
94% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol., 23 June 2023
Sec. Cell Physiology
Volume 14 - 2023 | https://doi.org/10.3389/fphys.2023.1179315
This article is part of the Research TopicRole of angiotensin-converting enzyme in myeloid immune functionsView all 6 articles
 Ron Danziger1,2
Ron Danziger1,2 Dieu-Trang Fuchs1
Dieu-Trang Fuchs1 Yosef Koronyo1
Yosef Koronyo1 Altan Rentsendorj1Julia Sheyn1
Altan Rentsendorj1Julia Sheyn1 Eric Y. Hayden3
Eric Y. Hayden3 David B. Teplow3
David B. Teplow3 Keith L. Black1
Keith L. Black1 Sebastien Fuchs4Kenneth E. Bernstein5,6
Sebastien Fuchs4Kenneth E. Bernstein5,6 Maya Koronyo-Hamaoui1,2,5*
Maya Koronyo-Hamaoui1,2,5*This review examines the role of angiotensin-converting enzyme (ACE) in the context of Alzheimer’s disease (AD) and its potential therapeutic value. ACE is known to degrade the neurotoxic 42-residue long alloform of amyloid β-protein (Aβ42), a peptide strongly associated with AD. Previous studies in mice, demonstrated that targeted overexpression of ACE in CD115+ myelomonocytic cells (ACE10 models) improved their immune responses to effectively reduce viral and bacterial infection, tumor growth, and atherosclerotic plaque. We further demonstrated that introducing ACE10 myelomonocytes (microglia and peripheral monocytes) into the double transgenic APPSWE/PS1ΔE9 murine model of AD (AD+ mice), diminished neuropathology and enhanced the cognitive functions. These beneficial effects were dependent on ACE catalytic activity and vanished when ACE was pharmacologically blocked. Moreover, we revealed that the therapeutic effects in AD+ mice can be achieved by enhancing ACE expression in bone marrow (BM)-derived CD115+ monocytes alone, without targeting central nervous system (CNS) resident microglia. Following blood enrichment with CD115+ ACE10-monocytes versus wild-type (WT) monocytes, AD+ mice had reduced cerebral vascular and parenchymal Aβ burden, limited microgliosis and astrogliosis, as well as improved synaptic and cognitive preservation. CD115+ ACE10-versus WT-monocyte-derived macrophages (Mo/MΦ) were recruited in higher numbers to the brains of AD+ mice, homing to Aβ plaque lesions and exhibiting a highly Aβ-phagocytic and anti-inflammatory phenotype (reduced TNFα/iNOS and increased MMP-9/IGF-1). Moreover, BM-derived ACE10-Mo/MΦ cultures had enhanced capability to phagocytose Aβ42 fibrils, prion-rod-like, and soluble oligomeric forms that was associated with elongated cell morphology and expression of surface scavenger receptors (i.e., CD36, Scara-1). This review explores the emerging evidence behind the role of ACE in AD, the neuroprotective properties of monocytes overexpressing ACE and the therapeutic potential for exploiting this natural mechanism for ameliorating AD pathogenesis.
A hallmark of early pathological changes in Alzheimer’s Disease (AD) is accumulation of amyloid β-protein (Aβ) in the brain and retina (Selkoe, 2001; Hardy and Selkoe, 2002; Selkoe, 2008; Shankar et al., 2008; Koronyo-Hamaoui et al., 2011; La Morgia et al., 2016; Koronyo et al., 2017; Shi et al., 2020; Koronyo et al., 2023). The amyloidogenic 42-residue long alloform (Aβ42) is particularly important due to its ability to rapidly assemble into highly synaptotoxic soluble oligomers as well as aggregate into insoluble fibrils and form plaques (Li et al., 2020). In particular, the Aβ42 oligomers (oAβ42) exert downstream effects on cell signaling pathways that ultimately result in functional-structural synaptic and neuronal abnormalities (Shankar et al., 2007; Li et al., 2020). One potential mechanism for this is via the binding of oligomeric Aβ to the cellular prion protein receptor (PrPC), which forms a complex with the metabotropic glutamate receptor 5 (mGluR5) at the cell surface. This subsequently results in synergistic co-activation of Pyk2 (Protein rich tyrosine kinase 2) and Fyn (Fyn proto-oncogene, Src Family Tyrosine Kinase). Fyn then phosphorylates tau which results in neuronal dysfunction (Brody and Strittmatter, 2018). Another proposed mechanism implicates the N-methyl-D-aspartate (NMDAR) type glutamate receptor, which at low frequency synaptic stimulation enhances long term depression (LTD) via a calcineurin/cofilin dependent pathway that leads to spine retraction. In experimental models, soluble Aβ enhances this pathway and ultimately leads to dendritic spine loss (Shankar et al., 2007). It has also been shown that accumulation of Aβ42 may lead to reactive oxygen species (ROS) generation (Cenini et al., 2010). One proposed mechanism is that Aβ42 activates caspase signaling pathways in cerebral cortical neurons and leads to upregulation of pathways involved in mitochondrial fission. This also leads to the reduction of mitochondrial membrane potentials resulting in increased ROS generation with subsequent activation of mitophagy and eventual neuronal apoptosis (Han et al., 2017). These cellular pathways, together with other pathological processes such as chronic neuroinflammation and vascular deficiencies are hypothesized to reduce synaptic plasticity and result in the debilitating cognitive deficits that characterize this devastating illness (Walsh et al., 2002; Shankar and Walsh, 2009; Tu et al., 2014; Umeda et al., 2015; Yu et al., 2019). Given the injurious role that soluble Aβ42 oligomers play in AD pathophysiology, there has been considerable effort to target and remove them as a strategy to prevent disease progression (Viola and Klein, 2015; Honig et al., 2018; Alexander et al., 2021; Mintun et al., 2021).
Aβ peptides are catabolized by peptidases that can convert these peptides to shorter, less pathologically active forms (Mukherjee et al., 2000; Kanemitsu et al., 2003; Hemming and Selkoe, 2005; Yin et al., 2006). One of which is angiotensin-converting enzyme (ACE), a zinc dipeptide carboxypeptidase containing two proteolytically active homologous regions, designated the N- and C-domains (Wei et al., 1991; Bernstein et al., 2013). ACE is expressed ubiquitously by endothelium as well as cortical neurons and is well known for its role in conversion of angiotensin II to angiotensin I and maintenance of hemodynamic stability (Kehoe et al., 2009). ACE can also be expressed by circulating monocytes, tissue macrophages and microglia (Bernstein et al., 2014; Cao et al., 2020a). On monocytes, most ACE is membrane bound but some can be released into the extracellular space by protease activity at the cell surface (Wei et al., 1991; Bernstein et al., 2013). There now exists evidence that ACE may play a pivotal role in cleavage of Aβ by promoting degradation of naturally occurring Aβ42 and Aβ40 peptides (Hemming and Selkoe, 2005) and the conversion of Aβ42 into shorter, less pathogenic alloforms (Zou et al., 2007), and thus has a protective role against the accumulation of synaptotoxic and neurotoxic oligomeric and fibril forms. More recent studies have demonstrated that macrophages that express higher levels of ACE degrade extracellular Aβ42 faster and more efficiently than WT macrophages (Koronyo-Hamaoui et al., 2020).
In addition, there is emerging evidence that certain microglial subtypes, and moreover peripherally-derived monocytes and macrophages (Mo/MΦ), once believed to have purely detrimental effects in AD pathophysiology, now may play a neuroprotective role, home to sites of amyloid deposition and are crucial in modulating chronic inflammation and facilitating Aβ phagocytosis and degradation (Butovsky et al., 2006; Simard et al., 2006; Butovsky et al., 2007; Deane et al., 2009; Koronyo-Hamaoui et al., 2009; Marques et al., 2009; El Khoury, 2010; Lebson et al., 2010; Michaud et al., 2013; Koronyo et al., 2015; Keren-Shaul et al., 2017; Zuroff et al., 2017; Rentsendorj et al., 2018; Doustar et al., 2020; Koronyo-Hamaoui et al., 2020; Dvir-Szternfeld et al., 2022). Enhanced expression of ACE in CD115+ myelomonocytic cells (ACE10 or ACE10/10 model) improve their ability to mediate an effective immune response to remove viral and bacterial infections, cancer growth, and atherosclerotic plaques (Shen et al., 2007; Okwan-Duodu et al., 2010). Importantly, it has been demonstrated that targeted overexpression of ACE in CD115+ myelomonocytic cells (i.e., monocytes, macrophages, microglia) in the double-transgenic APPSWE/PS1ΔE9 murine models of AD (AD+ mice), increased cerebral Aβ clearance, curbed neuroinflammation, and strikingly, preserved memory and learning functions (Bernstein et al., 2014). These effects were dependent on ACE catalytic activity, which was lost when ACE was pharmacologically blocked by the ACE inhibitor ramipril. Notably, adoptive transfer of young WT, and moreover, ACE10/10 bone marrow (BM)-derived CD115+ monocytes to the peripheral blood of AD+ mice, enhanced their recruitment to cerebral Aβ lesion sites, reduced parenchymal and vascular amyloidosis, and restored synaptic integrity (Koronyo-Hamaoui et al., 2020). These ACE10/10 monocytes exhibited neuroprotective properties with improved phagocytosis of Aβ42 fibrils and oligomers as well as reduced production of pro-inflammatory cytokines such as tumor necrosis factor α (TNFα) and inducible nitric oxide synthase (iNOS), with elevated production of matrix metallopeptidase 9 (MMP-9) and neurotrophic Insulin-like growth factor 1 (IGF-1). Consequently, these AD+ mice had a marked reduction in brain amyloid plaque area and soluble Aβ42 levels, which was reflected in improved cognitive testing and preserved spatial memory and learning capacity.
The mechanism by which ACE expression regulates the secretion of these cytokines is not well understood but there is emerging evidence that ACE overexpression aids in metabolic reprograming associated with the polarization of not only monocytes, but all myeloid cells. ACE enhances the metabolic state of these cells by increasing ATP, Krebs cycle intermediates, and electron transport chain proteins (Cao et al., 2020b). ACE also has an important function in antigen presenting cells (APCs) as it functions as a peptidase involved in peptide trimming and enhances major histocompatibility complex (MHC) class 1 epitope presentation. Furthermore, ACE changes MHC class II presentation of peptide epitopes via unclear mechanisms hence driving CD4 differentiation towards different phenotypes which in turn alters the cytokine profile present in the inflammatory milieu (Bernstein et al., 2018).
This review explores the emerging evidence behind the role of ACE in AD, the neuroprotective properties of monocytes overexpressing ACE and the therapeutic potential for exploiting this natural mechanism for amyloid clearance.
There now exists extensive histological, biochemical and genetic evidence for the role of ACE in AD. In an analysis of parenchymal tissue by quantitative immunofluorescence, frontal cortex samples from patients with AD without cerebral amyloid angiopathy (CAA) were compared to those with more severe CAA as well as controls (Miners et al., 2008). In AD patients, neuronal and perivascular ACE expression was increased and correlated with parenchymal Aβ levels in a statistically significant way. Perivascular ACE levels were elevated in patients with CAA in particular. This suggests that ACE may be upregulated in cases of increased Aβ deposition as its activity confers an important role in amyloid degradation. It also raises the possibility that this natural mechanism of raising ACE in response to Aβ is not sufficient to clear vascular Aβ in AD patients and requires further enhancement. In another postmortem analysis, ACE levels but not activity was reduced in the cerebrospinal fluid (CSF) of AD patients and correlated with increased Braak staging, a marker of topographic progression of AD (Miners et al., 2009). These findings are thought-provoking as increasing Braak staging denotes neurofibrillary tangle involvement progressing through the transentorhinal, limbic and then neocortical regions (Braak and Braak, 1991). Increased Braak staging has been correlated with the degree and severity of memory decline (Grober et al., 1999). As AD progresses through the brain, ACE activity may increase as a protective mechanism to promote amyloid clearance. This was shown by CSF studies in living patients that demonstrated increased ACE activity in patients with mild cognitive impairment (MCI) and AD when compared to healthy controls (He et al., 2006; Jochemsen et al., 2014).
While these studies show a correlation between ACE levels and disease severity, they do not solidify a causal relationship between ACE activity and amyloid clearance. There are indeed alternate hypotheses that suggest the renin angiotensin aldosterone system (RAAS) plays a key role in neuroinflammation and cerebrovascular attenuation and may itself be implicated in the pathogenesis of AD (Mateos et al., 2011; Kehoe et al., 2019). This could imply that elevated ACE in the CSF is merely a byproduct of CNS RAAS upregulation in AD rather than ACE itself being selectively upregulated to clear amyloidogenic protein. However, Genome Wide Association Studies (GWAS) indicate ACE as risk factor for AD (Meng et al., 2006; Marioni et al., 2019; Xin et al., 2021) and GWAS data on ACE polymorphisms show a correlation between low ACE activity and increased AD severity (Meng et al., 2006; Marioni et al., 2019). In one study of living patients with subjective memory complaints or diagnosed AD, ACE protein levels as well as Aβ42 levels were measured in the CSF. Both ACE protein levels and activity were lower in the CSF of patients with diagnosed AD. Lower CSF ACE protein levels were associated with lower CSF Aβ42 levels which suggests more accumulation of Aβ in the brain parenchyma (Jochemsen et al., 2014). Again, from this study, it can be postulated that ACE levels and activity are directly correlated with amyloid clearance.
Given the discussion so far, ACE is functionally able to degrade pathogenic amyloid protein and human tissue studies demonstrate that ACE may serve a protective role in combatting cerebral Aβ accumulation. It stands to reason that if this mechanism is deranged by decreasing ACE activity in the brain, the amyloidogenic proteins may be able to freely accumulate. Perhaps a genetic aberration may result in this derangement. The ACE gene is located on chromosome 17q23. Several studies have attempted to elucidate ACE gene polymorphisms that result in increased susceptibility to AD. One large recent meta-analysis of 65 studies involving 82 cohorts of differing ethnic composition and comprising of 47,000 genotyped cases and controls implicated the rs1799752 polymorphisms with susceptibility for development of AD (Xin et al., 2021). This is the insertion/deletion (I/D) variant of 287-bp intron 16 and is the most common polymorphism of the ACE gene. Carriers of the D allele have higher levels of plasma ACE compared to I homozygotes and almost 50% of variability in plasma ACE levels are dependent on this polymorphism in the human population. When analyzing association of each allele with risk of AD, the I allele was shown to confer an increased susceptibility to AD compared to the D allele. However, an ethnic difference was also noted with a significant association only found in North European populations but not in East Asian populations or those of South European descent. Other meta-analysis studies have implicated other insertion/deletion variants of intron 16 that may reduce plasma levels of ACE and contribute to pathogenesis of disease (Lehmann et al., 2005; Bertram et al., 2007). For example, GWAS analysis has revealed that the single nucleotide combination of rs4343 and rs4351, two adjacent polymorphisms in the DCP1/ACE gene that are linked to the same Insertion variant, may result in up to a 45-fold increased risk of developing AD (Meng et al., 2006; Marioni et al., 2019).
It is crucial to consider more broadly the role of microglia and monocytes in AD pathophysiology before launching into a discussion of the important role of monocytes overexpressing ACE. While traditionally conceptualized as a purely neurodegenerative disease, recent advances have demonstrated the neuroinflammatory basis of AD, which predominantly includes activated brain-resident microglia and reactive astrocytes but also infiltrating cells from the peripheral immune system and the subsequent pro-inflammatory milieu and neuronal damage that ensues (Prinz and Priller, 2017; Rossi et al., 2021). Systemic inflammation may contribute to the neuroinflammation implicated in the neurodegenerative cascade of AD (Zhang et al., 2013). Additionally, microglia may be primed by the toxic environment in the AD brain to then differentiate towards a more maladaptive inflammatory phenotype (Cunningham, 2013). Microglia exert these detrimental effects by aggregating around Aβ plaques and secreting high levels of pro-inflammatory cytokines such as TNFα and IL-6 which result in significant neurotoxicity (Xu et al., 2016).
While monocytes and microglia can have the aforementioned deleterious effects in AD pathophysiology, there is emerging evidence for the neuroprotective and anti-inflammatory roles that subpopulations of these pleomorphic cells may have (Morgan et al., 2005; Koronyo-Hamaoui et al., 2009; Lebson et al., 2010; Michaud et al., 2013; Koronyo et al., 2015; Rentsendorj et al., 2018; Li et al., 2020). Recent in vivo imaging studies have demonstrated the capacity for circulating peripheral immune cells to directly clear cerebral Aβ (Michaud et al., 2013). Using two photon microscopy in murine models of AD it was demonstrated that patrolling Ly6Clo monocytes crawl on the luminal walls of Aβ-positive veins, phagocytose Aβ and then circulate back into the bloodstream. This finding is particularly important as CAA is critical in AD pathogenesis and is correlated with worsening cognition (Charidimou et al., 2017). Perivascular and parenchymal Aβ exist in a state of equilibrium. Hence reduction of perivascular Aβ may result in an efflux of parenchymal Aβ to restore this equilibrium. This could potentially result in a decreased burden of parenchymal Aβ and contribute to reduced neurodegeneration (Deane et al., 2009; Marques et al., 2009; Pimentel-Coelho and Rivest, 2012).
In a 2015 study the ways in which peripheral monocytes can be harnessed for Aβ clearance was explored (Koronyo et al., 2015). It was shown that weekly immunization of AD+ mice with glatiramer acetate (GA) resulted in enhanced recruitment of peripheral monocytes to Aβ plaques, decreased burden (45%–50% reduction) of these plaques and retained cognition. In this experiment CD115+- monocytes were injected once monthly into the peripheral blood of AD+ mice with or without weekly GA injection and compared to a cohort treated with GA injection alone as well as an age matched control. Every treatment group exhibited a substantial decrease in cognitive deficits assessed by Barnes Maze testing, increased synaptic perseveration, and marked reduction in cerebral Aβ levels and astrogliosis. Fluorescently labelled peripherally engrafted monocytes were localized around cerebral amyloid plaques and engulfed Aβ. This effect was enhanced in the GA immunized mice and was associated with increased IL-10 and MMP-9 levels. Further in vitro studies using cultured BM demonstrated that GA treated macrophages could phagocytose fibrillar Aβ42 (fAβ42) and exhibited increased expression of scavenger receptors CD36, SCARA1 and MMP9, all of which has been shown to facilitate Aβ phagocytosis (Coraci et al., 2002; Yin et al., 2006; Frenkel et al., 2013). These results were significant as they indicate that peripheral monocytes, even without ACE overexpression, play a critical role in phagocytosis and degradation of pathogenic Aβ that may result in synaptic preservation, reduced astrogliosis and cognitive improvement.
In another study, the specific mechanisms by which BM-derived macrophages protected synapses against pathogenic Aβ was explored (Li et al., 2020). Firstly, it was found that oAβ42, more-so than fAβ42 inhibited neuritic arborization, induced loss of excitatory VGluT1/PSD95 synapses and caused functional alterations in neurons. When co-cultured with fAβ42 and oAβ42, BM-derived macrophages were able to remove these pathogenic species. Fibrils were cleared via CD36/EEA1+ early endosomal proteolysis while oligomers were removed via extracellular/MMP-9 enzymatic degradation. In vivo analysis revealed enhanced entorhinal cortex and hippocampal preservation of excitatory VGluT1/PSD95 synapses in GA immunized and CD115+-monocyte-grafted AD+ mice compared to AD+ mice control. A 2022 study using genetic fate-mapping demonstrated that 6% of amyloid plaque associated macrophages are derived from circulating hematopoietic originating monocytes rather than brain native microglia which are yolk sac derived (Yan et al., 2022). Additionally peripheral monocytes enriched the choroid plexus, meninges and perivascular spaces more so in aged AD mice than WT control. Splenectomy reduced circulating monocytes, thereby reducing amyloid plaque associated macrophages of a peripheral origin and resulted in increased amyloid plaque burden. Thus, despite making up a small amount of plaque associated myelomonocytes, those of a peripheral origin clearly play a significant role in tempering AD pathogenesis.
Microglia too, can have a protective effect in AD. A recent study using single cell transcriptomic techniques uncovered a rare microglia sub-population termed disease associated microglia (DAM), involved in restricting cerebral Aβ plaque formation. Histologically these DAM cells were frequently located near Aβ plaques in both human and mice brains. Immunohistochemical staining of both mice and human AD brains show DAMs with intracellular phagocytosed Aβ, suggesting their ability to absorb Aβ peptides and a protective role in AD. In normal physiology, DAM activation is tightly controlled via checkpoint mediated inhibition. Regulation occurs via a 2-step process. In the first step a Trem2-independent process occurs which involves reduced expression of homeostatic microglia checkpoint genes such as Cx3cr1 and P2ry12/P2ry12 and upregulation of B2m, Apoe and Tyrobp. This intermediate DAM is then further activated by a Trem2-dependent process involving regulation of phagocytic and lipid metabolism genes such as CSt7 and Lpl. It is feasible that the inhibition of some of these microglia specific inhibitory checkpoints (such as Cx3cr1) which in turn disinhibits DAM activation could prove to be an important therapeutic target in the future (Keren-Shaul et al., 2017).
A parallel study identified a similar subpopulation of microglia, termed neurodegeneration-associated microglia (MGnD) controlled by TREM2-APOE pathway and post-transcriptionally regulated by microRNA(miR)-155 in the proximity of plaques, implying their detrimental role in neurodegeneration (Krasemann et al., 2017). Ultimately further studies are needed into these DAM or MGnD microglia, particularly when it comes to the inciting triggers in the AD brain microenvironment that skew microglia towards either of these unique phenotypes.
As discussed, ACE is an important zinc peptidase capable of degrading Aβ. In addition, microglia and monocytes play a crucial inflammatory role in AD and themselves are capable of clearing Aβ aggregates via mechanisms involving cellular uptake and enzymatic degradation. It is apparent that even in the absence of ACE overexpression, monocytes play an important role in mitigating AD pathology. Prior studies have demonstrated that the adoptive transfer of BM-derived CD115+ monocyte subsets (WT for ACE expression) from young donor mice (8–10 weeks old) into 10-month-old AD+ mice leads to diminished neuropathology and preserved cognitive function (Koronyo et al., 2015).
In humans and mice, ACE is expressed on monocytes and is naturally upregulated in multiple disease processes. In particular, macrophages and myeloid derived giant cells express abundant ACE in many granulomatous diseases (for example,: sarcoidosis, leprosy, tuberculosis, histoplasmosis and Wegener’s granulomatosis) (Lieberman, 1975; Williams and Williams, 1983; Brice et al., 1995; Baudin, 2002; Bernstein et al., 2018). Hence it was long postulated that ACE may improve the antibacterial effectiveness of macrophages. In mice, ACE expression in neutrophils, macrophages and dendritic cells rapidly increases following Staphylococcus aureus or Listeria infection (Okwan-Duodu et al., 2010). ACE is also upregulated in non-infectious disease processes such as atherosclerotic disease. Macrophages identified in both early and late-stage human atherosclerotic material were found to produce abundant ACE (Diet et al., 1996; Ohishi et al., 1997). It is now known that ACE overexpression not only improves bacterial defenses but actually enhances the adaptive and innate immune response via a diverse array of mechanisms. This includes not only improved antibacterial defenses but also antigen processing, enhanced antitumor responses and improved antibody production (Bernstein et al., 2013; Bernstein et al., 2018; Koronyo-Hamaoui et al., 2014; Cao et al., 2023).
Further studies have explored the unique immunomodulatory functions of myelomonocytes overexpressing ACE in the context of AD and discovered that these cells possess uniquely enhanced neuroprotective properties and an enhanced capacity to clear Aβ (Bernstein et al., 2014). This phenomenon has been examined using the ACE10 mouse model. In this model, gene targeting is used to inactivate the intrinsic ACE promoter and subsequently place ACE expression under the c-fms promoter. ACE is then overexpressed in the myelomonocytic lineage. Homozygous ACE10/10 mice lack ACE expression by any tissue that does not recognize the c-fms promoter including endothelial cells. While the heterozygous ACE10/WT mice retain endogenous ACE expression and have increased ACE expression by myelomonocytes (Figure 1A). In situations where the immune response is challenged directly by tumor or bacterial infection these mice demonstrated an increased immune response with substantially enhanced monocyte recruitment to sites of inflammation (Shen et al., 2007; Okwan-Duodu et al., 2010).
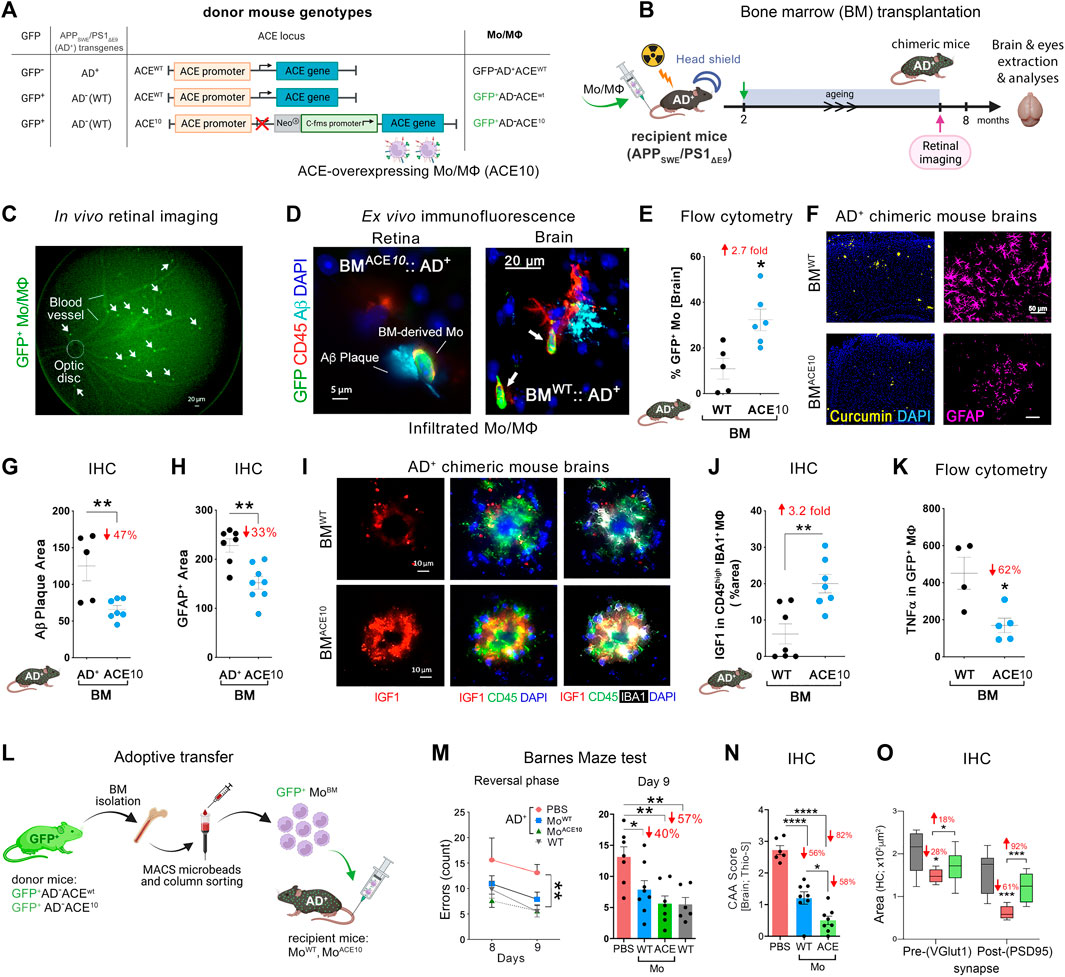
FIGURE 1. Neuroprotection by ACE-overexpressing monocytes and macrophages in APPSWE/PS1ΔE9 (AD+) mice. (A) Description of the genetic composition for the 3 groups of bone marrow (BM) donor mice at the GFP, APPSWE/PS1ΔE9 (AD+), and ACE loci: GFP−AD+ACEWT, GFP+AD−ACEWT, or GFP+AD−ACE10. In the ACE10 mouse models, ACE expression is controlled by the c-fms (CD115) promoter that substituted the endogenous ACE promoter. This results in the over expression of ACE in monocytes and macrophages (Mo/MΦ). (B) BM transplantation experiments: 2-month-old double transgenic AD+ recipient mice underwent irradiation with head shielding to prevent brain irradiation and inflammation. Their BM was then reconstituted by an intravenous injection of 5 million BM cells from one of the three types of donors. This BM transplantation generated BMAD+, BMWT, and BMACE10 chimeric AD+ mice. These mice underwent retinal imaging at the age of 7 months, followed by perfusion and harvest of eyes and brains for analysis. (C) A representative live fluorescence image of a BM-chimeric AD+ mouse retina, demonstrating the feasibility to noninvasively detect in vivo infiltrating GFP+ Mo/MΦ in the retina. [(D), left] Fluorescence micrograph of a BMACE10::AD+ chimeric mouse retina (previously in vivo imaged) showing the homing of GFP+/CD45hi ACE10 Mo/MΦ to the site of retinal 4G8+-Aβ deposit. Scale bar = 5 μm. [(D), right] Infiltrating Mo/MΦ (GFP+/CD45hi; arrows) at the 6E10+-Aβ plaque site are also detected in the BMWT::AD+ chimeric mouse brain. Scale bar = 20 μm. (E) Quantitative flow cytometry analysis of cerebral infiltrating GFP+-Mo/MΦ in BMACE10 versus BMWT chimeric AD+ mice. (F) Micrographs of curcumin+-Aβ plaques (yellow) or GFAP+ astrocytes (pink) in the cortices of BMWT versus BMACE10 AD+ chimeric mice. Scale bar = 50 μm. (G) Quantitative immunohistochemical (IHC) analysis of 6E10+-Aβ plaque area in the cortex of BMACE10 versus BMAD+ AD+-chimeric mice. (H) Quantitative IHC analysis of cortical GFAP+ astrocytosis area in AD+-chimeric mice. (I) Representative fluorescent micrographs of cortical brain regions stained against IGF-1 (red) combined with myeloid cell markers, IBA1 (white) and CD45 (green), and nuclei (DAPI, blue) in BMWT versus BMACE10 AD+-chimeric mice. Scale bars = 10 μm. Brain-infiltrating CD45hi IBA1+ ACE10-versus WT-Mo/MΦ highly expressed IGF-1 in AD+-chimeric mice. (J). Quantitative IHC analysis of % IGF-1+ area colocalized with Mo/MΦ in BMWT versus BMACE10 AD+-chimeric mice. (K) Quantitative flow cytometry analysis of TNFα expression levels in infiltrating ACE10-versus WT-MΦ (GFP+ CD11b+ CD45hi Ly6Cint-hi F4/80hi) in chimeric AD+ mice. (L) An additional study involved adoptive transfer of 5-6 million ACE10-or WT-CD115+ monocytes (MoBM) from young donor mice into the peripheral blood of 8-month-old AD+ mice. These inflammatory monocytes were isolated from the BM of GFP+ donor mice and enriched in the tube using the CD115-positive MACS column sorting procedure. ACE10- (MoACE10) or WT- (MoWT) BM were then injected into the tail vein of recipient AD+ mice. At the age of 11 months, mice underwent behavioral testing. (M) Barnes maze cognitive test: average errors count during the reversal phase, days 8 and 9. Statistics: (left) two-way ANOVA and Bonferroni’s post-test; (right) Day 9 memory test by one-way ANOVA and Bonferroni’s post test. (N) Severity of vascular Aβ deposits is defined as CAA score assessed with thioflavin-S (Thio-S+) staining in AD+ mice brain. (O) Quantitative IHC analysis of hippocampal pre-(VGluT1+) and post-(PSD95+) synaptic areas in MoACE10-treated mice compared to age- and gender-matched PBS-injected control AD+ and naïve WT mice. Illustration in panels A, B and L were created with Biorender.com, as well as images and graphs were used as original or modified from (Koronyo-Hamaoui et al., 2020) by permission of Oxford University Press.
In an important 2014 study (Bernstein et al., 2014) ACE10/10 mice were crossed with double-transgenic AD+ mice. This is an established AD mouse model whereby these mice overproduce amyloidogenic Aβ42 peptides and show progressive memory deficits. AD+ mice with WT ACE alleles (AD+ACEWT/WT), heterozygous for the ACE10 allele (AD+ACE10/WT) and homozygous for this allele (AD+ACE10/10), as well as WT mice (AD–ACEWT/WT) were compared. Histological quantification of hippocampal and cortical Aβ plaques demonstrated a marked reduction in plaque burden in AD+ mice with the ACE10 allele(s), Plaque area was significantly reduced in AD+ACE10/WT mice versus AD+ACEWT/WT mice. This reduction was greater in AD+ACE10/10 mice, which showed a reduced plaque burden of 69%–79% compared with AD+ACEWT/WT mice (p < 0.0001). Histological analysis also revealed substantially less perivascular Aβ deposition for AD+ACE10/WT mice compared to either AD+ACEWT/WT or AD+ACE10/10 mice. Interestingly AD+ACE10/10 mice lack ACE expression in vascular endothelium. This highlights the critical importance of both intra parenchymal and endothelial ACE in clearance of Aβ. Soluble amyloidogenic Aβ42 was also reduced in mice overexpressing ACE in the myelomonocytic lineage (Bernstein et al., 2014).
Astrogliosis is an important marker of AD pathophysiology that may lead to neuronal damage and the devastating cognitive effects of the disease (Wyss-Coray and Mucke, 2002). Interestingly, AD+ACE10/WT and AD+ACE10/10 mice also saw a significant reduction of 45%–75% in astrogliosis compared to AD+ACEWT/WT mice at 7 months. This was maintained at 13 months with a 50% reduction in astrogliosis in AD+ACE10/10 mice compared to AD+ACEWT/WT mice. This marked decrease in astrogliosis was reflected in cognitive testing. Barnes maze testing in 11- and 12-month-old mice with at least one ACE10 allele showed intact maze solving capabilities not statistically different from non-transgenic AD–ACEWT/WT control mice. Thus, the presence of even a single ACE10 allele allows for reduction of amyloidogenic Aβ42, reduction in vascular and parenchymal Aβ burden, reduced astrogliosis and microgliosis and thus preserved cognition.
ACE enriched monocytes appears to play a pivotal role in tempering AD pathogenesis. In another 2020 study, the selective contribution of monocytes, but not microglia, overexpressing ACE was further explored (Koronyo-Hamaoui et al., 2020). Two approaches were applied in AD+ mice recipients. In one approach, BM from GFP+ACE10/10 donor mice was transplanted into AD+ mice whereby the recipients were lead head shielded from BM suppressive radiation to prevent extraneous infiltration of peripheral immune cells induced by the irradiation (Figures 1B–K). In the other approach adoptive transfer of CD115+- monocytes from GFP+ACE10/10 young donor mice into the peripheral blood of recipient AD+ mice took place; this procedure enriched by ∼5-folds the population of circulating CD115+- monocytes in the blood of the recipient mice (Figures 1L–O). These in vivo studies revealed the reduction in parenchymal and vascular Aβ deposits by 47%–82%, soluble Aβ42 levels by 31%, and GFAP+ astrogliosis cell number and area by 33% in AD+ mice that had received BM transplantation or adoptive transfer of GFP+ACE10/10 marrow compared to GFP+ACEWT/WT marrow control (Figures 1G,H). As in the BM transplantation experiment described above (Koronyo-Hamaoui et al., 2020), not only is there clearly less plaque burden but so too a reduction in amyloidogenic Aβ42 and a significant reduction in astrogliosis. These ACE overexpressing monocytes took on a neuroprotective phenotype and surrounded brain and retinal amyloid plaques. These cells expressed 3.2-fold higher insulin-like growth factor-1 (IGF-1), a protein with anti-inflammatory functions implicated in neuroprotection in AD (George et al., 2017; Arjunan et al., 2023), while expressing 60% less TNFα, implicated in harmful neurotoxic effects in AD (Chang et al., 2017; Liddelow et al., 2017) (Figures 1J,K). This was true even in subsequent in vitro studies with ACE10/10 bone marrow-derived macrophages that exhibited distinct Aβ-phagocytic, anti-inflammatory, neuroprotective immune profiles with high IGF-1, and scavenger receptors (i.e., CD36, Scara-1 and MMP-9) as well as low iNOS and an elongated cell morphology even after direct exposure to Aβ42 soluble oligomers (Koronyo-Hamaoui et al., 2020).
Given the evident reduction in astrogliosis and neuroinflammation, the implications of these for preservation of cognition was explored in vivo after adoptive transfer of CD115+ monocytes as described above. Barnes Maze tests comparing diseased AD+ mice with blood enrichment of either CD115+-ACE10/10 or CD115+-ACEWT/WT monocytes, and healthy WT controls demonstrated preservation of spatial learning and memory capacity in AD+ mice following CD115+-ACE10/10 monocyte adoptive transfer, with virtually no difference compared to the healthy WT mice of similar age (Figure 1M). This was also reflected in immunohistochemical analysis whereby these mice had preservation of pre- and post-synaptic marker densities, far fewer hippocampal and total brain plaques and lower CAA scores compared to diseased controls (Figures 1N,O). These phenotypic changes in ACE10/10 monocytes and macrophages were not restricted to cytokine expression or changes in cell morphology. Quantitative immunocytochemistry (ICC) analysis of primary macrophage cultures (Figure 2A) demonstrated a 2-fold increase in the surface binding of soluble oAβ42 by ACE10/10 macrophages (Figure 2C) with a significant increase in Scara1 (Figure 2B), TREM2, CD163, and CD36 (Figure 2D) surface scavenger expression. In previous studies, these receptors have been associated with increased Aβ uptake (Hickman et al., 2008; Frenkel et al., 2013; Kim et al., 2017). ACE10/10 macrophages even demonstrated enhanced endosomal-lysosomal and extracellular degradation of Aβ42 (Figure 2E). Furthermore, using the ACE inhibitor lisinopril it was confirmed that ACE catalytic activity is crucial for clearing fAβ42. Fibrillar Aβ42 uptake was markedly reduced in ACE10/10 monocytes pretreated with lisinopril compared to control not treated ACE10/10 monocytes (Figure 2G). In addition to highlighting ACE catalytic activity, the in vitro analysis further solidified the neuroprotective effects these ACE10/10 monocytes have in response to fAβ42. The immune profile of these cells was analyzed using sensitive MSD (Meso Scale Discovery) multiplex inflammatory assay. Stimulation of both WT and ACE10/10 monocytes with fAβ42 resulted in increased expression of TNFα, that was however increased to a much lower extent in ACE10/10 monocytes (Figure 2H). This was also true for other important pro-inflammatory cytokines such as IL-6, CXCL1 and IL-1β. This suggests that ACE10/10 monocytes, when exposed to Aβ42, shifts the inflammatory milieu towards a more neuroprotective state with a less profound increase in destructive inflammatory cytokines when compared to the WT monocytes exposed to the same Aβ42 levels. It is possible that the ACE overexpressing monocytes, not only increase expression of this important proteolytic enzyme but also exhibit a distinct anti-inflammatory and neuroprotective phenotype that may prove to be a powerful tool in ameliorating AD pathogenesis. The proposed mechanism for the neuroprotective role of ACE10 monocytes in AD is summarized in Figure 3.
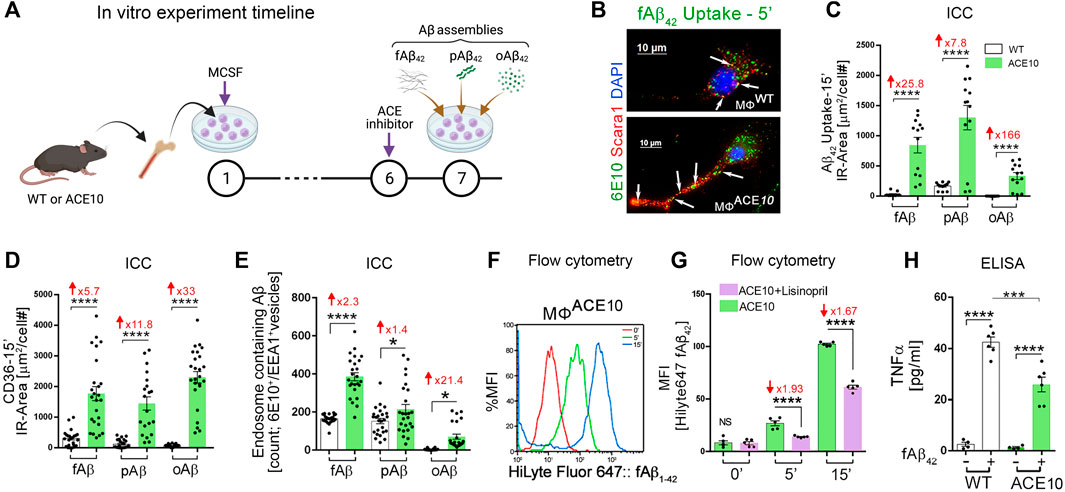
FIGURE 2. Enhanced clearance of Aβ42 assemblies by ACE-overexpressing macrophages. (A) In vitro experimental procedure: BM was isolated from young WT and ACE10 mice (8–12 weeks old) and cultured for 6 days with macrophage colony stimulating factor (MCSF)-enriched media, generating primary macrophage cultures (MΦWT and MΦACE10, respectively). On Day 7, MΦ were either unstimulated (Un) or stimulated by one of the 3 Aβ42 forms: fibrils (fAβ42), prion rod-like structures (pAβ42), or cross-linked oligomers (oAβ42). MΦACE10 were analyzed and compared to control MΦWT for cell surface Aβ42 binding and receptor expression as well as uptake of Aβ42 species at incremental time points from 5 to 15 min (incubated at 37°C). Additional experimental groups were pretreated at day 6 with the ACE inhibitor lisinopril, blocking its two catalytic domains. (B) Representative fluorescence micrograph of MΦWT and MΦACE10 cells that were incubated for 5 min with 250 nM fibril Aβ42 and later immunolabelled for Scara1 (red), human Aβ (6E10, green; white arrows indicate intracellular Aβ within vesicles in these phagocytic cells), and nuclei (DAPI, blue). (C) Quantitative immunocytochemistry (ICC) analysis of 6E10+ IR area (μm2/cell #) following intracellular uptake of the 3 Aβ42 species (fAβ42, pAβ42, or oAβ42) by MΦACE10 versus MΦWT in a period of 15 min. (D) Quantitative ICC of cell surface CD36 scavenger receptor expression (μm2/cell #) in MΦACE10 versus MΦWT following 15-min exposed to the three types of Aβ42 species. (E) Quantitative analysis of colocalized 6E10+ within EEA1+ endosomes in MΦWT versus MΦACE10 following exposure to the three types of Aβ42 species. (F) Representative flow cytometry peak analysis in % MFI of HiLyte Fluor 647-labelled fAβ42 uptake by MΦACE10 at the 0-, 5- and 15-min time points. (G) MFI analysis of HiLyte Fluor 647-labelled fAβ42 uptake by MΦACE10 at the 0-, 5- and 15-min time points, with and without lisinopril pretreatment. (H) MSD biochemical assay determined TNFα levels in MΦWT versus MΦACE10, either unstimulated (−) or stimulated (+) with Aβ42 fibrils [100 nM] for 24 h. MFI, Median Fluorescence Intensity. Illustration in panel A created with Biorender.com and other panels modified or adapted from (Koronyo-Hamaoui et al., 2020) by permission of Oxford University Press.
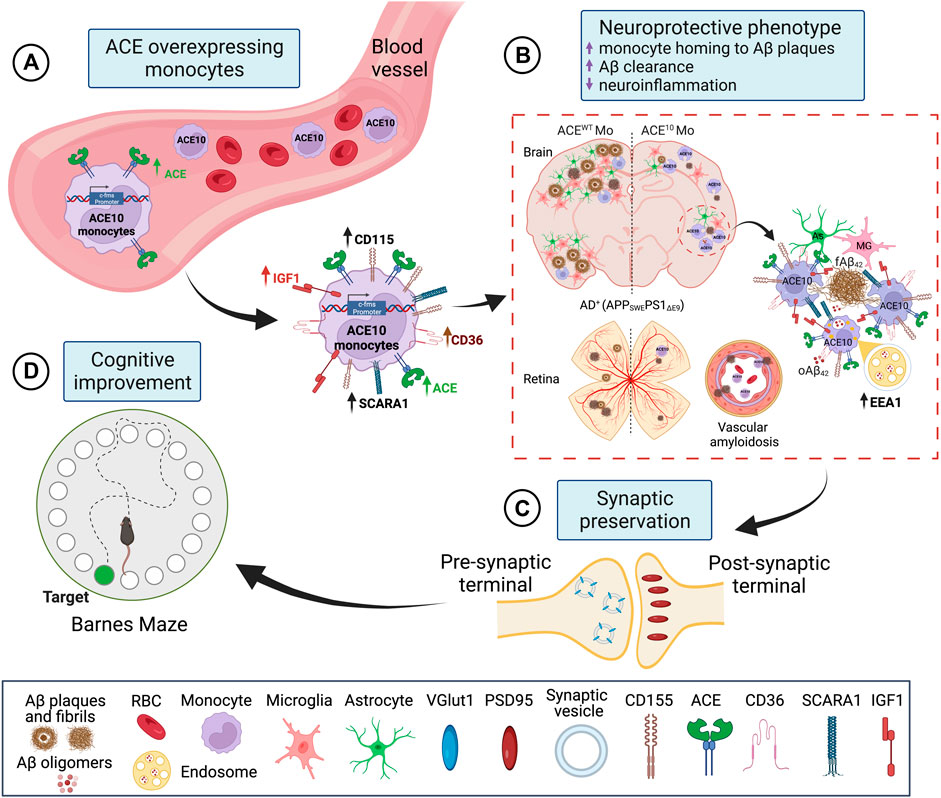
FIGURE 3. Proposed mechanism for neuroprotective role of ACE overexpression in monocytes in AD. (A) ACE overexpression under the activation of the c-fms promotor in CD115+ monocytes is named ACE10 model. CD115+ ACE10-monocytes exhibit a neuroprotective phenotype with lower expression of pro-inflammatory mediators such as iNOS and TNFα and increased expression of the neurotrophic factor IGF-1, Aβ-degrading enzymes (ACE, MMP-9), and surface scavenger receptors as CD36 and Scara-1 that facilitate phagocytosis of Aβ. (B) CD115+ ACE10-monocytes are recruited in larger numbers than WT monocytes to AD-like brains and retinas, resulting in neuro-immunomodulation. The ACE10 monocyte-derived macrophages aggregate at areas of amyloid plaque deposition and facilitate the clearance of Aβ oligomers and plaques. This is concomitant with reduced microgliosis and astrogliosis. Notably, in the ACE10 versus WT macrophages, there is increased presence of Aβ assemblies within EEA1 endosomes, suggesting enhanced intracellular lysosomal Aβ degradation machinery in ACE10 macrophages. (C) Murine models of AD treated with ACE10 monocytes show reduced AD-related pathology, preservation of excitatory synapses, and (D) improved cognitive functions. Created with Biorender.com.
In recent years histological, biochemical, and genetic studies have demonstrated the association between ACE activity and the development of AD pathophysiology. While the biochemical enzymatic ability of ACE in degrading pathogenic Aβ has been well described, more recent studies demonstrate that ACE overexpression induces a phenotypic change in circulating monocytes and tissue macrophages. The circulating monocytes can home to regions of plaque deposition and possess a unique neuroprotective phenotype with an enhanced ability to phagocytose and clear pathogenic Aβ. This represents an extraordinary opportunity to exploit this natural mechanism of ameliorating AD disease progression. More studies are needed to determine ways in which endogenous ACE can be upregulated in patients suffering from this debilitating illness.
RD, DTF, and MK-H: draft manuscript and figure preparation. RD, DTF, YK, AR, JS, EH, DT, KB, SF, KB, and MK-H: manuscript editing. MK-H: study supervision. All authors contributed to the article and approved the submitted version.
This study was supported by the National Institute on Aging of the National Institutes of Health under Award Numbers R01 AG055865, R01 AG056478, and R01 AG075998 (MK-H), and R01 AG042195 (DT), A2013328S00 BrightFocus Foundation Award (MK-H), and The Coins for Alzheimer’s Research Trust—CART Fund (MK-H). This study is also supported by Cedars-Sinai’s Jona Goldrich Center for Alzheimer’s and Memory Disorders (KB), as well as The Saban, The Gordon, The Marciano and The Wilstein Private Foundations (MK-H), and by The National Center for Advancing Translational Sciences through CTSI Grant UL1TR000124 (MK-H).
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alexander, G. C., Knopman, D. S., Emerson, S. S., Ovbiagele, B., Kryscio, R. J., Perlmutter, J. S., et al. (2021). Revisiting FDA approval of aducanumab. N. Engl. J. Med. 385, 769–771. doi:10.1056/NEJMp2110468
Arjunan, A., Sah, D. K., Woo, M., and Song, J. (2023). Identification of the molecular mechanism of insulin-like growth factor-1 (IGF-1): A promising therapeutic target for neurodegenerative diseases associated with metabolic syndrome. Cell. Biosci. 13, 16. doi:10.1186/s13578-023-00966-z
Baudin, B. (2002). New aspects on angiotensin-converting enzyme: From gene to disease. Clin. Chem. Lab. Med. 40, 256–265. doi:10.1515/CCLM.2002.042
Bernstein, K. E., Khan, Z., Giani, J. F., Cao, D. Y., Bernstein, E. A., and Shen, X. Z. (2018). Angiotensin-converting enzyme in innate and adaptive immunity. Nat. Rev. Nephrol. 14, 325–336. doi:10.1038/nrneph.2018.15
Bernstein, K. E., Koronyo, Y., Salumbides, B. C., Sheyn, J., Pelissier, L., Lopes, D. H., et al. (2014). Angiotensin-converting enzyme overexpression in myelomonocytes prevents Alzheimer's-like cognitive decline. J. Clin. Invest. 124, 1000–1012. doi:10.1172/JCI66541
Bernstein, K. E., Ong, F. S., Blackwell, W. L., Shah, K. H., Giani, J. F., Gonzalez-Villalobos, R. A., et al. (2013). A modern understanding of the traditional and nontraditional biological functions of angiotensin-converting enzyme. Pharmacol. Rev. 65, 1–46. doi:10.1124/pr.112.006809
Bertram, L., Mcqueen, M. B., Mullin, K., Blacker, D., and Tanzi, R. E. (2007). Systematic meta-analyses of alzheimer disease genetic association studies: The AlzGene database. Nat. Genet. 39, 17–23. doi:10.1038/ng1934
Braak, H., and Braak, E. (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 82, 239–259. doi:10.1007/BF00308809
Brice, E. A., Friedlander, W., Bateman, E. D., and Kirsch, R. E. (1995). Serum angiotensin-converting enzyme activity, concentration, and specific activity in granulomatous interstitial lung disease, tuberculosis, and COPD. Chest 107, 706–710. doi:10.1378/chest.107.3.706
Brody, A. H., and Strittmatter, S. M. (2018). Synaptotoxic signaling by amyloid beta oligomers in Alzheimer’s disease through prion protein and mGluR5. Adv Pharmacol. 82, 293–323. doi:10.1016/bs.apha.2017.09.007
Butovsky, O., Koronyo-Hamaoui, M., Kunis, G., Ophir, E., Landa, G., Cohen, H., et al. (2006). Glatiramer acetate fights against Alzheimer's disease by inducing dendritic-like microglia expressing insulin-like growth factor 1. Proc. Natl. Acad. Sci. U. S. A. 103, 11784–11789. doi:10.1073/pnas.0604681103
Butovsky, O., Kunis, G., Koronyo-Hamaoui, M., and Schwartz, M. (2007). Selective ablation of bone marrow-derived dendritic cells increases amyloid plaques in a mouse Alzheimer's disease model. Eur. J. Neurosci. 26, 413–416. doi:10.1111/j.1460-9568.2007.05652.x
Cao, D., Khan, Z., Li, X., Saito, S., Bernstein, E. A., Victor, A. R., et al. (2023). Macrophage angiotensin-converting enzyme (ACE) reduces atherosclerosis by increasing PPARα and fundamentally changing lipid metabolism. Cardiovasc Res. doi:10.1093/cvr/cvad082
Cao, D. Y., Saito, S., Veiras, L. C., Okwan-Duodu, D., Bernstein, E. A., Giani, J. F., et al. (2020a). Role of angiotensin-converting enzyme in myeloid cell immune responses. Cell. Mol. Biol. Lett. 25, 31. doi:10.1186/s11658-020-00225-w
Cao, D. Y., Spivia, W. R., Veiras, L. C., Khan, Z., Peng, Z., Jones, A. E., et al. (2020b). ACE overexpression in myeloid cells increases oxidative metabolism and cellular ATP. J. Biol. Chem. 295, 1369–1384. doi:10.1074/jbc.RA119.011244
Cenini, G., Cecchi, C., Pensalfini, A., Bonini, S. A., Ferrari-Toninelli, G., Liguri, G., et al. (2010). Generation of reactive oxygen species by beta amyloid fibrils and oligomers involves different intra/extracellular pathways. Amino Acids 38, 1101–1106. doi:10.1007/s00726-009-0319-7
Chang, R., Yee, K. L., and Sumbria, R. K. (2017). Tumor necrosis factor alpha Inhibition for Alzheimer's Disease. J. Cent. Nerv. Syst. Dis. 9, 9278. doi:10.1177/1179573517709278
Charidimou, A., Boulouis, G., Gurol, M. E., Ayata, C., Bacskai, B. J., Frosch, M. P., et al. (2017). Emerging concepts in sporadic cerebral amyloid angiopathy. Brain 140, 1829–1850. doi:10.1093/brain/awx047
Coraci, I. S., Husemann, J., Berman, J. W., Hulette, C., Dufour, J. H., Campanella, G. K., et al. (2002). CD36, a class B scavenger receptor, is expressed on microglia in Alzheimer's disease brains and can mediate production of reactive oxygen species in response to beta-amyloid fibrils. Am. J. Pathol. 160, 101–112. doi:10.1016/s0002-9440(10)64354-4
Cunningham, C. (2013). Microglia and neurodegeneration: The role of systemic inflammation. Glia 61, 71–90. doi:10.1002/glia.22350
Deane, R., Bell, R. D., Sagare, A., and Zlokovic, B. V. (2009). Clearance of amyloid-beta peptide across the blood-brain barrier: Implication for therapies in Alzheimer's disease. CNS Neurol. Disord. Drug Targets 8, 16–30. doi:10.2174/187152709787601867
Diet, F., Pratt, R. E., Berry, G. J., Momose, N., Gibbons, G. H., and Dzau, V. J. (1996). Increased accumulation of tissue ACE in human atherosclerotic coronary artery disease. Circulation 94, 2756–2767. doi:10.1161/01.cir.94.11.2756
Doustar, J., Rentsendorj, A., Torbati, T., Regis, G. C., Fuchs, D. T., Sheyn, J., et al. (2020). Parallels between retinal and brain pathology and response to immunotherapy in old, late-stage Alzheimer's disease mouse models. Aging Cell. 19, 13246. doi:10.1111/acel.13246
Dvir-Szternfeld, R., Castellani, G., Arad, M., Cahalon, L., Colaiuta, S. P., Keren-Shaul, H., et al. (2022). Alzheimer's disease modification mediated by bone marrow-derived macrophages via a TREM2-independent pathway in mouse model of amyloidosis. Nat. Aging 2, 60–73. doi:10.1038/s43587-021-00149-w
El Khoury, J. (2010). Neurodegeneration and the neuroimmune system. Nat. Med. 16, 1369–1370. doi:10.1038/nm1210-1369
Frenkel, D., Wilkinson, K., Zhao, L., Hickman, S. E., Means, T. K., Puckett, L., et al. (2013). Scara1 deficiency impairs clearance of soluble amyloid-beta by mononuclear phagocytes and accelerates Alzheimer's-like disease progression. Nat. Commun. 4, 2030. doi:10.1038/ncomms3030
George, C., Gontier, G., Lacube, P., Francois, J. C., Holzenberger, M., and Aid, S. (2017). The Alzheimer's disease transcriptome mimics the neuroprotective signature of IGF-1 receptor-deficient neurons. Brain 140, 2012–2027. doi:10.1093/brain/awx132
Grober, E., Dickson, D., Sliwinski, M. J., Buschke, H., Katz, M., Crystal, H., et al. (1999). Memory and mental status correlates of modified Braak staging. Neurobiol. Aging 20, 573–579. doi:10.1016/s0197-4580(99)00063-9
Han, X. J., Hu, Y. Y., Yang, Z. J., Jiang, L. P., Shi, S. L., Li, Y. R., et al. (2017). Amyloid beta-42 induces neuronal apoptosis by targeting mitochondria. Mol. Med. Rep. 16, 4521–4528. doi:10.3892/mmr.2017.7203
Hardy, J., and Selkoe, D. J. (2002). The amyloid hypothesis of Alzheimer's disease: Progress and problems on the road to therapeutics. Science 297, 353–356. doi:10.1126/science.1072994
He, M., Ohrui, T., Maruyama, M., Tomita, N., Nakayama, K., Higuchi, M., et al. (2006). ACE activity in CSF of patients with mild cognitive impairment and Alzheimer disease. Neurology 67, 1309–1310. doi:10.1212/01.wnl.0000238102.04582.ec
Hemming, M. L., and Selkoe, D. J. (2005). Amyloid beta-protein is degraded by cellular angiotensin-converting enzyme (ACE) and elevated by an ACE inhibitor. J. Biol. Chem. 280, 37644–37650. doi:10.1074/jbc.M508460200
Hickman, S. E., Allison, E. K., and El Khoury, J. (2008). Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer's disease mice. J. Neurosci. 28, 8354–8360. doi:10.1523/JNEUROSCI.0616-08.2008
Honig, L. S., Vellas, B., Woodward, M., Boada, M., Bullock, R., Borrie, M., et al. (2018). Trial of solanezumab for mild dementia due to Alzheimer's disease. N. Engl. J. Med. 378, 321–330. doi:10.1056/NEJMoa1705971
Jochemsen, H. M., Teunissen, C. E., Ashby, E. L., Van Der Flier, W. M., Jones, R. E., Geerlings, M. I., et al. (2014). The association of angiotensin-converting enzyme with biomarkers for Alzheimer's disease. Alzheimers Res. Ther. 6, 27. doi:10.1186/alzrt257
Kanemitsu, H., Tomiyama, T., and Mori, H. (2003). Human neprilysin is capable of degrading amyloid beta peptide not only in the monomeric form but also the pathological oligomeric form. Neurosci. Lett. 350, 113–116. doi:10.1016/s0304-3940(03)00898-x
Kehoe, P. G., Al Mulhim, N., Zetterberg, H., Blennow, K., and Miners, J. S. (2019). Cerebrospinal fluid changes in the renin-angiotensin system in Alzheimer's disease. J. Alzheimers Dis. 72, 525–535. doi:10.3233/JAD-190721
Kehoe, P. G., Miners, S., and Love, S. (2009). Angiotensins in Alzheimer's disease - friend or foe? Trends Neurosci. 32, 619–628. doi:10.1016/j.tins.2009.07.006
Keren-Shaul, H., Spinrad, A., Weiner, A., Matcovitch-Natan, O., Dvir-Szternfeld, R., Ulland, T. K., et al. (2017). A unique microglia type associated with restricting development of Alzheimer's disease. Cell. 169, 1276–1290. doi:10.1016/j.cell.2017.05.018
Kim, S. M., Mun, B. R., Lee, S. J., Joh, Y., Lee, H. Y., Ji, K. Y., et al. (2017). TREM2 promotes Aβ phagocytosis by upregulating C/EBPα-dependent CD36 expression in microglia. Sci. Rep. 7, 11118. doi:10.1038/s41598-017-11634-x
Koronyo, Y., Biggs, D., Barron, E., Boyer, D. S., Pearlman, J. A., Au, W. J., et al. (2017). Retinal amyloid pathology and proof-of-concept imaging trial in Alzheimer's disease. JCI Insight 2, 93621. doi:10.1172/jci.insight.93621
Koronyo, Y., Rentsendorj, A., Mirzaei, N., Regis, G. C., Sheyn, J., Shi, H., et al. (2023). Retinal pathological features and proteome signatures of Alzheimer's disease. Acta Neuropathol. 145, 409–438. doi:10.1007/s00401-023-02548-2
Koronyo, Y., Salumbides, B. C., Sheyn, J., Pelissier, L., Li, S., Ljubimov, V., et al. (2015). Therapeutic effects of glatiramer acetate and grafted CD115⁺ monocytes in a mouse model of Alzheimer's disease. Brain 138, 2399–2422. doi:10.1093/brain/awv150
Koronyo-Hamaoui, M., Ko, M. K., Koronyo, Y., Azoulay, D., Seksenyan, A., Kunis, G., et al. (2009). Attenuation of AD-like neuropathology by harnessing peripheral immune cells: Local elevation of IL-10 and MMP-9. J. Neurochem. 111, 1409–1424. doi:10.1111/j.1471-4159.2009.06402.x
Koronyo-Hamaoui, M., Koronyo, Y., Ljubimov, A. V., Miller, C. A., Ko, M. K., Black, K. L., et al. (2011). Identification of amyloid plaques in retinas from Alzheimer's patients and noninvasive in vivo optical imaging of retinal plaques in a mouse model. Neuroimage 54 (1), S204–S217. doi:10.1016/j.neuroimage.2010.06.020
Koronyo-Hamaoui, M., Shah, K., Koronyo, Y., Bernstein, E., Giani, J. F., Janjulia, T., et al. (2014). ACE overexpression in myelomonocytic cells: effect on a mouse model of Alzheimer’s disease. Curr. Hypertens. Rep. 16 (7), 444.
Koronyo-Hamaoui, M., Sheyn, J., Hayden, E. Y., Li, S., Fuchs, D. T., Regis, G. C., et al. (2020). Peripherally derived angiotensin converting enzyme-enhanced macrophages alleviate Alzheimer-related disease. Brain 143 (1), 336–358. doi:10.1093/brain/awz364
Krasemann, S., Madore, C., Cialic, R., Baufeld, C., Calcagno, N., El Fatimy, R., et al. (2017). The TREM2-APOE pathway drives the transcriptional phenotype of dysfunctional microglia in neurodegenerative diseases. Immunity 47, 566–581. doi:10.1016/j.immuni.2017.08.008
La Morgia, C., Ross-Cisneros, F. N., Koronyo, Y., Hannibal, J., Gallassi, R., Cantalupo, G., et al. (2016). Melanopsin retinal ganglion cell loss in Alzheimer disease. Ann. Neurol. 79, 90–109. doi:10.1002/ana.24548
Lebson, L., Nash, K., Kamath, S., Herber, D., Carty, N., Lee, D. C., et al. (2010). Trafficking CD11b-positive blood cells deliver therapeutic genes to the brain of amyloid-depositing transgenic mice. J. Neurosci. 30, 9651–9658. doi:10.1523/JNEUROSCI.0329-10.2010
Lehmann, D. J., Cortina-Borja, M., Warden, D. R., Smith, A. D., Sleegers, K., Prince, J. A., et al. (2005). Large meta-analysis establishes the ACE insertion-deletion polymorphism as a marker of Alzheimer's disease. Am. J. Epidemiol. 162, 305–317. doi:10.1093/aje/kwi202
Li, S., Hayden, E. Y., Garcia, V. J., Fuchs, D. T., Sheyn, J., Daley, D. A., et al. (2020). Activated bone marrow-derived macrophages eradicate alzheimer's-related Aβ42 oligomers and protect synapses. Front. Immunol. 11, 49. doi:10.3389/fimmu.2020.00049
Liddelow, S. A., Guttenplan, K. A., Clarke, L. E., Bennett, F. C., Bohlen, C. J., Schirmer, L., et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. doi:10.1038/nature21029
Lieberman, J. (1975). Elevation of serum angiotensin-converting-enzyme (ACE) level in sarcoidosis. Am. J. Med. 59, 365–372. doi:10.1016/0002-9343(75)90395-2
Marioni, R. E., Harris, S. E., Zhang, Q., Mcrae, A. F., Hagenaars, S. P., Hill, W. D., et al. (2019). Correction: GWAS on family history of Alzheimer's disease. Transl. Psychiatry 9, 161. doi:10.1038/s41398-019-0498-2
Marques, M. A., Kulstad, J. J., Savard, C. E., Green, P. S., Lee, S. P., Craft, S., et al. (2009). Peripheral amyloid-beta levels regulate amyloid-beta clearance from the central nervous system. J. Alzheimers Dis. 16, 325–329. doi:10.3233/JAD-2009-0964
Mateos, L., Ismail, M. A., Gil-Bea, F. J., Leoni, V., Winblad, B., Bjorkhem, I., et al. (2011). Upregulation of brain renin angiotensin system by 27-hydroxycholesterol in Alzheimer's disease. J. Alzheimers Dis. 24, 669–679. doi:10.3233/JAD-2011-101512
Meng, Y., Baldwin, C. T., Bowirrat, A., Waraska, K., Inzelberg, R., Friedland, R. P., et al. (2006). Association of polymorphisms in the Angiotensin-converting enzyme gene with Alzheimer disease in an Israeli Arab community. Am. J. Hum. Genet. 78, 871–877. doi:10.1086/503687
Michaud, J. P., Bellavance, M. A., Prefontaine, P., and Rivest, S. (2013). Real-time in vivo imaging reveals the ability of monocytes to clear vascular amyloid beta. Cell. Rep. 5, 646–653. doi:10.1016/j.celrep.2013.10.010
Miners, J. S., Ashby, E., Van Helmond, Z., Chalmers, K. A., Palmer, L. E., Love, S., et al. (2008). Angiotensin-converting enzyme (ACE) levels and activity in Alzheimer's disease, and relationship of perivascular ACE-1 to cerebral amyloid angiopathy. Neuropathol. Appl. Neurobiol. 34, 181–193. doi:10.1111/j.1365-2990.2007.00885.x
Miners, S., Ashby, E., Baig, S., Harrison, R., Tayler, H., Speedy, E., et al. (2009). Angiotensin-converting enzyme levels and activity in Alzheimer's disease: Differences in brain and CSF ACE and association with ACE1 genotypes. Am. J. Transl. Res. 1, 163–177.
Mintun, M. A., Lo, A. C., Duggan Evans, C., Wessels, A. M., Ardayfio, P. A., Andersen, S. W., et al. (2021). Donanemab in early Alzheimer's disease. N. Engl. J. Med. 384, 1691–1704. doi:10.1056/NEJMoa2100708
Morgan, D., Gordon, M. N., Tan, J., Wilcock, D., and Rojiani, A. M. (2005). Dynamic complexity of the microglial activation response in transgenic models of amyloid deposition: Implications for alzheimer therapeutics. J. Neuropathol. Exp. Neurol. 64, 743–753. doi:10.1097/01.jnen.0000178444.33972.e0
Mukherjee, A., Song, E., Kihiko-Ehmann, M., Goodman, J. P., Pyrek, J. S., Estus, S., et al. (2000). Insulysin hydrolyzes amyloid beta peptides to products that are neither neurotoxic nor deposit on amyloid plaques. J. Neurosci. 20, 8745–8749. doi:10.1523/JNEUROSCI.20-23-08745.2000
Ohishi, M., Ueda, M., Rakugi, H., Naruko, T., Kojima, A., Okamura, A., et al. (1997). Enhanced expression of angiotensin-converting enzyme is associated with progression of coronary atherosclerosis in humans. J. Hypertens. 15, 1295–1302. doi:10.1097/00004872-199715110-00014
Okwan-Duodu, D., Datta, V., Shen, X. Z., Goodridge, H. S., Bernstein, E. A., Fuchs, S., et al. (2010). Angiotensin-converting enzyme overexpression in mouse myelomonocytic cells augments resistance to Listeria and methicillin-resistant Staphylococcus aureus. J. Biol. Chem. 285, 39051–39060. doi:10.1074/jbc.M110.163782
Pimentel-Coelho, P. M., and Rivest, S. (2012). The early contribution of cerebrovascular factors to the pathogenesis of Alzheimer's disease. Eur. J. Neurosci. 35, 1917–1937. doi:10.1111/j.1460-9568.2012.08126.x
Prinz, M., and Priller, J. (2017). The role of peripheral immune cells in the CNS in steady state and disease. Nat. Neurosci. 20, 136–144. doi:10.1038/nn.4475
Rentsendorj, A., Sheyn, J., Fuchs, D. T., Daley, D., Salumbides, B. C., Schubloom, H. E., et al. (2018). A novel role for osteopontin in macrophage-mediated amyloid-beta clearance in Alzheimer's models. Brain Behav. Immun. 67, 163–180. doi:10.1016/j.bbi.2017.08.019
Rossi, B., Santos-Lima, B., Terrabuio, E., Zenaro, E., and Constantin, G. (2021). Common peripheral immunity mechanisms in multiple sclerosis and Alzheimer's disease. Front. Immunol. 12, 639369. doi:10.3389/fimmu.2021.639369
Selkoe, D. J. (2001). Alzheimer's disease results from the cerebral accumulation and cytotoxicity of amyloid beta-protein. J. Alzheimers Dis. 3, 75–80. doi:10.3233/jad-2001-3111
Selkoe, D. J. (2008). Soluble oligomers of the amyloid beta-protein impair synaptic plasticity and behavior. Behav. Brain Res. 192, 106–113. doi:10.1016/j.bbr.2008.02.016
Shankar, G. M., Bloodgood, B. L., Townsend, M., Walsh, D. M., Selkoe, D. J., and Sabatini, B. L. (2007). Natural oligomers of the Alzheimer amyloid-beta protein induce reversible synapse loss by modulating an NMDA-type glutamate receptor-dependent signaling pathway. J. Neurosci. 27, 2866–2875. doi:10.1523/JNEUROSCI.4970-06.2007
Shankar, G. M., Li, S., Mehta, T. H., Garcia-Munoz, A., Shepardson, N. E., Smith, I., et al. (2008). Amyloid-beta protein dimers isolated directly from Alzheimer's brains impair synaptic plasticity and memory. Nat. Med. 14, 837–842. doi:10.1038/nm1782
Shankar, G. M., and Walsh, D. M. (2009). Alzheimer's disease: Synaptic dysfunction and abeta. Mol. Neurodegener. 4, 48. doi:10.1186/1750-1326-4-48
Shen, X. Z., Li, P., Weiss, D., Fuchs, S., Xiao, H. D., Adams, J. A., et al. (2007). Mice with enhanced macrophage angiotensin-converting enzyme are resistant to melanoma. Am. J. Pathol. 170, 2122–2134. doi:10.2353/ajpath.2007.061205
Shi, H., Koronyo, Y., Rentsendorj, A., Regis, G. C., Sheyn, J., Fuchs, D. T., et al. (2020). Identification of early pericyte loss and vascular amyloidosis in Alzheimer's disease retina. Acta Neuropathol. 139, 813–836. doi:10.1007/s00401-020-02134-w
Simard, A. R., Soulet, D., Gowing, G., Julien, J. P., and Rivest, S. (2006). Bone marrow-derived microglia play a critical role in restricting senile plaque formation in Alzheimer's disease. Neuron 49, 489–502. doi:10.1016/j.neuron.2006.01.022
Tu, S., Okamoto, S., Lipton, S. A., and Xu, H. (2014). Oligomeric Aβ-induced synaptic dysfunction in Alzheimer's disease. Mol. Neurodegener. 9, 48. doi:10.1186/1750-1326-9-48
Umeda, T., Ramser, E. M., Yamashita, M., Nakajima, K., Mori, H., Silverman, M. A., et al. (2015). Intracellular amyloid beta oligomers impair organelle transport and induce dendritic spine loss in primary neurons. Acta Neuropathol. Commun. 3, 51. doi:10.1186/s40478-015-0230-2
Viola, K. L., and Klein, W. L. (2015). Amyloid beta oligomers in Alzheimer's disease pathogenesis, treatment, and diagnosis. Acta Neuropathol. 129, 183–206. doi:10.1007/s00401-015-1386-3
Walsh, D. M., Klyubin, I., Fadeeva, J. V., Cullen, W. K., Anwyl, R., Wolfe, M. S., et al. (2002). Naturally secreted oligomers of amyloid beta protein potently inhibit hippocampal long-term potentiation in vivo. Nature 416, 535–539. doi:10.1038/416535a
Wei, L., Alhenc-Gelas, F., Corvol, P., and Clauser, E. (1991). The two homologous domains of human angiotensin I-converting enzyme are both catalytically active. J. Biol. Chem. 266, 9002–9008. doi:10.1016/s0021-9258(18)31543-6
Williams, G. T., and Williams, W. J. (1983). Granulomatous inflammation-a review. J. Clin. Pathol. 36, 723–733. doi:10.1136/jcp.36.7.723
Wyss-Coray, T., and Mucke, L. (2002). Inflammation in neurodegenerative disease-a double-edged sword. Neuron 35, 419–432. doi:10.1016/s0896-6273(02)00794-8
Xin, X. Y., Lai, Z. H., Ding, K. Q., Zeng, L. L., and Ma, J. F. (2021). Angiotensin-converting enzyme polymorphisms AND Alzheimer's disease susceptibility: An updated meta-analysis. PLoS One 16, 0260498. doi:10.1371/journal.pone.0260498
Xu, L., He, D., and Bai, Y. (2016). Microglia-mediated inflammation and neurodegenerative disease. Mol. Neurobiol. 53, 6709–6715. doi:10.1007/s12035-015-9593-4
Yan, P., Kim, K. W., Xiao, Q., Ma, X., Czerniewski, L. R., Liu, H., et al. (2022). Peripheral monocyte-derived cells counter amyloid plaque pathogenesis in a mouse model of Alzheimer's disease. J. Clin. Invest. 132, 152565. doi:10.1172/JCI152565
Yin, K. J., Cirrito, J. R., Yan, P., Hu, X., Xiao, Q., Pan, X., et al. (2006). Matrix metalloproteinases expressed by astrocytes mediate extracellular amyloid-beta peptide catabolism. J. Neurosci. 26, 10939–10948. doi:10.1523/JNEUROSCI.2085-06.2006
Yu, L., Petyuk, V. A., Tasaki, S., Boyle, P. A., Gaiteri, C., Schneider, J. A., et al. (2019). Association of cortical beta-amyloid protein in the absence of insoluble deposits with alzheimer disease. JAMA Neurol. 76, 818–826. doi:10.1001/jamaneurol.2019.0834
Zhang, R., Miller, R. G., Madison, C., Jin, X., Honrada, R., Harris, W., et al. (2013). Systemic immune system alterations in early stages of Alzheimer's disease. J. Neuroimmunol. 256, 38–42. doi:10.1016/j.jneuroim.2013.01.002
Zou, K., Yamaguchi, H., Akatsu, H., Sakamoto, T., Ko, M., Mizoguchi, K., et al. (2007). Angiotensin-converting enzyme converts amyloid beta-protein 1-42 (Abeta(1-42)) to Abeta(1-40), and its inhibition enhances brain Abeta deposition. J. Neurosci. 27, 8628–8635. doi:10.1523/JNEUROSCI.1549-07.2007
Keywords: microglia, monocytes, macrophages, innate immune cells, degrading peptidase, pro-inflammatory cytokines, neurodegenerative diseases, neuroscience
Citation: Danziger R, Fuchs D-T, Koronyo Y, Rentsendorj A, Sheyn J, Hayden EY, Teplow DB, Black KL, Fuchs S, Bernstein KE and Koronyo-Hamaoui M (2023) The effects of enhancing angiotensin converting enzyme in myelomonocytes on ameliorating Alzheimer’s-related disease and preserving cognition. Front. Physiol. 14:1179315. doi: 10.3389/fphys.2023.1179315
Received: 03 March 2023; Accepted: 07 June 2023;
Published: 23 June 2023.
Edited by:
Tuantuan Zhao, Mayo Clinic, United StatesReviewed by:
Yanyan Zheng, Mayo Clinic, United StatesCopyright © 2023 Danziger, Fuchs, Koronyo, Rentsendorj, Sheyn, Hayden, Teplow, Black, Fuchs, Bernstein and Koronyo-Hamaoui. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Maya Koronyo-Hamaoui, bWF5YS5rb3JvbnlvQGNzbWMuZWR1
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.