 Joanne G. Ma
Joanne G. Ma Jamie I. Vandenberg
Jamie I. Vandenberg Chai-Ann Ng
Chai-Ann Ng
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 27 November 2023
Sec. Cardiac Electrophysiology
Volume 14 - 2023 | https://doi.org/10.3389/fphys.2023.1294741
This article is part of the Research TopicMethods and Applications in Cardiac Electrophysiology - application to inherited arrhythmiasView all 8 articles
Advances in next-generation sequencing have been exceptionally valuable for identifying variants in medically actionable genes. However, for most missense variants there is insufficient evidence to permit definitive classification of variants as benign or pathogenic. To overcome the deluge of Variants of Uncertain Significance, there is an urgent need for high throughput functional assays to assist with the classification of variants. Advances in parallel planar patch clamp technologies has enabled the development of automated high throughput platforms capable of increasing throughput 10- to 100-fold compared to manual patch clamp methods. Automated patch clamp electrophysiology is poised to revolutionize the field of functional genomics for inheritable cardiac ion channelopathies. In this review, we outline i) the evolution of patch clamping, ii) the development of high-throughput automated patch clamp assays to assess cardiac ion channel variants, iii) clinical application of these assays and iv) where the field is heading.
Cardiac myocyte action potentials, generated by the delicate balance of inward (INa, ICa, If) and outward (IKAch, IK1, Ito, IKur, IKr, IKs) currents through the sarcolemma, are critical for coordinating the contractions of the myocardium (Grant, 2009). Disruptions, genetic or acquired, to ion channel function increase the risk of abnormal heart rhythms. Ion channelopathies arising from genetic variants in cardiac ion channel genes can lead to fatal arrhythmias without overt structural abnormalities in the heart. This is believed to be responsible for 40% of sudden cardiac deaths in young healthy individuals (Bagnall et al., 2016).
The discovery of cardiac ion channel genes underpinning Long QT Syndrome (LQTS; MIM 192500) in the 1990s–KCNQ1 (KVLQT1) (Wang Q. et al., 1996), KCNH2 (KV11.1/hERG) (Curran et al., 1995), SCN5A (NaV1.5) (Wang et al., 1995)—were a major breakthrough in our understanding of congenital arrhythmia syndromes. At least 20 genes have now been implicated in various inheritable arrhythmia syndromes, including LQTS, Brugada Syndrome (BrS; MIM 601144), Catecholaminergic Polymorphic Ventricular Tachycardia (MIM 604772), Short QT Syndrome (MIM 609620), Idiopathic Ventricular Fibrillation (MIM 603829), Progressive Cardiac Conduction Disease (MIM 113900), and Timothy Syndrome (MIM 601005). There is not however a simple relationship between diseases and ion channel mutations, with many patients showing oligogenic and polygenic inheritance patterns, whereby more than one genetic variant contributes to disease, and pleiotropy, where one gene can be responsible for different diseases (Cerrone et al., 2019).
The rapid evolution of genomic sequencing technology (van Dijk et al., 2014) and the introduction of high-throughput next-generation sequencing (Metzker, 2010) have made genetic testing more affordable and accessible. This has had clinical benefits for patients with genetic arrhythmogenic conditions, such as detecting or confirming diagnoses, guiding medical interventions and management, and aiding cascade screening (Musunuru et al., 2020). However, identifying a variant in a disease-associated gene does not necessarily mean that the variant is the cause of the disease in that individual. From large scale genome sequencing projects such as ExAC/gnomAD (Lek et al., 2016; Chen et al., 2022), it is now clear that rare missense variants occur in all individuals and the majority of these are benign. Thus, the American College of Medical Genetics and Association for Molecular Pathology (ACMG/AMP) developed guidelines to help interpret whether variants should be classified as pathogenic, benign or ‘Variants of Uncertain Significance’ (VUS) (Richards et al., 2015). Because most variants in rare diseases occur in a very small number of individuals it is often difficult to obtain sufficient clinical evidence to enable definitive evidence for pathogenicity on clinical data alone. Consequently, approximately half of all variants in genes implicated in cardiac diseases are classified as VUS (Anderson et al., 2022). Functional assays hold great promise to help ease the burden of VUS and appropriately calibrated functional assays can provide up to strong functional evidence for variant classification (Brnich et al., 2020). This is especially valuable for reporting secondary findings in medically actionable genes, of which 34/73 relate to cardiovascular phenotypes (Miller et al., 2021a).
There are numerous comprehensive reviews of inherited arrhythmia syndromes, including (Kapplinger et al., 2009; Kapplinger et al., 2010; Gray and Behr, 2016; Wilde and Amin, 2018; Schwartz et al., 2020; Wilde et al., 2022b; Remme, 2023). Here, we focus on recent developments in the functional characterization of genetic variants involved in inheritable arrhythmia syndromes.
In the 1940s, Hodgkin and Huxley proposed that ionic mechanisms underlie the initiation and propagation of neuronal action potentials (Hodgkin and Huxley, 1952; The Nobel Prize in Physiology or Medicine, 1963). The first cellular electrical recordings from heart tissues were performed in the late 1940s using microelectrodes (Coraboeuf, 1949; Coraboeuf and Weidmann, 1949), which set in motion the extraordinary progress that led to our understanding of the molecular mechanisms underlying cell electrophysiology and cardiac rhythms. The next big breakthrough occurred in the mid-1970s when Neher and Sakmann developed the patch-clamp technique that enabled the detection of single channel currents (Neher and Sakmann, 1976; The Nobel Prize in Physiology or Medicine, 1991). The patch clamp technique involves the formation of a giga-ohm seal between the cell plasma membrane and a glass micropipette containing electrolyte solution, thus isolating a membrane patch electrically (Hamill et al., 1981). The ion channels located in this membrane patch enable the movement of selected ionic currents into the micropipette for recording and determining conductance by an electrode. Variations of this technique have been used in electrophysiology studies, including whole-cell which detects the collective current across the entire cell membrane; inside-out where a cell patch is retracted into the bath solution allowing control of the cytoplasmic environment; and outside-out where conversely the cell patch is retracted and resealed, forming an independent patch facing the bath solution allowing control of the extracellular environment (Hamill et al., 1981). This technique remains the gold standard for in vitro electrophysiology however, it yields low throughput, even with a highly skilled experimentalist.
Over the last two decades there has been considerable interest in developing automated platforms to improve the throughput of patch clamp techniques (González et al., 1999). Ion channels play a crucial role in excitable tissues and are important therapeutic targets. In addition to screening for therapeutic efficacy, patch clamping has proven to be invaluable for toxicity studies and specifically screening for inadvertent drug block of hERG channels that can lead to potentially life-threatening cardiac arrhythmias (Fermini and Fossa, 2003). As such, considerable investments in automated patch clamp (APC) systems were driven by pharmaceutical companies.
The pioneering development of the planar patch clamp, in 2002 (Fertig et al., 2002), allowed the simultaneous parallel patching of cells and is at the core of many APC used today. The use of a planar glass chip and suction below the chip to draw in the cell, forming a seal, then simultaneously rupturing the cell membrane to achieve electrical access to each cell in a 384-well format (Figure 1) can increase the output by two orders of magnitude compared to manual patch clamping. In addition to increasing throughput, APC offer many advantages including cell handling facilities, ability to clamp current or voltage, temperature control which is important for temperature-dependent channels and compounds, and internal solution exchange that enables activation or inhibition by compounds added intracellularly. Furthermore, APC is applicable to various cell expression systems used in cardiac electrophysiology experiments (Table 1), including human-induced pluripotent stem cell-derived cardiomyocytes (hiPSC-CMs) (Ma et al., 2011; Becker et al., 2013; Obergrussberger et al., 2016; Rajamohan et al., 2016; Li et al., 2019; Potet et al., 2020; Rapedius et al., 2022; Melgari et al., 2023). There are, however, some limitations. Most notably there is less consistency in high quality spatial and temporal voltage control which necessitates more stringent quality control. Failure to do this may result in significant variations in apparent IC50 values measured for the same drug on different days or different systems (Chambers et al., 2016; Kramer et al., 2020). The importance of stringent quality control is discussed in more detail below in reference to the analysis of gating defects in ion channel variants. A summary of the technical specifications of the latest APC systems available is shown in Table 1 and more in depth reviews of the application and history of APC can be found in Liu et al. (2019) and Bell and Fermini (2021).
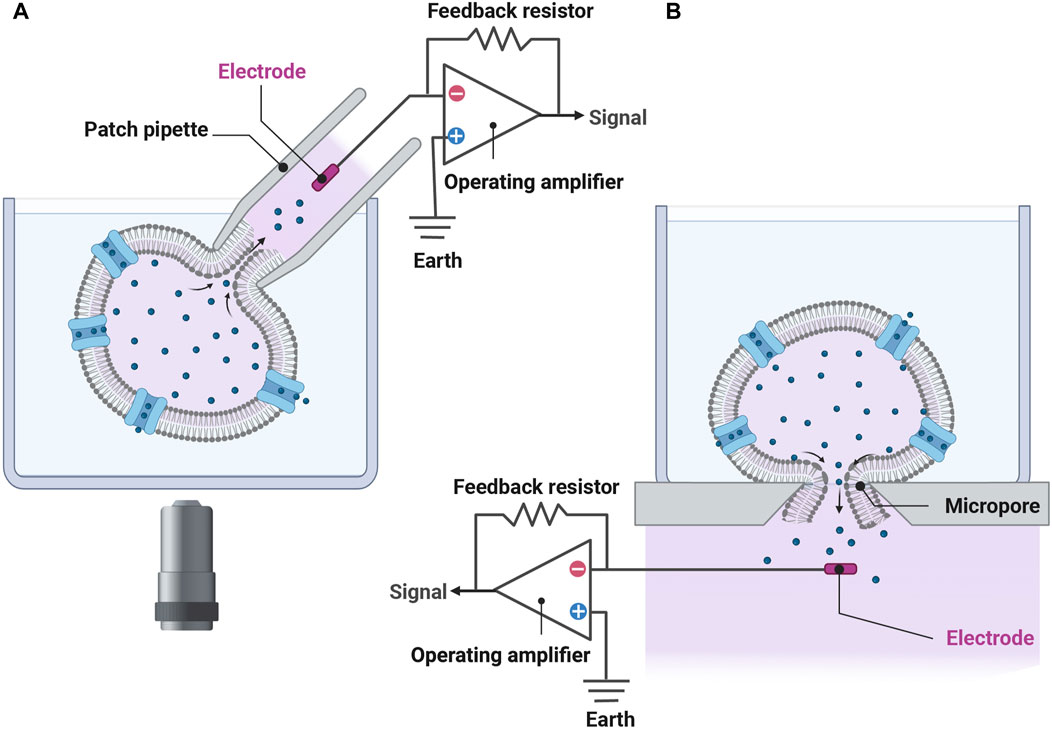
FIGURE 1. The transition from the conventional patch-clamp system to a planar patch-clamp system has enabled simultaneous patching of cells and underlies many of the APC used today. (A) In conventional whole-cell patch-clamp recordings, a pipette filled with electrolyte solution contains an electrode enabling the experimenter to apply various stimuli and measure the cell’s response. It takes considerable skill to attach the pipette to the cell using micromanipulators and a microscope. (B) In planar patch-clamp technology, cells are drawn down and adhered to a micropore using suction, acting similar to a pipette tip. Here, the cell is ruptured enabling stimulation and measurement of electrical activities. Chips used in modern APC platforms can record up to 768 cells in parallel (see Table 1). No micromanipulation is necessary. Figure developed using Biorender.
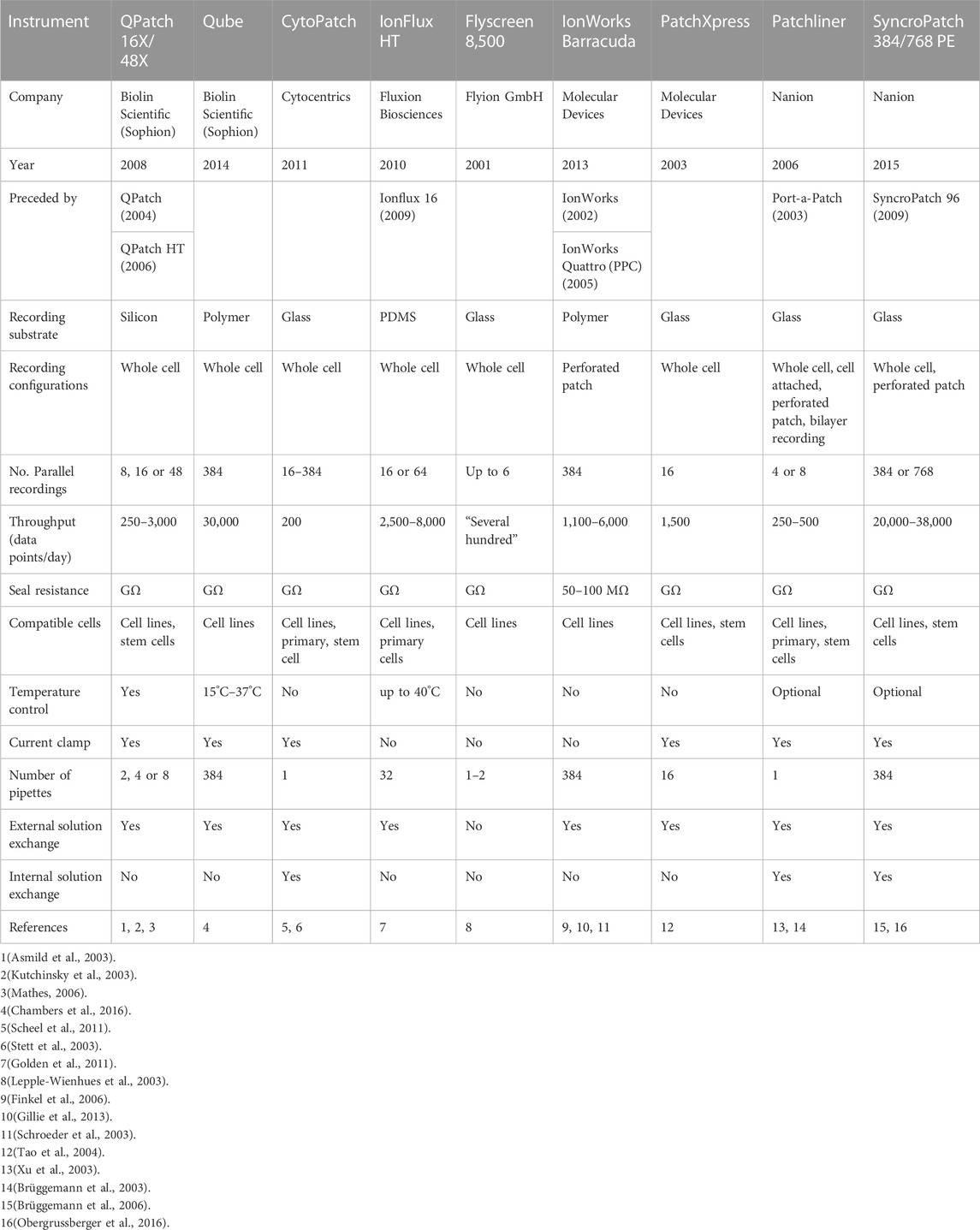
TABLE 1. Various APCs have been developed over time, improving on prior models. The most current model of each series is detailed.
Deleterious ion channel gene variants only impact their respective ionic current. Consequently, it is possible to assess the functional effect of ion channel variants in heterologous expression systems. Traditionally, these have employed manual patch clamp techniques (Bennett et al., 1995; Morais Cabral et al., 1998; Wang et al., 2000; Marx et al., 2002). More recently, APC platforms have been utilized for the characterization of large panels of ion channel variants (Vanoye et al., 2018; Glazer et al., 2020b; Kozek et al., 2020; Kuenze et al., 2020; Ng et al., 2020; Ng et al., 2021; Jiang et al., 2022; Ng et al., 2022; O’Neill et al., 2022; Vanoye et al., 2022; Ma et al., 2023; Thomson et al., 2023). APC has the potential to increase the throughput for variant functional assessment by two orders of magnitude compared to manual patch clamp. However, it is important to consider what effects of the variant can be assayed (e.g., Loss-Of-Function; LOF or Gain-Of-Function; GOF) and the potential sources of errors or challenges that can arise during these measurements when we design any high throughput APC assay.
Trafficking-defective variants are the main cause of LOF in ion channel diseases. Distinguished by their minimal presence at the cell surface membrane, they are often caused by protein misfolding consequent to the inappropriate exposure of hydrophobic residues and subsequent aggregation with other misfolded proteins (Asher et al., 2006). Because of defective trafficking, these variants result in little or no assayable current during electrophysiological analyses (Delisle et al., 2004). As such, these variants are easily assayed in APC platforms. In the case of hERG channels, current density measured for a range of trafficking defective variants by APC, has been validated using ELISA assays (Ng et al., 2020), Western blot (Ng et al., 2022) and a massively parallel trafficking assay (Ng et al., 2022).
However, there is considerable variability in protein expression from cell-to-cell due to the stochastic nature of transcription and translation within each cell (Figure 2A) (Lachaud et al., 2022). Thus, it is necessary to undertake large numbers of measurements to ensure the average measurement is a true approximation of the population mean (Figure 2) (Jiang et al., 2022). In heterologous expression systems, there is the added complication that plasmids may be incorporated into random locations within the genome, and these may be associated with altered or inconsistent expression (Mizuguchi et al., 2000). This problem can be overcome by using a genomic landing pad for targeted gene expression, such as the Flp-In recombinase cassette, as this will ensure that all variant lines have the plasmid expressed in the same region of the genome (Ward et al., 2011). One can also utilize a tetracycline-induced expression element (Ng et al., 2021) to reduce the risk of the inserted plasmid being epigenetically silenced (Ríos-Pérez et al., 2021) and reduce the problem of constitutively expressed plasmids leading to reduced cell viability due to cell toxicity. The generation of stable cell lines does take longer (typically ∼3 weeks for antibiotic selection and expansion of positive clones) than using transient transfection (typically 48 h) but the reduction in variability between different transfections and being able to store stable lines for replicate experiments saves time and resources during data acquisition.
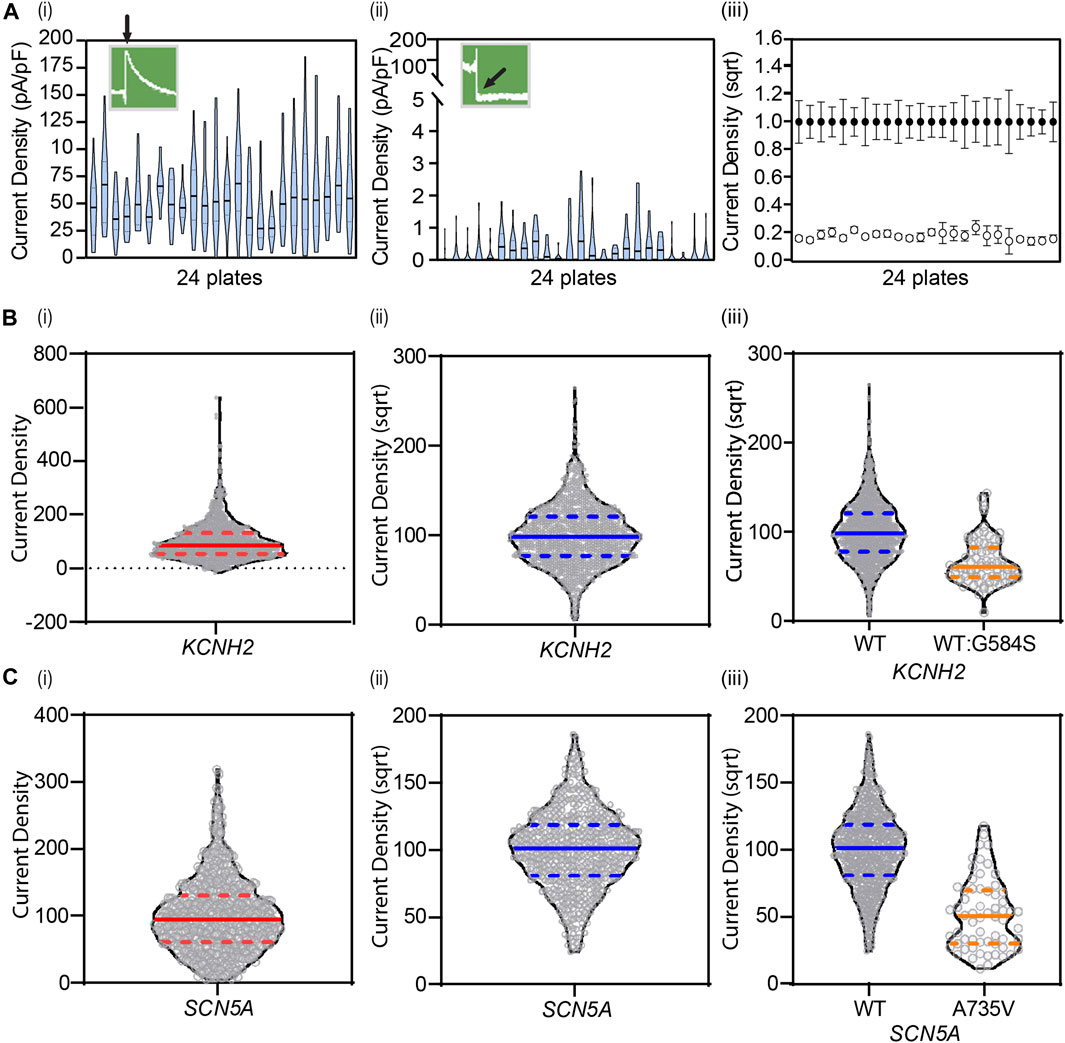
FIGURE 2. Cell-to-cell variability necessitates large numbers of replicate measurements to obtain an accurate estimate of the mean value. (A) Violin plots of KCNH2 WT (i) and negative (ii) control lines show experimental cell-to-cell variability, plate-to-plate variability across 24 plates, and overall dynamic range of the assay revealing clear separation between positive and negative controls (iii). Data are shown as mean ± 95% CI. (B) Square-root transformations enable gaussian-distributed data for analysis as seen with KCNH2 before (i) and after (ii) transformation (N = 1,198), and (C) SCN5A before (i) and after (ii) (N = 609). Transformations are applied to all data, including variants, enabling distinction of any changes in current density (iii). Solid lines denote median. Dashed lines denote quartiles. (A) Adapted from Jiang et al.(2022), with permission from Elsevier.
The application of Flp-In recombinase in high-throughput assays has been utilized by many researchers in a broad range of fields, including GPCRs (Ward et al., 2011) as well as sodium (Pablo et al., 2023), calcium (Pan et al., 2018; Baez-Nieto et al., 2022; Zhang et al., 2022) and potassium (Ng et al., 2021) ion channels. It is also possible to use IRES plasmids in combination with the Flp-In recombinase cassette so that two subunits, e.g., KCNE1+KCNQ1 to recapitulate IKs or mutant + WT alleles to mimic heterozygous expression, are co-expressed from the same locus in all cells (Ng et al., 2021; Jiang et al., 2022). More recently, Jensen et al. (2020) have developed a double Flp-In system which when combined with IRES cassettes in theory could permit co-expression of 4 different subunits, although such an approach has not yet been published.
The conformation of voltage-gated ion channels (e.g., closed, open, deactivated, inactivated and recovery states) are precisely regulated by membrane potential (Grant, 2009). Thus, to assess the impact of a variant that expresses sufficient current density (i.e., does not have a significant trafficking defect) require a thorough analysis of both the steady-state distribution between closed, open and inactivated states as well as the kinetics of transitions between these states. The specific protocols used will depend on the channel being studied and are typically outlined in the supplementary materials of the relevant papers (e.g., KCNQ1+KCNE1 (Vanoye et al., 2018); KCNH2 (Ng et al., 2020); SCN5A (Glazer et al., 2020b). Although less common, abnormal gating is still an important cause for both LOF and GOF variants that result in arrhythmia syndromes. For example, in KV11.1-N633S channels, enhanced inactivation can result in reduced current density (Ng et al., 2020) and in NaV1.5-R1632H channels, delayed recovery from inactivation reduces current density during standard depolarization steps (Glazer et al., 2020b).
The accuracy of the measurement of voltage-dependent gating parameters relies heavily on the quality of temporal and spatial voltage clamp during recordings (Hamill et al., 1981). The quality of the voltage clamp is influenced by the size of the currents being recorded, i.e., the larger the current the more difficult it is to ensure effective spatial and temporal control of voltage. Experimentally, one way to limit this problem is to adjust ion concentrations in the recording solutions to reduce the driving force and consequently amplitude of current. The accuracy of voltage clamp control is also critically dependent on the access resistance between the cell and the recording patch electrode or across the ‘hole’ in a planar patch clamp recording. There is a positive correlation between access resistance values and voltage errors in patch clamp measurements. For example, when measuring INa with current amplitudes of 10 nA, compared to current amplitudes of 1 nA, series resistances of 2 and 5 MΩ have been demonstrated to shift V50 of activation by −7 and −11 mV and reduce the slope factor by a factor of 1.5 and 1.8, respectively (Montnach et al., 2021). It is therefore recommended to use low access resistance plates for APC recordings. However, this needs to be balanced against the fact that if the holes are larger (therefore giving a lower access resistance) it is harder to achieve high quality seals, especially with smaller cells. Also, given that the changes to gating kinetics caused by a variant will be similar irrespective of the level of current expressed in each cell, it is advisable to apply more stringent quality control criteria (Figure 3; e.g., minimum seal resistance, maximum series resistance, time to peak current, and minimum and maximum current amplitudes) for the analysis of gating characteristics to produce more reliable data (Rapedius et al., 2022).
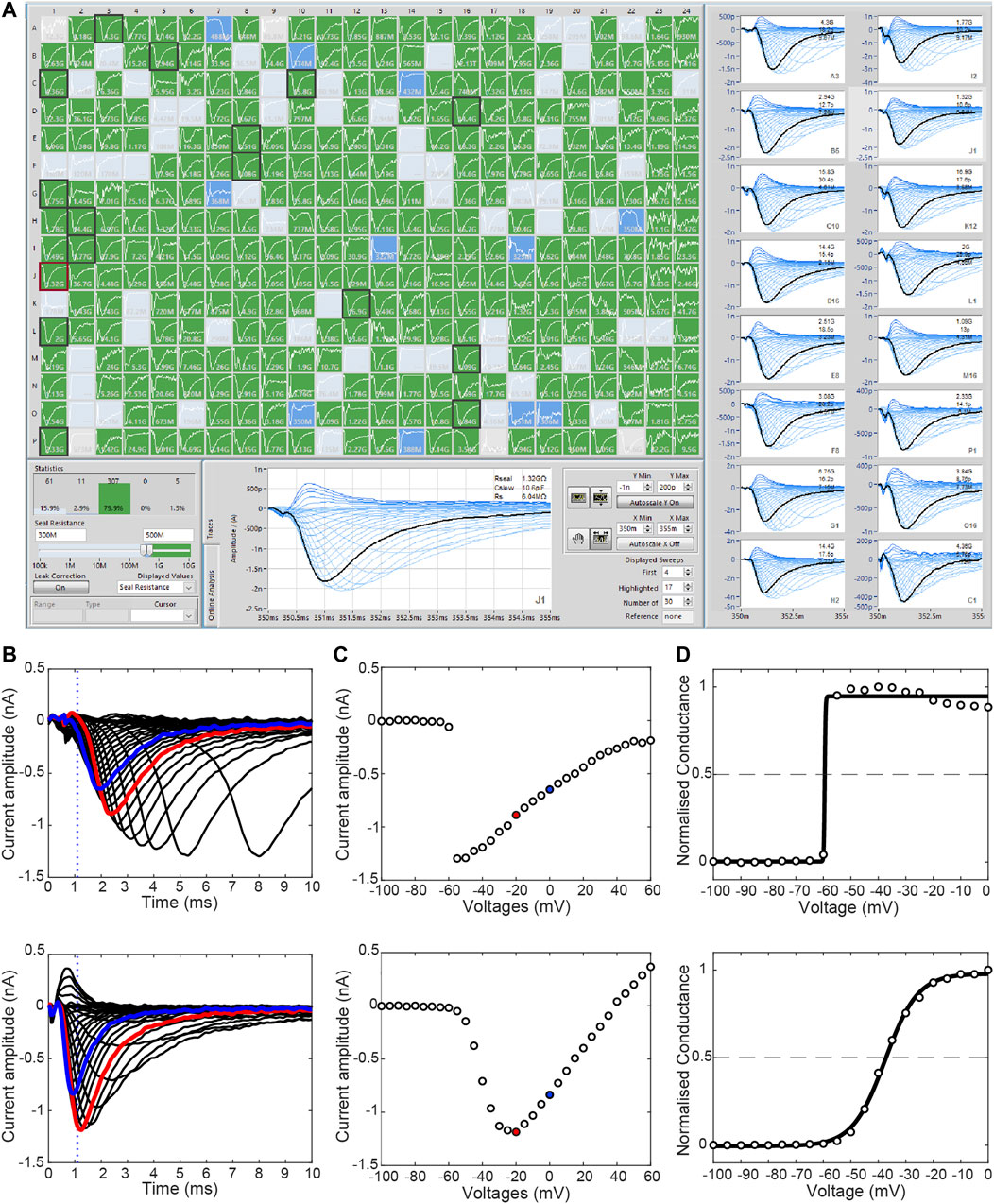
FIGURE 3. Stringent quality control is required to remove bad quality recordings. (A) The SyncroPatch 384 PE offers high throughput APC of mammalian cell lines. A minimum seal resistance of 500 MΩ is adequate to produce a high-quality seal (green wells). Cells with seal resistant <300 MΩ (grey) and between 300 and 500 MΩ (blue) indicate lower quality seals. (B) Current traces illustrating the activation of two WT SCN5A HEK293 cells (−100 mV to +60 mV, Δ5 mV). The top row shows an example of current recorded with poor voltage control whilst the bottom row shows recordings from a cell with good quality voltage control. Due to the rapid kinetics of sodium channel, a “time to peak” measurement can be used to identify poorly clamped cells. In this instance, time to peak is applied at 0 mV (blue) and dotted line represents 1.1 ms post-activation. (C) Respective current-voltage curves revealing steep jump in current in the poorly clamped cell (top). (D) Steady-state activation and Boltzmann function of respective cells reveal the poorly clamp cell (top) have visibly severely impacted V50 (voltage required to activate half of the cells’ channels) and slope, which is typically used for comparison between cells. Red indicates −20 mV sweep used for current density measurements in this case.
Changes in ion selectivity can be assayed by measuring differences in reversal potential between variants and WT channels. Such measurements do not enable precise quantification of the change in ion selectivity but are generally sufficient to identify that it will cause abnormal function (Ng et al., 2020). As with measuring gating defects, it is also important to ensure you have high quality voltage clamp control when measuring reversal potentials.
Some trafficking-defective proteins result in only partially reduced expression at the cell surface. These proteins may also have abnormal gating (Gui et al., 2010; Balijepalli et al., 2012; Kanters et al., 2015; Perry et al., 2016), which may exacerbate the effect of the trafficking defect or in some cases reduce the impact of the gating defect. Thus, it is sensible to design protocols that assess current density under physiologically-relevant voltage ranges that can provide an integrated assessment of both trafficking and gating defects. For example, IKr tail current density at −50 mV (peak current during repolarization in cardiac myocytes) after a 1s depolarization step to +40 mV (to mimic cardiac action potential) will take into account the effect of trafficking defects, slow or reduced activation during the depolarization step and enhanced inactivation at −50 mV during the repolarization phase. Similarly for INa, the peak current density measured during a depolarization step from a holding potential of −90 mV (to mimic the cardiac resting membrane potential) can reveal reduced channel availability due to hyperpolarized inactivation that would otherwise have not been observed when using a holding potential of −120 mV. For example, Ma et al. showed that SCN5A variants E1784K, R1632H and R1632C produce a more marked reduction in current density when holding cells at −90 mV compared to −120 mV relative to what is seen for WT channels (Ma et al., 2023) concordant with previous manual patch clamp studies for E1784K (Makita et al., 2008), R1632H (Benson et al., 2003), and R1632C (Nakajima et al., 2015). An alternative approach is to use parameters derived from the analysis of steady-state activation/inactivation and kinetics of gating transitions to adjust in silico ion channel models that can then be incorporated into in silico models of the cardiac action potential (Clancy et al., 2003; Zhao et al., 2009; Romero et al., 2015; Kernik et al., 2020; Aghasafari et al., 2021).
The most common cardiac ion channelopathy is the Long QT Syndrome (LQTS), which is characterized by a prolonged QT-interval on the surface electrocardiogram (Roden, 2008). LQTS is associated with early after depolarizations and Torsades de Pointes, a form of polymorphic ventricular tachycardia that leads to ventricular fibrillation and sudden cardiac death. According to the latest guidelines (Adler et al., 2020), KCNQ1, KCNH2 and SCN5A are the only ion channel genes definitively associated with LQTS. In addition, KCNQ1 and KCNH2 are the only two ion channel genes definitively associated with Short QT Syndrome, and SCN5A is the only gene definitively implicated in Brugada Syndrome (BrS). To date, specific APC assays have been developed for assessing the effect of genetic variants found in KCNQ1, KCNH2 and SCN5A.
The cardiac voltage-gated potassium channel KV7.1, encoded by KCNQ1, consists of four identical pore-forming subunits, each with six transmembrane helices and a pore loop. It co-assembles with the β-subunit KCNE1 to form the channel complex that passes the slow delayed rectifier potassium current (IKs) in the heart (Barhanin et al., 1996; Sanguinetti et al., 1996). KCNE1 is essential in modulating the slow activation kinetics of IKs. The KCNQ1-KCNE1 complex also does not inactivate on the timescale relevant to cardiac repolarization, when compared to channels encoded by KCNQ1 alone (Pusch et al., 1998). KCNQ1-KCNE1 channels were the first ion channel investigated using the SyncroPatch 384 PE APC system (Vanoye et al., 2018). CHO cells were doubly transfected with KCNQ1 plasmids (either WT or one of 78 variants) coupled to GFP and KCNE1 plasmid coupled to RFP. Transfected cells were then quantified using flow cytometry to determine transfection efficiency, before being analyzed for current density and voltage-dependence of activation. As LQTS is an autosomal dominant condition, a subset of 56 KCNQ1 variants (coupled to eGFP) were co-transfected with WT KCNQ1 (coupled to mScarlet), in a CHO cell line stably expressing KCNE1, to investigate possible dominant-negative effect of variants. The authors used the data from this assay as strong evidence for pathogenicity, providing evidence for the reclassification of 23/35 VUS as likely pathogenic (Vanoye et al., 2018). The application of this assay (as well as KCHN2 and SCN5A assays) in the comprehensive eMERGE-III sequencing study of participants without known cardiac indications provided evidence to support the reclassification of 5/30 VUSs tested in vitro (Glazer et al., 2022).
β1-Adrenergic receptor (β1-AR) activation impacts intracellular cAMP levels leading to protein kinase A (PKA) activation, resulting in phosphorylation of the N-terminal region of KCNQ1. This leads to faster channel activation and, ultimately, shortening of the cardiac action potential (Dixit et al., 2020). Explicitly, LQTS-associated KCNQ1 variants can prolong the action potential duration disruptions to the rate-dependent shortening typically instigated by β-adrenergic stimulation. Assaying β-adrenergic regulation of IKs in heterologous systems however is not straight forward as it requires co-expression of AKAP9 (Yotiao) scaffolding protein in the heterologous expression system (Marx et al., 2002; Dvir et al., 2014). A-kinase anchoring protein 9 (AKAP9) binds to the regulatory subunit of PKA, protein phosphatase 1, phosphodiesterase and adenylate cyclase; thereby providing the scaffold for adrenergic regulation of IKs function (Marx et al., 2002; Kurokawa et al., 2003). To date, no APC assays incorporating AKAP9 have been developed, thus, one cannot be certain that variants with a normal IKs current density in these assays do not affect β-adrenergic regulation.
The cardiac voltage-gated potassium channel KV11.1, encoded by KCNH2, contains four identical pore-forming subunits with 6 transmembrane helices. In addition, it has large cytoplasmic PAS domain at its N-terminal and cyclic nucleotide binding homology domain at its C-terminal (Vandenberg et al., 2012), which are both important for maintaining the protein stability needed for trafficking (Ke et al., 2013; Anderson et al., 2014), and regulation of the slow channel activation, fast inactivation and slow deactivation (Morais Cabral et al., 1998; Wang et al., 1998; Aydar and Palmer, 2001; Vandenberg et al., 2012; Ng et al., 2014). The current passed by KV11.1 potassium channels is known as the rapid delayed rectifier potassium current, IKr. Loss of IKr can be due to four mechanisms: reduced synthesis, defective trafficking, defective gating, or altered ion permeation (Delisle et al., 2004). Defective protein trafficking is the principle underlying cause of KCNH2-related long QT syndrome (Anderson et al., 2014). In the first APC assay for KCNH2 variants, the function of 23 homozygous KCNH2 variants were first assessed and compared to expression measured using an ELISA assay (Ng et al., 2020). This was followed with an analysis of 30 heterozygous KCNH2 variants expressed using bicistronic plasmid to assess the dominant-negative effect in stably expressed Flp-In HEK293 cells (Ng et al., 2020). The use of stably integrated doxycycline-inducible Flp-In HEK293 allowed the isogenic comparison between the functional effect of variant and WT to prevent random insertion thereby ensuring reliability in current density measurement. The use of stable cell lines also allowed generation of a biobank of KCNH2 variant cell lines (Ng et al., 2021; Ng et al., 2022). Current density of KCNH2 variants quantified by APC in heterozygous expression system, when sufficiently large N numbers were obtained, not only represents the trafficking phenotype when it was compared to ELISA (Ng et al., 2020), but also allowed for assessment of gating defects and changes to ion permeability (Ng et al., 2020).
This KCNH2 assay has also been used to cross validate a massively parallel trafficking assay for KCNH2 (Kozek et al., 2020). In a follow up study, Ng et al. (2022) investigated 458 single-nucleotide missense variants in exon 2 of KCNH2, the largest APC experiment performed to date. The APC data generated for these 458 KCNH2 variants was used as the benchmark for validating an independently acquired massively parallel trafficking assay dataset for exon 2 of KCNH2 (Ng et al., 2022), a known hotspot for causing KCNH2-related long QT syndrome. In this study 42% of variants showed >50% reduction in protein trafficking, of which 65% exerted a dominant negative effect when co-expressed with WT (Ng et al., 2022). A separate massively parallel trafficking assay dataset for the S3 to S5 transmembrane helices has also been generated, which included 51 clinically identified variants, and the trafficking results for them were cross-validated against the peak tail current density data measured by the KCNH2 assay, which found that 4/6 were correctly classified as functionally normal and 42/44 correctly classified as LOF (Ullah et al., 2022).
Before these assays can be used to provide evidence to support classification of variants as pathogenic or benign, it is essential that they are formally assessed according to the criteria published by the ClinGen Sequence Variant Interpretation (SVI) Working Group (Brnich et al., 2020). To date, the KCNH2 APC assay is the only assay that has been formally calibrated using clinically-verified pathogenic and benign variant controls to determine the sensitivity and specificity of the assay (Jiang et al., 2022). From the performance of the assay, the odds of pathogenicity of the KCNH2 assay was determined to have the equivalent strength of moderate evidence level for both normal and abnormal protein function, enabling the reclassification of 16% of VUSs assessed as likely pathogenic (Jiang et al., 2022). Recently, this assay has been updated to provide strong evidence strength using 30 benign and 30 pathogenic variant controls (Thomson et al., 2023).
The cardiac voltage-gated sodium channel NaV1.5, encoded by SCN5A, contains four pore-forming domains, but unlike the cardiac potassium channels, these four domains are part of a single polypeptide (de Lera Ruiz and Kraus, 2015). Typically, LOF mutations in SCN5A result in BrS, whereas gain of function mutations results in LQTS. However there are many other syndromes associated with SCN5A variants including sudden infant death syndrome, dilated cardiomyopathy, progressive cardiac conduction disorder, sick sinus syndrome, atrial fibrillation, early repolarization syndrome, and idiopathic ventricular fibrillation (Amin et al., 2010; Moreau et al., 2015; Gray and Behr, 2016; Liu et al., 2016; Remme, 2023). Moreover, some SCN5A variants can cause mixed phenotypes, referred to as “sodium channel overlap syndrome” (Bezzina et al., 1999; Makita et al., 2008; Wilde and Amin, 2018). Loss of sodium channel activity results in decreased cardiac excitability and reduced electrical conduction velocity, thereby increasing the risk of cardiac arrhythmias (Wilde and Amin, 2018). LQTS3, the second most prevalent SCN5A-associated disease (accounts for 5%–10% of LQTS patients) is caused by gating-defective SCN5A variants that cause GOF via a loss of inactivation (Liu et al., 2016; Wilde and Amin, 2018). The resulting increased late INa delays repolarization which leads to the prolongation of the QT interval on the surface ECG.
The first APC assay for SCN5A variants incorporated an mCherry-blasticidinR fusion protein, forming SCN5A:IRES:mCherry:blasticidinR (Glazer et al., 2020a; Glazer et al., 2020b). This enabled the selection of blasticidin-resistance present on the SCN5A plasmid and quantification through flow cytometry for mCherry-positive cells (or Blue Fluorescent Protein/iCasp for non-integrated cells) to determine the proportion of cells with successful plasmid integration. Using this SCN5A-BrS APC assay, the data supported reclassification of 61/83 VUS (Glazer et al., 2020b). The feasibility in applying this approach in the clinical setting was recently demonstrated in the eMERGE-III for VUS reclassification (Glazer et al., 2022). More recently, Ma et al. (2023) have optimized an SCN5A APC assay for assessing LOF in BrS-associated variants that satisfies the recommendations from the ClinGen SVI Working Group (Brnich et al., 2020) to establish clinical grade evidence for assistance with classification of VUS. The odds of Pathogenicity (OddsPath) scores for this assay indicate that the assay can achieve strong evidence levels for both normal (BS3) and abnormal (PS3) protein function (Ma et al., 2023).
On the other end of the disease spectrum, an SCN5A-LQTS (GOF) assay is not yet available. A major reason for this is the very small size of the late sodium current (approximately 0.2% of peak of WT). Thus, it is not possible to design a single assay that can record both GOF and LOF, as the high Na+ concentrations required to measure GOF in the late sodium current (Wang D. W. et al., 1996; Keller et al., 2003; Tester et al., 2010; Ma et al., 2023; Stutzman et al., 2023) will generally create voltage clamp errors when trying to measure peak sodium currents. GOF defects are primarily a result of failures in channel inactivation resulting in a late/persistent current (Wang D. W. et al., 1996; Keller et al., 2003; Ruan et al., 2007; Makita et al., 2008; Olesen et al., 2012; Moreau et al., 2013; Peters et al., 2016; Veltmann et al., 2016; Li et al., 2021; Stutzman et al., 2023) or caused by a change in the overlap of the voltage dependence of steady-state activation and inactivation resulting in a window current (Wang D. W. et al., 1996; Moreau et al., 2013; Moreau et al., 2015; Peters et al., 2016; Peters et al., 2021; Stutzman et al., 2023). Additional mechanisms that can contribute include faster recovery from inactivation (Clancy et al., 2003) and the presence of gating pore currents (Moreau et al., 2015; Peters et al., 2021). Thus, any APC assay designed to detect SCN5A GOF will have to be able to assay these diverse range of mechanisms.
Though traditionally thought to form functional monomers, dominant-negative effects in NaV1.5 have been reported in vitro (Keller et al., 2005; Hoshi et al., 2014; O’Neill et al., 2022) and in vivo (Doisne et al., 2021). Interactions between NaV1.5 α-subunits was suggested to cause dominant-negative effects observed in trafficking-defective (Clatot et al., 2012), and gating-defective variants (Clatot et al., 2018). They corroborated this with co-immunoprecipitation, protein crosslinking, western blots, single molecule pull-down and electrophysiological analysis (Clatot et al., 2012; Clatot et al., 2017). However, naturally, NaV1.5 proteins form macromolecular complexes with NaV1.5-interacting proteins that can modulate its trafficking and gating (Marchal and Remme, 2022). The impact of SCN5A variants on these interactions are not yet clear but, altered protein-protein interactions within NaV1.5 complexes have been associated with GOF and LOF defects, predominantly at the intercalated discs and lateral membrane regions, respectively. Additionally, variants in the interacting-proteins (e.g., NaV1.5 β-subunits, Ankyrin-B, Caveolin-3) have also been associated with sodium channelopathies. Hence, whether these dominant-negative effects are truly dominant-negative or are a result of altered subdomain-specific NaV1.5-interacting proteins will require further investigation.
Beyond the channel-specific limitations discussed above, there are other limitations that may be classified as biological and experimental. Normal function gathered from in vitro assays does not guarantee there will be no functional defects in vivo (Watanabe et al., 2011). For example, functional results can be influenced by the isoform used (Tan et al., 2005; Wang et al., 2007), mRNA splicing, protein interactions that may not be present in heterologous expression systems (Watanabe et al., 2009), transcriptional factors that may impact gene and variant expression (Barc et al., 2022), and non-coding variants that may impact expression in native cells but not in the heterologous expression system. This can be addressed by studying variants in iPSC-derived cardiac myocytes, but at the expense of much lower throughput, (Sendfeld et al., 2019). Furthermore, though genotype-phenotype studies can identify LOF and GOF variants associated with specific diseases, there are variants that may overlap several diseases, such as in the case for SCN5A-E1784K and CACNA1C-E1115K (Makita et al., 2008; Kashiwa et al., 2023). Many of the assays developed to date have also not studied enough benign variant controls to be able to formally fulfil the ClinGen SVI working group criteria for validation of functional assays (Brnich et al., 2020). The strength of functional evidence for any given assay is dependent on i) the assay’s ability to accurately distinguish normal from abnormal function (Jiang et al., 2022) and ii) the number of control variants available to test. So, if there are very few benign missense variants (e.g., because the gene is very small) then it may not be possible to achieve more than moderate evidence strength (Jiang et al., 2022). Similarly, if a disease is very rare, then there may be an insufficient number of definitely (likely) pathogenic variants available to calibrate the assay to achieve more than moderate evidence strength. Furthermore, not all assays have incorporated the potential for dominant-negative effects, and difficulties in co-expression of multiple sub-units in the correct stoichiometry (Zou et al., 2022) need to be considered. There are also no assays yet developed for channels with more complex stoichiometries, e.g., L-type calcium channels which require co-expression of multiple subunits (Dolphin, 2016).
At an experimental level, temperature control can also be an issue, which is important to study ion channel kinetics of temperature-sensitive variants (Vandenberg et al., 2006; Abdelsayed et al., 2015; Lei et al., 2019; Jones, 2022; Ren et al., 2022; Kriegeskorte et al., 2023). Whilst temperature can be preset on most APC systems (e.g., Biolin Scientific, Fluxion and Nanion Technologies, Table 1), most APC assays for cardiac ion channels have been conducted at room temperature (Vanoye et al., 2018; Glazer et al., 2020b; Ng et al., 2021) due to an inherent instability of cell membranes at physiological temperatures leading to lower success rates. Temperature can have varying impacts on different properties. For example, increased temperature can exacerbate trafficking defects but also reduces inactivation in hERG channels but this may vary between variants. Analyzing the effect of temperature would also be especially valuable for fever-inducible disorders such as BrS (Dumaine et al., 1999; Keller et al., 2005; Peters et al., 2016) but at the moment this remains challenging for APC systems.
The widespread use of fluoride as the primary anion in internal solutions have benefited patch clamp studies through the formation of CaF2 crystals at the site of membrane rupture, improving the seal resistance and cell stability. In cardiac potassium channels, the use of fluoride did not alter biophysical properties and was shown to be significantly superior to alternatives, K-gluconate and KCl, for patch success rates (Zeng et al., 2008; Rapedius et al., 2022). Whilst in some neuronal sodium channels (NaV1.9, NaV1.7), fluoride has been reported to impact gating (Meadows et al., 2002; Rugiero et al., 2003; Coste et al., 2004; Jarecki et al., 2008) but not in others (NaV1.8) (Coste et al., 2004). The recent release of Fluoride-free APC chips could overcome these issues, however they are associated with a reduction of seal quality, e.g., from 77.9% with seal >1 GΩ to 35.9% > 1 GΩ in the fluoride free solutions (Rapedius et al., 2022).
Sudden cardiac death accounts for ∼80% of cardiac deaths in young, healthy individuals (Doolan et al., 2004; Bagnall et al., 2016). Approximately a third are from primary arrhythmogenic disorders (Doolan et al., 2004; Bagnall et al., 2016). Therefore, genetic tests can be incredibly valuable for variant discovery in arrhythmogenic diseases and for familial screening and therapy (Tester and Ackerman, 2007; Gladding et al., 2010). Identifying a variant, however, does not necessarily equate to finding the cause of the disease. An additional complication is that some diseases can be caused by variants in different genes (e.g., LQTS in KCNQ1, KCNH2, and SCN5A; Table 2) and management will vary depending on which gene is implicated (Wilde et al., 2022a). Robust APC assays can be useful to identify which variants alter function and are therefore most likely to be the true cause of disease.
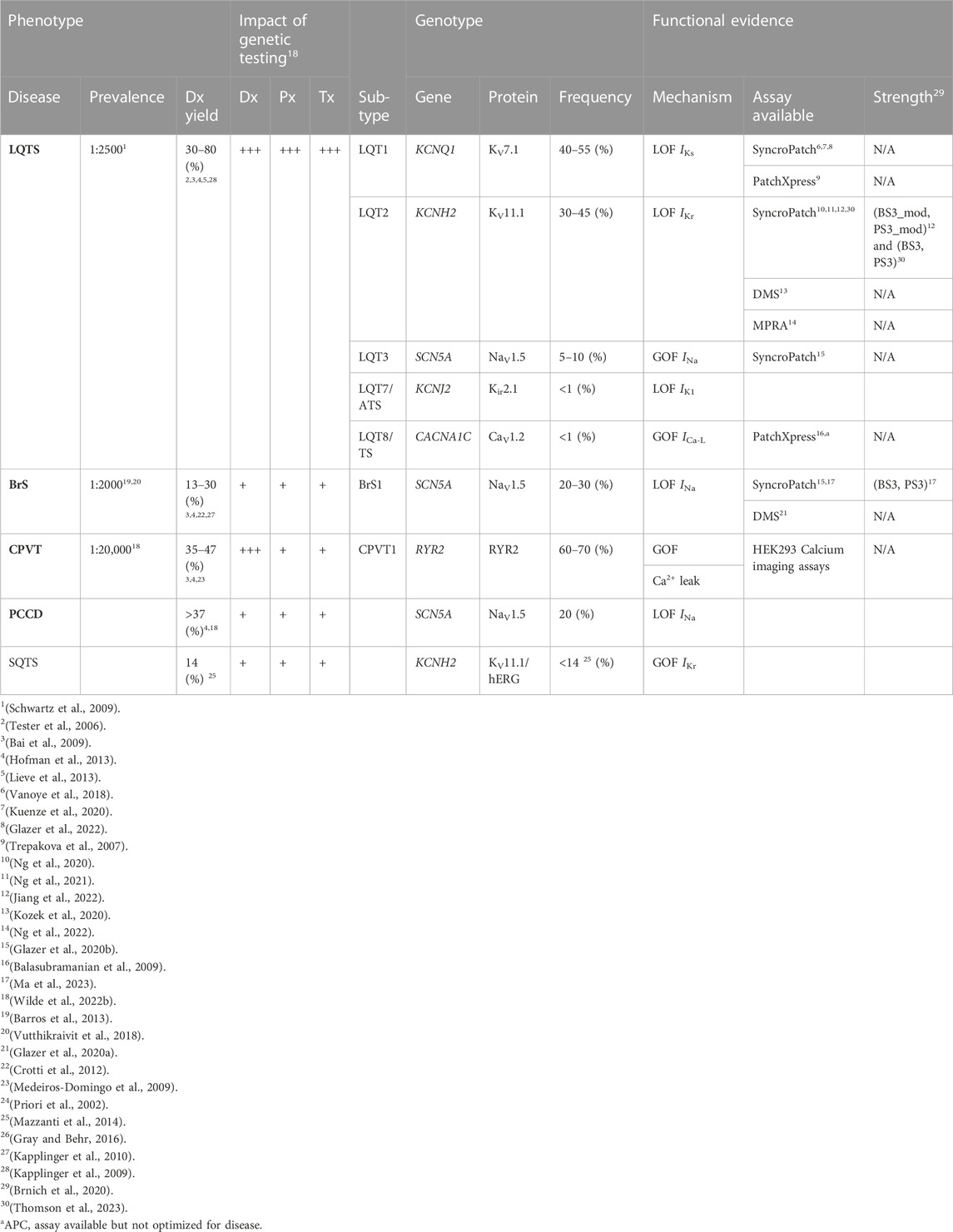
TABLE 2. Summary of common cardiac channelopathy-associated genes. LQTS, Long QT Syndrome; CPVT, Catecholaminergic Polymorphic Ventricular Tachycardia. BrS: Brugada Syndrome; CCD, Cardiac Conduction Disease; SUDS, Sudden Unexplained Death Syndrome; SIDS, Sudden Infant Death Syndrome; ATS, Andersen-Tawil Syndrome; Dx, diagnostic; Px, prognostic; Tx, therapeutic/treatment; Recommended, indicated or useful (+++). Can be recommended or useful (++). May be considered or useful (+).
In addition to finding variants in patients with clinical evidence for a genetic condition, there are an increasing number of patients who are having genome sequencing for other conditions and a rare, variant is identified in a cardiac ion channel gene. In theory, such variants could have the potential to significantly increase the risk of cardiac arrhythmias. In 2013, the ACMG devised a list of genes identifying highly penetrant genetic disorders amenable to medical intervention (Green et al., 2013). Secondary findings, defined as variants in the list of medically actionable genes that are determined to be disease-causing, are required to be reported irrespective of whether it was the original reason for seeking genetic testing (Kalia et al., 2017; Miller et al., 2021b). Regulated by the Secondary Findings Maintenance Working Group, the list currently contains 73 genes, of which 34 are associated with cardiovascular phenotypes including the aforementioned genes KCNQ1, KCNH2 and SCN5A (Miller et al., 2021a).
Variant classifications are guided by ACMG/AMP’s evidence strength-dependent, multi-tiered criterion (Richards et al., 2015) and have been adopted by 95% of laboratories (Niehaus et al., 2019). Classifications are used to determine clinical risks however, a limitation of this system is when insufficient clinical phenotype data are available to determine pathogenicity (Kroncke et al., 2020). ClinVar contains interpretations for more than half a million variants (Landrum et al., 2018; Landrum et al., 2019). However, concordance rates for cardiac genes was only 62% (Amendola et al., 2020) and approximately half of all variants uncovered in genes implicated in inheritable cardiac disease are classified as VUS (Anderson et al., 2022). Unfortunately, the classification where there is most concordance is VUS and, alarmingly, discordances that would influence clinical recommendations were found in 11% of variants when the Sequence and Diagnostic Yield working group evaluated variant classifications in medically actionable genes across 8 laboratories (Amendola et al., 2020). Functional assays have the potential to help resolve much of this discordance. However, as noted by Harrison and colleagues (Harrison et al., 2017), there has been very inconsistent application of evidence from functional assays between genetic testing laboratories. Consequently, a detailed ClinGen framework for the application of functional evidence was released (Table 3) (Brnich et al., 2020) and it has been shown that high throughput APC functional assays for cardiac ion channels can be designed to meet a strong level of evidence for assessment of variants (Jiang et al., 2022; Ma et al., 2023; Thomson et al., 2023).
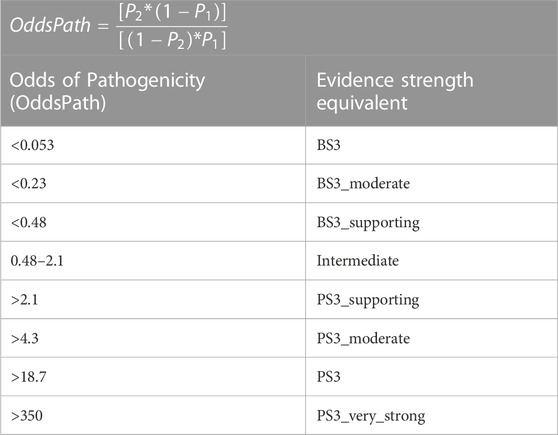
TABLE 3. The level of evidence applicable for each functional assay is determined by an ‘Odds of Pathogenicity’ score (Reproduced from Brnich et al. (2020), licensed under CC-BY 4.0). Functional evidence BS3: well established functional evidence shows no deleterious effect. PS3: Well-established functional studies show a deleterious effect (Richards et al., 2015).
For many ion channels, functional activity requires co-expression of multiple subunits. It is possible to co-express two subunits from a single plasmid by including an Internal Ribosome Entry Site (IRES) between the two cDNA sequences. This approach has been used to co-express beta β-subunits with α- or other β-subunits for sodium (Fletcher et al., 2011; Nakajima et al., 2015) and potassium channels (Ng et al., 2020; Ng et al., 2021). However, the expression between the genes using IRES may not be optimal as the gene located after the IRES is typically expressed at lower levels (Mizuguchi et al., 2000; Bochkov and Palmenberg, 2006). Recently, a modified Flp-In HEK293 was developed to enable the expression of 2 genes at equal expression by introducing a second plasmid that has puromycin antibiotic resistance (in addition to hygromycin on the first plasmid) (Ward et al., 2011; Jensen et al., 2020). By incorporating IRES into this modified Flp-In HEK293, up to 4 genes can be expressed in a single Flp-In HEK293 cell line. This will facilitate the efficient generation of multi-subunit channel complexes, such as the L-type calcium channel (CaV1.2, ICa,L) which is crucial for the plateau phase of the cardiac action potential and requires co-expression of CACNA1C, CACNB2B, and CACNA2D1. CaV1.2 GOF variants can cause Timothy Syndrome, a multi-organ dysfunction associated with LQTS (LQTS8) and sudden cardiac death (Splawski et al., 2004). While CaV1.2 LOF variants have been associated with BrS (1%–2%) (Gray and Behr, 2016; Cerrone et al., 2022; Nakano and Shimizu, 2022) and SQTS (Templin et al., 2011), although this association is not yet classified as definitive (Wilde et al., 2022b). This strategy can also be employed to generate stable cell lines that co-expressed both KCNQ1/KCNE1 and the A-kinase adaptor protein, AKAP9, which is necessary to enable modulation by adrenergic signaling (Dvir et al., 2014).
In addition to using APC to determine the effect of variants on channel function, it is possible to simultaneously test the effect of drugs on each variant. This approach is likely to be particularly important for GOF mutations. For example, a recent in vitro study comparing the efficacy of sodium channel blockers found phentytoin, an anti-seizure medication, to be more effective at rescuing the LQT3 phenotype than the commonly recommended medication, mexiletine, for SCN5A-F1760C (Stutzman et al., 2023). Similarly, one can also envisage screening GOF CACNA1C variants in Timothy syndrome patients to identify the optimal drug for suppressing the late ICa,L without reducing peak ICa,L. This approach, however, need not be limited to GOF variants. For example, APC methods have provided important preliminary screening data to identify LOF variants in CFTR that are amenable to rescue with Trikaftor (Brüggemann et al., 2017).
In recent years there has been tremendous progress in developing high throughput APC assays to assess the functional effects of cardiac ion channel variants. One can envisage that within a few years all presently known clinically occurring variants in these genes will have been characterized. Whilst it is unlikely that APC assays will enable prospective characterization of all potential missense variants in an ion channel, this could be achieved with higher throughput multiplexed assays of variant effect (MAVE) assays (Findlay et al., 2018), including, e.g., fluorescence based trafficking assays and cell survival assays combined with deep mutational scanning (Glazer et al., 2020a; Kozek et al., 2020; Fayer et al., 2021; Coyote-Maestas et al., 2022; Ng et al., 2022). In this context, APC assays have been very useful for validating the results from these techniques (Glazer et al., 2020a; Kozek et al., 2020; Ng et al., 2022). Ultimately, this could facilitate the development of databases containing functional data for all potential variants that clinicians could access immediately upon finding any new variants in an ion channel gene. One could also envisage APC functional genomics assays being combined with drug screening to enable identification of optimal drug therapy for each specific variant.
JM: Writing–original draft, Writing–review and editing. JV: Writing–original draft, Writing–review and editing. C-AN: Writing–original draft, Writing–review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The work in the authors’ laboratory was funded by a grant from the Medical Research Future Fund–Genomics Health Futures Mission (#MRF2016760 to JV and C-AN), a NSW Health CVD Senior Scientist Grant 2019 (to JV) and an Research Training Program Scholarship 2022 to JM
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Abdelsayed M., Peters C. H., Ruben P. C. (2015). Differential thermosensitivity in mixed syndrome cardiac sodium channel mutants. J. Physiol. 593 (18), 4201–4223. doi:10.1113/jp270139
Adler A., Novelli V., Amin A. S., Abiusi E., Care M., Nannenberg E. A., et al. (2020). An international, multicentered, evidence-based reappraisal of genes reported to cause congenital long QT syndrome. Circulation 141 (6), 418–428. doi:10.1161/CIRCULATIONAHA.119.043132
Aghasafari P., Yang P. C., Kernik D. C., Sakamoto K., Kanda Y., Kurokawa J., et al. (2021). A deep learning algorithm to translate and classify cardiac electrophysiology. Elife 10, e68335. doi:10.7554/eLife.68335
Amendola L. M., Muenzen K., Biesecker L. G., Bowling K. M., Cooper G. M., Dorschner M. O., et al. (2020). Variant classification concordance using the ACMG-AMP variant interpretation guidelines across nine genomic implementation research studies. Am. J. Hum. Genet. 107 (5), 932–941. doi:10.1016/j.ajhg.2020.09.011
Amin A. S., Asghari-Roodsari A., Tan H. L. (2010). Cardiac sodium channelopathies. Pflugers Arch. 460 (2), 223–237. doi:10.1007/s00424-009-0761-0
Anderson C. L., Kuzmicki C. E., Childs R. R., Hintz C. J., Delisle B. P., January C. T. (2014). Large-scale mutational analysis of Kv11.1 reveals molecular insights into type 2 long QT syndrome. Nat. Commun. 5, 5535. doi:10.1038/ncomms6535
Anderson C. L., Munawar S., Reilly L., Kamp T. J., January C. T., Delisle B. P., et al. (2022). How functional genomics can keep pace with VUS identification. Front. Cardiovasc Med. 9, 900431. doi:10.3389/fcvm.2022.900431
Asher G., Reuven N., Shaul Y. (2006). 20S proteasomes and protein degradation "by default. Bioessays 28 (8), 844–849. doi:10.1002/bies.20447
Asmild M., Oswald N., Krzywkowski K. M., Friis S., Jacobsen R. B., Reuter D., et al. (2003). Upscaling and automation of electrophysiology: toward high throughput screening in ion channel drug discovery. Recept. Channels 9 (1), 49–58. doi:10.3109/10606820308258
Aydar E., Palmer C. (2001). Functional characterization of the C-terminus of the human ether-à-go-go-related gene K(+) channel (HERG). J. Physiol. 534 (Pt 1), 1–14. doi:10.1111/j.1469-7793.2001.t01-3-00001.x
Baez-Nieto D., Allen A., Akers-Campbell S., Yang L., Budnik N., Pupo A., et al. (2022). Analysing an allelic series of rare missense variants of CACNA1I in a Swedish schizophrenia cohort. Brain 145 (5), 1839–1853. doi:10.1093/brain/awab443
Bagnall R. D., Weintraub R. G., Ingles J., Duflou J., Yeates L., Lam L., et al. (2016). A prospective study of sudden cardiac death among children and young adults. N. Engl. J. Med. 374 (25), 2441–2452. doi:10.1056/NEJMoa1510687
Bai R., Napolitano C., Bloise R., Monteforte N., Priori S. G. (2009). Yield of genetic screening in inherited cardiac channelopathies: how to prioritize access to genetic testing. Circ. Arrhythm. Electrophysiol. 2 (1), 6–15. doi:10.1161/circep.108.782888
Balasubramanian B., Imredy J. P., Kim D., Penniman J., Lagrutta A., Salata J. J. (2009). Optimization of Cav1.2 screening with an automated planar patch clamp platform. J. Pharmacol. Toxicol. Methods 59 (2), 62–72. doi:10.1016/j.vascn.2009.02.002
Balijepalli S. Y., Lim E., Concannon S. P., Chew C. L., Holzem K. E., Tester D. J., et al. (2012). Mechanism of loss of Kv11.1 K+ current in mutant T421M-Kv11.1-expressing rat ventricular myocytes: interaction of trafficking and gating. Circulation 126 (24), 2809–2818. doi:10.1161/CIRCULATIONAHA.112.118018
Barc J., Tadros R., Glinge C., Chiang D. Y., Jouni M., Simonet F., et al. (2022). Genome-wide association analyses identify new Brugada syndrome risk loci and highlight a new mechanism of sodium channel regulation in disease susceptibility. Nat. Genet. 54 (3), 232–239. doi:10.1038/s41588-021-01007-6
Barhanin J., Lesage F., Guillemare E., Fink M., Lazdunski M., Romey G. (1996). K(V)LQT1 and lsK (minK) proteins associate to form the I(Ks) cardiac potassium current. Nature 384 (6604), 78–80. doi:10.1038/384078a0
Barros M. A., Fernandes H. F., Barros C. M., Motta F. J., Canalle R., Rey J. A., et al. (2013). Brugada syndrome in a family with a high mortality rate: a case report. J. Med. Case Rep. 7, 78. doi:10.1186/1752-1947-7-78
Becker N., Stoelzle S., Göpel S., Guinot D., Mumm P., Haarmann C., et al. (2013). Minimized cell usage for stem cell-derived and primary cells on an automated patch clamp system. J. Pharmacol. Toxicol. Methods 68 (1), 82–87. doi:10.1016/j.vascn.2013.03.009
Bell D. C., Fermini B. (2021). Use of automated patch clamp in cardiac safety assessment: past, present and future perspectives. J. Pharmacol. Toxicol. Methods 111, 107114. doi:10.1016/j.vascn.2021.107114
Bennett P. B., Yazawa K., Makita N., George A. L. (1995). Molecular mechanism for an inherited cardiac arrhythmia. Nature 376 (6542), 683–685. doi:10.1038/376683a0
Benson D. W., Wang D. W., Dyment M., Knilans T. K., Fish F. A., Strieper M. J., et al. (2003). Congenital sick sinus syndrome caused by recessive mutations in the cardiac sodium channel gene (SCN5A). J. Clin. Invest. 112 (7), 1019–1028. doi:10.1172/jci18062
Bezzina C., Veldkamp M. W., van Den Berg M. P., Postma A. V., Rook M. B., Viersma J. W., et al. (1999). A single Na(+) channel mutation causing both long-QT and Brugada syndromes. Circ. Res. 85 (12), 1206–1213. doi:10.1161/01.res.85.12.1206
Bochkov Y. A., Palmenberg A. C. (2006). Translational efficiency of EMCV IRES in bicistronic vectors is dependent upon IRES sequence and gene location. Biotechniques 41 (3), 283–284. doi:10.2144/000112243
Brnich S. E., Abou Tayoun A. N., Couch F. J., Cutting G. R., Greenblatt M. S., Heinen C. D., et al. (2020). Recommendations for application of the functional evidence PS3/BS3 criterion using the ACMG/AMP sequence variant interpretation framework. Genome Med. 12 (1), 3. doi:10.1186/s13073-019-0690-2
Brüggemann A., Friis S., Strassmeier T., Rapedius M., Goetze T., Rinke I., et al. (2017). Characterization of CFTR activators and inhibitors by the use of a planar patch clamp system. Biophysical J. 112 (3), 411a. doi:10.1016/j.bpj.2016.11.2561
Brüggemann A., George M., Klau M., Beckler M., Steindl J., Behrends J. C., et al. (2003). High quality ion channel analysis on a chip with the NPC technology. Assay. Drug Dev. Technol. 1 (5), 665–673. doi:10.1089/154065803770381020
Brüggemann A., Stoelzle S., George M., Behrends J. C., Fertig N. (2006). Microchip technology for automated and parallel patch-clamp recording. Small 2 (7), 840–846. doi:10.1002/smll.200600083
Cerrone M., Costa S., Delmar M. (2022). The genetics of Brugada syndrome. Annu. Rev. Genomics Hum. Genet. 23, 255–274. doi:10.1146/annurev-genom-112921-011200
Cerrone M., Remme C. A., Tadros R., Bezzina C. R., Delmar M. (2019). Beyond the one gene-one disease paradigm: complex genetics and pleiotropy in inheritable cardiac disorders. Circulation 140 (7), 595–610. doi:10.1161/CIRCULATIONAHA.118.035954
Chambers C., Witton I., Adams C., Marrington L., Kammonen J. (2016). High-throughput screening of NaV1.7 modulators using a giga-seal automated patch clamp instrument. ASSAY Drug Dev. Technol. 14 (2), 93–108. doi:10.1089/adt.2016.700
Chen S., Francioli L. C., Goodrich J. K., Collins R. L., Kanai M., Wang Q., et al. (2022). A genome-wide mutational constraint map quantified from variation in 76,156 human genomes. bioRxiv, 2022.2003.2020.485034. doi:10.1101/2022.03.20.485034
Clancy C. E., Tateyama M., Liu H., Wehrens X. H. T., Kass R. S. (2003). Non-equilibrium gating in cardiac Na+ channels: an original mechanism of arrhythmia. Circulation 107 (17), 2233–2237. doi:10.1161/01.CIR.0000069273.51375.BD
Clatot J., Hoshi M., Wan X., Liu H., Jain A., Shinlapawittayatorn K., et al. (2017). Voltage-gated sodium channels assemble and gate as dimers. Nat. Commun. 8 (1), 2077. doi:10.1038/s41467-017-02262-0
Clatot J., Zheng Y., Girardeau A., Liu H., Laurita K. R., Marionneau C., et al. (2018). Mutant voltage-gated Na(+) channels can exert a dominant negative effect through coupled gating. Am. J. Physiol. Heart Circ. Physiol. 315 (5), H1250-H1257–h1257. doi:10.1152/ajpheart.00721.2017
Clatot J., Ziyadeh-Isleem A., Maugenre S., Denjoy I., Liu H., Dilanian G., et al. (2012). Dominant-negative effect of SCN5A N-terminal mutations through the interaction of Na(v)1.5 α-subunits. Cardiovasc Res. 96 (1), 53–63. doi:10.1093/cvr/cvs211
Coraboeuf E. (1949). Potentiels d'action du muscle cardiaque obtenus A l'aide de microelectrodes intracellulaires: presence d'une inversion de potentiel. CR Soc. Biol. Paris 143, 1360–1361.
Coraboeuf E., Weidmann S. (1949). Potentiel de repos et potentiels d’action du muscle cardiaque mesurés à l’aide d’èlectrodes intracellulaires. Comptes Rendus Des Seances De La Soc. De Biol. De Ses Fil. 143 (19-2), 1329–1331.
Coste B., Osorio N., Padilla F., Crest M., Delmas P. (2004). Gating and modulation of presumptive NaV1.9 channels in enteric and spinal sensory neurons. Mol. Cell. Neurosci. 26 (1), 123–134. doi:10.1016/j.mcn.2004.01.015
Coyote-Maestas W., Nedrud D., He Y., Schmidt D. (2022). Determinants of trafficking, conduction, and disease within a K+ channel revealed through multiparametric deep mutational scanning. eLife 11, e76903. doi:10.7554/eLife.76903
Crotti L., Marcou C. A., Tester D. J., Castelletti S., Giudicessi J. R., Torchio M., et al. (2012). Spectrum and prevalence of mutations involving BrS1- through BrS12-susceptibility genes in a cohort of unrelated patients referred for Brugada syndrome genetic testing: implications for genetic testing. J. Am. Coll. Cardiol. 60 (15), 1410–1418. doi:10.1016/j.jacc.2012.04.037
Curran M. E., Splawski I., Timothy K. W., Vincent G. M., Green E. D., Keating M. T. (1995). A molecular basis for cardiac arrhythmia: HERG mutations cause long QT syndrome. Cell 80 (5), 795–803. doi:10.1016/0092-8674(95)90358-5
de Lera Ruiz M., Kraus R. L. (2015). Voltage-gated sodium channels: structure, function, pharmacology, and clinical indications. J. Med. Chem. 58 (18), 7093–7118. doi:10.1021/jm501981g
Delisle B. P., Anson B. D., Rajamani S., January C. T. (2004). Biology of cardiac arrhythmias: ion channel protein trafficking. Circ. Res. 94 (11), 1418–1428. doi:10.1161/01.RES.0000128561.28701.ea
Dixit G., Dabney-Smith C., Lorigan G. A. (2020). The membrane protein KCNQ1 potassium ion channel: functional diversity and current structural insights. Biochim. Biophys. Acta Biomembr. 1862 (5), 183148. doi:10.1016/j.bbamem.2019.183148
Doisne N., Grauso M., Mougenot N., Clergue M., Souil C., Coulombe A., et al. (2021). In vivo dominant-negative effect of an SCN5A Brugada syndrome variant. Front. Physiol. 12, 661413. doi:10.3389/fphys.2021.661413
Dolphin A. C. (2016). Voltage-gated calcium channels and their auxiliary subunits: physiology and pathophysiology and pharmacology. J. Physiology 594 (19), 5369–5390. doi:10.1113/JP272262
Doolan A., Langlois N., Semsarian C. (2004). Causes of sudden cardiac death in young Australians. Med. J. Aust. 180 (3), 110–112. doi:10.5694/j.1326-5377.2004.tb05830.x
Dumaine R., Towbin J. A., Brugada P., Vatta M., Nesterenko D. V., Nesterenko V. V., et al. (1999). Ionic mechanisms responsible for the electrocardiographic phenotype of the Brugada syndrome are temperature dependent. Circ. Res. 85 (9), 803–809. doi:10.1161/01.res.85.9.803
Dvir M., Strulovich R., Sachyani D., Ben-Tal Cohen I., Haitin Y., Dessauer C., et al. (2014). Long QT mutations at the interface between KCNQ1 helix C and KCNE1 disrupt I(KS) regulation by PKA and PIP₂. J. Cell Sci. 127 (Pt 18), 3943–3955. doi:10.1242/jcs.147033
Fayer S., Horton C., Dines J. N., Rubin A. F., Richardson M. E., McGoldrick K., et al. (2021). Closing the gap: systematic integration of multiplexed functional data resolves variants of uncertain significance in BRCA1, TP53, and PTEN. Am. J. Hum. Genet. 108 (12), 2248–2258. doi:10.1016/j.ajhg.2021.11.001
Fermini B., Fossa A. A. (2003). The impact of drug-induced QT interval prolongation on drug discovery and development. Nat. Rev. Drug Discov. 2 (6), 439–447. doi:10.1038/nrd1108
Fertig N., Blick R. H., Behrends J. C. (2002). Whole cell patch clamp recording performed on a planar glass chip. Biophysical J. 82 (6), 3056–3062. doi:10.1016/S0006-3495(02)75646-4
Findlay G. M., Daza R. M., Martin B., Zhang M. D., Leith A. P., Gasperini M., et al. (2018). Accurate classification of BRCA1 variants with saturation genome editing. Nature 562 (7726), 217–222. doi:10.1038/s41586-018-0461-z
Finkel A., Wittel A., Yang N., Handran S., Hughes J., Costantin J. (2006). Population patch clamp improves data consistency and success rates in the measurement of ionic currents. J. Biomol. Screen 11 (5), 488–496. doi:10.1177/1087057106288050
Fletcher E. V., Kullmann D. M., Schorge S. (2011). Alternative splicing modulates inactivation of type 1 voltage-gated sodium channels by toggling an amino acid in the first S3-S4 linker. J. Biol. Chem. 286 (42), 36700–36708. doi:10.1074/jbc.M111.250225
Gillie D. J., Novick S. J., Donovan B. T., Payne L. A., Townsend C. (2013). Development of a high-throughput electrophysiological assay for the human ether-à-go-go related potassium channel hERG. J. Pharmacol. Toxicol. Methods 67 (1), 33–44. doi:10.1016/j.vascn.2012.10.002
Gladding P. A., Evans C. A., Crawford J., Chung S. K., Vaughan A., Webster D., et al. (2010). Posthumous diagnosis of long QT syndrome from neonatal screening cards. Heart rhythm. 7 (4), 481–486. doi:10.1016/j.hrthm.2009.12.023
Glazer A. M., Davogustto G. E., Shaffer C. M., Vanoye C. G., Desai R. R., Farber-Eger E., et al. (2022). Arrhythmia variant associations and reclassifications in the eMERGE-III sequencing study. Circulation 0 (0), 877–891. doi:10.1161/CIRCULATIONAHA.121.055562
Glazer A. M., Kroncke B. M., Matreyek K. A., Yang T., Wada Y., Shields T., et al. (2020a). Deep mutational scan of an SCN5A voltage sensor. Circ. Genom Precis. Med. 13 (1), e002786. doi:10.1161/circgen.119.002786
Glazer A. M., Wada Y., Li B., Muhammad A., Kalash O. R., O'Neill M. J., et al. (2020b). High-throughput reclassification of SCN5A variants. Am. J. Hum. Genet. 107 (1), 111–123. doi:10.1016/j.ajhg.2020.05.015
Golden A. P., Li N., Chen Q., Lee T., Nevill T., Cao X., et al. (2011). IonFlux: a microfluidic patch clamp system evaluated with human ether-à-go-go related gene channel physiology and pharmacology. ASSAY Drug Dev. Technol. 9 (6), 608–619. doi:10.1089/adt.2010.0362
González J. E., Oades K., Leychkis Y., Harootunian A., Negulescu P. A. (1999). Cell-based assays and instrumentation for screening ion-channel targets. Drug Discov. Today 4 (9), 431–439. doi:10.1016/S1359-6446(99)01383-5
Grant A. O. (2009). Cardiac ion channels. Circulation Arrhythmia Electrophysiol. 2 (2), 185–194. doi:10.1161/CIRCEP.108.789081
Gray B., Behr E. R. (2016). New insights into the genetic basis of inherited arrhythmia syndromes. Circ. Cardiovasc. Genet. 9 (6), 569–577. doi:10.1161/CIRCGENETICS.116.001571
Green R. C., Berg J. S., Grody W. W., Kalia S. S., Korf B. R., Martin C. L., et al. (2013). ACMG recommendations for reporting of incidental findings in clinical exome and genome sequencing. Genet. Med. 15 (7), 565–574. doi:10.1038/gim.2013.73
Gui J., Wang T., Jones R. P., Trump D., Zimmer T., Lei M. (2010). Multiple loss-of-function mechanisms contribute to SCN5A-related familial sick sinus syndrome. PLoS One 5 (6), e10985. doi:10.1371/journal.pone.0010985
Hamill O. P., Marty A., Neher E., Sakmann B., Sigworth F. J. (1981). Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 391 (2), 85–100. doi:10.1007/bf00656997
Harrison S. M., Dolinsky J. S., Knight Johnson A. E., Pesaran T., Azzariti D. R., Bale S., et al. (2017). Clinical laboratories collaborate to resolve differences in variant interpretations submitted to ClinVar. Genet. Med. 19 (10), 1096–1104. doi:10.1038/gim.2017.14
Hodgkin A. L., Huxley A. F. (1952). A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 117 (4), 500–544. doi:10.1113/jphysiol.1952.sp004764
Hofman N., Tan H. L., Alders M., Kolder I., de Haij S., Mannens M. M. A. M., et al. (2013). Yield of molecular and clinical testing for arrhythmia syndromes: report of 15 years' experience. Circulation 128 (14), 1513–1521. doi:10.1161/CIRCULATIONAHA.112.000091
Hoshi M., Du X. X., Shinlapawittayatorn K., Liu H., Chai S., Wan X., et al. (2014). Brugada syndrome disease phenotype explained in apparently benign sodium channel mutations. Circ. Cardiovasc. Genet. 7 (2), 123–131. doi:10.1161/CIRCGENETICS.113.000292
Jarecki B. W., Sheets P. L., Jackson Ii J. O., Cummins T. R. (2008). Paroxysmal extreme pain disorder mutations within the D3/S4–S5 linker of Nav1.7 cause moderate destabilization of fast inactivation. J. Physiology 586 (17), 4137–4153. doi:10.1113/jphysiol.2008.154906
Jensen O., Ansari S., Gebauer L., Müller S. F., Lowjaga K. A. A. T., Geyer J., et al. (2020). A double-Flp-in method for stable overexpression of two genes. Sci. Rep. 10 (1), 14018. doi:10.1038/s41598-020-71051-5
Jiang C., Richardson E., Farr J., Hill A. P., Ullah R., Kroncke B. M., et al. (2022). A calibrated functional patch-clamp assay to enhance clinical variant interpretation in KCNH2-related long QT syndrome. Am. J. Hum. Genet. 109 (7), 1199–1207. doi:10.1016/j.ajhg.2022.05.002
Jones D. K. (2022). Hysteretic hERG channel gating current recorded at physiological temperature. Sci. Rep. 12 (1), 5950. doi:10.1038/s41598-022-10003-7
Kalia S. S., Adelman K., Bale S. J., Chung W. K., Eng C., Evans J. P., et al. (2017). Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2016 update (ACMG SF v2.0): a policy statement of the American College of Medical Genetics and Genomics. Genet. Med. 19 (2), 249–255. doi:10.1038/gim.2016.190
Kanters J. K., Skibsbye L., Hedley P. L., Dembic M., Liang B., Hagen C. M., et al. (2015). Combined gating and trafficking defect in Kv11.1 manifests as a malignant long QT syndrome phenotype in a large Danish p.F29L founder family. Scand. J. Clin. Laboratory Investigation 75 (8), 699–709. doi:10.3109/00365513.2015.1091090
Kapplinger J. D., Tester D. J., Alders M., Benito B., Berthet M., Brugada J., et al. (2010). An international compendium of mutations in the SCN5A-encoded cardiac sodium channel in patients referred for Brugada syndrome genetic testing. Heart rhythm. 7 (1), 33–46. doi:10.1016/j.hrthm.2009.09.069
Kapplinger J. D., Tester D. J., Salisbury B. A., Carr J. L., Harris-Kerr C., Pollevick G. D., et al. (2009). Spectrum and prevalence of mutations from the first 2,500 consecutive unrelated patients referred for the FAMILION long QT syndrome genetic test. Heart rhythm. 6 (9), 1297–1303. doi:10.1016/j.hrthm.2009.05.021
Kashiwa A., Makiyama T., Kohjitani H., Maurissen T. L., Ishikawa T., Yamamoto Y., et al. (2023). Disrupted Ca(V)1.2 selectivity causes overlapping long QT and Brugada syndrome phenotypes in the CACNA1C-E1115K iPS cell model. Heart rhythm. 20 (1), 89–99. doi:10.1016/j.hrthm.2022.08.021
Ke Y., Ng C. A., Hunter M. J., Mann S. A., Heide J., Hill A. P., et al. (2013). Trafficking defects in PAS domain mutant Kv11.1 channels: roles of reduced domain stability and altered domain-domain interactions. Biochem. J. 454 (1), 69–77. doi:10.1042/bj20130328
Keller D. I., Acharfi S., Delacrétaz E., Benammar N., Rotter M., Pfammatter J.-P., et al. (2003). A novel mutation in SCN5A, delQKP 1507–1509, causing long QT syndrome:: role of Q1507 residue in sodium channel inactivation. J. Mol. Cell. Cardiol. 35 (12), 1513–1521. doi:10.1016/j.yjmcc.2003.08.007
Keller D. I., Rougier J. S., Kucera J. P., Benammar N., Fressart V., Guicheney P., et al. (2005). Brugada syndrome and fever: genetic and molecular characterization of patients carrying SCN5A mutations. Cardiovasc Res. 67 (3), 510–519. doi:10.1016/j.cardiores.2005.03.024
Kernik D. C., Yang P. C., Kurokawa J., Wu J. C., Clancy C. E. (2020). A computational model of induced pluripotent stem-cell derived cardiomyocytes for high throughput risk stratification of KCNQ1 genetic variants. PLoS Comput. Biol. 16 (8), e1008109. doi:10.1371/journal.pcbi.1008109
Kozek K. A., Glazer A. M., Ng C.-A., Blackwell D., Egly C. L., Vanags L. R., et al. (2020). High-throughput discovery of trafficking-deficient variants in the cardiac potassium channel KV11.1. Heart rhythm. 17 (12), 2180–2189. doi:10.1016/j.hrthm.2020.05.041
Kramer J., Himmel H. M., Lindqvist A., Stoelzle-Feix S., Chaudhary K. W., Li D., et al. (2020). Cross-site and cross-platform variability of automated patch clamp assessments of drug effects on human cardiac currents in recombinant cells. Sci. Rep. 10 (1), 5627. doi:10.1038/s41598-020-62344-w
Kriegeskorte S., Bott R., Hampl M., Korngreen A., Hausmann R., Lampert A. (2023). Cold and warmth intensify pain-linked sodium channel gating effects and persistent currents. J. Gen. Physiol. 155 (9), e202213312. doi:10.1085/jgp.202213312
Kroncke B. M., Smith D. K., Zuo Y., Glazer A. M., Roden D. M., Blume J. D. (2020). A Bayesian method to estimate variant-induced disease penetrance. PLOS Genet. 16 (6), e1008862. doi:10.1371/journal.pgen.1008862
Kuenze G., Vanoye C. G., Desai R. R., Adusumilli S., Brewer K. R., Woods H., et al. (2020). Allosteric mechanism for KCNE1 modulation of KCNQ1 potassium channel activation. eLife 9, e57680. doi:10.7554/eLife.57680
Kurokawa J., Chen L., Kass R. S. (2003). Requirement of subunit expression for cAMP-mediated regulation of a heart potassium channel. Proc. Natl. Acad. Sci. U. S. A. 100 (4), 2122–2127. doi:10.1073/pnas.0434935100
Kutchinsky J., Friis S., Asmild M., Taboryski R., Pedersen S., Vestergaard R. K., et al. (2003). Characterization of potassium channel modulators with QPatch automated patch-clamp technology: system characteristics and performance. Assay. Drug Dev. Technol. 1 (5), 685–693. doi:10.1089/154065803770381048
Lachaud Q., Aziz M. H. N., Burton F. L., Macquaide N., Myles R. C., Simitev R. D., et al. (2022). Electrophysiological heterogeneity in large populations of rabbit ventricular cardiomyocytes. Cardiovasc Res. 118 (15), 3112–3125. doi:10.1093/cvr/cvab375
Landrum M. J., Chitipiralla S., Brown G. R., Chen C., Gu B., Hart J., et al. (2019). ClinVar: improvements to accessing data. Nucleic Acids Res. 48 (D1), D835-D844–D844. doi:10.1093/nar/gkz972
Landrum M. J., Lee J. M., Benson M., Brown G. R., Chao C., Chitipiralla S., et al. (2018). ClinVar: improving access to variant interpretations and supporting evidence. Nucleic Acids Res. 46 (D1), D1062-D1067–d1067. doi:10.1093/nar/gkx1153
Lei C. L., Clerx M., Beattie K. A., Melgari D., Hancox J. C., Gavaghan D. J., et al. (2019). Rapid characterization of hERG channel kinetics II: temperature dependence. Biophys. J. 117 (12), 2455–2470. doi:10.1016/j.bpj.2019.07.030
Lek M., Karczewski K. J., Minikel E. V., Samocha K. E., Banks E., Fennell T., et al. (2016). Analysis of protein-coding genetic variation in 60,706 humans. Nature 536 (7616), 285–291. doi:10.1038/nature19057
Lepple-Wienhues A., Ferlinz K., Seeger A., Schäfer A. (2003). Flip the tip: an automated, high quality, cost-effective patch clamp screen. Recept. Channels 9 (1), 13–17. doi:10.3109/10606820308257
Li W., Luo X., Ulbricht Y., Wagner M., Piorkowski C., El-Armouche A., et al. (2019). Establishment of an automated patch-clamp platform for electrophysiological and pharmacological evaluation of hiPSC-CMs. Stem Cell Res. 41, 101662. doi:10.1016/j.scr.2019.101662
Li Z., Jin X., Wu T., Zhao X., Wang W., Lei J., et al. (2021). Structure of human Na(v)1.5 reveals the fast inactivation-related segments as a mutational hotspot for the long QT syndrome. Proc. Natl. Acad. Sci. U. S. A. 118 (11), e2100069118. doi:10.1073/pnas.2100069118
Lieve K. V., Williams L., Daly A., Richard G., Bale S., Macaya D., et al. (2013). Results of genetic testing in 855 consecutive unrelated patients referred for long QT syndrome in a clinical laboratory. Genet. Test. Mol. Biomarkers 17 (7), 553–561. doi:10.1089/gtmb.2012.0118
Liu C., Li T., Chen J. (2019). Role of high-throughput electrophysiology in drug discovery. Curr. Protoc. Pharmacol. 87 (1), e69. doi:10.1002/cpph.69
Liu M., Yang K. C., Dudley S. C. (2016). “Chapter sixteen - cardiac sodium channel mutations: why so many phenotypes?,” in Current topics in membranes. Editors R. J. French, and S. Y. Noskov (Cambridge: Academic Press), 513–559.
Ma G., O’Neill M., Richardson E., Thomson K., Ingles J., Roden D., et al. (2023). Utility of a high-throughput electrophysiology assay to determine pathogenicity of SCN5A variants associated with Brugada syndrome. Heart, Lung Circulation 32, S131–S132. doi:10.1016/j.hlc.2023.06.763
Ma J., Guo L., Fiene S. J., Anson B. D., Thomson J. A., Kamp T. J., et al. (2011). High purity human-induced pluripotent stem cell-derived cardiomyocytes: electrophysiological properties of action potentials and ionic currents. Am. J. Physiology-Heart Circulatory Physiology 301 (5), H2006–H2017. doi:10.1152/ajpheart.00694.2011
Makita N., Behr E., Shimizu W., Horie M., Sunami A., Crotti L., et al. (2008). The E1784K mutation in SCN5A is associated with mixed clinical phenotype of type 3 long QT syndrome. J. Clin. Invest. 118 (6), 2219–2229. doi:10.1172/jci34057
Marchal G. A., Remme C. A. (2022). Subcellular diversity of Nav1.5 in cardiomyocytes: distinct functions, mechanisms and targets. J. Physiol. 601, 941–960. doi:10.1113/jp283086
Marx S. O., Kurokawa J., Reiken S., Motoike H., D'Armiento J., Marks A. R., et al. (2002). Requirement of a macromolecular signaling complex for beta adrenergic receptor modulation of the KCNQ1-KCNE1 potassium channel. Science 295 (5554), 496–499. doi:10.1126/science.1066843
Mathes C. (2006). QPatch: the past, present and future of automated patch clamp. Expert Opin. Ther. Targets 10 (2), 319–327. doi:10.1517/14728222.10.2.319
Mazzanti A., Kanthan A., Monteforte N., Memmi M., Bloise R., Novelli V., et al. (2014). Novel insight into the natural history of short QT syndrome. J. Am. Coll. Cardiol. 63 (13), 1300–1308. doi:10.1016/j.jacc.2013.09.078
Meadows L. S., Chen Y. H., Powell A. J., Clare J. J., Ragsdale D. S. (2002). Functional modulation of human brain Nav1.3 sodium channels, expressed in mammalian cells, by auxiliary β1, β2 and β3 subunits. Neuroscience 114 (3), 745–753. doi:10.1016/S0306-4522(02)00242-7
Medeiros-Domingo A., Bhuiyan Z. A., Tester D. J., Hofman N., Bikker H., van Tintelen J. P., et al. (2009). The RYR2-encoded ryanodine receptor/calcium release channel in patients diagnosed previously with either catecholaminergic polymorphic ventricular tachycardia or genotype negative, exercise-induced long QT syndrome: a comprehensive open reading frame mutational analysis. J. Am. Coll. Cardiol. 54 (22), 2065–2074. doi:10.1016/j.jacc.2009.08.022
Melgari D., Calamaio S., Frosio A., Prevostini R., Anastasia L., Pappone C., et al. (2023). Automated patch-clamp and induced pluripotent stem cell-derived cardiomyocytes: a synergistic approach in the study of Brugada syndrome. Int. J. Mol. Sci. [Online] 24 (7), 6687. doi:10.3390/ijms24076687
Metzker M. L. (2010). Sequencing technologies — the next generation. Nat. Rev. Genet. 11 (1), 31–46. doi:10.1038/nrg2626
Miller D. T., Lee K., Chung W. K., Gordon A. S., Herman G. E., Klein T. E., et al. (2021a). ACMG SF v3.0 list for reporting of secondary findings in clinical exome and genome sequencing: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 23 (8), 1381–1390. doi:10.1038/s41436-021-01172-3
Miller D. T., Lee K., Gordon A. S., Amendola L. M., Adelman K., Bale S. J., et al. (2021b). Recommendations for reporting of secondary findings in clinical exome and genome sequencing, 2021 update: a policy statement of the American College of Medical Genetics and Genomics (ACMG). Genet. Med. 23 (8), 1391–1398. doi:10.1038/s41436-021-01171-4
Mizuguchi H., Xu Z., Ishii-Watabe A., Uchida E., Hayakawa T. (2000). IRES-dependent second gene expression is significantly lower than cap-dependent first gene expression in a bicistronic vector. Mol. Ther. 1 (4), 376–382. doi:10.1006/mthe.2000.0050
Montnach J., Lorenzini M., Lesage A., Simon I., Nicolas S., Moreau E., et al. (2021). Computer modeling of whole-cell voltage-clamp analyses to delineate guidelines for good practice of manual and automated patch-clamp. Sci. Rep. 11 (1), 3282. doi:10.1038/s41598-021-82077-8
Morais Cabral J. H., Lee A., Cohen S. L., Chait B. T., Li M., Mackinnon R. (1998). Crystal structure and functional analysis of the HERG potassium channel N terminus: a eukaryotic PAS domain. Cell 95 (5), 649–655. doi:10.1016/s0092-8674(00)81635-9
Moreau A., Gosselin-Badaroudine P., Boutjdir M., Chahine M. (2015). Mutations in the voltage sensors of domains I and II of Nav1.5 that are associated with arrhythmias and dilated cardiomyopathy generate gating pore currents. Front. Pharmacol. 6, 301. doi:10.3389/fphar.2015.00301
Moreau A., Krahn A., Gosselin-Badaroudine P., Klein G., Christé G., Vincent Y., et al. (2013). Sodium overload due to a persistent current that attenuates the arrhythmogenic potential of a novel LQT3 mutation. Front. Pharmacol. 4, 126. doi:10.3389/fphar.2013.00126
Musunuru K., Hershberger R. E., Day S. M., Klinedinst N. J., Landstrom A. P., Parikh V. N., et al. (2020). Genetic testing for inherited cardiovascular diseases: a scientific statement from the American heart association. Circulation Genomic Precis. Med. 13 (4), e000067. doi:10.1161/HCG.0000000000000067
Nakajima T., Kaneko Y., Saito A., Ota M., Iijima T., Kurabayashi M. (2015). Enhanced fast-inactivated state stability of cardiac sodium channels by a novel voltage sensor SCN5A mutation, R1632C, as a cause of atypical Brugada syndrome. Heart rhythm. 12 (11), 2296–2304. doi:10.1016/j.hrthm.2015.05.032
Nakano Y., Shimizu W. (2022). Brugada syndrome as a major cause of sudden cardiac death in asians. JACC Asia 2 (4), 412–421. doi:10.1016/j.jacasi.2022.03.011
Neher E., Sakmann B. (1976). Single-channel currents recorded from membrane of denervated frog muscle fibres. Nature 260 (5554), 799–802. doi:10.1038/260799a0
Ng C. A., Farr J., Young P., Windley M. J., Perry M. D., Hill A. P., et al. (2021). Heterozygous KCNH2 variant phenotyping using Flp-In HEK293 and high-throughput automated patch clamp electrophysiology. Biol. Methods Protoc. 6 (1), bpab003. doi:10.1093/biomethods/bpab003
Ng C.-A., Perry M. D., Liang W., Smith N. J., Foo B., Shrier A., et al. (2020). High-throughput phenotyping of heteromeric human ether-à-go-go-related gene potassium channel variants can discriminate pathogenic from rare benign variants. Heart rhythm. 17 (3), 492–500. doi:10.1016/j.hrthm.2019.09.020
Ng C. A., Phan K., Hill A. P., Vandenberg J. I., Perry M. D. (2014). Multiple interactions between cytoplasmic domains regulate slow deactivation of Kv11.1 channels. J. Biol. Chem. 289 (37), 25822–25832. doi:10.1074/jbc.M114.558379
Ng C.-A., Ullah R., Farr J., Hill A. P., Kozek K. A., Vanags L. R., et al. (2022). A massively parallel assay accurately discriminates between functionally normal and abnormal variants in a hotspot domain of KCNH2. Am. J. Hum. Genet. 109 (7), 1208–1216. doi:10.1016/j.ajhg.2022.05.003
Niehaus A., Azzariti D. R., Harrison S. M., DiStefano M. T., Hemphill S. E., Senol-Cosar O., et al. (2019). A survey assessing adoption of the ACMG-AMP guidelines for interpreting sequence variants and identification of areas for continued improvement. Genet. Med. 21 (8), 1699–1701. doi:10.1038/s41436-018-0432-7
Obergrussberger A., Brüggemann A., Goetze T. A., Rapedius M., Haarmann C., Rinke I., et al. (2016). Automated patch clamp meets high-throughput screening: 384 cells recorded in parallel on a planar patch clamp module. J. Lab. Autom. 21 (6), 779–793. doi:10.1177/2211068215623209
Olesen M. S., Yuan L., Liang B., Holst A. G., Nielsen N., Nielsen J. B., et al. (2012). High prevalence of long QT syndrome-associated SCN5A variants in patients with early-onset lone atrial fibrillation. Circ. Cardiovasc Genet. 5 (4), 450–459. doi:10.1161/CIRCGENETICS.111.962597
O’Neill M. J., Muhammad A., Li B., Wada Y., Hall L., Solus J. F., et al. (2022). Dominant negative effects of SCN5A missense variants. Genet. Med. 24, 1238–1248. doi:10.1016/j.gim.2022.02.010
Pablo J. L. B., Cornett S. L., Wang L. A., Jo S., Brünger T., Budnik N., et al. (2023). Scanning mutagenesis of the voltage-gated sodium channel NaV1.2 using base editing. Cell Rep. 42 (6), 112563. doi:10.1016/j.celrep.2023.112563
Pan J. Q., Baez-Nieto D., Allen A., Wang H.-R., Cottrell J. R. (2018). “Developing high-throughput assays to analyze and screen electrophysiological phenotypes,” in Phenotypic screening: methods and protocols. Editor B. Wagner (New York, NY: Springer New York), 235–252.
Perry M. D., Ng C. A., Phan K., David E., Steer K., Hunter M. J., et al. (2016). Rescue of protein expression defects may not be enough to abolish the pro-arrhythmic phenotype of long QT type 2 mutations. J. Physiol. 594 (14), 4031–4049. doi:10.1113/jp271805
Peters C. H., Abdelsayed M., Ruben P. C. (2016). Triggers for arrhythmogenesis in the Brugada and long QT 3 syndromes. Prog. Biophys. Mol. Biol. 120 (1-3), 77–88. doi:10.1016/j.pbiomolbio.2015.12.009
Peters S., Thompson B. A., Perrin M., James P., Zentner D., Kalman J. M., et al. (2021). Arrhythmic phenotypes are a defining feature of dilated cardiomyopathy-associated SCN5A variants: a systematic review. Circ. Genom Precis. Med. 0 (0), CIRCGEN121003432. doi:10.1161/CIRCGEN.121.003432
Potet F., Egecioglu D. E., Burridge P. W., George A. L. (2020). GS-967 and eleclazine block sodium channels in human induced pluripotent stem cell-derived cardiomyocytes. Mol. Pharmacol. 98 (5), 540–547. doi:10.1124/molpharm.120.000048
Priori S. G., Napolitano C., Gasparini M., Pappone C., Della Bella P., Giordano U., et al. (2002). Natural history of Brugada syndrome: insights for risk stratification and management. Circulation 105 (11), 1342–1347. doi:10.1161/hc1102.105288
Pusch M., Magrassi R., Wollnik B., Conti F. (1998). Activation and inactivation of homomeric KvLQT1 potassium channels. Biophys. J. 75 (2), 785–792. doi:10.1016/s0006-3495(98)77568-x
Rajamohan D., Kalra S., Duc Hoang M., George V., Staniforth A., Russell H., et al. (2016). Automated electrophysiological and pharmacological evaluation of human pluripotent stem cell-derived cardiomyocytes. Stem Cells Dev. 25 (6), 439–452. doi:10.1089/scd.2015.0253
Rapedius M., Obergrussberger A., Humphries E. S. A., Scholz S., Rinke-Weiss I., Goetze T. A., et al. (2022). There is no F in APC: using physiological fluoride-free solutions for high throughput automated patch clamp experiments. Front. Mol. Neurosci. 15, 982316. doi:10.3389/fnmol.2022.982316
Remme C. A. (2023). SCN5A channelopathy: arrhythmia, cardiomyopathy, epilepsy and beyond. Philosophical Trans. R. Soc. B Biol. Sci. 378 (1879), 20220164. doi:10.1098/rstb.2022.0164
Ren M., Randolph A. L., Alvarez-Baron C., Guo D., Tran P. N., Thiebaud N., et al. (2022). Experimental factors that impact CaV1.2 channel pharmacology-Effects of recording temperature, charge carrier, and quantification of drug effects on the step and ramp currents elicited by the "step-step-ramp" voltage protocol. PLoS One 17 (11), e0276995. doi:10.1371/journal.pone.0276995
Richards S., Aziz N., Bale S., Bick D., Das S., Gastier-Foster J., et al. (2015). Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular Pathology. Genet. Med. 17 (5), 405–424. doi:10.1038/gim.2015.30
Ríos-Pérez E. B., Liu F., Stevens-Sostre W. A., Eichel C. A., Silignavong J., Robertson G. A. (2021). A stable cell line inducibly expressing hERG1a/1b heteromeric channels. J. Pharmacol. Toxicol. Methods 110, 107081. doi:10.1016/j.vascn.2021.107081
Roden D. M. (2008). Clinical practice. Long-QT syndrome. N. Engl. J. Med. 358 (2), 169–176. doi:10.1056/NEJMcp0706513
Romero L., Trenor B., Yang P. C., Saiz J., Clancy C. E. (2015). In silico screening of the impact of hERG channel kinetic abnormalities on channel block and susceptibility to acquired long QT syndrome. J. Mol. Cell Cardiol. 87, 271–282. doi:10.1016/j.yjmcc.2015.08.015
Ruan Y., Liu N., Bloise R., Napolitano C., Priori S. G. (2007). Gating properties of SCN5A mutations and the response to mexiletine in long-QT syndrome type 3 patients. Circulation 116 (10), 1137–1144. doi:10.1161/CIRCULATIONAHA.107.707877
Rugiero F., Mistry M., Sage D., Black J. A., Waxman S. G., Crest M., et al. (2003). Selective expression of a persistent tetrodotoxin-resistant Na+ current and NaV1.9 subunit in myenteric sensory neurons. J. Neurosci. 23 (7), 2715–2725. doi:10.1523/JNEUROSCI.23-07-02715.2003
Sanguinetti M. C., Curran M. E., Zou A., Shen J., Spector P. S., Atkinson D. L., et al. (1996). Coassembly of K(V)LQT1 and minK (IsK) proteins to form cardiac I(Ks) potassium channel. Nature 384 (6604), 80–83. doi:10.1038/384080a0
Scheel O., Himmel H., Rascher-Eggstein G., Knott T. (2011). Introduction of a modular automated voltage-clamp platform and its correlation with manual human ether-à-go-go related gene voltage-clamp data. ASSAY Drug Dev. Technol. 9 (6), 600–607. doi:10.1089/adt.2010.0352
Schroeder K., Neagle B., Trezise D. J., Worley J. (2003). IonWorks™ ht: a new high-throughput electrophysiology measurement platform. J. Biomol. Screen. 8 (1), 50–64. doi:10.1177/1087057102239667
Schwartz P. J., Ackerman M. J., Antzelevitch C., Bezzina C. R., Borggrefe M., Cuneo B. F., et al. (2020). Inherited cardiac arrhythmias. Nat. Rev. Dis. Prim. 6 (1), 58. doi:10.1038/s41572-020-0188-7
Schwartz P. J., Stramba-Badiale M., Crotti L., Pedrazzini M., Besana A., Bosi G., et al. (2009). Prevalence of the congenital long-QT syndrome. Circulation 120 (18), 1761–1767. doi:10.1161/circulationaha.109.863209
Sendfeld F., Selga E., Scornik F. S., Pérez G. J., Mills N. L., Brugada R. (2019). Experimental models of Brugada syndrome. Int. J. Mol. Sci. 20 (9), 2123. doi:10.3390/ijms20092123
Splawski I., Timothy K. W., Sharpe L. M., Decher N., Kumar P., Bloise R., et al. (2004). Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119 (1), 19–31. doi:10.1016/j.cell.2004.09.011
Stett A., Burkhardt C., Weber U., van Stiphout P., Knott T. (2003). CYTOCENTERING: a novel technique enabling automated cell-by-cell patch clamping with the CYTOPATCH chip. Recept Channels 9 (1), 59–66. doi:10.3109/10606820308254
Stutzman M. J., Gao X., Kim M., Ye D., Zhou W., Tester D. J., et al. (2023). Functional characterization and identification of a therapeutic for a novel SCN5A-F1760C variant causing type 3 long QT syndrome refractory to all guideline-directed therapies. Heart rhythm. 20, 709–717. doi:10.1016/j.hrthm.2023.01.032
Tan B. H., Valdivia C. R., Rok B. A., Ye B., Ruwaldt K. M., Tester D. J., et al. (2005). Common human SCN5A polymorphisms have altered electrophysiology when expressed in Q1077 splice variants. Heart rhythm. 2 (7), 741–747. doi:10.1016/j.hrthm.2005.04.021
Tao H., Santa Ana D., Guia A., Huang M., Ligutti J., Walker G., et al. (2004). Automated tight seal electrophysiology for assessing the potential hERG liability of pharmaceutical compounds. ASSAY Drug Dev. Technol. 2 (5), 497–506. doi:10.1089/adt.2004.2.497
Templin C., Ghadri J. R., Rougier J. S., Baumer A., Kaplan V., Albesa M., et al. (2011). Identification of a novel loss-of-function calcium channel gene mutation in short QT syndrome (SQTS6). Eur. Heart J. 32 (9), 1077–1088. doi:10.1093/eurheartj/ehr076
Tester D. J., Ackerman M. J. (2007). Postmortem long QT syndrome genetic testing for sudden unexplained death in the young. J. Am. Coll. Cardiol. 49 (2), 240–246. doi:10.1016/j.jacc.2006.10.010
Tester D. J., Valdivia C., Harris-Kerr C., Alders M., Salisbury B. A., Wilde A. A. M., et al. (2010). Epidemiologic, molecular, and functional evidence suggest A572D-SCN5A should not be considered an independent LQT3-susceptibility mutation. Heart rhythm. 7 (7), 912–919. doi:10.1016/j.hrthm.2010.04.014
Tester D. J., Will M. L., Haglund C. M., Ackerman M. J. (2006). Effect of clinical phenotype on yield of long QT syndrome genetic testing. J. Am. Coll. Cardiol. 47 (4), 764–768. doi:10.1016/j.jacc.2005.09.056
The Nobel Prize in Physiology or Medicine (1963). The Nobel prize in Physiology or medicine 1963. NobelPrize.org: Nobel Prize Outreach AB 2023. Available: https://www.nobelprize.org/prizes/medicine/1963/summary/(Accessed Thu. August 17, 2023).
The Nobel Prize in Physiology or Medicine (1991). The Nobel prize in Physiology or medicine. NobelPrize.org. Available: https://www.nobelprize.org/prizes/medicine/1991/summary/.
Thomson K. L., Jiang C., Richardson E., Westphal D. S., Burkard T., Wolf C. M., et al. (2023). Clinical interpretation of KCNH2 variants using a robust PS3/BS3 functional patch clamp assay. medRxiv, 2023.2010.2008.23296707. doi:10.1101/2023.10.08.23296707
Trepakova E. S., Malik M. G., Imredy J. P., Penniman J. R., Dech S. J., Salata J. J. (2007). Application of PatchXpress planar patch clamp technology to the screening of new drug candidates for cardiac KCNQ1/KCNE1 (I ks) activity. ASSAY Drug Dev. Technol. 5 (5), 617–627. doi:10.1089/adt.2007.091
Ullah R., Ng C. A., Vanags L., Mitchell D., Vandenberg J., Kroncke B. (2022). Abstract 12815: high throughput variant identification in the S3-S5 helices of KCNH2 (hERG). Circulation 146 (Suppl. l_1), A12815. doi:10.1161/circ.146.suppl_1.12815
Vandenberg J. I., Perry M. D., Perrin M. J., Mann S. A., Ke Y., Hill A. P. (2012). hERG K(+) channels: structure, function, and clinical significance. Physiol. Rev. 92 (3), 1393–1478. doi:10.1152/physrev.00036.2011
Vandenberg J. I., Varghese A., Lu Y., Bursill J. A., Mahaut-Smith M. P., Huang C. L. (2006). Temperature dependence of human ether-a-go-go-related gene K+ currents. Am. J. Physiol. Cell Physiol. 291 (1), C165–C175. doi:10.1152/ajpcell.00596.2005
van Dijk E. L., Auger H., Jaszczyszyn Y., Thermes C. (2014). Ten years of next-generation sequencing technology. Trends Genet. 30 (9), 418–426. doi:10.1016/j.tig.2014.07.001
Vanoye C. G., Desai R. R., Fabre K. L., Gallagher S. L., Potet F., DeKeyser J.-M., et al. (2018). High-throughput functional evaluation of KCNQ1 decrypts variants of unknown significance. Circulation Genomic Precis. Med. 11 (11), e002345. doi:10.1161/CIRCGEN.118.002345
Vanoye C. G., Desai R. R., Ji Z., Adusumilli S., Jairam N., Ghabra N., et al. (2022). High-throughput evaluation of epilepsy-associated KCNQ2 variants reveals functional and pharmacological heterogeneity. JCI Insight 7 (5), e156314. doi:10.1172/jci.insight.156314
Veltmann C., Barajas-Martinez H., Wolpert C., Borggrefe M., Schimpf R., Pfeiffer R., et al. (2016). Further insights in the most common SCN5A mutation causing overlapping phenotype of long QT syndrome, Brugada syndrome, and conduction defect. J. Am. Heart Assoc. 5 (7), e003379. doi:10.1161/JAHA.116.003379
Vutthikraivit W., Rattanawong P., Putthapiban P., Sukhumthammarat W., Vathesatogkit P., Ngarmukos T., et al. (2018). Worldwide prevalence of Brugada syndrome: a systematic review and meta-analysis. Acta Cardiol. Sin. 34 (3), 267–277. doi:10.6515/ACS.201805_34(3).20180302B
Wang D. W., Desai R. R., Crotti L., Arnestad M., Insolia R., Pedrazzini M., et al. (2007). Cardiac sodium channel dysfunction in sudden infant death syndrome. Circulation 115 (3), 368–376. doi:10.1161/CIRCULATIONAHA.106.646513
Wang D. W., Makita N., Kitabatake A., Balser J. R., George A. L. (2000). Enhanced Na(+) channel intermediate inactivation in Brugada syndrome. Circ. Res. 87 (8), E37–E43. doi:10.1161/01.res.87.8.e37
Wang D. W., Yazawa K., George A. L., Bennett P. B. (1996a). Characterization of human cardiac Na+ channel mutations in the congenital long QT syndrome. Proc. Natl. Acad. Sci. U. S. A. 93 (23), 13200–13205. doi:10.1073/pnas.93.23.13200
Wang J., Trudeau M. C., Zappia A. M., Robertson G. A. (1998). Regulation of deactivation by an amino terminal domain in human ether-à-go-go-related gene potassium channels. J. Gen. Physiol. 112 (5), 637–647. doi:10.1085/jgp.112.5.637
Wang Q., Curran M. E., Splawski I., Burn T. C., Millholland J. M., VanRaay T. J., et al. (1996b). Positional cloning of a novel potassium channel gene: KVLQT1 mutations cause cardiac arrhythmias. Nat. Genet. 12 (1), 17–23. doi:10.1038/ng0196-17
Wang Q., Shen J., Splawski I., Atkinson D., Li Z., Robinson J. L., et al. (1995). SCN5A mutations associated with an inherited cardiac arrhythmia, long QT syndrome. Cell 80 (5), 805–811. doi:10.1016/0092-8674(95)90359-3
Ward R. J., Alvarez-Curto E., Milligan G. (2011). Using the Flp-In™ T-Rex™ system to regulate GPCR expression. Methods Mol. Biol. 746, 21–37. doi:10.1007/978-1-61779-126-0_2
Watanabe H., Darbar D., Kaiser D. W., Jiramongkolchai K., Chopra S., Donahue B. S., et al. (2009). Mutations in sodium channel β1-and β2-subunits associated with atrial fibrillation. Circ. Arrhythm. Electrophysiol. 2 (3), 268–275. doi:10.1161/circep.108.779181
Watanabe H., Yang T., Stroud D. M., Lowe J. S., Harris L., Atack T. C., et al. (2011). Striking in vivo phenotype of a disease-associated human SCN5A mutation producing minimal changes in vitro. Circulation 124 (9), 1001–1011. doi:10.1161/circulationaha.110.987248
Weerakoon P., Culurciello E., Klemic K. G., Sigworth F. J. (2009). An integrated patch-clamp potentiostat with electrode compensation. IEEE Trans. Biomed. Circuits Syst. 3 (2), 117–125. doi:10.1109/tbcas.2008.2005419
Wilde A. A. M., Amin A. S. (2018). Clinical spectrum of SCN5A mutations: long QT syndrome, Brugada syndrome, and cardiomyopathy. JACC Clin. Electrophysiol. 4 (5), 569–579. doi:10.1016/j.jacep.2018.03.006
Wilde A. A. M., Amin A. S., Postema P. G. (2022a). Diagnosis, management and therapeutic strategies for congenital long QT syndrome. Heart 108 (5), 332–338. doi:10.1136/heartjnl-2020-318259
Wilde A. A. M., Semsarian C., Márquez M. F., Sepehri Shamloo A., Ackerman M. J., Ashley E. A., et al. (2022b). European heart rhythm association (EHRA)/Heart rhythm society (HRS)/Asia pacific heart rhythm society (APHRS)/Latin American heart rhythm society (LAHRS) expert consensus statement on the state of genetic testing for cardiac diseases. EP Eur. 24 (8), 1307–1367. doi:10.1093/europace/euac030
Xu J., Guia A., Rothwarf D., Huang M., Sithiphong K., Ouang J., et al. (2003). A benchmark study with SealChip™ planar patch-clamp technology. ASSAY Drug Dev. Technol. 1 (5), 675–684. doi:10.1089/154065803770381039
Zeng H., Penniman J. R., Kinose F., Kim D., Trepakova E. S., Malik M. G., et al. (2008). Improved throughput of PatchXpress hERG assay using intracellular potassium fluoride. ASSAY Drug Dev. Technol. 6 (2), 235–241. doi:10.1089/adt.2007.116
Zhang Y.-L., Moran S. P., Allen A., Baez-Nieto D., Xu Q., Wang L. A., et al. (2022). Novel fluorescence-based high-throughput FLIPR assay utilizing membrane-tethered genetic calcium sensors to identify T-type calcium channel modulators. ACS Pharmacol. Transl. Sci. 5 (3), 156–168. doi:10.1021/acsptsci.1c00233
Zhao J. T., Hill A. P., Varghese A., Cooper A. A., Swan H., Laitinen-Forsblom P. J., et al. (2009). Not all hERG pore domain mutations have a severe phenotype: G584S has an inactivation gating defect with mild phenotype compared to G572S, which has a dominant negative trafficking defect and a severe phenotype. J. Cardiovasc Electrophysiol. 20 (8), 923–930. doi:10.1111/j.1540-8167.2009.01468.x
Keywords: cardiac electrophysiology, inheritable arrhythmia, automated patch clamp, SCN5A, KCNH2, KCNQ1, variant classification, functional genomics
Citation: Ma JG, Vandenberg JI and Ng C-A (2023) Development of automated patch clamp assays to overcome the burden of variants of uncertain significance in inheritable arrhythmia syndromes. Front. Physiol. 14:1294741. doi: 10.3389/fphys.2023.1294741
Received: 15 September 2023; Accepted: 13 November 2023;
Published: 27 November 2023.
Edited by:
Isabelle Baró, INSERM U1087 Institut du Thorax, FranceReviewed by:
Jerome Montnach, INSERM U1087 Institut du Thorax, FranceCopyright © 2023 Ma, Vandenberg and Ng. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Jamie I. Vandenberg, ai52YW5kZW5iZXJnQHZpY3RvcmNoYW5nLmVkdS5hdQ==
†These authors share senior authorship
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.