 Rashida Lathan
Rashida Lathan
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol. , 14 May 2024
Sec. Renal Physiology and Pathophysiology
Volume 15 - 2024 | https://doi.org/10.3389/fphys.2024.1296504
This article is part of the Research Topic Advances In Understanding Tissue Damage to Develop New Repair Strategies View all 6 articles
We propose that the key initiators of renal fibrosis are myofibroblasts which originate from four predominant sources—fibroblasts, pericytes, endothelial cells and macrophages. Increased accumulation of renal interstitial myofibroblasts correlates with an increase in collagen, fibrillar proteins, and fibrosis severity. The canonical TGF-
Classically, the process of fibrosis in the kidney is broken down into five distinct stages.
Initially, an epithelial response to inflammatory monocytes and macrophages occurs. Secondly, there is the production of inflammatory cytokines (notably TGF-
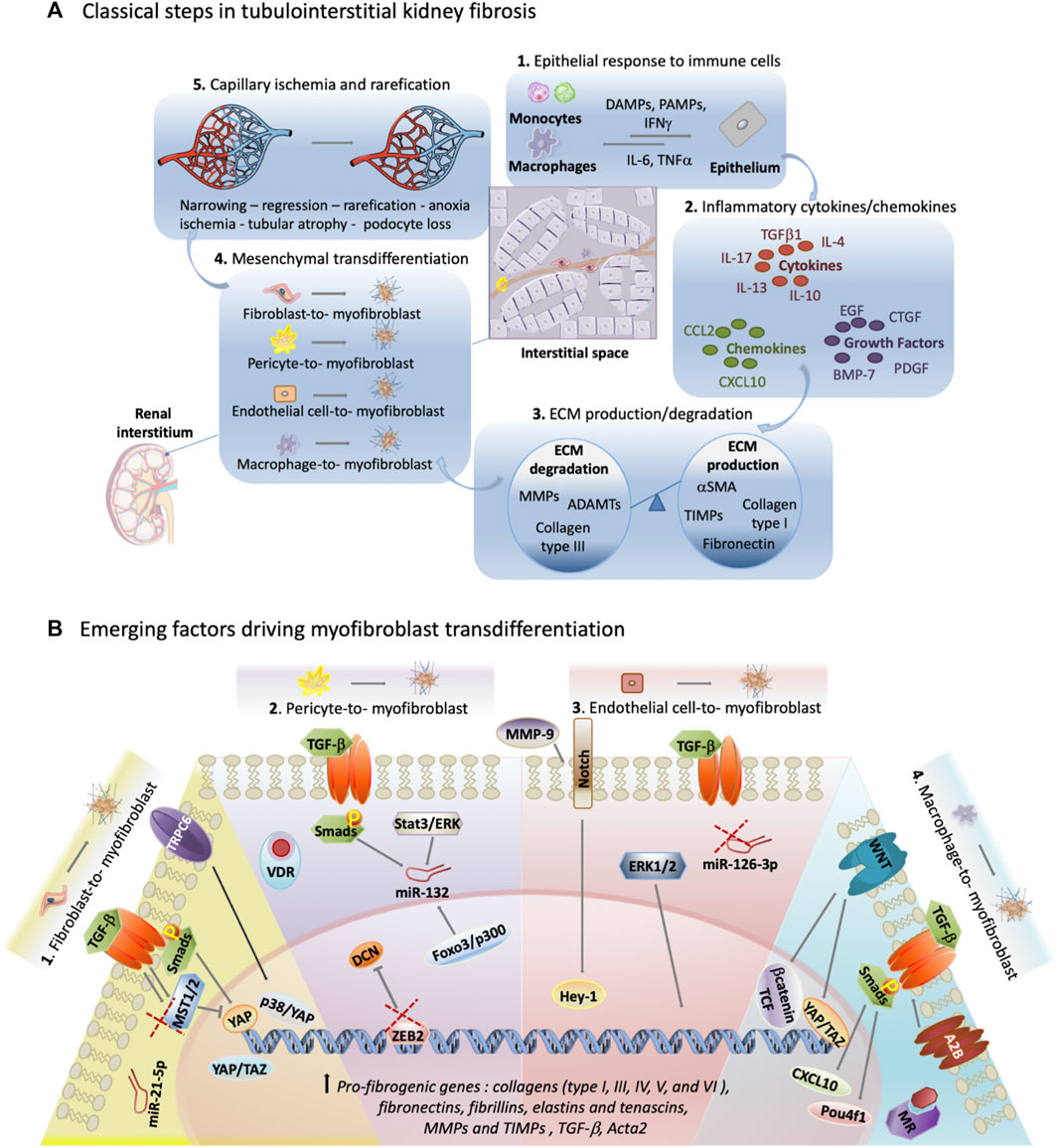
Figure 1. Exploring unconventional targets in myofibroblast transdifferentiation outside classical TGF-
The first and fourth stages of renal fibrosis introduce cells with the potential to differentiate into pathological myofibroblasts. The rise in active myofibroblast numbers within the interstitial space correlates positively with fibrosis severity (Sun et al., 2016). This review highlights myofibroblast transformation, with a focus on the cell of origin and the less-explored molecular pathways driving this change.
As the main producer of fibrotic ECM, myofibroblasts are distinguished from quiescent fibroblasts by elevated levels of alpha-smooth muscle actin (α-SMA) (Vierhout et al., 2021). Throughout the fibrotic process in the kidney, myofibroblasts generate collagens (type I, III, IV, V, and VI) and produce fibrillar matrix components such as fibronectins, fibrillins, and deposit connective matrix elastins and tenascins, as well as smaller molecules like proteoglycans and matricellular proteins such as MMPs and TIMPs (Klingberg et al., 2013). Protein matrices aid in tissue degradation, blood flow inhibition, and structural stiffening. Myofibroblast release of cytokines and chemokines like IL-1α, IL-1
While subject to controversy, fibroblasts, pericytes, endothelial cells, and macrophages collectively contribute significantly to myofibroblast formation in the renal interstitium during fibrosis. The ongoing debate over myofibroblast origin is fueled by differing methodologies used for cell fate tracing (Humphreys et al., 2010), and is further complicated by the impact of renal pathology on myofibroblast origin. LeBleu et al. reported that the contribution of epithelial-to-mesenchymal transition (EMT) to myofibroblasts is less than 5%, indicating a considerably lower role for this transition compared to demonstrated transformations of fibroblasts, pericytes, endothelial cells, and macrophages (Oldfield et al., 2001; Iwano et al., 2002; LeBleu et al., 2013; Falke et al., 2015). In several studies, renal perivascular fibroblasts have been shown to contribute to the myofibroblast pool during fibrosis, as much as 94% as determined by P0-Cre lineage-labelling (Asada et al., 2011). Pericytes, constituting approximately 2.5%–5% of all kidney cells also destabilize capillaries, leading to peritubular capillary rarefaction (Schrimpf and Duffield, 2011). In rat angiotensin II-induced kidney injury, pericyte transformation contributed to approximately 87% of myofibroblast formation (Faulkner et al., 2005). Conversely, in mice with unilateral ureteral obstruction (UUO), pericyte transformation accounted for either 6% or 0% of myofibroblast formation (LeBleu et al., 2013). Endothelial-to-mesenchymal transition (EndoMT) determined by double labelling with pan-endothelial marker, platelet endothelial cell adhesion molecule 1 (PECAM-1), αSMA, and Tie2-YFP lineage-traced cells in rodent models of kidney disease discovered that the EndoMT process contributed to ∼40% of all myofibroblasts (Zeisberg et al., 2008). Circulating monocyte-derived macrophages play a key role in renal injury, secreting pro-inflammatory cytokines and undergoing myofibroblast transformation. Studies on obstructive nephropathy reveal their contribution to the myofibroblast pool, ranging from 35% to as much as 90%, as observed through red fluorescent transgene tracing controlled by Acta2 and identification using F4/80 and α-SMA markers (LeBleu et al., 2013; Vierhout et al., 2021).
Transforming growth factor beta (TGF-
Canonical TGF-
TGF-β/Smad pathway targets remain ineffective or have unintended impacts in renal fibrosis (Klinkhammer et al., 2017). Emerging molecular targets beyond the canonical TGF-
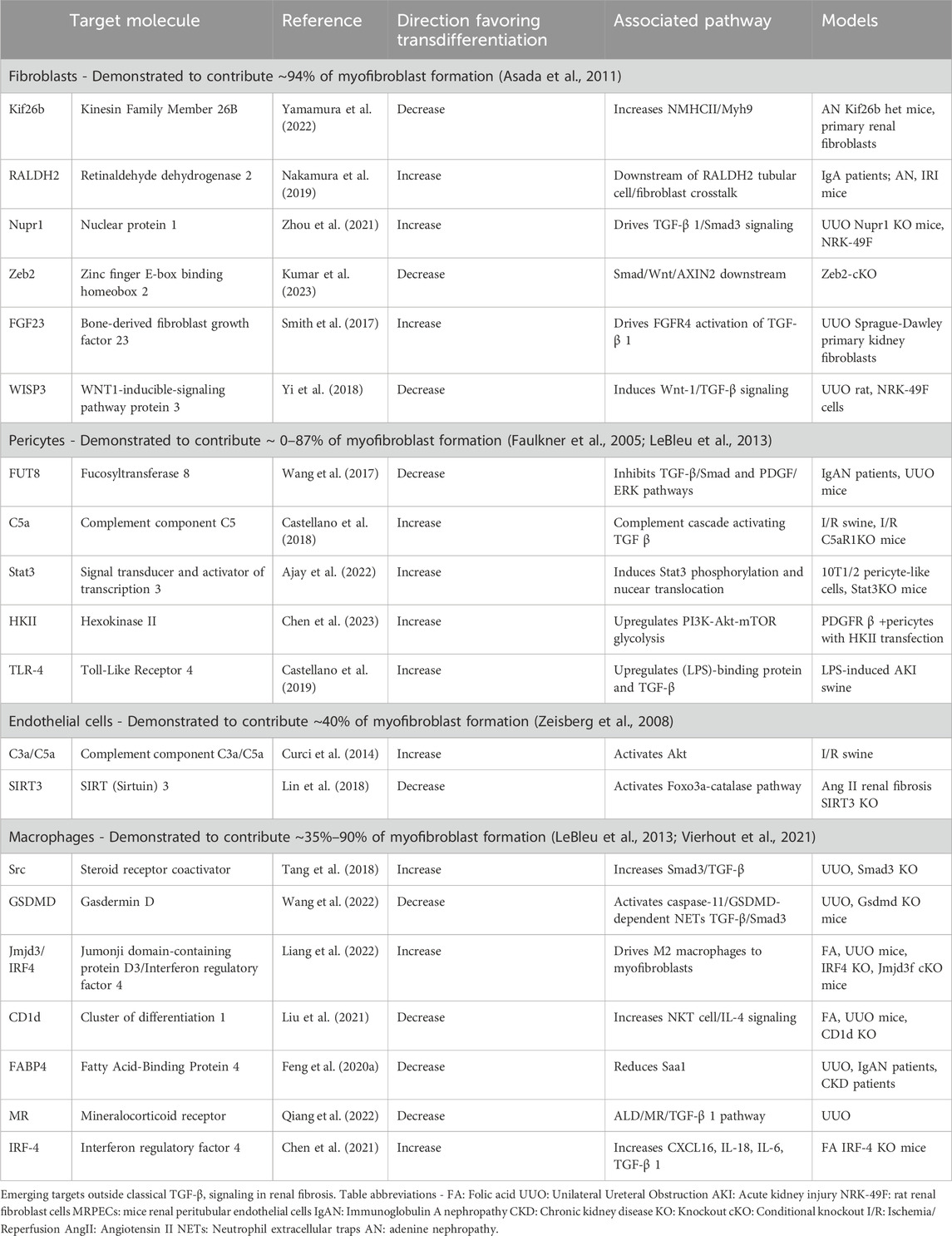
Table 1. Emerging targets outside classical TGF-β signaling in renal fibrosis.
The yes-associated protein (YAP) signaling pathway has been shown in renal models to regulate fibrosis (An et al., 2022). YAP is part of the Hippo signaling pathway known to control cell growth, proliferation and organ size (Piccolo et al., 2014). It also plays a role in promoting fibroblast activation and myofibroblast differentiation induced by Smad/TGF-
Associated with YAP molecular signaling, another molecule emerges-transcriptional coactivator with PDZ-binding motif (TAZ), a transcriptional coactivator also within the Hippo pathway. Recent studies conducted in both mouse and human tissues have unveiled a notable pattern: following kidney transplant injury and in mouse models of UUO and ischemia-reperfusion injury (IRI) injury, the levels of YAP and TAZ in myofibroblasts surge dramatically. However, when myofibroblast YAP/TAZ is in short supply, fibrosis in unilateral ureteral obstruction-induced kidneys is diminished. Conversely, an excess of fibroblast YAP/TAZ intensifies fibrotic injury.
Furthermore, canonical transient receptor potential channel 6 (TRPC6), a nonselective cation channel, has been linked to TGF-
Lastly, TGF-
Studies utilizing pathway and gene expression analysis implicate miR-132 in pericyte transformation under TGF-
Zinc finger E-box binding homeobox 2 (ZEB2), a promoter region with binding sites for FOXD1, exerts inhibitory control over both transcription factor, Zeb2 and decorin (DCN). An intriguing aspect of DCN, a proteoglycan, is that it sequesters TGF-
In a comprehensive study investigating the impact of inflammation-induced upregulation of vitamin D receptors (VDR) in the kidney, a significant link was established between VDR activators and the conversion of pericytes into myofibroblasts. This study examined the influence of VDR activators in the context of hypoxia or in the presence of TGF-
In UUO mice induced for kidney fibrosis, matrix metalloproteinase 9 (MMP-9) knockouts have reduced EndoMT, evidenced by decreased histological VE-cadherin and α-SMA colocalization compared to wildtype controls. Primary endothelial cells (MRPECs) treated with recombinant TGF-
While ERK1/2 signaling is crucial in development and homeostasis, modest pharmacological intervention to prevent inhibition of ERK1/2 signaling may halt endothelial-to-myofibroblast transformation (Cao et al., 2019). Mice lacking global ERK1 and containing endothelial-specific ERK2 (Erk1−/− Erk2iEC−/−) by two and 3 weeks of age undergo myofibroblast transition of endothelial cells as indicated by VE cadherin + αSMA + expression in the kidney. By 5 weeks of age, these mice succumb to organ failure, primarily a result of fibrosis of the heart. An increase of EndoMT was replicated through siRNA inhibition of ERK1/2 signaling of human endothelial cells (HUVECs) when compared to wildtype controls. RNA Seq analysis of these siRNA treated cells and scrambled controls indicated a gene hub that consisted of a TGF-
MiRNA-driven myofibroblast transformation is evident in endothelial cells, as indicated by a study highlighting the regulatory role of miR-126-3p. Endothelial derived myofibroblasts were traced by YFP under a Cdh5 promoter in mice which were induced to kidney fibrosis by unilateral ureteral obstruction. Five days post-operation, YFP + cells differentiated within the glomeruli, capillaries, and blood vessel intima, and represented 9% of αSMA + cells. Examination via RT-qPCR and in situ hybridization of kidney sections revealed a significant downregulation of miR-126-3p in fibrotic mouse kidneys compared to healthy kidneys. This downregulation was also observed in fibrotic human kidneys compared to normal kidneys. Diminished levels of miR-126-3p are evident in diverse injury contexts, including human renal IRI and myocardial infarction, indicating its prospective utility as a valuable molecular biomarker for disease monitoring and a potential target for intervention (Jordan et al., 2021).
Studies utilizing chromatin immunoprecipitation (ChIP) techniques have identified POU Class 4 Homeobox 1 (Pou4f1) as a downstream target of Smad3 and a regulator of macrophage-to-myofibroblast transition (MMT). Microarray analysis further revealed Pou4f1 as a pivotal node in a fibrogenic gene network that promotes TGF-
The TGF-
In a concurrent signaling cascade, the A2B adenosine receptor, notable for its ability to initiate G protein signaling that sets in motion a multitude of intracellular processes, such as alterations in gene expression, ion channel activity, and metabolism, showcases the potential to amplify the TGF-
Another receptor pathway-the aldosterone mineralocorticoid receptor (MR) has been shown to aide the transition of M1 macrophages to myofibroblasts upon stimulation of TGF-
Lastly, analyzing the transcriptome of macrophages treated with TGF-
It is important to recognize that often the mechanism driving alterations in myofibroblast cells involves paracrine cross-talk. Tubular cells, when damaged during renal fibrosis, transition to a secretory phenotype and generate fibrogenic agents such as sonic hedgehog (Shh), Wnt ligands, and TGF-
Since the hallmark of successful renal fibrosis resolution in disease models includes a decrease in myofibroblasts (Sun and Kisseleva, 2015), comprehending myofibroblast generation dynamics is key in identifying therapeutic targets in renal fibrotic diseases. In order to realistically carry out myofibroblast transdifferentiation inhibition it is reasonable to either alter the secretory phenotype of kidney resident cells or to block circulating recruitment into the renal interstitium of bone marrow derived macrophages fated for MMT (Kok et al., 2014; Meng et al., 2014; Tampe and Zeisberg, 2014). Targeting the TGF-
Expanding the search for targets outside of the classical TGF-
RL: Conceptualization, Data curation, Writing–original draft, Writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. The following funding sources are acknowledged: UK Regenerative Medicine Platform (Medical Research Council) award and CSO/Kidney Research UK post-doctoral training fellowship (CSO_PDF/2018/2).
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Adler S. G., Schwartz S., Williams M. E., Arauz-Pacheco C., Bolton W. K., Lee T., et al. (2010). Phase 1 study of anti-CTGF monoclonal antibody in patients with diabetes and microalbuminuria. Clin. J. Am. Soc. Nephrol. 5 (8), 1420–1428. doi:10.2215/CJN.09321209
Ajay A. K., Zhao L., Vig S., Fujiwara M., Thakurela S., Jadhav S., et al. (2022). Deletion of STAT3 from Foxd1 cell population protects mice from kidney fibrosis by inhibiting pericytes trans-differentiation and migration. Cell Rep. 38 (10), 110473. doi:10.1016/j.celrep.2022.110473
An Y., Ren Y., Wang J., Zang J., Gao M., Wang H., et al. (2022). MST1/2 in PDGFRα(+) cells negatively regulates TGF-β-induced myofibroblast accumulation in renal fibrosis. Am. J. Physiol. Ren. Physiol. 322 (5), F512–f526. doi:10.1152/ajprenal.00367.2021
Asada N., Takase M., Nakamura J., Oguchi A., Asada M., Suzuki N., et al. (2011). Dysfunction of fibroblasts of extrarenal origin underlies renal fibrosis and renal anemia in mice. J. Clin. Investig. 121 (10), 3981–3990. doi:10.1172/JCI57301
Baum J., Duffy H. S. (2011). Fibroblasts and myofibroblasts: what are we talking about? J. Cardiovasc Pharmacol. 57 (4), 376–379. doi:10.1097/FJC.0b013e3182116e39
Bijkerk R., de Bruin R. G., van Solingen C., van Gils J. M., Duijs J. M., van der Veer E. P., et al. (2016). Silencing of microRNA-132 reduces renal fibrosis by selectively inhibiting myofibroblast proliferation. Kidney Int. 89 (6), 1268–1280. doi:10.1016/j.kint.2016.01.029
Black L. M., Lever J. M., Agarwal A. (2019). "Renal inflammation and fibrosis: a double-edged sword. J. Histochem Cytochem 67 (9), 663–681. doi:10.1369/0022155419852932
Cao L., Yuan X., Bao F., Lv W., He Z., Tang J., et al. (2019). Downregulation of HSPA2 inhibits proliferation via ERK1/2 pathway and endoplasmic reticular stress in lung adenocarcinoma. Ann. Transl. Med. 7 (20), 540. doi:10.21037/atm.2019.10.16
Cardenas A., Toledo C., Oyarzun C., Sepulveda A., Quezada C., Guillen-Gomez E., et al. (2013). Adenosine A(2B) receptor-mediated VEGF induction promotes diabetic glomerulopathy. Lab. Investig. 93 (1), 135–144. doi:10.1038/labinvest.2012.143
Castellano G., Franzin R., Stasi A., Divella C., Sallustio F., Pontrelli P., et al. (2018). Complement activation during ischemia/reperfusion injury induces pericyte-to-myofibroblast transdifferentiation regulating peritubular capillary lumen reduction through pERK signaling. Front. Immunol. 9, 1002. doi:10.3389/fimmu.2018.01002
Castellano G., Stasi A., Franzin R., Sallustio F., Divella C., Spinelli A., et al. (2019). LPS-binding protein modulates acute renal fibrosis by inducing pericyte-to-myofibroblast trans-differentiation through TLR-4 signaling. Int. J. Mol. Sci. 20 (15), 3682. doi:10.3390/ijms20153682
Chen L., Li X., Deng Y., Chen J., Huang M., Zhu F., et al. (2023). The PI3K-Akt-mTOR pathway mediates renal pericyte-myofibroblast transition by enhancing glycolysis through HKII. J. Transl. Med. 21 (1), 323. doi:10.1186/s12967-023-04167-7
Chen M., Wen X., Gao Y., Liu B., Zhong C., Nie J., et al. (2021). IRF-4 deficiency reduces inflammation and kidney fibrosis after folic acid-induced acute kidney injury. Int. Immunopharmacol. 100, 108142. doi:10.1016/j.intimp.2021.108142
Curci C., Castellano G., Stasi A., Divella C., Loverre A., Gigante M., et al. (2014). Endothelial-to-mesenchymal transition and renal fibrosis in ischaemia/reperfusion injury are mediated by complement anaphylatoxins and Akt pathway. Nephrol. Dial. Transpl. 29 (4), 799–808. doi:10.1093/ndt/gft516
Derynck R., Zhang Y. E. (2003). Smad-dependent and Smad-independent pathways in TGF-beta family signalling. Nature 425 (6958), 577–584. doi:10.1038/nature02006
Duffield J. S. (2014). Cellular and molecular mechanisms in kidney fibrosis. J. Clin. Investig. 124 (6), 2299–2306. doi:10.1172/JCI72267
El-Atifi M., Dreyfus M., Berger F., Wion D. (2015). Expression of CYP2R1 and VDR in human brain pericytes: the neurovascular vitamin D autocrine/paracrine model. Neuroreport 26 (5), 245–248. doi:10.1097/WNR.0000000000000328
Falke L. L., Gholizadeh S., Goldschmeding R., Kok R. J., Nguyen T. Q. (2015). Diverse origins of the myofibroblast—implications for kidney fibrosis. Nat. Rev. Nephrol. 11 (4), 233–244. doi:10.1038/nrneph.2014.246
Faulkner J. L., Szcykalski L. M., Springer F., Barnes J. L. (2005). Origin of interstitial fibroblasts in an accelerated model of angiotensin II-induced renal fibrosis. Am. J. Pathol. 167 (5), 1193–1205. doi:10.1016/S0002-9440(10)61208-4
Feng Y., Guo F., Mai H., Liu J., Xia Z., Zhu G., et al. (2020a). Pterostilbene, a bioactive component of blueberries, alleviates renal interstitial fibrosis by inhibiting macrophage-myofibroblast transition. Am. J. Chin. Med. 48 (7), 1715–1729. doi:10.1142/S0192415X20500858
Feng Y., Guo F., Xia Z., Liu J., Mai H., Liang Y., et al. (2020b). Inhibition of fatty acid-binding protein 4 attenuated kidney fibrosis by mediating macrophage-to-myofibroblast transition. Front. Immunol. 11, 566535. doi:10.3389/fimmu.2020.566535
Fetting J. L., Guay J. A., Karolak M. J., Iozzo R. V., Adams D. C., Maridas D. E., et al. (2014). FOXD1 promotes nephron progenitor differentiation by repressing decorin in the embryonic kidney. Development 141 (1), 17–27. doi:10.1242/dev.089078
Garber K. (2009). Companies waver in efforts to target transforming growth factor beta in cancer. J. Natl. Cancer Inst. 101 (24), 1664–1667. doi:10.1093/jnci/djp462
Hathaway C. K., Gasim A. M., Grant R., Chang A. S., Kim H. S., Madden V. J., et al. (2015). Low TGFβ1 expression prevents and high expression exacerbates diabetic nephropathy in mice. Proc. Natl. Acad. Sci. U. S. A. 112 (18), 5815–5820. doi:10.1073/pnas.1504777112
Humphreys B. D. (2018). Mechanisms of renal fibrosis. Annu. Rev. Physiol. 80, 309–326. doi:10.1146/annurev-physiol-022516-034227
Humphreys B. D., Lin S. L., Kobayashi A., Hudson T. E., Nowlin B. T., Bonventre J. V., et al. (2010). Fate tracing reveals the pericyte and not epithelial origin of myofibroblasts in kidney fibrosis. Am. J. Pathol. 176 (1), 85–97. doi:10.2353/ajpath.2010.090517
Iwano M., Plieth D., Danoff T. M., Xue C., Okada H., Neilson E. G. (2002). Evidence that fibroblasts derive from epithelium during tissue fibrosis. J. Clin. Investig. 110 (3), 341–350. doi:10.1172/JCI15518
Jiang S., Gu L., Hu Y., Ren Y., Yang Z., Chai C., et al. (2022). Inhibition of TRPC6 suppressed TGFβ-induced fibroblast-myofibroblast transdifferentiation in renal interstitial NRK-49F cells. Exp. Cell Res. 421 (1), 113374. doi:10.1016/j.yexcr.2022.113374
Jordan N. P., Tingle S. J., Shuttleworth V. G., Cooke K., Redgrave R. E., Singh E., et al. (2021). MiR-126-3p is dynamically regulated in endothelial-to-mesenchymal transition during fibrosis. Int. J. Mol. Sci. 22 (16), 8629. doi:10.3390/ijms22168629
Klingberg F., Hinz B., White E. S. (2013). The myofibroblast matrix: implications for tissue repair and fibrosis. J. Pathol. 229 (2), 298–309. doi:10.1002/path.4104
Klinkhammer B. M., Goldschmeding R., Floege J., Boor P. (2017). Treatment of renal fibrosis-turning challenges into opportunities. Adv. Chronic Kidney Dis. 24 (2), 117–129. doi:10.1053/j.ackd.2016.11.002
Kok H. M., Falke L. L., Goldschmeding R., Nguyen T. Q. (2014). Targeting CTGF, EGF and PDGF pathways to prevent progression of kidney disease. Nat. Rev. Nephrol. 10 (12), 700–711. doi:10.1038/nrneph.2014.184
Kumar S., Fan X., Rasouly H. M., Sharma R., Salant D. J., Lu W. (2023). ZEB2 controls kidney stromal progenitor differentiation and inhibits abnormal myofibroblast expansion and kidney fibrosis. JCI Insight 8 (1), e158418. doi:10.1172/jci.insight.158418
Lacouture M. E., Morris J. C., Lawrence D. P., Tan A. R., Olencki T. E., Shapiro G. I., et al. (2015). Cutaneous keratoacanthomas/squamous cell carcinomas associated with neutralization of transforming growth factor β by the monoclonal antibody fresolimumab (GC1008). Cancer Immunol. Immunother. 64 (4), 437–446. doi:10.1007/s00262-015-1653-0
Lamouille S., Xu J., Derynck R. (2014). Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 15 (3), 178–196. doi:10.1038/nrm3758
LeBleu V. S., Taduri G., O'Connell J., Teng Y., Cooke V. G., Woda C., et al. (2013). Origin and function of myofibroblasts in kidney fibrosis. Nat. Med. 19 (8), 1047–1053. doi:10.1038/nm.3218
Li J., Bertram J. F. (2010). Review: endothelial-myofibroblast transition, a new player in diabetic renal fibrosis. Nephrol. Carlt. 15 (5), 507–512. doi:10.1111/j.1440-1797.2010.01319.x
Li N., Wang Z., Gao F., Lei Y., Li Z. (2020). Melatonin ameliorates renal fibroblast-myofibroblast transdifferentiation and renal fibrosis through miR-21-5p regulation. J. Cell Mol. Med. 24 (10), 5615–5628. doi:10.1111/jcmm.15221
Liang H., Liu B., Gao Y., Nie J., Feng S., Yu W., et al. (2022). Jmjd3/IRF4 axis aggravates myeloid fibroblast activation and m2 macrophage to myofibroblast transition in renal fibrosis. Front. Immunol. 13, 978262. doi:10.3389/fimmu.2022.978262
Lim J. H., Yook J. M., Oh S. H., Jeon S. J., Noh H. W., Jung H. Y., et al. (2021). Paricalcitol improves hypoxia-induced and TGF-β1-induced injury in kidney pericytes. Int. J. Mol. Sci. 22 (18), 9751. doi:10.3390/ijms22189751
Lin J. R., Zheng Y. J., Zhang Z. B., Shen W. L., Li X. D., Wei T., et al. (2018). Suppression of endothelial-to-mesenchymal transition by SIRT (sirtuin) 3 alleviated the development of hypertensive renal injury. Hypertension 72 (2), 350–360. doi:10.1161/HYPERTENSIONAHA.118.10482
Liu B., Jiang J., Liang H., Xiao P., Lai X., Nie J., et al. (2021). Natural killer T cell/IL-4 signaling promotes bone marrow-derived fibroblast activation and M2 macrophage-to-myofibroblast transition in renal fibrosis. Int. Immunopharmacol. 98, 107907. doi:10.1016/j.intimp.2021.107907
Liu F., Zhuang S. (2019). New therapies for the treatment of renal fibrosis. Adv. Exp. Med. Biol. 1165, 625–659. doi:10.1007/978-981-13-8871-2_31
Liu M., Ning X., Li R., Yang Z., Yang X., Sun S., et al. (2017). Signalling pathways involved in hypoxia-induced renal fibrosis. J. Cell Mol. Med. 21 (7), 1248–1259. doi:10.1111/jcmm.13060
Loeffler I., Wolf G. (2015). Epithelial-to-Mesenchymal transition in diabetic nephropathy: fact or fiction? Cells 4 (4), 631–652. doi:10.3390/cells4040631
Maarouf O. H., Ikeda Y., Humphreys B. D. (2015). Wnt signaling in kidney tubulointerstitium during disease. Histol. Histopathol. 30 (2), 163–171. doi:10.14670/HH-30.163
Mack M., Yanagita M. (2015). Origin of myofibroblasts and cellular events triggering fibrosis. Kidney Int. 87 (2), 297–307. doi:10.1038/ki.2014.287
Mariasegaram M., Tesch G. H., Verhardt S., Hurst L., Lan H. Y., Nikolic-Paterson D. J. (2010). Lefty antagonises TGF-beta1 induced epithelial-mesenchymal transition in tubular epithelial cells. Biochem. Biophys. Res. Commun. 393 (4), 855–859. doi:10.1016/j.bbrc.2010.02.098
Meng X. M., Nikolic-Paterson D. J., Lan H. Y. (2014). Inflammatory processes in renal fibrosis. Nat. Rev. Nephrol. 10 (9), 493–503. doi:10.1038/nrneph.2014.114
Meng X. M., Nikolic-Paterson D. J., Lan H. Y. (2016). TGF-β: the master regulator of fibrosis. Nat. Rev. Nephrol. 12 (6), 325–338. doi:10.1038/nrneph.2016.48
Moustakas A., Heldin C. H. (2005). Non-Smad TGF-beta signals. J. Cell Sci. 118 (Pt 16), 3573–3584. doi:10.1242/jcs.02554
Nakamura J., Sato Y., Kitai Y., Wajima S., Yamamoto S., Oguchi A., et al. (2019). Myofibroblasts acquire retinoic acid-producing ability during fibroblast-to-myofibroblast transition following kidney injury. Kidney Int. 95 (3), 526–539. doi:10.1016/j.kint.2018.10.017
Neelisetty S., Alford C., Reynolds K., Woodbury L., Nlandu-Khodo S., Yang H., et al. (2015). Renal fibrosis is not reduced by blocking transforming growth factor-β signaling in matrix-producing interstitial cells. Kidney Int. 88 (3), 503–514. doi:10.1038/ki.2015.51
Oldfield M. D., Bach L. A., Forbes J. M., Nikolic-Paterson D., McRobert A., Thallas V., et al. (2001). Advanced glycation end products cause epithelial-myofibroblast transdifferentiation via the receptor for advanced glycation end products (RAGE). J. Clin. Investig. 108 (12), 1853–1863. doi:10.1172/JCI11951
Piccolo S., Dupont S., Cordenonsi M. (2014). The biology of YAP/TAZ: hippo signaling and beyond. Physiol. Rev. 94 (4), 1287–1312. doi:10.1152/physrev.00005.2014
Qiang P., Hao J., Yang F., Han Y., Chang Y., Xian Y., et al. (2022). Esaxerenone inhibits the macrophage-to-myofibroblast transition through mineralocorticoid receptor/TGF-β1 pathway in mice induced with aldosterone. Front. Immunol. 13, 948658. doi:10.3389/fimmu.2022.948658
Qin W., Chung A. C., Huang X. R., Meng X. M., Hui D. S., Yu C. M., et al. (2011). TGF-β/Smad3 signaling promotes renal fibrosis by inhibiting miR-29. J. Am. Soc. Nephrol. 22 (8), 1462–1474. doi:10.1681/ASN.2010121308
Ren S., Duffield J. S. (2013). Pericytes in kidney fibrosis. Curr. Opin. Nephrol. Hypertens. 22 (4), 471–480. doi:10.1097/MNH.0b013e328362485e
Ricard N., Scott R. P., Booth C. J., Velazquez H., Cilfone N. A., Baylon J. L., et al. (2019). Endothelial ERK1/2 signaling maintains integrity of the quiescent endothelium. J. Exp. Med. 216 (8), 1874–1890. doi:10.1084/jem.20182151
Roman J., Mutsaers S. E. (2018). Epigenetic control of CXCL10: regulating the counterregulator in idiopathic pulmonary fibrosis. Am. J. Respir. Cell Mol. Biol. 58 (4), 419–420. doi:10.1165/rcmb.2017-0389ED
Schrimpf C., Duffield J. S. (2011). Mechanisms of fibrosis: the role of the pericyte. Curr. Opin. Nephrol. Hypertens. 20 (3), 297–305. doi:10.1097/MNH.0b013e328344c3d4
Smith E. R., Holt S. G., Hewitson T. D. (2017). FGF23 activates injury-primed renal fibroblasts via FGFR4-dependent signalling and enhancement of TGF-β autoinduction. Int. J. Biochem. Cell Biol. 92, 63–78. doi:10.1016/j.biocel.2017.09.009
Smith S. W., Schrimpf C., Parekh D. J., Venkatachalam M., Duffield J. S. (2012). Kidney pericytes: a novel therapeutic target in interstitial fibrosis. Histol. Histopathol. 27 (12), 1503–1514. doi:10.14670/HH-27.1503
Sun M., Kisseleva T. (2015). Reversibility of liver fibrosis. Clin. Res. Hepatol. Gastroenterol. 39 (Suppl. 1), S60–S63. doi:10.1016/j.clinre.2015.06.015
Sun Y. B., Qu X., Caruana G., Li J. (2016). The origin of renal fibroblasts/myofibroblasts and the signals that trigger fibrosis. Differentiation 92 (3), 102–107. doi:10.1016/j.diff.2016.05.008
Tager A. M., Kradin R. L., LaCamera P., Bercury S. D., Campanella G. S., Leary C. P., et al. (2004). Inhibition of pulmonary fibrosis by the chemokine IP-10/CXCL10. Am. J. Respir. Cell Mol. Biol. 31 (4), 395–404. doi:10.1165/rcmb.2004-0175OC
Tampe D., Zeisberg M. (2014). Potential approaches to reverse or repair renal fibrosis. Nat. Rev. Nephrol. 10 (4), 226–237. doi:10.1038/nrneph.2014.14
Tang P. M., Zhang Y. Y., Xiao J., Tang P. C., Chung J. Y., Li J., et al. (2020). Neural transcription factor Pou4f1 promotes renal fibrosis via macrophage-myofibroblast transition. Proc. Natl. Acad. Sci. U. S. A. 117 (34), 20741–20752. doi:10.1073/pnas.1917663117
Tang P. M., Zhou S., Li C. J., Liao J., Xiao J., Wang Q. M., et al. (2018). The proto-oncogene tyrosine protein kinase Src is essential for macrophage-myofibroblast transition during renal scarring. Kidney Int. 93 (1), 173–187. doi:10.1016/j.kint.2017.07.026
Torres A., Munoz K., Nahuelpan Y., Ap R. S., Mendoza P., Jara C., et al. (2020). Intraglomerular monocyte/macrophage infiltration and macrophage-myofibroblast transition during diabetic nephropathy is regulated by the A(2B) adenosine receptor. Cells 9 (4), 1051. doi:10.3390/cells9041051
Vierhout M., Ayoub A., Naiel S., Yazdanshenas P., Revill S. D., Reihani A., et al. (2021). Monocyte and macrophage derived myofibroblasts: is it fate? A review of the current evidence. Wound Repair Regen. 29 (4), 548–562. doi:10.1111/wrr.12946
Wang N., Deng Y., Liu A., Shen N., Wang W., Du X., et al. (2017). Novel mechanism of the pericyte-myofibroblast transition in renal interstitial fibrosis: core fucosylation regulation. Sci. Rep. 7 (1), 16914. doi:10.1038/s41598-017-17193-5
Wang S., Meng X. M., Ng Y. Y., Ma F. Y., Zhou S., Zhang Y., et al. (2016). TGF-β/Smad3 signalling regulates the transition of bone marrow-derived macrophages into myofibroblasts during tissue fibrosis. Oncotarget 7 (8), 8809–8822. doi:10.18632/oncotarget.6604
Wang Y., Li Y., Chen Z., Yuan Y., Su Q., Ye K., et al. (2022). GSDMD-dependent neutrophil extracellular traps promote macrophage-to-myofibroblast transition and renal fibrosis in obstructive nephropathy. Cell Death Dis. 13 (8), 693. doi:10.1038/s41419-022-05138-4
Wei J., Xu Z., Yan X. (2022). The role of the macrophage-to-myofibroblast transition in renal fibrosis. Front. Immunol. 13, 934377. doi:10.3389/fimmu.2022.934377
Wight T. N., Potter-Perigo S. (2011). The extracellular matrix: an active or passive player in fibrosis? Am. J. Physiol. Gastrointest. Liver Physiol. 301 (6), G950–G955. doi:10.1152/ajpgi.00132.2011
Wu C. F., Chiang W. C., Lai C. F., Chang F. C., Chen Y. T., Chou Y. H., et al. (2013). Transforming growth factor β-1 stimulates profibrotic epithelial signaling to activate pericyte-myofibroblast transition in obstructive kidney fibrosis. Am. J. Pathol. 182 (1), 118–131. doi:10.1016/j.ajpath.2012.09.009
Yamamura Y., Iwata Y., Furuichi K., Kato T., Yamamoto N., Horikoshi K., et al. (2022). Kif26b contributes to the progression of interstitial fibrosis via migration and myofibroblast differentiation in renal fibroblast. FASEB J. 36 (11), e22606. doi:10.1096/fj.202200355R
Yang J., Wang M., Zhu F., Sun J., Xu H., Chong Lee Shin O. L., et al. (2019). Putative endothelial progenitor cells do not promote vascular repair but attenuate pericyte-myofibroblast transition in UUO-induced renal fibrosis. Stem Cell Res. Ther. 10 (1), 104. doi:10.1186/s13287-019-1201-5
Yang M., Liu J. W., Zhang Y. T., Wu G. (2021). The role of renal macrophage, AIM, and TGF-β1 expression in renal fibrosis progression in IgAN patients. Front. Immunol. 12, 646650. doi:10.3389/fimmu.2021.646650
Yang X., Letterio J. J., Lechleider R. J., Chen L., Hayman R., Gu H., et al. (1999). Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-beta. EMBO J. 18 (5), 1280–1291. doi:10.1093/emboj/18.5.1280
Yang Y., Duan W., Jin Z., Yi W., Yan J., Zhang S., et al. (2013). JAK2/STAT3 activation by melatonin attenuates the mitochondrial oxidative damage induced by myocardial ischemia/reperfusion injury. J. Pineal Res. 55 (3), 275–286. doi:10.1111/jpi.12070
Yi Y., Ma J., Jianrao L., Wang H., Zhao Y. (2018). WISP3 prevents fibroblast-myofibroblast transdifferentiation in NRK-49F cells. Biomed. Pharmacother. 99, 306–312. doi:10.1016/j.biopha.2018.01.005
Zeisberg E. M., Potenta S. E., Sugimoto H., Zeisberg M., Kalluri R. (2008). Fibroblasts in kidney fibrosis emerge via endothelial-to-mesenchymal transition. J. Am. Soc. Nephrol. 19 (12), 2282–2287. doi:10.1681/ASN.2008050513
Zhao Y., Qiao X., Tan T. K., Zhao H., Zhang Y., Liu L., et al. (2017). Matrix metalloproteinase 9-dependent Notch signaling contributes to kidney fibrosis through peritubular endothelial-mesenchymal transition. Nephrol. Dial. Transpl. 32 (5), 781–791. doi:10.1093/ndt/gfw308
Zhou R., Liao J., Cai D., Tian Q., Huang E., Lü T., et al. (2021). Nupr1 mediates renal fibrosis via activating fibroblast and promoting epithelial-mesenchymal transition. Faseb J. 35 (3), e21381. doi:10.1096/fj.202000926RR

Keywords: fibrosis, myofibroblasts, fibroblast transition, extracellular matrix, kidney, renal fibrosis, TGF-beta, transdifferentiation
Citation: Lathan R (2024) Exploring unconventional targets in myofibroblast transdifferentiation outside classical TGF-
Received: 19 September 2023; Accepted: 22 April 2024;
Published: 14 May 2024.
Edited by:
Yanhan Dong, University of North Carolina at Chapel Hill, United StatesReviewed by:
Haofei Wang, University of North Carolina at Chapel Hill, United StatesCopyright © 2024 Lathan. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Rashida Lathan, cmFzaGlkYS5sYXRoYW5AZ2xhc2dvdy5hYy51aw==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.