
95% of researchers rate our articles as excellent or good
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.
Find out more
REVIEW article
Front. Physiol. , 23 May 2023
Sec. Cell Physiology
Volume 14 - 2023 | https://doi.org/10.3389/fphys.2023.1146834
This article is part of the Research Topic Kallikrein-Kinin System: Insights into a Multifunctional System View all 5 articles
 Aleksandr Shamanaev
Aleksandr Shamanaev S. Kent DickesonIvan Ivanov
S. Kent DickesonIvan Ivanov Maxim Litvak
Maxim Litvak Mao-Fu SunSunil Kumar
Mao-Fu SunSunil Kumar Quifang ChengPriyanka SrivastavaTracey Z. He
Quifang ChengPriyanka SrivastavaTracey Z. He David Gailani*
David Gailani*Patients with the inherited disorder hereditary angioedema (HAE) suffer from episodes of soft tissue swelling due to excessive bradykinin production. In most cases, dysregulation of the plasma kallikrein-kinin system due to deficiency of plasma C1 inhibitor is the underlying cause. However, at least 10% of HAE patients have normal plasma C1 inhibitor activity levels, indicating their syndrome is the result of other causes. Two mutations in plasma protease zymogens that appear causative for HAE with normal C1 inhibitor activity have been identified in multiple families. Both appear to alter protease activity in a gain-of-function manner. Lysine or arginine substitutions for threonine 309 in factor XII introduces a new protease cleavage site that results in formation of a truncated factor XII protein (Δ-factor XII) that accelerates kallikrein-kinin system activity. A glutamic acid substitution for lysine 311 in the fibrinolytic protein plasminogen creates a consensus binding site for lysine/arginine side chains. The plasmin form of the variant plasminogen cleaves plasma kininogens to release bradykinin directly, bypassing the kallikrein-kinin system. Here we review work on the mechanisms of action of the FXII-Lys/Arg309 and Plasminogen-Glu311 variants, and discuss the clinical implications of these mechanisms.
The term hereditary angioedema (HAE) encompasses a group of inherited disorders characterized by episodic swelling involving mucosal, submucosal and/or subcutaneous tissues. Edema primarily involves the face, oropharynx, hands, genitals and gastrointestinal tract and may be associated with pain (Kaplan and Joseph, 2010; Schmaier, 2019; Bova et al., 2020; Busse and Christiansen, 2020; Lumry and Settipane, 2020). It is estimated that one in 50,000–100,000 individuals have the condition. Tissue swelling in HAE is usually not associated with itching (urticaria), which helps to distinguish it from more common forms of angioedema induced by histamine release from mast cells (Maurer and Magerl, 2021; Farkas et al., 2022a). Instead, the symptoms of HAE are due primarily to dysregulated formation of the peptide bradykinin that promotes vasodilatation and increases vascular permeability (Fields et al., 1983; Kaplan and Joseph, 2010; Schmaier, 2016; De Maat et al., 2018; Schmaier, 2019; Bova et al., 2020; Busse and Christiansen, 2020; Lumry and Settipane, 2020; Wedner, 2020). Bradykinin is normally generated through the activity of the plasma kallikrein-kinin system (KKS).
The KKS is comprised of three blood plasma proteins that are synthesized in the liver. Prekallikrein (PK) and factor XII (FXII) are zymogens of the trypsin-like proteases plasma kallikrein (PKa) and FXIIa, respectively (Long et al., 2016; Schmaier, 2016; Srivastava and Gailani, 2020; Girolami et al., 2021). Conversion of PK to PKa, and FXII to FXIIa, involves internal proteolytic cleavage in both zymogens (Figure 1A). As PK is a substrate for FXIIa, and FXII a substrate for PKa, PK, and FXII reciprocally activate each other when mixed in solution (Figure 1A) (Ivanov et al., 2017; Ivanov et al., 2019; Srivastava and Gailani, 2020). Low levels of proteolytic activity intrinsic to zymogen FXII and PK may sustain reciprocal activation in plasma (Ivanov et al., 2017; Ivanov et al., 2020). The third KKS component, high-molecular-weight kininogen (HK), is a 110-kDa glycoprotein that contains the nine amino acid bradykinin sequence within its D4 domain (Figure 1B) (Ponczek, 2021; Dickeson et al., 2022; Kaplan et al., 2022). PKa cleaves HK after Lys362 and Arg371, releasing the nine amino acid bradykinin peptide. The physiologic and pathologic effects of bradykinin are mediated primarily through the G-protein-coupled B2 receptor, which is constitutively expressed on cell surfaces in many tissues (Lau et al., 2020; Marceau et al., 2020).
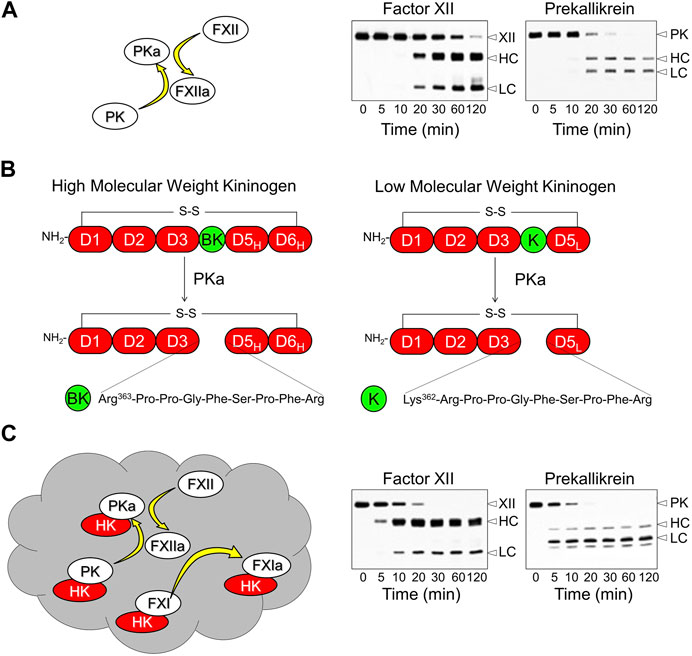
FIGURE 1. The Kallikrein-Kinin System and Contact Activation. (A) In the absence of a surface, factor XII (FXII) and prekallikrein (PK) will reciprocally convert each other to plasma kallikrein (PKa) and factor XIIa (FXIIa) in the absence of a surface. This process involves internal proteolytic cleavage of each protein creating a heavy chain (HC) and catalytic light chain (LC) that remain connected by a disulfide bond. The western blots in the right hand panels of the image are from a time course experiment in which 200 nM FXII and 200 nM PK were incubated together. Blots were run under reducing conditions and were developed with polyclonal antibodies to FXII (left) or PK (right) (B) Schematic diagrams of kininogens. High molecular weight kininogen (HK, left) contains six domains (D1-D6) with the bradykinin (BK) sequence within domain 4. The D1-D4 domains of low molecular weight kininogen (LK, right) are identical to those of HK, but the D5 domain is shorter and there is no D6 domain. PKa cleaves HK at two locations to release the nine amino acid bradykinin peptide from HK. It releases BK weakly from LK because of the absence of D6, which contains a binding site for PKa. LK is cleaved preferentially by tissue kallikreins releasing the ten amino acid kallidin (K) peptide. Kallidin is also called lysyl-bradykinin. (C) Contact activation on a negatively charged surface (gray cloud) involves autoactivation of FXII, subsequent reciprocal activation of FXII and PK, and FXIIa activation of factor XI (FXI). HK facilitates PK and FXI binding to the surface. Note in the western blots showing FXII and PK cleavage on a contact surface that the reaction proceeds faster than in the blots in panel (A). Adapted from Shamanaev A et al. Recent advances in factor XII structure and function. Curr Opin Hematol. 2022; 29:233–243, and used with permission.
Reciprocal activation of PK and FXII is enhanced by a process called contact activation, which occurs when KKS components bind to certain macromolecules or surfaces (Figure 1C) (Colman and Schmaier, 1997; Long et al., 2016; Schmaier, 2016; Petersen et al., 2022). A variety of organic (e.g., nucleic acids, glycosaminoglycans) and inorganic (e.g., polyphosphates, silicates) substances support plasma contact activation (Colman and Schmaier, 1997; Long et al., 2016; Schmaier, 2016; Tillman and Gailani, 2018). Most have a negative surface charge. During contact activation, FXII is autocatalytically converted to FXIIa, initiating more rapid reciprocal activation with PK than occurs in solution (Silverberg et al., 1980a; Long et al., 2016; Girolami et al., 2021). Most PK in plasma circulates as a complex with HK (Mandle et al., 1976; Thompson et al., 1979). HK, in addition to serving as a substrate for PKa, is a cofactor that facilitate PK surface-binding, and may alter PK conformation to make it a better substrate for FXIIa (Wiggins et al., 1977).
The main KKS regulator in plasma is the serpin C1-inhibitor (C1-INH) (Farkas et al., 2022b; Karnaukhova, 2022), encoded by the SERPING1 gene. C1-INH inhibits PKa and FXIIa, placing a limit on the rate of basal reciprocal turnover of PK and FXII in plasma. This likely maintains bradykinin production within a physiologic range that contributes to setting normal vascular tone and permeability (Iwaki and Castellino, 2006; Revenko et al., 2011). The vascular endothelium likely supports basal bradykinin generation. The components of the KKS assemble on multi-component receptors on vascular endothelial cells comprised of the urokinase plasminogen activator receptor, cytokeratin 1 and the gC1q receptor (Schmaier et al., 1988; Joseph et al., 1999). Binding of KKS proteins to a contact surface at least partially overcomes the capacity of C1-INH to restrict PK and FXII activation, increasing bradykinin production. Local surface-induced contact activation may generate bradykinin at injury sites, promoting vascular leak, tissue edema and pain sensation. It is widely assumed that surface-mediated enhancement of KKS activity is important in HAE, although evidence to support this hypothesis is relatively meager, and the nature of the surfaces involved are not certain.
HK is encoded by the Kng1 gene. The protein is organized into 6 domains (D1 through D6, Figure 1B, left). D4 contains the bradykinin sequence, D5 is involved in surface-binding, and D6 contains a binding site for PK (Silverberg et al., 1980b; Colman and Schmaier, 1997; Ponczek, 2021; Dickeson et al., 2022; Kaplan et al., 2022). Kng1 also encodes an alternatively spliced mRNA for the plasma protein low-molecular-weight kininogen (LK, Figure 1B, right) (Müller-Esterl et al., 1982; Maier et al., 1983; Sueiras-Diaz et al., 1994; Dickeson et al., 2022; Kaplan et al., 2022). The D1 through D4 domains of LK are identical to those of HK, but D5 is shorter and there is no D6. Because of this, LK interacts weakly with surfaces and with PK and PKa. Consequently, LK is a poor substrate for PKa (Müller-Esterl et al., 1982; Maier et al., 1983; Sueiras-Diaz et al., 1994; Ivanov et al., 2020), and is thought to be primarily a substrate for tissue kallikreins, which cleave it after Met361 and Arg371 to release the decapeptide lysyl-bradykinin (also called kallidin, Figure 1B), another potent B2 receptor agonist (Lau et al., 2020; Marceau et al., 2020). For our discussion, it is important to recognize that LK is present in plasma at up to four times the concentration of HK (2.4 versus 0.6 μM, respectively) and is, therefore, a greater potential source for vasoactive kinins than is HK.
Hypothetically, bradykinin-triggered angioedema could be caused by inherited or acquired conditions that increase kinin formation, increase kinin half-lives, or increase tissue sensitivity to kinins (Table 1, left column). Most patients with HAE have reduced plasma C1-INH activity (5%–30% of normal) due to reduced C1-INH protein (type 1) or a dysfunctional C1-INH variant (type 2) (Table 1, right column) (Kaplan and Joseph, 2010; Schmaier, 2019; Bova et al., 2020; Busse and Christiansen, 2020; Lumry and Settipane, 2020; Farkas et al., 2022b). In these patients, angioedema typically responds to C1-INH infusion (Kaplan and Joseph, 2010; Schmaier, 2019; Bova et al., 2020; Busse and Christiansen, 2020; Lumry and Settipane, 2020; Valerieva and Longhurst, 2022), or drugs that neutralize PK/PKa or FXII/FXIIa (Cohn et al., 2020; Davoine et al., 2020; Busse and Kaplan, 2022), consistent with KKS hyperactivity as an underlying cause. Patients with physical findings and histories consistent with HAE, but with normal C1-INH levels (HAEnC1), were first reported in the year 2000, and may represent 10% or more of all HAE patients (Bork et al., 2000; Santacroce et al., 2021). Six mutations in genes unrelated to C1-INH have been identified in HAEnC1 patients (Table 1, right column) that are assumed to be causative for, or contributory to, angioedema (Dewald and Bork, 2006; Bork et al., 2015; Bafunno et al., 2018; Dewald, 2018; Bork et al., 2019; Ariano et al., 2020; Bork et al., 2021; Veronez et al., 2021). Here we discuss work on mechanisms by which two mutations, one in FXII that changes Thr309 to lysine or arginine (Ivanov et al., 2019) and one in plasminogen changing Lys311 to glutamic acid (Dickeson et al., 2022), contribute to HAE.
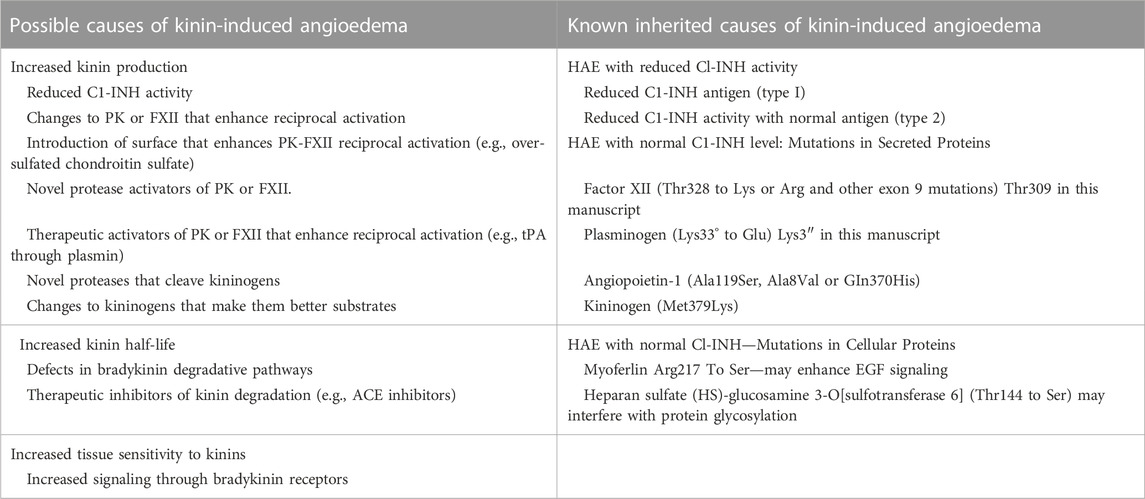
TABLE 1. Mechanisms underlying hereditary angioedema.
FXII is an 80-kDa polypeptide encoded by the F12 gene that is synthesized primarily in hepatocytes (Cool et al., 1985; de Maat and Maas, 2016; Maas and Renné, 2018; Shamanaev et al., 2022a; Shamanaev et al., 2022b). Figure 2 shows the amino acid sequence and predicted domain structures for human FXII (Cool et al., 1985; Shamanaev et al., 2022a; Shamanaev et al., 2022b). The N-terminal isoleucine of the protein in plasma is designated residue 1. From the N-terminus, FXII contains a fibronectin type 2 (FN2), first epidermal growth factor (EGF1), fibronectin type 1 (FN1), second epidermal growth factor (EGF2), and kringle (KNG) domain, a proline-rich region (PRR), and a trypsin-like protease domain. Conversion of FXII to FXIIa requires proteolytic cleavage after Arg353, creating a heavy chain (amino acids 1–353) and light chain (amino acids 354–596) that remain connected by the Cys340-Cys467 disulfide bond. The heavy chain has several functions. First, when FXII is in solution (not bound to a surface), elements of the heavy chain maintain the protein in a “closed” conformation that is relatively resistant to activation by PKa (de Maat et al., 2019; Ivanov et al., 2019; Clark et al., 2020; Shamanaev et al., 2022a; Shamanaev et al., 2022b). Second, during contact activation, the heavy chain mediates FXII surface-binding, opening the protein conformation to facilitate activation (de Maat et al., 2019; Shamanaev et al., 2022a; Shamanaev et al., 2022b). Third, once FXII is converted to FXIIa, the heavy chain keeps the protease associated with the surface (de Maat and Maas, 2016; Maas and Renné, 2018; Shamanaev et al., 2022a; Shamanaev et al., 2022b), where it efficiently activates FXII (autoactivation) and PK.
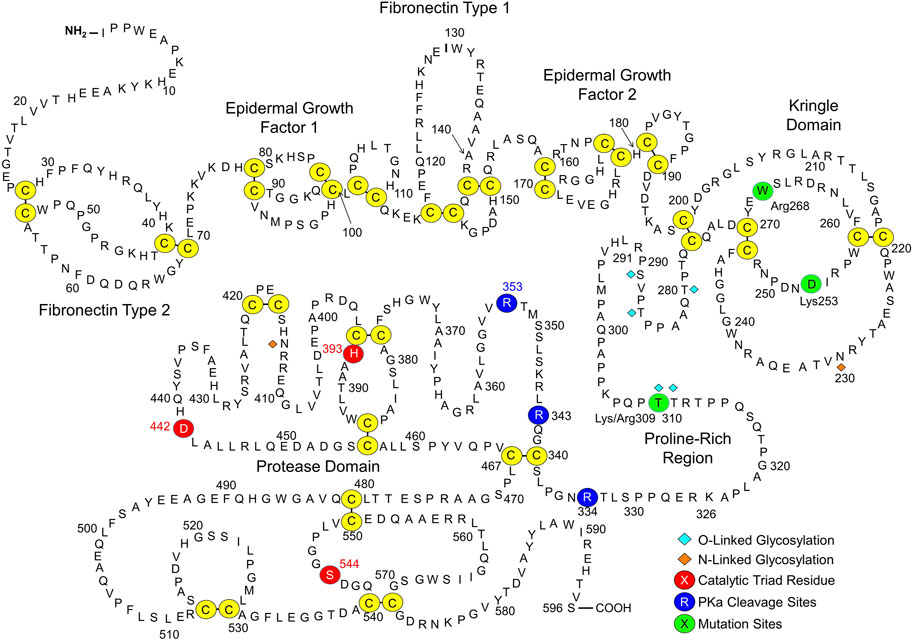
FIGURE 2. Factor XII. Shown is the amino acid sequence and domain structure of human plasma FXII. The FXII heavy chain contains fibronectin type 2, epidermal growth factor 1, fibronectin type 1, epidermal growth factor 2 and kringle domains, and a proline-rich region. The protease domain is a trypsin-like catalytic unit. Disulfide bonds between cysteine residues are shown in yellow. The catalytic triad (His393, Asp442, and Ser544) is indicated by red circles. Cleavage sites for PKa (Arg334, Arg343, Arg353) are indicated by dark blue circles. Adapted from Shamanaev A et al. Recent advances in factor XII structure and function. Curr Opin Hematol. 2022; 29:233–243, and used with permission.
In 2006, Dewald and Bork described single base pair changes in exon 9 of the F12 gene in some HAEnC1 patients (Dewald and Bork, 2006). The mutations result in replacement of Thr309 in the PRR (Thr328 if counting from the initiator methionine on the signal peptide) with either lysine or arginine (Figure 2). The substitution disrupts an N-linked glycosylation site at residue 309 (Björkqvist et al., 2015) and may affect an adjacent glycosylation site at Thr310. Lys/Arg309 and a few rarer F12 exon 9 mutations have been identified in more than 150 families with HAE (Dewald and Bork, 2006; Bork et al., 2015; Santacroce et al., 2021; Veronez et al., 2021). Introducing a basic amino acid into a protein sequence may create a novel cleavage site for trypsin-like proteases. In 2016 de Maat et al. showed that the fibrinolytic protease plasmin cleaves FXII-Lys/Arg309 variants after residue 309 (Figure 3A) (de Maat et al., 2016). Subsequently, we reported that the coagulation proteases thrombin and factor XIa cleave FXII-Lys/Arg309 at the same site (Figure 3A) (Ivanov et al., 2019), perhaps explaining why patients with Lys/Arg309 substitutions often have attacks of angioedema following trauma that activates the coagulation mechanism (Müller-Esterl et al., 1982). Cleavage after Lys/Arg309 separates most of the heavy chain from the protease domain, resulting in a truncated FXII (ΔFXII, Figure 3B) that cannot bind properly to surfaces (Ivanov et al., 2019).
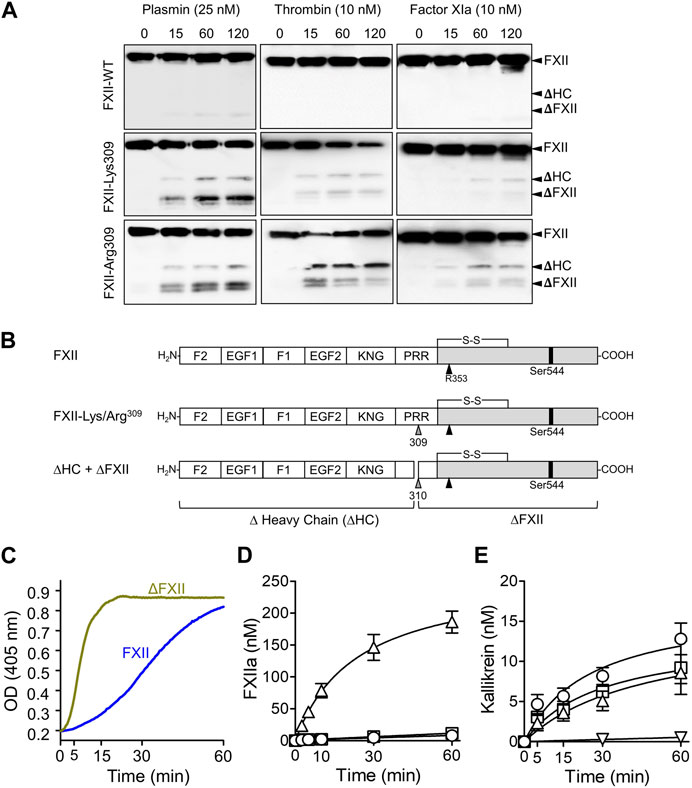
FIGURE 3. ΔFXII. (A) Non-reducing Western blots for time courses of FXII species (200 nM) cleavage by 25 nM plasmin (left column), 10 nM thrombin (center) or 10 nM factor XIa. Wild type FXII (top row), FXII-Lys309 (middle row), FXII-Lys309 (bottom row). Blots were developed with a mixture of monoclonal IgGs to the FXII heavy and light chains. Positions of standards for FXII (FXII), the heavy (HC) and light (LC) chains of FXIIa, the heavy chain of FXII-Lys309 or FXII-Arg309 cleaved after residue 309 (ΔHC) and FXII residues Thr310 to Ser596 (ΔFXII). (B) Schematic diagrams of wild type FXII and FXII with lysine or arginine replacement of Thr309 (gray arrow). Cleavage after Lys/Arg309 creates two proteins, Δ-heavy chain (ΔHC) and ΔFXII. (C) FXII-PK Reciprocal activation. PK (60 nM) was mixed with 12.5 nM FXII or ∆FXII, and 200 nM chromogenic substrate S-2302 at 37°C. Changes in optical density (OD) at 405 nm were continuously monitored. The signals are created by the activities of kallikrein and FXIIa. (D) Activation of 200 nM FXII (O), the full-length FXII precursor used to generate ∆FXII (•), or ∆FXII (△) by PKa (10 nM) at 37°C. FXIIa activity was measured by chromogenic assay. (E) PK (60 nM) was incubated with 50 pM FXIIa (O), βFXIIa (□) or ΔFXIIa-3C (△), or no FXIIa (▽) at 37°C. At various times PKa generation was determined by chromogenic assay. (A–D) are from Ivanov et al. (2019), and (C) is from Shamanaev et al. (2022a).
As shown in Figure 1, mixing PK and FXII in solution leads to reciprocal activation of both proteases, and the rates of both reactions are increased when a surface is added (Srivastava and Gailani, 2020; Shamanaev et al., 2022a; Shamanaev et al., 2022b). Replacing FXII with ΔFXII accelerates reciprocal activation with PK in the absence of a surface (Figure 3C) (Ivanov et al., 2019; Shamanaev et al., 2022a; Shamanaev et al., 2022b). Indeed, ΔFXII accelerates reciprocal activation with PK to a degree similar to FXII variants that lack heavy chain regulatory function (Shamanaev et al., 2022a; Shamanaev et al., 2022b). This suggests that ΔFXII is activated faster than FXII by PKa, that PK is activated more rapidly by ΔFXIIa than by FXIIa, or a combination of the processes. ΔFXII is activated by PKa at least 15-fold more rapidly than is FXII (Figure 3D) (Ivanov et al., 2019), consistent with absence of heavy chain regulatory function that normally maintains FXII in a closed conformation (Ivanov et al., 2019; Shamanaev et al., 2022a). In contrast, ΔFXIIa and full-length FXIIa activate PK at roughly similar rates, indicating the heavy chain is not required for PK activation in the absence of a surface (Figure 3E) (Ivanov et al., 2019). Interestingly, adding surface to reactions containing ΔFXII and PK does not accelerate activation as in reactions with full-length FXII, probably because FXII requires its heavy chain to bind to a surface (Ivanov et al., 2019).
These observations support a model in which FXII truncation results in a protein that is activated at a supraphysiologic rate by PKa. The greater amount of activated FXIIa protease generated (ΔFXIIa, in this case), in turn, accelerates PK activation. Accelerated reciprocal activation mediated by ΔFXII may overwhelm the capacity of C1-INH at physiologic concentrations to control the reaction, resulting in a surface-independent increase in bradykinin. Consistent with this hypothesis, adding ΔFXII, but not full-length FXII or the full-length precursor of ΔFXII (FXII-Lys/Arg309), to human plasma results in rapid HK cleavage (Figure 4A, top row) and bradykinin release (Figure 4B) (Ivanov et al., 2019). Running the plasma reactions in the presence of a surface (silica) accelerates HK cleavage with full-length FXII and FXII-Lys/Arg309, but does not change the effect of ΔFXII (Figure 4A, bottom row), which cannot bind to the surface. HK cleavage occurs rapidly after intravenous infusion of ΔFXII into wild type C57Bl/6 mice, but does not occur after infusion of FXII or FXII-Lys/Arg309 (Figure 4C). HK cleavage does not occur after infusing ΔFXII into mice lacking PK (Figure 4D, left panel), and ΔFXII does not induce bradykinin generation in normal human plasma that contains the potent PKa inhibitor KV999272 (Figure 4B), supporting the hypothesis that ΔFXII causes marked acceleration of the plasma KKS.
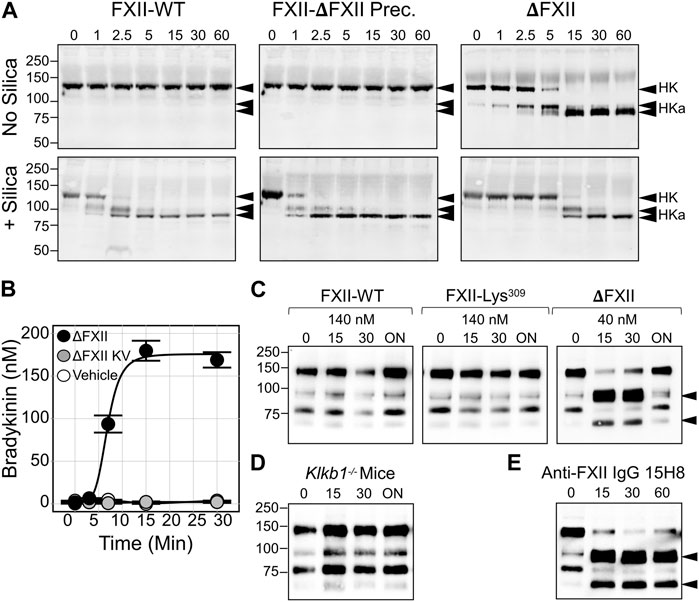
FIGURE 4. Effects of ΔFXII on the KKS. (A) HK cleavage in human plasma. Shown are western blots of human FXII-deficient plasma supplemented with FXII-WT. The full-length precursor of ∆FXII or ∆FXII (400 nM) in the absence (−) or presence (+) of a silica-based reagent that induces contact activation. At indicated times samples were removed into non-reducing sample buffer. Western blots were probed with goat anti-human HK IgG (HK). Positions of standards for HK, and the two bands of cleaved HK (HKa), are shown on the right. Positions of molecular mass standards in kilodaltons are shown to the left of the images. (B) Bradykinin generation in normal plasma after addition of 160 nM ∆FXII (•), ∆FXII and the PKa inhibitor (KV999272 10 nM; •), or vehicle (O). Bradykinin was measured by ELISA. (C) FXII-WT, FXII-Lys309 or ∆FXII were administered intravenously to wild-type C57Bl/6 mice to an estimated final plasma concentration of 140 nM or 40 nM. Shown are non-reducing western blots of plasma collected 0, 15, 30 min, or ∼18 h (ON, for overnight) after FXII infusion. Blots were developed with anti-murine HK IgG (anti-mHK). (D) As in (C), except that 40 nM ∆FXII was infused into Klkb1 null (PK deficient mice). (E) As in (C), except that FXII-deficient mice replete with 140 nM FXII-WT were treated with monoclonal antibody 15H8, which binds to the human FXII FN2 domain. For (C,E), the positions of bands indicating cleaved HK are indicated by black arrows. (A–E) are from Ivanov et al. (2019). (B) is from Dickeson et al. (2022).
Angioedema caused by ΔFXII may be one example of a broader phenomenon involving loss of intrinsic regulation of FXII activation via removal or disruption of the heavy chain, rather than loss of extrinsic regulation due to C1-INH deficiency. Scheffel et al. described a FXII Trp268 to arginine substitution in patients with a novel autoinflammatory syndrome (Scheffel et al., 2020). Hofman et al. showed that Arg268 disrupts the closed conformation of FXII, facilitating rapid activation and truncation within the KNG domain (Figure 2) (Hofman et al., 2020). Zamolodchikov et al. described an alternatively spliced FXII mRNA expressed in neurons that encodes a truncated FXII (the first amino acid is FXII residue 297) found in cerebrospinal fluid of patients with Alzheimer disease or multiple sclerosis (Figure 2) (Zamolodchikov et al., 2019). Like ΔFXII, this truncated protein is rapidly activated by PKa. de Maat and co-workers reported that truncation of wild type human FXII within the PRR by neutrophil elastase or cathepsin K results in proteins that are rapidly activated by PKa (Figure 2) (de Maat et al., 2019).
Novel inhibitors directed at FXII and FXIIa are being developed to treat or prevent thrombotic disorders (Fredenburgh and Weitz, 2021; Kluge et al., 2022). Because individuals lacking FXII do not have obvious abnormalities related to the deficiency, a long-acting compound such as an antibody could hypothetically be used to safely block FXII activation on a long-term basis. Antibodies to the FXII heavy chain can be used to specifically block surface-induced FXII activation, reducing factor XI activation by FXIIa, and subsequent thrombin generation. Indeed, this approach was effective in a baboon thrombosis model using a monoclonal anti-FXII IgG (15H8) that binds to the FXII FN2 domain (Figure 2) (Matafonov et al., 2014). However, while 15H8 blocked contact activation, it also disrupted the regulatory activity of the heavy chain that limits FXII activation, leading to a surge in PK activation and HK cleavage in vivo, comparable to what is observed with ΔFXII infusion (Figure 4E) (Ivanov et al., 2019). Antithrombotic strategies targeting the FXII(a) protease domain would, therefore, seem preferable, because they would not increase surface-independent FXII activation and the associated risk of angioedema.
Plasminogen is the 90 kDa zymogen of the protease plasmin (Novokhatny, 2008; Law et al., 2012; Law et al., 2013). It is encoded by the PLG gene. From the N-terminus, full-length human Glu-plasminogen (Figure 5A) contains a PAN domain, five kringle domains (KNG1-KNG-5), and a trypsin-like protease domain. The KNG1, KNG2, KNG4 and KNG5 domains of plasminogen contain Asp-X-Asp/Glu motifs that bind side chains of lysine and arginine residues (Novokhatny, 2008; Law et al., 2012; Law et al., 2013). In Glu-plasminogen, these motifs are involved in intramolecular binding interactions that maintain the zymogen in a closed conformation that is activated slowly by the plasminogen activator tPA. The FXII KNG domain seems to contribute to a closed conformation by a similar mechanism (Shamanaev et al., 2022a; Shamanaev et al., 2022b). Glu-plasminogen binding to fibrin, like FXII binding to a surface, results in conformational changes that expose the activation cleavage site, increasing the rate of activation by tPA (Urano et al., 2018).
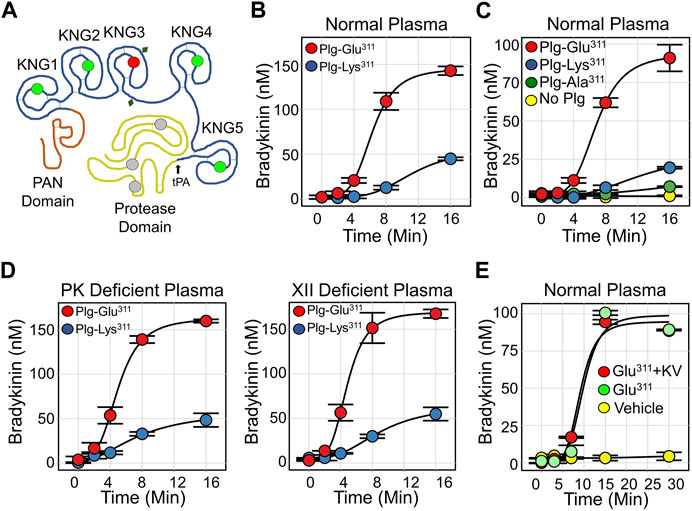
FIGURE 5. Plasmin and Bradykinin Production. (A) Diagram of human plasminogen (PLG), showing the N-terminal PAN domain, five kringle domains (KNG1-5) and the protease domain. Locations of lysine/arginine-binding Asp-X-Asp/Glu motifs are indicated by green circles, and the location of Lys311 is indicated by the red circle. The catalytic triad is indicated in gray and glycosylation sites by the diamonds. The site at which tPA cleaves PLG to generate plasmin is indicated by a black arrow. (B) Bradykinin generation in normal plasma supplemented with 600 nM (final concentration) Plg-Lys311 (blue) or Plg-Glu311 (red) after addition of tPA to 50 nM. (C) Bradykinin generation in normal plasma supplemented with 600 nM Plg-Lys311 (blue), Plg-Ala311 (green), Plg-Glu311 (red) or vehicle (yellow) after addition of tPA to 50 nM. (D) Bradykinin generation in PK-deficient plasma (left) or FXII-deficient (right) plasma supplemented with 600 nM (final concentration) Plg-Lys311 (blue) or Plg-Glu311 (red) after addition of tPA to 50 nM. (E) Bradykinin generation in normal plasma supplemented with 600 nM Plg-Glu311 (red, green), or vehicle (yellow) in response to tPA (50 nM) in the absence (green) or presence (red) of 10 μM KV999272. (B–E) are from Dickeson et al. (2022).
Plasmin contributes to multiple processes including degradative reactions, tissue remodeling and inflammation (Maas, 2019; Heissig et al., 2020; Rahman and Krause, 2020; Keragala and Medcalf, 2021). It is a promiscuous protease that cleaves numerous plasma proteins, including the three components of the KKS. Plasmin has long been recognized as a FXII activator (Kaplan and Austen, 1971), although its ability to cleave the protein after Arg353 is relatively weak when compared to PKa (Kaplan and Austen, 1971; de Maat et al., 2016; Dickeson et al., 2022). It can also slowly convert PK to PKa (Dickeson et al., 2022). While plasmin readily cleaves HK (Marcos-Contreras et al., 2016; Henderson et al., 2021; Dickeson et al., 2022); however, it is not clear that bradykinin is a major product of the reaction. FXII activation by plasmin appears to be at least 20 to 50-fold slower than with PKa (Joseph et al., 2017; Dickeson et al., 2022). The lysine analog ε-aminocaproic acid (Amicar) inhibits plasmin cleavage of HK, indicating that binding interactions between the Asp-X-Asp/Glu motifs on plasmin KNG domains and lysine/arginine residues on HK are involved in the interaction (Kleniewski and Donaldson, 1987; Dickeson et al., 2022).
In 2018 Bork et al. and Dewald et al. described a point mutation in the PLG genes of two HAEnC1 patients that replaces Glu-plasminogen Lys311 with glutamic acid (Lys330 if counting from the initiator methionine on the signal peptide) (Bork et al., 2015; Dewald, 2018). The substitution has been found in over 150 individuals from more than 30 families on three continents, suggesting a wide distribution (Bork et al., 2018; Santacroce et al., 2021; Veronez et al., 2021). Lys311 is in the KNG3 domain (Figure 5A), the only one of the five plasminogen KNG domains lacking an Asp-X-Asp/Glu motif (Law et al., 2012; Law et al., 2013). It is Lys311 that disrupts what would otherwise be an intact Asp-X-Asp/Glu motif (Asp-X-Lys), and the Glu311 substitution in HAE patients creates a new lysine/arginine binding site (Asp-X-Glu). This suggests Glu311 is a gain-of-function mutation. In collaboration with Ruby Law, James Whisstock and Adam Quek of Monash University, we studied wild type and mutant plasminogens (Plg-Lys311 and Plg-Glu311, respectively) to determine how Plg-Glu311 might cause angioedema (Dickeson et al., 2022).
When tPA is added to normal plasma supplemented with recombinant plasminogen, bradykinin generation is much greater with Plg-Glu311 than with Plg-Lys311 (Figure 5B) (Ivanov et al., 2020). Activity associated with Glu311 cannot be attributed specifically to loss of Lys311, as plasminogen with an Ala311 substitution did not enhance bradykinin release (Figure 5C). The effect of Plg-Glu311 is also observed in plasmas lacking FXII or PK (Figure 5D), indicating that bradykinin is somehow produced independently of the KKS. In studies using purified proteins, Plg-Glu311 and Plg-Lys311 are activated at comparable rates by tPA. Both zymogens are also weakly activated by PKa and FXIIa (Dickeson et al., 2022). Furthermore, the active plasmin forms (Plm-Glu311 and Plm-Lys311) both weakly catalyze FXII and PK activation. Taken together, these data suggest the Glu311 substitution confers a gain-of-function that facilitates bradykinin release independently of the KKS. Indeed, a potent PKa inhibitor (KV999272) has no effect on kinin formation in tPA-treated plasma containing Plg-Glu311 (Figure 5E), while blocking kinin formation induced by addition of ΔFXII (Figure 4B).
HK and LK contain disulfide bonds (Cys10-Cys596 and Cys10-Cys389, respectively) that connect their N- and C-termini (Figure 1B) (Ponczek, 2021; Dickeson et al., 2022; Kaplan et al., 2022). PKa cleavage of the HK Arg371-Ser372 peptide bond leads to a pronounced shift in HK migration on non-reducing SDS-PAGE (Figure 6A, left panel), consistent with a change in conformation from a circular to more linear extended form (Ivanov et al., 2020). Cleavage of the Lys362-Arg363 bond then releases bradykinin, causing a second subtler downward shift. Western blots using an anti-bradykinin IgG indicate the BK sequence remains associated with HK after one cleavage, and is released by the second (Figure 6A, right panel). LK cleavage by PKa causes a slight upward shift in migration (Figure 6B, left panel) and relatively high PKa concentrations are required to observe cleavage (Figure 6B, right panel) (Dickeson et al., 2022). While both Plm-Lys311 and Plm-Glu311 readily cleave HK (Figure 6C, top panels), western blots indicate bradykinin is released by Plm-Glu311 more rapidly than by Plm-Lys311 (Figure 6C, bottom panels). A similar shift to that observed with PKa occurs when LK is incubated with Plm-Lys311 or Plm-Glu311 (Figure 6D).
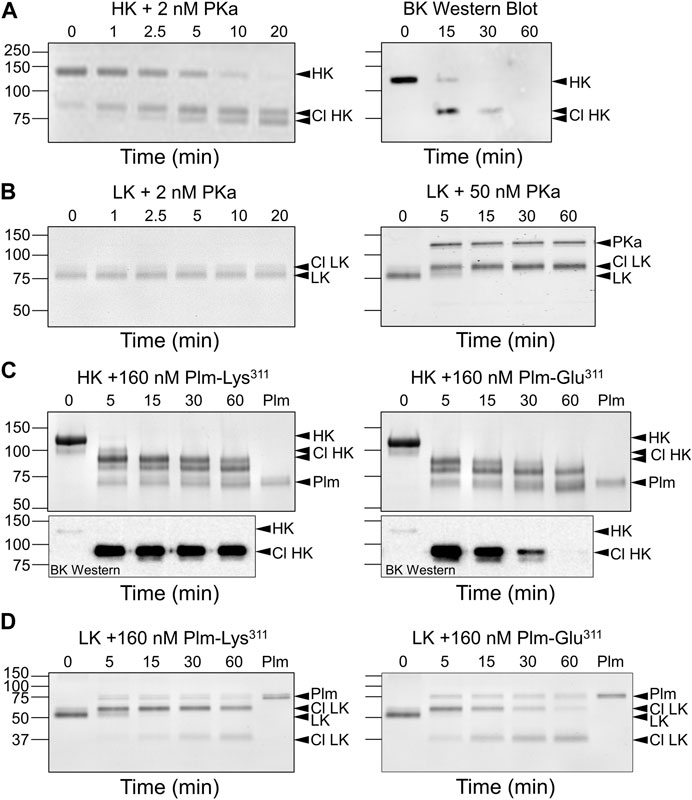
FIGURE 6. Kininogen Cleavage by Kallikrein and Plasmin. Coomassie Blue-stained SDS-polyacrylamide gels of time courses of PKa (A,B) or plasmin (C,D) cleavage of HK (A,C) or LK (B,D). At indicated times, samples were removed into non-reducing sample buffer, size-fractionated on 10% SDS-PAGE, followed by staining. (A) Human plasma-derived HK (200 nM) was incubated with PKa (2 nM) at 37°C. The image on the right is a Western blot of a similar reaction using an antibody to bradykinin. (B) Human plasma-derived LK (200 nM) was incubated with 2 nM (left) or 50 nM (right) PKa at 37°C. (C) Top Panels. Human plasma-derived HK (800 nM) incubated with 160 nM Plm-Lys311 (left) or Plm-Glu311 (right). Bottom Panels. Western blot of samples from reactions similar to those in the Top Panels using an antibody to bradykinin. (D) Human plasma-derived LK (200 nM) incubated with 160 nM Plm-Lys311 or Plm-Glu311. For all panels positions of molecular mass standards in kDa are indicated on the left. Positions of standards for uncleaved HK or LK, cleaved forms of HK (Cl HK) or (Cl LK), and kallikrein (PKa) or plasmin (Plm) are indicated on the right. All images are from Dickeson et al. (2022).
We measured kinin production from HK and LK incubated with plasmin using an ELISA that detects bradykinin and lysyl-bradykinin comparably (Dickeson et al., 2022). In all cases, mass spectroscopic analysis confirmed that the released peptide was bradykinin. Consistent with published literature, PKa releases bradykinin from HK 50–100-fold faster than from LK (Figure 7A). Bradykinin release from HK catalyzed by wild type Plm-Lys311 is at least 50-fold slower than in reactions with PKa (Figure 7B), while PKa and Plm-Lys311 release bradykinin from LK at similar rates (Figure 7C). Interestingly, Plm-Lys311 releases bradykinin ∼3-4 fold faster from LK than from HK (Figure 7D). Given that LK is two to four times more abundant in plasma than HK, these findings suggest that kinin release from kininogens catalyzed by wild type plasmin would primarily come from LK.
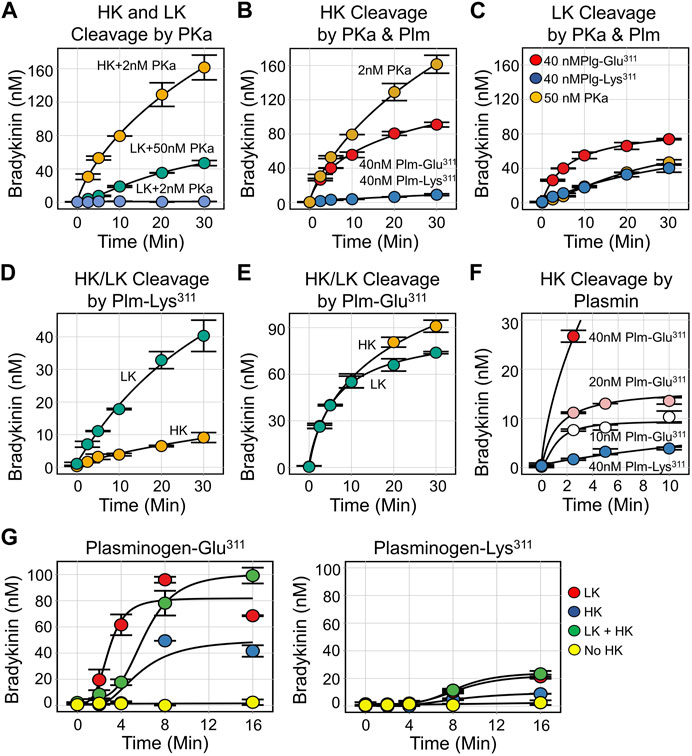
FIGURE 7. Kinin Generation by Kallikrein and Plasmin. For all reactions, samples were collected at indicated time points and bradykinin concentration determined by ELISA. (A) Plasma-derived HK (200 nM) incubated with 2 nM PKa (orange), plasma-derived LK (200 nM) incubated with 50 nM PKa (green) or 2 nM PKa (blue). (B) Plasma-derived HK (200 nM) incubated with 2 nM PKa (orange), 40 nM Plm-Glu311 (red), or 40 nM Plm-Lys311 (blue). (C) Plasma-derived LK (200 nM) incubated with 50 nM PKa (orange), 40 nM Plm-Glu311 (red), or 40 nM Plm-Lys311 (blue). (D) Plasma derived HK (orange) or LK (green), 200 nM, incubated with 40 nM Plm-Lys311. (E) Plasma derived HK (orange) or LK (green), 200 nM, incubated with 40 nM Plm-Glu311. (F) Plasma-derived HK (200 nM) incubated with 40 nM (red), 20 nM (pink), or 10 nM (white) Plm-Glu311 or 40 nM Plg-Lys311 (blue). (G) Plasma from a patient deficient in HK and LK was supplemented with 600 nM plasma-derived HK (blue), 2.3 μM plasma-derived LK (red), HK, and LK (green) or vehicle (yellow). Plm-Glu311 (left) or Plm-Lys311 (right) were added to a final concentration of 600 nM, and tPA (50 nM) was added to activate plasminogen. All images are from Dickeson et al. (2022).
The initial rates of bradykinin release catalyzed by Plm-Glu311 is at least 10-fold faster for HK (Figure 7B), and 2-3-fold faster for LK (Figure 7C), than with Plm-Lys311 (Dickeson et al., 2022). Indeed, with Plm-Glu311, bradykinin release is comparable for HK and LK (Figure 7E). For HK, peak bradykinin generation roughly correlates with the Plm-Glu311 concentration (Figure 7F), suggesting a stochiometric interaction rather than a process with classic Michaelis-Menten kinetics. Plasma from a person deficient in HK and LK was supplemented with physiologic concentrations of HK (640 nM), LK (2.3 μM) or both. Plg-Lys311 or Plg-Glu311 was added, followed by tPA to generate plasmin (Figure 7G). For both Plg-Lys311 and Plg-Glu311, bradykinin generation was greater in plasma containing LK than in plasma containing HK, consistent with LK being the main target for plasmin. Bradykinin generation was substantially greater with Plg-Glu311 than Plg-Lys311.
The data presented suggest Plm-Glu311 causes angioedema by a mechanism with features that distinguish it from HAE due to C1-INH deficiency or FXII-Lys/Arg309. In brief, the Glu311 substitution converts plasmin into a kininogenase that is more efficient than Plg-Lys311, with the capacity to release bradykinin at comparable rates from HK and LK. Recently, Hintze et al. confirmed that Plm-Glu311 releases bradykinin from HK more rapidly than Plm-Lys311 (Hintze et al., 2023). The higher plasma concentration of LK suggests it, and not HK, is the major kinin source during angioedema in carriers of Plg-Glu311. The KKS does not appear to be necessary for bradykinin generation in this disorder. These features may provide insight into some interesting clinical observations.
The dependence of the proposed mechanism on fibrinolysis suggests that bradykinin production would be greatest in tissues where fibrinolysis is most active. There is a predilection for oral-lingual angioedema in Plg-Glu311 carriers (Bork et al., 2018; Bork et al., 2020a). Tongue swelling occurs in 80% of symptomatic patients, and this is often the only manifestation of HAE (Bork et al., 2018; Bork et al., 2020a; Bork et al., 2020b). Edema of the larynx, extremities, or GI tract is less common, and erythema marginatum, a prodromal rash associated with C1-INH deficiency, is rare in Plg-Glu311 carriers. Observations of patients with bleeding disorders suggest that intrinsic fibrinolytic activity is higher in the oral cavity than in most other tissues (Gailani et al., 2023). The predilection for oral-lingual edema in Plg-Glu311 patients may, therefore, reflect the normally brisk plasminogen activation in the mouth. Angiotensin-converting enzyme (ACE) inhibitors inhibit bradykinin degradation and can trigger oral-lingual angioedema in some patients (Carucci et al., 2020; Wilkerson and Winters, 2022). Fibrinolytic inhibitors such as tranexamic acid have been effective in treating ACE-inhibitor induced angioedema (Beauchêne et al., 2018), while recent studies suggest that C1-INH infusion is no better than placebo in this situation (Perza et al., 2020; Strassen et al., 2022). Taken as a whole, these observations suggest that basal bradykinin generation is normally greater in the oropharynx than in other tissues, and that plasmin may contribute significantly to bradykinin formation in this area of the body.
Clinical observations, and the work presented here, also raise the possibility that some of the therapeutic options commonly used to treat HAE patients with low C1-INH, including C1-INH infusion and PKa inhibition, may be less effective in patients with Plg-Glu311 than in other forms of HAE. Consistent with this notion, Bork and colleagues reported that the mean duration of angioedema episodes in patients with Plg-Glu311 was substantially longer after C1-INH infusion (mean duration decrease from 48.2 +/−32.5 h untreated to 31.5 +/−8.6 h treated) than with the B2 receptor antagonist icatibant (mean duration decrease from 44.7+/−28.6 h untreated compared with to 4.3 +/−2.6 h treated) (Bork et al., 2020b). While investigations into optimal treatments for HAE based on underlying mutations are at an early phase, available mechanistic and clinical data suggest that strategies targeting the KKS will not be satisfactory for all patient types.
The AS, KD, II, ML, MS, SK, QC, PS, and TH of this manuscript contributed to the generation of the data in three publications that are the basis for this review paper. AS and DG wrote the manuscript. All authors listed have made a substantial, direct, and intellectual contribution to the work and approved it for publication.
The work was supported by the following grant R35 HL140025 from the National Heart, Lung and Blood Institute (United States), and funds from the Ernest W. Goodpasture chair in Experimental Pathology for Translational Research from Vanderbilt University.
The authors wish to acknowledge generous support from the Ernest W. Goodpasture chair in Experimental Pathology for Translational Research, and award R35 HL140025 from the National Heart, Lung and Blood Institute.
DG is a consultant for pharmaceutical companies developing therapies that target plasma proteases to prevent or treat thrombotic disorders. These activities do not relate directly to the topic of this review article.
The remaining authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article, or claim that may be made by its manufacturer, is not guaranteed or endorsed by the publisher.
Ariano, A., D'Apolito, M., Bova, M., Bellanti, F., Loffredo, S., D'Andrea, G., et al. (2020). A myoferlin gain-of-function variant associates with a new type of hereditary angioedema. Allergy 75 (11), 2989–2992. doi:10.1111/all.14454
Bafunno, V., Firinu, D., D'Apolito, M., Cordisco, G., Loffredo, S., Leccese, A., et al. (2018). Mutation of the angiopoietin-1 gene (ANGPT1) associates with a new type of hereditary angioedema. J. Allergy Clin. Immunol. 141 (3), 1009–1017. doi:10.1016/j.jaci.2017.05.020
Beauchêne, C., Martins-Héricher, J., Denis, D., Martin, L., and Maillard, H. (2018). Tranexamic acid as first-line emergency treatment for episodes of bradykinin-mediated angioedema induced by ACE inhibitors. Rev. Med. Interne 39 (10), 772–776. doi:10.1016/j.revmed.2018.04.014
Björkqvist, J., de Maat, S., Lewandrowski, U., Di Gennaro, A., Oschatz, C., Schönig, K., et al. (2015). Defective glycosylation of coagulation factor XII underlies hereditary angioedema type III. J. Clin. Invest. 125 (8), 3132–3146. doi:10.1172/JCI77139
Bork, K., Barnstedt, S. E., Koch, P., and Traupe, H. (2000). Hereditary angioedema with normal C1-inhibitor activity in women. Lancet 56 (9225), 213–217. doi:10.1016/S0140-6736(00)02483-1
Bork, K., Machnig, T., Wulff, K., Witzke, G., Prusty, S., and Hardt, J. (2020). Clinical features of genetically characterized types of hereditary angioedema with normal C1 inhibitor: A systematic review of qualitative evidence. Orphanet J. Rare Dis. 15 (1), 289. doi:10.1186/s13023-020-01570-x
Bork, K., Wulff, K., Möhl, B. S., Steinmüller-Magin, L., Witzke, G., Hardt, J., et al. (2021). Novel hereditary angioedema linked with a heparan sulfate 3-O-sulfotransferase 6 gene mutation. J. Allergy Clin. Immunol. 148 (4), 1041–1048. doi:10.1016/j.jaci.2021.01.011
Bork, K., Wulff, K., Rossmann, H., Steinmüller-Magin, L., Braenne, I., Witzke, G., et al. (2019). Hereditary angioedema cosegregating with a novel kininogen 1 gene mutation changing the N-terminal cleavage site of bradykinin. Allergy 74 (12), 2479–2481. doi:10.1111/all.13869
Bork, K., Wulff, K., Steinmüller-Magin, L., Braenne, I., Staubach-Renz, P., Witzke, G., et al. (2018). Hereditary angioedema with a mutation in the plasminogen gene. Allergy 73 (2), 442–450. doi:10.1111/all.13270
Bork, K., Wulff, K., Witzke, G., and Hardt, J. (2015). Hereditary angioedema with normal C1-INH with versus without specific F12 gene mutations. Allergy 70 (8), 1004–1012. doi:10.1111/all.12648
Bork, K., Wulff, K., Witzke, G., Machnig, T., and Hardt, J. (2020). Treatment of patients with hereditary angioedema with the c.988A>G (p.Lys330Glu) variant in the plasminogen gene. Orphanet J. Rare Dis. 15 (1), 52. doi:10.1186/s13023-020-1334-8
Bova, M., Suffritti, C., Bafunno, V., Loffredo, S., Cordisco, G., Del Giacco, S., et al. (2020). Impaired control of the contact system in hereditary angioedema with normal C1-inhibitor. Allergy 75 (6), 1394–1403. doi:10.1111/all.14160
Busse, P., and Kaplan, A. (2022). Specific targeting of plasma kallikrein for treatment of hereditary angioedema: A revolutionary decade. Allergy Clin. Immunol. Pract. 10 (3), 716–722. doi:10.1016/j.jaip.2021.11.011
Busse, P. J., and Christiansen, S. C. (2020). Hereditary angioedema. N. Eng. J. Med. 382, 1136–1148. doi:10.1056/NEJMra1808012
Carucci, L., Bova, M., Petraroli, A., Ferrara, A. L., Sutic, A., de Crescenzo, G., et al. (2020). Angiotensin-converting enzyme inhibitor-associated angioedema: From bed to bench. J. Investig. Allergol. Clin. Immunol. 30 (4), 272–280. doi:10.18176/jiaci.0458
Clark, C. C., Hofman, Z. L. M., Sanrattana, W., den Braven, L., de Maat, S., and Maas, C. (2020). The fibronectin type II domain of factor XII ensures zymogen quiescence. Thromb. Haemost. 120 (3), 400–411. doi:10.1055/s-0039-3402760
Cohn, D. M., Viney, N. J., Fijen, L. M., Schneider, E., Alexander, V. J., Xia, S., et al. (2020). Antisense inhibition of prekallikrein to control hereditary angioedema. N. Engl. J. Med. 383 (13), 1242–1247. doi:10.1056/NEJMoa1915035
Colman, R. W., and Schmaier, A. H. (1997). Contact system: A vascular biology modulator with anticoagulant, profibrinolytic, antiadhesive, and proinflammatory attributes. Blood 90 (10), 3819–3843. doi:10.1182/blood.v90.10.3819
Cool, D. E., Edgell, C. J., Louie, G. V., Zoller, M. J., Brayer, G. D., and MacGillivray, R. T. (1985). Characterization of human blood coagulation factor XII cDNA. Prediction of the primary structure of factor XII and the tertiary structure of beta-factor XIIa. J. Biol. Chem. 260 (25), 13666–13676. doi:10.1016/s0021-9258(17)38776-8
Davoine, C., Bouckaert, C., Fillet, M., and Pochet, L. (2020). Factor XII/XIIa inhibitors: Their discovery, development, and potential indications. Eur. J. Med. Chem. 208, 112753. doi:10.1016/j.ejmech.2020.112753
de Maat, S., Björkqvist, J., Suffritti, C., Wiesenekker, C. P., Nagtegaal, W., Koekman, A., et al. (2016). Plasmin is a natural trigger for bradykinin production in patients with hereditary angioedema with factor XII mutations. J. Allergy Clin. Immunol. 138 (5), 1414–1423. doi:10.1016/j.jaci.2016.02.021
de Maat, S., Clark, C. C., Boertien, M., Parr, N., Sanrattana, W., Hofman, Z. L. M., et al. (2019). Factor XII truncation accelerates activation in solution. J. Thromb. Haemost. 17 (1), 183–194. doi:10.1111/jth.14325
De Maat, S., Hofman, Z. L. M., and Maas, C. (2018). Hereditary angioedema: The plasma contact system out of control. J. Thromb. Haemost. 16 (9), 1674–1685. doi:10.1111/jth.14209
de Maat, S., and Maas, C. (2016). Factor XII: Form determines function. J. Thromb. Haemost. 14 (8), 1498–1506. doi:10.1111/jth.13383
Dewald, G., and Bork, K. (2006). Missense mutations in the coagulation factor XII (Hageman factor) gene in hereditary angioedema with normal C1 inhibitor. Biochem. Biophys. Res. Commun. 343 (4), 1286–1289. doi:10.1016/j.bbrc.2006.03.092
Dewald, G. (2018). A missense mutation in the plasminogen gene, within the plasminogen kringle 3 domain, in hereditary angioedema with normal C1 inhibitor. Biochem. Biophys. Res. Commun. 498 (1), 193–198. doi:10.1016/j.bbrc.2017.12.060
Dickeson, S. K., Kumar, S., Sun, M. F., Mohammed, B. M., Phillips, D. R., Whisstock, J. C., et al. (2022). A mechanism for hereditary angioedema caused by a lysine 311-to-glutamic acid substitution in plasminogen. Blood 139 (18), 2816–2829. doi:10.1182/blood.2021012945
Farkas, H., Balla, Z., and Riedl, M. A. (2022a). Differentiating histaminergic and nonhistaminergic angioedema with or without urticaria. J. Allergy Clin. Immunol. 150 (6), 1405–1409. doi:10.1016/j.jaci.2022.10.016
Farkas, H., Germenis, A. E., and Longhurst, H. (2022b). Editorial: C1 inhibitor deficiency and angioedema. Front. Allergy 3, 1065400. doi:10.3389/falgy.2022.1065400
Fields, T., Ghebrehiwet, B., and Kaplan, A. P. (1983). Kinin formation in hereditary angioedema plasma: Evidence against kinin derivation from C2 and in support of "spontaneous" formation of bradykinin. J. Allergy Clin. Immunol. 72, 54–60. doi:10.1016/0091-6749(83)90052-0
Fredenburgh, J. C., and Weitz, J. I. (2021). New anticoagulants: Moving beyond the direct oral anticoagulants. J. Thromb. Haemost. 19 (1), 20–29. doi:10.1111/jth.15126
Gailani, D., Tillman, B. F., and Wheeler, A. P. (2023). “Rare coagulation factor deficiencies,” in Hematology: Basic principles and practice. Editors R. Hoffman, E. J. Benz, L. E. Silberstein, H. E. Heslop, J. I. Weitz, M. E. Salamaet al. 8th ed (Philadelphia, PA: Elsevier), 2125–2143.
Girolami, J. P., Bouby, N., Richer-Giudicelli, C., and Alhenc-Gelas, F. (2021). Kinins and kinin receptors in cardiovascular and renal diseases. Pharm. (Basel) 14 (3), 240. doi:10.3390/ph14030240
Heissig, B., Salama, Y., Takahashi, S., Osada, T., and Hattori, K. (2020). The multifaceted role of plasminogen in inflammation. Cell Signal 75, 109761. doi:10.1016/j.cellsig.2020.109761
Henderson, M. W., Sparkenbaugh, E. M., Wang, S., Ilich, A., Noubouossie, D. F., Mailer, R., et al. (2021). Plasmin-mediated cleavage of high-molecular-weight kininogen contributes to acetaminophen-induced acute liver failure. Blood 138 (3), 259–272. doi:10.1182/blood.2020006198
Hintze, S., Möhl, B. S., Beyerl, J., Wulff, K., Wieser, A., Bork, K., et al. (2023). Mutant plasminogen in hereditary angioedema is bypassing FXII/kallikrein to generate bradykinin. Front. Physiol. 13, 1090732. doi:10.3389/fphys.2022.1090732
Hofman, Z. L. M., Clark, C. C., Sanrattana, W., Nosairi, A., Parr, N. M. J., Živkovic, M., et al. (2020). A mutation in the kringle domain of human factor XII that causes autoinflammation, disturbs zymogen quiescence, and accelerates activation. J. Biol. Chem. 295 (2), 363–374. doi:10.1074/jbc.RA119.009788
Ivanov, I., Matafonov, A., Sun, M. F., Cheng, Q., Dickeson, S. K., Verhamme, I. M., et al. (2017). Proteolytic properties of single-chain factor XII: A mechanism for triggering contact activation. Blood 129 (11), 1527–1537. doi:10.1182/blood-2016-10-744110
Ivanov, I., Matafonov, A., Sun, M. F., Mohammed, B. M., Cheng, Q., Dickeson, S. K., et al. (2019). A mechanism for hereditary angioedema with normal C1 inhibitor: An inhibitory regulatory role for the factor XII heavy chain. Blood 133 (10), 1152–1163. doi:10.1182/blood-2018-06-860270
Ivanov, I., Verhamme, I. M., Sun, M. F., Mohammed, B., Cheng, Q., Matafonov, A., et al. (2020). Protease activity in single-chain prekallikrein. Blood 135 (8), 558–567. doi:10.1182/blood.2019002224
Iwaki, T., and Castellino, F. J. (2006). Plasma levels of bradykinin are suppressed in factor XII-deficient mice. Thromb. Haemost. 95 (6), 1003–1010. doi:10.1160/TH06-03-0128
Joseph, K., Ghebrehiwet, B., and Kaplan, A. P. (1999). Cytokeratin 1 and gC1qR mediate high molecular weight kininogen binding to endothelial cells. Clin. Immunol. 92, 246–255. doi:10.1006/clim.1999.4753
Joseph, K., Tholanikunnel, B. G., and Kaplan, A. P. (2017). Cytokine and estrogen stimulation of endothelial cells augments activation of the prekallikrein-high molecular weight kininogen complex: Implications for hereditary angioedema. J. Allergy Clin. Immunol. 140 (1), 170–176. doi:10.1016/j.jaci.2016.09.032
Kaplan, A. P., and Austen, K. F. (1971). A prealbumin activator of prekallikrein. II. Derivation of activators of prekallikrein from active Hageman factor by digestion with plasmin. J. Exp. Med. 133 (4), 696–712. doi:10.1084/jem.133.4.696
Kaplan, A. P., Joseph, K., and Ghebrehiwet, B. (2022). The complex role of kininogens in hereditary angioedema. Front. Allergy 3, 952753. doi:10.3389/falgy.2022.952753
Kaplan, A. P., and Joseph, K. (2010). The bradykinin forming cascade and its role in hereditary angioedema. Ann. allergy Asthma Immunol. 104, 193–204. doi:10.1016/j.anai.2010.01.007
Karnaukhova, E. (2022). C1-Inhibitor: Structure, functional diversity and therapeutic development. Curr. Med. Chem. 29 (3), 467–488. doi:10.2174/0929867328666210804085636
Keragala, C. B., and Medcalf, R. L. (2021). Plasminogen: An enigmatic zymogen. Blood 137 (21), 2881–2889. doi:10.1182/blood.2020008951
Kleniewski, J., and Donaldson, V. H. (1987). Comparison of human high molecular weight kininogen digestion by plasma kallikrein and by plasmin. A revised method of purification of high molecular weight kininogen. J. Lab. Clin. Med. 109 (4), 469–479.
Kluge, K. E., Seljeflot, I., Arnesen, H., Jensen, T., Halvorsen, S., and Helseth, R. (2022). Coagulation factors XI and XII as possible targets for anticoagulant therapy. Thromb. Res. 214, 53–62. doi:10.1016/j.thromres.2022.04.013
Lau, J., Rousseau, J., Kwon, D., Bénard, F., and Lin, K. S. (2020). A systematic review of molecular imaging agents targeting bradykinin B1 and B2 receptors. Pharm. (Basel). 13 (8), 199. doi:10.3390/ph13080199
Law, R. H., Abu-Ssaydeh, D., and Whisstock, J. C. (2013). New insights into the structure and function of the plasminogen/plasmin system. Curr. Opin. Struct. Biol. 23 (6), 836–841. doi:10.1016/j.sbi.2013.10.006
Law, R. H., Caradoc-Davies, T., Cowieson, N., Horvath, A. J., Quek, A. J., Encarnacao, J. A., et al. (2012). The X-ray crystal structure of full-length human plasminogen. Cell Rep. 1 (3), 185–190. doi:10.1016/j.celrep.2012.02.012
Long, A. T., Kenne, E., Jung, R., Fuchs, T. A., and Renné, T. (2016). Contact system revisited: An interface between inflammation, coagulation, and innate immunity. J. Thromb. Haemost. 14 (3), 427–437. doi:10.1111/jth.13235
Lumry, W. R., and Settipane, R. A. (2020). Hereditary angioedema: Epidemiology and burden of disease. Allergy Asthma Proc. 41 (6), S08–S13. doi:10.2500/aap.2020.41.200050
Maas, C. (2019). Plasminflammation - an emerging pathway to bradykinin production. Front. Immunol. 10, 2046. doi:10.3389/fimmu.2019.02046
Maas, C., and Renné, T. (2018). Coagulation factor XII in thrombosis and inflammation. Blood 131 (17), 1903–1909. doi:10.1182/blood-2017-04-569111
Maier, M., Austen, K. F., and Spragg, J. (1983). Purification of single-chain human low-molecular-weight kininogen and demonstration of its cleavage by human urinary kallikrein. Proc. Natl. Acad. Sci. U. S. A. 80 (13), 336–346. doi:10.1016/0003-2697(83)90307-x
Mandle, R. J., Colman, R. W., and Kaplan, A. P. (1976). Identification of prekallikrein and high-molecular-weight kininogen as a complex in human plasma. Proc. Natl. Acad. Sci. 73, 4179–4183. doi:10.1073/pnas.73.11.4179
Marceau, F., Bachelard, H., Bouthillier, J., Fortin, J. P., Morissette, G., Bawolak, M. T., et al. (2020). Bradykinin receptors: Agonists, antagonists, expression, signaling, and adaptation to sustained stimulation. Int. Immunopharmacol. 82, 106305. doi:10.1016/j.intimp.2020.106305
Marcos-Contreras, O. A., Martinez de Lizarrondo, S., Bardou, I., Orset, C., Pruvost, M., Anfray, A., et al. (2016). Hyperfibrinolysis increases blood-brain barrier permeability by a plasmin- and bradykinin-dependent mechanism. Blood 128 (20), 2423–2434. doi:10.1182/blood-2016-03-705384
Matafonov, A., Leung, P. Y., Gailani, A. E., Grach, S. L., Puy, C., Cheng, Q., et al. (2014). Factor XII inhibition reduces thrombus formation in a primate thrombosis model. Blood 123 (11), 1739–1746. doi:10.1182/blood-2013-04-499111
Maurer, M., and Magerl, M. (2021). Differences and similarities in the mechanisms and clinical expression of bradykinin-mediated vs. mast cell-mediated angioedema. Clin. Rev. Allergy Immunol. 61 (1), 40–49. doi:10.1007/s12016-021-08841-w
Müller-Esterl, W., Vohle-Timmermann, M., Boos, B., and Dittman, B. (1982). Purification and properties of human low molecular weight kininogen. Biochim. Biophys. Acta 706 (2), 145–152. doi:10.1016/0167-4838(82)90480-0
Novokhatny, V. (2008). Structure and activity of plasmin and other direct thrombolytic agents. Thromb. Res. 122 (3), S3–S8. doi:10.1016/j.thromres.2008.06.018
Perza, M., Koczirka, S., and Nomura, J. T. (2020). C1 esterase inhibitor for ace-inhibitor angioedema: A case series and literature review. J. Emerg. Med. 58 (3), e121–e127. doi:10.1016/j.jemermed.2019.10.031
Petersen, R. S., Fijen, L. M., Levi, M., and Cohn, D. M. (2022). Hereditary angioedema: The clinical picture of excessive contact activation. Semin. Thromb. Hemost. 2022, 1758820. (in press). doi:10.1055/s-0042-1758820
Ponczek, M. B. (2021). High molecular weight kininogen: A review of the structural literature. Int. J. Mol. Sci. 22 (24), 13370. doi:10.3390/ijms222413370
Rahman, F. A., and Krause, M. P. (2020). PAI-1, the plasminogen system, and skeletal muscle. Int. J. Mol. Sci. 21 (19), 7066. doi:10.3390/ijms21197066
Revenko, A. S., Gao, D., Crosby, J. R., Bhattacharjee, G., Zhao, C., May, C., et al. (2011). Selective depletion of plasma prekallikrein or coagulation factor XII inhibits thrombosis in mice without increased risk of bleeding. Blood 118 (19), 5302–5311. doi:10.1182/blood-2011-05-355248
Santacroce, R., D'Andrea, G., Maffione, A. B., Margaglione, M., and d'Apolito, M. (2021). The genetics of hereditary angioedema: A review. J. Clin. Med. 10 (9), 2023. doi:10.3390/jcm10092023
Scheffel, J., Mahnke, N. A., Hofman, Z. L. M., Maat, S., Wu, J., Bonnekoh, H., et al. (2020). Cold-induced urticarial autoinflammatory syndrome related to factor XII activation. Nat. Commun. 11 (1), 179. doi:10.1038/s41467-019-13984-8
Schmaier, A. H., Kuo, A., Lundberg, D., Murry, S., and Cines, D. B. (1988). The expression of high molecular weight kininogen on human umbilical vein endothelial cells. J. Biol. Chem. 263, 16327–16333. doi:10.1016/s0021-9258(18)37596-3
Schmaier, A. H. (2016). The contact activation and kallikrein/kinin systems: Pathophysiologic and physiologic activities. J. Thromb. Haemost. 14 (1), 28–39. doi:10.1111/jth.13194
Schmaier, A. H. (2019). The hereditary angioedema syndromes. J. Clin. Invest. 129 (1), 66–68. doi:10.1172/JCI125378
Shamanaev, A., Ivanov, I., Sun, M. F., Litvak, M., Srivastava, P., Mohammed, B. M., et al. (2022a). Model for surface-dependent factor XII activation: The roles of factor XII heavy chain domains. Blood Adv. 6 (10), 3142–3154. doi:10.1182/bloodadvances.2021005976
Shamanaev, A., Litvak, M., and Gailani, D. (2022b). Recent advances in factor XII structure and function. Curr. Opin. Hematol. 29 (5), 233–243. doi:10.1097/MOH.0000000000000727
Silverberg, M., Dunn, J., Garen, K., and Kaplan, A. P. (1980a). Autoactivation of human Hageman factor. Demonstration utilizing a synthetic substrate. J. Biol. Chem. 255, 7281–7286. doi:10.1016/s0021-9258(20)79699-7
Silverberg, M., Nicoll, J., and Kaplan, A. P. (1980b). The mechanism by which the light chain of cleaved HMW-kininogen augments the activation of prekallikrein, factor XI and Hageman factor. Thromb. Res. 20, 173–189. doi:10.1016/0049-3848(80)90383-7
Srivastava, P., and Gailani, D. (2020). The rebirth of the contact pathway: A new therapeutic target. Curr. Opin. Hematol. 27 (5), 311–319. doi:10.1097/MOH.0000000000000603
Strassen, U., Bas, M., Wirth, M., Wirth, M., Gröger, M., Stelter, K., et al. (2022). Efficacy of human C1 esterase inhibitor concentrate for treatment of ACE-inhibitor induced angioedema. Am. J. Emerg. Med. 64, 121–128. doi:10.1016/j.ajem.2022.12.001
Sueiras-Diaz, J., Jones, D. M., Ashworth, D., Horton, J., Evans, D. M., and Szelke, M. (1994). Cleavage of human kininogen fragments at Met-Lys by human tissue kallikrein. Braz J. Med. Biol. Res. 27 (8), 1935–1942.
Thompson, R. E., Mandle, R. J., and Kaplan, A. P. (1979). Studies of binding of prekallikrein and Factor XI to high molecular weight kininogen and its light chain. Proc. Natl. Acad. Sci. 76, 4862–4866. doi:10.1073/pnas.76.10.4862
Tillman, B., and Gailani, D. (2018). Inhibition of factors XI and XII for prevention of thrombosis induced by artificial surfaces. Semin. Thromb. Hemost. 44 (1), 60–69. doi:10.1055/s-0037-1603937
Urano, T., Castellino, F. J., and Suzuki, Y. (2018). Regulation of plasminogen activation on cell surfaces and fibrin. J. Thromb. Haemost. 16 (8), 1487–1497. doi:10.1111/jth.14157
Valerieva, A., and Longhurst, H. J. (2022). Treatment of hereditary angioedema-single or multiple pathways to the rescue. Front. Allergy 3, 952233. doi:10.3389/falgy.2022.952233
Veronez, C. L., Csuka, D., Sheikh, F. R., Zuraw, B. L., Farkas, H., and Bork, K. (2021). The expanding spectrum of mutations in hereditary angioedema. J. Allergy Clin. Immunol. Pract. 9 (6), 2229–2234. doi:10.1016/j.jaip.2021.03.008
Wedner, H. J. (2020). Hereditary angioedema: Pathophysiology (HAE type I, HAE type II, and HAE nC1-INH). Allergy Asthma Proc. 41 (6), S14–S17. doi:10.2500/aap.2020.41.200081
Wiggins, R. L., Bouma, B. N., Cochrane, C. G., and Griffin, J. H. (1977). Role of high-molecular-weight kininogen in surface-binding and activation of coagulation Factor XI and prekallikrein. Proc. Natl. Acad. Sci. 77, 4636–4640. doi:10.1073/pnas.74.10.4636
Wilkerson, R. G., and Winters, M. E. (2022). Angiotensin-converting enzyme inhibitor-induced angioedema. Emerg. Med. Clin. North Am. 40 (1), 79–98. doi:10.1016/j.emc.2021.09.004
Keywords: hereditary angioedema, kallikrein-kinin system, kallikrein, factor XII, plasminogen
Citation: Shamanaev A, Dickeson SK, Ivanov I, Litvak M, Sun M-F, Kumar S, Cheng Q, Srivastava P, He TZ and Gailani D (2023) Mechanisms involved in hereditary angioedema with normal C1-inhibitor activity. Front. Physiol. 14:1146834. doi: 10.3389/fphys.2023.1146834
Received: 17 January 2023; Accepted: 12 May 2023;
Published: 23 May 2023.
Edited by:
Zia Shariat-Madar, University of Mississippi, United StatesReviewed by:
Peter Meinke, LMU Munich University Hospital, GermanyCopyright © 2023 Shamanaev, Dickeson, Ivanov, Litvak, Sun, Kumar, Cheng, Srivastava, He and Gailani. This is an open-access article distributed under the terms of the Creative Commons Attribution License (CC BY). The use, distribution or reproduction in other forums is permitted, provided the original author(s) and the copyright owner(s) are credited and that the original publication in this journal is cited, in accordance with accepted academic practice. No use, distribution or reproduction is permitted which does not comply with these terms.
*Correspondence: David Gailani, ZGF2ZS5nYWlsYW5pQHZ1bWMub3Jn
Disclaimer: All claims expressed in this article are solely those of the authors and do not necessarily represent those of their affiliated organizations, or those of the publisher, the editors and the reviewers. Any product that may be evaluated in this article or claim that may be made by its manufacturer is not guaranteed or endorsed by the publisher.
Research integrity at Frontiers
Learn more about the work of our research integrity team to safeguard the quality of each article we publish.