 Ghazi-Abdullah Saroya1
Ghazi-Abdullah Saroya1 Erica Siismets1,2Max Hu1,3Christopher Panaretos1
Erica Siismets1,2Max Hu1,3Christopher Panaretos1 Adam Rice1†Kurt Reynolds4,5Chengji J. Zhou4,5
Adam Rice1†Kurt Reynolds4,5Chengji J. Zhou4,5 Vesa Kaartinen1*
Vesa Kaartinen1*- 1Department of Biologic and Materials Sciences, University of Michigan School of Dentistry, Ann Arbor, MI, United States
- 2Oral Health Sciences PhD Program, University of Michigan School of Dentistry, Ann Arbor, MI, United States
- 3College of Literature, Sciences and the Arts, University of Michigan, Ann Arbor, MI, United States
- 4School of Medicine, Institute for Pediatric Regenerative Medicine, Shriners Hospitals for Children-Northern California, University of California at Davis, Sacramento, CA, United States
- 5Department of Biochemistry and Molecular Medicine, School of Medicine, University of California at Davis, Sacramento, CA, United States
The secondary palate forms from two lateral primordia called the palatal shelves which form a contact in the midline, become adherent at the fusing interface (medial edge epithelia, MEE) and subsequently fuse. The gene encoding transforming growth factor-ß3 (Tgfb3) is strongly and specifically expressed in MEE cells. Our previous study suggested that Tgfb3 expression is controlled via upstream cis-regulatory elements in and around the neighboring Ift43 gene. Another study suggested that the canonical Wnt signaling via ß-Catenin is responsible for the MEE-specific Tgfb3 gene expression, since deletion of the Ctnnb1 gene by a commonly used Keratin 14-Cre (K14Cre) mouse line almost completely abolished Tgfb3 expression in the MEE resulting in cleft palate. Here, we wanted to analyze whether Tcf/Lef consensus binding sites located in the previously identified regions of the Ift43 gene are responsible for the spatiotemporal control of Tgfb3 expression during palatogenesis. We show that contrary to the previous report, deletion of the Ctnnb1 gene in basal MEE cells by the K14Cre driver (the same K14Cre mouse line was used as in the previous study referenced above) does not affect the MEE-specific Tgfb3 expression or TGFß3-dependent palatal epithelial fusion. All mutant embryos showed a lack of palatal rugae accompanied by other craniofacial defects, e.g., a narrow snout and a small upper lip, while only a small subset (<5%) of Ctnnb1 mutants displayed a cleft palate. Moreover, the K14Cre:Ctnnb1 embryos showed reduced levels and altered patterns of Shh expression. Our present data imply that epithelial ß-catenin may not be required for MEE-specific Tgfb3 expression or palatal epithelial fusion.
Introduction
Cleft palate, a common birth defect in humans, results from a failure in palatogenesis (Bush and Jiang, 2012; Lan et al., 2015). During this process, bilateral palatal primordia called palatal shelves grow out from the maxillary process of the first pharyngeal arch, elevating and fusing in the midline to form an intact secondary palate. Developing palatal shelves are mostly composed of the neural crest-derived mesenchyme and ectoderm-derived surface epithelium. The oral epithelium and developing palatal shelves are covered by a thin, one-cell layer thick tissue called the periderm. The periderm plays an important role in preventing aberrant fusions during oral development and must be eliminated at sites where fusion occurs, including epithelial tips of prefusion palatal shelves (Lan et al., 2015).
Palatal shelf growth and patterning are governed by complex reciprocal signaling processes between palatal mesenchymal and epithelial cells. Sonic hedgehog (Shh) expressed specifically in the oral epithelium in developing rugae is critical for controlling appropriate development of the size and shape of the palatal shelves, particularly in the anterior palate, by signaling to the adjacent mesenchyme (Bush and Jiang, 2012; Lane and Kaartinen, 2014).
During palatal shelf fusion, the palatal periderm must be eliminated before the underlying basal epithelial cells in the midline can become adherent (Hu et al., 2015; Richardson et al., 2017). How exactly this elimination takes place is currently controversial. It was initially thought that the periderm cells just slough off, undergo programmed cell death, or migrate toward the nasal and oral epithelia (Yoshida et al., 2012; Hu et al., 2015; Richardson et al., 2017). However, our recent studies suggest that the squamous periderm cells rapidly dedifferentiate into cuboidal epithelial cells (Saroya et al., 2023) incorporated into the midline seam, which soon disappears via mechanisms involving cell migration, convergence, and protrusion, allowing mesenchymal continuity necessary for appropriate fusion (Kim et al., 2015; Teng et al., 2022).
Tgfb3 has been shown to be strongly and specifically expressed in both the basal (Fitzpatrick et al., 1990; Pelton et al., 1990) and peridermal (Lane et al., 2014) medial edge epithelium (MEE). Tgfb3 expression is prerequisite for successful palatal fusion, since mice lacking Tgfb3 display cleft palate with 100% penetrance (Kaartinen et al., 1995; Proetzel et al., 1995).
While signaling processes downstream of TGFβ3 are relatively well-understood (Bush and Jiang, 2012), very little is known about mechanisms governing Tgfb3 expression in MEE cells of prefusion palatal shelves. It has been suggested that mutations in the Foxe1 gene result in reduced Tgfb3 expression in the MEE and that Tgfb3 is a direct target of Foxe1 (Venza et al., 2011). Another study showed that deletion of the Ctnnb1 gene by using the K14Cre mouse line, which induces recombination in the basal MEE cells (Lane et al., 2014; Saroya et al., 2023), completely eliminates Tgfb3 expression in the MEE subsequently leading to a failure in palatal epithelial fusion (He et al., 2011).
To further characterize mechanisms targeting Tgfb3 expression to the MEE, we used the Tgfb3 BAC reporter lines (Lane et al., 2014) together with tissue-specific gene deletion and expression analyses. These assays suggested that Tgfb3 expression in MEE cells recombined with K14Cre may not be controlled by canonical Wnt signaling. Instead, the K14Cre:Ctnnb1 mutants displayed several previously reported phenotypes and expected alterations in Shh expression.
Experimental procedures
Animal care
This study was carried out in accordance with the recommendations of the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All the experiments involving animals described in this study were approved by the Animal Care and Use Committee of the University of Michigan-Ann Arbor (protocol number: PRO00004320).
Mouse lines
Generation of the #291 Tgfb3 reporter line has been described earlier (Lane et al., 2014). Epithelium-specific Ctnnb1 mutants with the Tgfb3 reporter were obtained by crossing mice heterozygous for the floxed Ctnnb1 and carrying the epithelial K14Cre driver (Andl et al., 2004) with homozygous floxed Ctnnb1 (Ctnnb1F/F) mice (Brault et al., 2001) carrying the #291 reporter (Lane et al., 2014). Ctnnb1F and mTmG reporter mice were obtained from the Jackson Laboratories. The mouse lines were maintained in a mixed (C57BL/6, 129SvJ and Black Swill) genetic background.
LacZ staining
Samples were briefly fixed in formaldehyde, then incubated in a solution of potassium ferrocyanide, ferricyanide, and X-Gal to develop color (Nagy et al., 2007). Samples were fixed overnight, imaged on the Leica M165FC stereomicroscope with DP73 and software.
DAPI dilactate staining
Samples were fixed overnight in formaldehyde, incubated in a 5 μg/mL solution of DAPI dilactate in PBS (Sandell et al., 2018) and imaged on fluorescence stereomicroscope as outlined above.
Histology and immunohistochemistry
Samples were dissected, fixed in 4% PFA, dehydrated in alcohol, and embedded in paraffin wax. 8 μm sections were stained with Hematoxylin and Eosin. Sections were viewed and documented using an Olympus BX51 microscope and an Olympus DP71 digital camera. For immunohistochemistry, tissues were fixed overnight in 4% paraformaldehyde, processed through sucrose gradient to OCT compound and embedded for cryo-sectioning. Sections were stained with β-Catenin (Cell Signaling #9587; dilution 1:200) and ZO1 (Invitrogen 33-9,100; dilution 1:200) primary antibodies and antibody binding was visualized with Alexafluor-488 and Alexafluor-594 -conjugated secondary antibodies (Life Technologies) on slides mounted with Prolong Diamond antifade/DAPI (ThermoFisher). Images were acquired by using a Leica DMi8 microscope controlled by Leica Application Suite X, 3.7.4.23463 software.
In Situ hybridization
ISH probes were made by performing in vitro RNA synthesis using digoxigenin-labeled ribonucleotides, RNA polymerase, and a linearized plasmid containing a cloned cDNA fragment of Tgfb3 or Shh as previously described (Moorman et al., 2001). The staining procedure was performed on thin sections or whole tissue pieces by first treating with proteinase, briefly fixing with formaldehyde, and incubating the tissue with the digoxigenin-labeled RNA probe. An alkaline phosphatase-conjugated anti-digoxigenin antibody was applied to bind to the RNA probe, and the staining pattern was visualized by incubation with the BM purple chromogenic substrate solution.
qRT-PCR
Palatal shelves were dissected from embryos and total RNA was isolated with the RNeasy kit (Qiagen Cat # 74104). Reverse transcription was performed with the RT2 First Strand kit (Qiagen Cat # 330401). QPCR was performed with Taqman probes for Tgfb3 (F-ccctggacaccaattactgc/R-tcaatataaagggggcgtaca), Shh (F-tccactgttctgtgaaagcag/R-gggacgtaagtccttcacca), and Actb (F-tgacaggatgcagaaggaga/R-cgctcaggaggagcaatg) as the housekeeping gene. 30mL reactions were quantified using Applied Biosystems ABI7300PCR and ViiA7 detection systems and software.
Micro-CT
Micro-CT scanning was performed with the µCT100 system (Scanco Medical, Bassersdorf, Switzerland) and scan settings: 12 µm voxel, 55 kVp, 109 μA, 0.5 mm AL filter, and 500 ms integration time. Image rendering and distance measurements were performed with the Microview software (Parallax Innovations). The landmarks used were published in Ho et al. (2015).
Statistical analysis
The Mann-Whitney non-parametric test was used to detect group differences for the qPCR results in Figure 2I (n = 10) and Figure 4C (n = 10). A multiple comparison-adjusted T-test was used to detect group differences for the micro-CT scanning measurements in Figure 3S (n = 3). An asterisk indicates a significant group difference with a false positive probability less than five percent (p < 0.05).
Results
K14Cre:Ctnnb1 mutant mice do not show a clear reduction in Tgfb3 expression
It was previously reported that epithelial-specific K14Cre:Ctnnb1 mutants show a nearly complete loss of Tgfb3 expression in medial edge epithelial cells, which results in a failure in palatogenesis (He et al., 2011). To better understand the mechanisms by which canonical Wnt signaling regulates Tgfb3 expression, we first tested whether epithelial-specific K14Cre:Ctnnb1 mutants display reduced or absent Tgfb3-lacZ BAC reporter activity. TheTgfb3-LacZ-BAC #291 (Figure 1) covers about 190-kb of mouse genomic sequence around the Tgfb3 gene and recapitulates the specific pattern of the endogenous Tgfb3 expression in the palatal epithelium (Lane et al., 2014). The commonly used K14Cre mouse line (Andl et al., 2004) induced efficient recombination of the mTmG reporter in the basal MEE cells, but not in periderm cells (Figure 1D). Similarly, immunostaining for β-Catenin implied that the Ctnnb1 gene is inactivate in the basal MEE of K14Cre:Ctnnb1 mutant embryos (Figure 1). Surprisingly, theTgfb3-LacZ-BAC #291 displayed similar reporter activity in the MEE of the control and K14Cre:Ctnnb1 mutant embryos (Figure 1). Next, we compared endogenous Tgfb3 expression between control and K14Cre:Ctnnb1 mutant mice to determine whether Tgfb3 expression was unaffected in Ctnnb1 mutants, or whether regulation of the reporter BAC differed from that of the endogenous Tgfb3 gene (Figure 2). Similar to the results of the reporter assay, in situ hybridization analyses did not reveal notable differences in Tgfb3 expression in the prefusion palatal epithelium between control and mutant samples. Prefusion palatal shelf tissues were harvested from control and mutant embryos and subjected to quantitative real-time PCR (n = 10). Results demonstrated that instead of being lost or dramatically reduced, overall Tgfb3 mRNA levels were marginally increased in palatal tissues of K14Cre:Ctnnb1 mutant embryos when compared to those of control littermates.
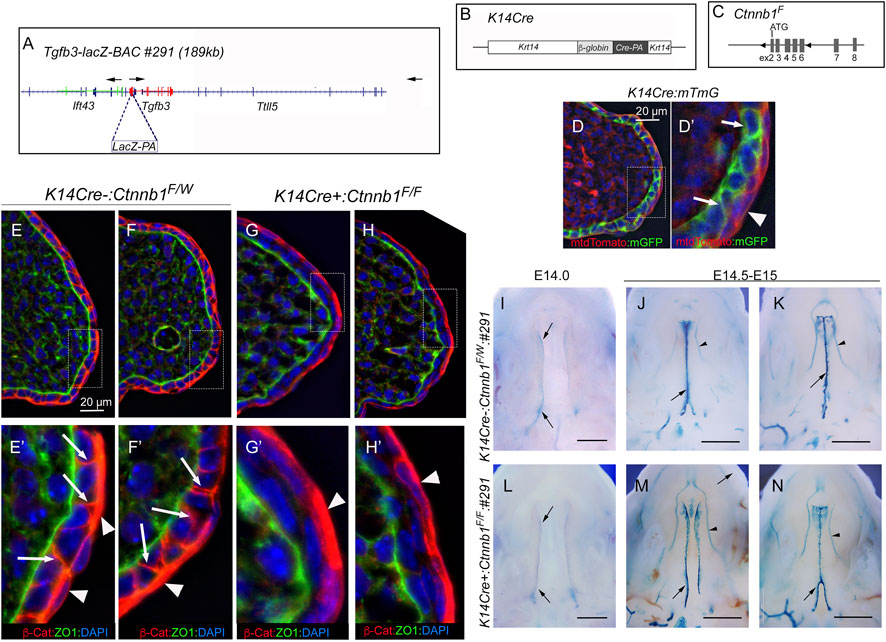
FIGURE 1. Tgfb3 lacZ reporter mice do not reveal differences in the MEE between K14Cre:Ctnnb1 mutant and control embryos. (A) Schematic presentation showing LacZ insertion into exon 1 of the Tgfb3 gene in BAC RP24-299H18. Arrows show the position and orientation of the Ift43, Tgfb3, and Ttll5 genes. (B) Schematic presentation of the K14Cre transgene. (C) Schematic presentation of the conditional Ctnnb1 allele. LoxP sites surround exons 2-6 of the Ctnnb1 gene. (D) K14Cre-induced recombination of the mTmG reporter cassette in pre-fusion palatal shelves. (D′) shows the high-power image of the field illustrated with a hatched box in (D). Basal epithelial cells recombined with K14Cre show membrane-bound green fluorescence [white arrows in (D′)]. K14Cre does not recombine in the palatal periderm (red membrane-bound fluorescence highlighted with a white arrowhead). Counterstaining with DAPI (blue nuclear fluorescence). Scale bar, 20 µm. (E–H) Immunostaining demonstrates a loss of ß-Catenin in mutants [(G, H); pre-fusion palatal shelves at E14.5; two separate embryos shown] when compared to control littermates [(E, F); two separate embryos shown]. (E′, F′, G′, H′) show the high-power images of the fields illustrated with hatched boxes in (E-H). White arrows in (E′, F′) point to adherent junctions of palatal basal epithelial cells. Similar staining cannot be seen in mutants (G′, H′). White arrowheads in (E′–H′) point to palatal periderm cells that are not recombined by K14Cre. ZO1 immunostaining was used to highlight the basement membrane. Counterstaining with DAPI (blue nuclear fluorescence), ß-cat, ß-catenin. Scale bar, 20 µm. (I–N) Wholemount LacZ staining showing similar ß-galactosidase activity in the MEE of K14Cre:Ctnnb1 mutant embryos (L–N) compared that of controls (I–K) at E14.0 (I, J, L) and E14.5-E15.0 (J, K, M, N). Black arrows, positive reporter activity in the MEE; black arrowheads, reporter activity in blood vessels. Scale bars in (I, L); 500 μm; (J, K, M, N); 1 mm.
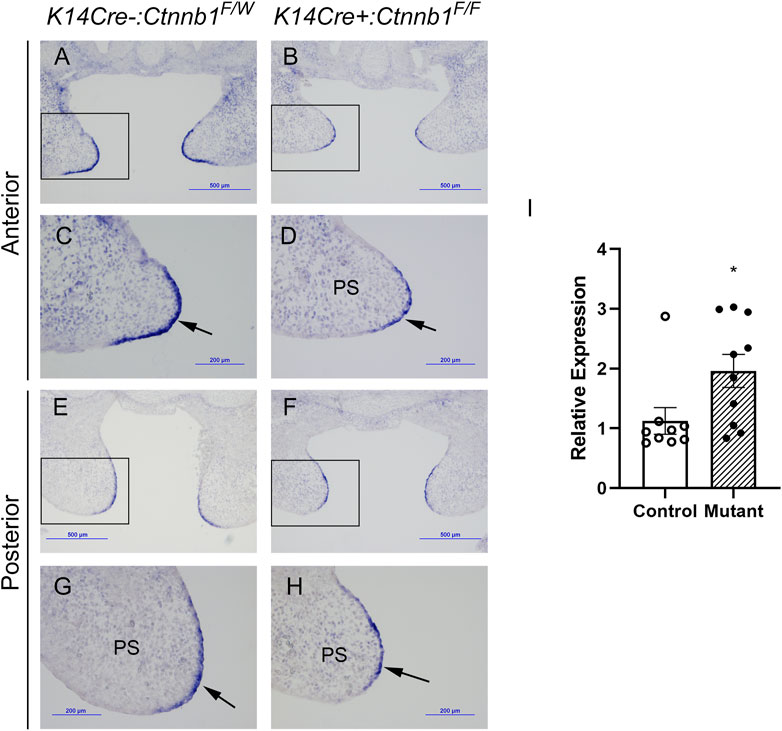
FIGURE 2. MEE-specific Tgfb3 expression does not differ between control and K14Cre:Ctnnb1 mutant embryos at E14.5. (A–H) In situ hybridization for Tgfb3 on frontal sections of epithelium-specific Ctnnb1 mutant (B, D, F, H) and control (A, C, E, G) embryos; Boxed area in (A, B, E, F), are shown as high-power images in (C, D, G, H), respectively. Black arrows point to the positive Tgfb3 signal in the MEE. (I) Quantitation of Tgfb3 mRNA using qRT-PCR. Control, open circles, and white column; mutant, closed circles and hatched column. Column height depicts the mean; error bar depicts the SEM. *Non-parametric test showed a group difference at the p < 0.05 significance level (n = 10).
K14Cre:Ctnnb1 mutants show several craniofacial defects while the palatal epithelial fusion is not affected
Next, we compared palatal and other craniofacial tissue tissues between control and mutant embryos. Consistent with the previous report (He et al., 2011), palatal rugae (Figures 3B’,D’) and whisker follicles (data not shown) were missing in Ctnnb1 mutants. Mutants also displayed open eyelids and smaller upper lips compared to controls (Figures 3K–N). Despite these defects, most of the mutants showed seemingly normal closure of the secondary palate (Figures 3A–D). Serial sectioning of control and mutant tissues confirmed that palate fusion in K14Cre:Ctnnb1 mutants was indistinguishable from that of controls throughout the anterior-posterior axis (Figures 3E–J). Among 64 K14Cre:Ctnnb1 mutants analyzed, only three of them displayed cleft palate. Micro-CT analysis showed that regardless of the cleft palate phenotype, the maxilla of K14Cre:Ctnnb1 mutant embryos appeared wider and shorter than those of controls (Figures 3O–S). In addition, this analysis demonstrated that even in the embryo which displayed a normally fused palate, the palatine bones were smaller than those in controls.
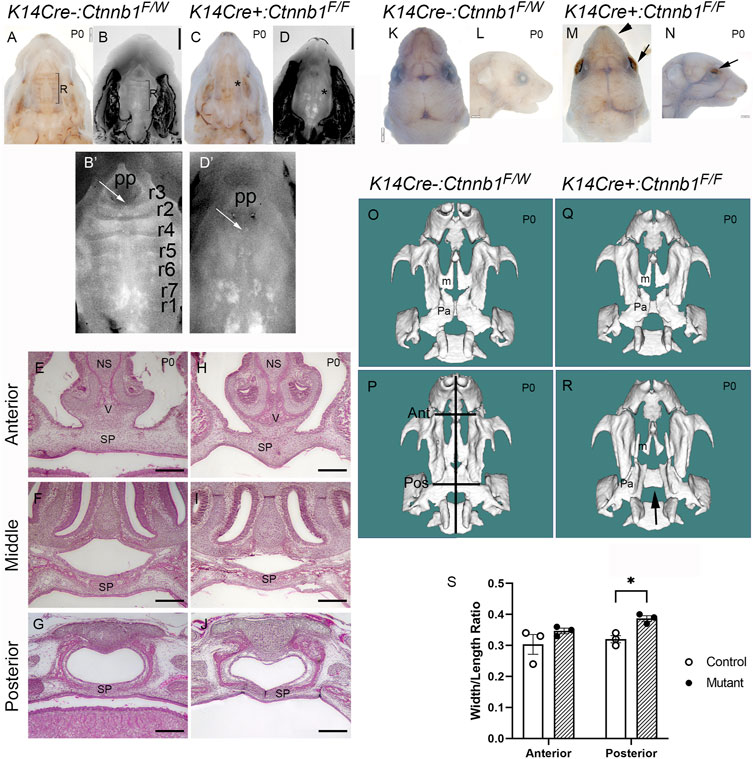
FIGURE 3. Craniofacial defects observed in K14Cre:Ctnnb1 mutant embryos. (A–D) Bright field (A, C) and DAPI dilactate (B, D) images showing fused palate of control (A, B) and K14Cre:Ctnnb1 mutant newborn mice (C, D). R in (A, B) depicts the rugae, * in (C, D) illustrates the absence of rugae. (B′, D′) magnified images shown in (B,D). White arrows point to incisive foramen; pp primary palate. Rugae in (B′) have been numbered as shown in ref (Baek et al., 2011). (E–J) H&E staining of frontal sections showing fused palate of control (E–G) and K14Cre:Ctnnb1 mutant (H–J) newborn mice (NS, nasal septum, SP, secondary Palate; V, Vomer) Scale bars, 500 µm. (K–N) Superior (K, M) and lateral (L, N) views of control (K, L) and K14Cre:Ctnnb1 mutant (M, N) newborn mice. Black arrows (M, N) point to open eyelids, black arrowhead in M points to abnormal upper lip. (O–R) CT scan renderings of control (O, P) and K14Cre:Ctnnb1 mutant (R–S) newborn mice. A mutant in (R) shows fused palate, while a mutant in (S) shows cleft palate (m, Maxillary bones; p, palatine bones; black arrows (S, T) point highlight the cleft palate. Black lines in P show the landmarks used for measurements in S; Ant, anterior; Pos, posterior).(S) Ratio of head length to anterior or posterior maxilla width. Control, open circles; mutant, closed circles. Column height depicts the mean; error bar depicts the SEM. *Multiple comparison-adjusted T-test showed a group difference at the p < 0.05 significance level (n = 3).
Shh expression levels are reduced in K14Cre:Ctnnb1 mutants
Ctnnb1 mutants displayed absence of rugae, of which development is dependent on Shh expression (Lee et al., 2011), and which specifically express Shh during normal palate development (Rice et al., 2006). Consistent with the lack of rugae, the K14Cre:Ctnnb1 mutant embryos failed to show characteristic Shh-staining patterns on the oral side of the developing hard palate (Figure 4), while the posterior oral palate displayed Shh-positive taste buds comparable to those of controls. Additionally, aberrant Shh-positive spots reminiscent of taste buds could be seen in the mutant hard palate. Control embryos displayed positive Shh staining in the primary palate, while comparable staining could not be seen in mutants. Concordant with the in situ hybridization findings, q-PCR showed that Shh expression levels were reduced in mutant palatal tissues harvested from prefusion palatal shelves when compared to controls.
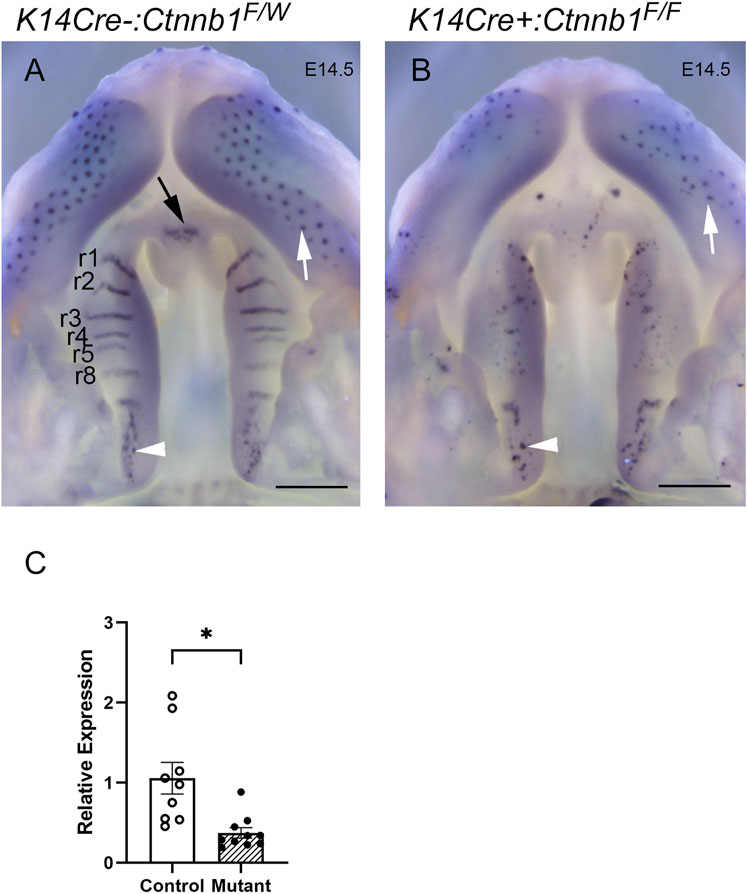
FIGURE 4. Shh expression in control and K14Cre:Ctnnb1 mutant embryos at E14.5. Wholemount in situ hybridization for Shh in control (A) and K14Cre:Ctnb1 mutant (B) embryos at E14.5. Black arrow (A) points to the positive signal in the nasal septum, white arrowheads (A, B) point to positive staining in taste buds in posterior palatal shelves, and white arrows (A, B) point to positive staining in hair follicles. Scale bars; 500 µm. (C) Quantitation of Shh mRNA using qRT-PCR. Control, open circles, and white column; mutant, closed circles and hatched column. Column height depicts the mean; error bar depicts the SEM. *Non-parametric test showed a group difference at the p < 0.05 significance level (n = 10).
Discussion
Palatogenesis is a complex developmental process involving reciprocal interactions between the mesenchyme and epithelium to govern tightly coordinated palatal shelf growth, elevation and fusion (Lan et al., 2015). Many previous studies have shown that Tgfb3 is strongly and specifically expressed in the MEE of the prefusion palatal shelves and that it plays a critical role in successful palatal epithelial fusion (Fitzpatrick et al., 1990; Pelton et al., 1990; Kaartinen et al., 1995; Proetzel et al., 1995). However, very little is known about molecular mechanisms responsible for spatiotemporal control of Tgfb3 expression during palatogenesis.
A previous study proposed that K14Cre:Ctnnb1 mice lacking ß-Catenin in basal epithelial cells develop cleft palate and display complete absence of Tgfb3 expression in the basal and periderm MEE. Using BAC transgenesis, we previously identified several distant cis-regulatory regions, which contributed the palate-specific Tgfb3 expression (Lane et al., 2014). With the help of these previously generated reporter lines, we aimed to further explore the mechanisms by which canonical Wnt signaling regulates Tgfb3 expression in MEE cells. Moreover, we aimed to clarify if indeed, the loss of Ctnnb1 in the basal MEE could have influenced Tgfb3 expression in the peridermal MEE, i.e., in cells in which K14Cre does not show the recombinase activity (Saroya et al., 2023). Our current results show that neither the Tgfb3 reporter activity nor the endogenous Tgfb3 expression levels were significantly affected in K14Cre:Ctnnb1 mutants. Concordant with appropriate Tgfb3 expression, palatal epithelial fusion occurred normally in most of the K14Cre:Ctnnb1 mutants, while some (<5%) displayed cleft palate. However, the K14Cre:Ctnnb1 mutant embryos consistently showed other phenotypes described in the previous study (He et al., 2011), e.g., a nearly complete absence of palatal rugae and notable reduction in Shh expression. Mutants also showed other craniofacial defects, including a narrow snout, small upper lip, open eyelids, and hind limb defects. Differences between the previous (He et al., 2011) and present studies could be attributed to variations in the genetic backgrounds of the mice used or discrepancies in the recombination efficiency within the K14Cre driver lines.
The role of Wnt signaling in rugae development and palate patterning is well-established (Lin et al., 2011), and many studies have examined interactions between Wnt and Hedgehog signaling processes in rugae patterning and palate morphogenesis (Lin et al., 2011; Sagai et al., 2017; Seo et al., 2018). Our studies agree with these earlier studies and confirm the critical role of canonical Wnt signaling in the regulation of Shh expression during palate formation and rugae patterning. Previous studies have shown that Shh expressed in the palatal epithelium acts on the mesenchyme, where it is required for appropriate growth and patterning of palatal shelves (Rice et al., 2004). The cleft palate observed with low penetrance in our K14Cre:Ctnnb1 mutants may possibly be caused by altered Shh expression affecting maxillary and palatal shelf growth.
Our present data suggest that, at least in our outbred background, epithelial ß-catenin may not be required for MEE-specific Tgfb3 expression and epithelial fusion of the secondary palate.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was reviewed and approved by University of Michigan IACUC.
Author contributions
G-AS, execution of experiments, manuscript preparations; ES, study design, execution of experiments; MH, study design, execution of experiments; CP, execution of experiments, manuscript preparations; AR, execution of experiments, study design; KR, execution of experiments; CZ, study design; VK, study design, manuscript preparation, overall supervision. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication. All authors contributed to the article and approved the submitted version.
Funding
This study was supported by a grant from the National Institute of Dental and Craniofacial Research, National Institutes of Health (2R01DE013085, 1R56DE026464 to VK).
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Andl, T., Ahn, K., Kairo, A., Chu, E. Y., Wine-Lee, L., Reddy, S. T., et al. (2004). Epithelial Bmpr1a regulates differentiation and proliferation in postnatal hair follicles and is essential for tooth development. Development 131 (10), 2257–2268. doi:10.1242/dev.01125
Baek, J. A., Lan, Y., Liu, H., Maltby, K. M., Mishina, Y., and Jiang, R. (2011). Bmpr1a signaling plays critical roles in palatal shelf growth and palatal bone formation. Dev. Biol. 350 (2), 520–531. doi:10.1016/j.ydbio.2010.12.028
Brault, V., Moore, R., Kutsch, S., Ishibashi, M., Rowitch, D. H., McMahon, A. P., et al. (2001). Inactivation of the beta-catenin gene by Wnt1-Cre-mediated deletion results in dramatic brain malformation and failure of craniofacial development. Development 128 (8), 1253–1264. doi:10.1242/dev.128.8.1253
Bush, J. O., and Jiang, R. (2012). Palatogenesis: Morphogenetic and molecular mechanisms of secondary palate development. Development 139 (2), 231–243. doi:10.1242/dev.067082
Fitzpatrick, D. R., Denhez, F., Kondaiah, P., and Akhurst, R. J. (1990). Differential expression of TGF beta isoforms in murine palatogenesis. Development 109 (3), 585–595. doi:10.1242/dev.109.3.585
He, F., Xiong, W., Wang, Y., Li, L., Liu, C., Yamagami, T., et al. (2011). Epithelial Wnt/β-catenin signaling regulates palatal shelf fusion through regulation of Tgfβ3 expression. Dev. Biol. 350 (2), 511–519. doi:10.1016/j.ydbio.2010.12.021
Ho, T. V., Iwata, J., Ho, H. A., Grimes, W. C., Park, S., Sanchez-Lara, P. A., et al. (2015). Integration of comprehensive 3D microCT and signaling analysis reveals differential regulatory mechanisms of craniofacial bone development. Dev. Biol. 400 (2), 180–190. doi:10.1016/j.ydbio.2015.02.010
Hu, L., Liu, J., Li, Z., Ozturk, F., Gurumurthy, C., Romano, R. A., et al. (2015). TGFβ3 regulates periderm removal through ΔNp63 in the developing palate. J. Cell. Physiol. 230 (6), 1212–1225. doi:10.1002/jcp.24856
Kaartinen, V., Voncken, J. W., Shuler, C., Warburton, D., Bu, D., Heisterkamp, N., et al. (1995). Abnormal lung development and cleft palate in mice lacking TGF-beta 3 indicates defects of epithelial-mesenchymal interaction. Nat. Genet. 11 (4), 415–421. doi:10.1038/ng1295-415
Kim, S., Lewis, A. E., Singh, V., Ma, X., Adelstein, R., and Bush, J. O. (2015). Convergence and extrusion are required for normal fusion of the Mammalian secondary palate. PLoS Biol. 13 (4), e1002122. doi:10.1371/journal.pbio.1002122
Lan, Y., Xu, J., and Jiang, R. (2015). Cellular and molecular mechanisms of palatogenesis. Curr. Top. Dev. Biol. 115, 59–84. doi:10.1016/bs.ctdb.2015.07.002
Lane, J., and Kaartinen, V. (2014). Signaling networks in palate development. Wiley Interdiscip. Rev. Syst. Biol. Med. 6 (3), 271–278. doi:10.1002/wsbm.1265
Lane, J., Yumoto, K., Pisano, J., Azhar, M., Thomas, P. S., and Kaartinen, V. (2014). Control elements targeting Tgfb3 expression to the palatal epithelium are located intergenically and in introns of the upstream Ift43 gene. Front. Physiol. 5, 258. doi:10.3389/fphys.2014.00258
Lee, J. M., Miyazawa, S., Shin, J. O., Kwon, H. J., Kang, D. W., Choi, B. J., et al. (2011). Shh signaling is essential for rugae morphogenesis in mice. Histochem Cell. Biol. 136 (6), 663–675. doi:10.1007/s00418-011-0870-7
Lin, C., Fisher, A. V., Yin, Y., Maruyama, T., Veith, G. M., Dhandha, M., et al. (2011). The inductive role of Wnt-beta-Catenin signaling in the formation of oral apparatus. Dev. Biol. 356 (1), 40–50. doi:10.1016/j.ydbio.2011.05.002
Moorman, A. F., Houweling, A. C., de Boer, P. A., and Christoffels, V. M. (2001). Sensitive nonradioactive detection of mRNA in tissue sections: Novel application of the whole-mount in situ hybridization protocol. JHistochemCytochem 49 (1), 1–8. doi:10.1177/002215540104900101
Nagy, A., Gertsenstein, M., Vintersten, K., and Behringer, R. (2007). Staining frozen mouse embryo sections for {beta}-Galactosidase (lacZ) activity. CSH Protoc. 2007, pdb.prot4726. doi:10.1101/pdb.prot4726
Pelton, R. W., Hogan, B. L., Miller, D. A., and Moses, H. L. (1990). Differential expression of genes encoding TGFs beta 1, beta 2, and beta 3 during murine palate formation. Dev. Biol. 141 (2), 456–460. doi:10.1016/0012-1606(90)90401-4
Proetzel, G., Pawlowski, S. A., Wiles, M. V., Yin, M., Boivin, G. P., Howles, P. N., et al. (1995). Transforming growth factor-beta 3 is required for secondary palate fusion. Nat. Genet. 11 (4), 409–414. doi:10.1038/ng1295-409
Rice, R., Connor, E., and Rice, D. P. (2006). Expression patterns of Hedgehog signalling pathway members during mouse palate development. Gene Expr. Patterns 6 (2), 206–212. doi:10.1016/j.modgep.2005.06.005
Rice, R., Spencer-Dene, B., Connor, E. C., Gritli-Linde, A., McMahon, A. P., Dickson, C., et al. (2004). Disruption of Fgf10/Fgfr2b-coordinated epithelial-mesenchymal interactions causes cleft palate. J. Clin. Invest. 113 (12), 1692–1700. doi:10.1172/JCI20384
Richardson, R., Mitchell, K., Hammond, N. L., Mollo, M. R., Kouwenhoven, E. N., Wyatt, N. D., et al. (2017). p63 exerts spatio-temporal control of palatal epithelial cell fate to prevent cleft palate. PLoS Genet. 13 (6), e1006828. doi:10.1371/journal.pgen.1006828
Sagai, T., Amano, T., Maeno, A., Kimura, T., Nakamoto, M., Takehana, Y., et al. (2017). Evolution of Shh endoderm enhancers during morphological transition from ventral lungs to dorsal gas bladder. Nat. Commun. 8, 14300. doi:10.1038/ncomms14300
Sandell, L., Inman, K., and Trainor, P. (2018). DAPI staining of whole-mount mouse embryos or fetal organs. Cold Spring Harb. Protoc. 2018 (10), pdb.prot094029. doi:10.1101/pdb.prot094029
Saroya, G., Hu, J., Hu, M., Panaretos, C., Mann, J., Kim, S., et al. (2023). Periderm fate during palatogenesis: TGF-β and periderm dedifferentiation. J. Dent. Res. 102(4):459–466. doi:10.1177/00220345221146454
Seo, H., Amano, T., Seki, R., Sagai, T., Kim, J., Cho, S. W., et al. (2018). Upstream enhancer elements of Shh regulate oral and dental patterning. J. Dent. Res. 97, 1055–1063. doi:10.1177/0022034518758642
Teng, T., Teng, C. S., Kaartinen, V., and Bush, J. O. (2022). A unique form of collective epithelial migration is crucial for tissue fusion in the secondary palate and can overcome loss of epithelial apoptosis. Development 149 (10), dev200181. doi:10.1242/dev.200181
Venza, I., Visalli, M., Parrillo, L., De Felice, M., Teti, D., and Venza, M. (2011). MSX1 and TGF-beta3 are novel target genes functionally regulated by FOXE1. Hum. Mol. Genet. 20 (5), 1016–1025. doi:10.1093/hmg/ddq547
Yoshida, M., Shimono, Y., Togashi, H., Matsuzaki, K., Miyoshi, J., Mizoguchi, A., et al. (2012). Periderm cells covering palatal shelves have tight junctions and their desquamation reduces the polarity of palatal shelf epithelial cells in palatogenesis. Genes. cells. 17 (6), 455–472. doi:10.1111/j.1365-2443.2012.01601.x
Keywords: beta-Catenin, TGF-beta3, canonical Wnt signal pathway, palatogenesis, cleft palate (CP)
Citation: Saroya G-A, Siismets E, Hu M, Panaretos C, Rice A, Reynolds K, Zhou CJ and Kaartinen V (2023) Canonical Wnt signaling is not required for Tgfb3 expression in the basal medial edge epithelium during palatogenesis. Front. Physiol. 14:704406. doi: 10.3389/fphys.2023.704406
Received: 02 May 2021; Accepted: 28 April 2023;
Published: 12 May 2023.
Edited by:
Pierfrancesco Pagella, Linköping University, SwedenReviewed by:
Katherine Ann Fantauzzo, University of Colorado Anschutz Medical Campus, United StatesJean-Pierre Saint-Jeannet, New York University, United States
Copyright © 2023 Saroya, Siismets, Hu, Panaretos, Rice, Reynolds, Zhou and Kaartinen. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Vesa Kaartinen, dmVzYWtAdW1pY2guZWR1
†Present addresses: Adam Rice, Konikoff Dental Associates Lynnhaven, Virginia Beach, VA, United States