
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 28 July 2023
Sec. Mitochondrial Research
Volume 14 - 2023 | https://doi.org/10.3389/fphys.2023.1217815
This article is part of the Research TopicEnzyme-based TheranosticsView all 7 articles
 Noymar Luque-Campos1,2,3
Noymar Luque-Campos1,2,3 Ricardo Riquelme4
Ricardo Riquelme4 Luis Molina5
Luis Molina5 Gisela Canedo-Marroquín2,6
Gisela Canedo-Marroquín2,6 Ana María Vega-Letter7
Ana María Vega-Letter7 Patricia Luz-Crawford1,2,3*
Patricia Luz-Crawford1,2,3* Felipe A. Bustamante-Barrientos1,2,3*
Felipe A. Bustamante-Barrientos1,2,3*Mitochondrial dysfunction is a central event in the pathogenesis of several degenerative brain disorders. It entails fission and fusion dynamics disruption, progressive decline in mitochondrial clearance, and uncontrolled oxidative stress. Many therapeutic strategies have been formulated to reverse these alterations, including replacing damaged mitochondria with healthy ones. Spontaneous mitochondrial transfer is a naturally occurring process with different biological functions. It comprises mitochondrial donation from one cell to another, carried out through different pathways, such as the formation and stabilization of tunneling nanotubules and Gap junctions and the release of extracellular vesicles with mitochondrial cargoes. Even though many aspects of regulating these mechanisms still need to be discovered, some key enzymatic regulators have been identified. This review summarizes the current knowledge on mitochondrial dysfunction in different neurodegenerative disorders. Besides, we analyzed the usage of mitochondrial transfer as an endogenous revitalization tool, emphasizing the enzyme regulators that govern this mechanism. Going deeper into this matter would be helpful to take advantage of the therapeutic potential of mitochondrial transfer.
Multiple mechanisms have been described in the onset and progression of degenerative brain disorders, such as defective protein quality control, intra- and extracellular propagation of peptide aggregates, disturbs in synaptic activity, and mitochondrial dysfunction (de Vrij et al., 2004; Selfridge et al., 2013; Marsh and Alifragis, 2018; Balupuri et al., 2020; Monteiro et al., 2020). Mitochondria are essential organelles for oxidative metabolism, and their loss of functionality is frequently associated with overall energetic failure (Liesa and Shirihai, 2013; Fang et al., 2019). Mitochondria’s integrity is regulated through three fundamental mechanisms like fusion, fission, and mitophagy. Their proper functioning acts as a protective barrier against oxidative stress resulting from mitochondrial damage (Ma et al., 2020). Here, we summarize the current evidence regarding the deregulation of these processes in different neurodegenerative disorders, including 1) Alzheimer’s disease (AD), 2) Parkinson’s disease (PD), 3) Huntington’s disease (HD), 4) Frontotemporal dementia (FTD), and 5) Amyotrophic lateral sclerosis (ALS).
Based on the above, the donation of mitochondria has recently moved into the center of attention, given its potential to replace damaged mitochondria with healthy ones (Hayakawa et al., 2016; Peruzzotti-Jametti et al., 2020). The magnitude of the evidence has led to the development of several therapeutic strategies, including minocycline (Investigators, 2006; Quintero et al., 2006; Antonenko et al., 2010; Howard et al., 2020), polyphenols, such as curcumin and resveratrol (Quintero et al., 2006; Sandoval-Acuna et al., 2014; Naoi et al., 2019; Pannu and Bhatnagar, 2019), vitamins (Etminan et al., 2005; Hiller et al., 2018; Marie et al., 2021), coenzyme Q10 (Shults et al., 2002), monoamine oxidase B inhibitors (Guay, 2006; Carradori et al., 2016), creatine (Matthews et al., 1999; Investigators, 2006; Investigators, 2008) and mitoquinone (MitoQ) (Doughan and Dikalov, 2007; Snow et al., 2010; Aimaiti et al., 2021) (For a more description, the reader is redirected to Table 1). Unfortunately, none of these approaches comprehensively addresses mitochondrial dysfunction, but rather its consequences. Therefore, we summarize the described mitochondrial transfer (MT) regulators, part of a complex enzymatic network that still represents a question mark. Furthermore, we analyzed the relevance of identifying “help-me” signals to modulate the activation of MT-associated enzymes endogenously.
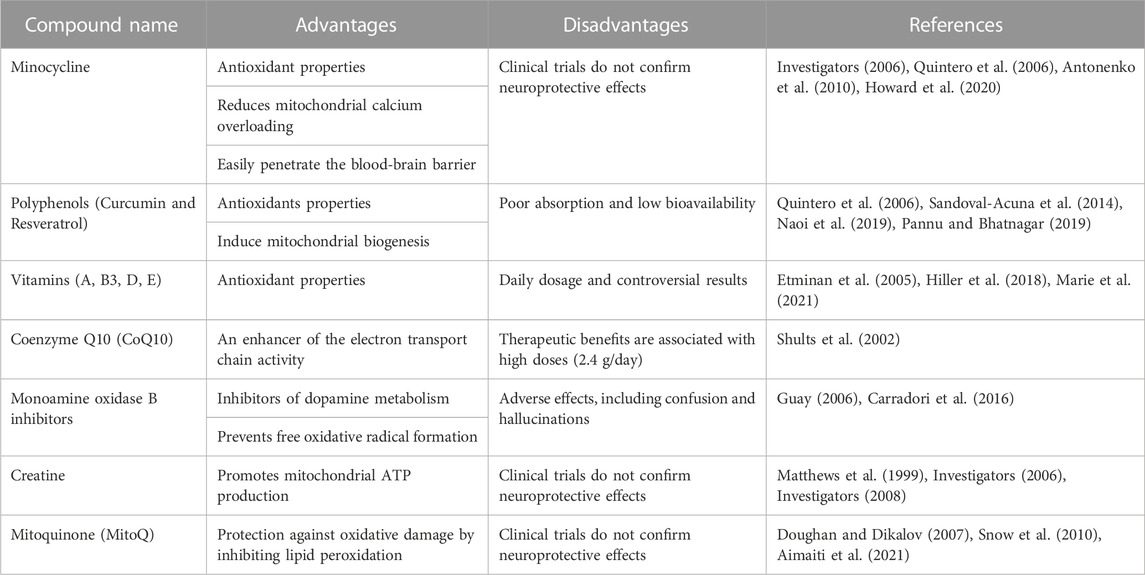
TABLE 1. Current therapeutic strategies against mitochondrial dysfunction in neurodegenerative diseases.
Etiological aspects of here referred degenerative brain disorders, and their association with mitochondrial defects, are detailed in Table 2.
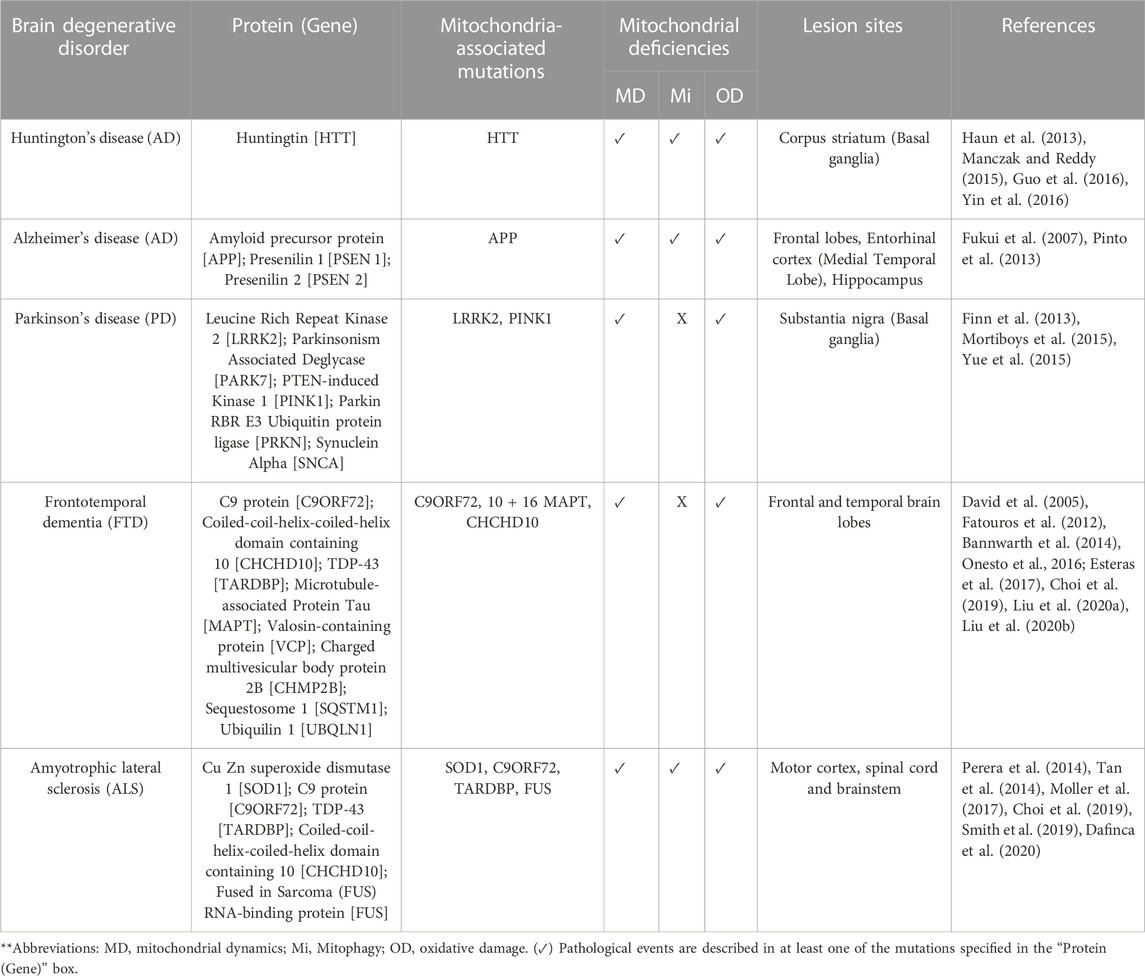
TABLE 2. Neurodegenerative-related mutations and their association with mitochondrial dysfunction.
Mitochondrial dynamics are multistep transitions that modify mitochondria’s spatial organization and functionality (Palikaras et al., 2015; Tilokani et al., 2018). Fission involves the exact and highly controlled fragmentation of impaired mitochondria, while fusion refers to the mechanism for constructing mitochondrial biomass from smaller mitochondrial units (Tilokani et al., 2018). The GTPase dynamic-related protein 1 (Drp-1) and mitochondrial fission protein 1 (Fis1) are essential enzymatic regulators in fission dynamics, controlling the constriction of mitochondrial membrane portions and inhibiting the fusion machinery (Tilokani et al., 2018).
Experiments performed in HD models such as R6/1 and HdhQ111/Q111 mouse neurons exhibit the same findings. Mice-derived R6/1 striatal neurons exhibit excessive Drp-1-dependent mitochondrial fragmentation and accumulation of oxygen free radicals (Cherubini et al., 2020). At the same time, HdhQ111/Q111 neurons express severe downregulation of fusion regulators like OPA1 and Mfn1/2 but also significant upregulation of fission markers, including Drp1 and Fis1 (Manczak and Reddy, 2015). Guo et al. studies complement these observations, whose findings reveal that striatal and spiny neurons carrying mutant huntingtin protein have significant mitochondrial fragmentation, increased motor deficit, and striatal disturbances in different HD mice models (Guo et al., 2016). Based on evidence suggesting that the pool of deficient huntingtin proteins is receptive to post-translational modifications (Haun et al., 2013), authors have proposed that molecular mechanisms such as S-nitrosylation aggravate mitochondrial fragmentation and provoke severe disorganization of dendritic spines by increasing its affinity with fission regulators (Haun et al., 2013). These observations have been convincingly demonstrated in BACHD rats-expressing human full-length huntingtin (Q97) and post-mortem brain samples (Haun et al., 2013).
In PD, nigral neurons have been broadly studied due to their dense mitochondrial biomass and oxidative activity, which make them especially susceptible to the consequences of mitochondrial dysfunction (Macdonald et al., 2018). Neurons exposed to toxic environmental substances like pesticides display considerable mitochondrial fragmentation and ATP depletion, correlating with the appearance of PD-like phenotypes in vitro (Chen et al., 2017). At the same time, mitochondria derived from LrrKG 2019S knock-in mice expressing a familial PD-related mutation are characterized by staying arrested in fission (Yue et al., 2015).
FTD-causing mutations, including C9orf72, 10 + 16 MAPT, and CHCHD10, have been connected with abnormalities in mitochondrial dynamics (Fatouros et al., 2012; Bannwarth et al., 2014; Onesto et al., 2016; Liu et al., 2020a; Liu et al., 2020b). Motor and mechanosensory neurons from C. elegans worms-expressing human Tau transgenes showed a significant redistribution of their mitochondrial network from the distal to the proximal region of axons (Fatouros et al., 2012), correlating with disturbances in the presynaptic region and locomotor deficiencies. Remarkably, Tau aggregation inhibitors reduce detergent-insoluble Tau aggregates, delay the accumulation of neuronal deficits, and promote a moderate improvement of locomotor abilities (Fatouros et al., 2012). Likewise, human fibroblasts carrying the C9orf72 mutation sustain abnormalities in mitochondrial fragmentation with a severe loss of ultrastructural features, the so-called cristae pattern (Onesto et al., 2016). At the same time, CHCHD10S55L dopaminergic neurons have the same pathological findings accompanied by marked swelling (Anderson et al., 2019). Consistently, CHCHD10 mutation was reported since it provokes inhibition of mitochondrial fusion through dissociating OPA1-mitofilin complexes (Liu et al., 2020a).
An elevated number of FTD-related cases underlie the C9orf72 gene mutation, representing many genetic ALS cases (Ferrari et al., 2011). Additionally, ALS-related mutations might include those over Cu, Zn superoxide dismutase 1 (SOD1), CHCHD10, and TDP-43, which also cause various mitochondrial disturbances (Tan et al., 2014; Davis et al., 2018; Choi et al., 2019; Dafinca et al., 2020). In SOD1 mutant mice, the deregulation in IP3R/VDAC complexes has been associated with abnormalities in the interaction between the endoplasmic reticulum and mitochondria, leading to abnormal calcium dynamics, extensive mitochondrial fragmentation, and damage of ultrastructural features (Tan et al., 2014; Smith et al., 2019). Mice expressing SOD1A4V, SOD1G73R, and SOD1G93A mutations develop disorganization of mitochondrial networks in different types of neurons (Moller et al., 2017). By comparison, experiments conducted by Choi et al. support that C9orf72 mutants replicate most of these pathogenic features, such as glutamate-induced synaptic excitotoxicity and subsequent activation of apoptotic pathways (Choi et al., 2019; Dafinca et al., 2020).
Mitophagy implies an autophagic pathway to recycle and reuse mitochondrial constituents (Palikaras et al., 2015). Although mitophagy undergoes a natural decline as aging progresses, numerous studies link the formation of protein aggregates and the dysregulation of intracellular pathways associated with mitophagy.
Experiments conducted by Tammineni et al. show that Aβ aggregates disturb the expression of specific molecular adaptors that regulate the identification, transport, and positioning of mitochondria through neuronal axonal projections (Tammineni et al., 2017), which is recognized as an event that precedes synaptic dysfunction. In AD, Aβ peptides and hyperphosphorylated forms of Tau aggregates provoke the downregulation of mitophagy-related proteins and deficiencies in lysosomal functionality, thus causing the accumulation of damaged mitochondria (Reddy and Oliver, 2019). Moreover, the long-term dysregulation of mitophagy exacerbates the appearance of mitochondrial DNA damage and provokes irreversible activation of apoptotic pathways (Fang et al., 2016).
In HD models, brain and skeletal muscle cells express elevated co-localization between autophagic/ubiquitination markers and mitochondrial constituents (Pinho et al., 2020). YAC128 and R6/2 mice carrying HD-like mutations acquire behavioral and motor abnormalities, which correlate with severe mitochondrial fragmentation and mitophagy in striatal and spiny neurons. Remarkably, blocking the interaction between mutant huntingtin and valosin-containing protein (VCP), a member of the AAA (+) ATPase family of chaperone-line proteins, reduces the mitochondrial translocation of the later, improves the mitochondrial organization in neurons and partially rescues disturbances in the behavior and motor abilities (Guo et al., 2016).
Lastly, ALS-associated TDP-43 mutations were recently documented by provoking evident deregulation of autophagy up-stream regulators like the AMP-activated protein kinase (AMPK) pathway (Perera et al., 2014; Smith et al., 2019); while mutations related to optineurin disrupt the recruitment of the autophagic machinery to the outer mitochondrial membrane and suspend its incorporation into autophagosome structures (Wong and Holzbaur, 2014).
Oxidative stress results from the uncontrolled generation of reactive oxygen species (ROS) like superoxide radicals (O2•-), hydrogen peroxide (H2O2), and hydroxyl radicals (•OH), thus exceeding the antioxidant defenses of cells and tissues (Pizzino et al., 2017). ROS are predominantly generated through mitochondrial respiration, obtaining O2•- molecules which are subsequently metabolized using intrinsic ROS scavenging enzymes, including superoxide dismutase (SOD), catalase, and glutathione peroxidase (Ray et al., 2012). Then, the coordinated metabolization of O2•- via SOD gives rise to the generation of H2O2, while radical hydroxyl molecules are generated through Fenton’s reaction, which entails the reaction between iron (Fe2+) and hydrogen peroxide (Fe2+ + H2O2 > Fe3+ + •OH + OH-) (Ray et al., 2012).
Numerous environmental and chemical stressors can exacerbate ROS production, including radiation, anti-blastic drugs, and exposure to heavy metals, among many others (Pizzino et al., 2017). Likewise, brain tissue differs from the rest, given its higher metabolic activity (Watts et al., 2018), providing a more significant potential to generate damage due to oxidative stress. Moreover, there is a natural age-related decline in mitochondrial functionality, which favors the accumulation of damaged mitochondria that generate high ROS amounts but lower ATP content (Hamilton et al., 2001). The following section focuses on how neurodegenerative conditions accelerate the functional decline of mitochondria, predisposing cells, and tissue to oxidative damage.
Experiments conducted in different research models, such as yeast (Bulteau et al., 2012), Drosophila (Greene et al., 2003), worms (Ren et al., 2019), zebrafish (Flinn et al., 2009), mice (Fukui et al., 2007; Pinto et al., 2013; Choi et al., 2019), and rats (Hoglinger et al., 2005; Escobar-Khondiker et al., 2007; Orozco-Ibarra et al., 2018), or even samples obtained from patients (Mahad et al., 2009; Mao and Reddy, 2010; Domercq et al., 2011; Mao et al., 2013; Sanders et al., 2014; Esteras et al., 2017; Igoillo-Esteve et al., 2020; Pistono et al., 2020), support that the expression and functioning of electron transport chain complexes are deregulated during brain degeneration (Hoglinger et al., 2005).
In AD, the metabolic profile seems to be a crucial determinant in generating amyloidogenic derivates since oxidative stress is tightly associated with the intensification in the Aβ peptide (AβPP) processing into Aβ (Gabuzda et al., 1994; Leuner et al., 2012). Experiments performed in cytoplasmic hybrids cells (or Cybrids) reveal that cytosolic constituents isolated from AD samples can replicate AD-like pathologic features once transplanted into healthy non-nucleated cells (Khan et al., 2000). These cells showed robust activation of proteolytic enzymes involved in regulating apoptotic pathways, increased DNA oxidative damage, poor ATP production, and strong activation of the amyloidogenic pathway, restored by antioxidant agents (Khan et al., 2000). The impact of the electron transport chain is evidenced through in vivo experiments crossing COXI mice carrying cytochrome c oxidase deficiencies and AD mice expressing human amyloid precursor protein (APP). Compared with AD mice, COXI/AD mice maintain reduced levels of the oxidative stress marker 8-hydroxy-2-deoxyguanosine (8-OHdG) and several Aβ deposits. At the same time, their mitochondrial DNA stability in the cortex and the hippocampus is significantly higher (Fukui et al., 2007; Pinto et al., 2013).
Most PD sporadic presentations are closely linked to defects in the expression and functionality of the mitochondrial complex I in the substantia nigra and the prefrontal cortex (Schapira et al., 1990; Parker et al., 2008). As expected, this deficiency type is primarily connected with energetic failure and progressive ROS accumulation (Greenamyre et al., 2001). Post-mortem analyses in the brain of PD-diagnosed patients support that nigral neurons carry extensive mitochondrial DNA damage (Sanders et al., 2014), and their cerebrospinal fluid contained elevated amounts of antioxidant co-enzyme Q10 and oxidized nucleosides (Isobe et al., 2010), revealing the presence of DNA lesions. Although most evidence attributes these alterations to the downregulation of mitochondrial complex I, experiments conducted by Schapira et al. in the early 1990s reported that PD patients do not display differences in the total protein and mitochondrial biomass neurons residing in the substantia nigra; however, their functionality at the level of complex I, is reduced (Schagger, 1995). Numerous investigations currently support these findings. For example, Drosophila (Greene et al., 2003) and zebrafish embryos (Flinn et al., 2009) carrying Parkin mutations show reduced functionality in complex I, while PINK1 mutants develop dysfunction in mitochondrial complexes I and III (Flinn et al., 2013). Although evidence consistently points to complex I dysfunction, the pathological contribution of complexes III subunit UQCR2, complex IV, and V subunit ATP5A have also been reported in a LrrkG2019S knock-in mouse (Mortiboys et al., 2015; Yue et al., 2015).
Striatal cells exposed to 3-nitro propionic acid, a chemical method for inducing HD-like striatal degeneration, proved to generate suppression of mitochondrial complex II and reduced ATP levels (Orozco-Ibarra et al., 2018). Notably, the isolation of mitochondria from neuronal synaptic terminals revealed variations in the protein expression of mitochondrial complexes, encompassing the upregulation of the complex II 70 kDa subunit (Hamilton et al., 2017), while neurons carrying huntingtin mutations (HdhQ111/Q111) exhibit dysregulation of different mitochondrial-encoded electron transport chain subunits (Manczak and Reddy, 2015; Yin et al., 2016).
Comparably, neuron-like cells obtained via differentiation of FTD patient-derived iPSCs underline the consequences of the 10 + 16 MAPT mutation, which disrupts the mitochondrial membrane potential, reduces the activity of complex I, increase ROS generation and provoke cell death (Esteras et al., 2017). In agreement, six-month-old CamKII; (GR)80 mice express a GGGGCC repeat expansion within a non-coding region in the gene encoding for C9orf72. These mice evidence poor mitochondrial complexes I and V activity, severe DNA damage, and neuronal degeneration in the prefrontal, parietal, and occipital cortex. Also, these lesions correlate with synaptic abnormalities and FTD-like behavior (Choi et al., 2019). In contrast, mutations on TDP-43 provoke an aggressive phenotype characterized by abnormalities in the assembly and activity of complex I. This leads to severe neurotoxicity involving degeneration of cortical neurons and motor tracts, which is efficiently rescued by blocking the entrance of mutated TDP-43 protein into mitochondria (Wang et al., 2016). Similar observations were performed in P301L Tau transgenic mice. Proteomic and functional analyses showed that the generation of Tau aggregates destabilizes the activity of cytosolic upstream regulators for mitochondrial functions and antioxidant defenses (David et al., 2005). In agreement with both C9orf72 and TDP-43 mutants, these mice sustain the poor activity of mitochondrial complex I and low ATP content (David et al., 2005). In turn, the authors reported the dysregulation of different detoxifying enzymes, including glutathione reductase, glutathione peroxidase, and superoxide dismutase (David et al., 2005), pointing out that the enzymatic network that maintains mitochondrial homeostasis is altered at different levels.
Lastly, several ALS-associated mutations, including those shared with FTD, develop extensive oxidative damage. SOD1 mutated proteins are progressively stored at the mitochondrial intermembrane space, forming protein aggregates that lead to the deregulation of mitochondrial respiratory complexes and increased ROS generation (Smith et al., 2019). A comparable phenotype is observed in cells carrying mutations on the nuclear ribonucleoprotein P2, or FUS. These mutations are linked to uncontrolled ROS production and poor ATP biosynthesis. However, the mechanism breaks the interaction between the endoplasmic reticulum and mitochondria (Smith et al., 2019). On the other hand, neurons derived from C9orf72 transgenic mice sustain low activity in mitochondrial complexes I and V and severe DNA damage. As expected, these alterations converge in the rapid activation of apoptosis (Choi et al., 2019).
Altogether this data supports that the onset and progression of degenerative brain disorders entail enzymatic deregulation at different levels. The resolution of these defects has been focused through various methodologies, including i) nutritional supplements (Bobadilla et al., 2021; Dubey et al., 2021); ii) enzymatic replacement therapy based on soluble enzymes or even enzyme-loaded nanoparticles (Dickson et al., 2007; Del Grosso et al., 2019; Sato and Okuyama, 2020); and iii) mono-drug schemes (Andreux et al., 2013; Singh et al., 2021). Nevertheless, none of these methods can integrally correct mitochondrial dysfunction since they are directed against specific defects.
The following section discusses the potential of naturally occurring mitochondrial transfer as a therapeutic strategy expected to re-establish the functionality of mitochondria in neural cells undergoing energetic distress. By contrast with other therapeutic approaches, mitochondrial transfer offers the complete replacement of impaired mitochondria.
A growing body of evidence highlights the benefits of naturally occurring mitochondrial transfer, given its potential to restore mitochondrial dysfunction and energetic balance in neighboring cells.
Defined as the naturally occurring transfer of mitochondrial units from a healthy donor to a specific cell acceptor with damaged mitochondria. Donor cells deliver mitochondria through different cellular mechanisms, including i) the formation of transient tunneling nanotubes (TNTs), which originate as filopodia-like cell membrane projections that extend, contact, and fuse with targeted cells (Gerdes et al., 2007; Caicedo et al., 2017); ii) the opening of intercellular channels located laterally in the membrane known as Gap junctions, by means cells exchange a variety of molecules as small-sized mitochondrial constituents or even energetic nucleotides (Li et al., 2019); and iii) the release of extracellular vesicles (EVs) that carry mitochondrial constituents of different sizes (Hayakawa et al., 2016; Peruzzotti-Jametti and Pluchino, 2018). For example, microvesicles ranging between 500 and 900 nm can transport entire mitochondrial units (Hayakawa et al., 2016). In comparison, those oscillating between 50 and 100 nm can carry small mitochondrial microdomains containing respiratory complexes and other integral membrane elements (Peruzzotti-Jametti et al., 2020). Figure 1.
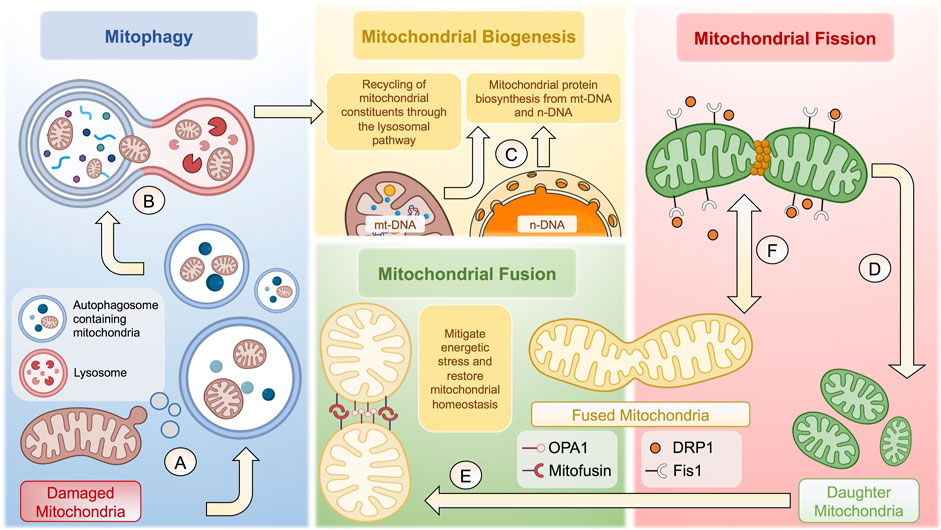
FIGURE 1. Cellular and molecular mechanisms behind mitochondrial homeostasis. (A) Damaged membrane microdomains of mitochondria are packaged in autophagosomes to follow the lysosomal pathway. (B) Autophagosomes containing mitochondria fuse with lysosomes to form autophagolysosomes to degrade mitochondria. (C) Once degraded, mitochondrial constituents are recycled to potentiate mitochondrial biogenesis together with proteins synthetized from n-DNA and/or mt-DNA. (D) Mitochondrial fission is tightly regulated through Fis1-dependent recruitment of DRP1, which generates a constriction ring to give rise to two daughter mitochondria. (E) Resulting mitochondria can undergo mitochondrial fusion to increase the mitochondrial biomass and thus mitigate energetic stress through OPA1 and Mitofusin proteins. (F) The balance between mitochondrial fusion and fission is highly dynamic, depending of various factors, including the bioenergetic status and cell-specific functions, among many others. The complexity of the regulation of mitochondrial homeostasis is not fully depicted. The figure was created in BioRender.com.
Most mechanisms behind regulating mitochondrial transfer and their crosstalk are still poorly understood (Figure 2). In addition, current enzymatic functions are detailed in Table 3.
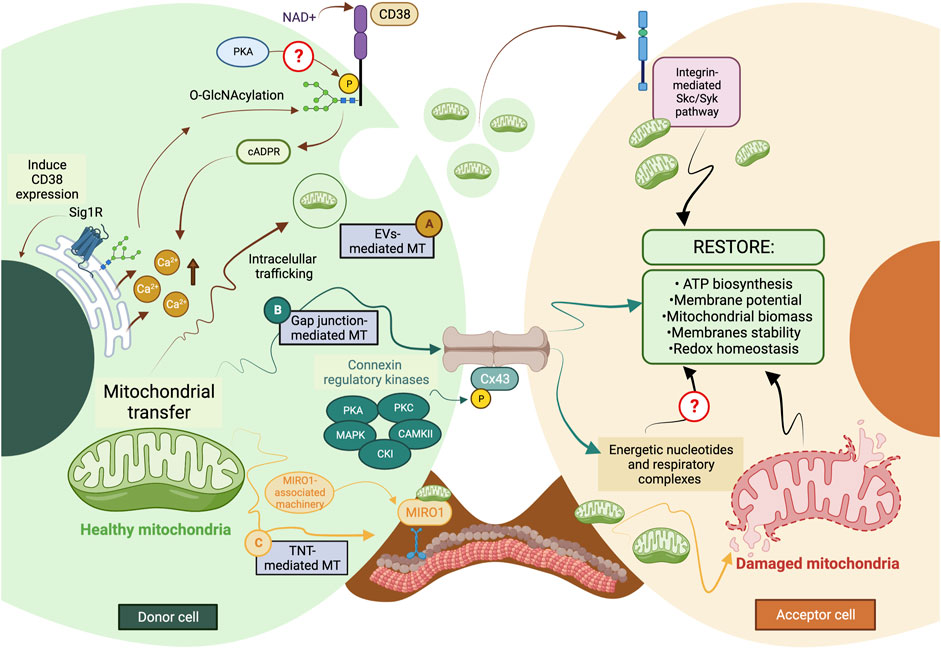
FIGURE 2. The current model of cellular and molecular mechanisms behind mitochondrial transfer. (A) EVs-mediated MT is regulated through the CD38/cADPR signaling, inducing calcium release from the endoplasmic reticulum. The CD38 activity can be post-translationally modulated through O-GlcNAcylation and phosphorylation, and its expression is induced in a Sig1R-dependent manner. (B) Connexins’ protein expression, assembly, and stabilization are precisely controlled through different kinases, such as PKA, PKC, MAPK, CK1, and CAMKII. Gap junctions-mediated MT is predicted to facilitate the donation of low-weight molecules comprising energetic nucleotides and mitochondrial respiratory complexes. (C) The formation of TNTs relies on the molecular interaction between MIRO1 and accessory motor enzymes like dynein, kinesins, and myosins. TNT-mediated MT entails donating complete mitochondrial units, whose subcellular fate is still the subject of discussion. The complexity of the exocytic pathway is not fully depicted. Question marks denote mechanisms not currently described. MT, mitochondrial transfer; PKA, protein kinase A; PKC, protein kinase C; MAPK, mitogen-activated protein kinase (MAPK); CK1, casein kinase 1; and CAMKII, Ca2+/calmodulin-dependent kinase II. The figure was created in BioRender.com.
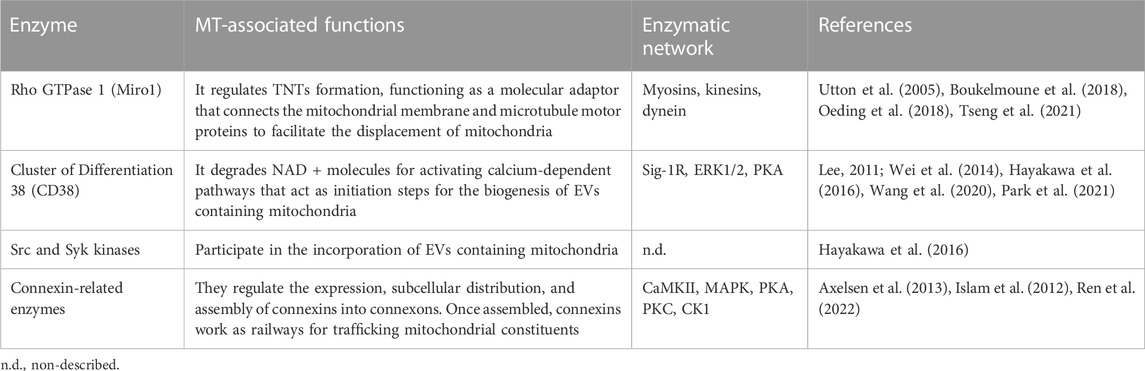
TABLE 3. Current roles of mitochondrial transfer-related enzymes.
During TNT’s formation, mitochondrial Rho GTPase 1, also known as MIRO1, mediates the polymerization of actin filaments and the subsequent establishment of intercellular structures connecting the donor and acceptor cells (Lopez-Domenech et al., 2018). This GTPase functions as a molecular adaptor that binds the mitochondrial outer membrane and microtubule motor proteins, thus facilitating the displacement of mitochondria along microtubules (Lopez-Domenech et al., 2018). MIRO1 gain-and-loss of function experiments resulted in changes in MT’s efficiency and repair capacity (Boukelmoune et al., 2018; Tseng et al., 2021).
CD38 (Cluster of Differentiation 38) is a multifunctional ectoenzyme that controls extracellular nucleotide homeostasis and intracellular calcium oscillations (Hogan et al., 2019). CD38 degrades NAD + molecules as a cyclase to produce cyclic ADP-ribose (cADPR) molecules, a potent second messenger for calcium mobilization (Hogan et al., 2019). The release of EVs containing mitochondrial units and constituents depends on the CD38/cADPR axis (Hayakawa et al., 2016). Furthermore, cellular states like oxygen-glucose deprivation and reoxygenation induce changes in the post-translational modification of mitochondrial proteins via O-GlcNAcylation (O-GlcNAc), increasing the release of EVs containing mitochondria in a CD38-dependent manner (Park et al., 2021). Consistently, the genetic inhibition of CD38 reduces mitochondrial transfer in vitro and in vivo (Hayakawa et al., 2016); defects in the endoplasmic reticulum–Golgi traffic resulted in the release of mitochondria with reduced membrane potential and mt-DNA (Park et al., 2021). Since O-GlcNAc has been previously implicated in protein quality control (Collins and Chatham, 2020), it is tempting to speculate whether mitochondria undergo a pre-selection before being charged and released. Little is known about the internalization of EVs carrying mitochondria, but it entails the activation of the integrin-mediated Src/Syk signaling pathway (Hayakawa et al., 2016) (Figure 2).
MIRO1-mediated MT relies on the synchronized interaction of many enzymes involving myosins, dynein, and the kinesin superfamily proteins (KIFs). A proposal of their interaction during MT is detailed in Figure 2, while their molecular mechanisms are summarized in Table 3.
Myosins are molecular motors that produce mechanical energy through ATP consumption, comprising a superfamily classified into 18 classes (Foth et al., 2006). These proteins contain C-terminal light chain-associated domain, which is regulated through phosphorylation to modulate myosin’s activity (Sata et al., 1997). Furthermore, myosins contain motor domains that catalyze the hydrolysis of ATP. The direct interaction between myosin and MIRO1 facilitates actin-based mitochondrial transport (Oeding et al., 2018).
KIFs are motor proteins whose classification relies on the location of their motor domains, comprising N- (N-terminal), C- (C-terminal), and M- (Middle portion) kinesins (Rath and Kozielski, 2012). These enzymes participate in various cellular processes such as intracellular traffic dynamics, cell division, and the anterograde transport of mitochondria through axons (Hirokawa et al., 2009). Mechanistically, KIFs intervene in the binding of motor proteins and mitochondria, and their loss of function, specifically on the kinesin-related protein 5 (KIF5), disturbs the traffic of mitochondria and correlates with abnormalities in their subcellular location (Utton et al., 2005).
Dynein is a minus-end-direct motor enzyme whose ATP consumption displaces mitochondria across the cell body, distributed in axonal and cytoplasmic isoforms (Gibbons and Rowe, 1965). Dynein functionality is subject to the cofactor dynactin, a 23-subunit complex that supports the retrograde transport of cellular constituents, including mitochondria (Urnavicius et al., 2015). The genetic inhibition of dynein ameliorates its binding to mitochondria and gives rise to cytoplasmic inclusions that interfere with mitochondria’s transport in neurons (Chen et al., 2014).
The topology of CD38 is still a matter of discussion. It is predicted to be a type-II transmembrane protein with a catalytic C-terminal domain pointing to the extracellular media (Wei et al., 2014). This presents a dilemma because the NAD substrate is stored intracellularly. Cells co-express different types of CD38, which differ in the subcellular distribution and structurally (Zhao et al., 2012). In this hypothesis, some catalytic domains should be oriented outside the cell, while others should be disposed to the cytoplasm. In this scheme, the catalytic C-terminal domain takes relevance since it contains multiple serine (Ser) residues, among which a phosphorylation site for protein kinase A (PKA) is predicted to exist (Lee, 2011; Wei et al., 2014).
Sigma-1 receptor (Sig-1R), one of two sigma receptor subtypes, is a 223-amino-acid-long trans-membrane chaperone at the endoplasmic reticulum (Zhemkov et al., 2021). Mechanistically, the activation of Sig-1R upregulates the expression of CD38 through extracellular regulated protein kinases 1/2 (ERK1/2), facilitating the CD38-driven MT. Knocking down CD38 abolishes Sig-1R-induced MT (Wang et al., 2020).
The stabilization and maintenance of Gap junctions entail the synchronic displacement of connexins along the plasma membrane, laterally assembled into connexin hemichannels, known as connexons (each one formed by six connexins) (Zimmermann, 1984). Gap junctions’ formation is coordinated through several phosphatases and kinases (Solan and Lampe, 2016). The specific phosphorylation of serine (Ser) and tyrosine (Tyr) residues in connexins instruct their correct displacement and assembly into connexons (Solan and Lampe, 2016). Some sites of phosphorylation and their specific kinase include 1) Ser244 and Ser314 for Ca2+/calmoduline-dependent kinase II (CaMKII); 2) Tyr247 and Tyr265 for Src kinase; 3) Ser255, Ser279, and Ser282 for Mitogen-activated protein kinase (MAPK); 4) Ser262 for protein kinase C (PKC); 5) Ser325, Ser328, and Ser330 for casein kinase 1 (CK1); and 6) Ser364, Ser365, Ser368, Ser369, Ser372, and Ser373 for PKA and PKC [Reviewed in (Axelsen et al., 2013)]. Their possible interactions are proposed in Figure 2.
Different types of cells employ spontaneous mitochondrial transfer in the central and peripheral nervous systems, including various subtypes of neurons (Li et al., 2019; English et al., 2020; van der Vlist et al., 2022), astrocytes (Hayakawa et al., 2016; English et al., 2020), neural and endothelial progenitor cells (Hayakawa et al., 2018), and nervous tissue-residing immune cells (Luz-Crawford et al., 2019; Court et al., 2020). Although the incorporation mechanisms remain poorly detailed, host cells recover their survival and mitochondrial functionality by increasing oxygen consumption and ATP generation (Hayakawa et al., 2016). Cells carrying mitochondrial impairment incorporate approximately 40% more biomass than healthy acceptor cells (English et al., 2020); current evidence suggests that donor cells can transfer between 5% and 13% of their whole mitochondrial biomass (Hayakawa et al., 2018; Gao et al., 2019), allowing them to remain viable. Both proteomic and gene ontology determinations support that adult neural progenitors can transfer mitochondrial complexes encoded in mitochondrial and nuclear genomes (Peruzzotti-Jametti et al., 2020). The purification and subsequent intravenous administration of mitochondria increase the expression of antioxidant enzymes, reduce lipid peroxidation, limit the generation of oxygen and nitrogen-derived reactive species in oxygen-deprivation conditions, and increase neuronal survival (Zhang et al., 2019).
In the host tissue, MT modulates pro-angiogenic properties, endothelial permeability, neuronal survival in front of oxygen and glucose deficiency, and inflammatory pain resolution, as well as the restriction of reactive astrogliosis and the induction of adult neurogenesis (Bond et al., 2015; Hayakawa et al., 2018; Zhang et al., 2019; van der Vlist et al., 2022). Even neuroglial transmitophagy represents an MT-based mechanism through which astrocytes internalize and degrade axonal mitochondria (Lampinen et al., 2022), which can induce neuroprotection in pathological contexts (Hayakawa et al., 2016; English et al., 2020). Analogously, this mechanism is also employed by cone photoreceptors. These retinal neurons can transfer injured mitochondria from the cone to Müller glia to be degraded (Hutto et al., 2023).
The therapeutic potential of MT in degenerative brain disorders is still a growing field of research. Mice expressing 1-Methyl-4-phenil-1,2,3,6-tetrahydropyridine (MTPT)-induced PD-like phenotype respond positively to mitochondria’s administration, improving their locomotor and behavioral abilities (Zhang et al., 2019). Zhang et al. tested the impact of muscle-derived mitochondria once intraventricularly injected into rats’ brains. They showed that mitochondria confer strong resistance against oxidative stress and significantly increase mitochondrial biomass after injury (Zhang et al., 2019). In addition, AD mice intravenously injected with mitochondria improve their cognitive performance, which correlates with a significant increase in neurons’ survival and further restriction of reactive astrogliosis (Nitzan et al., 2019). These effects are subjected to Gap-junctions-mediated MT. Activating the GJA1-20K/connexin 43 (Cx43) axis rescues the dendrite length and promotes mitochondrial protein expression in neurons after brain injury through the spontaneous transfer of mitochondria from astrocytes (Ren et al., 2022). Consistently, the loss of MT from astrocytes to neurons has also been documented in toxic-induced cognitive impairment and leukodystrophies (Gao et al., 2019; English et al., 2020).
Differences in preserving or declining the integrity of certain neuroanatomic regions through brain degeneration remain a long-standing question (Morrison et al., 1998; Mrdjen et al., 2019). Cellular and molecular differences between CNS-residing cells seem to explain the vulnerability or resistance against stressor factor, such as secretory and biosynthetic demands, oxidative stress, misfolding and aggregation of proteins, calcium fluxes, glucose and oxygen restriction, and nervous and/or peripheral inflammatory responses (Mrdjen et al., 2019). Moreover, aging gradually impairs the capacity of cells to deal with these stressors (Mrdjen et al., 2019).
The neuroanatomical perspective takes strength due to the specific pattern of cell decline for each disorder, involving corticocortical neurons in AD, cingulate and insular neurons in FTD, upper and lower motor neurons in ALS, striatal neurons in HD, and nigrostriatal neurons in PD (Morrison et al., 1998; Nana et al., 2019). In this regard, the differential expression of neurofilament proteins and neuron-specific receptors has been proposed as major vulnerability mechanisms. For example, glutamatergic neurons expressing N-methyl-D-aspartate (NMDA) receptors are highly vulnerable to voltage-gated Ca2+ channel-dependent excitotoxicity (Morrison et al., 1998).
Although the chronic course of degenerative brain disorders compromises other brain regions, there are areas with high resistance that remain intact even in advanced stages (Mrdjen et al., 2019). Thus, stimulating endogenous MT could represent a complementary method to induce the regeneration of diseased brain regions from healthy ones. Even it may cover some technical aspects associated with the exogenous administration of mitochondria, a research field with promising advances (McCully et al., 2009; Masuzawa et al., 2013; Cowan et al., 2016; Emani et al., 2017; Shi et al., 2017).
Biodistribution studies show that nervous cells store significant amount of mitochondria after being intravenously administered, increasing ATP biosynthesis, restoring the energetic failure and thus promoting survival in recipient cells (Shi et al., 2017). Nevertheless, some experimental considerations should be covered when studying the therapeutic potential of isolated mitochondria.
Mitochondria’s onion-like organization plays a fundamental role in maintaining mitochondria’s functionality, favoring the establishment of cristae-like structures that increase the reaction surface. Mitochondrial swelling is a morphological feature resulting from the opening of the permeability transition pore, the entry of water, and then the disorganization of cristae structures (Javadov et al., 2018), representing a hallmark of mitochondrial dysfunction. Mitochondria are isolated using centrifugation steps aimed at separating them from the rest of cellular components (Caicedo et al., 2015), and to be subsequently added to cell cultures, and injected directly into the tissue or circulation (McCully et al., 2009; Caicedo et al., 2015; Cowan et al., 2016; Kaza et al., 2017; Shi et al., 2017). Since these environments do not preserve the osmolarity of the intracellular microenvironment given that calcium oscillates in micromolar concentrations (Valentine et al., 2018), it is interesting to speculate about how intact remain mitochondria once transferred outside the cell. Current evidence show that isolated mitochondria remain functional and conserve their ultrastructural organization (Caicedo et al., 2015; Peruzzotti-Jametti et al., 2020). However, our knowledge on the route they follow after being incorporated into cells is largely unknown. Perhaps, these mitochondria become dysfunctional on the way to be incorporated but still contribute to the energetic failure in recipient cells by acting as a substrate for the recycling of components needed to initiate mitochondrial biogenesis.
Following the line of structural integrity, immune system activation arises as a possible pitfall. Despite MT induces anti-inflammatory responses in different tissues (Luz-Crawford et al., 2019; van der Vlist et al., 2022), the loss of mitochondria’s integrity could derive in the release of mitochondrial DNA (mt-DNA). Several pathological conditions including oxidative damage, genotoxic stress, activation of pro-inflammatory factors and mitochondrial dysfunctions favor the release of mt-DNA (Kim et al., 2023). Once released, mt-DNA is recognized as a strong agonist of innate immunity, activating pro-inflammatory pathways such as endosomal localized TLR9, cytosolic cGAS-STING, and cytosolic inflammasome AIM2/NLRP3 (Riley and Tait, 2020). Therefore, experimental approaches that poorly preserve mitochondrial integrity through the isolation step could generate results conditioned by the activation of the immune system, but not associated with the incorporation of functional mitochondria.
Spontaneous mitochondrial transfer occurs through the formation of TNTs and connexons and the release of EVs. The deepening into MT-associated molecular and cellular mechanisms, and the development of strategies aimed at modulating them could represent an alternative to the osmolarity and immune considerations since bypass the exposure of mitochondria to the extracellular medium.
Most pharmacologic and genetic approaches aimed to modulate the MT-related enzymatic machinery are oriented to its inhibition since states of overactivation correlate with the appearance of pathological features (Braicu et al., 2019; Nassal et al., 2020). This represents a limitation given the need to enhance its function to induce mitochondrial transfer, calling to identify new mechanisms to activate endogenous MT.
Neural cells employ various “help-me” signals (i.e., cell-derived molecules released when cells undergo different types of damage, including mitochondrial damage) able to modulate neuroprotective mechanisms, such as regulating neurogenic and angiogenic pathways. These mechanisms include the release of mitochondrial debris, growth factors, chemokines, and cytokines, which have been extensively reviewed in the literature (Xing and Lo, 2017). Once released, these extracellular signals recruit neighboring cells, potentiate neuroprotection and promote endogenous brain regeneration (Xing and Lo, 2017). There is no evidence regarding variations in the secretome of cells with energy failure, not at least in neural cells. However, mitochondrial damage-associated molecular patterns (DAMPs) are conserved in mammals (Nakahira et al., 2015; Grazioli and Pugin, 2018). These patterns involve the release of mt-DNA and mitochondrial proteins from damaged cells, mediating the activation of diverse cellular mechanisms in their environment or even distant niches as they enter circulation (Nakahira et al., 2015; Grazioli and Pugin, 2018). On the other hand, experiments performed in epithelial, adipose, and fibroblast cells suffering dysfunction in oxidative phosphorylation revealed significant variations in their secretome (Llobet et al., 2017; Garrido-Perez et al., 2020). Molecules associated with focal adhesion, complement, and coagulation cascades, extracellular matrix receptors, glucose transporters, intracellular trafficking proteins, and hormones related to regulating appetite and satiety in the hypothalamus presented significant variations (Llobet et al., 2017; Garrido-Perez et al., 2020; Sturm et al., 2023). Besides, the secretion of cytokines and metabokines also suffers modifications in cells with age-related mitochondrial dysfunction. The progressive decline in mitochondria’s functionality is closely associated with a hypermetabolic state presenting significant mt-DNA instability and elevated secretion of growth differentiation factors (Sturm et al., 2023). Lastly, ROS overproduction represents a critical stimulus. It has been described that rotenone-induced oxidative damage potentiates MT efficiency, which is inhibited through administrating ROS scavenger molecules (Jiang et al., 2016; Burt et al., 2019). In this regard, NADPH oxidase-2-derived superoxide radicals are critical. Marlein et al. reported that blocking the generation of superoxide species abolishes MT in myeloid leukemia blasts (Marlein et al., 2017).
Based on the above data, an exciting projection regarding MT-related enzymes could rely on identifying “help-me” signals from cells undergoing bioenergetic stress. Indeed, there is little evidence regarding the regenerative potential of MT in degenerative brain disorders. However, both in vitro and in vivo determinations, associated or not with the nervous system, suggest that this is a promising tool to promote the endogenous revitalization of damaged tissue. Going deeper into identifying “help-me” signals could put us one step ahead of genetic or pharmacological approaches to modulate MT-related enzymes.
NL-C and FB-B designed and drafted the manuscript. RR, LM, GC-M, and AV-L revised the draft and prepared the table. NL-C prepared the figure. PL-C and FB-B validated the manuscript. The contribution of NL-C remains equal. All authors contributed to the article and approved the submitted version.
This work was supported by FONDECYT Postdoctorado 3220204 (FB-B); PAI ANID SA77210076 (GC-M); and ANID-Basal funding for Scientific and Technological Center of Excellence, IMPACT (FB210024).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Aimaiti, M., Wumaier, A., Aisa, Y., Zhang, Y., Xirepu, X., Aibaidula, Y., et al. (2021). Acteoside exerts neuroprotection effects in the model of Parkinson's disease via inducing autophagy: Network pharmacology and experimental study. Eur. J. Pharmacol. 903, 174136. doi:10.1016/j.ejphar.2021.174136
Anderson, C. J., Bredvik, K., Burstein, S. R., Davis, C., Meadows, S. M., Dash, J., et al. (2019). ALS/FTD mutant CHCHD10 mice reveal a tissue-specific toxic gain-of-function and mitochondrial stress response. Acta Neuropathol. 138, 103–121. doi:10.1007/s00401-019-01989-y
Andreux, P. A., Houtkooper, R. H., and Auwerx, J. (2013). Pharmacological approaches to restore mitochondrial function. Nat. Rev. Drug Discov. 12, 465–483. doi:10.1038/nrd4023
Antonenko, Y. N., Rokitskaya, T. I., Cooper, A. J., and Krasnikov, B. F. (2010). Minocycline chelates Ca2+, binds to membranes, and depolarizes mitochondria by formation of Ca2+-dependent ion channels. J. Bioenerg. Biomembr. 42, 151–163. doi:10.1007/s10863-010-9271-1
Axelsen, L. N., Calloe, K., Holstein-Rathlou, N. H., and Nielsen, M. S. (2013). Managing the complexity of communication: Regulation of gap junctions by post-translational modification. Front. Pharmacol. 4, 130. doi:10.3389/fphar.2013.00130
Balupuri, A., Choi, K. E., and Kang, N. S. (2020). Aggregation mechanism of Alzheimer's amyloid β-peptide mediated by α-Strand/α-Sheet structure. Int. J. Mol. Sci. 21, 1094. doi:10.3390/ijms21031094
Bannwarth, S., Ait-El-Mkadem, S., Chaussenot, A., Genin, E. C., Lacas-Gervais, S., Fragaki, K., et al. (2014). A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain 137, 2329–2345. doi:10.1093/brain/awu138
Bobadilla, M., Garcia-Sanmartin, J., and Martinez, A. (2021). Natural food supplements reduce oxidative stress in primary neurons and in the mouse brain, suggesting applications in the prevention of neurodegenerative diseases. Antioxidants (Basel) 10, 46. doi:10.3390/antiox10010046
Bond, A. M., Ming, G. L., and Song, H. (2015). Adult mammalian neural stem cells and neurogenesis: Five decades later. Cell Stem Cell 17, 385–395. doi:10.1016/j.stem.2015.09.003
Boukelmoune, N., Chiu, G. S., Kavelaars, A., and Heijnen, C. J. (2018). Mitochondrial transfer from mesenchymal stem cells to neural stem cells protects against the neurotoxic effects of cisplatin. Acta Neuropathol. Commun. 6, 139. doi:10.1186/s40478-018-0644-8
Braicu, C., Buse, M., Busuioc, C., Drula, R., Gulei, D., Raduly, L., et al. (2019). A comprehensive review on MAPK: A promising therapeutic target in cancer. Cancers (Basel) 11, 1618. doi:10.3390/cancers11101618
Bulteau, A. L., Planamente, S., Jornea, L., Dur, A., Lesuisse, E., Camadro, J. M., et al. (2012). Changes in mitochondrial glutathione levels and protein thiol oxidation in yfh1 yeast cells and the lymphoblasts of patients with Friedreich's ataxia. Biochim. Biophys. Acta 1822, 212–225. doi:10.1016/j.bbadis.2011.11.003
Burt, R., Dey, A., Aref, S., Aguiar, M., Akarca, A., Bailey, K., et al. (2019). Activated stromal cells transfer mitochondria to rescue acute lymphoblastic leukemia cells from oxidative stress. Blood 134, 1415–1429. doi:10.1182/blood.2019001398
Caicedo, A., Aponte, P. M., Cabrera, F., Hidalgo, C., and Khoury, M. (2017). Artificial mitochondria transfer: Current challenges, advances, and future applications. Stem Cells Int. 2017, 7610414. doi:10.1155/2017/7610414
Caicedo, A., Fritz, V., Brondello, J. M., Ayala, M., Dennemont, I., Abdellaoui, N., et al. (2015). MitoCeption as a new tool to assess the effects of mesenchymal stem/stromal cell mitochondria on cancer cell metabolism and function. Sci. Rep. 5, 9073. doi:10.1038/srep09073
Carradori, S., Gidaro, M. C., Petzer, A., Costa, G., Guglielmi, P., Chimenti, P., et al. (2016). Inhibition of human monoamine oxidase: Biological and molecular modeling studies on selected natural flavonoids. J. Agric. Food Chem. 64, 9004–9011. doi:10.1021/acs.jafc.6b03529
Chen, T., Tan, J., Wan, Z., Zou, Y., Afewerky, H. K., Zhang, Z., et al. (2017). Effects of commonly used pesticides in China on the mitochondria and ubiquitin-proteasome system in Parkinson's disease. Int. J. Mol. Sci. 18, 2507. doi:10.3390/ijms18122507
Chen, X. J., Xu, H., Cooper, H. M., and Liu, Y. (2014). Cytoplasmic dynein: A key player in neurodegenerative and neurodevelopmental diseases. Sci. China Life Sci. 57, 372–377. doi:10.1007/s11427-014-4639-9
Cherubini, M., Lopez-Molina, L., and Gines, S. (2020). Mitochondrial fission in Huntington's disease mouse striatum disrupts ER-mitochondria contacts leading to disturbances in Ca(2+) efflux and Reactive Oxygen Species (ROS) homeostasis. Neurobiol. Dis. 136, 104741. doi:10.1016/j.nbd.2020.104741
Choi, S. Y., Lopez-Gonzalez, R., Krishnan, G., Phillips, H. L., Li, A. N., Seeley, W. W., et al. (2019). C9ORF72-ALS/FTD-associated poly(GR) binds Atp5a1 and compromises mitochondrial function in vivo. Nat. Neurosci. 22, 851–862. doi:10.1038/s41593-019-0397-0
Collins, H. E., and Chatham, J. C. (2020). Regulation of cardiac O-GlcNAcylation: More than just nutrient availability. Biochim. Biophys. Acta Mol. Basis Dis. 1866, 165712. doi:10.1016/j.bbadis.2020.165712
Court, A. C., Le-Gatt, A., Luz-Crawford, P., Parra, E., Aliaga-Tobar, V., Batiz, L. F., et al. (2020). Mitochondrial transfer from MSCs to T cells induces Treg differentiation and restricts inflammatory response. EMBO Rep. 21, e48052. doi:10.15252/embr.201948052
Cowan, D. B., Yao, R., Akurathi, V., Snay, E. R., Thedsanamoorthy, J. K., Zurakowski, D., et al. (2016). Intracoronary delivery of mitochondria to the ischemic heart for cardioprotection. PLoS One 11, e0160889. doi:10.1371/journal.pone.0160889
Dafinca, R., Barbagallo, P., Farrimond, L., Candalija, A., Scaber, J., Ababneh, N. A., et al. (2020). Impairment of mitochondrial calcium buffering links mutations in C9ORF72 and TARDBP in iPS-derived motor neurons from patients with ALS/FTD. Stem Cell Rep. 14, 892–908. doi:10.1016/j.stemcr.2020.03.023
David, D. C., Hauptmann, S., Scherping, I., Schuessel, K., Keil, U., Rizzu, P., et al. (2005). Proteomic and functional analyses reveal a mitochondrial dysfunction in P301L tau transgenic mice. J. Biol. Chem. 280, 23802–23814. doi:10.1074/jbc.M500356200
Davis, S. A., Itaman, S., Khalid-Janney, C. M., Sherard, J. A., Dowell, J. A., Cairns, N. J., et al. (2018). TDP-43 interacts with mitochondrial proteins critical for mitophagy and mitochondrial dynamics. Neurosci. Lett. 678, 8–15. doi:10.1016/j.neulet.2018.04.053
De Vrij, F. M., Fischer, D. F., van Leeuwen, F. W., and Hol, E. M. (2004). Protein quality control in Alzheimer's disease by the ubiquitin proteasome system. Prog. Neurobiol. 74, 249–270. doi:10.1016/j.pneurobio.2004.10.001
Del Grosso, A., Galliani, M., Angella, L., Santi, M., Tonazzini, I., Parlanti, G., et al. (2019). Brain-targeted enzyme-loaded nanoparticles: A breach through the blood-brain barrier for enzyme replacement therapy in krabbe disease. Sci. Adv. 5, eaax7462. doi:10.1126/sciadv.aax7462
Dickson, P., Mcentee, M., Vogler, C., Le, S., Levy, B., Peinovich, M., et al. (2007). Intrathecal enzyme replacement therapy: Successful treatment of brain disease via the cerebrospinal fluid. Mol. Genet. Metab. 91, 61–68. doi:10.1016/j.ymgme.2006.12.012
Domercq, M., Alberdi, E., Sanchez-Gomez, M. V., Ariz, U., Perez-Samartin, A., and Matute, C. (2011). Dual-specific phosphatase-6 (Dusp6) and ERK mediate AMPA receptor-induced oligodendrocyte death. J. Biol. Chem. 286, 11825–11836. doi:10.1074/jbc.M110.153049
Doughan, A. K., and Dikalov, S. I. (2007). Mitochondrial redox cycling of mitoquinone leads to superoxide production and cellular apoptosis. Antioxid. Redox Signal 9, 1825–1836. doi:10.1089/ars.2007.1693
Dubey, P., Reddy, S., Boyd, S., Bracamontes, C., Sanchez, S., Chattopadhyay, M., et al. (2021). Effect of nutritional supplementation on oxidative stress and hormonal and lipid profiles in PCOS-affected females. Nutrients 13, 2938. doi:10.3390/nu13092938
Emani, S. M., Piekarski, B. L., Harrild, D., Del Nido, P. J., and Mccully, J. D. (2017). Autologous mitochondrial transplantation for dysfunction after ischemia-reperfusion injury. J. Thorac. Cardiovasc Surg. 154, 286–289. doi:10.1016/j.jtcvs.2017.02.018
English, K., Shepherd, A., Uzor, N. E., Trinh, R., Kavelaars, A., and Heijnen, C. J. (2020). Astrocytes rescue neuronal health after cisplatin treatment through mitochondrial transfer. Acta Neuropathol. Commun. 8, 36. doi:10.1186/s40478-020-00897-7
Escobar-Khondiker, M., Hollerhage, M., Muriel, M. P., Champy, P., Bach, A., Depienne, C., et al. (2007). Annonacin, a natural mitochondrial complex I inhibitor, causes tau pathology in cultured neurons. J. Neurosci. 27, 7827–7837. doi:10.1523/JNEUROSCI.1644-07.2007
Esteras, N., Rohrer, J. D., Hardy, J., Wray, S., and Abramov, A. Y. (2017). Mitochondrial hyperpolarization in iPSC-derived neurons from patients of FTDP-17 with 10+16 MAPT mutation leads to oxidative stress and neurodegeneration. Redox Biol. 12, 410–422. doi:10.1016/j.redox.2017.03.008
Etminan, M., Gill, S. S., and Samii, A. (2005). Intake of vitamin E, vitamin C, and carotenoids and the risk of Parkinson's disease: A meta-analysis. Lancet Neurol. 4, 362–365. doi:10.1016/S1474-4422(05)70097-1
Fang, E. F., Hou, Y., Palikaras, K., Adriaanse, B. A., Kerr, J. S., Yang, B., et al. (2019). Mitophagy inhibits amyloid-beta and tau pathology and reverses cognitive deficits in models of Alzheimer's disease. Nat. Neurosci. 22, 401–412. doi:10.1038/s41593-018-0332-9
Fang, E. F., Scheibye-Knudsen, M., Chua, K. F., Mattson, M. P., Croteau, D. L., and Bohr, V. A. (2016). Nuclear DNA damage signalling to mitochondria in ageing. Nat. Rev. Mol. Cell Biol. 17, 308–321. doi:10.1038/nrm.2016.14
Fatouros, C., Pir, G. J., Biernat, J., Koushika, S. P., Mandelkow, E., Mandelkow, E. M., et al. (2012). Inhibition of tau aggregation in a novel Caenorhabditis elegans model of tauopathy mitigates proteotoxicity. Hum. Mol. Genet. 21, 3587–3603. doi:10.1093/hmg/dds190
Ferrari, R., Kapogiannis, D., Huey, E. D., and Momeni, P. (2011). FTD and ALS: A tale of two diseases. Curr. Alzheimer Res. 8, 273–294. doi:10.2174/156720511795563700
Flinn, L. J., Keatinge, M., Bretaud, S., Mortiboys, H., Matsui, H., De Felice, E., et al. (2013). TigarB causes mitochondrial dysfunction and neuronal loss in PINK1 deficiency. Ann. Neurol. 74, 837–847. doi:10.1002/ana.23999
Flinn, L., Mortiboys, H., Volkmann, K., Koster, R. W., Ingham, P. W., and Bandmann, O. (2009). Complex I deficiency and dopaminergic neuronal cell loss in parkin-deficient zebrafish (Danio rerio). Brain 132, 1613–1623. doi:10.1093/brain/awp108
Foth, B. J., Goedecke, M. C., and Soldati, D. (2006). New insights into myosin evolution and classification. Proc. Natl. Acad. Sci. U. S. A. 103, 3681–3686. doi:10.1073/pnas.0506307103
Fukui, H., Diaz, F., Garcia, S., and Moraes, C. T. (2007). Cytochrome c oxidase deficiency in neurons decreases both oxidative stress and amyloid formation in a mouse model of Alzheimer's disease. Proc. Natl. Acad. Sci. U. S. A. 104, 14163–14168. doi:10.1073/pnas.0705738104
Gabuzda, D., Busciglio, J., Chen, L. B., Matsudaira, P., and Yankner, B. A. (1994). Inhibition of energy metabolism alters the processing of amyloid precursor protein and induces a potentially amyloidogenic derivative. J. Biol. Chem. 269, 13623–13628. doi:10.1016/s0021-9258(17)36875-8
Gao, L., Zhang, Z., Lu, J., and Pei, G. (2019). Mitochondria are dynamically transferring between human neural cells and alexander disease-associated GFAP mutations impair the astrocytic transfer. Front. Cell Neurosci. 13, 316. doi:10.3389/fncel.2019.00316
Garrido-Perez, N., Vela-Sebastian, A., Lopez-Gallardo, E., Emperador, S., Iglesias, E., Meade, P., et al. (2020). Oxidative phosphorylation dysfunction modifies the cell secretome. Int. J. Mol. Sci. 21, 3374. doi:10.3390/ijms21093374
Gerdes, H. H., Bukoreshtliev, N. V., and Barroso, J. F. (2007). Tunneling nanotubes: A new route for the exchange of components between animal cells. FEBS Lett. 581, 2194–2201. doi:10.1016/j.febslet.2007.03.071
Gibbons, I. R., and Rowe, A. J. (1965). Dynein: A protein with adenosine triphosphatase activity from cilia. Science 149, 424–426. doi:10.1126/science.149.3682.424
Grazioli, S., and Pugin, J. (2018). Mitochondrial damage-associated molecular patterns: From inflammatory signaling to human diseases. Front. Immunol. 9, 832. doi:10.3389/fimmu.2018.00832
Greenamyre, J. T., Sherer, T. B., Betarbet, R., and Panov, A. V. (2001). Complex I and Parkinson's disease. IUBMB Life 52, 135–141. doi:10.1080/15216540152845939
Greene, J. C., Whitworth, A. J., Kuo, I., Andrews, L. A., Feany, M. B., and Pallanck, L. J. (2003). Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc. Natl. Acad. Sci. U. S. A. 100, 4078–4083. doi:10.1073/pnas.0737556100
Guay, D. R. (2006). Rasagiline (TVP-1012): A new selective monoamine oxidase inhibitor for Parkinson's disease. Am. J. Geriatr. Pharmacother. 4, 330–346. doi:10.1016/j.amjopharm.2006.12.001
Guo, X., Sun, X., Hu, D., Wang, Y. J., Fujioka, H., Vyas, R., et al. (2016). VCP recruitment to mitochondria causes mitophagy impairment and neurodegeneration in models of Huntington's disease. Nat. Commun. 7, 12646. doi:10.1038/ncomms12646
Hamilton, J., Brustovetsky, T., and Brustovetsky, N. (2017). Oxidative metabolism and Ca(2+) handling in striatal mitochondria from YAC128 mice, a model of Huntington's disease. Neurochem. Int. 109, 24–33. doi:10.1016/j.neuint.2017.01.001
Hamilton, M. L., van Remmen, H., Drake, J. A., Yang, H., Guo, Z. M., Kewitt, K., et al. (2001). Does oxidative damage to DNA increase with age? Proc. Natl. Acad. Sci. U. S. A. 98, 10469–10474. doi:10.1073/pnas.171202698
Haun, F., Nakamura, T., Shiu, A. D., Cho, D. H., Tsunemi, T., Holland, E. A., et al. (2013). S-nitrosylation of dynamin-related protein 1 mediates mutant huntingtin-induced mitochondrial fragmentation and neuronal injury in Huntington's disease. Antioxid. Redox Signal 19, 1173–1184. doi:10.1089/ars.2012.4928
Hayakawa, K., Chan, S. J., Mandeville, E. T., Park, J. H., Bruzzese, M., Montaner, J., et al. (2018). Protective effects of endothelial progenitor cell-derived extracellular mitochondria in brain endothelium. Stem Cells 36, 1404–1410. doi:10.1002/stem.2856
Hayakawa, K., Esposito, E., Wang, X., Terasaki, Y., Liu, Y., Xing, C., et al. (2016). Transfer of mitochondria from astrocytes to neurons after stroke. Nature 535, 551–555. doi:10.1038/nature18928
Hiller, A. L., Murchison, C. F., Lobb, B. M., O'Connor, S., O'Connor, M., and Quinn, J. F. (2018). A randomized, controlled pilot study of the effects of vitamin D supplementation on balance in Parkinson's disease: Does age matter? PLoS One 13, e0203637. doi:10.1371/journal.pone.0203637
Hirokawa, N., Noda, Y., Tanaka, Y., and Niwa, S. (2009). Kinesin superfamily motor proteins and intracellular transport. Nat. Rev. Mol. Cell Biol. 10, 682–696. doi:10.1038/nrm2774
Hogan, K. A., Chini, C. C. S., and Chini, E. N. (2019). The multi-faceted ecto-enzyme CD38: Roles in immunomodulation, cancer, aging, and metabolic diseases. Front. Immunol. 10, 1187. doi:10.3389/fimmu.2019.01187
Hoglinger, G. U., Lannuzel, A., Khondiker, M. E., Michel, P. P., Duyckaerts, C., Feger, J., et al. (2005). The mitochondrial complex I inhibitor rotenone triggers a cerebral tauopathy. J. Neurochem. 95, 930–939. doi:10.1111/j.1471-4159.2005.03493.x
Howard, R., Zubko, O., Bradley, R., Harper, E., Pank, L., O'Brien, J., et al. (2020). Minocycline at 2 different dosages vs placebo for patients with mild alzheimer disease: A randomized clinical trial. JAMA Neurol. 77, 164–174. doi:10.1001/jamaneurol.2019.3762
Hutto, R. A., Rutter, K. M., Giarmarco, M. M., Parker, E. D., Chambers, Z. S., and Brockerhoff, S. E. (2023). Cone photoreceptors transfer damaged mitochondria to Muller glia. Cell Rep. 42, 112115. doi:10.1016/j.celrep.2023.112115
Igoillo-Esteve, M., Oliveira, A. F., Cosentino, C., Fantuzzi, F., Demarez, C., Toivonen, S., et al. (2020). Exenatide induces frataxin expression and improves mitochondrial function in Friedreich ataxia. JCI Insight 5, e134221. doi:10.1172/jci.insight.134221
Investigators, N. N.-P. (2008). A pilot clinical trial of creatine and minocycline in early Parkinson disease: 18-month results. Clin. Neuropharmacol. 31, 141–150. doi:10.1097/WNF.0b013e3181342f32
Investigators, N. N.-P. (2006). A randomized, double-blind, futility clinical trial of creatine and minocycline in early Parkinson disease. Neurology 66, 664–671. doi:10.1212/01.wnl.0000201252.57661.e1
Isobe, C., Abe, T., and Terayama, Y. (2010). Levels of reduced and oxidized coenzyme Q-10 and 8-hydroxy-2'-deoxyguanosine in the cerebrospinal fluid of patients with living Parkinson's disease demonstrate that mitochondrial oxidative damage and/or oxidative DNA damage contributes to the neurodegenerative process. Neurosci. Lett. 469, 159–163. doi:10.1016/j.neulet.2009.11.065
Javadov, S., Chapa-Dubocq, X., and Makarov, V. (2018). Different approaches to modeling analysis of mitochondrial swelling. Mitochondrion 38, 58–70. doi:10.1016/j.mito.2017.08.004
Jiang, D., Gao, F., Zhang, Y., Wong, D. S., Li, Q., Tse, H. F., et al. (2016). Mitochondrial transfer of mesenchymal stem cells effectively protects corneal epithelial cells from mitochondrial damage. Cell Death Dis. 7, e2467. doi:10.1038/cddis.2016.358
Kaza, A. K., Wamala, I., Friehs, I., Kuebler, J. D., Rathod, R. H., Berra, I., et al. (2017). Myocardial rescue with autologous mitochondrial transplantation in a porcine model of ischemia/reperfusion. J. Thorac. Cardiovasc Surg. 153, 934–943. doi:10.1016/j.jtcvs.2016.10.077
Khan, S. M., Cassarino, D. S., Abramova, N. N., Keeney, P. M., Borland, M. K., Trimmer, P. A., et al. (2000). Alzheimer's disease cybrids replicate beta-amyloid abnormalities through cell death pathways. Ann. Neurol. 48, 148–155. doi:10.1002/1531-8249(200008)48:2<148::aid-ana3>3.0.co;2-7
Kim, J., Kim, H. S., and Chung, J. H. (2023). Molecular mechanisms of mitochondrial DNA release and activation of the cGAS-STING pathway. Exp. Mol. Med. 55, 510–519. doi:10.1038/s12276-023-00965-7
Lampinen, R., Belaya, I., Saveleva, L., Liddell, J. R., Rait, D., Huuskonen, M. T., et al. (2022). Neuron-astrocyte transmitophagy is altered in Alzheimer's disease. Neurobiol. Dis. 170, 105753. doi:10.1016/j.nbd.2022.105753
Lee, H. C. (2011). Cyclic ADP-ribose and NAADP: Fraternal twin messengers for calcium signaling. Sci. China Life Sci. 54, 699–711. doi:10.1007/s11427-011-4197-3
Leuner, K., Schutt, T., Kurz, C., Eckert, S. H., Schiller, C., Occhipinti, A., et al. (2012). Mitochondrion-derived reactive oxygen species lead to enhanced amyloid beta formation. Antioxid. Redox Signal 16, 1421–1433. doi:10.1089/ars.2011.4173
Li, H., Wang, C., He, T., Zhao, T., Chen, Y. Y., Shen, Y. L., et al. (2019). Mitochondrial transfer from bone marrow mesenchymal stem cells to motor neurons in spinal cord injury rats via gap junction. Theranostics 9, 2017–2035. doi:10.7150/thno.29400
Liesa, M., and Shirihai, O. S. (2013). Mitochondrial dynamics in the regulation of nutrient utilization and energy expenditure. Cell Metab. 17, 491–506. doi:10.1016/j.cmet.2013.03.002
Liu, T., Woo, J. A., Bukhari, M. Z., Lepochat, P., Chacko, A., Selenica, M. B., et al. (2020a). CHCHD10-regulated OPA1-mitofilin complex mediates TDP-43-induced mitochondrial phenotypes associated with frontotemporal dementia. FASEB J. 34, 8493–8509. doi:10.1096/fj.201903133RR
Liu, Y. T., Huang, X., Nguyen, D., Shammas, M. K., Wu, B. P., Dombi, E., et al. (2020b). Loss of CHCHD2 and CHCHD10 activates OMA1 peptidase to disrupt mitochondrial cristae phenocopying patient mutations. Hum. Mol. Genet. 29, 1547–1567. doi:10.1093/hmg/ddaa077
Llobet, L., Bayona-Bafaluy, M. P., Pacheu-Grau, D., Torres-Perez, E., Arbones-Mainar, J. M., Navarro, M. A., et al. (2017). Pharmacologic concentrations of linezolid modify oxidative phosphorylation function and adipocyte secretome. Redox Biol. 13, 244–254. doi:10.1016/j.redox.2017.05.026
Lopez-Domenech, G., Covill-Cooke, C., Ivankovic, D., Halff, E. F., Sheehan, D. F., Norkett, R., et al. (2018). Miro proteins coordinate microtubule- and actin-dependent mitochondrial transport and distribution. EMBO J. 37, 321–336. doi:10.15252/embj.201696380
Luz-Crawford, P., Hernandez, J., Djouad, F., Luque-Campos, N., Caicedo, A., Carrere-Kremer, S., et al. (2019). Mesenchymal stem cell repression of Th17 cells is triggered by mitochondrial transfer. Stem Cell Res. Ther. 10, 232. doi:10.1186/s13287-019-1307-9
Ma, K., Chen, G., Li, W., Kepp, O., Zhu, Y., and Chen, Q. (2020). Mitophagy, mitochondrial homeostasis, and cell fate. Front. Cell Dev. Biol. 8, 467. doi:10.3389/fcell.2020.00467
Macdonald, R., Barnes, K., Hastings, C., and Mortiboys, H. (2018). Mitochondrial abnormalities in Parkinson's disease and Alzheimer's disease: Can mitochondria be targeted therapeutically? Biochem. Soc. Trans. 46, 891–909. doi:10.1042/BST20170501
Mahad, D. J., Ziabreva, I., Campbell, G., Lax, N., White, K., Hanson, P. S., et al. (2009). Mitochondrial changes within axons in multiple sclerosis. Brain 132, 1161–1174. doi:10.1093/brain/awp046
Manczak, M., and Reddy, P. H. (2015). Mitochondrial division inhibitor 1 protects against mutant huntingtin-induced abnormal mitochondrial dynamics and neuronal damage in Huntington's disease. Hum. Mol. Genet. 24, 7308–7325. doi:10.1093/hmg/ddv429
Mao, P., Manczak, M., Shirendeb, U. P., and Reddy, P. H. (2013). MitoQ, a mitochondria-targeted antioxidant, delays disease progression and alleviates pathogenesis in an experimental autoimmune encephalomyelitis mouse model of multiple sclerosis. Biochim. Biophys. Acta 1832, 2322–2331. doi:10.1016/j.bbadis.2013.09.005
Mao, P., and Reddy, P. H. (2010). Is multiple sclerosis a mitochondrial disease? Biochim. Biophys. Acta 1802, 66–79. doi:10.1016/j.bbadis.2009.07.002
Marie, A., Darricau, M., Touyarot, K., Parr-Brownlie, L. C., and Bosch-Bouju, C. (2021). Role and mechanism of vitamin A metabolism in the pathophysiology of Parkinson's disease. J. Park. Dis. 11, 949–970. doi:10.3233/JPD-212671
Marlein, C. R., Zaitseva, L., Piddock, R. E., Robinson, S. D., Edwards, D. R., Shafat, M. S., et al. (2017). NADPH oxidase-2 derived superoxide drives mitochondrial transfer from bone marrow stromal cells to leukemic blasts. Blood 130, 1649–1660. doi:10.1182/blood-2017-03-772939
Marsh, J., and Alifragis, P. (2018). Synaptic dysfunction in Alzheimer's disease: The effects of amyloid beta on synaptic vesicle dynamics as a novel target for therapeutic intervention. Neural Regen. Res. 13, 616–623. doi:10.4103/1673-5374.230276
Masuzawa, A., Black, K. M., Pacak, C. A., Ericsson, M., Barnett, R. J., Drumm, C., et al. (2013). Transplantation of autologously derived mitochondria protects the heart from ischemia-reperfusion injury. Am. J. Physiol. Heart Circ. Physiol. 304, H966–H982. doi:10.1152/ajpheart.00883.2012
Matthews, R. T., Ferrante, R. J., Klivenyi, P., Yang, L., Klein, A. M., Mueller, G., et al. (1999). Creatine and cyclocreatine attenuate MPTP neurotoxicity. Exp. Neurol. 157, 142–149. doi:10.1006/exnr.1999.7049
Mccully, J. D., Cowan, D. B., Pacak, C. A., Toumpoulis, I. K., Dayalan, H., and Levitsky, S. (2009). Injection of isolated mitochondria during early reperfusion for cardioprotection. Am. J. Physiol. Heart Circ. Physiol. 296, H94–H105. doi:10.1152/ajpheart.00567.2008
Moller, A., Bauer, C. S., Cohen, R. N., Webster, C. P., and De Vos, K. J. (2017). Amyotrophic lateral sclerosis-associated mutant SOD1 inhibits anterograde axonal transport of mitochondria by reducing Miro1 levels. Hum. Mol. Genet. 26, 4668–4679. doi:10.1093/hmg/ddx348
Monteiro, K. L. C., Alcantara, M., De Aquino, T. M., and Da Silva-Junior, E. F. (2020). Tau protein aggregation in Alzheimer's disease: Recent advances in the development of novel therapeutic agents. Curr. Pharm. Des. 26, 1682–1692. doi:10.2174/1381612826666200414164038
Morrison, B. M., Hof, P. R., and Morrison, J. H. (1998). Determinants of neuronal vulnerability in neurodegenerative diseases. Ann. Neurol. 44, S32–S44. doi:10.1002/ana.410440706
Mortiboys, H., Furmston, R., Bronstad, G., Aasly, J., Elliott, C., and Bandmann, O. (2015). UDCA exerts beneficial effect on mitochondrial dysfunction in LRRK2(G2019S) carriers and in vivo. Neurology 85, 846–852. doi:10.1212/WNL.0000000000001905
Mrdjen, D., Fox, E. J., Bukhari, S. A., Montine, K. S., Bendall, S. C., and Montine, T. J. (2019). The basis of cellular and regional vulnerability in Alzheimer's disease. Acta Neuropathol. 138, 729–749. doi:10.1007/s00401-019-02054-4
Nakahira, K., Hisata, S., and Choi, A. M. (2015). The roles of mitochondrial damage-associated molecular patterns in diseases. Antioxid. Redox Signal 23, 1329–1350. doi:10.1089/ars.2015.6407
Nana, A. L., Sidhu, M., Gaus, S. E., Hwang, J. L., Li, L., Park, Y., et al. (2019). Neurons selectively targeted in frontotemporal dementia reveal early stage TDP-43 pathobiology. Acta Neuropathol. 137, 27–46. doi:10.1007/s00401-018-1942-8
Naoi, M., Wu, Y., Shamoto-Nagai, M., and Maruyama, W. (2019). Mitochondria in neuroprotection by phytochemicals: Bioactive polyphenols modulate mitochondrial apoptosis system, function and structure. Int. J. Mol. Sci. 20, 2451. doi:10.3390/ijms20102451
Nassal, D., Gratz, D., and Hund, T. J. (2020). Challenges and opportunities for therapeutic targeting of calmodulin kinase II in heart. Front. Pharmacol. 11, 35. doi:10.3389/fphar.2020.00035
Nitzan, K., Benhamron, S., Valitsky, M., Kesner, E. E., Lichtenstein, M., Ben-Zvi, A., et al. (2019). Mitochondrial transfer ameliorates cognitive deficits, neuronal loss, and gliosis in Alzheimer's disease mice. J. Alzheimers Dis. 72, 587–604. doi:10.3233/JAD-190853
Oeding, S. J., Majstrowicz, K., Hu, X. P., Schwarz, V., Freitag, A., Honnert, U., et al. (2018). Identification of Miro1 and Miro2 as mitochondrial receptors for myosin XIX. J. Cell Sci. 131, jcs219469. doi:10.1242/jcs.219469
Onesto, E., Colombrita, C., Gumina, V., Borghi, M. O., Dusi, S., Doretti, A., et al. (2016). Gene-specific mitochondria dysfunctions in human TARDBP and C9ORF72 fibroblasts. Acta Neuropathol. Commun. 4, 47. doi:10.1186/s40478-016-0316-5
Orozco-Ibarra, M., Garcia-Morales, J., Calvo-Silva, F. J., Fernandez-Valverde, F., and Serrano-Garcia, N. (2018). Striatal mitochondria response to 3-nitropropionic acid and fish oil treatment. Nutr. Neurosci. 21, 132–142. doi:10.1080/1028415X.2016.1237074
Palikaras, K., Lionaki, E., and Tavernarakis, N. (2015). Balancing mitochondrial biogenesis and mitophagy to maintain energy metabolism homeostasis. Cell Death Differ. 22, 1399–1401. doi:10.1038/cdd.2015.86
Pannu, N., and Bhatnagar, A. (2019). Resveratrol: From enhanced biosynthesis and bioavailability to multitargeting chronic diseases. Biomed. Pharmacother. 109, 2237–2251. doi:10.1016/j.biopha.2018.11.075
Park, J. H., Nakamura, Y., Li, W., Hamanaka, G., Arai, K., Lo, E. H., et al. (2021). Effects of O-GlcNAcylation on functional mitochondrial transfer from astrocytes. J. Cereb. Blood Flow. Metab. 41, 1523–1535. doi:10.1177/0271678X20969588
Parker, W. D., Parks, J. K., and Swerdlow, R. H. (2008). Complex I deficiency in Parkinson's disease frontal cortex. Brain Res. 1189, 215–218. doi:10.1016/j.brainres.2007.10.061
Perera, N. D., Sheean, R. K., Scott, J. W., Kemp, B. E., Horne, M. K., and Turner, B. J. (2014). Mutant TDP-43 deregulates AMPK activation by PP2A in ALS models. PLoS One 9, e95549. doi:10.1371/journal.pone.0095549
Peruzzotti-Jametti, L., Bernstock, J. D., Manferrari, G., Rogall, R., Fernandez-Vizarra, E., Williamson, J. C., et al. (2020). Neural stem cells traffic functional mitochondria via extracellular vesicles to correct mitochondrial dysfunction in target cells. bioRxiv, 2020.01.29.923441.
Peruzzotti-Jametti, L., and Pluchino, S. (2018). Targeting mitochondrial metabolism in neuroinflammation: Towards a therapy for progressive multiple sclerosis. Trends Mol. Med. 24, 838–855. doi:10.1016/j.molmed.2018.07.007
Pinho, B. R., Duarte, A. I., Canas, P. M., Moreira, P. I., Murphy, M. P., and Oliveira, J. M. A. (2020). The interplay between redox signalling and proteostasis in neurodegeneration: In vivo effects of a mitochondria-targeted antioxidant in Huntington's disease mice. Free Radic. Biol. Med. 146, 372–382. doi:10.1016/j.freeradbiomed.2019.11.021
Pinto, M., Pickrell, A. M., Fukui, H., and Moraes, C. T. (2013). Mitochondrial DNA damage in a mouse model of Alzheimer's disease decreases amyloid beta plaque formation. Neurobiol. Aging 34, 2399–2407. doi:10.1016/j.neurobiolaging.2013.04.014
Pistono, C., Monti, M. C., Boiocchi, C., Berzolari, F. G., Osera, C., Mallucci, G., et al. (2020). Response to oxidative stress of peripheral blood mononuclear cells from multiple sclerosis patients and healthy controls. Cell Stress Chaperones 25, 81–91. doi:10.1007/s12192-019-01049-0
Pizzino, G., Irrera, N., Cucinotta, M., Pallio, G., Mannino, F., Arcoraci, V., et al. (2017). Oxidative stress: Harms and benefits for human health. Oxid. Med. Cell Longev. 2017, 8416763. doi:10.1155/2017/8416763
Quintero, E. M., Willis, L., Singleton, R., Harris, N., Huang, P., Bhat, N., et al. (2006). Behavioral and morphological effects of minocycline in the 6-hydroxydopamine rat model of Parkinson's disease. Brain Res. 1093, 198–207. doi:10.1016/j.brainres.2006.03.104
Rath, O., and Kozielski, F. (2012). Kinesins and cancer. Nat. Rev. Cancer 12, 527–539. doi:10.1038/nrc3310
Ray, P. D., Huang, B. W., and Tsuji, Y. (2012). Reactive oxygen species (ROS) homeostasis and redox regulation in cellular signaling. Cell Signal 24, 981–990. doi:10.1016/j.cellsig.2012.01.008
Reddy, P. H., and Oliver, D. M. (2019). Amyloid beta and phosphorylated tau-induced defective autophagy and mitophagy in Alzheimer's disease. Cells 8, 488. doi:10.3390/cells8050488
Ren, D., Zheng, P., Zou, S., Gong, Y., Wang, Y., Duan, J., et al. (2022). GJA1-20K enhances mitochondria transfer from astrocytes to neurons via cx43-TnTs after traumatic brain injury. Cell Mol. Neurobiol. 42, 1887–1895. doi:10.1007/s10571-021-01070-x
Ren, J., Yuan, L., Wang, W., Zhang, M., Wang, Q., Li, S., et al. (2019). Tricetin protects against 6-OHDA-induced neurotoxicity in Parkinson's disease model by activating Nrf2/HO-1 signaling pathway and preventing mitochondria-dependent apoptosis pathway. Toxicol. Appl. Pharmacol. 378, 114617. doi:10.1016/j.taap.2019.114617
Riley, J. S., and Tait, S. W. (2020). Mitochondrial DNA in inflammation and immunity. EMBO Rep. 21, e49799. doi:10.15252/embr.201949799
Sanders, L. H., Mccoy, J., Hu, X., Mastroberardino, P. G., Dickinson, B. C., Chang, C. J., et al. (2014). Mitochondrial DNA damage: Molecular marker of vulnerable nigral neurons in Parkinson's disease. Neurobiol. Dis. 70, 214–223. doi:10.1016/j.nbd.2014.06.014
Sandoval-Acuna, C., Ferreira, J., and Speisky, H. (2014). Polyphenols and mitochondria: An update on their increasingly emerging ROS-scavenging independent actions. Arch. Biochem. Biophys. 559, 75–90. doi:10.1016/j.abb.2014.05.017
Sata, M., Stafford, W. F., 3R. D., Mabuchi, K., and Ikebe, M. (1997). The motor domain and the regulatory domain of myosin solely dictate enzymatic activity and phosphorylation-dependent regulation, respectively. Proc. Natl. Acad. Sci. U. S. A. 94, 91–96. doi:10.1073/pnas.94.1.91
Sato, Y., and Okuyama, T. (2020). Novel enzyme replacement therapies for neuropathic mucopolysaccharidoses. Int. J. Mol. Sci. 21, 400. doi:10.3390/ijms21020400
Schagger, H. (1995). Quantification of oxidative phosphorylation enzymes after blue native electrophoresis and two-dimensional resolution: normal complex I protein amounts in Parkinson's disease conflict with reduced catalytic activities. Electrophoresis 16, 763–770. doi:10.1002/elps.11501601125
Schapira, A. H., Cooper, J. M., Dexter, D., Clark, J. B., Jenner, P., and Marsden, C. D. (1990). Mitochondrial complex I deficiency in Parkinson's disease. J. Neurochem. 54, 823–827. doi:10.1111/j.1471-4159.1990.tb02325.x
Selfridge, J. E., E, L., Lu, J., and Swerdlow, R. H. (2013). Role of mitochondrial homeostasis and dynamics in Alzheimer's disease. Neurobiol. Dis. 51, 3–12. doi:10.1016/j.nbd.2011.12.057
Shi, X., Zhao, M., Fu, C., and Fu, A. (2017). Intravenous administration of mitochondria for treating experimental Parkinson's disease. Mitochondrion 34, 91–100. doi:10.1016/j.mito.2017.02.005
Shults, C. W., Oakes, D., Kieburtz, K., Beal, M. F., Haas, R., Plumb, S., et al. (2002). Effects of coenzyme Q10 in early Parkinson disease: Evidence of slowing of the functional decline. Arch. Neurol. 59, 1541–1550. doi:10.1001/archneur.59.10.1541
Singh, A., Faccenda, D., and Campanella, M. (2021). Pharmacological advances in mitochondrial therapy. EBioMedicine 65, 103244. doi:10.1016/j.ebiom.2021.103244
Smith, E. F., Shaw, P. J., and De Vos, K. J. (2019). The role of mitochondria in amyotrophic lateral sclerosis. Neurosci. Lett. 710, 132933. doi:10.1016/j.neulet.2017.06.052
Snow, B. J., Rolfe, F. L., Lockhart, M. M., Frampton, C. M., O'Sullivan, J. D., Fung, V., et al. (2010). A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson's disease. Mov. Disord. 25, 1670–1674. doi:10.1002/mds.23148
Solan, J. L., and Lampe, P. D. (2016). Kinase programs spatiotemporally regulate gap junction assembly and disassembly: Effects on wound repair. Semin. Cell Dev. Biol. 50, 40–48. doi:10.1016/j.semcdb.2015.12.010
Sturm, G., Karan, K. R., Monzel, A. S., Santhanam, B., Taivassalo, T., Bris, C., et al. (2023). OxPhos defects cause hypermetabolism and reduce lifespan in cells and in patients with mitochondrial diseases. Commun. Biol. 6, 22. doi:10.1038/s42003-022-04303-x
Tammineni, P., Ye, X., Feng, T., Aikal, D., and Cai, Q. (2017). Impaired retrograde transport of axonal autophagosomes contributes to autophagic stress in Alzheimer's disease neurons. Elife 6, e21776. doi:10.7554/eLife.21776
Tan, W., Pasinelli, P., and Trotti, D. (2014). Role of mitochondria in mutant SOD1 linked amyotrophic lateral sclerosis. Biochim. Biophys. Acta 1842, 1295–1301. doi:10.1016/j.bbadis.2014.02.009
Tilokani, L., Nagashima, S., Paupe, V., and Prudent, J. (2018). Mitochondrial dynamics: Overview of molecular mechanisms. Essays Biochem. 62, 341–360. doi:10.1042/EBC20170104
Tseng, N., Lambie, S. C., Huynh, C. Q., Sanford, B., Patel, M., Herson, P. S., et al. (2021). Mitochondrial transfer from mesenchymal stem cells improves neuronal metabolism after oxidant injury in vitro: The role of Miro1. J. Cereb. Blood Flow. Metab. 41, 761–770. doi:10.1177/0271678X20928147
Urnavicius, L., Zhang, K., Diamant, A. G., Motz, C., Schlager, M. A., Yu, M., et al. (2015). The structure of the dynactin complex and its interaction with dynein. Science 347, 1441–1446. doi:10.1126/science.aaa4080
Utton, M. A., Noble, W. J., Hill, J. E., Anderton, B. H., and Hanger, D. P. (2005). Molecular motors implicated in the axonal transport of tau and alpha-synuclein. J. Cell Sci. 118, 4645–4654. doi:10.1242/jcs.02558
Valentine, M. L., Cardenas, A. E., Elber, R., and Baiz, C. R. (2018). Physiological calcium concentrations slow dynamics at the lipid-water interface. Biophys. J. 115, 1541–1551. doi:10.1016/j.bpj.2018.08.044
van der Vlist, M., Raoof, R., Willemen, H., Prado, J., Versteeg, S., Martin Gil, C., et al. (2022). Macrophages transfer mitochondria to sensory neurons to resolve inflammatory pain. Neuron 110, 613–626 e9. doi:10.1016/j.neuron.2021.11.020
Wang, W., Wang, L., Lu, J., Siedlak, S. L., Fujioka, H., Liang, J., et al. (2016). The inhibition of TDP-43 mitochondrial localization blocks its neuronal toxicity. Nat. Med. 22, 869–878. doi:10.1038/nm.4130
Wang, Y., Ni, J., Gao, T., Gao, C., Guo, L., and Yin, X. (2020). Activation of astrocytic sigma-1 receptor exerts antidepressant-like effect via facilitating CD38-driven mitochondria transfer. Glia 68, 2415–2426. doi:10.1002/glia.23850
Watts, M. E., Pocock, R., and Claudianos, C. (2018). Brain energy and oxygen metabolism: Emerging role in normal function and disease. Front. Mol. Neurosci. 11, 216. doi:10.3389/fnmol.2018.00216
Wei, W., Graeff, R., and Yue, J. (2014). Roles and mechanisms of the CD38/cyclic adenosine diphosphate ribose/Ca(2+) signaling pathway. World J. Biol. Chem. 5, 58–67. doi:10.4331/wjbc.v5.i1.58
Wong, Y. C., and Holzbaur, E. L. (2014). Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc. Natl. Acad. Sci. U. S. A. 111, E4439–E4448. doi:10.1073/pnas.1405752111
Xing, C., and Lo, E. H. (2017). Help-me signaling: Non-cell autonomous mechanisms of neuroprotection and neurorecovery. Prog. Neurobiol. 152, 181–199. doi:10.1016/j.pneurobio.2016.04.004
Yin, X., Manczak, M., and Reddy, P. H. (2016). Mitochondria-targeted molecules MitoQ and SS31 reduce mutant huntingtin-induced mitochondrial toxicity and synaptic damage in Huntington's disease. Hum. Mol. Genet. 25, 1739–1753. doi:10.1093/hmg/ddw045
Yue, M., Hinkle, K. M., Davies, P., Trushina, E., Fiesel, F. C., Christenson, T. A., et al. (2015). Progressive dopaminergic alterations and mitochondrial abnormalities in LRRK2 G2019S knock-in mice. Neurobiol. Dis. 78, 172–195. doi:10.1016/j.nbd.2015.02.031
Zhang, Z., Ma, Z., Yan, C., Pu, K., Wu, M., Bai, J., et al. (2019). Muscle-derived autologous mitochondrial transplantation: A novel strategy for treating cerebral ischemic injury. Behav. Brain Res. 356, 322–331. doi:10.1016/j.bbr.2018.09.005
Zhao, Y. J., Lam, C. M., and Lee, H. C. (2012). The membrane-bound enzyme CD38 exists in two opposing orientations. Sci. Signal 5, ra67. doi:10.1126/scisignal.2002700
Zhemkov, V., Ditlev, J. A., Lee, W. R., Wilson, M., Liou, J., Rosen, M. K., et al. (2021). The role of sigma 1 receptor in organization of endoplasmic reticulum signaling microdomains. Elife 10, e65192. doi:10.7554/eLife.65192
Keywords: degenerative brain disorders, mitochondrial dysfunction, fission and fusion, mitophagy, oxidative damage, mitochondrial transfer, cellular therapy, enzymes
Citation: Luque-Campos N, Riquelme R, Molina L, Canedo-Marroquín G, Vega-Letter AM, Luz-Crawford P and Bustamante-Barrientos FA (2023) Exploring the therapeutic potential of the mitochondrial transfer-associated enzymatic machinery in brain degeneration. Front. Physiol. 14:1217815. doi: 10.3389/fphys.2023.1217815
Received: 10 May 2023; Accepted: 12 July 2023;
Published: 28 July 2023.
Edited by:
Ru Huang, Hainan University, ChinaReviewed by:
Agnese De Mario, University of Padua, ItalyCopyright © 2023 Luque-Campos, Riquelme, Molina, Canedo-Marroquín, Vega-Letter, Luz-Crawford and Bustamante-Barrientos. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Patricia Luz-Crawford, cGF0cmljaWEubHV6Y0BnbWFpbC5jb20=; Felipe A. Bustamante-Barrientos, ZmJ1c3RhbWFudGUuMTk5MEBnbWFpbC5jb20=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.