 Kay-Pong Yip
Kay-Pong Yip Luisa Ribeiro-Silva1
Luisa Ribeiro-Silva1 Timo Rieg
Timo Rieg James S. K. Sham
James S. K. Sham- 1Department of Molecular Pharmacology and Physiology, Morsani College of Medicine, University of South Florida, Tampa, FL, United States
- 2Hypertension and Kidney Research Center, Morsani College of Medicine, University of South Florida, Tampa, FL, United States
- 3James A. Haley Veterans’ Hospital, Tampa, FL, United States
- 4Division of Pulmonary and Critical Care Medicine, Johns Hopkins University School of Medicine, Baltimore, MD, United States
Arginine vasopressin (AVP) induces an increase in intracellular Ca2+ concentration ([Ca2+]i) with an oscillatory pattern in isolated perfused kidney inner medullary collecting duct (IMCD). The AVP-induced Ca2+ mobilization in inner medullary collecting ducts is essential for apical exocytosis and is mediated by the exchange protein directly activated by cyclic adenosine monophosphate (Epac). Murine principal kidney cortical collecting duct cells (mpkCCD) is the cell model used for transcriptomic and phosphoproteomic studies of AVP signaling in kidney collecting duct. The present study examined the characteristics of Ca2+ mobilization in mpkCCD cells, and utilized mpkCCD as a model to investigate the Epac-induced intracellular and intra-organellar Ca2+ mobilization. Ca2+ mobilization in cytosol, endoplasmic reticulum lumen, and mitochondrial matrix were monitored with a Ca2+ sensitive fluorescent probe and site-specific Ca2+ sensitive biosensors. Fluorescence images of mpkCCD cells and isolated perfused inner medullary duct were collected with confocal microscopy. Cell permeant ligands of ryanodine receptors (RyRs) and inositol 1,4,5 trisphosphate receptors (IP3Rs) both triggered increase of [Ca2+]i and Ca2+ oscillations in mpkCCD cells as reported previously in IMCD. The cell permeant Epac-specific cAMP analog Me-cAMP/AM also caused a robust Ca2+ mobilization and oscillations in mpkCCD cells. Using biosensors to monitor endoplasmic reticulum (ER) luminal Ca2+ and mitochondrial matrix Ca2+, Me-cAMP/AM not only triggered Ca2+ release from ER into cytoplasm, but also shuttled Ca2+ from ER into mitochondria. The Epac-agonist induced synchronized Ca2+ spikes in cytosol and mitochondrial matrix, with concomitant declines in ER luminal Ca2+. Me-cAMP/AM also effectively triggered store-operated Ca2+ entry (SOCE), suggesting that Epac-agonist is capable of depleting ER Ca2+ stores. These Epac-induced intracellular and inter-organelle Ca2+ signals were mimicked by the RyR agonist 4-CMC, but they were distinctly different from IP3R activation. The present study hence demonstrated that mpkCCD cells retain all reported features of Ca2+ mobilization observed in isolated perfused IMCD. It further revealed information on the dynamics of Epac-induced RyR-dependent Ca2+ signaling and ER-mitochondrial Ca2+ transfer. ER-mitochondrial Ca2+ coupling may play a key role in the regulation of ATP and reactive oxygen species (ROS) production in the mitochondria along the nephron. Our data suggest that mpkCCD cells can serve as a renal cell model to address novel questions of how mitochondrial Ca2+ regulates cytosolic Ca2+ signals, inter-organellar Ca2+ signaling, and renal tubular functions.
Introduction
Physiological concentration of arginine vasopressin (AVP) induces intracellular Ca2+ mobilization in form of oscillation in isolated perfused rat inner medullary collecting duct (IMCD) (Yip, 2002). Confocal fluorescence microscopy revealed that each IMCD cells have their own unique oscillatory frequency and amplitude. Such Ca2+ mobilization is essential for the associated apical exocytosis, as intracellular Ca+2 chelators inhibit both AVP-induced Ca2+ mobilization and apical exocytosis in perfused IMCD. AVP exerts its actions via binding of the V2-receptors to stimulate adenylate cyclase and cAMP production in IMCD cells (Knepper and Inoue, 1997), the latter is mediated by adenylyl cyclase 6 (Rieg et al., 2010). It has traditionally been thought that cAMP activates the protein kinase A (PKA)-dependent signaling pathway to mediate AVP-regulated osmotic water permeability of IMCD. However, our previous study found that PKA inhibitors did not prevent AVP-induced Ca2+ mobilization and oscillation. Instead, the cAMP analog 8-pCPT-2′-O-Me-cAMP, which specifically activates exchange protein directly activated by cAMP (Epac) but not PKA, triggered intracellular Ca2+ mobilization and apical exocytosis of aquaporin-2 (AQP2) in perfused IMCD (Yip, 2006). Moreover, flash photolysis of caged cADP-ribose (an endogenous ligand of ryanodine receptors) activated Ca2+ oscillations resembling AVP-induced Ca2+ response (Yip and Sham, 2011). Previous studies showed that Ca2+ release from ryanodine receptors (RyRs) is essential in AVP-mediated AQP2 trafficking (Chou et al., 2000; Yip, 2002), and the process is independent of the phosphoinositol signaling pathway (Chou et al., 1998). AVP could also trigger Ca2+ influx via the store-operated Ca2+ entry (SOCE) mechanism. It was concluded that AVP-induced Ca2+ oscillation in IMCD is mediated by an Epac-dependent mechanism through the interplay of Ca2+ release from ryanodine receptors and a Ca2+ influx mechanism involving SOCE (Yip and Sham, 2011). Epac-induced Ca2+ release from RYRs-gated Ca2+ stores have been reported in other cell types. In cardiac myocytes, Epac-activation enhances RYR activity through protein kinase Cepsilon and Ca2+/calmodulain kinase II (CaMKII)-dependent phosphorylation of RYRs (Pereira et al., 2007; Oestreich et al., 2009), leading to SR Ca2+ leak and arrhythmia (Pereira et al., 2013; Li et al., 2017; Pereira et al., 2017). Epac-induced activation of RYRs also causes membrane hyperpolarization and relaxation of mesenteric arteries through Ca2+-sensitive K+ channel activation (Roberts et al., 2013).
Murine principal kidney cortical collecting duct (mpkCCD) cells are commonly used cell model used for transcriptomic and phosphoproteomic studies of AVP-signaling in kidney collecting ducts (Rinschen et al., 2010; Huling et al., 2012; Sandoval et al., 2013; Yang et al., 2022; Park et al., 2023). It is assumed that mpkCCD cell retains the feature of intact collecting duct cell in AVP-induced signaling events of AQP2 trafficking. We have demonstrated cAMP-dependent vectorial trafficking and exocytosis of AQP2 tagged with photoactivable fluorescent protein in mpkCCD cells at real time (Yip et al., 2015). It has also been shown in mpkCCD cells that Wnt5A, an endogenous ligand of the non-canonical branch of the Wnt pathway, is capable of inducing AQP2 apical expression and trafficking via basolateral Fzd receptors-mediated Ca2+ mobilization without activation of cAMP/PKA signal pathway (Ando et al., 2016). These observations highlight the potential of targeting Ca2+ pathways to ameliorate polyuria associated with nephrogenic diabetes insipidus (Mortensen et al., 2020). However, there is no information on the mechanisms underlying the dynamics of intracellular Ca2+ mobilization in mpkCCD cells. It is also unclear whether mpkCCD cells retain the specific properties of Ca2+ mobilization observed in perfused IMCD. In the present study, we sought to verify mpkCCD cells as a reliable model representing collecting duct cells for the study of the intracellular Ca2+ stores, the mechanisms of Ca2+ release, and extracellular Ca2+ influx. Moreover, special emphasis has been placed on the Epac-induced temporal relationship of Ca2+ dynamics in the cytosol, endoplasmic reticulum (ER) and mitochondria. Our results demonstrate that mpkCCD cells display similar characteristics of intracellular Ca2+ mobilization observed in intact cells of collecting duct, and that the Epac agonist triggered intracellular Ca2+ mobilization and oscillation are mediated by RyR-gated Ca2+ release and SOCE associated with reciprocal decrease of Ca2+ content in the ER. Moreover, the Epac agonist can effectively shuttle ER luminal Ca2+ to both the cytosol and mitochondrial matrix.
Materials and methods
Cell culture
Experiments were performed on a male mouse CCD principal cell line (mpkCCDC14, kindly provided by Dr. Douglas Eaton, Emory University) grown in AVP-free culture medium. Cells were maintained in a 1:1 mixture of DMEM/Ham’s F12 medium with phenol red (Gibco), supplemented with dexamethasone (50 nM), triiodothyronine (1 nM), selenium (60 nM), insulin (5 μg/mL), mouse EGF (10 ng/mL), transferrin (5 μg/mL), and 2% fetal calf serum in a humidified atmosphere with 5% CO2 at 37°C. mpkCCD cells between 20 and 30 passages were grown on collagen coated glass bottom dish prior to the experiments.
Monitoring of cytosolic Ca2+
mpkCCD cells grown on collagen coated glass bottom dish (MatTek) were loaded with cell permeant Ca2+ sensitive fluorescence probe (Cal-520/AM, 5 μM, AAT Bioquest) in phenol red free medium (1:1 mixture of DMEM/Ham’s F12 medium, Gibco) for 30 min at 37°C, followed by 20 min for de-esterification. Fluorescent images were collected with a Leica TCS SP5 confocal imaging system using water immersion objective lens (×63, N.A. 1.2) equipped with environmental chamber. Cal-520 was excited at 488 nm, and the emission was collected with a spectral window of 495–530 nm at 1 Hz. The spatial and temporal variations of [Ca2+]i in individual cells were measured from the stored images with Leica Application Suite Advanced Fluorescence software as reported previously (Yip and Sham, 2011). Store-operated calcium entry (SOCE) was induced by thapsigargin (10 µM) in calcium-free Hanks’ Balanced Salt Solution (Gibco) following by re-addition of 2 mM Ca2+ in the extracellular buffering solution.
Monitoring of calcium in ER and mitochondria with biosensors
To monitor ER [Ca2+] ([Ca2+]ER) or mitochondrial [Ca2+] ([Ca2+]MITO) simultaneously with cytosolic [Ca2+]i, mpkCCD cells were transfected with either ER Ca2+ biosensor R-CEPIA1er (Addgene Plasmid #58216, λex: 543 nm, λem: 560–600 nm) or mitochondrial Ca2+ biosensor mito-RCaMP1h (Addgene Plasmid #105013, λex: 543 nm, λem: 560–600 nm) (Suzuki et al., 2014). Cells were seeded at 6 × 104 cells/cm2 on collagen coated glass bottom dishes for 24 h before transfection. Cells were transfected with Lipofectamine (0.5 µg DNA/1 × 105 cells) for 24 h according to manufacturer’s instruction. Studies were performed in transfected cells from 48 to 72 h after transfection. Cytosolic [Ca2+]i was monitored simultaneously with cell permeant Ca2+ sensitive fluorescence probe (Cal-520/AM) in the transfected cells incubating with phenol red free medium (1:1 mixture of DMEM/Ham’s F12 medium, Gibco). To monitor ER-mitochondrial Ca2+ transfer in mpkCCD cells, cells were co-transfected with the ER Ca2+ biosensor (G-CEPIA1er, Addgene Plasmid #58215, λex: 488 nm, λem: 510–540 nm) and mitochondrial biosensor mito-RCaMP1h. Fluorescent images were collected with the respective laser lines for excitation and spectral windows for emission using the Lecia TCS SP5 imaging system.
Perfusion of rat inner medullary collection duct (IMCD)
All animal experimentation was conducted in accordance with the National Institutes of Health Guide for Care and Use of Laboratory Animals (National Institute of Health, Bethesda, MD) and was approved by the University of South Florida Institutional Animal Care and Use Committee (PROTOCOL #R3982). IMCDs were isolated from male Sprague-Dawley rats and perfused as described previously (Yip, 2002). Cytosolic [Ca2+]i in perfused IMCD was monitored with fluo-4/AM (5 μM, Invitrogen) in individual IMCD cells. Confocal fluorescent images of IMCD were collected and analyzed as reported previously (Yip, 2002).
Chemicals
6-Bnz-cAMP-AM, Me-cAMP-AM (8-pCPT-2′-O-Me-cAMP-AM), and ESI-09 were purchased from Biolog (Germany). Ryanodine, SKF-96365, and Xestospongin C were purchased from MilliporeSigma (Burlington, MA). Bt3-Ins(1,3,5)P3/AM was purchased from SiChem. ATP, 4-CMC (4-Chloro-m-cresol), H-89, and thapsigargin were from Sigma-Aldrich (St. Louis, MO).
Data analysis
Time series of fluorescence emission variations in individual mpkCCD cells were extracted and normalized with respective to the base line from stored XYT images. Time series of Cal-520 emission from individual cells were sampled at 1 Hz for spectral analysis. Each time series was subjected to linear trend removal. 512 or 1,024 data points were used to calculate the power spectrum with an algorithm based on Fast Fourier Transform (Yip et al., 1991). Results were reported as mean ± standard error. Statistical significance was calculated by using student’s t tests for paired or unpaired data and considered significant when p < 0.05.
Results
Intracellular Ca2+ mobilization in mpkCCD cells
RyRs are mainly expressed in the sarcoplasmic reticulum of skeletal, cardiac and smooth muscle cells, whereas inositol 1,4,5 trisphosphate receptors (IP3Rs) are the predominant Ca2+ release channels of the ER in non-excitable cells. We have previously shown that endogenous agonist of RyRs and IP3Rs triggered Ca2+ oscillations in individual cells of perfused IMCD, indicating both functional RyR-gated and IP3R-gated intracellular Ca2+ stores are present in IMCD cells (Yip and Sham, 2011). To test whether these Ca2+ stores are intact in mpkCCD cells, changes in [Ca2+]i were monitored with Ca2+ sensitive fluorescence probe when cells were stimulated with 4-CMC (a cell permeant RyR agonist) or Bt3-Ins(1,3,5)P3/AM (a cell permeant agonist of IP3R). Ryanodine was used as blocker of RyRs, and Xestospongin C was used for IP3Rs. Both agonists triggered robust intracellular Ca2+ mobilization and oscillations in mpkCCD cells (Figures 1A, B). The 4-CMC-induced Ca2+ response was almost instantaneous, compared to the delayed Bt3-Ins(1,3,5)P3/AM triggered Ca2+ response (∼100–150 s). The 4-CMC-induced Ca2+ transient was completed at 800 s, whereas the IP3-triggered response was more sustained. The delayed IP3–induced Ca2+ response in the Ca2+ responses were possibly related to the rate of membrane permeation and de-esterification of Bt3-Ins(1,3,5)P3/AM. The differences in the kinetic profiles triggered by the two stores could be due to differences in the potency of the agonists, inactivation kinetics of the receptors, and the depletion or replenishment of the SR Ca2+ stores. Both 4-CMC and Bt3-Ins(1,3,5)P3/AM-induced Ca2+ responses were completely abolished in the presence of their respective receptor blockers. These observations confirmed that mpkCCD cells possess both functional RyR- and IP3R-gated Ca2+ stores as in IMCD.
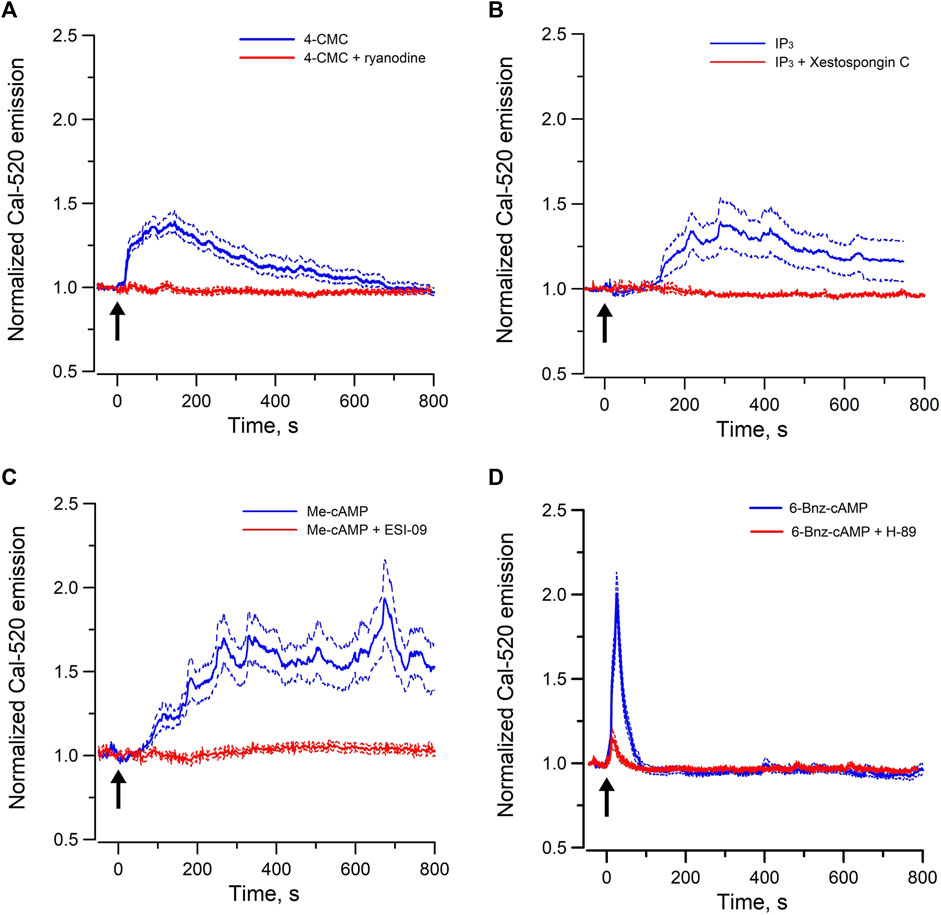
FIGURE 1. Mean normal time courses of Ca2+ mobilization in mpkCCD cells induced by (A) cell permeant ryanodine receptor agonist (50 µM 4-CMC, 54 cells/4 dishes), and (B) cell permeant IP3 receptor agonist (200 µM Bt3-Ins(1,3,5)P3/AM, 49 cells/4 dishes), (C) cell permeant Epac-agonist (40 µM Me-cAMP/AM, 92 cells/6 dishes), and (D) cell permeant PKA-agonist (40 µM 6-Bnz-cAMP/AM, 95 cells/6 dishes). Ryanodine (50 μM, 26 cells/3 dishes), Xestospongin C (10 μM, 35 cells/3 dishes), ESI-09 (25 μM, 36 cells/3 dishes), and H-89 (10 μM, 31 cells/3 dishes) were used as the corresponding receptor blockers or antagonists. Arrow (↑) indicates application of agonist in each time course. Dash lines are standard error.
To examine the Epac-dependent Ca2+ mobilization and oscillation (Yip, 2006), mpkCCD cells were stimulated with Me-cAMP/AM. Me-cAMP/AM is a cell permeant cAMP analog which activates specifically Epac but not PKA. Me-cAMP/AM triggered larger and sustained intracellular Ca2+ mobilization and oscillations in mpkCCD cells compared to those induced by 4-CMC and IP3 (Figure 1C). The Ca2+ response was blocked by ESI-09, an inhibitor of Epac1 and Epac2. These two Epac isoforms are expressed in IMCD and mpkCCD cells (Li et al., 2008; Kortenoeven et al., 2012). In contrast, 6-Bnz-cAMP/AM, a cell permeant cAMP analog which activates PKA but not Epac, triggered only a brief transient Ca2+ mobilization without Ca2+ oscillation (Figure 1D). The brief Ca2+ transient was effectively attenuated by the PKA inhibitor H-89. These observations suggested that activation of the Epac-dependent signal pathway elicits sustained Ca2+ mobilization and oscillation in mpkCCD cells. Spectral analysis was further applied to individual time series of Ca2+ signals of individual mpkCCD cells to characterize the oscillatory frequencies and the power of the Ca2+ oscillations. The mean power spectral density induced by Me-cAMP/AM, 4-CMC, and Bt3-Ins(1,3,5)P3/AM, were shown in Figure 2. All three mean power spectra had broad distribution over a range of frequencies. Most of the oscillations were confined in frequency range of 0.007–0.1 Hz, which are consistent with observations from intact IMCD cells of perfused IMCD (Yip, 2002; Yip and Sham, 2011). The power of Me-cAMP/AM, 4-CMC, and Ins(1,3,5)P3/AM-induced Ca2+ oscillations were similar, except Me-cAMP/AM induced Ca2+ oscillation had more power at the higher frequencies (0.03–0.1 Hz frequency range).
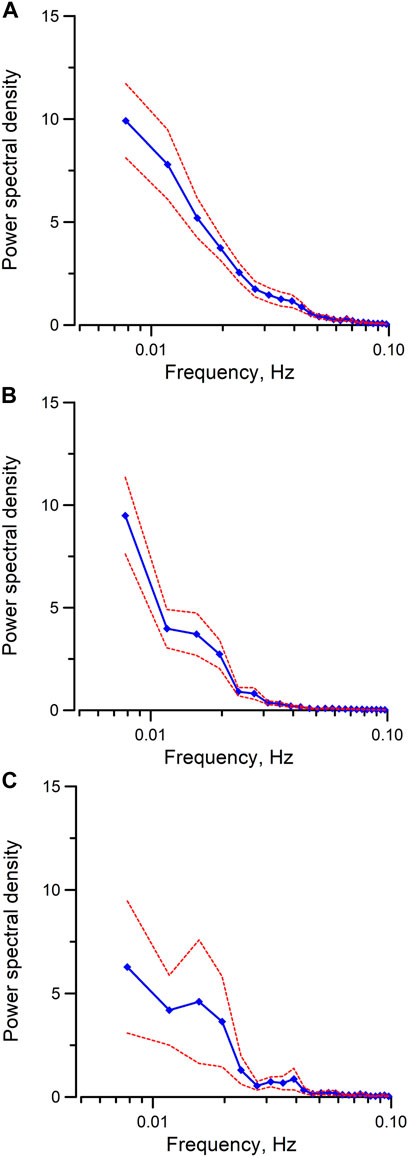
FIGURE 2. Mean power spectral spectra of cytosolic Ca2+ oscillations induced by (A) Me-cAMP/AM, (B) 4-CMC, (C) Bt3-Ins(1,3,5)P3/AM in mpkCCD cells. The same time series presented in Figure 1 were used for spectral analysis. By integrating the spectral power density from 0.03 Hz to 0.1 Hz in each individual power spectrum, the mean integrated spectral power density is significantly higher (p < 0.05) when cytosolic calcium oscillations were induced by Me-cAMP/AM than those induced by 4-CMC, or by Bt3-Ins(1,3,5)P3/AM. Dash lines are standard error.
Epac and RyR-agonist mobilize ER Ca2+ for cytosolic Ca2+ oscillation and ER-mitochondrial Ca2+ transfer in mpkCCD cells
To further characterize Epac-dependent activation of ER Ca2+ stores in mpkCCD cells, the temporal variations of cytosolic Ca2+ and ER luminal Ca2+ were monitored simultaneously with the Ca2+ sensitive-fluorescence probe Cal-520/AM and the ER luminal Ca2+ biosensor R-CEPIA1er, respectively. Me-cAMP/AM triggered an increase of cytosolic Ca2+ which was associated with a synchronized decrease in ER luminal Ca2+ content (Figure 3A). The oscillations in cytosolic Ca2+ were mirror images of those in ER luminal Ca2+. These observations suggested that the Epac-induced increase of cytosolic Ca2+ was due to release of Ca2+ from ER intracellular Ca2+ stores, and the cyclic variations in luminal ER Ca2+ content were likely due to ER Ca2+ depletion and refilling by Ca2+ uptake via the sacroplasmic/endoplasmic reticular Ca2+-ATPase (SERCA). RyR-agonist 4-CMC triggered similar response in mpkCCD cells (Figure 3D), indicating that Ca2+ release from RyR-gated stores generates cytosolic and ER Ca2+ signals comparable to those of Epac activation, congruent with reports that Epac triggered Ca2+ release via RyRs in IMCD and cardiomyocytes (Yip, 2006; Valli et al., 2018).
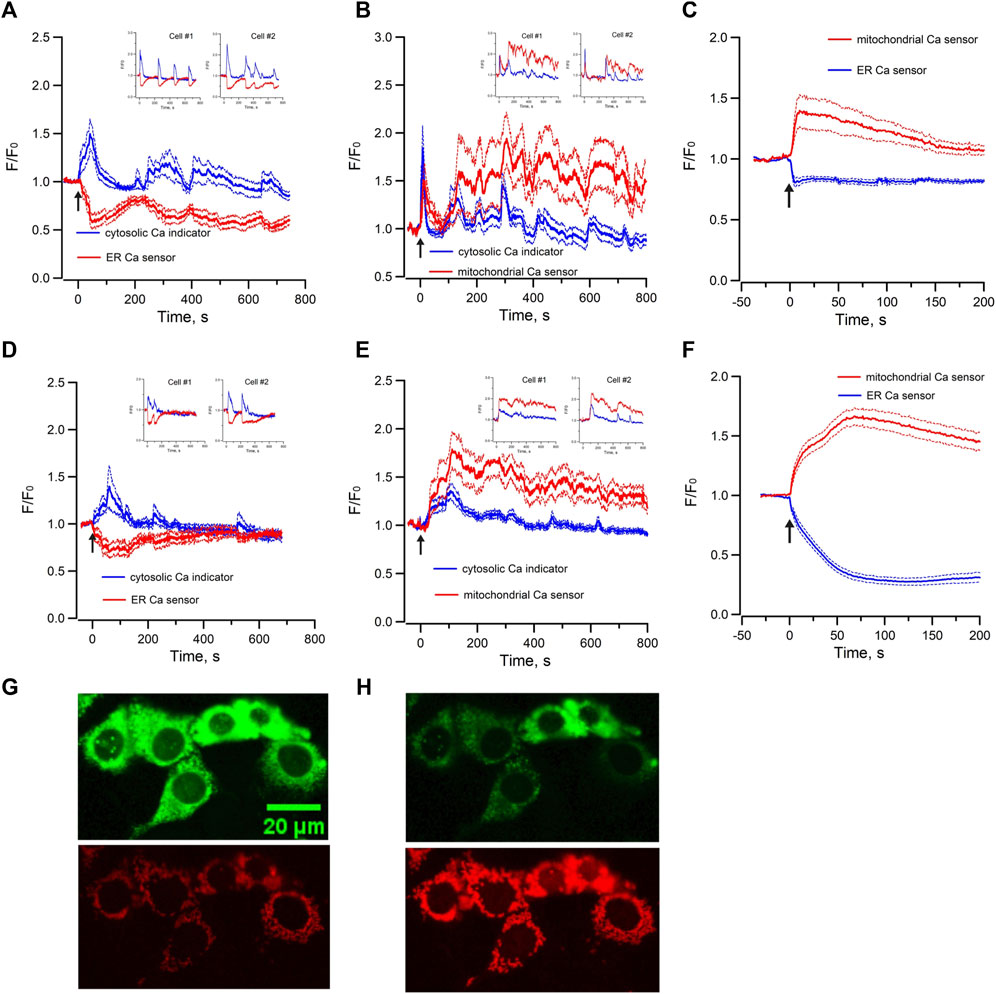
FIGURE 3. Mean normalized time courses of simultaneous changes in (A) cytosolic Ca2+ and ER luminal Ca2+ (15 cells/3 dishes), (B) cytosolic Ca2+ and mitochondrial matrix Ca2+ (20 cells/3 dishes) and (C) ER luminal Ca2+ and mitochondrial matrix Ca2+ (36 cells/7 dishes) induced by 40 µM Me-cAMP/AM in mpkCCD cells. Corresponding changes induced by 50 µM 4-CMC are shown in (D) cytosolic Ca2+ and ER luminal Ca2+ (13 cells/3 dishes), (E) cytosolic Ca2+ and mitochondrial matrix Ca2+ (21 cells/3 dishes), and (F) ER luminal Ca2+ and mitochondrial matrix Ca2+ 55 cells/9 dishes). mpkCCD cells expressing both ER biosensor (green) and mitochondrial biosensor (red) before (G) and after (H) exposure to RyR agonist 4-CMC. The Arrow (↑) indicates application of agonist in each time course. Inserts are tracings from individual cells with multiple Ca2+ spikes. F/F0 is the fractional change in fluorescence emission of the fluorescent probe or biosensor. Dash lines are standard error.
Mitochondrial Ca2+ concentration is important for the regulation of mitochondrial functions, and it is regulated by local Ca2+ concentration in the proximity of Ca2+ release channels of ER (Rizzuto et al., 2012; Csordas et al., 2018). To test whether there is mitochondrial matrix Ca2+ uptake during Epac-mediated Ca2+ mobilization in mpkCCD cells, the variations of cytosolic Ca2+ and mitochondrial matrix Ca2+ were monitored simultaneously with Ca2+ sensitive fluorescence probe and the mitochondrial matrix Ca2+ biosensor mito-RCaMP1h. Application of Me-cAMP/AM to mpkCCD cells activated multiple synchronized Ca2+ spikes in the cytosol and mitochondrial matrix (Figure 3B). These observations suggested that the Epac-agonist not only triggers release of ER Ca2+ to the cytosol, but also shuttles ER Ca2+ into the mitochondria. RyR-agonist 4-CMC triggered a similar response with synchronized Ca2+ spikes in the cytosol and mitochondrial matrix (Figure 3E). To determine the temporal relationship between ER luminal Ca2+ and mitochondrial matrix Ca2+, the ER luminal Ca2+ biosensor G-CEPIA1er and the mitochondrial matrix Ca2+ biosensor mito-RCaMP1h were co-expressed in mpkCCD cells. Me-cAMP/AM triggered a decrease in ER luminal Ca2+, which was associated with a concomitant increase in mitochondrial matrix Ca2+ (Figure 3C). RyR-agonist 4-CMC triggered a similar response (Figure 3F), indicative of effective RyR-coupled ER-mitochondrial Ca2+ transfer in mpkCCD cells. Figures 3G, H are fluorescence images of ER (green) and mitochondria (red) demonstrating Ca2+ transfer from ER to mitochondria induced by RyR agonist 4-CMC.
Moreover, the Me-cAMP/AM induced cytosolic and mitochondrial Ca2+ responses were completely blocked by ryanodine but were unaffected by xestospongin C (Figures 4A, B), indicating that the Epac-agonist mediated Ca2+ release was mainly derived from RyR-gated Ca2+ store to trigger mitochondrial Ca2+ transfer.
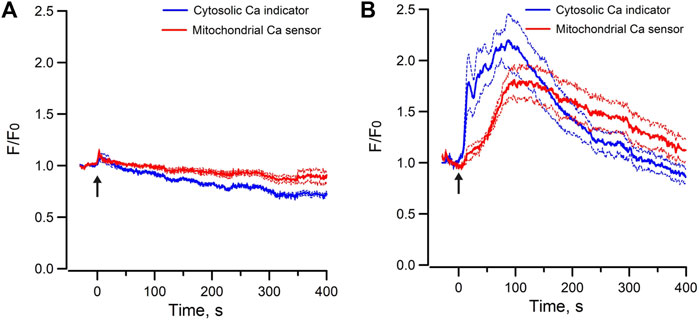
FIGURE 4. Mean normalized time courses of simultaneous changes in cytosolic Ca2+ and mitochondrial matrix Ca2+ induced by 40 µM Me-cAMP/AM in mpkCCD cells preincubated with (A) Ryanodine (50 μM, 19 cells/3 dishes), and (B) Xestospongin C (10 μM, 14 cells/3 dishes). Ryanodine but not Xestospongin C inhibits Epac-agonist induced cytosolic calcium mobilization and calcium uptake in mitochondria. Arrow (↑) indicates application of agonist in each time course. F/F0 is the fractional change in fluorescence emission of the fluorescent probe or biosensor. Dash lines are standard error.
ATP-mediated Ca2+ mobilization and Ca2+ transfer between ER and mitochondria
In contrast, exogenous application of Bt3-Ins(1,3,5)P3/AM only triggered decrease in ER luminal Ca2+ without concomitant increase of mitochondria Ca2+ in mpkCCD cells (Figure 5D). To test whether increasing the abundance of endogenous IP3 can induce Ca2+ transfer between ER and mitochondria, ATP was used to stimulate endogenous IP3 production via purinergic receptors expressed in mpkCCD cells (Wildman et al., 2009). Application of ATP elicited a transient increase in cytosolic [Ca2+] of less than 50 s in mpkCCD cells (Figure 5A). Of note, this was associated with a sustained decrease ER luminal Ca2+ (Figure 5A), and a more prolonged increase in mitochondrial [Ca2+] (Figure 5B). When ER luminal Ca2+ and mitochondrial matrix Ca2+ were monitored simultaneously, ATP triggered decrease in ER luminal Ca2+ and concomitant increase in mitochondrial matrix Ca2+ (Figure 5C). These observations suggest that endogenous activation of IP3Rs is capable of triggering ER-mitochondrial Ca2+ transfer in mpkCCD cells, even though Epac-induced Ca2+ response is independent of the IP3R-dependent mechanism. ATP-induced intracellular Ca2+ mobilization was also detected in perfused IMCD (Figure 5E), which is consistent with the observations on ATP mobilized intracellular Ca2+ in mpkCCD cells.
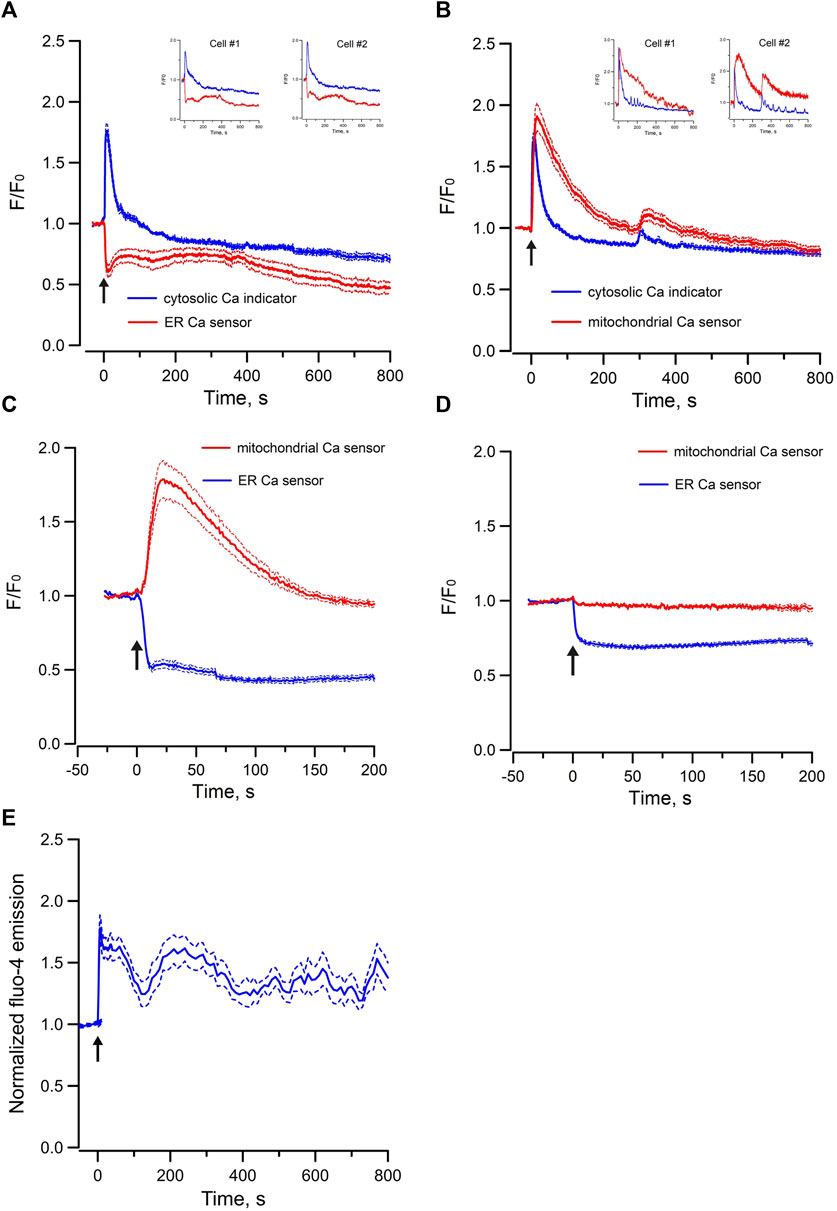
FIGURE 5. Mean normalized time courses of simultaneous changes in (A) cytosolic Ca2+ and ER luminal Ca2+ (15 cells/3 dishes), (B) cytosolic Ca2+ and mitochondrial matrix Ca2+ (9 cells/2 dishes), (C) ER luminal Ca2+ and mitochondrial matrix Ca2+ (32 cells/4 dishes) induced by 5 µM ATP, and (D) ER luminal Ca2+ and mitochondrial matrix Ca2+ induced by 200 µM Bt3-Ins(1,3,5)P3/AM in mpkCCD cells (45 cells/7 dishes). (E) mean normalized time course of cytosolic Ca2+ induced by 5 µM ATP in freshly isolated perfused rat IMCD (23 cells/3 tubules). The Arrow (↑) indicates application of agonist in each time course. Inserts are tracings from individual cells with multiple Ca2+ spikes. F/F0 is the fractional change in fluorescence emission of the fluorescent probe or biosensor. Dash lines are standard error.
Epac-mediated store-operated Ca2+ entry in mpkCCD cells
Since Epac-activation mobilizes ER Ca2+ to elicit cytosolic Ca2+ signals and ER-mitochondrial Ca2+ transfer, the reduction in ER luminal Ca2+ may activate SOCE to replenish ER Ca2+ stores. SOCE was first examined in mpkCCD by depleting ER Ca2+ stores using the SERCA inhibitor thapsigargin (10 µM) in the absence of extracellular Ca2+. Reintroduction of 2 mM Ca2+ in the external solution elicited robust Ca2+ entry, which was inhibited by the SOCE inhibitor SKF96365 (50 µM) (Figure 6A). Incubation of mpkCCD cells with the Epac-agonist Me-cAMP/AM in the absence of extracellular Ca2+ also activated SOCE similar to that induced by thapsigargin (Figure 6B). Robust Me-cAMP/AM induced Ca2+ entry was also observed in mpkCCD cells pretreated with xestospongin C; but the response was abolished in cells treated with both ryanodine and xestospongin C (Figure 6C). Moreover, Me-cAMP/AM-induced Ca2+ entry was observed in isolated perfused IMCD, and the effect was abolished by inhibition of SOCE using SKF96365 (Figure 6D). These results suggest that Epac activation is capable of inducing ER luminal Ca2+ depletion and SOCE in both mpkCCD and perfused IMCD.
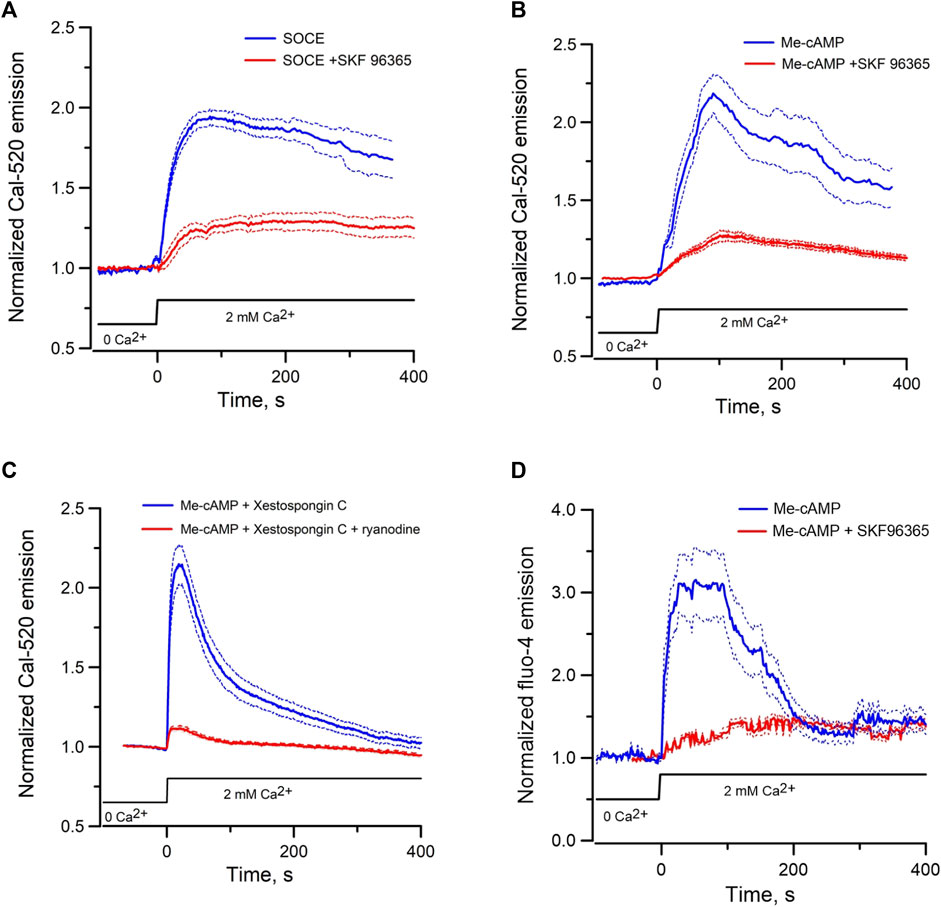
FIGURE 6. Mean normalized time courses of Ca2+ entry in mpkCCD cells and perfused IMCD triggered by re-addition of 2 mM of Ca2+. (A) Store-operated Ca2+ entry in the absence (69 cells/4 dishes) or presence (72 cells/3 dishes) of 50 µM SKF 96365 in mpkCCD cells, (B) Ca2+ entry induced by pre-incubation of mpkCCD cells with Epac-agonist (40 µM Me-cAMP/AM) in the absence (52 cells/3 dishes) or presence (59 cells/3 dishes) of SKF 96365, (C) Ca2+ entry induced by pre-incubation of mpkCCD cells with 40 µM Me-cAMP/AM in presence of 10 µM Xestospongin C (63 cells/3 dishes), or 10 µM Xestospongin +50 µM ryanodine (79 cell/3 dishes). (D) Ca2+ entry induced by pre-incubation of perfused IMCD with 40 µM Me-cAMP/AM in the absence (39 cells/4 tubules) or presence (27 cells/3 tubules) of SKF96369. Dash lines are standard error.
Discussion
Epac-dependent Ca2+ mobilization was associated with AVP-induced apical exocytosis in perfused IMCD. mpkCCD cells have been used as the cell model for transcriptomic and phosphoproteomic studies of AVP-signaling in the collecting duct (Rinschen et al., 2010; Huling et al., 2012; Sandoval et al., 2013; Yang et al., 2022; Park et al., 2023). We have demonstrated that mpkCCD cells retain the characteristics of Epac-dependent Ca2+ mobilization as in intact IMCD cells. Taking advantage of expressing ER and mitochondrial specific biosensor proteins in mpkCCD cells, the dynamic properties and relationship between cytosolic Ca2+, ER luminal Ca2+, and mitochondrial matrix Ca2+ were characterized. Epac-agonist mobilized Ca2+ from ER Ca2+ stores, depleted ER luminal Ca2+, and activated SOCE in mpkCCD cells. The oscillation of cytosolic Ca2+ triggered by Epac-agonist was entrained to Ca2+ oscillation in mitochondrial matrix, while 180° out-of-phase to the oscillation in ER luminal Ca2+. These observations indicated that Epac-mediated oscillatory Ca2+ signaling event is an integrated process which involves interplay of luminal ER Ca2+ release and refill, Ca2+ entry and efflux in mitochondrial matrix, and extracellular Ca2+ entry secondary to ER Ca2+ depletion in mpkCCD cells.
Time series of Ca2+ oscillation extracted from mpkCCD cells were analyzed in frequency domain using algorithm based on Fast Fourier Transform. Frequencies of Epac-dependent oscillations were detected in the range between 0.007 and 0.1 Hz, which is similar to the frequency ranges induced by cADP-ribose, an endogenous agonist of RyRs, observed in intact IMCD cells (Yip, 2002; Yip and Sham, 2011). Moreover, the sustained Me-cAMP-mediated Ca2+ oscillation in mpkCCD cells is similar to those observed in perfused IMCD triggered by caged cyclic-ADP-ribose (Yip and Sham, 2011). However, the 4-CMC-induced Ca2+ transient is more transient in the IMCD cells. The disparity in the kinetic profile of 4-CMC-triggered Ca2+ oscillations could be related to the differences in the agonist sensitivity, the activation/inactivation kinetics, sensitization of Ca2+-induced-Ca2+ release of the RyRs. Nevertheless, the complete inhibition of Epac-agonist-induced Ca2+ oscillation with ryanodine, but not by the IP3R-antagonist xestrospongin C, suggests that RyR is the primary Ca2+ source contributing to the Epac-induced Ca2+ oscillation in the mpkCCD cells.
It has been established that AVP regulates AQP2 shuttling through a cAMP-dependent pathway, and PKA has been considered as the only effector protein of cAMP for mediating AVP-regulated water permeability in kidney collecting ducts (Knepper and Inoue, 1997). It is now known that Epac is an important effector protein of cAMP. Epac1 and Epac2 isoforms are expressed in collecting duct and mpkCCD cells (Li et al., 2008; Kortenoeven et al., 2012). Our previous study found that Epac activation, but not PKA activation, mimics AVP in triggering Ca2+ mobilization and oscillations and induces apical shuttling of AQP2 in perfused IMCD (Yip, 2006). In the present study, PKA specific cAMP analog (6-Bnz-cAMP/AM) did not trigger Ca2+ oscillations in mpkCCD cells, while Epac-specific cAMP (Me-cAMP/AM) triggered long lasting Ca2+ oscillations. Moreover, long-term regulation of AQP2 by AVP in mpkCCD cells is mediated by Epac but not by PKA (Kortenoeven et al., 2012). Such evidence is consistent with an Epac-dependent signal pathway for regulation of collecting duct water permeability. It is also consistent that mice lacking Epac1 or Epac2 showed impaired urinary concentration ability and augmented urinary excretion of Na+ and urea (Cherezova et al., 2019). However, no defects in AVP-induced Ca2+ signaling in split-opened collecting ducts or changes in AQP2 protein abundance were observed. The urinary concentrating defect might be caused by reduced expression of the Na+/H+ exchanger isoform 3 (NHE3) (Cherezova et al., 2019). An inhibitory effect of Epac1 on NHE3 activity was previously shown in opossum kidney cells and mouse kidney slices (Honegger et al., 2006). A recent study reported compromised tight junctions in the collecting duct of Epac1 knockout mice in conjunction with a reduced papillary osmolarity (Sivertsen Asrud et al., 2020), suggesting Epac1 is involved in regulating paracellular permeability in the collecting duct. The reason for the discrepancies in the two knock-out mice studies is unclear but could be related to mouse dietary conditions or the existence of a different microbiome between different institutions.
Our data also show that Epac-agonist triggered Ca2+ oscillations resemble those activated by a RyR agonist in mpkCCD cells, suggesting that Epac-agonist mobilizes ER Ca2+ in mpkCCD cells via RyRs. Organelle-specific Ca2+-sensitive biosensors expressed in mpkCCD cells showed that the Epac agonist triggered synchronized Ca2+ spikes in cytosol and mitochondrial matrix, which are temporally correlated with reciprocal changes in ER luminal Ca2+. These observations indicated that agonist-induced Epac activation not only triggered release of Ca2+ into cytosol, but also transferred Ca2+ from ER to mitochondria.
The resting [Ca2+] of mitochondrial matrix is comparable to the resting cytosolic [Ca2+]. Mitochondrial Ca2+ uptake takes place at specialized microdomains where cytosolic [Ca2+] are high. Such microdomains are localized in the mitochondria-associated membranes (MAMs), where the endoplasmic reticulum membrane is within 10–30 nm from the outer mitochondrial membrane (Hajnoczky et al., 2002). Ca2+ released from ER enters the intermembrane space through voltage-dependent anion channels (VDACs) localized in the outer membrane, and then enters mitochondrial matrix through the mitochondrial Ca2+ uniporter (MCU) of the inner membrane (Patergnani et al., 2011; Giorgi et al., 2015). RyR agonist (4-CMC) triggered similar Ca2+ transfer from ER to mitochondria as Epac-agonist suggested that the Epac-induced ER-mitochondrial Ca2+ transfer is mediated by RyRs in MAMs of mpkCCD cells. IP3Rs and RyRs have been localized in MAMs (Hajnoczky et al., 2002; Chen et al., 2012; Bartok et al., 2019), but their distribution in mpkCCD cells or native renal collecting duct cells is unclear. The current study explored on this knowledge gap. Exogenous Bt3-Ins(1,3,5)P3/AM, which effectively activated cytosolic Ca2+ oscillation, only triggered a decrease in ER luminal Ca2+ but not concomitantly increased mitochondria Ca2+ in mpkCCD cells. It is possible that IP3Rs are less efficacious in facilitating ER-mitochondrial Ca2+ transfer in mpkCCD cells; or the cell permeant Bt3-Ins(1,3,5)P3/AM, had a poor access to the IP3Rs in MAMs. To test the latter hypothesis, the native agonist ATP was applied to increase endogenous IP3 abundance in mpkCCD cells. ATP triggered synchronized increase in cytosolic [Ca2+] with concomitant elevation in mitochondrial matrix [Ca2+] and depletion ER luminal Ca2+. ATP-induced Ca2+ transfer between ER and mitochondria was also visualized in cells co-expressed with ER and mitochondrial Ca2+ biosensors. These observations indicated that IP3Rs is capable of mediating Ca2+ transfer between ER and mitochondria in mpkCCD cells. However, the kinetics of ATP-induced cytosolic, endoplasmic, and mitochondrial Ca2+ responses are distinctly different from those induced by Epac or RyR agonists, distinguishing the Ca2+ signals activated by the two different agonists induced signaling pathways. Our studies also support that this mechanism is at play in vivo and it is noteworthy that 5 µM ATP also triggered Ca2+ mobilization and oscillations in isolated perfused IMCD (Figure 5E), confirming that purinergic receptor mediated Ca2+ mobilization is present in both intact IMCD cells and mpkCCD cells. Consistent with this, studies in acutely isolated connecting tubule/collecting duct of mice support that an acute increase in cytosolic [Ca2+] inhibits ENaC activity (Mamenko et al., 2011) mediated by P2Y2 receptor activation (Pochynyuk et al., 2008).
Mitochondrial Ca2+dynamics plays important roles in intracellular Ca2+signaling, cell metabolism, cell survival, and other cell-type specific functions (Rizzuto et al., 2012). As described above MCU supports cytoplasmic Ca2+ oscillations, SOCE and Ca2+-dependent gene expression in response to receptor-mediated stimulation (Samanta et al., 2014). It has been proposed that mitochondrial Ca2+ shuttling via MCU sustains the cytosolic Ca2+ signal by preventing Ca2+-dependent inactivation of IP3Rs and store-operated CRAC channels (Yoast et al., 2021). ER-mitochondrial Ca2+ transfer also stimulates Ca2+-sensitive dehydrogenases and respiratory chain components to promote oxidative phosphorylation, ATP, and ROS production (Territo et al., 2000; Territo et al., 2001; Hou et al., 2013). The physiological implications for the ER-mitochondrial Ca2+ transfer in the regulation of cellular functions in renal tubular cells remain to be determined.
In conclusion, mpkCCD cells retained all reported features of Epac-induced Ca2+ mobilization observed in isolated perfused IMCD. The temporal relationship between cytosolic Ca2+, ER luminal Ca2+, and mitochondrial matrix Ca2+ activated by Epac and RyR-agonists are highly compatible, but is distinctly different from those induced by IP3R stimulation. Furthermore, we have provided the first characterization of ER-mitochondrial Ca2+ transfer in mpkCCD cell, which can be used as a renal cell model to address novel questions of how mitochondrial Ca2+ regulates cytosolic Ca2+ signals, inter-organellar Ca2+ signaling, and other renal tubular functions.
Data availability statement
The raw data supporting the conclusions of this article will be made available by the authors, without undue reservation.
Ethics statement
The animal study was approved by IACUC University of South Florida. The study was conducted in accordance with the local legislation and institutional requirements.
Author contributions
K-PY, TR, and JS contributed to design and conception of the study. K-PY, LR-S, BC performed experiments. K-PY and JS interpreted results of experiments. K-PY wrote the first draft of the manuscript. K-PY, TR, BC, and JS wrote sections of the manuscript. All authors contributed to the article and approved the submitted version.
Funding
This work was supported by an American Heart Association Grant-In-Aid (K-PY). TR was supported the National Institute of Diabetes and Digestive and Kidney Diseases (NIDDK) Grant 1R01DK110621, Veterans Affairs Merit Review Award IBX004968A, and American Heart Association Transformational Research Award, 19TPA34850116. Financial support for this work was also provided by NIDDK Diabetic Complications Consortium (RRID SCR_001415, www.diacomp.org) Grants DK076169 and DK115255.
Conflict of interest
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Publisher’s note
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
References
Ando, F., Sohara, E., Morimoto, T., Yui, N., Nomura, N., Kikuchi, E., et al. (2016). Wnt5a induces renal AQP2 expression by activating calcineurin signalling pathway. Nat. Commun. 7, 13636. doi:10.1038/ncomms13636
Bartok, A., Weaver, D., Golenar, T., Nichtova, Z., Katona, M., Bansaghi, S., et al. (2019). IP3 receptor isoforms differently regulate ER-mitochondrial contacts and local calcium transfer. Nat. Commun. 10, 3726. doi:10.1038/s41467-019-11646-3
Chen, Y., Csordas, G., Jowdy, C., Schneider, T. G., Csordas, N., Wang, W., et al. (2012). Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca(2+) crosstalk. Circ. Res. 111, 863–875. doi:10.1161/CIRCRESAHA.112.266585
Cherezova, A., Tomilin, V., Buncha, V., Zaika, O., Ortiz, P. A., Mei, F., et al. (2019). Urinary concentrating defect in mice lacking Epac1 or Epac2. FASEB J. 33, 2156–2170. doi:10.1096/fj.201800435R
Chou, C. L., Rapko, S. I., and Knepper, M. A. (1998). Phosphoinositide signaling in rat inner medullary collecting duct. Am. J. Physiol. 274, F564–F572. doi:10.1152/ajprenal.1998.274.3.F564
Chou, C. L., Yip, K. P., Michea, L., Kador, K., Ferraris, J. D., Wade, J. B., et al. (2000). Regulation of aquaporin-2 trafficking by vasopressin in the renal collecting duct. Roles of ryanodine-sensitive Ca2+ stores and calmodulin. J. Biol. Chem. 275, 36839–36846. doi:10.1074/jbc.M005552200
Csordas, G., Weaver, D., and Hajnoczky, G. (2018). Endoplasmic reticulum-mitochondrial contactology: structure and signaling functions. Trends Cell Biol. 28, 523–540. doi:10.1016/j.tcb.2018.02.009
Giorgi, C., Missiroli, S., Patergnani, S., Duszynski, J., Wieckowski, M. R., and Pinton, P. (2015). Mitochondria-associated membranes: composition, molecular mechanisms, and physiopathological implications. Antioxid. Redox Signal 22, 995–1019. doi:10.1089/ars.2014.6223
Hajnoczky, G., Csordas, G., and Yi, M. (2002). Old players in a new role: mitochondria-associated membranes, VDAC, and ryanodine receptors as contributors to calcium signal propagation from endoplasmic reticulum to the mitochondria. Cell Calcium 32, 363–377. doi:10.1016/s0143416002001872
Honegger, K. J., Capuano, P., Winter, C., Bacic, D., Stange, G., Wagner, C. A., et al. (2006). Regulation of sodium-proton exchanger isoform 3 (NHE3) by PKA and exchange protein directly activated by cAMP (EPAC). Proc. Natl. Acad. Sci. U. S. A. 103, 803–808. doi:10.1073/pnas.0503562103
Hou, T., Zhang, X., Xu, J., Jian, C., Huang, Z., Ye, T., et al. (2013). Synergistic triggering of superoxide flashes by mitochondrial Ca2+ uniport and basal reactive oxygen species elevation. J. Biol. Chem. 288, 4602–4612. doi:10.1074/jbc.M112.398297
Huling, J. C., Pisitkun, T., Song, J. H., Yu, M. J., Hoffert, J. D., and Knepper, M. A. (2012). Gene expression databases for kidney epithelial cells. Am. J. Physiol. Ren. Physiol. 302, F401–F407. doi:10.1152/ajprenal.00457.2011
Knepper, M. A., and Inoue, T. (1997). Regulation of aquaporin-2 water channel trafficking by vasopressin. Curr. Opin. Cell Biol. 9, 560–564. doi:10.1016/s0955-0674(97)80034-8
Kortenoeven, M. L., Trimpert, C., Van Den Brand, M., Li, Y., Wetzels, J. F., and Deen, P. M. (2012). In mpkCCD cells, long-term regulation of aquaporin-2 by vasopressin occurs independent of protein kinase A and CREB but may involve Epac. Am. J. Physiol. Ren. Physiol. 302, F1395–F1401. doi:10.1152/ajprenal.00376.2011
Li, M., Hothi, S. S., Salvage, S. C., Jeevaratnam, K., Grace, A. A., and Huang, C. L. (2017). Arrhythmic effects of Epac-mediated ryanodine receptor activation in Langendorff-perfused murine hearts are associated with reduced conduction velocity. Clin. Exp. Pharmacol. Physiol. 44, 686–692. doi:10.1111/1440-1681.12751
Li, Y., Konings, I. B., Zhao, J., Price, L. S., De Heer, E., and Deen, P. M. (2008). Renal expression of exchange protein directly activated by cAMP (Epac) 1 and 2. Am. J. Physiol. Ren. Physiol. 295, F525–F533. doi:10.1152/ajprenal.00448.2007
Mamenko, M., Zaika, O., Jin, M., O'Neil, R. G., and Pochynyuk, O. (2011). Purinergic activation of Ca2+-permeable TRPV4 channels is essential for mechano-sensitivity in the aldosterone-sensitive distal nephron. PLoS One 6, e22824. doi:10.1371/journal.pone.0022824
Mortensen, L. A., Bistrup, C., Jensen, B. L., and Hinrichs, G. R. (2020). A mini-review of pharmacological strategies used to ameliorate polyuria associated with X-linked nephrogenic diabetes insipidus. Am. J. Physiol. Ren. Physiol. 319, F746–F753. doi:10.1152/ajprenal.00339.2020
Oestreich, E. A., Malik, S., Goonasekera, S. A., Blaxall, B. C., Kelley, G. G., Dirksen, R. T., et al. (2009). Epac and phospholipase Cepsilon regulate Ca2+ release in the heart by activation of protein kinase Cepsilon and calcium-calmodulin kinase II. J. Biol. Chem. 284, 1514–1522. doi:10.1074/jbc.M806994200
Park, E., Yang, C. R., Raghuram, V., Deshpande, V., Datta, A., Poll, B. G., et al. (2023). Data resource: vasopressin-regulated protein phosphorylation sites in the collecting duct. Am. J. Physiol. Ren. Physiol. 324, F43–F55. doi:10.1152/ajprenal.00229.2022
Patergnani, S., Suski, J. M., Agnoletto, C., Bononi, A., Bonora, M., De Marchi, E., et al. (2011). Calcium signaling around mitochondria associated membranes (MAMs). Cell Commun. Signal 9, 19. doi:10.1186/1478-811X-9-19
Pereira, L., Bare, D. J., Galice, S., Shannon, T. R., and Bers, D. M. (2017). β-Adrenergic induced SR Ca2+ leak is mediated by an Epac-NOS pathway. J. Mol. Cell Cardiol. 108, 8–16. doi:10.1016/j.yjmcc.2017.04.005
Pereira, L., Cheng, H., Lao, D. H., Na, L., Van Oort, R. J., Brown, J. H., et al. (2013). Epac2 mediates cardiac β1-adrenergic-dependent sarcoplasmic reticulum Ca2+ leak and arrhythmia. Circulation 127, 913–922. doi:10.1161/CIRCULATIONAHA.12.148619
Pereira, L., Metrich, M., Fernandez-Velasco, M., Lucas, A., Leroy, J., Perrier, R., et al. (2007). The cAMP binding protein Epac modulates Ca2+ sparks by a Ca2+/calmodulin kinase signalling pathway in rat cardiac myocytes. J. Physiol. 583, 685–694. doi:10.1113/jphysiol.2007.133066
Pochynyuk, O., Bugaj, V., Rieg, T., Insel, P. A., Mironova, E., Vallon, V., et al. (2008). Paracrine regulation of the epithelial Na+ channel in the mammalian collecting duct by purinergic P2Y2 receptor tone. J. Biol. Chem. 283, 36599–36607. doi:10.1074/jbc.M807129200
Rieg, T., Tang, T., Murray, F., Schroth, J., Insel, P. A., Fenton, R. A., et al. (2010). Adenylate cyclase 6 determines cAMP formation and aquaporin-2 phosphorylation and trafficking in inner medulla. J. Am. Soc. Nephrol. 21, 2059–2068. doi:10.1681/ASN.2010040409
Rinschen, M. M., Yu, M. J., Wang, G., Boja, E. S., Hoffert, J. D., Pisitkun, T., et al. (2010). Quantitative phosphoproteomic analysis reveals vasopressin V2-receptor-dependent signaling pathways in renal collecting duct cells. Proc. Natl. Acad. Sci. U. S. A. 107, 3882–3887. doi:10.1073/pnas.0910646107
Rizzuto, R., De Stefani, D., Raffaello, A., and Mammucari, C. (2012). Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 13, 566–578. doi:10.1038/nrm3412
Roberts, O. L., Kamishima, T., Barrett-Jolley, R., Quayle, J. M., and Dart, C. (2013). Exchange protein activated by cAMP (Epac) induces vascular relaxation by activating Ca2+-sensitive K+ channels in rat mesenteric artery. J. Physiol. 591, 5107–5123. doi:10.1113/jphysiol.2013.262006
Samanta, K., Douglas, S., and Parekh, A. B. (2014). Mitochondrial calcium uniporter MCU supports cytoplasmic Ca2+ oscillations, store-operated Ca2+ entry and Ca2+-dependent gene expression in response to receptor stimulation. PLoS One 9, e101188. doi:10.1371/journal.pone.0101188
Sandoval, P. C., Slentz, D. H., Pisitkun, T., Saeed, F., Hoffert, J. D., and Knepper, M. A. (2013). Proteome-wide measurement of protein half-lives and translation rates in vasopressin-sensitive collecting duct cells. J. Am. Soc. Nephrol. 24, 1793–1805. doi:10.1681/ASN.2013030279
Sivertsen Asrud, K., Bjornstad, R., Kopperud, R., Pedersen, L., Van Der Hoeven, B., Karlsen, T. V., et al. (2020). Epac1 null mice have nephrogenic diabetes insipidus with deficient corticopapillary osmotic gradient and weaker collecting duct tight junctions. Acta Physiol. (Oxf) 229, e13442. doi:10.1111/apha.13442
Suzuki, J., Kanemaru, K., Ishii, K., Ohkura, M., Okubo, Y., and Iino, M. (2014). Imaging intraorganellar Ca2+ at subcellular resolution using CEPIA. Nat. Commun. 5, 4153. doi:10.1038/ncomms5153
Territo, P. R., French, S. A., Dunleavy, M. C., Evans, F. J., and Balaban, R. S. (2001). Calcium activation of heart mitochondrial oxidative phosphorylation: rapid kinetics of mVO2, nadh, and light scattering. J. Biol. Chem. 276, 2586–2599. doi:10.1074/jbc.M002923200
Territo, P. R., Mootha, V. K., French, S. A., and Balaban, R. S. (2000). Ca(2+) activation of heart mitochondrial oxidative phosphorylation: role of the F(0)/F(1)-ATPase. Am. J. Physiol. Cell Physiol. 278, C423–C435. doi:10.1152/ajpcell.2000.278.2.C423
Valli, H., Ahmad, S., Sriharan, S., Dean, L. D., Grace, A. A., Jeevaratnam, K., et al. (2018). Epac-induced ryanodine receptor type 2 activation inhibits sodium currents in atrial and ventricular murine cardiomyocytes. Clin. Exp. Pharmacol. Physiol. 45, 278–292. doi:10.1111/1440-1681.12870
Wildman, S. S., Boone, M., Peppiatt-Wildman, C. M., Contreras-Sanz, A., King, B. F., Shirley, D. G., et al. (2009). Nucleotides downregulate aquaporin 2 via activation of apical P2 receptors. J. Am. Soc. Nephrol. 20, 1480–1490. doi:10.1681/ASN.2008070686
Yang, H. H., Su, S. H., Ho, C. H., Yeh, A. H., Lin, Y. J., and Yu, M. J. (2022). Glucocorticoid receptor maintains vasopressin responses in kidney collecting duct cells. Front. Physiol. 13, 816959. doi:10.3389/fphys.2022.816959
Yip, K. P., Cha, B. J., Tse, C. M., Amin, M. E., and Amin, J. (2015). Functional expression of aquaporin-2 tagged with photoconvertible fluorescent protein in mpkCCD cells. Cell Physiol. Biochem. 36, 670–682. doi:10.1159/000430129
Yip, K. P. (2002). Coupling of vasopressin-induced intracellular Ca(2+) mobilization and apical exocytosis in perfused rat kidney collecting duct. J. Physiol. 538, 891–899. doi:10.1113/jphysiol.2001.012606
Yip, K. P. (2006). Epac-mediated Ca(2+) mobilization and exocytosis in inner medullary collecting duct. Am. J. Physiol. Ren. Physiol. 291, F882–F890. doi:10.1152/ajprenal.00411.2005
Yip, K. P., Holstein-Rathlou, N. H., and Marsh, D. J. (1991). Chaos in blood flow control in genetic and renovascular hypertensive rats. Am. J. Physiol. 261, F400–F408. doi:10.1152/ajprenal.1991.261.3.F400
Yip, K. P., and Sham, J. S. (2011). Mechanisms of vasopressin-induced intracellular Ca2+ oscillations in rat inner medullary collecting duct. Am. J. Physiol. Ren. Physiol. 300, F540–F548. doi:10.1152/ajprenal.00544.2009
Keywords: intracellular calcium stores, Epac, mitochondria-associated membrane, aquaporin-2, ryanodine receptor
Citation: Yip K-P, Ribeiro-Silva L, Cha B, Rieg T and Sham JSK (2023) Epac induces ryanodine receptor-dependent intracellular and inter-organellar calcium mobilization in mpkCCD cells. Front. Physiol. 14:1250273. doi: 10.3389/fphys.2023.1250273
Received: 29 June 2023; Accepted: 11 August 2023;
Published: 29 August 2023.
Edited by:
Carolyn Mary Ecelbarger, Georgetown University, United StatesReviewed by:
Victor Babich, Mercy College of Health Sciences, United StatesDavid Giovannucci, University of Toledo, United States
Copyright © 2023 Yip, Ribeiro-Silva, Cha, Rieg and Sham. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Kay-Pong Yip, ZHlpcEB1c2YuZWR1