 Hiroki Ueharu
Hiroki Ueharu Yuji Mishina
Yuji Mishina
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
MINI REVIEW article
Front. Physiol. , 18 May 2023
Sec. Craniofacial Biology and Dental Research
Volume 14 - 2023 | https://doi.org/10.3389/fphys.2023.1170511
This article is part of the Research Topic Animal Models and Transgenic Technology in Craniofacial Biology View all 6 articles
Cranial neural crest cells (NCCs) are the origin of the anterior part of the face and the head. Cranial NCCs are multipotent cells giving rise to bones, cartilage, adipose-tissues in the face, and neural cells, melanocytes, and others. The behavior of cranial NCCs (proliferation, cell death, migration, differentiation, and cell fate specification) are well regulated by several signaling pathways; abnormalities in their behavior are often reported as causative reasons for craniofacial anomalies (CFAs), which occur in 1 in 100 newborns in the United States. Understanding the pathological mechanisms of CFAs would facilitate strategies for identifying, preventing, and treating CFAs. Bone morphogenetic protein (BMP) signaling plays a pleiotropic role in many cellular processes during embryonic development. We and others have reported that abnormalities in BMP signaling in cranial NCCs develop CFAs in mice. Abnormal levels of BMP signaling cause miscorrelation with other signaling pathways such as Wnt signaling and FGF signaling, which mutations in the signaling pathways are known to develop CFAs in mice and humans. Recent Genome-Wide Association Studies and exome sequencing demonstrated that some patients with CFAs presented single nucleotide polymorphisms (SNPs), missense mutations, and duplication of genes related to BMP signaling activities, suggesting that defects in abnormal BMP signaling in human embryos develop CFAs. There are still a few cases of BMP-related patients with CFAs. One speculation is that human embryos with mutations in coding regions of BMP-related genes undergo embryonic lethality before developing the craniofacial region as well as mice development; however, no reports are available that show embryonic lethality caused by BMP mutations in humans. In this review, we will summarize the recent advances in the understanding of BMP signaling during craniofacial development in mice and describe how we can translate the knowledge from the transgenic mice to CFAs in humans.
Craniofacial anomalies (CFAs) are birth defects affecting the shape of the head and the face. The incidence is approximately 1 in 100 newborns (Twigg and Wilkie, 2015; Sakai and Trainor, 2016; Wei et al., 2016; Kitami et al., 2018). Orofacial cleft (1:700) and craniosynostosis (1:2000) are the most common CFAs (Morriss-Kay and Wilkie, 2005; Nasreddine et al., 2021). Approximately 70% of patients with orofacial cleft and 85% of patients with craniosynostosis are non-syndromic, with no other abnormalities in the patients, and most have no other affected family members (Timberlake et al., 2016; Howe et al., 2018). Therefore, it has been unclear whether genetic or non-genetic reasons cause that, and caused by single gene mutation or combined effect of several genes. The current primary therapeutic option for orofacial cleft and craniosynostosis is invasive surgeries, which decrease their quality of life (Esparza and Hinojosa, 2008; Seruya et al., 2011; Shkoukani et al., 2014; Opriş et al., 2022; Spazzapan et al., 2022). Therefore, defining the molecular and cellular pathogenesis of CFAs could facilitate the strategies for the early identification, prevention, and treatment of these developmental diseases. Cranial neural crest cells (NCCs) are the origin of the face and the anterior part of the head (Mishina and Snider, 2014). Cranial NCCs are multipotent cells giving rise to osteoblasts, chondrocytes, adipocytes, melanocytes, odontoblasts, neural cells, glia, pituitary hormone-producing cells and others (Le Douarin et al., 2004; Ishii et al., 2012; Ueharu et al., 2017; Srinivasan and Toh, 2019). Defects of migration, proliferation, cell death, differentiation, and cell fate specification in cranial NCCs are often reported as causative reasons for CFAs (Yu et al., 2005; Teng et al., 2008; Gu et al., 2014). Thus, understanding the regulation mechanisms of cranial NCCs would provide new therapeutic options for CFAs. Two transgenic mice, Wnt1-Cre mice and P0-Cre mice, are largely used for tracing NCCs and NC-specific activation or deletion of targeted genes by the Cre-LoxP system (Danielian et al., 1998; Yamauchi et al., 1999). Both transgenic mice label cranial NCCs in a similar manner, but due to the small differences in the expression patterns, resulting mutant mice sometimes show phenotype differences between Wnt1-Cre mice and P0-Cre mice (Wang et al., 2011; Kulkarni et al., 2018; Zhang et al., 2022; Ueharu et al., 2023b). Detailed comparison between 2 Cre transgenes using R26-LacZ reporter mice revealed that Wnt1-Cre activity initially found at the midbrain region while that of P0-Cre is found at the hindbrain region (Chen et al., 2017). Wnt1 (not Wnt1-Cre) expression is more abundant in premigratory neural crest cells that post-migratory cells (Echelard et al., 1994), while P0-Cre proteins present at the 19 somite stage (Chen et al., 2017). Expression of endogenous P0 gene during neural crest development is not available. Taken together, these differences may result similar but unique phenotypes.
Bone morphogenetic proteins (BMPs) are members of the Transforming Growth Factor Beta superfamily. Upon BMP ligands (e.g., BMP2, BMP4, BMP7) binding, BMP receptors form hetero-multimers consisting of BMP type II receptors (BMPR2 and ACVR2A and ACVR2B) and BMP type I receptors (BMPR1A, BMPR1B, ACVR1, and ACVRL1) then transduce the signaling through phosphorylation of SMAD1/5/9 (SMAD-dependent pathway) or TAK1/p38 MAP kinases (SMAD-independent pathway) (Wu et al., 2016; Gomez-Puerto et al., 2019). The signaling level of BMPs is well regulated by a BMP antagonist Noggin and inhibitory SMAD6/7 (Figure 1).
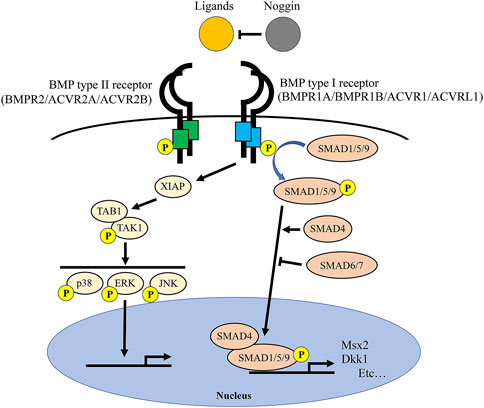
FIGURE 1. BMP signaling and BMP-related genes. Once BMP ligands bind to BMP receptor type 2 (BMPR2/ACVR2A/ACVR2B) and BMP receptor type 1 (BMPR1A/BMPR1B/ACVR1/ACVRL1) complexes, type 1 receptors are phosphorylated by type 2 receptors. Subsequently, BMP type 1 receptors phosphorylate SMAD1/5/9 proteins that transduce signals to the nucleus with SMAD4 protein to alter expressions of downstream target genes (e.g., Msx2 and Dkk1). Inhibitory SMAD6 and SMAD7 prevent the phosphorylation and thus nuclear transition of SMAD1/5/9 proteins. Noggin is an extracellular antagonist for the BMP ligands to suppress BMP signaling.
Bone morphogenetic proteins (BMPs) were originally discovered as bone inducers (Urist, 1965). Interestingly, recent reports published by our group and others demonstrated that BMP signaling plays pleiotropic roles in embryogenesis, including craniofacial development, by regulating proliferation, cell death, differentiation, and cell fate specification, in addition to osteogenesis (Komatsu et al., 2013; Graf et al., 2016; Grafe et al., 2018; Yang et al., 2021a). Many studies have shown that loss of function mutation or gain of function mutation of BMPs, BMP receptors, and downstream target genes for BMP signaling in mice develops CFAs; however, only a few case reports of human patients with BMP mutations are available. In this review, we will describe that transgenic mice targeted for BMP signaling and BMP-downstream genes that develop CFAs. We will further discuss a potential reason of the discrepancy in why it is rare to see human CFAs patients with BMP mutations while many transgenic mice develop CFAs.
Orofacial cleft (cleft lip, cleft palate, and midfacial cleft) is the most common CFAs that show splitting lip and/or splitting shelves of the mouth (palate) (Shkoukani et al., 2014). Because of the abnormal structures, patients with an orofacial cleft frequently have difficulties feeding, swallowing, and breathing (Warren et al., 1992; Reid, 2004; Duarte et al., 2016). Serial invasive surgeries after birth are the only therapeutic options (Srinivasan and Toh, 2019), which are burdens for the patients and increase medical expenses; thus understanding molecular mechanisms of pathological craniofacial development could facilitate the strategies for early identification and prevention of orofacial cleft, which will provide us novel therapeutic options such as taking a drug to the pregnant mothers to prevent developing orofacial cleft, in addition to surgeries.
The Pierre-Robin sequence is a birth defect characterized by an underdeveloped lower jaw (Hsieh and Woo, 2019). The underdeveloped lower jaw results in bringing the tongue back and preventing closure of the palate. As a result of the smaller jaw, approximately 85% of Patients with the Pierre-Robin sequence develop a cleft palate (Hsieh and Woo, 2019). Bmp7-deficient mice (Bmp7Δ/Δ mice) develop cleft palates with the shorter Meckel’s cartilage (Kouskoura et al., 2013). Their ex vivo culture of palatal shelves showed that palatal shelves from Bmp7Δ/Δ mice retain the ability to fuse when placed in close proximity (Kouskoura et al., 2013). This fact may suggest that the shorter Meckel’s cartilage by Bmp7 deletion could develop a cleft palate by mimicking the Pierre-Robin sequence. However, ex vivo culture of the mutant embryos after removal of the mandibular, the tongue and the brain show poor shelf elevation and failure to fuse, suggesting directing involvement of BMP7 function in palatial shelf elevation (Kouskoura et al., 2013). Palatal mesenchyme-specific Noggin expressing mice (Osr2-Cre;pMes-Noggin mice) develop cleft palate with lower proliferation and suppressed osteogenic condensation at the palate (Li et al., 2021). On the other hand, neural crest (NC)-specific deletion of Bmp2 in mice (Wnt1-Cre;Bmp2fl/fl mice) develops cleft palate as a consequence of the failure of tongue descent (Chen et al., 2019). Wnt1-Cre;Bmp2fl/fl mice showed lower cell proliferation in the mandibular and the Meckel’s cartilage, but not in the anterior and posterior palate, than that in control mice, along with the failure of the tongue descent. These studies suggest that downregulation of BMP signaling causes lower proliferation and suppressed differentiation in the craniofacial tissues that causes miscoordinations of craniofacial development, resulting in developing orofacial cleft, but its etiology may not be same as that of the Pierre-Robin Sequence.
On the other hand, NC-specific expression of constitutively activated Bmpr1a (caBmpr1a, the kinase activity is ligand-independent due to the Q233D mutation) in mice (Wnt1-Cre;pMes-caBmpr1a mice) also develop cleft lip and cleft palate. Wnt1-Cre;pMes-caBmpr1a mice showed reduced proliferation and ectopic cartilage formation at the palatal mesenchyme (Li et al., 2013). NC-specific deletion of Tak1 in mice (Wnt1-Cre;Tak1fl/fl mice) develops cleft palate association with a higher level of fibroblast growth factor (FGF) signaling that is a known causative reason for Apert syndrome, a subset of patients with the syndrome develops cleft palate (Song et al., 2013; Willie et al., 2022). We found that NC-specific expression of caBmpr1a with deletion of Tak1 in mice (P0-Cre;caBmpr1afl/+;Tak1fl/fl mice) develops cleft palate while P0-Cre;caBmpr1afl/+ mice and P0-Cre;Tak1fl/fl mice did not develop a cleft palate. (Liu et al., 2018). Although the phenotype differences between us and other laboratories might be developed by the difference of Wnt1-Cre mice and P0-Cre mice, it is reasonable to speculate that unbalance between the SMAD-dependent and SMAD-independent pathways in cranial NCCs may be a reason for an orofacial cleft in P0-Cre;caBmpr1afl/+;Tak1fl/fl mice. We recently reported that two transgenic mouse lines expressing constitutively activated Acvr1 (caAcvr1, Acvr1 with the Q207D mutation) in NCCs in mice (P0-Cre;caAcvr1-L35 line and P0-Cre;caAcvr1-A11 line) develops midfacial defects including orofacial cleft (Yang et al., 2021a; Yang et al., 2021b). Interestingly, the facial phenotypes between P0-Cre;caAcvr1-L35 line and P0-Cre;caAcvr1-A11 line are different. There are still questions why three transgenic mice, i.e., caBmpr1a mice, caAcvr1-L35 mice, and caAcvr1-A11 mice crossed with P0-Cre mice, showed different phenotypes. We found that the phosphorylation level of SMAD1/5/9 differs between caAcvr1-L35 mice and caAcvr1-A11 mice (Yang et al., 2021b), which may be a reason for similar but distinct phenotypes between these two lines. Another possibility is that because these mice have been generated through random transgenesis, their expression patterns may be different depending on the genomic locus where the transgenic constructs are integrated. The third possibility is intrinsic differences between type 1 receptors. For example, ACVR1 is known to bind to activins without signal transduction, while BMPR1A does not bind to activins (Alessi Wolken et al., 2018). Phosphorylation of Smad1/5/9 is the common downstream event but there may be unique functions of each receptor for signal transduction, which is poorly understood.
Taken together, craniofacial tissues coordinate to develop the craniofacial region, and abnormal SMAD-dependent pathways and SMAD-independent pathways disrupt the orchestration, causing CFAs such as orofacial cleft.
Craniosynostosis is another common CFAs characterized by abnormal shapes of the skull and the face caused by premature fusions of cranial sutures. Cranial sutures consist of mesenchymal tissues housing stem/progenitor cells to support the growth of infant’s bones. Premature fusion of cranial sutures results in an imbalance of growth between the skull and brain leading to an increase of intracranial pressure and may secondarily develop neurologic issues such as deafness (Swanson and Mishina, 2021). It has been thought that excess osteogenic differentiation of suture mesenchymal cells causes craniosynostosis. As the name implies, BMP signaling prompts osteogenic differentiation. Many scientists focused on the relation between craniosynostosis and BMP signaling; however, recent studies have shown that BMP signaling develops craniosynostosis by disruptions of cranial NCCs and suture stem cells, in addition to prompt osteogenic differentiation of osteoblasts.
In 1993, a gain of function mutation of MSX2, a downstream target of BMP signaling, was found in human patients with the Boston-type craniosynostosis (Jabs et al., 1993). Higher Msx2 expression at the middle of the sagittal suture in mice developed overgrown parietal bones (Liu et al., 1999). Interestingly, NC-specific deletions (cKO) of three out of four alleles in Msx1/Msx2 in mice (Wnt1-Cre;Msx1cKO/+;Msx2cKO/cKO or Wnt1-Cre;Msx1cKO/cKO;Msx2cKO/+) develop large calvarial defects in the frontal bones as expected; however, NC-specific deletions of all four alleles in Msx1/Msx2 in mice (Wnt1-Cre;Msx1cKO/cKO;Msx2cKO/cKO) develop ectopic bones at the calvaria (Roybal et al., 2010). Tamoxifen-inducible deletions of Msx1/Msx2 in mice (Cagg-CreER™; Msx1cKO/cKO;Msx2cKO/cKO mice) demonstrated that Msx1 and Msx2 at E10.5 to E11.5 are required to suppress the ectopic bone formation while those at E11.5 to E12.5 are required to prompt calvarial formation (Roybal et al., 2010). These results suggest that MSX proteins during calvarial development control two distinct biological processes; one is to suppress the cell fate of neural crest cells towards osteogenic differentiation and the other is to prompt osteogenic differentiation once the progenitor cells commit to osteogenic lineage.
An antagonist of BMP signaling Noggin is expressed in the patent sutures (Warren et al., 2003). Elevation of FGF signaling, a causative reason for some craniosynostosis such as Crouzon syndrome and Apert syndrome, suppressed Noggin expression in the coronal suture in mice (Warren et al., 2003). Inhibition of Smad7 expression in the calvaria results in premature fusion of the coronal suture (Zhou et al., 2014). Smad6 and Smad7 are known as inhibitory Smads inhibiting signaling pathways of the TGFβ superfamily (Massagué and Chen, 2000). Protein levels of FGF10 and phosphorylated ERK1/2, a transducer of FGF signaling, were elevated by siSmad7-treated suture cells, in which TGFβ signaling level was augmented while BMP signaling level was not examined in this experiment (Zhou et al., 2014). We previously reported that P0-Cre;caBmpr1a mice showed elevation of FGF signaling; however, unlikely the case of Crouzon syndrome, the elevated FGF signaling does not directly involve in craniosynostosis (Komatsu et al., 2013). Suggestively, it is reported that during calvarial development BMP signaling negatively regulates levels of FGF signaling (Maruyama et al., 2010; Maruyama et al., 2017). When BMP signaling was enhanced in Axin2 expressing cells, resulting mice show premature suture fusion with ectopic cartilage, but FGF signaling is suppressed (Maruyama et al., 2017). Taken together, these suggest that augmented BMP signaling is a primary reason for craniosynostosis in these mouse models. However, there is a sharp contrast about the levels of FGF signaling between two models, suggesting that mechanisms of how BMP signaling regulates FGF signaling is developmental stage and cell type-specific manner. In our P0-Cre;caBmpr1a mice, an elevation of BMP signaling in neural crest cells starts around E8.0-E8.5 (Komatsu et al., 2013; Ueharu et al., 2023b); however, Maruyama et al. designed to elevate BMP signaling in Axin2-expressing suture stem cells during late embryonic stage to newborn stage by Doxycycline, and they observed elevated BMP signaling activity at postnatal day 3 (Maruyama et al., 2017). The differences in cell type and developmental stage between the two animal models may generate different outcomes in FGF signaling.
We reported that NC-specific expression of caBmpr1a in mice (P0-Cre;caBmpr1a mice) develops premature fusion of the anterior frontal suture and the naso-premaxillary suture, which leads to craniosynostosis (Komatsu et al., 2013; Pan et al., 2017; Kramer et al., 2018; Liu et al., 2018; Ueharu et al., 2023b). We found elevated cell death in cranial NCCs in P0-Cre;caBmpr1a mice, and inhibition of p53-induced cell death partially rescued premature suture fusion (Komatsu et al., 2013; Hayano et al., 2015; Ueharu et al., 2023a; Ueharu et al., 2023b). Of note, ectopic cartilage is developed only in the sutures which prematurely fused, and during the fusion process, the ectopic cartilage is replaced into bone nodules (Ueharu et al., 2023b). It is reported that global knockout of Axin2 with heterozygous Fgfr1 develops premature suture fusion in the presence of ectopic cartilage in sutures (Maruyama et al., 2010). Together with a follow up report, it is suggested that an imbalance between BMP and FGF signaling may alter cell fate of cranial suture mesenchymal cells to develop ectopic cartilage leading to premature fusion of cranial sutures (Maruyama et al., 2017). Thus, we propose that augmentation of BMP signaling in cranial NCCs prompts them towards chondrogenic fate and that results in premature suture fusion through endochondral ossification (Figure 2). In the future, it is an essential effort to identify how cell death and ectopic cartilage formation cooperatively or independently causes craniosynostosis. Interestingly, the premature suture fusion patterns between P0-Cre;caBmpr1a mice and Wnt1-Cre;caBmpr1a mice are different, which the anterior frontal suture commonly causes premature fusion in both P0-Cre;caBmpr1a mice and Wnt1-Cre;caBmpr1a mice while the naso-premaxillary suture causes premature fusion only in P0-Cre;caBmpr1a mice (Ueharu et al., 2023b). As we discussed, P0-Cre mice and Wnt1-Cre mice showed similar but not identical recombination patterns. Therefore, utilizing the two transgenic mice could facilitate to identify the pathological mechanisms for craniosynostosis by BMP signaling. On the other hand, Bmpr1a cKO mice (Axin2Cre−Dox;Bmpr1afl/fl) develop craniosynostosis (Maruyama et al., 2021). Self-renewal and osteogenic capability of suture mesenchymal stem cells were dramatically reduced in Bmpr1a cKO mice. These results suggest that defects of stem cells in sutures are one of the reasons for craniosynostosis rather than excess osteogenic differentiation by BMP signaling. The insights are letting us shift the strategies to the next step, which is how the stemness of suture stem cells is controlled, and whether we can control their stemness in a timely manner by genetic and epigenetic methods.
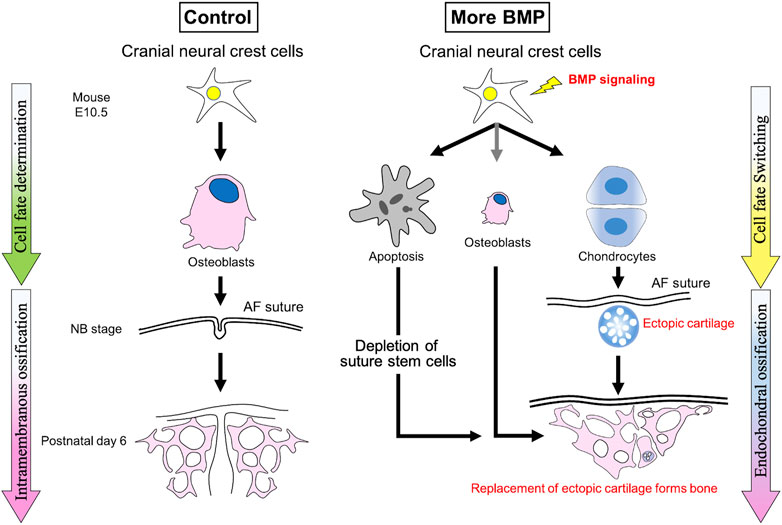
FIGURE 2. Augmentation of BMP signaling in cranial neural crest cells develops craniosynostosis through endochondral ossification due to cell fate switching towards chondrogenic fate. Enhanced BMP signaling alters cell fate of cranial neural crest cells towards chondrogenic fate at early embryonic stages along with excess cell death leading to ectopic cartilage formation in the sutures, where cause premature suture fusion. These data suggest that the premature fusion of cranial sutures is caused by endochondral ossification prompted by ectopically formed cartilage.
Taken together, both loss of and gain of function mutations of BMP signaling develop craniosynostosis and orofacial cleft. These results suggest that BMP signaling plays a critical role in calvarial development depending on stages of development by fine-tuning in proliferation, cell death, differentiation, and cell fate specification of cranial NCCs.
We described that many of BMP signaling-targeted transgenic mice develop CFAs. However, there are few reports regarding BMP-related CFAs in human. Here, we describe human cases of BMP-related CFAs and discuss how knowledge from animal models can help understanding etiology of BMP-related CFAs in human.
Duplication of human chromosome 10 q22.3q23.2, which includes BMPR1A, develops a hypertelorism (van Bon et al., 2011). Micro deletion of 20p12.3, including BMP2, develops cleft lip and palate (Sahoo et al., 2011). BMP4 polymorphisms are found in patients with cleft lips w/wo cleft palate (Li et al., 2017). Those reports may suggest that defects in BMP signaling develop CFAs in humans. In cases of mice, global loss of function mutations and gain of function mutations in BMP ligands, BMP receptors, and BMP-related genes causes embryonic lethal at the early embryonic stage (Mishina et al., 1995; Winnier et al., 1995; Zhang and Bradley, 1996; Mishina et al., 1999). It is reasonable to speculate that a significant change in BMP signaling activity may also cause lethality in human subjects. Recently, it has been reported that homozygous missense mutation of BMPR1A (results in amino acid substitution of BMPR1AR406L) develops brachycephaly with unilateral coronal craniosynostosis in humans with a slight elevation of phospho-SMAD levels (Russell et al., 2019). Mutations in ACVRL1 (ACVRL1V228I, kinase domain) and ACVR2A (ACVR2AT63A, BMP7 binding site) are found in patients with lambdoid craniosynostosis (Timberlake et al., 2022). Despite a lack of experimental evidence, it is possible to speculate that these two mutations alter levels of BMP signaling. These results suggest that slight elevation or slight suppression of BMP signaling in humans develop CFAs otherwise they cause embryonic lethal at an early embryonic stage.
Recent reports focusing on patients with non-syndromic craniosynostosis showed that single nucleotide polymorphisms (SNPs) were found in a Smad6 exon and a putative enhancer region of BMP2, and frameshift mutations in Smad6 were found (Justice et al., 2012; Komatsu and Mishina, 2016; Timberlake et al., 2016; Timberlake et al., 2018). SMAD6 is an inhibitory SMAD, that inhibits the phosphorylation of SMAD1/5/9 (Estrada et al., 2011). As discussed above, patients with unilateral coronal craniosynostosis slightly elevated the phospho-SMAD1/5/9 level (Russell et al., 2019). Based on the knowledge from animal studies, it is reasonable to speculate that mutations in regulatory sequences may slightly alter levels of wild-type proteins, which may lead to pathologic conditions. From that point of view, the fact that a limited portion of human subjects with the SNPs in the putative enhancer region of BMP2 develops CFA while all patients who also have mutations in a SMAD6 exon develop craniosynostosis eloquently demonstrate small increase of BMP signaling is critically involved in etiology of CFAs in human.
There are still uninvaded niches, especially, why missense or nonsense mutations in genes related to BMP signaling are rarely found in patients with CFAs. In the future, it is an essential effort to analyze BMP mutations in preterm and stillbirth babies. It is also essential to identify gene mutations in patients with non-syndromic CFAs. Genome-wide association studies (GWAS) and exome sequencing are powerful tools to determine whether they have mutations in BMP ligands, BMP receptors, Smads, noncanonical signaling of BMP signaling, and downstream target genes of BMP signaling, especially identifying SNPs in regulatory regions. We also need to examine whether these mutations change signaling levels of BMP signaling by testing the phosphorylation of SMAD1/5/9 and non-SMAD signaling levels, which is less understood at this moment. These efforts could provide novel therapeutic options for non-syndromic patients with CFAs.
HU and YM contribute to the conception and design of the review. HU wrote the manuscript draft and YM reviewed and edited the manuscript. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
This work was supported by grants from the NIH (R01DE020843 and R01DE027662 to YM).
We thank Benton Swanson, Rafael Correia Cavalcante, and Karen Zapien-Guerra for critical reading.
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Alessi Wolken, D. M., Idone, V., Hatsell, S. J., Yu, P. B., and Economides, A. N. (2018). The obligatory role of Activin A in the formation of heterotopic bone in Fibrodysplasia Ossificans Progressiva. Bone 109, 210–217. doi:10.1016/j.bone.2017.06.011
Chen, G., Ishan, M., Yang, J., Kishigami, S., Fukuda, T., Scott, G., et al. (2017). Specific and spatial labeling of P0-Cre versus Wnt1-Cre in cranial neural crest in early mouse embryos. Genesis 55, e23034. doi:10.1002/dvg.23034
Chen, Y., Wang, Z., Chen, Y., and Zhang, Y. (2019). Conditional deletion of Bmp2 in cranial neural crest cells recapitulates Pierre Robin sequence in mice. Cell. Tissue Res. 376, 199–210. doi:10.1007/s00441-018-2944-5
Danielian, P. S., Muccino, D., Rowitch, D. H., Michael, S. K., and McMahon, A. P. (1998). Modification of gene activity in mouse embryos in utero by a tamoxifen-inducible form of Cre recombinase. Curr. Biol. 8, 1323–1326. doi:10.1016/s0960-9822(07)00562-3
Duarte, G. A., Ramos, R. B., and Cardoso, M. C. (2016). Feeding methods for children with cleft lip and/or palate: A systematic review. Braz J. Otorhinolaryngol. 82, 602–609. doi:10.1016/j.bjorl.2015.10.020
Esparza, J., and Hinojosa, J. (2008). Complications in the surgical treatment of craniosynostosis and craniofacial syndromes: Apropos of 306 transcranial procedures. Childs Nerv. Syst. 24, 1421–1430. doi:10.1007/s00381-008-0691-8
Echelard, Y., Vassileva, G., and McMahon, A. P. (1994). Cis-acting regulatory sequences governing Wnt-1 expression in the developing mouse CNS. Development 120, 2213–2224. doi:10.1242/dev.120.8.2213
Estrada, K. D., Retting, K. N., Chin, A. M., and Lyons, K. M. (2011). Smad6 is essential to limit BMP signaling during cartilage development. J. Bone Min. Res. 26, 2498–2510. doi:10.1002/jbmr.443
Gomez-Puerto, M. C., Iyengar, P. V., García de Vinuesa, A., Ten Dijke, P., and Sanchez-Duffhues, G. (2019). Bone morphogenetic protein receptor signal transduction in human disease. J. Pathol. 247, 9–20. doi:10.1002/path.5170
Graf, D., Malik, Z., Hayano, S., and Mishina, Y. (2016). Common mechanisms in development and disease: BMP signaling in craniofacial development. Cytokine Growth Factor Rev. 27, 129–139. doi:10.1016/j.cytogfr.2015.11.004
Grafe, I., Alexander, S., Peterson, J. R., Snider, T. N., Levi, B., Lee, B., et al. (2018). TGF-Β family signaling in mesenchymal differentiation. Cold Spring Harb. Perspect. Biol. 10, a022202. doi:10.1101/cshperspect.a022202
Gu, S., Wu, W., Liu, C., Yang, L., Sun, C., Ye, W., et al. (2014). BMPRIA mediated signaling is essential for temporomandibular joint development in mice. PLoS One 9, e101000. doi:10.1371/journal.pone.0101000
Hayano, S., Komatsu, Y., Pan, H., and Mishina, Y. (2015). Augmented BMP signaling in the neural crest inhibits nasal cartilage morphogenesis by inducing p53-mediated apoptosis. Development 142, 1357–1367. doi:10.1242/dev.118802
Howe, L. J., Lee, M. K., Sharp, G. C., Davey Smith, G., St Pourcain, B., Shaffer, J. R., et al. (2018). Investigating the shared genetics of non-syndromic cleft lip/palate and facial morphology. PLoS Genet. 14, e1007501. doi:10.1371/journal.pgen.1007501
Hsieh, S. T., and Woo, A. S. (2019). Pierre robin sequence. Clin. Plast. Surg. 46, 249–259. doi:10.1016/j.cps.2018.11.010
Ishii, M., Arias, A. C., Liu, L., Chen, Y. B., Bronner, M. E., and Maxson, R. E. (2012). A stable cranial neural crest cell line from mouse. Stem Cells Dev. 21, 3069–3080. doi:10.1089/scd.2012.0155
Jabs, E. W., Müller, U., Li, X., Ma, L., Luo, W., Haworth, I. S., et al. (1993). A mutation in the homeodomain of the human MSX2 gene in a family affected with autosomal dominant craniosynostosis. Cell. 75, 443–450. doi:10.1016/0092-8674(93)90379-5
Justice, C. M., Yagnik, G., Kim, Y., Peter, I., Jabs, E. W., Erazo, M., et al. (2012). A genome-wide association study identifies susceptibility loci for nonsyndromic sagittal craniosynostosis near BMP2 and within BBS9. Nat. Genet. 44, 1360–1364. doi:10.1038/ng.2463
Kitami, K., Kitami, M., Kaku, M., Wang, B., and Komatsu, Y. (2018). BRCA1 and BRCA2 tumor suppressors in neural crest cells are essential for craniofacial bone development. PLoS Genet. 14, e1007340. doi:10.1371/journal.pgen.1007340
Komatsu, Y., and Mishina, Y. (2016). An epistatic explanation. Elife 5, e21162. doi:10.7554/eLife.21162
Komatsu, Y., Yu, P. B., Kamiya, N., Pan, H., Fukuda, T., Scott, G. J., et al. (2013). Augmentation of Smad-dependent BMP signaling in neural crest cells causes craniosynostosis in mice. J. Bone Min. Res. 28, 1422–1433. doi:10.1002/jbmr.1857
Kouskoura, T., Kozlova, A., Alexiou, M., Blumer, S., Zouvelou, V., Katsaros, C., et al. (2013). The etiology of cleft palate formation in BMP7-deficient mice. PLoS One 8, e59463. doi:10.1371/journal.pone.0059463
Kramer, K., Yang, J., Swanson, W. B., Hayano, S., Toda, M., Pan, H., et al. (2018). Rapamycin rescues BMP mediated midline craniosynostosis phenotype through reduction of mTOR signaling in a mouse model. Genesis 56, e23220. doi:10.1002/dvg.23220
Kulkarni, A. K., Louie, K. W., Yatabe, M., Ruellas, A. C. O., Mochida, Y., Cevidanes, L. H. S., et al. (2018). A ciliary protein EVC2/LIMBIN plays a critical role in the skull base for mid-facial development. Front. Physiol. 9, 1484. doi:10.3389/fphys.2018.01484
Le Douarin, N. M., Creuzet, S., Couly, G., and Dupin, E. (2004). Neural crest cell plasticity and its limits. Development 131, 4637–4650. doi:10.1242/dev.01350
Li, L., Wang, Y., Lin, M., Yuan, G., Yang, G., Zheng, Y., et al. (2013). Augmented BMPRIA-mediated BMP signaling in cranial neural crest lineage leads to cleft palate formation and delayed tooth differentiation. PLoS One 8, e66107. doi:10.1371/journal.pone.0066107
Li, N., Liu, J., Liu, H., Wang, S., Hu, P., Zhou, H., et al. (2021). Altered BMP-Smad4 signaling causes complete cleft palate by disturbing osteogenesis in palatal mesenchyme. J. Mol. Histol. 52, 45–61. doi:10.1007/s10735-020-09922-4
Li, Y. H., Yang, J., Zhang, J. L., Liu, J. Q., Zheng, Z., and Hu, D. H. (2017). BMP4 rs17563 polymorphism and nonsyndromic cleft lip with or without cleft palate: A meta-analysis. Med. Baltim. 96, e7676. doi:10.1097/MD.0000000000007676
Liu, X., Hayano, S., Pan, H., Inagaki, M., Ninomiya-Tsuji, J., Sun, H., et al. (2018). Compound mutations in Bmpr1a and Tak1 synergize facial deformities via increased cell death. Genesis 56, e23093. doi:10.1002/dvg.23093
Liu, Y. H., Tang, Z., Kundu, R. K., Wu, L., Luo, W., Zhu, D., et al. (1999). Msx2 gene dosage influences the number of proliferative osteogenic cells in growth centers of the developing murine skull: A possible mechanism for MSX2-mediated craniosynostosis in humans. Dev. Biol. 205, 260–274. doi:10.1006/dbio.1998.9114
Maruyama, T., Jiang, M., Abbott, A., Yu, H. I., Huang, Q., Chrzanowska-Wodnicka, M., et al. (2017). Rap1b is an effector of Axin2 regulating crosstalk of signaling pathways during skeletal development. J. Bone Min. Res. 32, 1816–1828. doi:10.1002/jbmr.3171
Maruyama, T., Mirando, A. J., Deng, C. X., and Hsu, W. (2010). The balance of WNT and FGF signaling influences mesenchymal stem cell fate during skeletal development. Sci. Signal 3, ra40. doi:10.1126/scisignal.2000727
Maruyama, T., Stevens, R., Boka, A., DiRienzo, L., Chang, C., Yu, H. I., et al. (2021). BMPR1A maintains skeletal stem cell properties in craniofacial development and craniosynostosis. Sci. Transl. Med. 13, eabb4416. doi:10.1126/scitranslmed.abb4416
Massagué, J., and Chen, Y. G. (2000). Controlling TGF-beta signaling. Genes. Dev. 14, 627–644. doi:10.1101/gad.14.6.627
Mishina, Y., and Snider, T. N. (2014). Neural crest cell signaling pathways critical to cranial bone development and pathology. Exp. Cell. Res. 325, 138–147. doi:10.1016/j.yexcr.2014.01.019
Mishina, Y., Crombie, R., Bradley, A., and Behringer, R. R. (1999). Multiple roles for activin-like kinase-2 signaling during mouse embryogenesis. Dev. Biol. 213, 314–326. doi:10.1006/dbio.1999.9378
Mishina, Y., Suzuki, A., Ueno, N., and Behringer, R. R. (1995). Bmpr encodes a type I bone morphogenetic protein receptor that is essential for gastrulation during mouse embryogenesis. Genes. Dev. 9, 3027–3037. doi:10.1101/gad.9.24.3027
Morriss-Kay, G. M., and Wilkie, A. O. (2005). Growth of the normal skull vault and its alteration in craniosynostosis: Insights from human genetics and experimental studies. J. Anat. 207, 637–653. doi:10.1111/j.1469-7580.2005.00475.x
Nasreddine, G., El Hajj, J., and Ghassibe-Sabbagh, M. (2021). Orofacial clefts embryology, classification, epidemiology, and genetics. Mutat. Res. Rev. Mutat. Res. 787, 108373. doi:10.1016/j.mrrev.2021.108373
Opriş, D., Băciuţ, G., Bran, S., Dinu, C., Armencea, G., Opriş, H., et al. (2022). The quality of life after cleft lip and palate surgery. Med. Pharm. Rep. 95, 461–466. doi:10.15386/mpr-2472
Pan, H., Zhang, H., Abraham, P., Komatsu, Y., Lyons, K., Kaartinen, V., et al. (2017). BmpR1A is a major type 1 BMP receptor for BMP-Smad signaling during skull development. Dev. Biol. 429, 260–270. doi:10.1016/j.ydbio.2017.06.020
Reid, J. (2004). A review of feeding interventions for infants with cleft palate. Cleft Palate Craniofac J. 41, 268–278. doi:10.1597/02-148.1
Roybal, P. G., Wu, N. L., Sun, J., Ting, M. C., Schafer, C. A., and Maxson, R. E. (2010). Inactivation of Msx1 and Msx2 in neural crest reveals an unexpected role in suppressing heterotopic bone formation in the head. Dev. Biol. 343, 28–39. doi:10.1016/j.ydbio.2010.04.007
Russell, B. E., Rigueur, D., Weaver, K. N., Sund, K., Basil, J. S., Hufnagel, R. B., et al. (2019). Homozygous missense variant in BMPR1A resulting in BMPR signaling disruption and syndromic features. Mol. Genet. Genomic Med. 7, e969. doi:10.1002/mgg3.969
Sahoo, T., Theisen, A., Sanchez-Lara, P. A., Marble, M., Schweitzer, D. N., Torchia, B. S., et al. (2011). Microdeletion 20p12.3 involving BMP2 contributes to syndromic forms of cleft palate. Am. J. Med. Genet. A 155a, 1646–1653. doi:10.1002/ajmg.a.34063
Sakai, D., and Trainor, P. A. (2016). Face off against ROS: Tcof1/Treacle safeguards neuroepithelial cells and progenitor neural crest cells from oxidative stress during craniofacial development. Dev. Growth Differ. 58, 577–585. doi:10.1111/dgd.12305
Seruya, M., Oh, A. K., Boyajian, M. J., Posnick, J. C., Myseros, J. S., Yaun, A. L., et al. (2011). Long-term outcomes of primary craniofacial reconstruction for craniosynostosis: A 12-year experience. Plast. Reconstr. Surg. 127, 2397–2406. doi:10.1097/PRS.0b013e318213a178
Shkoukani, M. A., Lawrence, L. A., Liebertz, D. J., and Svider, P. F. (2014). Cleft palate: A clinical review. Birth Defects Res. C Embryo Today 102, 333–342. doi:10.1002/bdrc.21083
Song, Z., Liu, C., Iwata, J., Gu, S., Suzuki, A., Sun, C., et al. (2013). Mice with Tak1 deficiency in neural crest lineage exhibit cleft palate associated with abnormal tongue development. J. Biol. Chem. 288, 10440–10450. doi:10.1074/jbc.M112.432286
Spazzapan, P., Kocar, M., Eberlinc, A., Haber, B., and Velnar, T. (2022). Craniofacial reconstructions in children with craniosynostosis. J. Integr. Neurosci. 21, 106. doi:10.31083/j.jin2104106
Srinivasan, A., and Toh, Y. C. (2019). Human pluripotent stem cell-derived neural crest cells for tissue regeneration and disease modeling. Front. Mol. Neurosci. 12, 39. doi:10.3389/fnmol.2019.00039
Swanson, W. B., and Mishina, Y. (2021). “The Biology of the sutures of the skull,” in The sutures of the skull: Anatomy, embryology, imaging, and surgery. Editors M. Turgut, R. S. Tubbs, A. T. Turgut, and A. S. Dumont (Cham: Springer International Publishing), 171–199.
Teng, L., Mundell, N. A., Frist, A. Y., Wang, Q., and Labosky, P. A. (2008). Requirement for Foxd3 in the maintenance of neural crest progenitors. Development 135, 1615–1624. doi:10.1242/dev.012179
Timberlake, A. T., Choi, J., Zaidi, S., Lu, Q., Nelson-Williams, C., Brooks, E. D., et al. (2016). Two locus inheritance of non-syndromic midline craniosynostosis via rare SMAD6 and common BMP2 alleles. Elife 5, e20125. doi:10.7554/eLife.20125
Timberlake, A. T., Kiziltug, E., Jin, S. C., Nelson-Williams, C., Loring, E., Allocco, A., et al. (2022). De novo mutations in the BMP signaling pathway in lambdoid craniosynostosis. Hum. Genet. 142, 21–32. doi:10.1007/s00439-022-02477-2
Timberlake, A. T., Wu, R., Nelson-Williams, C., Furey, C. G., Hildebrand, K. I., Elton, S. W., et al. (2018). Co-occurrence of frameshift mutations in SMAD6 and TCF12 in a child with complex craniosynostosis. Hum. Genome Var. 5, 14. doi:10.1038/s41439-018-0014-x
Twigg, S. R., and Wilkie, A. O. (2015). A genetic-pathophysiological framework for craniosynostosis. Am. J. Hum. Genet. 97, 359–377. doi:10.1016/j.ajhg.2015.07.006
Ueharu, H., Pan, H., Hayano, S., Zapien-Guerra, K., Yang, J., and Mishina, Y. (2023a). Augmentation of bone morphogenetic protein signaling in cranial neural crest cells in mice deforms skull base due to premature fusion of intersphenoidal synchondrosis. Genesis 61, e23509. doi:10.1002/dvg.23509
Ueharu, H., Pan, H., Liu, X., Ishii, M., Pongetti, J., Kulkarni, A. K., et al. (2023b). Augmentation of BMP signaling in cranial neural crest cells leads to premature cranial sutures fusion through endochondral ossification in mice. JBMR Plus 7, e10716. doi:10.1002/jbm4.10716
Ueharu, H., Yoshida, S., Kikkawa, T., Kanno, N., Higuchi, M., Kato, T., et al. (2017). Gene tracing analysis reveals the contribution of neural crest-derived cells in pituitary development. J. Anat. 230, 373–380. doi:10.1111/joa.12572
Urist, M. R. (1965). Bone: Formation by autoinduction. Science 150, 893–899. doi:10.1126/science.150.3698.893
van Bon, B. W., Balciuniene, J., Fruhman, G., Nagamani, S. C., Broome, D. L., Cameron, E., et al. (2011). The phenotype of recurrent 10q22q23 deletions and duplications. Eur. J. Hum. Genet. 19, 400–408. doi:10.1038/ejhg.2010.211
Winnier, G., Blessing, M., Labosky, P. A., and Hogan, B. L. (1995). Bone morphogenetic protein-4 is required for mesoderm formation and patterning in the mouse. Genes. Dev. 9, 2105–2116. doi:10.1101/gad.9.17.2105
Wang, S. K., Komatsu, Y., and Mishina, Y. (2011). Potential contribution of neural crest cells to dental enamel formation. Biochem. Biophys. Res. Commun. 415, 114–119. doi:10.1016/j.bbrc.2011.10.026
Warren, D. W., Drake, A. F., and Davis, J. U. (1992). Nasal airway in breathing and speech. Cleft Palate Craniofac J. 29, 511–519. doi:10.1597/1545-1569_1992_029_0511_naibas_2.3.co_2
Warren, S. M., Brunet, L. J., Harland, R. M., Economides, A. N., and Longaker, M. T. (2003). The BMP antagonist noggin regulates cranial suture fusion. Nature 422, 625–629. doi:10.1038/nature01545
Wei, X., Hu, M., Mishina, Y., and Liu, F. (2016). Developmental regulation of the growth plate and cranial synchondrosis. J. Dent. Res. 95, 1221–1229. doi:10.1177/0022034516651823
Willie, D., Holmes, G., Jabs, E. W., and Wu, M. (2022). Cleft palate in Apert syndrome. J. Dev. Biol. 10, 33. doi:10.3390/jdb10030033
Wu, M., Chen, G., and Li, Y. P. (2016). TGF-β and BMP signaling in osteoblast, skeletal development, and bone formation, homeostasis and disease. Bone Res. 4, 16009. doi:10.1038/boneres.2016.9
Yamauchi, Y., Abe, K., Mantani, A., Hitoshi, Y., Suzuki, M., Osuzu, F., et al. (1999). A novel transgenic technique that allows specific marking of the neural crest cell lineage in mice. Dev. Biol. 212, 191–203. doi:10.1006/dbio.1999.9323
Yang, J., Kitami, M., Pan, H., Nakamura, M. T., Zhang, H., Liu, F., et al. (2021a). Augmented BMP signaling commits cranial neural crest cells to a chondrogenic fate by suppressing autophagic β-catenin degradation. Sci. Signal 14, eaaz9368. doi:10.1126/scisignal.aaz9368
Yang, J., Toda Nakamura, M., Hallett, S. A., Ueharu, H., Zhang, H., Kelley, K., et al. (2021b). Generation of a new mouse line with conditionally activated signaling through the BMP receptor, ACVR1: A tool to characterize pleiotropic roles of BMP functions. Genesis 59, e23419. doi:10.1002/dvg.23419
Yu, H. M., Jerchow, B., Sheu, T. J., Liu, B., Costantini, F., Puzas, J. E., et al. (2005). The role of Axin2 in calvarial morphogenesis and craniosynostosis. Development 132, 1995–2005. doi:10.1242/dev.01786
Zhang, H., and Bradley, A. (1996). Mice deficient for BMP2 are nonviable and have defects in amnion/chorion and cardiac development. Development 122, 2977–2986. doi:10.1242/dev.122.10.2977
Zhang, H., Louie, K. W., Kulkarni, A. K., Zapien-Guerra, K., Yang, J., and Mishina, Y. (2022). The posterior Part Influences the anterior part of the mouse cranial base development. JBMR Plus 6, e10589. doi:10.1002/jbm4.10589
Keywords: craniofacial development, BMP, transgenic animal, orofacial cleft, craniosynostosis
Citation: Ueharu H and Mishina Y (2023) BMP signaling during craniofacial development: new insights into pathological mechanisms leading to craniofacial anomalies. Front. Physiol. 14:1170511. doi: 10.3389/fphys.2023.1170511
Received: 21 February 2023; Accepted: 10 May 2023;
Published: 18 May 2023.
Edited by:
Junichi Iwata, University of Texas Health Science Center at Houston, United StatesReviewed by:
Wei Hsu, Harvard University, United StatesCopyright © 2023 Ueharu and Mishina. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: Hiroki Ueharu, aGlyb2tpdUB1bWljaC5lZHU=; Yuji Mishina, bWlzaGluYUB1bWljaC5lZHU=
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.