.lfs.2019.116828
Luo P., Zhang Q., Zhong T. Y., Chen J. Y., Zhang J. Z., Tian Y., et al. (2022). Celastrol mitigates inflammation in sepsis by inhibiting the PKM2-dependent Warburg effect. Mil. Med. Res. 9 (1), 22. doi:10.1186/s40779-022-00381-4
Medzhitov R., Schneider D. S., Soares M. P. (2012). Disease tolerance as a defense strategy. Science 335 (6071), 936–941. doi:10.1126/science.1214935
Naito M., Bomsztyk K., Zager R. A. (2008). Endotoxin mediates recruitment of RNA polymerase II to target genes in acute renal failure. J. Am. Soc. Nephrol. 19 (7), 1321–1330. doi:10.1681/ASN.2007121368
Pagliarini D. J., Calvo S. E., Chang B., Sheth S. A., Vafai S. B., Ong S. E., et al. (2008). A mitochondrial protein compendium elucidates complex I disease biology. Cell 134 (1), 112–123. doi:10.1016/j.cell.2008.06.016
Parikh S. M., Yang Y., He L., Tang C., Zhan M., Dong Z. (2015). Mitochondrial function and disturbances in the septic kidney. Semin. Nephrol. 35 (1), 108–119. doi:10.1016/j.semnephrol.2015.01.011
Peerapornratana S., Manrique-Caballero C. L., Gómez H., Kellum J. A. (2019). Acute kidney injury from sepsis: current concepts, epidemiology, pathophysiology, prevention and treatment. Kidney Int. 96 (5), 1083–1099. doi:10.1016/j.kint.2019.05.026
Podrini C., Cassina L., Boletta A. (2020). Metabolic reprogramming and the role of mitochondria in polycystic kidney disease. Cell Signal 67, 109495. doi:10.1016/j.cellsig.2019.109495
Poston J. T., Koyner J. L. (2019). Sepsis associated acute kidney injury. Bmj 364, k4891. doi:10.1136/bmj.k4891
Schafer J. F. (1971). Tolerance to plant disease. Annu. Rev. Phytopathology 9 (1), 235–252. doi:10.1146/annurev.py.09.090171.001315
Schneider D. S., Ayres J. S. (2008). Two ways to survive infection: what resistance and tolerance can teach us about treating infectious diseases. Nat. Rev. Immunol. 8 (11), 889–895. doi:10.1038/nri2432
Schuler A., Wulf D. A., Lu Y., Iwashyna T. J., Escobar G. J., Shah N. H., et al. (2018). The impact of acute organ dysfunction on long-term survival in sepsis. Crit. Care Med. 46 (6), 843–849. doi:10.1097/CCM.0000000000003023
Singer M., Deutschman C. S., Seymour C. W., Shankar-Hari M., Annane D., Bauer M., et al. (2016). The third international consensus definitions for sepsis and septic shock (Sepsis-3). Jama 315 (8), 801–810. doi:10.1001/jama.2016.0287
Smith J. A., Stallons L. J., Collier J. B., Chavin K. D., Schnellmann R. G. (2015). Suppression of mitochondrial biogenesis through toll-like receptor 4-dependent mitogen-activated protein kinase kinase/extracellular signal-regulated kinase signaling in endotoxin-induced acute kidney injury. J. Pharmacol. Exp. Ther. 352 (2), 346–357. doi:10.1124/jpet.114.221085
Smith J. A., Stallons L. J., Schnellmann R. G. (2014). Renal cortical hexokinase and pentose phosphate pathway activation through the EGFR/Akt signaling pathway in endotoxin-induced acute kidney injury. Am. J. Physiol. Ren. Physiol. 307 (4), F435–F444. doi:10.1152/ajprenal.00271.2014
Stanski N. L., Cvijanovich N. Z., Fitzgerald J. C., Bigham M. T., Wong H. R.Genomics of Pediatric Septic Shock Investigators (2020). Severe acute kidney injury is independently associated with mortality in children with septic shock. Intensive Care Med. 46 (5), 1050–1051. doi:10.1007/s00134-020-05940-8
Sun J., Zhang J., Tian J., Virzì G. M., Digvijay K., Cueto L., et al. (2019). Mitochondria in sepsis-induced AKI. J. Am. Soc. Nephrol. 30 (7), 1151–1161. doi:10.1681/ASN.2018111126
Toro J., Manrique-Caballero C. L., Gomez H. (2021). Metabolic reprogramming and host tolerance: a novel concept to understand sepsis-associated AKI. J. Clin. Med. 10 (18), 4184. doi:10.3390/jcm10184184
Tran M., Tam D., Bardia A., Bhasin M., Rowe G. C., Kher A., et al. (2011). PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J. Clin. Invest. 121 (10), 4003–4014. doi:10.1172/JCI58662
Vander Heiden M. G., Cantley L. C., Thompson C. B. (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324 (5930), 1029–1033. doi:10.1126/science.1160809
van der Poll T., van de Veerdonk F. L., Scicluna B. P., Netea M. G. (2017). The immunopathology of sepsis and potential therapeutic targets. Nat. Rev. Immunol. 17 (7), 407–420. doi:10.1038/nri.2017.36
van der Slikke E. C., Star B. S., van Meurs M., Henning R. H., Moser J., Bouma H. R. (2021). Sepsis is associated with mitochondrial DNA damage and a reduced mitochondrial mass in the kidney of patients with sepsis-AKI. Crit. Care 25 (1), 36. doi:10.1186/s13054-020-03424-1
Waltz P., Carchman E., Gomez H., Zuckerbraun B. (2016). Sepsis results in an altered renal metabolic and osmolyte profile. J. Surg. Res. 202 (1), 8–12. doi:10.1016/j.jss.2015.12.011
Wu J., Rong S., Zhou J., Yuan W. (2021). The role and mechanism of PKM2 in the development of LPS-induced acute kidney injury. Histol. Histopathol. 36 (8), 845–852. doi:10.14670/HH-18-343
Xiong W., Xiong Z., Song A., Lei C., Ye C., Zhang C. (2021). Relieving lipid accumulation through UCP1 suppresses the progression of acute kidney injury by promoting the AMPK/ULK1/autophagy pathway. Theranostics 11 (10), 4637–4654. doi:10.7150/thno.56082
Xu C., Hong Q., Zhuang K., Ren X., Cui S., Dong Z., et al. (2023). Regulation of pericyte metabolic reprogramming restricts the AKI to CKD transition. Metabolism 145, 155592. doi:10.1016/j.metabol.2023.155592
Xu S., Jia P., Fang Y., Jin J., Sun Z., Zhou W., et al. (2022). Nuclear farnesoid X receptor attenuates acute kidney injury through fatty acid oxidation. Kidney Int. 101 (5), 987–1002. doi:10.1016/j.kint.2022.01.029
Yang L., Xie M., Yang M., Yu Y., Zhu S., Hou W., et al. (2014). PKM2 regulates the Warburg effect and promotes HMGB1 release in sepsis. Nat. Commun. 5, 4436. doi:10.1038/ncomms5436
Yang X., Okamura D. M., Lu X., Chen Y., Moorhead J., Varghese Z., et al. (2017). CD36 in chronic kidney disease: novel insights and therapeutic opportunities. Nat. Rev. Nephrol. 13 (12), 769–781. doi:10.1038/nrneph.2017.126
Zarbock A., Nadim M. K., Pickkers P., Gomez H., Bell S., Joannidis M., et al. (2023). Sepsis-associated acute kidney injury: consensus report of the 28th Acute Disease Quality Initiative workgroup. Nat. Rev. Nephrol. 19, 401–417. doi:10.1038/s41581-023-00683-3
Keywords: sepsis-associated acute kidney injury, metabolic reprogramming, renal tubular epithelial cells, oxidative phosphorylation, glycolysis
Citation: Wang T, Huang Y, Zhang X, Zhang Y and Zhang X (2024) Advances in metabolic reprogramming of renal tubular epithelial cells in sepsis-associated acute kidney injury. Front. Physiol. 15:1329644. doi: 10.3389/fphys.2024.1329644
Received: 29 October 2023; Accepted: 09 January 2024;
Published: 19 January 2024.
Edited by:
Charles Mobbs, Icahn School of Medicine at Mount Sinai, United StatesReviewed by:
Zhenzhen Sun, Nanjing Children’s Hospital, ChinaYinghui Huang, Army Medical University, China
Copyright © 2024 Wang, Huang, Zhang, Zhang and Zhang. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Xiangcheng Zhang, emh4YzAzMThAMTYzLmNvbQ==