
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 12 January 2024
Sec. Renal Physiology and Pathophysiology
Volume 14 - 2023 | https://doi.org/10.3389/fphys.2023.1344271
This article is part of the Research Topic Renal physiology: Epithelial Cell Mechanics View all 8 articles
 Wenbin Tang1,2
Wenbin Tang1,2 Qingqing Wei2*
Qingqing Wei2*Kidney injury and repair are accompanied by significant disruptions in metabolic pathways, leading to renal cell dysfunction and further contributing to the progression of renal pathology. This review outlines the complex involvement of various energy production pathways in glucose, lipid, amino acid, and ketone body metabolism within the kidney. We provide a comprehensive summary of the aberrant regulation of these metabolic pathways in kidney injury and repair. After acute kidney injury (AKI), there is notable mitochondrial damage and oxygen/nutrient deprivation, leading to reduced activity in glycolysis and mitochondrial bioenergetics. Additionally, disruptions occur in the pentose phosphate pathway (PPP), amino acid metabolism, and the supply of ketone bodies. The subsequent kidney repair phase is characterized by a metabolic shift toward glycolysis, along with decreased fatty acid β-oxidation and continued disturbances in amino acid metabolism. Furthermore, the impact of metabolism dysfunction on renal cell injury, regeneration, and the development of renal fibrosis is analyzed. Finally, we discuss the potential therapeutic strategies by targeting renal metabolic regulation to ameliorate kidney injury and fibrosis and promote kidney repair.
The kidney consumes a large amount of energy for its function to maintain body homeostasis by filtering blood, excreting waste products, and balancing electrolytes (Ronco et al., 2019; Singh, 2023). The energy metabolism in the kidney involves multiple pathways to actively generate ATPs and other metabolites essential for renal functions. Renal cells from different segments of the nephron may preferentially utilize one metabolic pathway over another, depending on their specific functions and energy requirements (Ronco et al., 2019). In normal kidneys, the energy sources primarily include glucose, fatty acids, amino acids, and ketone bodies (Gewin, 2021; Rojas-Morales et al., 2021).
Meanwhile, the kidney is an organ that is highly susceptible to acute injury, which can result from various risk factors such as ischemia, nephrotoxins, sepsis, or rhabdomyolysis (Zuk and Bonventre, 2016). During acute kidney injury (AKI), the renal cells, especially the proximal tubular cells, suffer from significant cell death associated with the deprivation of energy supply and/or dysregulation of energy metabolism, leading to acute loss of renal function (Basile et al., 2012). Following AKI, the kidney may undergo an adaptive repair to a complete recovery or a maladaptive repair to progress to chronic kidney disease (CKD). The maladaptive repair is featured by metabolic reprogramming in the kidney, shifting the major energy production from citric acid cycle/oxidative phosphorylation to glycolysis (Ferenbach and Bonventre, 2015; Wen et al., 2021). This metabolic reprogramming exerts divergent effects on different renal cells and further promotes tubular cell degeneration, inflammation, and fibrosis (Ding et al., 2017; Wei et al., 2019; Yang et al., 2023a; Xu et al., 2023). Overall, the dysregulation of metabolism in kidney injury and repair not only affects the kidney energy homeostasis through ATP production dysfunction but also influences various cellular functions via dual-functional enzymes and regulatory metabolites derived from different metabolic pathways.
In this review, we will introduce the typical energy metabolic pathways in the kidney and delve into the aberrant energy metabolism regulation in AKI and maladaptive repair. After exploring the distinct pathological roles of dysregulation of various metabolisms and the associated signaling pathways in renal cells, we will discuss the potential therapeutic strategies for AKI treatment and the prevention of its progression to CKD.
With glucose as the foundational fuel to generate ATP, carbohydrate catabolism is the predominant energy production mechanism in most renal cells except proximal tubular cells (Gewin, 2021). The catabolic pathways include the oxidative phosphorylation and citric acid cycle and the glycolysis pathway. The pentose phosphate pathway (PPP) is an alternative anabolism pathway from glycolysis, which is critical for NADPH synthesis and oxidative stress suppression.
Glycolysis is a central pathway in glucose catabolism and catalyzed by a series of enzymes located in the cytosol (Figure 1) (Janson and Tischler, 2018). Under aerobic conditions, this process yields pyruvate and ATP from glucose, while under anaerobic conditions, it results in the formation of lactate and ATP. In the presence of oxygen, the end product, pyruvate, can enter the mitochondria and be further converted to acetyl coenzyme A (acetyl-CoA), serving as a starting metabolite in the citric acid cycle. However, under anaerobic conditions, the accumulation of lactate may lead to acidosis.
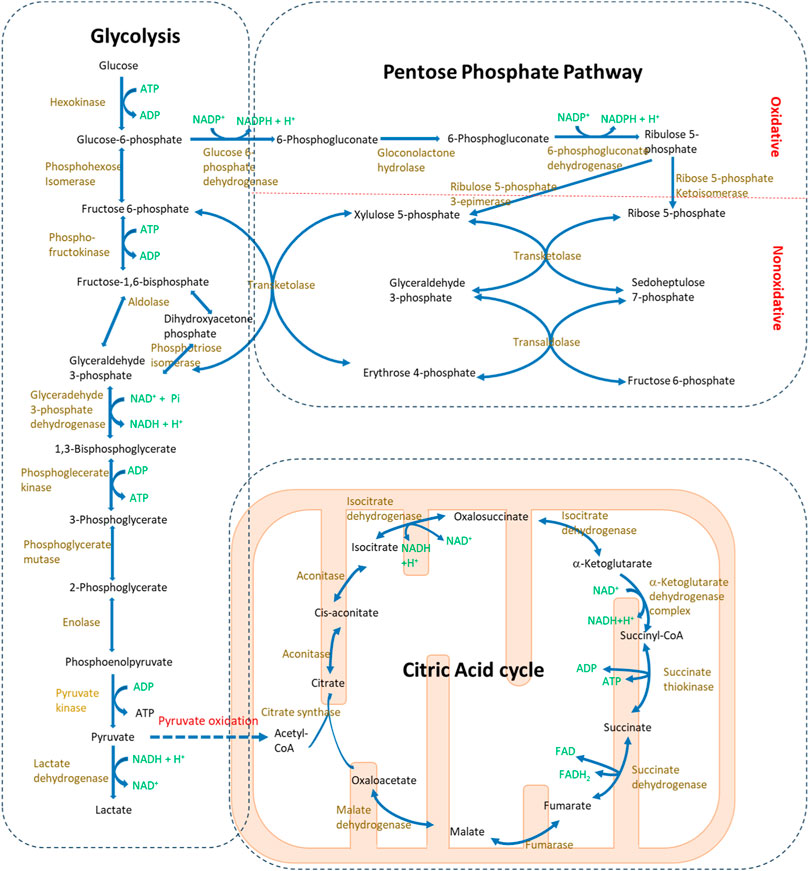
FIGURE 1. Glucose metabolism pathways in mammalian cells. Glucose serves as the fundamental substrate for bioenergy production, primarily through glycolysis and the citric acid cycle. During glycolysis, glucose is catabolized into pyruvate in the cytosol. Under anaerobic conditions, pyruvate is further converted to lactate. Conversely, in the presence of oxygen, pyruvate enters the mitochondria where it undergoes the citric acid cycle. Additionally, the pentose phosphate pathway, a branch metabolic pathway from glycolysis, is responsible for producing NADPH and supplying base metabolites for nucleotide synthesis.
Under normal conditions, the glycolysis level is not evenly distributed in the kidney. The inner medulla papilla and distal convoluted tubules are the major renal compartments showing high glycolytic activity (Ross et al., 1986). Furthermore, in the glomerulus, anaerobic glycolysis serves as the principal energy source for podocytes, playing a pivotal role in maintaining the function of the glomerular filtration barrier even with dysfunction of mitochondrial metabolism (Ozawa et al., 2015; Brinkkoetter et al., 2019). Although glycolysis levels are typically low in proximal tubular cells, a recent study indicates its crucial role in controlling phosphate homeostasis (Zhou et al., 2022a; Zhou et al., 2023). The blood phosphate can be sensed by proximal tubular cells, resulting in an increase in the glycolysis level. This process is characterized by enhanced activity of glyceraldehyde 3-phosphate dehydrogenase in the glycolysis pathway, which is coupled with the glycerol-3-phosphate dehydrogenase 1 activation through NAD/NADH balance modulation, leading to glycerol-3-phosphate (G-3-P) production. G-3-P is transported to the bone through blood, where it regulates the bone production of FGF23. This, in turn, provides feedback control over systemic phosphate levels by decreasing phosphate reabsorption in the proximal tubules and reducing intestinal phosphate uptake (Zhou et al., 2023).
The citric acid cycle, in conjunction with oxidative phosphorylation, represents the most efficient bioenergetic pathway for ATP production to ensure the renal cell function (Janson and Tischler, 2018). The entire reaction takes place in mitochondria (Figure 1). Acetyl-CoA, derived from pyruvate, enters the cycle and reacts with oxaloacetate to form citrate. Following a series of 10 enzymatically catalyzed reactions, the cycle concludes with the regeneration of oxaloacetate. During oxidative phosphorylation, the respiratory chain complexes receive electrons from the products of the citric acid cycle, facilitating the transport of protons from the mitochondrial matrix to the intermembrane space. The proton gradient between the intermembrane space and matrix drives the ATP production catalyzed by ATP synthase. Of note, mitochondria also produce a significant amount of reactive oxygen species (ROS) during oxidative phosphorylation (Tirichen et al., 2021), which plays a crucial role not only in transducing cellular signals but also in inducing oxidative stress and cellular damage in renal diseases.
The kidney cells predominantly depend on mitochondrial bioenergetics to meet the substantial energy demands required for solute reabsorption functions (Bhargava and Schnellmann, 2017). Notably, both proximal tubular cells and distal convoluted tubular cells are abundant in mitochondria (McCormick and Ellison, 2015; Bhargava and Schnellmann, 2017). Proximal tubular cells, in particular, heavily rely on the citric acid cycle and oxidative phosphorylation for energy supply, owing to their low glycolytic activity. Thus, they are most susceptible to oxygen deprivation in pathological conditions. In addition, the presence of high glucose can especially suppress mitochondrial respiration through the Crabtree effect (Darshi et al., 2023).
PPP is an anabolism pathway that branches from glycolysis (Figure 1) (Murray et al., 2012). Like glycolysis, the reactions of PPP occur in the cytosol; however, unlike glycolysis, PPP does not produce ATP. PPP can be divided into two phases: the irreversible oxidative phase and the reversible nonoxidative phase. The oxidative phase generates NADPH, which is essential for maintaining glutathione levels crucial for detoxification. The nonoxidative phase, on the other hand, produces ribose, vital for nucleotide and nucleic acid synthesis. PPP and glycolysis are intricately linked through shared metabolites, and alterations in the dynamics of one pathway inevitably influence the metabolic flux in the other.
Fatty acid β-oxidation is a pivotal process in lipid metabolism that converts free fatty acids into acetyl-CoA, which then enters the citric acid cycle and undergoes oxidative phosphorylation (Murray et al., 2012). These free fatty acids are transported to the kidney via the bloodstream and must be activated prior to β-oxidation. The entire β-oxidation process is aerobic and takes place in the mitochondria. Furthermore, peroxisomes assist in breaking down very-long-chain fatty acids before they are oxidized in the mitochondria. Collectively, fatty acid β-oxidation, when followed by the citric acid cycle and oxidative phosphorylation, provides the highest ATP yield compared to other energy substrates. Hence, it is the most favored energy metabolism pathway for proximal tubular cells (Jang et al., 2020a; Gao and Chen, 2022).
When fatty acid oxidation occurs at a high rate, ketone bodies are synthesized from acetyl-CoA by hepatocytes (Murray et al., 2012). These ketone bodies include acetoacetate, β-hydroxybutyrate, and acetone. Among them, acetoacetate and β-hydroxybutyrate are transported via the bloodstream to other organs as energy sources. Within the kidney, these ketone bodies are primarily reabsorbed by renal proximal tubular cells from the renal filtrate and are then oxidized into acetyl-CoA (Ferrier et al., 1992). The physiological concentration of ketone bodies supports kidney health during injury by aiding renal cells to survive from starvation conditions (Tajima et al., 2019; Rojas-Morales et al., 2021; Rojas-Morales et al., 2022; Liu and Yan, 2023). However, in pathological conditions such as diabetes, the overproduction of ketone bodies can lead to ketoacidosis, which is frequently associated with AKI (Orban et al., 2014; Huang et al., 2022).
Proteins undergo constant degradation and synthesis to sustain regular cellular functions. As the backbone of proteins, some amino acids are re-used during this protein turnover, while other extra amino acids undergo deamination. Their carbohydrate skeletons can either enter the citric acid cycle as substrates for energy production or be utilized to synthesis glucoses and fatty acids (Murray et al., 2012). The kidney plays a critical role in managing the amino acid reservoir for protein turnover by either absorbing or releasing specific amino acids such as glutamine, proline, serine, and cystine (Garibotto et al., 2010). It uptakes approximately 30% glutamine, 60% proline, 100% citrulline, 100% S-adenosylhomocysteine, and 90% cysteinylglycine from the bloodstream while releasing 100% of serine and cysteine, 50% arginine, 50% tyrosine, and 5%–20% lysine into the bloodstream. The nitrogen resultant from deamination will be converted into urea in the liver, which will be cleared out of the body by the kidney. Thus, the blood urea nitrogen level is an important index to monitor kidney function.
Among the twenty amino acids necessary for protein synthesis, nine are essential amino acids that cannot be synthesized by mammalian cells and must be acquired through diet (Murray et al., 2012). In recent years, the importance of maintaining a balance in the metabolism of branched-chain amino acids (BCAAs)—which include the three essential amino acids, namely, leucine, valine, and isoleucine—and aromatic amino acids (AAAs) (such as phenylalanine, tryptophan, tyrosine, and histidine, with three of them being essential) has gained attention in kidney injury and repair as emerging research studies show that their deficiency can cause malnutrition and progression of kidney diseases (Piret et al., 2021a; Barba et al., 2021; Mahbub et al., 2021; Shan et al., 2023). However, the specific roles and functions of these amino acids remain largely unexplored.
AKI is characterized by a rapid decline in renal function. As a major renal disease, AKI is associated with high mortality in clinics. The leading causes for AKI encompass renal ischemia due to severe cardiovascular conditions or surgeries, nephrotoxicity resulting from toxins or medications (e.g., the chemotherapy drug cisplatin and myoglobin due to rhabdomyolysis), and sepsis. Although the pathophysiology of AKI varies due to its underlying causes, proximal tubular cells are usually identified as the primary site of injury. The lethal and sub-lethal injury of proximal tubular cells has attracted considerable research attention because of their essential role in reabsorption and their heavy reliance on energy production (Basile et al., 2012). Nevertheless, recent findings highlight the crucial role of other renal cells, including inflammatory and endothelial cells, in the progression of AKI (Zuk and Bonventre, 2016).
In recent years, emerging studies have underscored the significance of metabolic dysregulation in the pathophysiology of AKI, which has been explored in comprehensive studies using systematic metabolomic profiling with either mass spectrometry or NMR spectroscopy (Portilla et al., 2006; Wei et al., 2014; Jouret et al., 2016; Huang et al., 2018; Ping et al., 2019; Standage et al., 2021; Davidson et al., 2022; Lim et al., 2023). These studies, undertaken by our team and a few other research groups, consistently highlight mitochondrial dysfunction as a common feature in different AKI models. The mitochondrial dysfunction leads to suppression of fatty acid β-oxidation, the citric acid cycle, and oxidative phosphorylation (Figure 2). In ischemia/reperfusion (I/R)-induced AKI, a disturbance in glycolysis is evident, due to both the insufficient glucose supply from the bloodstream and impaired gluconeogenesis in kidney proximal tubules (Wei et al., 2014; Legouis et al., 2020; Scantlebery et al., 2021) (Figure 2). However, PPP regulation is complicated depending on the original insults (Smith et al., 2014; Bushau-Sprinkle et al., 2020; Scantlebery et al., 2021). Additionally, dysregulation in inflammation-related tryptophan catabolism, osmolality, and impaired purine metabolism have also been reported in ischemic AKI (Wei et al., 2014; Jouret et al., 2016; Huang et al., 2018; Davidson et al., 2022). In sepsis-induced AKI, notable dysregulation in the metabolism of BCAAs has been observed (Standage et al., 2021). The specific roles and impacts of these energy production pathways in AKI have been the subject of extensive research studies and are summarized below.
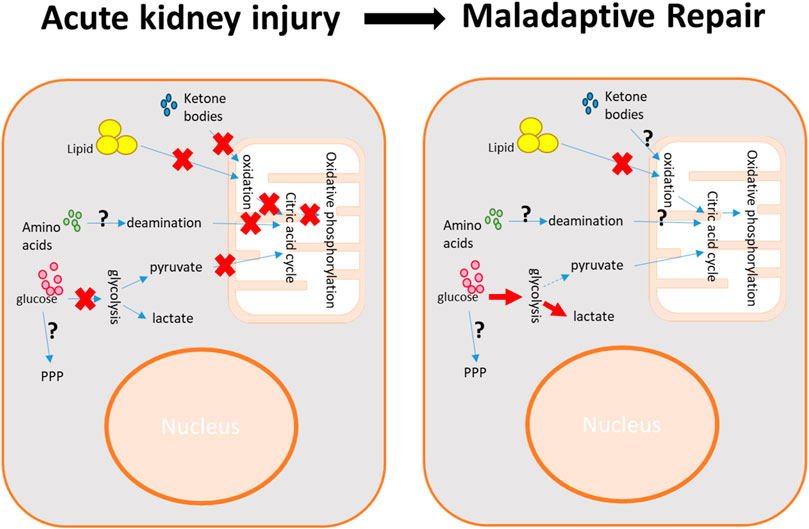
FIGURE 2. Metabolic impairment in kidney injury and reprogramming in kidney repair. During acute kidney injury, the deprivation of nutrients and oxygen results in a reduction in energy production, primarily marked by the suppression of glycolysis and mitochondrial bioenergetics. The maladaptive kidney repair is characterized by a shift in energy production from mitochondrial bioenergetics to glycolysis. The impairment of pathways such as the pentose phosphate pathway (PPP), amino acid metabolism, and ketone body metabolism varies depending on the specific pathological conditions.
During AKI, the renal tubular cells, especially the proximal tubular cells, suffer from severe mitochondrial damage, associated with the loss of some major enzymes for mitochondrial bioenergetics (Jin et al., 2021). Furthermore, the renal vascular impairment, resulting from the endothelial dysfunction, adversely affects the oxygen delivery efficiency and, consequently, the mitochondrial function (Zuk and Bonventre, 2016). Thus, the energy production via fatty acid β-oxidation, the citric acid cycle, and oxidative phosphorylation is significantly suppressed. Given that the proximal tubule is the major renal compartment with high-energy production demand from mitochondria, in AKI resulting from various injurious factors, the accumulation of lipid droplets in the kidney has been commonly observed (Iwaki et al., 2019; Jang et al., 2020b; Bugarski et al., 2021; Xu et al., 2022; Yang et al., 2023b; Chen et al., 2023; Wang et al., 2023). Therefore, promoting mitochondrial biogenesis has been considered a major therapeutic strategy for AKI treatment (Clark and Parikh, 2020; Pabla and Bajwa, 2022).
Peroxisome proliferator-activated receptors (PPARs) are ligand-activated transcription factors that regulate the gene expression related to lipid metabolism and are crucial in regulating mitochondrial biogenesis (Gao and Gu, 2022). The activation of PPARs promotes fatty acid oxidation, enhancing the mitochondrial bioenergy production. The function of various PPAR isoforms has been extensively investigated in AKI. In ischemic AKI, the overexpression of PPARα has been shown to protect mice from renal injury (Li et al., 2009). In addition, PPARα prevents sepsis-induced AKI by promoting fatty acid metabolism, which in turn helps suppress inflammation (Iwaki et al., 2019). Both Krüppel-like factor 15 and ERK1/2 have been reported to regulate the transcription of PPARα (Collier et al., 2016; Piret et al., 2021b). In cisplatin-induced AKI, PPARα translocates from the nucleus to mitochondria, binding to cyclophilin D, which further suppresses its transcription activity and reduces fatty acid metabolism (Jang et al., 2020a). Another isoform PPARγ shows similar fatty acid metabolism regulation function, and its agonists have been reported to exert protective effects in experimental AKI models (Doi et al., 2007; Reel et al., 2013; Zhang et al., 2016; Chen et al., 2017; Singh et al., 2019; Liu et al., 2020; Sharma and Patial, 2022). Furthermore, PPARG coactivator 1 alpha (PGC1α) is a co-activator of PPAR isoforms and a critical regulator in mitochondrial biogenesis (Lynch et al., 2018). PGC1α is suppressed in AKI (Portilla et al., 2002; Tran et al., 2011) and its suppression or deficiency worsens the renal injury, while its induction in renal tubular cells ameliorates ischemic AKI (Tran et al., 2011; Ruiz-Andres et al., 2016; Tran et al., 2016; Fontecha-Barriuso et al., 2019). Despite the central role of the PPAR/PGC1α pathway in AKI, a recent clinical study of the PPARδ agonist ASP1128 did not show significant renal beneficial effects on reducing AKI incidence or severity after major cardiac surgery as expected (van Till et al., 2023). Overall, their therapeutic potential and effectiveness in clinical settings may vary, indicating a need for further development and research of new activating chemicals.
Since the depletion of functional mitochondria is a major pathological event in AKI, in recent years, the transplantation of mitochondria to scavenge the renal cells has been examined in AKI therapy (Doulamis et al., 2020; Jabbari et al., 2020; Pabla and Bajwa, 2022; Rossi et al., 2023). Initial studies by two independent research groups have demonstrated that mitochondrial delivery can attenuate ischemic AKI in both rat and swine models (Doulamis et al., 2020; Jabbari et al., 2020; Rossi et al., 2023). No significant safety issue has been detected by mitochondrial transplantation (Doulamis et al., 2020). However, there are still challenges to be addressed. Ensuring the viability of the transplanted mitochondria remains a significant concern, as their functionality is crucial for the therapeutic benefit. Additionally, the efficacy of mitochondrial transplantation in AKI scenarios other than I/R is yet to be established.
Notably, hypoxia, another key pathological condition in AKI, limits the mitochondrial bioenergetics even in the presence of functional mitochondria. Paradoxically, excessive oxygen consumption by mitochondria may further aggravate renal hypoxia. Thus, it raises a question whether the kidney can be rescued simply by increasing mitochondrial bioenergetics. Kidney oxygen consumption is directly proportional to the glomerular filtration rate and sodium reabsorption (Redfors et al., 2010). Zhou (2023) highlights an alternative strategy for AKI treatment: reducing renal oxygen consumption by inhibiting sodium reabsorption in renal tubular cells. While this strategy has shown promise in animal models, its clinical effectiveness remains controversial. Thus, it underscores further investigation to elucidate how to maintain a balance of renal oxygen delivery, mitochondrial function, and oxygen consumption during AKI therapy.
Although the total glycolysis level is suppressed in AKI, the mitochondrial dysfunction, especially in proximal tubules, shifts the balance of energy production toward glycolysis. However, the pathological function of glycolysis is complicated due to its close connection with other metabolic pathways (Figure 1), and its impact varies across different renal cells (renal tubular cells vs. inflammatory cells) and AKI conditions. Glycolysis can be beneficial as it helps prevent ATP depletion in renal tubular cells. Meanwhile, the suppression of glycolysis may shift the energy production to other pathways such as citric acid cycle and PPP. For instance, preconditioning treatments with meclizine, enarodustat, or AMPK activators, which enhance glycolysis in proximal tubular cells, have been shown to ameliorate ischemic AKI both in vitro and in vivo (Kishi et al., 2015; Lieberthal et al., 2016; Ito et al., 2020). Additionally, dichloroacetate treatment or knockout of its target pyruvate dehydrogenase kinase 4, which shifts metabolism from glycolysis to the citric acid cycle and oxidative phosphorylation, has been effective in protecting mice from cisplatin-induced nephropathy (Galgamuwa et al., 2016; Oh et al., 2017). In contrast, in AKI induced by sepsis, the inhibition of glycolysis with agents like 2-deoxyglucose has been observed to have a protective effect in mice (Ji et al., 2021; Tan et al., 2021). One possibility is that glycolysis may enhance inflammation to promote M1 macrophage polarization (Takakura and Zandi-Nejad, 2019; Yang et al., 2023c). Intriguingly, AMPK activation still shows protection in sepsis-induced AKI, possibly due to its role in enhancing oxidative phosphorylation as well (Jin et al., 2020). Furthermore, tubular-specific knockout of pyruvate kinase M2 (PKM2), a key enzyme in glycolysis, has been reported to protect ischemic AKI by switching the metabolism to PPP (Zhou et al., 2019).
Disturbances in glycolysis during AKI result in altered levels of glycolytic metabolites, which can further influence renal injury and recovery. First, the acute loss of renal function leads to lactate accumulation, and the elevated serum lactate levels have been considered an index of AKI severity (Zhou et al., 2022b). One potential pathological role of lactate in AKI is to downregulate SIRT3 and p-AMPK, followed by autophagy inhibition (Tan et al., 2021). Furthermore, lactic acidosis has been considered to drive the development of CKD (Wesson et al., 2020). Pyruvate, the end product of anaerobic glycolysis and the initial metabolite for the citric acid cycle (Figure 1), decreases in the injured kidney (Zager et al., 2014). The renal protective effects of pyruvate have been identified in various AKI conditions, where it helps reduce oxidative stress and suppress inflammation (Salahudeen et al., 1991; Leelahavanichkul et al., 2008; Jun et al., 2018). In addition, fructose-1,6-bisphosphate, an intermediate metabolite in the glycolysis pathway (Figure 1), has been shown to mitigate kidney injury from I/R or cisplatin nephrotoxicity, although the underlying mechanism is not fully understood (Antunes et al., 2006; Azambuja et al., 2011).
Finally, some multifunctional enzymes in the glycolysis pathway, such as PKM2 and 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3), play crucial roles in regulating kidney injury through mechanisms beyond their primary metabolic functions. PKM2 is a rate-limiting enzyme to produce pyruvate in glycolysis. The specific knockout of PKM2 in renal tubular cells has been shown to protect mice from ischemic AKI and cisplatin-induced nephropathy (Zhou et al., 2019; Xie et al., 2023). While the exact mechanism through which PKM2 regulates ischemic AKI is not fully understood, its knockout seems to reduce oxidative stress and enhance PPP. In the kidneys injured by cisplatin, PKM2 is phosphorylated and translocates to mitochondria, leading to mitochondrial fragmentation and exacerbating tubular injury (Xie et al., 2023). PFKFB3, another enzyme in the glycolysis pathway, catalyzes the production of fructose 2,6-bisphosphate (F2,6P2), which in turn activates phosphofructokinase-1, a rate-limiting enzyme in glycolysis. PFKFB3 is significantly upregulated in the kidneys damaged by cisplatin, and its renal tubular-specific knockout or inhibition attenuates cisplatin-induced AKI (Wen et al., 2023). However, the detrimental role of PFKFB3 in kidney injury relies on the activation of CDK4 and the regulation of the cell cycle, rather than its metabolic activity. Considering the complexity of glycolysis-related metabolites and enzymes in different renal cells, much research is needed to examine the detailed function and mechanism of specific inhibitors or activators for glycolysis.
The activity of PPP in AKI varies depending on the initial insults of the kidney. In ischemic AKI, there is an increase in PPP-related gene expression 24 h post-injury (Scantlebery et al., 2021). In sepsis-induced AKI, the activity of glucose-6-phosphate dehydrogenase (G6PDH), a key enzyme in PPP, is significantly elevated (Smith et al., 2014). In the case of cisplatin-induced AKI, despite a decrease in intermediate metabolites, G6PDH activity is induced (Bushau-Sprinkle et al., 2020). PPP is unique in its ability to produce NADPH, which is critical for controlling oxidative stress and protection against kidney injury (Weng et al., 2018). The deficiency of G6PDH is directly associated with AKI in clinics (Owusu et al., 1972; Abdel Hakeem et al., 2016; Talwar et al., 2019). Furthermore, the induction of G6PDH or PPP activity has been shown to protect the kidneys from cisplatin- or I/R-induced AKI (Zhou et al., 2019; Bushau-Sprinkle et al., 2020). Notably, glucose can shuttle between PPP and glycolysis. Therefore, both the activation of hexokinase and the knockout of PKM2 have been reported to enhance PPP activity in the kidney (Smith et al., 2014; Zhou et al., 2019). However, whether the activation of PPP can regulate glycolysis is unclear. In addition, because PPP not only benefits the detoxification of oxidative stress but also provides basic metabolites for nucleotide synthesis, the detailed mechanism of PPP in AKI progression needs further exploration.
Various types of kidney injury lead to disturbances in amino acid metabolism, exhibiting different patterns depending on the nature of the injury. In ischemic AKI, there is a notable decrease in multiple amino acids, including glutamate, glutamine, tyrosine, proline, and methionine (Wei et al., 2014). Shan et al. (2023) have observed a significant reduction in plasma isoleucine levels following kidney I/R. In sepsis-induced AKI, a decrease in metabolites involved in BCAA metabolism has been detected (Standage et al., 2021). Following cisplatin treatment in mice, the urinary levels of alanine, leucine, and methionine have been significantly elevated, although the changes in their kidney levels have not been determined (Lim et al., 2023).
Our current understanding of the specific roles of individual amino acids in AKI is still evolving. Glycine has been extensively studied across various AKI conditions. Its administration has been shown to reduce free radical production and protect renal epithelial cells in both in vitro and in vivo models of ischemic AKI, as well as in lead- or cisplatin-induced nephrotoxicity (Heyman et al., 1991; Paller and Patten, 1992; Weinberg, 1992; Yin et al., 2002; Shafiekhani et al., 2019). However, this protective effect appears limited in milder injury conditions or chronic ischemia (Yin et al., 2002). Contrarily, Arora et al. (2014) have found that glycine administration can exacerbate ischemic AKI by activating the NMDA receptor. Moreover, glutamine supplementation has demonstrated universal protective effects in diverse AKI scenarios, including ischemic AKI, sepsis-induced AKI, cisplatin nephrotoxicity, and gentamycin-induced nephrotoxicity (Hu et al., 2012; Kim et al., 2015; Thomas et al., 2022; Zhan et al., 2022). L-Arginine deficiency has been identified in the kidney transplant recipients, and its supplementation protects rats from uranyl nitrate-induced AKI (Schramm et al., 2002). While BCAA metabolism has been reported to reduce aristolochic acid-induced kidney injury, potentially regulated by Krüppel-like factor 6 (Piret et al., 2021a), the mechanisms underlying these effects remain unclear. Overall, these findings underscore the complex and varied roles of amino acids in AKI, pointing to the need for much research to fully understand their functions and therapeutic potential in different AKI contexts.
While the overproduction of ketone bodies can lead to ketoacidosis in diabetic conditions, increasing the risk of AKI (Orban et al., 2014; Huang et al., 2022), the enhancement of plasma levels of ketone bodies within the normal range protects renal cells from AKI injury (Tajima et al., 2019; Rojas-Morales et al., 2022; Gui et al., 2023). In a recent study by Gui et al. (2023), calponin 2 has been found to increase in AKI resulting from I/R or cisplatin nephrotoxicity. The knockdown of calponin 2 attenuates kidney injury by upregulating hmgcs2, the key enzyme in ketogenesis, and increasing β-hydroxybutyrate levels in mice (Gui et al., 2023). In addition, both ketogenic diet and β-hydroxybutyrate administration have demonstrated efficacy in ameliorating I/R-induced kidney injury (Tajima et al., 2019; Rojas-Morales et al., 2022). Notably, ketone bodies may offer renal protection through mechanisms beyond just energy production. In the study by Tajima et al. (2019), β-hydroxybutyrate can restore the histone acetylation of the FOXO3 promoter, thereby suppressing pyroptosis through the induction of FOXO3 expression. All these findings highlight the needs of further exploration of the metabolic- and non-metabolic-related mechanisms of ketone bodies in AKI.
Following the acute injury phase, the kidney initiates self-repair processes to restore renal function. Clinically, many patients experience adaptive kidney repair with complete functional recovery within 1 week to 3 months, although the recovery rates can vary widely, ranging from 33% to 90% (Forni et al., 2017). However, a subset of patients may undergo maladaptive kidney repair, failing to achieve full functional restoration and progressing to CKD. As per the recent criteria proposed by the Acute Dialysis Quality Initiative (ADQI), patients who do not recover within 90 days are considered to have CKD (Forni et al., 2017). The kidney repair process involves multiple renal cell types, including the restoration of the renal vascular system through endothelial cell repair and regeneration, the re-establishment of functioning nephrons via renal tubular cell proliferation, and the modulation of the repair process by infiltrating and proliferating inflammatory cells (Ferenbach and Bonventre, 2015). In maladaptive repair, the disruption or renal vascular system results in hypoxia, nutrition deprivation, and oxidative stress accumulation, leading to cell cycle arrest in proliferating renal tubular cells. Concurrently, the infiltration of inflammatory cells may release more cytokines, which not only inhibit the renal cell repair but also promote myofibroblast activation and fibrosis development.
The disruption of kidney metabolism during the acute injury phase not only impacts the immediate functioning of the kidney but also influences the subsequent repair process. Meanwhile, kidney repair is featured by metabolic reprogramming, which regulates tubular degeneration, proliferation, and differentiation. This reprogramming includes the suppression of fatty acid β-oxidation, citric acid cycle, and oxidative phosphorylation, along with the induction of glycolysis. Although an increase in PPP has been observed in diabetic nephropathy, its rate-limiting enzyme G6PDH has been reported to decrease (Steer et al., 1985; Xu et al., 2005). Furthermore, the specific role of PPP activity and its associated enzymes in other kidney repair and fibrosis conditions remains unclear. Meanwhile, emerging studies are highlighting the critical role of amino acid metabolism in the kidney repair process (Kumar et al., 2012; Pillai et al., 2019; Ikeda, 2020; Barba et al., 2021; Lanzon et al., 2021; Prasad et al., 2023). Finally, the information of ketone bodies and glycogen in kidney repair is lacking, although the ketogenic diet has been reported to regulate fatty acid β-oxidation and suppress renal fibrosis (Qiu et al., 2023).
Metabolic reprogramming, particularly the shift in energy production from the citric acid cycle and oxidative phosphorylation to glycolysis, is a key pathological hallmark of maladaptive kidney repair (Schaub et al., 2021) (Figure 2). The upregulation of glycolysis has long been recognized in the repair and regeneration of proximal tubular cells following ischemic injury. This includes increased production of glycolytic end products and elevated levels of key glycolytic enzymes such as hexokinase 2 (HK2), 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase 3 (PFKFB3), and pyruvate kinase M2 (PKM2) (Lan et al., 2016). This metabolic shift was further validated by a recent study by Wang et al. (2022), which used high spatial resolution measurements to examine proximal tubular metabolism in situ within the kidney. In the same kidney with ischemic injury, the maladaptive repaired proximal tubules had significantly more lactate accumulation and less citric acid cycle activity compared to those repaired or healthy proximal tubules. Notably, the S3 segment of proximal tubules showed even greater lactate buildup compared to S1/S2 segments. An intriguing observation was that, compared to uninjured proximal tubules in the sham-operated kidneys, the healthy proximal tubules in the repaired kidneys still displayed higher lactate levels and lower concentrations of citric acid cycle metabolites, indicating some degree of prolonged metabolic reprogramming in the kidneys that have undergone repair.
The role of enhanced glycolysis in pathology has been a subject of intense study in recent years, yielding controversial results. In research using zebrafish models to investigate the energy metabolism, CXCL12 and MYC have been pinpointed to promote renal repair through the upregulation of glycolysis (Yakulov et al., 2018). However, the subsequent validation experiments using mouse models with CXCL12 or MYC knockout in renal tubules have failed to distinguish the renal injury and repair phase, leaving the conclusion obscure. In another study utilizing a PFKFB2 mutant knock-in mouse model, glycolysis has been suppressed, yet renal fibrosis has markedly increased, following ureteral obstruction or folic acid-induced injury (Lee et al., 2020). Conversely, data from our research and those of others have indicated that inhibiting glycolysis can significantly enhance renal repair and ameliorate renal fibrosis (Ding et al., 2017; Wei et al., 2019; Shen et al., 2020; Ye et al., 2021; Yang et al., 2023c; Yang et al., 2023d; Xu et al., 2023). Specifically, glycolysis inhibition can reduce macrophage infiltration and differentiation, fibroblast activation and proliferation, and the pericyte–fibroblast transition (Shen et al., 2020; Ye et al., 2021; Yang et al., 2023c; Yang et al., 2023d; Xu et al., 2023). The role of glycolysis in renal tubules is complicated. We have found that glycolysis inhibitors did not suppress the partial epithelial–mesenchymal transition in cultured proximal tubular cells but decreased renal tubular cell apoptosis in the obstructed kidneys (Wei et al., 2019). It is possible that the differentially injured proximal tubular cells may further regulate renal inflammation and fibroblast activation (Shen et al., 2020; Yang et al., 2023d; Kayhan et al., 2023). Overall, glycolysis inhibition may have divergent effects on different renal cells.
The increase in glycolysis levels in repaired kidneys is always associated with the dysregulation of lipid metabolism (Harzandi et al., 2021). This is particularly evident in proximal tubules, which rely heavily on fatty acid β-oxidation for energy production and contain abundant peroxisomes and mitochondria. These cells are especially susceptible to mitochondria damage and lipid metabolism dysregulation after kidney injury, resulting in lipid accumulation in the kidney and the presence of fatty acids in urine (Bataille et al., 2018; Jang et al., 2020a; Dhillon et al., 2021; Gewin, 2021; Gu et al., 2022; Rinaldi et al., 2022). Genome-wide transcript profiling has identified various patterns of aberrant expression of fatty acid metabolism regulators in proximal tubular cells across different types of fibrotic kidneys after ischemia/reperfusion, folic acid injury, or kidney transplant (Bataille et al., 2018; Dhillon et al., 2021; Rinaldi et al., 2022). In the folic acid-treated kidneys, estrogen-related receptor alpha, a nuclear receptor pivotal in regulating fatty acid oxidation, has been reported to promote proximal tubular cell lipid metabolism and differentiation (Dhillon et al., 2021). In I/R-induced kidney injury, αKlotho deficiency-induced lipid droplet accumulation in the kidney is associated with CKD transition (Wang et al., 2023). In addition, protein phosphatase 2Acα, which can inhibit fatty acid β-oxidation and simultaneously enhance glycolysis through the dephosphorylation of phospho-acetyl-CoA, is induced in the fibrotic kidneys (Gu et al., 2022). This enzyme induction suppresses kidney repair by increasing tubular cell death and fibrosis in the obstructed kidney. The enhanced fatty acid β-oxidation by PGC1α activator also inhibits the pericyte–myofibroblast transition to prevent AKI–CKD transition (Xu et al., 2023). However, our current understanding of the roles of these fatty acid metabolism-related genes in kidney repair remains incomplete. Additionally, how peroxisomes participate in this lipid metabolism regulation is unclear.
Amino acids, serving as fundamental components for protein synthesis, play a crucial role in kidney repair. Essential amino acids are transported to the kidney via the bloodstream and are absorbed by kidney cells through amino acid transporters (Broer, 2008). However, studies examining plasma concentrations of amino acids have yielded controversial results (Bednarek-Skublewska et al., 2002; Kumar et al., 2012; Ikeda, 2020; Lanzon et al., 2021; Mahbub et al., 2021). While most studies report a decrease in BCAAs in plasma (Bednarek-Skublewska et al., 2002; Kumar et al., 2012; Ikeda, 2020), Mahbub et al. (2021) have observed increased BCAAs in the blood of CKD patients, correlating strongly with a decline in the estimated glomerular filtration rate. One possible reason is that dialysis patients might experience nutritional loss during the treatment process (Ikeda, 2020). It is noteworthy that change patterns of BCAA and AAA are different (Mahbub et al., 2021). In a recent study by Pillai et al. (2019), the diet supplement effect of BCAA or AAA has been examined in the 5/6 nephrectomy rat model. Intriguingly, BCAA and AAA supplements have shown divergent impacts on kidney injury and fibrosis development. BCAA supplements promote fibrosis, while AAA supplements are protective for the kidneys. Conversely, another animal study has reported conflicting findings regarding the role of AAAs, indicating that a low AAA diet protects rats from adenine-induced nephrotoxicity, reducing proteinuria, fibrosis, and inflammation (Barba et al., 2021). Overall, our understanding of amino acid homeostasis in kidney repair remains quite limited.
In conclusion, the complex interplay of metabolic pathways plays a critical role in kidney injury and repair. The acute injury disturbs different metabolic pathways in the kidney due to mitochondrial damage, lack of nutrition, and oxygen deprivation. This metabolic dysfunction promotes kidney injury not only through energy and ATP depletion but also impacting various signaling pathways through different metabolites and multifunctional enzymes. Furthermore, it also affects the metabolic reprogramming in the kidney repair phase, leading to aberrant upregulation of glycolysis. Importantly, the therapeutic potential of targeting these metabolic pathways, ranging from mitochondrial bioenergetic production to modulation of glycolysis, PPP activity, and amino acid metabolism, opens new avenues for intervention. However, the differential responses in various kidney injury models highlight the necessity for exploration of injury-specific therapeutic approaches. Moreover, a deeper understanding of the underlying mechanism of metabolic dysregulation, including the roles of key enzymes and metabolites, is crucial. Future research focusing on unraveling the intricate molecular mechanisms and identifying novel therapeutic targets can effectively advance the treatment of AKI and prevent the transition from AKI to CKD.
WT: resources and writing–review and editing. QW: conceptualization, funding acquisition, resources, writing–original draft, and writing–review and editing.
The author(s) declare that financial support was received for the research, authorship, and/or publication of this article. QW was supported by a grant from NIH/NIDDK (1 R01 DK126763-01).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors, and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdel Hakeem G. L., Abdel Naeem E. A., Swelam S. H., El Morsi Aboul Fotoh L., El Mazary A. A., Abdel Fadil A. M., et al. (2016). Detection of occult acute kidney injury in glucose-6-phosphate dehydrogenase deficiency anemia. Mediterr. J. Hematol. Infect. Dis. 8 (1), e2016038. doi:10.4084/MJHID.2016.038
Antunes N., Martinusso C. A., Takiya C. M., da Silva A. J., de Ornellas J. F., Elias P. R., et al. (2006). Fructose-1,6 diphosphate as a protective agent for experimental ischemic acute renal failure. Kidney Int. 69 (1), 68–72. doi:10.1038/sj.ki.5000013
Arora S., Kaur T., Kaur A., Singh A. P. (2014). Glycine aggravates ischemia reperfusion-induced acute kidney injury through N-Methyl-D-Aspartate receptor activation in rats. Mol. Cell Biochem. 393 (1-2), 123–131. doi:10.1007/s11010-014-2052-0
Azambuja A. A., Lunardelli A., Nunes F. B., Gaspareto P. B., Donadio M. V., Poli de Figueiredo C. E., et al. (2011). Effect of fructose-1,6-bisphosphate on the nephrotoxicity induced by cisplatin in rats. Inflammation 34 (1), 67–71. doi:10.1007/s10753-010-9212-5
Barba C., Benoit B., Bres E., Chanon S., Vieille-Marchiset A., Pinteur C., et al. (2021). A low aromatic amino-acid diet improves renal function and prevent kidney fibrosis in mice with chronic kidney disease. Sci. Rep. 11 (1), 19184. doi:10.1038/s41598-021-98718-x
Basile D. P., Anderson M. D., Sutton T. A. (2012). Pathophysiology of acute kidney injury. Compr. Physiol. 2 (2), 1303–1353. doi:10.1002/cphy.c110041
Bataille A., Galichon P., Chelghoum N., Oumoussa B. M., Ziliotis M. J., Sadia I., et al. (2018). Increased fatty acid oxidation in differentiated proximal tubular cells surviving a reversible episode of acute kidney injury. Cell Physiol. Biochem. 47 (4), 1338–1351. doi:10.1159/000490819
Bednarek-Skublewska A., Swatowski A., Wawrzycki S., Baranowicz-Gaszczyk I., Ksiazek A. (2002). Concentrations of branched amino acids: isoleucine, valine, leucine in serum of hemodialysis patients during one-year observation. Ann. Univ. Mariae Curie Sklodowska Med. 57 (2), 322–329.
Bhargava P., Schnellmann R. G. (2017). Mitochondrial energetics in the kidney. Nat. Rev. Nephrol. 13 (10), 629–646. doi:10.1038/nrneph.2017.107
Brinkkoetter P. T., Bork T., Salou S., Liang W., Mizi A., Ozel C., et al. (2019). Anaerobic glycolysis maintains the glomerular filtration barrier independent of mitochondrial metabolism and dynamics. Cell Rep. 27 (5), 1551–1566. doi:10.1016/j.celrep.2019.04.012
Broer S. (2008). Amino acid transport across mammalian intestinal and renal epithelia. Physiol. Rev. 88 (1), 249–286. doi:10.1152/physrev.00018.2006
Bugarski M., Ghazi S., Polesel M., Martins J. R., Hall A. M. (2021). Changes in NAD and lipid metabolism drive acidosis-induced acute kidney injury. J. Am. Soc. Nephrol. 32 (2), 342–356. doi:10.1681/ASN.2020071003
Bushau-Sprinkle A., Barati M. T., Gagnon K. B., Khundmiri S. J., Kitterman K., Hill B. G., et al. (2020). NHERF1 loss upregulates enzymes of the pentose phosphate pathway in kidney cortex. Antioxidants (Basel) 9 (9), 862. doi:10.3390/antiox9090862
Chen D. Z., Chen L. Q., Lin M. X., Gong Y. Q., Ying B. Y., Wei D. Z. (2017). Esculentoside A inhibits LPS-induced acute kidney injury by activating PPAR-γ. Microb. Pathog. 110, 208–213. doi:10.1016/j.micpath.2017.06.037
Chen J., Zheng Q. Y., Wang L. M., Luo J., Chen K. H., He Y. N. (2023). Proteomics reveals defective peroxisomal fatty acid oxidation during the progression of acute kidney injury and repair. Heliyon 9 (7), e18134. doi:10.1016/j.heliyon.2023.e18134
Clark A. J., Parikh S. M. (2020). Mitochondrial metabolism in acute kidney injury. Semin. Nephrol. 40 (2), 101–113. doi:10.1016/j.semnephrol.2020.01.002
Collier J. B., Whitaker R. M., Eblen S. T., Schnellmann R. G. (2016). Rapid renal regulation of peroxisome proliferator-activated receptor γ coactivator-1α by extracellular signal-regulated kinase 1/2 in physiological and pathological conditions. J. Biol. Chem. 291 (52), 26850–26859. doi:10.1074/jbc.M116.754762
Darshi M., Tumova J., Saliba A., Kim J., Baek J., Pennathur S., et al. (2023). Crabtree effect in kidney proximal tubule cells via late-stage glycolytic intermediates. iScience 26 (4), 106462. doi:10.1016/j.isci.2023.106462
Davidson J. A., Robison J., Khailova L., Frank B. S., Jaggers J., Ing R. J., et al. (2022). Metabolomic profiling demonstrates evidence for kidney and urine metabolic dysregulation in a piglet model of cardiac surgery-induced acute kidney injury. Am. J. Physiol. Ren. Physiol. 323 (1), F20–F32. doi:10.1152/ajprenal.00039.2022
Dhillon P., Park J., Hurtado Del Pozo C., Li L., Doke T., Huang S., et al. (2021). The nuclear receptor ESRRA protects from kidney disease by coupling metabolism and differentiation. Cell Metab. 33 (2), 379–394.e8. doi:10.1016/j.cmet.2020.11.011
Ding H., Jiang L., Xu J., Bai F., Zhou Y., Yuan Q., et al. (2017). Inhibiting aerobic glycolysis suppresses renal interstitial fibroblast activation and renal fibrosis. Am. J. Physiol. Ren. Physiol. 313 (3), F561–F575. doi:10.1152/ajprenal.00036.2017
Doi S., Masaki T., Arakawa T., Takahashi S., Kawai T., Nakashima A., et al. (2007). Protective effects of peroxisome proliferator-activated receptor gamma ligand on apoptosis and hepatocyte growth factor induction in renal ischemia-reperfusion injury. Transplantation 84 (2), 207–213. doi:10.1097/01.tp.0000269614.21367.3f
Doulamis I. P., Guariento A., Duignan T., Kido T., Orfany A., Saeed M. Y., et al. (2020). Mitochondrial transplantation by intra-arterial injection for acute kidney injury. Am. J. Physiol. Ren. Physiol. 319 (3), F403–F413. doi:10.1152/ajprenal.00255.2020
Ferenbach D. A., Bonventre J. V. (2015). Mechanisms of maladaptive repair after AKI leading to accelerated kidney ageing and CKD. Nat. Rev. Nephrol. 11 (5), 264–276. doi:10.1038/nrneph.2015.3
Ferrier B., Martin M., Janbon B., Baverel G. (1992). Transport of beta-hydroxybutyrate and acetoacetate along rat nephrons: a micropuncture study. Am. J. Physiol. 262 (5 Pt 2), F762–F769. doi:10.1152/ajprenal.1992.262.5.F762
Fontecha-Barriuso M., Martin-Sanchez D., Martinez-Moreno J. M., Carrasco S., Ruiz-Andres O., Monsalve M., et al. (2019). PGC-1α deficiency causes spontaneous kidney inflammation and increases the severity of nephrotoxic AKI. J. Pathol. 249 (1), 65–78. doi:10.1002/path.5282
Forni L. G., Darmon M., Ostermann M., Oudemans-van Straaten H. M., Pettila V., Prowle J. R., et al. (2017). Renal recovery after acute kidney injury. Intensive Care Med. 43 (6), 855–866. doi:10.1007/s00134-017-4809-x
Galgamuwa R., Hardy K., Dahlstrom J. E., Blackburn A. C., Wium E., Rooke M., et al. (2016). Dichloroacetate prevents cisplatin-induced nephrotoxicity without compromising cisplatin anticancer properties. J. Am. Soc. Nephrol. 27 (11), 3331–3344. doi:10.1681/ASN.2015070827
Gao J., Gu Z. (2022). The role of peroxisome proliferator-activated receptors in kidney diseases. Front. Pharmacol. 13, 832732. doi:10.3389/fphar.2022.832732
Gao Z., Chen X. (2022). Fatty acid β-oxidation in kidney diseases: perspectives on pathophysiological mechanisms and therapeutic opportunities. Front. Pharmacol. 13, 805281. doi:10.3389/fphar.2022.805281
Garibotto G., Sofia A., Saffioti S., Bonanni A., Mannucci I., Verzola D. (2010). Amino acid and protein metabolism in the human kidney and in patients with chronic kidney disease. Clin. Nutr. 29 (4), 424–433. doi:10.1016/j.clnu.2010.02.005
Gewin L. S. (2021). Sugar or fat? Renal tubular metabolism reviewed in health and disease. Nutrients 13 (5), 1580. doi:10.3390/nu13051580
Gu M., Tan M., Zhou L., Sun X., Lu Q., Wang M., et al. (2022). Protein phosphatase 2Acα modulates fatty acid oxidation and glycolysis to determine tubular cell fate and kidney injury. Kidney Int. 102 (2), 321–336. doi:10.1016/j.kint.2022.03.024
Gui Y., Palanza Z., Gupta P., Li H., Pan Y., Wang Y., et al. (2023). Calponin 2 regulates ketogenesis to mitigate acute kidney injury. JCI Insight 8 (21), e170521. doi:10.1172/jci.insight.170521
Harzandi A., Lee S., Bidkhori G., Saha S., Hendry B. M., Mardinoglu A., et al. (2021). Acute kidney injury leading to CKD is associated with a persistence of metabolic dysfunction and hypertriglyceridemia. iScience 24 (2), 102046. doi:10.1016/j.isci.2021.102046
Heyman S. N., Rosen S., Silva P., Spokes K., Egorin M. J., Epstein F. H. (1991). Protective action of glycine in cisplatin nephrotoxicity. Kidney Int. 40 (2), 273–279. doi:10.1038/ki.1991.210
Hu Y. M., Pai M. H., Yeh C. L., Hou Y. C., Yeh S. L. (2012). Glutamine administration ameliorates sepsis-induced kidney injury by downregulating the high-mobility group box protein-1-mediated pathway in mice. Am. J. Physiol. Ren. Physiol. 302 (1), F150–F158. doi:10.1152/ajprenal.00246.2011
Huang H., van Dullemen L. F. A., Akhtar M. Z., Faro M. L., Yu Z., Valli A., et al. (2018). Proteo-metabolomics reveals compensation between ischemic and non-injured contralateral kidneys after reperfusion. Sci. Rep. 8 (1), 8539. doi:10.1038/s41598-018-26804-8
Huang J. X., Casper T. C., Pitts C., Myers S., Loomba L., Ramesh J., et al. (2022). Association of acute kidney injury during diabetic ketoacidosis with risk of microalbuminuria in children with type 1 diabetes. JAMA Pediatr. 176 (2), 169–175. doi:10.1001/jamapediatrics.2021.5038
Ikeda H. (2020). Cross-correlation of plasma concentrations of branched-chain amino acids: a comparison between healthy participants and patients with chronic kidney disease. Clin. Nutr. ESPEN 38, 201–210. doi:10.1016/j.clnesp.2020.04.014
Ito M., Tanaka T., Ishii T., Wakashima T., Fukui K., Nangaku M. (2020). Prolyl hydroxylase inhibition protects the kidneys from ischemia via upregulation of glycogen storage. Kidney Int. 97 (4), 687–701. doi:10.1016/j.kint.2019.10.020
Iwaki T., Bennion B. G., Stenson E. K., Lynn J. C., Otinga C., Djukovic D., et al. (2019). PPARα contributes to protection against metabolic and inflammatory derangements associated with acute kidney injury in experimental sepsis. Physiol. Rep. 7 (10), e14078. doi:10.14814/phy2.14078
Jabbari H., Roushandeh A. M., Rostami M. K., Razavi-Toosi M. T., Shokrgozar M. A., Jahanian-Najafabadi A., et al. (2020). Mitochondrial transplantation ameliorates ischemia/reperfusion-induced kidney injury in rat. Biochim. Biophys. Acta Mol. Basis Dis. 1866 (8), 165809. doi:10.1016/j.bbadis.2020.165809
Jang H. S., Noh M. R., Jung E. M., Kim W. Y., Southekal S., Guda C., et al. (2020a). Proximal tubule cyclophilin D regulates fatty acid oxidation in cisplatin-induced acute kidney injury. Kidney Int. 97 (2), 327–339. doi:10.1016/j.kint.2019.08.019
Jang H. S., Noh M. R., Kim J., Padanilam B. J. (2020b). Defective mitochondrial fatty acid oxidation and lipotoxicity in kidney diseases. Front. Med. (Lausanne) 7, 65. doi:10.3389/fmed.2020.00065
Janson L. W., Tischler M. E. (2018). The big picture: medical biochemistry. New York, NY: McGraw-Hill Education.
Ji R., Chen W., Wang Y., Gong F., Huang S., Zhong M., et al. (2021). The warburg effect promotes mitochondrial injury regulated by uncoupling protein-2 in septic acute kidney injury. Shock 55 (5), 640–648. doi:10.1097/SHK.0000000000001576
Jin K., Ma Y., Manrique-Caballero C. L., Li H., Emlet D. R., Li S., et al. (2020). Activation of AMP-activated protein kinase during sepsis/inflammation improves survival by preserving cellular metabolic fitness. FASEB J. 34 (5), 7036–7057. doi:10.1096/fj.201901900R
Jin X. B., Yu X. H., Su T. L., Yang D. N., Bai Y. T., Kong J. L., et al. (2021). Distributed deep fusion predictor for a multi-sensor system based on causality entropy. Entropy (Basel) 23 (2), 219. doi:10.3390/e23020219
Jouret F., Leenders J., Poma L., Defraigne J. O., Krzesinski J. M., de Tullio P. (2016). Nuclear magnetic resonance metabolomic profiling of mouse kidney, urine and serum following renal ischemia/reperfusion injury. PLoS One 11 (9), e0163021. doi:10.1371/journal.pone.0163021
Jun J. H., Song J. W., Shin E. J., Kwak Y. L., Choi N., Shim J. K. (2018). Ethyl pyruvate is renoprotective against ischemia-reperfusion injury under hyperglycemia. J. Thorac. Cardiovasc Surg. 155 (4), 1650–1658. doi:10.1016/j.jtcvs.2017.10.069
Kayhan M., Vouillamoz J., Rodriguez D. G., Bugarski M., Mitamura Y., Gschwend J., et al. (2023). Intrinsic TGF-β signaling attenuates proximal tubule mitochondrial injury and inflammation in chronic kidney disease. Nat. Commun. 14 (1), 3236. doi:10.1038/s41467-023-39050-y
Kim H. J., Park D. J., Kim J. H., Jeong E. Y., Jung M. H., Kim T. H., et al. (2015). Glutamine protects against cisplatin-induced nephrotoxicity by decreasing cisplatin accumulation. J. Pharmacol. Sci. 127 (1), 117–126. doi:10.1016/j.jphs.2014.11.009
Kishi S., Campanholle G., Gohil V. M., Perocchi F., Brooks C. R., Morizane R., et al. (2015). Meclizine preconditioning protects the kidney against ischemia-reperfusion injury. EBioMedicine 2 (9), 1090–1101. doi:10.1016/j.ebiom.2015.07.035
Kumar M. A., Bitla A. R., Raju K. V., Manohar S. M., Kumar V. S., Narasimha S. R. (2012). Branched chain amino acid profile in early chronic kidney disease. Saudi J. Kidney Dis. Transpl. 23 (6), 1202–1207. doi:10.4103/1319-2442.103560
Lan R., Geng H., Singha P. K., Saikumar P., Bottinger E. P., Weinberg J. M., et al. (2016). Mitochondrial pathology and glycolytic shift during proximal tubule atrophy after ischemic AKI. J. Am. Soc. Nephrol. 27 (11), 3356–3367. doi:10.1681/ASN.2015020177
Lanzon B., Martin-Taboada M., Castro-Alves V., Vila-Bedmar R., Gonzalez de Pablos I., Duberg D., et al. (2021). Lipidomic and metabolomic signature of progression of chronic kidney disease in patients with severe obesity. Metabolites 11 (12), 836. doi:10.3390/metabo11120836
Lee M., Harley G., Katerelos M., Gleich K., Sullivan M. A., Laskowski A., et al. (2020). Mutation of regulatory phosphorylation sites in PFKFB2 worsens renal fibrosis. Sci. Rep. 10 (1), 14531. doi:10.1038/s41598-020-71475-z
Leelahavanichkul A., Yasuda H., Doi K., Hu X., Zhou H., Yuen P. S., et al. (2008). Methyl-2-acetamidoacrylate, an ethyl pyruvate analog, decreases sepsis-induced acute kidney injury in mice. Am. J. Physiol. Ren. Physiol. 295 (6), F1825–F1835. doi:10.1152/ajprenal.90442.2008
Legouis D., Ricksten S. E., Faivre A., Verissimo T., Gariani K., Verney C., et al. (2020). Altered proximal tubular cell glucose metabolism during acute kidney injury is associated with mortality. Nat. Metab. 2 (8), 732–743. doi:10.1038/s42255-020-0238-1
Li S., Nagothu K. K., Desai V., Lee T., Branham W., Moland C., et al. (2009). Transgenic expression of proximal tubule peroxisome proliferator-activated receptor-alpha in mice confers protection during acute kidney injury. Kidney Int. 76 (10), 1049–1062. doi:10.1038/ki.2009.330
Lieberthal W., Tang M., Lusco M., Abate M., Levine J. S. (2016). Preconditioning mice with activators of AMPK ameliorates ischemic acute kidney injury in vivo. Am. J. Physiol. Ren. Physiol. 311 (4), F731–F739. doi:10.1152/ajprenal.00541.2015
Lim Y. J., Tonial N. C., Hartjes E. D., Haig A., Velenosi T. J., Urquhart B. L. (2023). Metabolomics for the identification of early biomarkers of nephrotoxicity in a mouse model of cisplatin-induced acute kidney injury. Biomed. Pharmacother. 163, 114787. doi:10.1016/j.biopha.2023.114787
Liu H., Yan L.-J. (2023). The role of ketone bodies in various animal models of kidney disease. Endocrines 4 (1), 236–249. doi:10.3390/endocrines4010019
Liu J., Zhao N., Shi G., Wang H. (2020). Geniposide ameliorated sepsis-induced acute kidney injury by activating PPARγ. Aging (Albany NY) 12 (22), 22744–22758. doi:10.18632/aging.103902
Lynch M. R., Tran M. T., Parikh S. M. (2018). PGC1α in the kidney. Am. J. Physiol. Ren. Physiol. 314 (1), F1–F8. doi:10.1152/ajprenal.00263.2017
Mahbub M. H., Yamaguchi N., Nakagami Y., Hase R., Takahashi H., Ishimaru Y., et al. (2021). Association of plasma branched-chain and aromatic amino acids with reduction in kidney function evaluated in apparently healthy adults. J. Clin. Med. 10 (22), 5234. doi:10.3390/jcm10225234
McCormick J. A., Ellison D. H. (2015). Distal convoluted tubule. Compr. Physiol. 5 (1), 45–98. doi:10.1002/cphy.c140002
Murray R. K., Bender D. A., Botham K. M., Kennelly P. J., Rodwell V. W., Weil P. A. (2012). Harper's illustrated biochemistry. London: McGraw Hill.
Oh C. J., Ha C. M., Choi Y. K., Park S., Choe M. S., Jeoung N. H., et al. (2017). Pyruvate dehydrogenase kinase 4 deficiency attenuates cisplatin-induced acute kidney injury. Kidney Int. 91 (4), 880–895. doi:10.1016/j.kint.2016.10.011
Orban J. C., Maiziere E. M., Ghaddab A., Van Obberghen E., Ichai C. (2014). Incidence and characteristics of acute kidney injury in severe diabetic ketoacidosis. PLoS One 9 (10), e110925. doi:10.1371/journal.pone.0110925
Owusu S. K., Addy J. H., Foli A. K., Janosi M., Konotey-Ahulu F. I., Larbi E. B. (1972). Acute reversible renal failure associated with glucose-6-phosphate-dehydrogenase deficiency. Lancet 1 (7763), 1255–1257. doi:10.1016/s0140-6736(72)90980-4
Ozawa S., Ueda S., Imamura H., Mori K., Asanuma K., Yanagita M., et al. (2015). Glycolysis, but not Mitochondria, responsible for intracellular ATP distribution in cortical area of podocytes. Sci. Rep. 5, 18575. doi:10.1038/srep18575
Pabla N., Bajwa A. (2022). Role of mitochondrial therapy for ischemic-reperfusion injury and acute kidney injury. Nephron 146 (3), 253–258. doi:10.1159/000520698
Paller M. S., Patten M. (1992). Protective effects of glutathione, glycine, or alanine in an in vitro model of renal anoxia. J. Am. Soc. Nephrol. 2 (8), 1338–1344. doi:10.1681/ASN.V281338
Pillai S. M., Herzog B., Seebeck P., Pellegrini G., Roth E., Verrey F. (2019). Differential impact of dietary branched chain and aromatic amino acids on chronic kidney disease progression in rats. Front. Physiol. 10, 1460. doi:10.3389/fphys.2019.01460
Ping F., Guo Y., Cao Y., Shang J., Yao S., Zhang J., et al. (2019). Metabolomics analysis of the renal cortex in rats with acute kidney injury induced by sepsis. Front. Mol. Biosci. 6, 152. doi:10.3389/fmolb.2019.00152
Piret S. E., Attallah A. A., Gu X., Guo Y., Gujarati N. A., Henein J., et al. (2021a). Loss of proximal tubular transcription factor Kruppel-like factor 15 exacerbates kidney injury through loss of fatty acid oxidation. Kidney Int. 100 (6), 1250–1267. doi:10.1016/j.kint.2021.08.031
Piret S. E., Guo Y., Attallah A. A., Horne S. J., Zollman A., Owusu D., et al. (2021b). Kruppel-like factor 6-mediated loss of BCAA catabolism contributes to kidney injury in mice and humans. Proc. Natl. Acad. Sci. U. S. A. 118 (23), e2024414118. doi:10.1073/pnas.2024414118
Portilla D., Dai G., McClure T., Bates L., Kurten R., Megyesi J., et al. (2002). Alterations of PPARalpha and its coactivator PGC-1 in cisplatin-induced acute renal failure. Kidney Int. 62 (4), 1208–1218. doi:10.1111/j.1523-1755.2002.kid553.x
Portilla D., Li S., Nagothu K. K., Megyesi J., Kaissling B., Schnackenberg L., et al. (2006). Metabolomic study of cisplatin-induced nephrotoxicity. Kidney Int. 69 (12), 2194–2204. doi:10.1038/sj.ki.5000433
Prasad G. V. R., Nash M. M., Yuan W., Beriault D., Yazdanpanah M., Connelly P. W. (2023). Plasma branched-chain amino acid concentrations and glucose homeostasis in kidney transplant recipients and candidates. Can. J. Kidney Health Dis. 10, 20543581231168085. doi:10.1177/20543581231168085
Qiu Y., Hu X., Xu C., Lu C., Cao R., Xie Y., et al. (2023). Ketogenic diet alleviates renal fibrosis in mice by enhancing fatty acid oxidation through the free fatty acid receptor 3 pathway. Front. Nutr. 10, 1127845. doi:10.3389/fnut.2023.1127845
Redfors B., Bragadottir G., Sellgren J., Sward K., Ricksten S. E. (2010). Acute renal failure is NOT an "acute renal success"--a clinical study on the renal oxygen supply/demand relationship in acute kidney injury. Crit. Care Med. 38 (8), 1695–1701. doi:10.1097/CCM.0b013e3181e61911
Reel B., Guzeloglu M., Bagriyanik A., Atmaca S., Aykut K., Albayrak G., et al. (2013). The effects of PPAR-gamma agonist pioglitazone on renal ischemia/reperfusion injury in rats. J. Surg. Res. 182 (1), 176–184. doi:10.1016/j.jss.2012.08.020
Rinaldi A., Lazareth H., Poindessous V., Nemazanyy I., Sampaio J. L., Malpetti D., et al. (2022). Impaired fatty acid metabolism perpetuates lipotoxicity along the transition to chronic kidney injury. JCI Insight 7 (18), e161783. doi:10.1172/jci.insight.161783
Rojas-Morales P., Leon-Contreras J. C., Sanchez-Tapia M., Silva-Palacios A., Cano-Martinez A., Gonzalez-Reyes S., et al. (2022). A ketogenic diet attenuates acute and chronic ischemic kidney injury and reduces markers of oxidative stress and inflammation. Life Sci. 289, 120227. doi:10.1016/j.lfs.2021.120227
Rojas-Morales P., Pedraza-Chaverri J., Tapia E. (2021). Ketone bodies for kidney injury and disease. Adv. Redox Res. 2, 100009. doi:10.1016/j.arres.2021.100009
Ronco C., Bellomo R., Kellum J. A., Ricci Z. (2019). Critical care nephrology. Philadelphia, PA: Elsevier.
Ross B. D., Espinal J., Silva P. (1986). Glucose metabolism in renal tubular function. Kidney Int. 29 (1), 54–67. doi:10.1038/ki.1986.8
Rossi A., Asthana A., Riganti C., Sedrakyan S., Byers L. N., Robertson J., et al. (2023). Mitochondria transplantation mitigates damage in an in vitro model of renal tubular injury and in an ex vivo model of DCD renal transplantation. Ann. Surg. 278 (6), e1313–e1326. doi:10.1097/SLA.0000000000006005
Ruiz-Andres O., Suarez-Alvarez B., Sanchez-Ramos C., Monsalve M., Sanchez-Nino M. D., Ruiz-Ortega M., et al. (2016). The inflammatory cytokine TWEAK decreases PGC-1α expression and mitochondrial function in acute kidney injury. Kidney Int. 89 (2), 399–410. doi:10.1038/ki.2015.332
Salahudeen A. K., Clark E. C., Nath K. A. (1991). Hydrogen peroxide-induced renal injury. A protective role for pyruvate in vitro and in vivo. J. Clin. Invest. 88 (6), 1886–1893. doi:10.1172/JCI115511
Scantlebery A. M., Tammaro A., Mills J. D., Rampanelli E., Kors L., Teske G. J., et al. (2021). The dysregulation of metabolic pathways and induction of the pentose phosphate pathway in renal ischaemia-reperfusion injury. J. Pathol. 253 (4), 404–414. doi:10.1002/path.5605
Schaub J. A., Venkatachalam M. A., Weinberg J. M. (2021). Proximal tubular oxidative metabolism in acute kidney injury and the transition to CKD. Kidney360 2 (2), 355–364. doi:10.34067/KID.0004772020
Schramm L., La M., Heidbreder E., Hecker M., Beckman J. S., Lopau K., et al. (2002). L-arginine deficiency and supplementation in experimental acute renal failure and in human kidney transplantation. Kidney Int. 61 (4), 1423–1432. doi:10.1046/j.1523-1755.2002.00268.x
Shafiekhani M., Ommati M. M., Azarpira N., Heidari R., Salarian A. A. (2019). Glycine supplementation mitigates lead-induced renal injury in mice. J. Exp. Pharmacol. 11, 15–22. doi:10.2147/JEP.S190846
Shan D., Wang Y. Y., Chang Y., Cui H., Tao M., Sheng Y., et al. (2023). Dynamic cellular changes in acute kidney injury caused by different ischemia time. iScience 26 (5), 106646. doi:10.1016/j.isci.2023.106646
Sharma V., Patial V. (2022). Peroxisome proliferator-activated receptor gamma and its natural agonists in the treatment of kidney diseases. Front. Pharmacol. 13, 991059. doi:10.3389/fphar.2022.991059
Shen Y., Jiang L., Wen P., Ye Y., Zhang Y., Ding H., et al. (2020). Tubule-derived lactate is required for fibroblast activation in acute kidney injury. Am. J. Physiol. Ren. Physiol. 318 (3), F689–F701. doi:10.1152/ajprenal.00229.2019
Singh A. P., Singh N., Pathak D., Bedi P. M. S. (2019). Estradiol attenuates ischemia reperfusion-induced acute kidney injury through PPAR-gamma stimulated eNOS activation in rats. Mol. Cell Biochem. 453 (1-2), 1–9. doi:10.1007/s11010-018-3427-4
Singh P. (2023). Reprogramming of energy metabolism in kidney disease. Nephron 147 (1), 61–64. doi:10.1159/000526308
Smith J. A., Stallons L. J., Schnellmann R. G. (2014). Renal cortical hexokinase and pentose phosphate pathway activation through the EGFR/Akt signaling pathway in endotoxin-induced acute kidney injury. Am. J. Physiol. Ren. Physiol. 307 (4), F435–F444. doi:10.1152/ajprenal.00271.2014
Standage S. W., Xu S., Brown L., Ma Q., Koterba A., Lahni P., et al. (2021). NMR-based serum and urine metabolomic profile reveals suppression of mitochondrial pathways in experimental sepsis-associated acute kidney injury. Am. J. Physiol. Ren. Physiol. 320 (5), F984–F1000. doi:10.1152/ajprenal.00582.2020
Steer K. A., Sochor M., McLean P. (1985). Renal hypertrophy in experimental diabetes. Changes in pentose phosphate pathway activity. Diabetes 34 (5), 485–490. doi:10.2337/diab.34.5.485
Tajima T., Yoshifuji A., Matsui A., Itoh T., Uchiyama K., Kanda T., et al. (2019). β-hydroxybutyrate attenuates renal ischemia-reperfusion injury through its anti-pyroptotic effects. Kidney Int. 95 (5), 1120–1137. doi:10.1016/j.kint.2018.11.034
Takakura A., Zandi-Nejad K. (2019). Lactate-induced activation of HCA2 improves survival in mice with sepsis. FASEB J. 33 (6), 7625–7634. doi:10.1096/fj.201801982R
Talwar M., Krishnamurthy S., Parameswaran N., Delhikumar C. G., Haridasan S., Srinivas B. H. (2019). Severe acute kidney injury owing to rhabdomyolysis and intravascular haemolysis in an 11-year-old child with G6PD deficiency. Paediatr. Int. Child. Health 39 (2), 150–153. doi:10.1080/20469047.2018.1439804
Tan C., Gu J., Li T., Chen H., Liu K., Liu M., et al. (2021). Inhibition of aerobic glycolysis alleviates sepsis-induced acute kidney injury by promoting lactate/Sirtuin 3/AMPK-regulated autophagy. Int. J. Mol. Med. 47 (3), 19. doi:10.3892/ijmm.2021.4852
Thomas K., Zondler L., Ludwig N., Kardell M., Luneburg C., Henke K., et al. (2022). Glutamine prevents acute kidney injury by modulating oxidative stress and apoptosis in tubular epithelial cells. JCI Insight 7 (21), e163161. doi:10.1172/jci.insight.163161
Tirichen H., Yaigoub H., Xu W., Wu C., Li R., Li Y. (2021). Mitochondrial reactive oxygen species and their contribution in chronic kidney disease progression through oxidative stress. Front. Physiol. 12, 627837. doi:10.3389/fphys.2021.627837
Tran M., Tam D., Bardia A., Bhasin M., Rowe G. C., Kher A., et al. (2011). PGC-1α promotes recovery after acute kidney injury during systemic inflammation in mice. J. Clin. Invest. 121 (10), 4003–4014. doi:10.1172/JCI58662
Tran M. T., Zsengeller Z. K., Berg A. H., Khankin E. V., Bhasin M. K., Kim W., et al. (2016). PGC1α drives NAD biosynthesis linking oxidative metabolism to renal protection. Nature 531 (7595), 528–532. doi:10.1038/nature17184
van Till J. W. O., Nojima H., Kameoka C., Hayashi C., Sakatani T., Washburn T. B., et al. (2023). The effects of peroxisome proliferator-activated receptor-delta modulator ASP1128 in patients at risk for acute kidney injury following cardiac surgery. Kidney Int. Rep. 8 (7), 1407–1416. doi:10.1016/j.ekir.2023.04.004
Wang G., Heijs B., Kostidis S., Mahfouz A., Rietjens R. G. J., Bijkerk R., et al. (2022). Analyzing cell-type-specific dynamics of metabolism in kidney repair. Nat. Metab. 4 (9), 1109–1118. doi:10.1038/s42255-022-00615-8
Wang Y., Ran L., Lan Q., Liao W., Wang L., Wang Y., et al. (2023). Imbalanced lipid homeostasis caused by membrane αKlotho deficiency contributes to the acute kidney injury to chronic kidney disease transition. Kidney Int. 104 (5), 956–974. doi:10.1016/j.kint.2023.08.016
Wei Q., Su J., Dong G., Zhang M., Huo Y., Dong Z. (2019). Glycolysis inhibitors suppress renal interstitial fibrosis via divergent effects on fibroblasts and tubular cells. Am. J. Physiol. Ren. Physiol. 316 (6), F1162–F1172. doi:10.1152/ajprenal.00422.2018
Wei Q., Xiao X., Fogle P., Dong Z. (2014). Changes in metabolic profiles during acute kidney injury and recovery following ischemia/reperfusion. PLoS One 9 (9), e106647. doi:10.1371/journal.pone.0106647
Weinberg J. M. (1992). Glutathione and glycine in acute renal failure. Ren. Fail 14 (3), 311–319. doi:10.3109/08860229209106635
Wen L., Li Y., Li S., Hu X., Wei Q., Dong Z. (2021). Glucose metabolism in acute kidney injury and kidney repair. Front. Med. (Lausanne) 8, 744122. doi:10.3389/fmed.2021.744122
Wen L., Wei Q., Livingston M. J., Dong G., Li S., Hu X., et al. (2023). PFKFB3 mediates tubular cell death in cisplatin nephrotoxicity by activating CDK4. Transl. Res. 253, 31–40. doi:10.1016/j.trsl.2022.10.001
Weng X. F., Li S. T., Song Q., Zhu Q., Song D. D., Qin Z. H., et al. (2018). Protective effect of nicotinamide adenine dinucleotide phosphate on renal ischemia-reperfusion injury. Kidney Blood Press Res. 43 (3), 651–663. doi:10.1159/000489620
Wesson D. E., Buysse J. M., Bushinsky D. A. (2020). Mechanisms of metabolic acidosis-induced kidney injury in chronic kidney disease. J. Am. Soc. Nephrol. 31 (3), 469–482. doi:10.1681/ASN.2019070677
Xie W., He Q., Zhang Y., Xu X., Wen P., Cao H., et al. (2023). Pyruvate kinase M2 regulates mitochondrial homeostasis in cisplatin-induced acute kidney injury. Cell Death Dis. 14 (10), 663. doi:10.1038/s41419-023-06195-z
Xu C., Hong Q., Zhuang K., Ren X., Cui S., Dong Z., et al. (2023). Regulation of pericyte metabolic reprogramming restricts the AKI to CKD transition. Metabolism 145, 155592. doi:10.1016/j.metabol.2023.155592
Xu S., Jia P., Fang Y., Jin J., Sun Z., Zhou W., et al. (2022). Nuclear farnesoid X receptor attenuates acute kidney injury through fatty acid oxidation. Kidney Int. 101 (5), 987–1002. doi:10.1016/j.kint.2022.01.029
Xu Y., Osborne B. W., Stanton R. C. (2005). Diabetes causes inhibition of glucose-6-phosphate dehydrogenase via activation of PKA, which contributes to oxidative stress in rat kidney cortex. Am. J. Physiol. Ren. Physiol. 289 (5), F1040–F1047. doi:10.1152/ajprenal.00076.2005
Yakulov T. A., Todkar A. P., Slanchev K., Wiegel J., Bona A., Gross M., et al. (2018). CXCL12 and MYC control energy metabolism to support adaptive responses after kidney injury. Nat. Commun. 9 (1), 3660. doi:10.1038/s41467-018-06094-4
Yang Q. H., Cai Y., Zhang Z., Dong C., Asara J. M., Shi H., et al. (2023c). Myeloid PFKFB3-mediated glycolysis promotes kidney fibrosis. Front. Immunol. 14, 1259434. doi:10.3389/fimmu.2023.1259434
Yang Q. H., Fan Y., Xiong M., Chen Y., Zhou Y., Liu X., et al. (2023a). Loss of renal tubular G9a benefits acute kidney injury by lowering focal lipid accumulation via CES1. EMBO Rep. 24 (6), e56128. doi:10.15252/embr.202256128
Yang Q. H., Huo E., Cai Y., Zhang Z., Dong C., Asara J. M., et al. (2023b). PFKFB3-Mediated glycolysis boosts fibroblast activation and subsequent kidney fibrosis. Cells 12 (16), 2081. doi:10.3390/cells12162081
Yang Q. H., Zhan X., Zhang C., Shi J., Wu J., Deng X., et al. (2023d). USP25-PKM2-glycolysis axis contributes to ischemia reperfusion-induced acute kidney injury by promoting M1-like macrophage polarization and proinflammatory response. Clin. Immunol. 251, 109279. doi:10.1016/j.clim.2023.109279
Ye Y., Xu L., Ding H., Wang X., Luo J., Zhang Y., et al. (2021). Pyruvate kinase M2 mediates fibroblast proliferation to promote tubular epithelial cell survival in acute kidney injury. FASEB J. 35 (7), e21706. doi:10.1096/fj.202100040R
Yin M., Zhong Z., Connor H. D., Bunzendahl H., Finn W. F., Rusyn I., et al. (2002). Protective effect of glycine on renal injury induced by ischemia-reperfusion in vivo. Am. J. Physiol. Ren. Physiol. 282 (3), F417–F423. doi:10.1152/ajprenal.00011.2001
Zager R. A., Johnson A. C., Becker K. (2014). Renal cortical pyruvate depletion during AKI. J. Am. Soc. Nephrol. 25 (5), 998–1012. doi:10.1681/ASN.2013070791
Zhan F., Wang X., Zhang J., Yi S., He P. (2022). Glutamine alleviates the renal dysfunction associated with gentamicin-induced acute kidney injury in Sprague-Dawley rats. Biotechnol. Appl. Biochem. 69 (1), 323–329. doi:10.1002/bab.2111
Zhang J., Zhang Y., Xiao F., Liu Y., Wang J., Gao H., et al. (2016). The peroxisome proliferator-activated receptor γ agonist pioglitazone prevents NF-κB activation in cisplatin nephrotoxicity through the reduction of p65 acetylation via the AMPK-SIRT1/p300 pathway. Biochem. Pharmacol. 101, 100–111. doi:10.1016/j.bcp.2015.11.027
Zhou H. L., Zhang R., Anand P., Stomberski C. T., Qian Z., Hausladen A., et al. (2019). Metabolic reprogramming by the S-nitroso-CoA reductase system protects against kidney injury. Nature 565 (7737), 96–100. doi:10.1038/s41586-018-0749-z
Zhou W., He Y., Hu L., Zhu Q., Lin Q., Hong X., et al. (2022b). Lactate level and lactate clearance for acute kidney injury prediction among patients admitted with ST-segment elevation myocardial infarction: a retrospective cohort study. Front. Cardiovasc Med. 9, 930202. doi:10.3389/fcvm.2022.930202
Zhou W., Simic P., Rhee E. P. (2022a). Fibroblast growth factor 23 regulation and acute kidney injury. Nephron 146 (3), 239–242. doi:10.1159/000517734
Zhou W., Simic P., Zhou I. Y., Caravan P., Vela Parada X., Wen D., et al. (2023). Kidney glycolysis serves as a mammalian phosphate sensor that maintains phosphate homeostasis. J. Clin. Invest. 133 (8), e164610. doi:10.1172/JCI164610
Zhou X. (2023). Reducing oxygen demand to alleviate acute kidney injury. Front. Biosci. (Landmark Ed. 28 (3), 62. doi:10.31083/j.fbl2803062
Keywords: acute kidney injury, maladaptive repair, oxidative phosphorylation, glycolysis, fatty acid β-oxidation, pentose phosphate pathway, amino acids, ketone bodies
Citation: Tang W and Wei Q (2024) The metabolic pathway regulation in kidney injury and repair. Front. Physiol. 14:1344271. doi: 10.3389/fphys.2023.1344271
Received: 25 November 2023; Accepted: 28 December 2023;
Published: 12 January 2024.
Edited by:
Francesca Di Sole, Des Moines University, United StatesReviewed by:
Takahiko Nakagawa, Shiga University of Medical Science, JapanCopyright © 2024 Tang and Wei. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Qingqing Wei, cXdlaUBhdWd1c3RhLmVkdQ==
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.