 Manoocher Soleimani
Manoocher Soleimani
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 08 November 2023
Sec. Renal Physiology and Pathophysiology
Volume 14 - 2023 | https://doi.org/10.3389/fphys.2023.1289388
This article is part of the Research Topic Insights in Renal Physiology and Pathophysiology: 2023 View all 10 articles
Tuberous Sclerosis Complex (TSC) is an autosomal dominant genetic disease caused by mutations in either TSC1 or TSC2 genes. Approximately, two million individuals suffer from this disorder worldwide. TSC1 and TSC2 code for the proteins harmartin and tuberin, respectively, which form a complex that regulates the mechanistic target of rapamycin complex 1 (mTORC1) and prevents uncontrollable cell growth. In the kidney, TSC presents with the enlargement of benign tumors (angiomyolipomas) and cysts whose presence eventually causes kidney failure. The factors promoting cyst formation and tumor growth in TSC are poorly understood. Recent studies on kidney cysts in various mouse models of TSC, including mice with principal cell- or pericyte-specific inactivation of TSC1 or TSC2, have identified a unique cystogenic mechanism. These studies demonstrate the development of numerous cortical cysts that are predominantly comprised of hyperproliferating A-intercalated (A-IC) cells that express both TSC1 and TSC2. An analogous cellular phenotype in cystic epithelium is observed in both humans with TSC and in TSC2+/− mice, confirming a similar kidney cystogenesis mechanism in TSC. This cellular phenotype profoundly contrasts with kidney cysts found in Autosomal Dominant Polycystic Kidney Disease (ADPKD), which do not show any notable evidence of A-IC cells participating in the cyst lining or expansion. RNA sequencing (RNA-Seq) and confirmatory expression studies demonstrate robust expression of Forkhead Box I1 (FOXI1) transcription factor and its downstream targets, including apical H+-ATPase and cytoplasmic carbonic anhydrase 2 (CAII), in the cyst epithelia of Tsc1 (or Tsc2) knockout (KO) mice, but not in Polycystic Kidney Disease (Pkd1) mutant mice. Deletion of FOXI1, which is vital to H+-ATPase expression and intercalated (IC) cell viability, completely inhibited mTORC1 activation and abrogated the cyst burden in the kidneys of Tsc1 KO mice. These results unequivocally demonstrate the critical role that FOXI1 and A-IC cells, along with H+-ATPase, play in TSC kidney cystogenesis. This review article will discuss the latest research into the causes of kidney cystogenesis in TSC with a focus on possible therapeutic options for this devastating disease.
TSC is caused by mutations in either TSC1 or TSC2 genes and affects multiple organs; including the kidney, heart, lung, and brain (Sampson and Harris, 1994; Crino et al., 2006; Henske et al., 2016; Rosset et al., 2017). TSC renal disease is characterized by the development and enlargement of cysts and benign tumors (angiomyolipomas) whose presence results in the decline of kidney function and eventually leads to end stage renal disease (Bissler et al., 2008; Henske et al., 2014; Bissler and Kingswood, 2018; Gallo-Bernal et al., 2022). The incidence of renal cell carcinoma (RCC) is increased in TSC, specifically affecting younger individuals afflicted with disease (Henske et al., 2016). Although the initiating event in TSC1 or TSC2 is well described, the factors that promote the kidney disease phenotype and progression are poorly understood.
Loss of function of TSC1 or TSC2 activates mTORC1, a ubiquitous protein kinase complex that integrates systemic signals, such as growth factors and cytokines, with local signals that sense the availability of nutrients (e.g., amino acids, glucose and oxygen), in order to regulate cell growth (Urbanska et al., 2013; Kim and Guan, 2019). The mTORC1 activation is the principal mechanism driving growth in benign tumors (angiomyolipomas) and cystogenesis in TSC kidneys (Bissler et al., 2008; Henske et al., 2014; Bissler and Kingswood, 2018; Gallo-Bernal et al., 2022) by initiating transcriptional, translational, and post-translational processes that promote a multitude of anabolic activities; including inhibiting cellular catabolic processes resulting in cell growth and proliferation (Sarbassov et al., 2005; Düvel et al., 2010; Howell et al., 2013).
The mTORC1 is under the control of the TSC1-TSC2 complex and the Ras-Homolog Enriched in Brain (RHEB). RHEB plays a vital role in regulation of growth and cell proliferation through the insulin/mTOR/S6 Kinase (S6K) signaling pathway (Sarbassov et al., 2005; Düvel et al., 2010; Howell et al., 2013; Urbanska et al., 2013; Kim and Guan, 2019). The TSC1-TSC2 complex inhibits mTORC1 by negatively regulating RHEB-GTPase (Fingar and Blenis, 2004; Kwiatkowski and Manning, 2005; Sarbassov et al., 2005; Düvel et al., 2010; Howell et al., 2013; Henske et al., 2014; Bissler and Kingswood, 2018). In the presence of inactivating mutations in TSC1 or TSC2, RHEB-GTPase is no longer under the inhibitory control of TSC1/TSC2, and in its GTP-bound form can activate mTORC1, which then phosphorylates the downstream elements, S6K and the eIF4E-binding proteins (4-EBP), leading to cell proliferation and growth (Fingar and Blenis, 2004; Kwiatkowski and Manning, 2005; Sancak et al., 2010; Ögmundsdóttir et al., 2012; Showkat et al., 2014).
Angiomyolipomas are benign kidney tumors consisting of fat, muscle, and blood vessels. They are usually accompanied by cysts and are subject to progressive growth and hemorrhage (Bissler et al., 2008; Henske et al., 2014; Bissler and Kingswood, 2018; Gallo-Bernal et al., 2022). Because of robust mTORC1 activation and its role in cell proliferation, mTORC1 inhibitors (e.g., sirolimus or everolimus) have been used in the treatment of kidney disease in TSC patients (Bissler et al., 2008; Henske et al., 2014). Unfortunately, a significant portion of individuals with TSC do not respond to this therapy. Further, kidney lesions can return to their baseline size when drugs are discontinued (Bissler et al., 2019a).
Kidney cysts are usually composed of cells that express intact TSC1 and TSC2 proteins in both mouse models, as well as in humans with TSC renal cystic disease (Onda et al., 1999; Bonsib et al., 2016). This occurrence is distinct from the angiomyolipomas which show a loss of TSC gene and function (Bonsib et al., 2016; Giannikou et al., 2016). These results may suggest that the factors and pathways that promote cyst growth and expansion are unique and different from those promoting the formation of angiomyolipomas in the kidneys of individuals with TSC.
While expressing phenotypically distinct cell types lining the cysts, both ADPKD and TSC kidney cysts display mTORC1 activation (Shillingford et al., 2006; Henske et al., 2014; Bissler and Kingswood, 2018). Yet, the role of mTORC1 activation in ADPKD cystogenesis remains conflicting. In humans with TSC, inhibition of mTORC1 or S6K profoundly blunts the overgrowth of cells, tumors, and cysts in the kidney, as long as patients remain on these inhibitors (Bissler et al., 2008; Henske et al., 2014; Bissler and Kingswood, 2018). In contrast, the inhibition of mTORC1 in humans with ADPKD did not exhibit a significant beneficial impact on kidney function and cyst volume (Serra et al., 2010). These results display contrasting effects of mTORC1 in cystogenesis in TSC vs. ADPKD. With the exception of mTORC1 inhibitors, there are no therapeutic (druggable) molecular targets to alleviate kidney cysts or tumors in TSC.
The first studies examining the identity of cells lining the cysts in TSC came from heterozygote Tsc2 (Tsc2+/−) mice, which showed a predominance of IC cells within the cyst epithelia (Onda et al., 1999). A more recent report on Tsc1 inactivation in mouse principal cells showed that kidney cysts exhibited a gradual loss of Aquaporin 2 (AQP-2)-positive cells in their epithelium (Chen et al., 2014). The disappearance of AQP-2-positive cells was attributed to the dedifferentiation of principal cells (Chen et al., 2014). However, there were no detailed characterizations of the non–AQP-2–expressing cells lining the cysts (Chen et al., 2014). Recently, we showed that in mice with either Tsc1 or Tsc2 deletion in kidney principal cells, the epithelia lining the cysts are predominantly comprised of cells exhibiting apical H+-ATPase and basolateral Cl−/HCO3− exchanger AE1 (SLC4A1) (Bissler et al., 2019b; Barone et al., 2021; Zahedi et al., 2022; Barone et al., 2023). These results convincingly identified the cells lining the kidney cysts in TSC as A-IC cells (Bissler et al., 2019b; Barone et al., 2021; Zahedi et al., 2022; Barone et al., 2023) and demonstrate that the loss of AQP-2 positive cells was due to the disappearance of principal cells and their replacement with A-intercalated cells in the cyst lining.
In addition to the apical H+-ATPase and basolateral AE1 expression, cells lining the cysts expressed the proliferation marker Proliferating Cell Nuclear Antigen (PCNA), along with the intact TSC locus (Bissler et al., 2019b; Barone et al., 2021). These results strongly support the view that the cyst epithelial cells are hyperproliferating A-IC cells that are genotypically normal and express both TSC1 and TSC2 (Bissler et al., 2019b; Barone et al., 2021). A similar cell phenotype was observed in kidney cysts in mice with pericyte-specific inactivation of TSC1 (Bissler et al., 2019b; Barone et al., 2021). Furthermore, an identical cell phenotype was observed in the cystic epithelium of humans with TSC, confirming a similar TSC kidney cystogenesis mechanism (Bissler et al., 2019b; Barone et al., 2021). The epithelial cells lining the cysts in individuals with ADPKD and in mutant Pkd1 mouse models do not show any notable evidence of IC cell presence or their participation in cyst expansion (Holthöfer et al., 1990; Shibazaki et al., 2008).
Figure 1 depicts double immunofluorescence labeling with H+-ATPase and AQP-2 in kidney cysts of mice with principal cell inactivation of Tsc1 (top panel), kidney-specific inactivation of Pkd1 (middle panel), and Tsc2 haploinsufficiency (Tsc2+/−). H+-ATPase shows abundant and widespread expression on the apical membrane of cells lining the cysts, with very few AQP-2 positive cells in Tsc1 KO mice (top panel). The H+-ATPase expression in Pkd1 mice is shown for comparison (middle panel) and indicates a completely different pattern, with few cells lining the cysts expressing H+-ATPase, whereas labeling with AQP-2 is very robust in cyst epithelia in Pkd1 mutant mice (middle panel). The expression of H+-ATPase was almost uniform in Tsc2+/− mice (bottom panel). Please see legend to Figure 1.
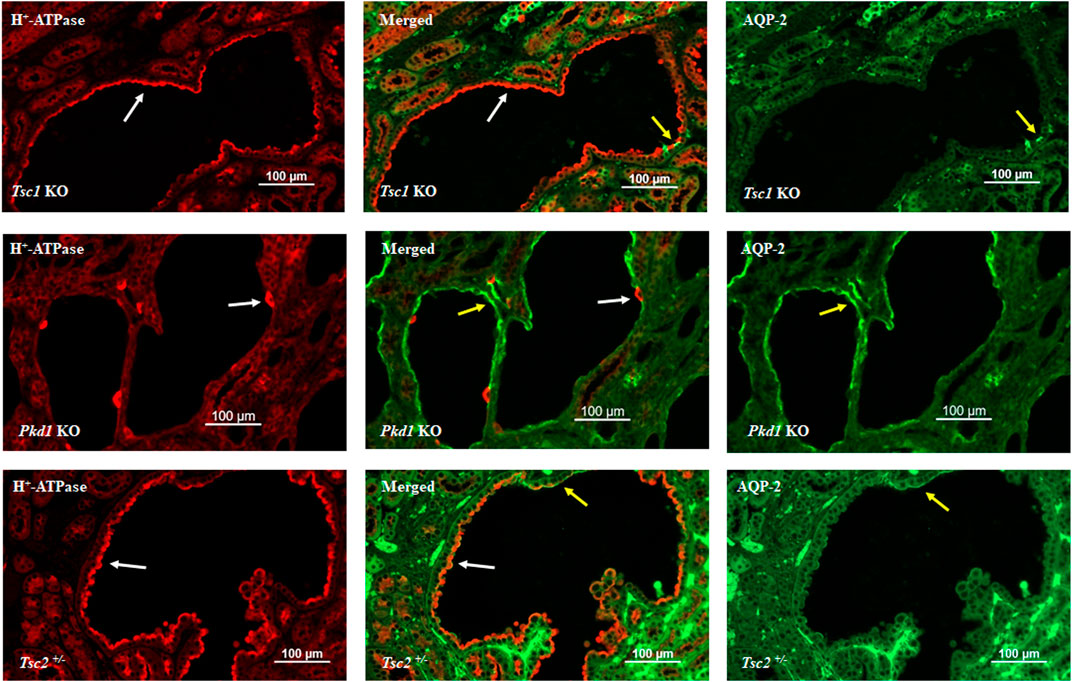
FIGURE 1. Double immunofluorescence labeling with H+-ATPase and AQP-2 in the kidney cysts of mice with principal cell-specific inactivation of Tsc1 (top panel), principal cell-specific inactivation of Pkd1 (middle panel), and Tsc2 haploinsufficiency (Tsc2+/−). As indicated, H+-ATPase shows abundant and widespread expression on the apical membrane of cells lining the cysts, with very few AQP-2 labeling in Tsc1 KO mice (top panel). The H+-ATPase expression in Pkd1 mice is shown for comparison (middle panel) and indicates a completely different labeling pattern. There are few H+-ATPase positive cells lining the cysts; whereas, AQP-2 labeling is very robust in Pkd1 cyst epithelia (middle panel). The expression of H+-ATPase was almost uniform in Tsc2+/− mice (bottom panel). Created with BioRender.com by Sharon Barone.
The presence of apical H+-ATPase in cells lining the cysts has brought new attention to the role of this molecule in TSC renal cystic disease (Bissler et al., 2019b; Barone et al., 2021; Zahedi et al., 2022; Barone et al., 2023). Vacuolar H+-ATPase (V H+-ATPase) is a large, multi-subunit H+ pump composed of V0 (membrane-spanning) and V1 (catalytic) complexes, and couples the energy of ATP hydrolysis to H+ translocation across plasma and intracellular membranes (Forgac, 2007; Hinton et al., 2009). Examination of the role of V H+-ATPase has revealed a crucial role for this pump in luminal acidification in several intracellular organelles, including late endosomes and lysosomes (Forgac, 2007; Hinton et al., 2009). Essential to mTORC1 activation is its recruitment to the lysosomal surface, which has brought additional attention to this organelle as a signaling hub since it incorporates many contributing signals required for cell proliferation and growth (Sancak et al., 2010; Ögmundsdóttir et al., 2012; Showkat et al., 2014).
Vacuolar H+-ATPase and mTORC1 have a reciprocal activating effect on each other in the lysosomal membrane. Recent studies have identified a network of signals/molecules that link mTORC1 to V H+-ATPase, a critical component of lysosomes. mTORC1 was able to enhance the expression of V H+-ATPases both in cells and mice (Peña-Llopis et al., 2011). The crosstalk between V H+-ATPase and mTORC1 on lysosome membranes is dependent on lysosomal biogenesis, which is regulated by Transcription factor EB (TFEB) (Peña-Llopis et al., 2011). Concurrently, increased H+-ATPase assembly and activity in the lysosomal membrane is necessary for amino acid-dependent mTORC1 recruitment (and activation) through interactions with the Rag GTPases, which are tethered to the lysosomal membrane by the Ragulator complex (Zoncu et al., 2011; Bar-Peled et al., 2013; Bar-Peled and Sabatini, 2014; Alesi et al., 2021). The inhibition or inactivation of H+-ATPase occurs through the decoupling of H+-ATPase from the Rag GTPases, leading to the inhibition of mTORC1 signaling (Forgac, 2007; Hinton et al., 2009; Nada et al., 2014). This inactivation leads to the neutralization of the luminal pH in lysosomes. It should be noted that the luminal lysosomal acidification consequent to the inward H+-ATPase-mediated H+ transport can simultaneously lead to the cytosolic alkalinization, which has further been implicated in the regulation of lysosomal perinuclear clustering (lysosomal topology) and mTORC1 (Chung et al., 2019). Taken together, these studies demonstrate the reciprocal stimulating effect of mTORC1 and H+-ATPase; mTORC1 enhances H+-ATPase expression and activity (Peña-Llopis et al., 2011; Alesi et al., 2021), while H+-ATPase plays a critical role in recruiting mTORC1 and sustaining its activation (Zoncu et al., 2011; Bar-Peled et al., 2013; Bar-Peled and Sabatini, 2014; Nada et al., 2014; Chung et al., 2019). Please see Figure 2 for more details.
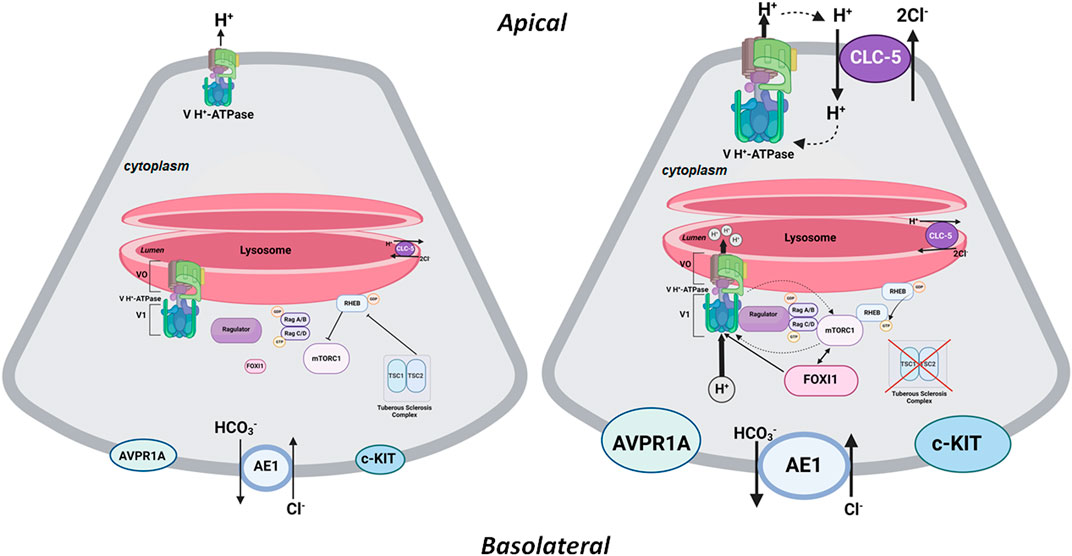
FIGURE 2. A schematic diagram depicting the interaction of mTORC1 and H+-ATPase on the lysosomal membrane. The left panel depicts a baseline state where the TSC1/TSC2 complex is intact and prevents RHEB from activating mTORC1. See additional description in the text under the heading, “The Central Role of FOXI1 in Kidney Cystogenesis in TSC.” The right panel depicts mTORC1 activation consequent to inactivating mutations or phosphorylation of TSC1 or TSC2. In its activated state, RHEB is released from the TSC1/TSC2 complex, resulting in the activation of mTORC1, which enhances the expression and assembly of V0 and V1 H+-ATPase subcomplexes (as depicted by bold arrows); therefore, resulting in increased H+-ATPase-mediate H+ transport. The feedback loop between mTORC1 and H+-ATPase also indicates the stimulatory effect of H+-ATPase on mTORC1 by enabling its recruitment and activation on the lysosomal membrane. In all mouse models of TSC examined thus far (Bissler et al., 2019b; Barone et al., 2021; Zahedi et al., 2022; Barone et al., 2023), mTORC1 is associated with enhanced expression of FOXI1 which activates H+-ATPase by enhancing the expression and activity of its subunits. Inhibition or inactivation of FOXI1 completely abrogates kidney cystogenesis and inhibits mTORC1 in TSC kidneys. The HCO3− exit via the basolateral Cl−/HCO3− exchange system (AE1; SLC4A1) is enhanced under an activated mTORC1 state. Created with BioRender.com by Sharon Barone.
The renal cyst epithelia in all mouse models of TSC display a robust mTORC1 activation as demonstrated by the presence of significantly elevated phospho-S6K levels (Bissler et al., 2019b; Barone et al., 2021). Active proliferation of A-IC cells in cyst epithelia was verified by a remarkable co-localization of PCNA and H+-ATPase in the same cells (Bissler et al., 2019b; Barone et al., 2021). These results indicate the activation of mTORC1 in A-IC cells lining the cysts and demonstrate that A-IC cells are the primary cells that are robustly proliferating in the epithelium of renal cysts.
V H+-ATPase is essential for the luminal acidification of intracellular organelles in all cells (Forgac, 2007; Hinton et al., 2009). This process includes acidifying the lumen of endosomes, lysosomes, and phagosomes; as well as several other intracellular organelles (Forgac, 2007; Hinton et al., 2009). In a few specialized cells, including kidney IC cells, pancreatic beta cells, and osteoclasts, V H+-ATPase plays two distinct roles. It pumps acid (H+) into the lumen of organelles, as well as across the plasma membrane into the extracellular compartment (Forgac, 2007; Hinton et al., 2009; Korolchuk et al., 2011; Soleimani and Rastegar, 2016; Do et al., 2022).
There is evidence supporting the recycling of V H+-ATPase between the plasma membrane and specialized intracellular vesicles (compartments) in kidney IC cells in response to either pH alteration or various stimuli, including several hormones, glucose, or signaling pathways (Carraro-Lacroix and Malnic, 2006; Brown et al., 2009; Alzamora et al., 2010; Gong et al., 2010; Ueno et al., 2014; Merkulova et al., 2015; Smith et al., 2016; Stransky et al., 2016; Eaton et al., 2021). One major pathway enhancing the trafficking of H+-ATPase from the specialized intracellular compartments to the plasma membrane is the activation of PKA by intracellular cAMP, which causes phosphorylation of several H+-ATPase subunits (Alzamora et al., 2010; Gong et al., 2010; Eaton et al., 2021). In A-IC cells, H+-ATPase plays an essential role in pumping H+ into the lumen of the collecting duct, thus regulating the systemic acid base balance (Soleimani and Rastegar, 2016; Bissler et al., 2019b; Barone et al., 2021; Do et al., 2022; Barone et al., 2023).
In addition to H+-ATPase, the electrogenic 2Cl−/H+ exchanger (CLC5) is also expressed on lysosomal membranes and works in tandem with H+-ATPase to maintain the luminal acidity of this crucial organelle in several nephron segments (Günther et al., 1998; Satoh et al., 2017). Both CLC5 and H+-ATPase are known to be expressed in A-IC cells (Chen et al., 2017; Park et al., 2018), and show co-localization on both the intracellular organelle membranes and the apical membrane of A-IC cells (Günther et al., 1998; Satoh et al., 2017). In kidneys of various mouse models of TSC (Tsc1 or Tsc2 KO in principal cells, Tsc1 KO in pericytes, as well in Tsc2+/−), V H+-ATPase and CLC5 show robust expression and remarkable co-localization on the apical membrane of A-IC cells lining the cysts (Barone et al., 2023).
The mode of transport of CLC5 is electrogenic and comprises the inward movement of 2Cl− in exchange for an outward movement of H+ from the lysosomal lumen (Günther et al., 1998; Satoh et al., 2017). The co-localization of V H+-ATPase and CLC5 in both the lysosomal membrane and the plasma membrane support the view that both molecules may traffic from the intracellular compartments to the plasma membrane in A-IC cells in TSC (Barone et al., 2023). It has been speculated that CLC5 co-localization with H+-ATPase on the apical membrane of A-IC cells could provide a mechanism for continued chloride secretion while maintaining the gradient for continuous H+-ATPase H+ secretion into the cyst lumen (Barone et al., 2023).
RNA sequencing (RNA-Seq) and confirmatory expression studies in our laboratories demonstrated robust expression of FOXI1 and its downstream targets, including H+-ATPase and cytoplasmic carbonic anhydrase 2 (CAII), in the cyst epithelia of Tsc1 (or Tsc2) knockout (KO) mice, but not in Pkd1 mutant mice (Barone et al., 2021; Barone et al., 2023). FOXI1 belongs to a large family of transcription factors that are important in cell-type specification during organogenesis (Benayoun et al., 2011). In the kidney, FOXI1 is exclusively expressed in IC cells and is necessary for expression of many H+-ATPase subunits in this nephron segment (Al-Awqati and Schwartz, 2004; Blomqvist et al., 2004; Vidarsson et al., 2009). In addition, FOXI1 is likely a major determinant for proper assembly of H+-ATPase subcomplexes and its activation at both the plasma membrane and the membranes of intracellular organelles.
Deletion of FOXI1, which is vital to H+-ATPase expression and IC cell development (Blomqvist et al., 2004), completely inhibited mTOR activation and abrogated the cyst burden in Tsc1 KO mice (Barone et al., 2021). These results unequivocally demonstrate the critical role that IC cells, along with their H+-ATPase and acid secreting machinery, play in TSC kidney cystogenesis. The collecting ducts of the FOXI1 null mice and the Tsc1/Foxi1 double KO (dKO) mice contained primitive cells that expressed both intercalated cell and principal cell transporters (Blomqvist et al., 2004; Barone et al., 2021). Deletion of FOXI1 resulted in profound suppression of several activated H+-ATPase subunits in the IC cells of Tsc1 KO mice (Barone et al., 2021).
Figure 2 is a schematic diagram that depicts the interaction of mTORC1 and H+-ATPase on the lysosomal membrane. The left panel depicts the state of mTORC1 in an inactivated state. As noted, the TSC1-TSC2 complex inhibits mTORC1 by negatively regulating RHEB-GTPase. Inactivating mutations in TSC1 or TSC2 remove the inhibitory effect of TSC complex on RHEB-GTPase leading to the activation of mTORC1, which then phosphorylates the downstream elements, S6K, and the eIF4E-binding proteins (4-EBP), enhancing cell proliferation and growth. The feedback loop between mTORC1 and H+-ATPase facilitates enhanced expression and assembly of H+-ATPase subcomplexes, and its activation by mTORC1. At the same time, the stimulatory effect of H+-ATPase on mTORC1 enables its recruitment and activation on lysosomal membrane. Unique to the kidney in TSC is the activation of FOXI1 transcription factor consequent to the activation of mTORC1 (Barone et al., 2021; Barone et al., 2023). FOXI1 is a master regulator of H+-ATPase subunits and its inactivation completely abrogated cystogenesis and mTORC1 activation in kidneys of TSC1 KO mice (Barone et al., 2021). As indicated the electrogenic Cl-transporter, CLC-5, is enhanced and translocated to the apical membrane of A-IC cells in addition to its original location on the lysosomal membrane. The CLC-5 maybe involved in movement of Cl- into the cyst lumen and expansion.
To discern the molecular basis of kidney cystogenesis in Tsc1 KO mice, we analyzed and contrasted the RNA transcriptomes in: 1) Tsc1 single KO mice which have numerous cysts and cyst adenomas; and compared it to 2) Tsc1/Foxi1 dKO mice that do not exhibit any kidney cysts. Our results identified the proto-oncogene c-KIT and the vasopressin receptor 1A (AVPR1A) as robustly enhanced transcripts in kidneys of Tsc1 KO mice (Zahedi et al., 2022; Zahedi et al., 2023). Both molecules exhibit enhanced expression at the mRNA and protein levels in kidney cysts in TSC1 KO mice. Both transcripts were completely suppressed in Tsc1/Foxi1 dKO mice (Zahedi et al., 2022; Zahedi et al., 2023). Both molecules are known to be expressed in A-IC cells under baseline conditions (Chen et al., 2017; Park et al., 2018).
Transfection of cultured m-IMCD cells with the full length FOX1 cDNA strongly induced the expression of c-KIT and AVPR1A (Zahedi et al., 2022; Zahedi et al., 2023). These results demonstrate that FOXI1 which is critical to the kidney cystogenesis in TSC (Barone et al., 2021), directly induces the expression of c-KIT and AVPR1A in cystic kidneys. Both genes, c-KIT (Hirota et al., 1998; Wiedmann and Caca, 2005; Larkin and Eisen, 2006; Bosbach et al., 2017; Lasota et al., 2019) and AVPR1A (Miller et al., 2013; Fenner, 2019; Zhao et al., 2019; Heidman et al., 2022), are known to play essential roles in enhancing cell proliferation in several uroepithelial cancers and other malignancies in part via mTORC1 activation. Both c-KIT and AVPR1A exhibit predominant expression in IC cells (Chen et al., 2017), with AVPR1A playing a crucial role in H+-ATPase stimulation in IC cells and acid secretion into the lumen of the collecting ducts (Yasuoka et al., 2013; Giesecke et al., 2019). This is in stark contrast with the cyst epithelia in ADPKD, which contain abundant principal cells with very few IC cells (Barone et al., 2021) and do not show any upregulation of c-KIT or AVPR1A (Zahedi et al., 2022; Zahedi et al., 2023).
c-KIT is a receptor tyrosine kinase that is expressed in several cell types and plays a critical role in enhancing cell proliferation and growth (Miettinen et al., 2005; de Toledo et al., 2023; Krimmer et al., 2023; Sternberg et al., 2023). Published reports indicate that increased activity of the kinase domain of c-KIT significantly contributes to the increased incidence of cancer consequent to uncontrolled cell proliferation. Such aberrant activity of c-KIT has been implicated in the pathogenesis of gastrointestinal stromal tumors (GISTs), mastocytosis, and hematological malignancies (Wiedmann and Caca, 2005; Bosbach et al., 2017; Lasota et al., 2019).
In the kidney, AVPR1A plays a key role secreting acid into the lumen of the collecting duct via stimulation of H+- secretion in A-IC cells (Yasuoka et al., 2013; Giesecke et al., 2019). Basolateral treatment of isolated perfused medullary collecting ducts with the AVPR1A agonist or vasopressin increased intracellular calcium levels in IC cells, enhanced apical abundance of H+-ATPase, and stimulated H+ secretion (Yasuoka et al., 2013; Giesecke et al., 2019). AVPR1A is essential in increasing cell proliferation in several tissues including prostate epithelial cells (Miller et al., 2013; Fenner, 2019; Zhao et al., 2019). The treatment of Castration-resistant prostate cancer (CRPC) cells with the AVPR1A ligand, arginine vasopressin (AVP), activated cAMP response element-binding protein (CREB) via the RAS-MAPK transduction cascade (Zhao et al., 2019). Inhibition by the selective AVPR1A antagonist, relcovaptan, decreased CRPC proliferation in mouse models (Miller et al., 2013). In addition, depletion of AVPR1A in CRPC significantly inhibited cell proliferation (Miller et al., 2013; Fenner, 2019).
The studies that are discussed in this review article demonstrate that in TSC renal cysts, A-IC cells that express both TSC1 and TSC2 constitute the cyst epithelium, and that FOXI1 plays a critical role in the process of cystogenesis. Currently, the only therapeutic option for TSC renal cystic disease is treatment with mTORC1 inhibitors, such as everolimus, which is fraught with many limitations, including resistance to treatment or the return of cysts and tumors to their original size upon discontinuation of therapy. Development of a thorough understanding of the process of renal cystogenesis in TSC should provide us with novel druggable targets for the treatment of this disorder. Delineating specific molecules and pathways that maybe critical to cyst expansion are part of the systemic approach aimed at understanding the mechanistic basis of TSC renal cystogenesis and developing novel and effective therapies for its treatment. The identification of c-KIT and AVPR1A as two differentially expressed genes with known effects on promotion of cell growth and mTORC1 activation in uroepithelial carcinomas and other malignancies is likely a first step toward the exciting discovery of new therapies for this devastating disease.
MS: Conceptualization, Visualization, Writing–original draft, Writing–review and editing.
The author(s) declare financial support was received for the research, authorship, and/or publication of this article. The author is supported by the United States Veterans Administration Department Merit Review Award (Grant Number: 2I01BX001000-10), Dialysis Clinic Inc. (Grant Number: DCI C-4149), and the National Institutes of Health (Grant Number: NIH/NHLBIT32HL007736; PI T. Resta). This research made use of the Fluorescence Microscopy and Cell Imaging Shared Resource, which is supported partially by the University of New Mexico (UNM) Comprehensive Cancer Center Support Grant NCIP30CA118100. The author is a Senior Clinician Scientist Investigator with the Department of Veterans Health Administration.
The critical review of this manuscript by Sharon Barone and Kamyar Zahedi is greatly appreciated. The Tsc2+/− mice were generous gifts from Dr. Jane Yu at the University of Cincinnati. Kidney sections from Pkd1 mutant mice were generous gifts from Dr. Stephen Somlo at Yale University.
The author declares that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
The author declared that they were an editorial board member of Frontiers, at the time of submission. This had no impact on the peer review process and the final decision.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Al-Awqati Q., Schwartz G. J. (2004). A fork in the road of cell differentiation in the kidney tubule. J. Clin. Invest. 113 (11), 1528–1530. doi:10.1172/JCI22029
Alesi N., Akl E. W., Khabibullin D., Liu H. J., Nidhiry A. S., Garner E. R., et al. (2021). TSC2 regulates lysosome biogenesis via a non-canonical RAGC and TFEB-dependent mechanism. Nat. Commun. 12 (1), 4245. doi:10.1038/s41467-021-24499-6
Alzamora R., Thali R. F., Gong F., Smolak C., Li H., Baty C. J., et al. (2010). PKA regulates vacuolar H+-ATPase localization and activity via direct phosphorylation of the a subunit in kidney cells. J. Biol. Chem. 285 (32), 24676–24685. doi:10.1074/jbc.M110.106278
Barone S., Brooks M., Zahedi K., Holliday L. S., Bissler J., Yu J. J., et al. (2023). Identification of an electrogenic 2Cl-/H+ exchanger, ClC5, as a chloride-secreting transporter candidate in kidney cyst epithelium in tuberous sclerosis. Am. J. Pathol. 193 (2), 191–200. doi:10.1016/j.ajpath.2022.10.007
Barone S., Zahedi K., Brooks M., Henske E. P., Yang Y., Zhang E., et al. (2021). Kidney intercalated cells and the transcription factor FOXi1 drive cystogenesis in tuberous sclerosis complex. Proc. Natl. Acad. Sci. U. S. A. 118 (6), e2020190118. doi:10.1073/pnas.2020190118
Bar-Peled L., Chantranupong L., Cherniack A. D., Chen W. W., Ottina K. A., Grabiner B. C., et al. (2013). A tumor suppressor complex with GAP activity for the Rag GTPases that signal amino acid sufficiency to mTORC1. Science 340 (6136), 1100–1106. doi:10.1126/science.1232044
Bar-Peled L., Sabatini D. M. (2014). Regulation of mTORC1 by amino acids. Trends Cell Biol. 24 (7), 400–406. doi:10.1016/j.tcb.2014.03.003
Benayoun B. A., Caburet S., Veitia R. A. (2011). Forkhead transcription factors: key players in health and disease. Trends Genet. 27 (6), 224–232. doi:10.1016/j.tig.2011.03.003
Bissler J. J., Budde K., Sauter M., Franz D. N., Zonnenberg B. A., Frost M. D., et al. (2019a). Effect of everolimus on renal function in patients with tuberous sclerosis complex: evidence from EXIST-1 and EXIST-2. Nephrol. Dial. Transpl. 34 (6), 1000–1008. doi:10.1093/ndt/gfy132
Bissler J. J., Kingswood J. C. (2018). Renal manifestation of tuberous sclerosis complex. Am. J. Med. Genet. C Semin. Med. Genet. 178 (3), 338–347. doi:10.1002/ajmg.c.31654
Bissler J. J., McCormack F. X., Young L. R., Elwing J. M., Chuck G., Leonard J. M., et al. (2008). Sirolimus for angiomyolipoma in tuberous sclerosis complex or lymphangioleiomyomatosis. N. Engl. J. Med. 358, 140–151. doi:10.1056/NEJMoa063564
Bissler J. J., Zadjali F., Bridges D., Astrinidis A., Barone S., Yao Y., et al. (2019b). Tuberous sclerosis complex exhibits a new renal cystogenic mechanism. Physiol. Rep. 7 (2), e13983. doi:10.14814/phy2.13983
Blomqvist S. R., Vidarsson H., Fitzgerald S., Johansson B. R., Ollerstam A., Brown R., et al. (2004). Distal renal tubular acidosis in mice that lack the forkhead transcription factor Foxi1. J. Clin. Invest. 113 (11), 1560–1570. doi:10.1172/JCI20665
Bonsib S. M., Boils C., Gokden N., Grignon D., Gu X., Higgins J. P., et al. (2016). Tuberous sclerosis complex: hamartin and tuberin expression in renal cysts and its discordant expression in renal neoplasms. Pathol. Res. Pract. 212 (11), 972–979. doi:10.1016/j.prp.2016.04.005
Bosbach B., Rossi F., Yozgat Y., Loo J., Zhang J. Q., Berrozpe G., et al. (2017). Direct engagement of the PI3K pathway by mutant KIT dominates oncogenic signaling in gastrointestinal stromal tumor. Proc. Natl. Acad. Sci. U. S. A. 114 (40), E8448–E8457. doi:10.1073/pnas.1711449114
Brown D., Paunescu T. G., Breton S., Marshansky V. (2009). Regulation of the V-ATPase in kidney epithelial cells: dual role in acid-base homeostasis and vesicle trafficking. J. Exp. Biol. 212 (11), 1762–1772. doi:10.1242/jeb.028803
Carraro-Lacroix L. R., Malnic G. (2006). Signaling pathways involved with the stimulatory effect of angiotensin II on vacuolar H+-ATPase in proximal tubule cells. Pflugers Arch. 452 (6), 728–736. doi:10.1007/s00424-006-0085-2
Chen L., Lee J. W., Chou C. L., Nair A. V., Battistone M. A., Păunescu T. G., et al. (2017). Transcriptomes of major renal collecting duct cell types in mouse identified by single-cell RNA-seq. Proc. Natl. Acad. Sci. U. S. A. 114 (46), E9989–E9998. doi:10.1073/pnas.1710964114
Chen Z., Dong H., Jia C., Song Q., Chen J., Zhang Y., et al. (2014). Activation of mTORC1 in collecting ducts causes hyperkalemia. J. Am. Soc. Nephrol. 25, 534–545. doi:10.1681/ASN.2013030225
Chung C. Y., Shin H. R., Berdan C. A., Ford B., Ward C. C., Olzmann J. A., et al. (2019). Covalent targeting of the vacuolar H+-ATPase activates autophagy via mTORC1 inhibition. Nat. Chem. Biol. 15 (8), 776–785. doi:10.1038/s41589-019-0308-4
Crino P. B., Nathanson K. L., Henske E. P. (2006). The tuberous sclerosis complex. N. Engl. J. Med. 355, 1345–1356. doi:10.1056/NEJMra055323
de Toledo M. A. S., Fu X., Maié T., Buhl E. M., Götz K., Schmitz S., et al. (2023). KIT D816V mast cells derived from induced pluripotent stem cells recapitulate systemic mastocytosis transcriptional profile. Int. J. Mol. Sci. 24 (6), 5275. doi:10.3390/ijms24065275
Do C., Vasquez P. C., Soleimani M. (2022). Metabolic alkalosis pathogenesis, diagnosis, and treatment: core curriculum 2022. Am. J. Kidney Dis. 80 (4), 536–551. doi:10.1053/j.ajkd.2021.12.016
Düvel K., Yecies J. L., Menon S., Raman P., Lipovsky A. I., Souza A. L., et al. (2010). Activation of a metabolic gene regulatory network downstream of mTOR complex 1. Mol. Cell 39 (2), 171–183. doi:10.1016/j.molcel.2010.06.022
Eaton A. F., Merkulova M., Brown D. (2021). The H+-ATPase (V-ATPase): from proton pump to signaling complex in health and disease. Am. J. Physiol. Cell Physiol. 320 (3), C392–C414. doi:10.1152/ajpcell.00442.2020
Fenner A. (2019). AVPR1A: a target in CRPC? Nat. Rev. Urol. 16 (9), 508. doi:10.1038/s41585-019-0218-y
Fingar D. C., Blenis J. (2004). Target of rapamycin (TOR): an integrator of nutrient and growth factor signals and coordinator of cell growth and cell cycle progression. Oncogene 23, 3151–3171. doi:10.1038/sj.onc.1207542
Forgac M. (2007). Vacuolar ATPases: rotary proton pumps in physiology and pathophysiology. Nat. Rev. Mol. Cell Biol. 8 (11), 917–929. doi:10.1038/nrm2272
Gallo-Bernal S., Kilcoyne A., Gee M. S., Paul E. (2022). Cystic kidney disease in tuberous sclerosis complex: current knowledge and unresolved questions. Pediatr. Nephrol. 38, 3253–3264. doi:10.1007/s00467-022-05820-x
Giannikou K., Malinowska I. A., Pugh T. J., Yan R., Tseng Y. Y., Oh C., et al. (2016). Whole exome sequencing identifies TSC1/TSC2 biallelic loss as the primary and sufficient driver event for renal angiomyolipoma development. PLoS Genet. 12 (8), e1006242. doi:10.1371/journal.pgen.1006242
Giesecke T., Himmerkus N., Leipziger J., Bleich M., Koshimizu T. A., Fähling M., et al. (2019). Vasopressin increases urinary acidification via V1a receptors in collecting duct intercalated cells. J. Am. Soc. Nephrol. 30 (6), 946–961. doi:10.1681/ASN.2018080816
Gong F., Alzamora R., Smolak C., Li H., Naveed S., Neumann D., et al. (2010). Vacuolar H+-ATPase apical accumulation in kidney intercalated cells is regulated by PKA and AMP-activated protein kinase. Am. J. Physiol. Ren. Physiol. 298 (5), F1162–F1169. doi:10.1152/ajprenal.00645.2009
Günther W., Lüchow A., Cluzeaud F., Vandewalle A., Jentsch T. J. (1998). ClC-5, the chloride channel mutated in Dent’s disease, colocalizes with the proton pump in endocytotically active kidney cells. Proc. Natl. Acad. Sci. U.S.A. 95 (14), 8075–8080. doi:10.1073/pnas.95.14.8075
Heidman L. M., Peinetti N., Copello V. A., Burnstein K. L. (2022). Exploiting dependence of castration-resistant prostate cancer on the arginine vasopressin signaling Axis by repurposing vaptans. Mol. Cancer Res. 20 (8), 1295–1304. doi:10.1158/1541-7786.MCR-21-0927
Henske E. P., Jóźwiak S., Kingswood J. C., Sampson J. R., Thiele E. A. (2016). Tuberous sclerosis complex. Nat. Rev. Dis. Prim. 2, 16035. doi:10.1038/nrdp.2016.35
Henske E. P., Rasooly R., Siroky B., Bissler J. (2014). Tuberous sclerosis complex, mTOR, and the kidney: report of an NIDDK-sponsored workshop. Am. J. Physiol. Ren. Physiol. 306, F279–F283. doi:10.1152/ajprenal.00525.2013
Hinton A., Bond S., Forgac M. (2009). V-ATPase functions in normal and disease processes. Pflugers Arch. 457 (3), 589–598. doi:10.1007/s00424-007-0382-4
Hirota S., Isozaki K., Moriyama Y., Hashimoto K., Nishida T., Ishiguro S., et al. (1998). Gain-of-function mutations of c-kit in human gastrointestinal stromal tumors. Science 279 (5350), 577–580. doi:10.1126/science.279.5350.577
Holthöfer H., Kumpulainen T., Rapola J. (1990). Polycystic disease of the kidney. Evaluation and classification based on nephron segment and cell-type specific markers. Lab. Invest. 62 (3), 363–369.
Howell J. J., Ricoult S. J., Ben-Sahra I., Manning B. D. (2013). A growing role for mTOR in promoting anabolic metabolism. Biochem. Soc. Trans. 41, 906–912. doi:10.1042/BST20130041
Kim J., Guan K. L. (2019). mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 21 (1), 63–71. doi:10.1038/s41556-018-0205-1
Korolchuk V. I., Saiki S., Lichtenberg M., Siddiqi F. H., Roberts E. A., Imarisio S., et al. (2011). Lysosomal positioning coordinates cellular nutrient responses. Nat. Cell Biol. 13 (4), 453–460. doi:10.1038/ncb2204
Krimmer S. G., Bertoletti N., Suzuki Y., Katic L., Mohanty J., Shu S., et al. (2023). Cryo-EM analyses of KIT and oncogenic mutants reveal structural oncogenic plasticity and a target for therapeutic intervention. Proc. Natl. Acad. Sci. U. S. A. 120 (13), e2300054120. doi:10.1073/pnas.2300054120
Kwiatkowski D. J., Manning B. D. (2005). Tuberous sclerosis: a GAP at the crossroads of multiple signaling pathways. Hum. Mol. Genet. 14, R251–R258. doi:10.1093/hmg/ddi260
Larkin J. M., Eisen T. (2006). Kinase inhibitors in the treatment of renal cell carcinoma. Crit. Rev. Oncol. Hematol. 60 (3), 216–226. doi:10.1016/j.critrevonc.2006.06.008
Lasota J., Kowalik A., Felisiak-Golabek A., Zięba S., Wang Z. F., Miettinen M. (2019). New mechanisms of mTOR pathway activation in KIT-mutant malignant GISTs. Appl. Immunohistochem. Mol. Morphol. 27 (1), 54–58. doi:10.1097/PAI.0000000000000541
Merkulova M., Păunescu T. G., Azroyan A., Marshansky V., Breton S., Brown D. (2015). Mapping the H(+) (V)-ATPase interactome: identification of proteins involved in trafficking, folding, assembly and phosphorylation. Sci. Rep. 5, 14827. doi:10.1038/srep14827
Miettinen M., Lasota J. (2005). KIT (CD117): a review on expression in normal and neoplastic tissues, and mutations and their clinicopathologic correlation. Appl. Immunohistochem. Mol. Morphol. 13 (3), 205–220. doi:10.1097/01.pai.0000173054.83414.22
Miller R. L., Sandoval P. C., Pisitkun T., Knepper M. A., Hoffert J. D. (2013). Vasopressin inhibits apoptosis in renal collecting duct cells. Am. J. Physiol. Ren. Physiol. 304 (2), F177–F188. doi:10.1152/ajprenal.00431.2012
Nada S., Mori S., Takahashi Y., Okada M. (2014). p18/LAMTOR1: a late endosome/lysosome-specific anchor protein for the mTORC1/MAPK signaling pathway. Methods Enzymol. 535, 249–263. doi:10.1016/B978-0-12-397925-4.00015-8
Ögmundsdóttir M. H., Heublein S., Kazi S., Reynolds B., Visvalingam S. M., Shaw M. K., et al. (2012). Proton-assisted amino acid transporter PAT1 complexes with Rag GTPases and activates TORC1 on late endosomal and lysosomal membranes. PloS One 7 (5), e36616. doi:10.1371/journal.pone.0036616
Onda H., Lueck A., Marks P. W., Warren H. B., Kwiatkowski D. J. (1999). Tsc2(+/-) mice develop tumors in multiple sites that express gelsolin and are influenced by genetic background. J. Clin. Invest. 104, 687–695. doi:10.1172/JCI7319
Park J., Shrestha R., Qiu C., Kondo A., Huang S., Werth M., et al. (2018). Single-cell transcriptomics of the mouse kidney reveals potential cellular targets of kidney disease. Science 360 (6390), 758–763. doi:10.1126/science.aar2131
Peña-Llopis S., Vega-Rubin-de-Celis S., Schwartz J. C., Wolff N. C., Tran T. A., Zou L., et al. (2011). Regulation of TFEB and V-ATPases by mTORC1. EMBO J. 30 (16), 3242–3258. doi:10.1038/emboj.2011.257
Rosset C., Netto C. B. O., Ashton-Prolla P. (2017). TSC1 and TSC2 gene mutations and their implications for treatment in Tuberous Sclerosis Complex: a review. Genet. Mol. Biol. 40, 69–79. doi:10.1590/1678-4685-GMB-2015-0321
Sampson J. R., Harris P. C. (1994). The molecular genetics of tuberous sclerosis. Hum. Mol. Genet. 3, 1477–1480. doi:10.1093/hmg/3.suppl_1.1477
Sancak Y., Bar-Peled L., Zoncu R., Markhard A. L., Nada S., Sabatini D. M. (2010). Ragulator-Rag complex targets mTORC1 to the lysosomal surface and is necessary for its activation by amino acids. Cell 141 (2), 290–303. doi:10.1016/j.cell.2010.02.024
Sarbassov D. D., Ali S. M., Sabatini D. M. (2005). Growing roles for the mTOR pathway. Curr Opin Cell Biol 17, 596–603. doi:10.1016/j.ceb.2005.09.009
Satoh N., Suzuki M., Nakamura M., Suzuki A., Horita S., Seki G., et al. (2017). Functional coupling of V-ATPase and CLC-5. World J. Nephrol. 6 (1), 14–20. doi:10.5527/wjn.v6.i1.14
Serra A. L., Poster D., Kistler A. D., Krauer F., Raina S., Young J., et al. (2010). Sirolimus and kidney growth in autosomal dominant polycystic kidney disease. N. Engl. J. Med. 363 (9), 820–829. doi:10.1056/NEJMoa0907419
Shibazaki S., Yu Z., Nishio S., Tian X., Thomson R. B., Mitobe M., et al. (2008). Cyst formation and activation of the extracellular regulated kinase pathway after kidney specific inactivation of Pkd1. Hum. Mol. Genet. 17 (11), 1505–1516. doi:10.1093/hmg/ddn039
Shillingford J. M., Murcia N. S., Larson C. H., Low S. H., Hedgepeth R., Brown N., et al. (2006). The mTOR pathway is regulated by polycystin-1, and its inhibition reverses renal cystogenesis in polycystic kidney disease. Proc. Natl. Acad. Sci. U. S. A. 103 (14), 5466–5471. doi:10.1073/pnas.0509694103
Showkat M., Beigh M. A., Andrabi K. I. (2014). mTOR signaling in protein translation regulation: implications in cancer genesis and therapeutic interventions. Mol. Biol. Int. 2014, 686984. doi:10.1155/2014/686984
Smith G. A., Howell G. J., Phillips C., Muench S. P., Ponnambalam S., Harrison M. A. (2016). Extracellular and luminal pH regulation by vacuolar H+-ATPase isoform expression and targeting to the plasma membrane and endosomes. J. Biol. Chem. 291 (16), 8500–8515. doi:10.1074/jbc.M116.723395
Soleimani M., Rastegar A. (2016). Pathophysiology of renal tubular acidosis: core curriculum 2016. Am. J. Kidney Dis. 68 (3), 488–498. doi:10.1053/j.ajkd.2016.03.422
Sternberg C. N., Davis I. D., Mardiak J., Szczylik C., Lee E., Wagstaff J., et al. (2023). Pazopanib in locally advanced or metastatic renal cell carcinoma: results of a randomized phase III trial. J. Clin. Oncol. 41 (11), 1957–1964. doi:10.1200/JCO.22.02622
Stransky L., Cotter K., Forgac M. (2016). The function of V-ATPases in cancer. Physiol. Rev. 96 (3), 1071–1091. doi:10.1152/physrev.00035.2015
Ueno K., Saito M., Nagashima M., Kojima A., Nishinoaki S., Toshima J. Y., et al. (2014). V-ATPase-dependent luminal acidification is required for endocytic recycling of a yeast cell wall stress sensor, Wsc1p. Biochem. Biophys. Res. Commun. 443 (2), 549–555. doi:10.1016/j.bbrc.2013.12.008
Urbanska M., Macias M., Skalecka A., Jaworski J. (2013). Beyond control of protein translation: what we have learned about the non-canonical regulation and function of mammalian target of rapamycin (mTOR). Malik Ar. Biochim. Biophys. Acta 1834 (7), 1434–1448. doi:10.1016/j.bbapap.2012.12.010
Vidarsson H., Westergren R., Heglind M., Blomqvist S. R., Breton S., Enerbäck S. (2009). The forkhead transcription factor Foxi1 is a master regulator of vacuolar H-ATPase proton pump subunits in the inner ear, kidney and epididymis. PLoS One 4 (2), e4471. doi:10.1371/journal.pone.0004471
Wiedmann M. W., Caca K. (2005). Molecularly targeted therapy for gastrointestinal cancer. Curr. Cancer Drug Targets 5 (3), 171–193. doi:10.2174/1568009053765771
Yasuoka Y., Kobayashi M., Sato Y., Zhou M., Abe H., Okamoto H., et al. (2013). The intercalated cells of the mouse kidney OMCD(is) are the target of the vasopressin V1a receptor axis for urinary acidification. Clin. Exp. Nephrol. 17 (6), 783–792. doi:10.1007/s10157-013-0783-y
Zahedi K., Barone S., Brooks M., Murray Stewart T., Casero R. A., Soleimani M. (2022). Renal transcriptome and metabolome in mice with principal cell-specific ablation of the Tsc1 gene: derangements in pathways associated with cell metabolism, growth and acid secretion. Int. J. Mol. Sci. 23 (18), 10601. doi:10.3390/ijms231810601
Zahedi K., Barone S., Zaidman N., Soleimani M. (2023). “Identification of transcripts critical to tsc-mTOR Axis dysregulation in tuberous sclerosis complex renal disease,” in American society of nephrology (ASN) kidney week (Berlin, Germany: Springer).
Zhao N., Peacock S. O., Lo C. H., Heidman L. M., Rice M. A., Fahrenholtz C. D., et al. (2019). Arginine vasopressin receptor 1a is a therapeutic target for castration-resistant prostate cancer. Sci. Transl. Med. 11 (498), eaaw4636. doi:10.1126/scitranslmed.aaw4636
Keywords: intercalated cells, kidney cysts, FOXI1, mTORC1, c-KIT, AVPR1A
Citation: Soleimani M (2023) Not all kidney cysts are created equal: a distinct renal cystogenic mechanism in tuberous sclerosis complex (TSC). Front. Physiol. 14:1289388. doi: 10.3389/fphys.2023.1289388
Received: 05 September 2023; Accepted: 18 October 2023;
Published: 08 November 2023.
Edited by:
Carolyn Mary Ecelbarger, Georgetown University, United StatesReviewed by:
Liming Chen, Huazhong University of Science and Technology, ChinaCopyright © 2023 Soleimani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Manoocher Soleimani, bXNvbGVpbWFuaUBzYWx1ZC51bm0uZWR1, bWFub29jaGVyLnNvbGVpbWFuaUB2YS5nb3Y=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.